
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCyclo-oligomerization of isocyanates with Na(PH2) or Na(OCP) as “P−” anion sources†
Dominikus
Heift
a,
Zoltán
Benkő
*ab,
Hansjörg
Grützmacher
*ac,
Andrew R.
Jupp
d and
Jose M.
Goicoechea
d
aDepartment of Chemistry and Applied Biosciences, ETH Zurich, CH-8093 Zurich, Switzerland. E-mail: hgruetzmacher@ethz.ch; benkoe@inorg.chem.ethz.ch
bBudapest University of Technology and Economics, 1111 Budapest, Hungary. E-mail: zbenko@mail.bme.hu
cLehn Institute of Functional Materials (LIFM), Sun Yat-Sen University, 510275 Guangzhou, China
dDepartment of Chemistry, University of Oxford, Chemistry Research Laboratory, 12 Mansfield Road, Oxford OX1 3TA, UK
First published on 13th May 2015
Abstract
We show that the 2-phosphaethynolate anion, OCP−, is a simple and efficient catalyst for the cyclotrimerization of isocyanates. This process proceeds step-wise and involves five-membered heterocycles, namely 1,4,2-diazaphospholidine-3,5-dionide anions and spiro-phosphoranides as detectable intermediates, both of which were also found to be involved in the catalytic conversion. These species can be considered as adducts of a phosphide anion with two and four isocyanate molecules, respectively, demonstrating that the OCP− anion acts as a formal “P−” source. The interconversion between these anionic species was found to be reversible, allowing them to serve as reservoirs for unique phosphorus-based living-catalysts for isocyanate trimerization.
Introduction
Rigid polyurethane foams, the reaction products of diisocyanates, polyols and water, are commonly applied as durable structural and insulating materials.1 The thermal and mechanical properties of these polymeric networks are crucially influenced by the degree of crosslinking, which can be achieved by incorporation of isocyanate cyclo-trimers. Lewis bases such as anions of simple inorganic and organic salts (e.g. fluorides, hydroxides, carboxylates, alkoxides),2 N-heterocyclic carbenes3 amine bases4 as well as acyclic5 or cyclic6–8 phosphanes are well known to catalyze the cyclo-oligomerization of isocyanates (A) to form uretdiones, B as cyclic dimers and isocyanurates C, or their isomers iminooxadiazinediones D, as trimers (Fig. 1). | ||
| Fig. 1 Isocyanates (A) and their oligomers (B–D). | ||
Interestingly, while phosphanes are known to catalyze the oligomerization of isocyanates, the reactivity of isocyanates towards phosphides (PR2−) or phosphido metal complexes has barely been investigated. In the latter systems, the insertion of isocyanates into the metal–phosphorus bond has been observed.9–11 Some yttrium–phosphido complexes were found to catalyze the cyclo-trimerization of isocyanates,9 however, relatively high catalyst loadings are required to reach full conversion.
Recently we have demonstrated that the dihydrogenphosphide sodium salt, Na(PH2), reacts with carbon monoxide in ethereal solution to form sodium phosphaethynolate Na(OCP).12 Alternatively, [K(18-crown-6)](OCP) can be prepared by refluxing K3P7 in presence of 18-crown-6 in DMF under an atmosphere of CO.13 Other CO-sources, like organic carbonates or iron pentacarbonyl, react likewise with Na(PH2) to give high yields of Na(OCP).12 Extending this observation, we investigated isocyanates as potential sources of CO. Screening of the reactions between Na(PH2) and isocyanates indeed indicated the formation of Na(OCP). However, we observed further reaction products depending on the substituent of the isocyanate, R–NCO, and the molar ratio of the starting materials R–NCO to Na(PH2). Specifically, the reactions between M(OCP) (M = Na, K) and isocyanates will be discussed which leads to spiro-cyclic anions with a σ4,λ5-P centre and Ψ-bipyramidal structure. Formally the anions are obtained when four equivalents of isocyanate wrap around a “P−” anion. Because this process is reversible, these anions serve as reservoirs for unique living-catalysts for isocyanate trimerization.
Results and discussion
The reaction of Dipp-NCO with Na(PH2)
Sodium phosphaethynolate Na(OCP) (Na[3]) was formed as the main reaction product (95%) within an hour when the rather bulky 2,6-diisopropylphenyl isocyanate (Dipp–NCO, 2a) and Na(PH2) (1) were refluxed in DME in a ratio of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. During this reaction we detected by 31P NMR spectroscopy two intermediates IM1 (δ31P: t, −150.0 ppm, 1JPH = 207 Hz) and IM2 (two isomers: δ31P: −70.0 ppm (d), 1JPH = 260 Hz; −83.0 ppm (d), 1JPH = 240 Hz) which were identified by comparison with literature data for closely related compounds (Scheme 1).14
:1. During this reaction we detected by 31P NMR spectroscopy two intermediates IM1 (δ31P: t, −150.0 ppm, 1JPH = 207 Hz) and IM2 (two isomers: δ31P: −70.0 ppm (d), 1JPH = 260 Hz; −83.0 ppm (d), 1JPH = 240 Hz) which were identified by comparison with literature data for closely related compounds (Scheme 1).14
 | ||
Scheme 1 (A) Reaction between Dipp–N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CO (2a) and Na(PH2) in refluxing DME. (B) Reaction between [K(18-crown-6)](OCP) and 2a in pyridine at room temperature. CO (2a) and Na(PH2) in refluxing DME. (B) Reaction between [K(18-crown-6)](OCP) and 2a in pyridine at room temperature. | ||
When an excess of 2a was added to Na(PH2) in refluxing DME, the color of the solution turned from yellow to orange and the 1,4,2-diazaphospholidine-3,5-dionide anion [4a]− (31P: δ = 117.0 ppm, UV-vis: λ = 464 nm) was formed in a clean reaction (Scheme 1). We repeated the reaction at 50 °C in THF using the dioxane adduct [Na(OCP)(dioxane)2.5] as starting material instead of Na(PH2), and again the clean formation of anion [4a]− was observed. With [K(18-crown-6)](OCP) (K[3]) and four equivalents of isocyanate 2a using pyridine as solvent, a different product K[5a] was observed which shows a 31P NMR resonance at δ = −42.7 ppm, typical for compounds containing a highly coordinated phosphorus centre. Indeed, an investigation of colorless single crystals with X-ray diffraction methods (see below and ESI† for details) reveals a spiro-anion with a 10-P-4 centre according to the Arduengo–Martin nomenclature.15,16 When these crystals were redissolved in pyridine, a 31P resonance at δ = 120.0 ppm indicated the formation of the five-membered heterocycle [4a]−, presumably through loss of two equivalents of the isocyanate. A single crystal of this compound as the potassium 18-crown-6 salt, [K(18-crown-6)][4a], was investigated by a X-ray diffraction study (see ESI† for details). Remarkably, a stable sodium spiro-anion salt Na[5a] is not obtained with the sodium phosphaethynolate, Na(OCP), and the sterically encumbered isocyanate 2a, but only with sterically less demanding isocyanates (vide infra). This observation indicates a marked influence of the counter cation, Na+vs. K+, on the outcome of the reactions between M(OCP) salts and isocyanates.
The structure of anion [4a]−, formally an adduct of a “P−” anion and two isocyanate molecules, was also elucidated by an X-ray diffraction study with a single crystal of the composition [Na(THF)2][4a] (Fig. 2). The PN2C2 ring of anion [4a]− is flat (maximum deviation from ring plane: 0.005 Å) and the bulky Dipp substituents are nearly perpendicular to the plane of the heterocycle. The sodium cations and the five-membered ring anions form a linear coordination polymer (for a picture see ESI†), in which the Na+ ions (in a tetrahedral coordination sphere) coordinate two THF molecules and two anions (one connected by O1, another by O2). Accordingly, the C1–O1 and C2–O2 bond lengths are identical within experimental error (1.245(3) and 1.254(3) Å). The C–N bonds are in the range of single bonds with the C2–N1 bond slightly elongated compared to the others. The P–C bond length [1.756(3) Å] indicates significant multiple bond character and suggests that the negative charge is delocalized over the PCO moiety. The relatively long P–N2 [1.779(2) Å] and N1–C2 [1.425(3) Å] distances which are in the range of P–N and C–N single bonds are in accord with the resonance structure shown in Scheme 1.
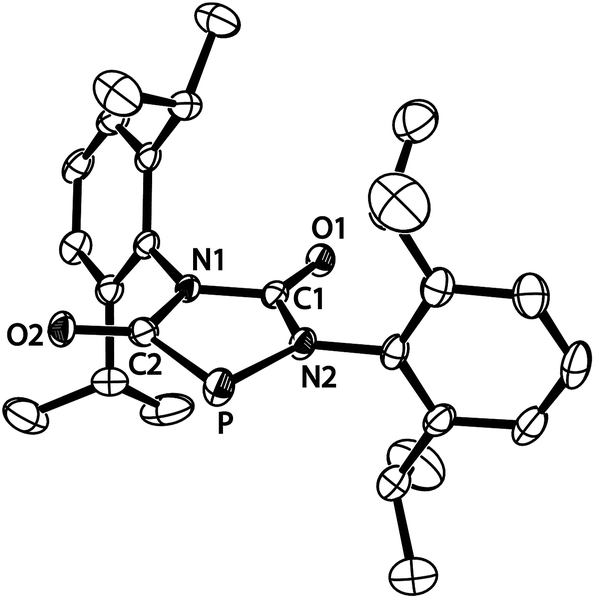 | ||
| Fig. 2 ORTEP plot of [4a]− (thermal ellipsoids are drawn at 50% probability). Hydrogen atoms have been omitted for clarity. Selected bond lengths (Å) and angles (°): C1–N1 1.378(3), N1–C2 1.425(3), C2–P 1.756(3), C2–O2 1.254(3), P–N2 1.779(2), N2–C1 1.347(3), C1–O1 1.245(3); C1–N1–C2 115.9(2), N1–C2–P 110.0(2), C2–P–N2 88.3(1), P–N2–C1 115.8(2), N2–C1–N1 110.1(2). | ||
The reaction of Na(OCP) with smaller isocyanates
The results obtained with 2,6-diisopropylphenyl isocyanate prompted us to study the reactivity between Na(OCP) (Na[3]) and sterically less demanding isocyanates. Na[3] was used as the phosphorus-containing reagent for further studies due to its relative ease of synthesis and handling.17The reaction of Na[3] with phenyl isocyanate 2b in THF at room temperature led to a mixture of compounds. The 31P NMR spectra of the solutions indicated the formation of two new phosphorus-containing species in addition to unreacted starting material Na[3] (Scheme 2). The minor component was identified as the six-membered ring anion [6b]− (δ31P: −16.9 ppm, ca. 15%), while the dominant product is the spiro-anion [5b]− (δ31P: −66.0 ppm) (ca. 85%). Full conversion of Na(OCP) could only be achieved by the addition of a larger excess of isocyanate (at least a ratio 1:8 is required). After a short time (ca. 5 minutes) colorless crystals precipitated from the solutions which were characterized as triphenyl isocyanurate 7b by NMR and IR spectroscopy as well as by X-ray crystallography. This suggests that the Na(OCP) acts as a catalyst for the isocyanate trimerization. Indeed, the addition of a catalytic amount (1 mol%) of Na(OCP) to phenyl isocyanate 2b at room temperature resulted in the selective trimerization to triphenyl isocyanurate 7b which was isolated in excellent yield (92%) and purity. In the 31P NMR spectrum of the reaction suspension only resonances corresponding to Na[5b] and Na[6b] were observed but no Na(OCP).
 | ||
| Scheme 2 Reaction of isocyanates with the phosphaethynolate ion. | ||
Similar reactions with two aliphatic isocyanates R–NCO [R = Cy (2c), nBu (2d)] were also investigated. These reactions again led to the formation of Na[5c,d], Na[6c,d] and 7c,d. Both sodium spiro-phosphoranides Na[5c,d] were unambiguously identified by X-ray diffraction studies with single crystals (see below). Formally, the anions [5a–d]− ([5a]− obtained as a potassium (18-crown-6) salt, vide supra) can be considered as adducts of four isocyanate moieties to a P− anion. The six-membered heterocyclic anions [6b–d]−, which formally consists of an (OCP)− anion and two isocyanate fragments, were identified by NMR and IR spectroscopy. The data compare very well with the ones of the recently reported anionic 4,6-diimino-1,3,5-diazaphosphinan-2-one, which is an adduct of an (OCP)− ion and two equivalents of dicyclohexylcarbodiimide (Cy–NCN–Cy).18
Varying the reaction conditions (solvent, temperature and concentration) did not allow us to obtain selectively only one of the compounds, Na[5b–d] or Na[6b–d], independently of the choice of substituent R = Ph, Cy or nBu. In all reactions the amount of anions [6b–d]− was much smaller (ca. 10–15%) than that of the corresponding spiro-phosphoranides [5b–d]−. All attempts to isolate Na[5b–d] or Na[6b–d] beside the isocyanurates 7b–d in larger amounts by fractional crystallization, chromatography or extraction failed. One reason may be the remarkably similar solubility in most common solvents. Another reason could be the aforementioned tendency of the spiro-anions [5a–d]− to decompose to 1,4,2-diazaphospholidine-3,5-dionide anions [4a–d]− and isocyanurates 7a–d, respectively. However, a few single crystals of the composition [(Na[5c](DME))∞] and [(Na[5d](THF))∞] could be obtained from the reaction solutions and the structures were determined by X-ray diffraction studies (Fig. 3).
 | ||
| Fig. 3 (a) ORTEP plot of the anion [5c]− (thermal ellipsoids are drawn at 50% probability, hydrogen atoms have been omitted for clarity). Selected bond lengths (Å) and angles (°): P–C1 1.860(7), C1–O1 1.209(7), C1–N1 1.380(7), N1–C2 1.424(7), C2–O2 1.255(6), C2–N2 1.305(7), N2–P 1.941(5), P–N4 1.917(6), N4–C16 1.318(7), C16–O4 1.252(7), C16–N3 1.423(8), N3–C15 1.365(7), C15–O3 1.221(7), C15–P 1.870(7); N2–P1–N4 170.8(2), N4–P1–C15 82.9(3), P1–C15–N3 112.8(5), C15–N3–C16 115.9(5), N3–C16–N4 111.24(6), P–C1–N1 112.1(5), C1–N1–C2 115.7(5), N1–C2–N2 112.4(6), C2–N2–P 113.6(4). Angle between the two ring planes: 69.8°. (b) ORTEP plot of [(Na[5c](DME))∞] (thermal ellipsoids are drawn at 50% probability, cyclohexyl groups and DMF molecules have been omitted for clarity). (c) ORTEP plot of anion [5d]− (thermal ellipsoids are drawn at 50% probability, hydrogen atoms have been omitted for clarity). Selected bond lengths (Å) and angles (°): P–N2 1.909(2), N2–C2 1.320(4), C2–O2 1.240(3), C2–N1 1.410(4), N1–C1 1.371(4), C1–O1 1.218(3), C1–P 1.888(3), P–C3 1.884(3), C3–O3 1.224(3), C3–N3 1.369(4), N3–C4 1.418(4), C4–O4 1.237(3), C4–N4 1.312(4), N4–P 1.892(2); P–N2–C2 115.9(2), N2–C2–N1 111.1(2), C2–N1–C1 117.1(2), N1–C1–P, C1–P–N2 83.0(1), C3–N3–C4 117.0(2), N3–C4–N4 110.6(2), C4–N4–P 117.5(2), N4–P–C3 83.0(1), P–C3–N3 111.8(2). Angle between the two ring planes: 72.1°. (d) ORTEP plot of [(Na[5d](THF))∞] (thermal ellipsoids are drawn at 50% probability, n-butyl groups and THF molecules have been omitted for clarity). | ||
Structural and computational studies
In both the structures of [(Na[5c](DME))∞] and of [(Na[5d](THF))∞], the sodium cations coordinate to one solvent molecule (DME or THF, respectively) and interact with the spiro-anions such that three-dimensional networks are formed. The crystal of [(Na[5c](DME))∞] contains only one enantiomer (Δ), but the crystal of [(Na[5d](THF))∞] is a racemic mixture of both enantiomers (see Fig. 3d). Because of their similarity only the structure of [(Na[5c](DME))∞] is discussed in detail. The spiro anion [5c]− consists of two identical and flat five-membered rings connected through the 10-P-4 phosphorus atom. The intersection-angle between the two ring planes is 69.8°.19 As expected on the basis of the VSEPR model, the lone pair and the carbonyl carbons occupy the equatorial positions in the pseudo-trigonal bipyramidal arrangement (Ψ-TBP) around the phosphorus atom. Due to their high electronegativity (cf. apicophilicity) the two nitrogen atoms N2 and N4 reside in the axial positions, forming a N2–P–N4 arrangement (α = 170°) which is compressed by 10° from linearity by the space-filling requirements of the lone pair [see the HOMO of the simplified anion [5e]− (R = Me) in Fig. 4a]. Compared to the monocyclic anion [4a]− (Fig. 2) the P–N bonds in [5c]− are considerably elongated (1.941(5) Å and 1.917(6) Å in [5c]−vs. 1.779(2) Å in anion [4a]−). Moreover, the phosphorus–nitrogen bond lengths in [5c]− are significantly longer than the axial P–N bonds in neutral phosphoranes.20 Note that the 31P NMR shift of [5a]− (δ31P = −43.0 ppm) is significantly more deshielded than the ones observed for [5b]− (δ31P = −64.0 ppm), [5c]− (δ31P = −86.0 ppm), and [5d]− (δ31P = −85.0 ppm) which we attribute to the on average even longer P–C and P–N bonds seen in [5a]− (see the ESI† for details). | ||
| Fig. 4 (a) The Kohn–Sham HOMO of anion [5e]−, (b) electrostatic potential map of [5e]−, (both at the B3LYP/aug-cc-pVDZ level) (c) resonance structure as main contributor to the electronic ground-state of anion [5e]−. | ||
Computations (at the B3LYP/aug-cc-pVDZ level) were performed for the model anion [5e]− where the substituents R correspond to Me. The weakening of the P–N bonds is reflected in the computed Wiberg bond order of 0.48. According to the natural bond orbital (NBO) calculations the P–N bond predominantly originates from the nitrogen atomic hybrid (with a contribution of 87%). The negative charges are mainly located on the oxygens and the P atom is positively charged (+0.80e) (see molecular electrostatic potential map in Fig. 4b). Therefore, anion [5]− is best described as a donor acceptor complex between a phosphenium cation, [PR2]+, and two terminal imino groups as parts of two anionic [CO–NR–C(O−) = NR] groups (Fig. 4c).
The lability of the P–N bond in the spiro anions was investigated with the partially fluorinated anion [5f]− using variable temperature 31P and 19F NMR spectroscopy. The sodium salt of [5f]− was prepared in situ in an exchange reaction between [5c]− and 2,6-difluorophenyl isocyanate (see below and ESI† for details).
The difluorophenyl groups were introduced as labels in order to evaluate the dynamic behaviour. At 233 K the 19F NMR spectrum of the in situ generated 2,6-difluorophenyl spiro-anion [5f]− shows four resonances: three singlets (−116.5, −117.4, −119.0 ppm) and a doublet (−118.7 ppm, JPF = 126 Hz). Each resonance corresponds to two pairwise identical 19F nuclei which are labelled A, B, C, D in the C2-symmetric anion [5f]− (Fig. 5). In the 31P NMR spectrum a triplet (−67.4 ppm, JPF = 126 Hz) is observed indicating through-space coupling between the central phosphorus nucleus and the fluorine nuclei FA (Fig. 5).21 By warming the solution to 283 K a dynamic phenomenon is observed. The 31P NMR spectrum now shows a quintet (−65.8 ppm, JPF = 61 Hz) resulting from the coupling with four equivalent fluorine atoms FA and FB, which appear as a broad singlet in the 19F NMR spectrum. The FC and FD nuclei remain non-equivalent (represented by two sharp singlets). Most likely, the exchange between FA and FB indicates a rotation of the 2,6-difluorophenyl rings at elevated temperature.
 | ||
| Fig. 5 Dynamic behaviour of anion [5f]−. | ||
We studied this dynamic process using DFT calculations. Relaxed scan calculations were performed by varying the torsional angle (C–C–N–P) = Θ which describes the tilt angle of the 2,6-F2C6H3 ring versus the P–N bond. In order to save computational time, in the calculations simplified model compounds were used instead of [5f]−: three of the four difluorophenyl groups were replaced by methyl substituents which results in the anions [5g]− and its isomer [5h]− (see Fig. 6). In [5g]− the computed rotation barrier around the bond between the 2,6-F2C6H3 substituent and the N atom in apical position at the phosphorus centre, CAr–NP, amounts only to 10.3 kcal mol−1, which explains the dynamic exchange of FA and FB in [5f]− at 283 K. In the activated complex in the transition state with Θ = 60°, the P–NP distance is 2.34 Å, which is significantly larger than that in the minimum (Θ = 0°, P–N = 2.13 Å). Hence the rotation around the N–C bond is facilitated by a partial breaking of this P–N bond.22,23 The rotation barrier around the CAr–N bond furthest from the phosphorus centre in [5h]− is much larger (28.5 kcal mol−1, Fig. 6), which is in accordance with the experimentally observed hindered exchange of FC and FD in [5f]−, which show separated 19F resonances up to 283 K (see Fig. 5).
 | ||
| Fig. 6 Potential energy profiles (at the B3LYP/6-31+G* level of theory) of the rotation around the N–CAr bonds. The filled curve in magenta corresponds to anion [5g]−, while the one in grey to anion [5h]−. The dotted magenta curve shows the variation of the P–N bond length in [5g]− with variation of the C–C–N–P torsional angle (Θ). | ||
DFT calculations on the reaction mechanisms
Experimental and theoretical studies have established that the oligomerization of isocyanates proceeds via sequential addition of isocyanates to the reactive species formed via nucleophilic addition of a Lewis base to the carbon centre of the isocyanate, R–NCO.5b,24–26 On this basis, we propose the reaction sequences shown in Fig. 7 and 8 for formation of the observed phosphorus species Na[4], Na[5], and Na[6] and isocyanurates 7, respectively. This mechanism was investigated with DFT methods using methyl isocyanate, Me–NCO, as a model reagent.
 | ||
| Fig. 7 Proposed reaction mechanism and Minimum Energy Reaction Pathway (MERP) at the B3LYP/aug-cc-pVDZ//B3LYP/6-31+G* level of theory. Intermediate [IIA]− is at a cross-point. The MERP shown in grey indicates the formation of the six-membered ring anion [6e]− [route (i)]. The MERP given in black leads to spiro-anion [5e]− [route (ii)]. Na+ was included as a counter cation in the computations but is not shown. | ||
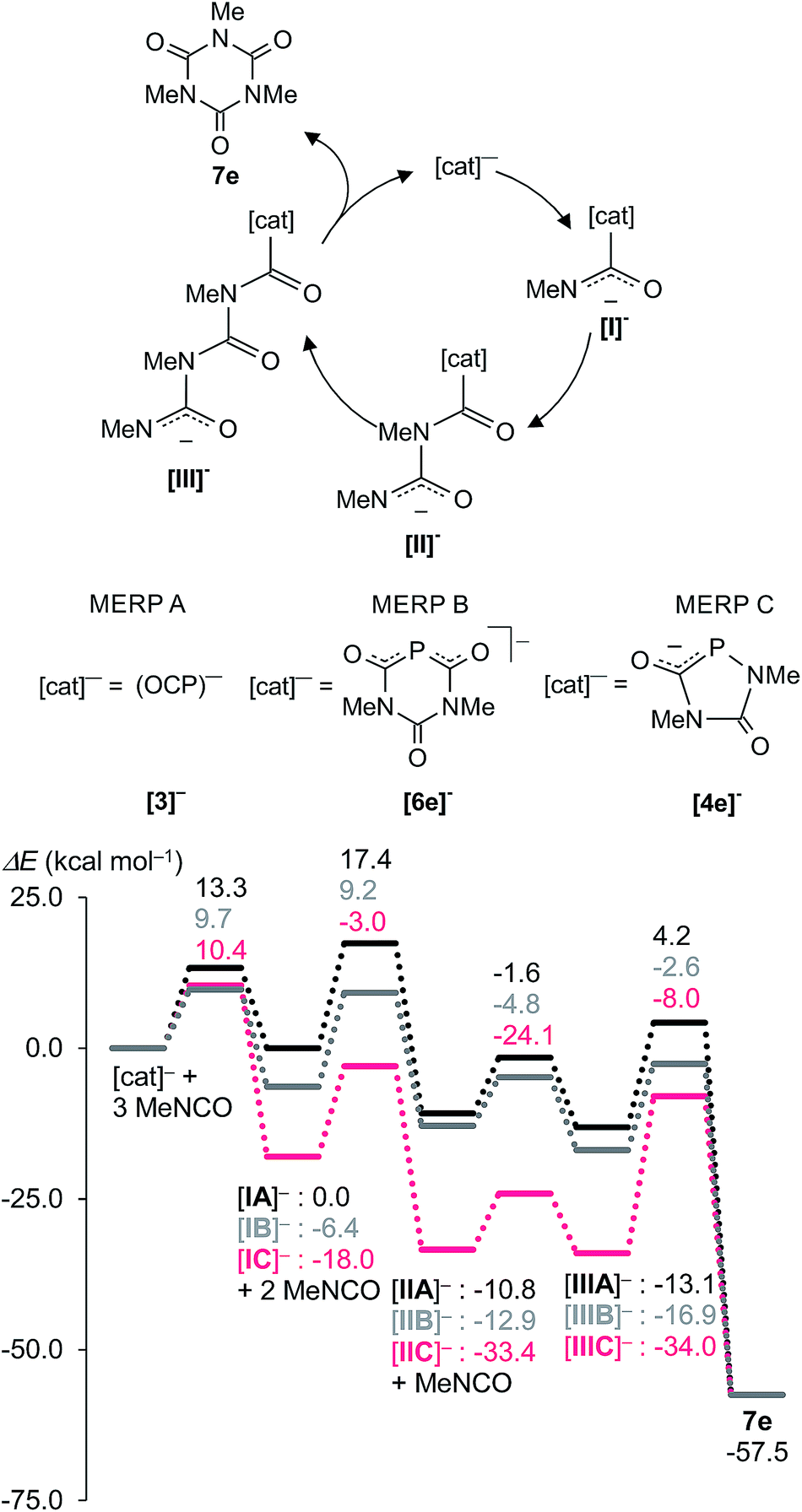 | ||
| Fig. 8 Proposed catalytic cycle leading to 7e on the MERPs A, B, and C computed at the B3LYP/aug-cc-pVDZ//B3LYP/6-31+G* level. MERP A (black) corresponds to the reaction catalysed by the (OCP)− anion [3]−, MERP B (grey) to the reaction catalyzed by the six-membered heterocyclic anion [6e]−, and MERP C (magenta) stands for the trimerization reaction catalyzed by the five-membered heterocyclic anion [4e]−. The notation of the intermediates: [I]−, [II]− and [III]− indicate adducts between [cat]− and one, two and three isocyanate molecules. Na+ was included as a counter cation in the computations but is not shown. | ||
The mechanism leading to the different phosphorus heterocycles is discussed first (Fig. 7). The initial reaction step is the attack of the nucleophilic P atom of the (OCP)− anion onto the carbon centre of an isocyanate molecule, forming the weakly bound adduct [IA]− which has the same energy as the starting materials. In an exothermic reaction (−10.8 kcal mol−1) the addition of a second isocyanate leads to [IIA]− and this reaction is associated with one of the highest activation barriers (Ea = 17.4 kcal mol−1). Intermediate [IIA]− is at the cross-road between two different reaction pathways (i) (marked in grey) and (ii) (shown in black) on the minimum energy reaction pathway (MERP) in Fig. 7. An intramolecular nucleophilic attack of the terminal dicoordinated nitrogen centre in [IIA]− on the carbon centre of the PCO unit leads to the six-membered ring [6e]− [route (i)]. Alternatively, an intramolecular nucleophilic attack of the same nitrogen centre on the phosphorus centre of the PCO moiety leads irreversibly under extrusion of CO to the five-membered heterocycle [4e]− [route (ii)]. The nucleophilic phosphorus centre in heterocycle [4e]− triggers the exothermic addition of two further isocyanate molecules – these steps are associated with Ea = 9 kcal mol−1 and 14.7 kcal mol−1, respectively – to give first [IC]− and then [IIC]− which isomerizes rapidly (Ea = 8.5 kcal mol−1) to the spiro-anion [5e]− in an almost thermoneutral process. This computational result is fully in accord with the experimentally observed dynamic behaviour seen with [5f]− (see above). Note, that the activation barrier Ea = 13.1 kcal mol−1 leading from intermediate [IIA]− at the bifurcation point of the MERP to heterocycle [4e]− – and from there on to [5e]− – is smaller than the one leading to [6e]− (Ea = 17.5 kcal mol−1), which is consistent with the observation that [6]− is detected in all experiments, albeit in low concentrations. Note further that all activation barriers on the MERP from [4e]− to [5e]− have moderate heights and indicate that the spiro-anions [5a–d]− may indeed decompose backwards to the five-membered heterocycles [4a–d]− as was observed experimentally. This endothermic process is facilitated by the strongly exothermic formation of isocyanurates (see below).
Fig. 8 shows the MERPs for the catalytic trimerization of methyl isocyanate promoted by either the (OCP)− anion (MERP A), the six-membered heterocycle [6e]− (MERP B), and the five-membered anionic heterocycle [4e]− (MERP C).
The reaction profiles for the trimerization reactions are comparable to those reported for isocyanate trimerizations catalyzed by neutral proazaphosphatranes.24 The trimerization reaction is thermodynamically strongly favoured by ΔHR = −57.5 kcal mol−1. In all cases the anionic [cat]− attacks the electrophilic carbon atom in Me–NCO and forms a weakly bound adduct [I]−. This further reacts sequentially with two further molecules of Me–NCO to give intermediates [II]− and [III]− which finally cyclizes to give the isocyanurate 7e under regeneration of the catalyst [cat]−. Note that the intermediate [IIA]− is also a key intermediate for the formation of the heterocycles [4e]− and [6e]−. The catalytic trimerization of Me–NCO with (OCP)− [3]− and the six-membered heterocycle [6e]−, (MERP A and B shown in black and grey in Fig. 8) proceed on two energetically closely related MERPs over moderate barrier heights which do not exceed 18 kcal mol−1. The first reaction step with the five-membered heterocycle 1,4,2-diazaphospholidine-2,5-dionide [4e]− as catalyst (MERP C) is more exothermic and places intermediate [IC]− 12 kcal mol−1 lower in energy than [IB]− (and 18 kcal mol−1 lower in energy than [IA]−). As a consequence, the final step in the reaction, which involves the release of isocyanurate 7e, costs 26 kcal mol−1 and indicates that [4e]− is a less efficient catalyst. Note also that the spiro-anion [5e]− is an inactive “reservoir” form of the catalytically active species [IIC]− and is “activated” by the cleavage of one P–N bond. Hence, the computations are in agreement with the experimental observation of the spiro-anions in course of the catalytic reactions.
“P−” transfer between 10-P-4 spiro-anions
A consequence of the proposed computed mechanisms shown in Fig. 7 and 8 is that the anionic heterocycles [4]− can be regenerated from the spiro-anions [5]− and an equivalent of isocyanate. This is accompanied by the formation of the isocyanate trimers (isocyanurates) as final products of a highly exothermic reaction (−57.5 kcal mol−1, see Fig. 8). This mechanism implies, that formally “P−” can be transferred from one spiro-anion to another when [5]− is reacted in excess with a different isocyanate. In order to prove this idea, we reacted a solution containing [5c]− [and small amounts of [6c]− but no Na(OCP)] with an excess of phenyl isocyanate (2b). Indeed, the clean formation of [5b]− was observed which is accompanied by a mixture of isocyanurates, (NCy)2(NPh)(CO)3 and (NPh)3(CO)3 as further products (Scheme 3). In the same way, the partially fluorinated derivative [5f]− was prepared in situ from [5c]− and an excess of 2,6-difluorophenyl isocyanate (see above Fig. 5).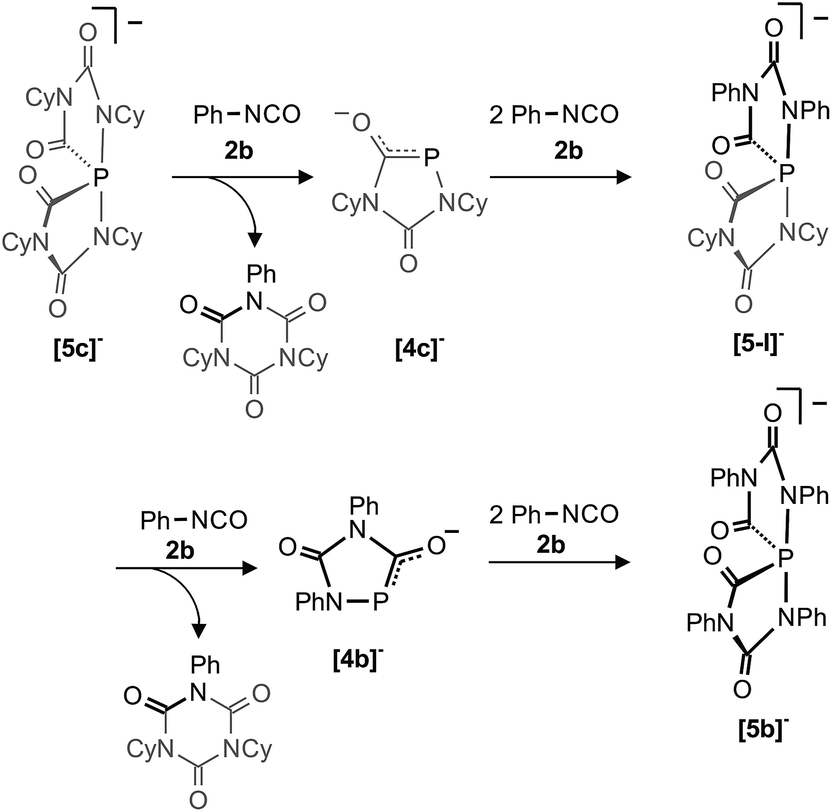 | ||
| Scheme 3 P− transfer from [5c]− to [5b]−. The heterocycles [4c]− and [4b]− as well as the spiro-anion [5-I]− were not observed under the reaction conditions but are likely intermediates. | ||
This transfer reaction involves formally the exchange of four equivalents of the cyclohexyl-substituted isocyanate Cy–NCO (2c shown in grey) with phenyl-substituted isocyanate Ph–NCO (2b shown in black) and a “P−” anion. We assume that in the first reaction step one P–N bond in [5c]− breaks to generate an intermediate like [IIC]− (see Fig. 7) in which the nucleophilic NR terminus reacts instantaneously with Ph–NCO to form a mixed isocyanurate and heterocycle [4c]− as an intermediate (see Fig. 8). The latter reacts with two further equivalents of Ph–NCO to form the mixed spiro-anion [5-I]− as an intermediate. In the reaction with Ph–NCO this again loses the mixed isocyanurate leading to heterocycle [4b]−. Finally this cycle reacts with Ph–NCO to the final product [5b]−. To the best of our knowledge, such an exchange reaction is without precedent and its observation strongly supports the assumptions and computations reported here. Note, that [5b]− is stable and does not react with the less reactive cyclohexyl isocyanate back to the spiro-anion [5c]−.
Conclusion and outlook
In conclusion, we have shown that Na(OCP) is an efficient catalyst for the trimerization of isocyanates. This process proceeds step-wise and involves five-membered heterocycles, namely 1,4,2-diazaphospholidine-3,5-dionide anions [4a–d]−, and spiro-phosphoranides [5a–d]− as detectable intermediates which are catalytically active. Furthermore six-membered anionic heterocycles [6b–d]− were detected and identified as active isocyanate trimerization catalysts. The spiro-phosphoranides [5]− are the latent reservoir species from which [4]− is generated as the active species. The isocyanate fragments in [5]− can be substituted by more reactive isocyanates. Furthermore, this fragmentation process of the spiro-anions can likely be controlled by the choice of the counter cations with smaller cations favouring close ion pairs and higher fragmentation rates. This is indicated by the stability of [5a]− with [K(18-crown-6)]+ as a separated counter cation while the spiro-anion was not detected with smaller cations like Na+.The findings reported here may find various applications. For example, preliminary experiments show that the reaction of Na(OCP) with industrially relevant diisocyanates, e.g. toluenediisocyanate, TDI, or methylenediphenyldiisocyanate, MDI, results rapidly in highly cross-linked materials, which are insoluble in most common solvents. On the other hand, in another preliminary experiment, the alkyl-substituted spiro-phosphoranide [5c]− gives a soluble spiro-phosphoranide which carries unreacted NCO groups in the side-chains in an exchange reaction with the aromatic diisocyanate TDI. With cyclohexane-1,4-dimethanol as the diol component this can be further reacted to a polyurethane, still soluble in THF, showing the typical 31P NMR shifts of the embedded spiro-phosphoranides. This polymer still shows catalytic activity. Consequently, the synthesis of polyurethanes with catalytically active cyclotrimerization sites can be envisioned which allow crosslinking of the polymer in the final step. This is a complementary method to the classical route where diisocyanates are first partially trimerized, which may impede the further processing. The reactions described here open new and promising possibilities and investigations along these lines are under way.
Acknowledgements
This work was supported by the Swiss National Science Foundation (SNF), the ETH Zurich, and Sun Yat-Sen University. The János Bolyai Research Fellowship (for Z.B.), the EPSRC and the University of Oxford (DTA studentship A.R.J.) are gratefully acknowledged.References
- Z. Wirpsza, Polyurethanes chemistry, technology and applications, Ellis Horwood, New York, 1993 Search PubMed.
- H. E. Reymore, P. S. Carleton, R. A. Kolakowski and A. A. R. Sayigh, J. Cell. Plast., 1975, 11, 328–344 CrossRef CAS.
- H. A. Duong, M. J. Cross and J. Louie, Org. Lett., 2004, 6, 4679–4681 CrossRef CAS PubMed.
- Y. Taguchi, I. Shibuya, M. Yasumoto, T. Tsuchiya and K. Yonemoto, Bull. Chem. Soc. Jpn., 1990, 63, 3486–3489 CrossRef CAS.
- (a) A. W. Hofmann, Chem. Ber., 1870, 3, 761–772 CrossRef CAS; (b) Z. Pusztai, G. Vlad, A. Bodor, I. T. Horváth, H. J. Laas, R. Halpaap and F. U. Richter, Angew. Chem., Int. Ed., 2006, 45, 107–110 CrossRef CAS PubMed.
- (a) S. M. Raders and J. G. Verkade, J. Org. Chem., 2010, 75, 5308–5311 CrossRef CAS PubMed; (b) J.-S. Tang and J. G. Verkade, Angew. Chem., Int. Ed., 1993, 32, 896–898 CrossRef.
- F. U. Richter, Chem.–Eur. J., 2009, 15, 5200–5202 CrossRef CAS PubMed.
- F. U. Richter, 1,4,2-Diazaphospholidine derivatives, WO 2010/037499A1, Bayer Materialscience AG, 2010.
- W. Y. Yi, J. Zhang, L. C. Hong, Z. X. Chen and X. G. Zhou, Organometallics, 2011, 30, 5809–5814 CrossRef CAS.
- A. J. Roering, S. E. Leshinski, S. M. Chan, T. Shalumova, S. N. MacMillan, J. M. Tanski and R. Waterman, Organometallics, 2010, 29, 2557–2565 CrossRef CAS.
- U. Segerer, J. Sieler and E. Hey-Hawkins, Organometallics, 2000, 19, 2445–2449 CrossRef CAS.
- F. F. Puschmann, D. Stein, D. Heift, C. Hendriksen, Z. A. Gál, H.-F. Grützmacher and H. Grützmacher, Angew. Chem., Int. Ed., 2011, 50, 8420–8423 CrossRef CAS PubMed.
- A. R. Jupp and J. M. Goicoechea, Angew. Chem., Int. Ed., 2013, 52, 10064–10067 CrossRef CAS PubMed.
- Similar 31P NMR chemical shifts and 1JPH coupling constants were reported for the closely related compounds methoxycarbonyl phosphane H3C–O–C(O)–PH2 (δ31P: −138.6 ppm, 1JPH = 218 Hz) and lithium methoxycarbonyl phosphanide [H3C–O–C(O)PH]Li (δ31P: −83.8 ppm, 1JPH = 219 Hz); see: G. Becker, G. Heckmann, K. Hübler and W. Schwarz, Z. Anorg. Allg. Chem., 1995, 621, 34–46 CrossRef CAS.
- C. W. Perkins, J. C. Martin, A. J. Arduengo, W. Lau, A. Alegria and J. Kochi, J. Am. Chem. Soc., 1980, 102, 7753 CrossRef CAS.
- A. A. Culley and A. J. Arduengo III, J. Am. Chem. Soc., 1984, 106, 1164–1165 CrossRef.
- In all reactions with the sterically less hindered and more reactive isocyanates the same species were obtained independently on the use of Na(PH2) or Na(OCP) as starting materials. In all the cases employing Na(PH2) the 31P NMR spectrum of the reaction mixtures indicated the formation of small amounts of Na(OCP) (δ31P: −388.9 ppm).
- D. Heift, Z. Benkő and H. Grützmacher, Angew. Chem., Int. Ed., 2014, 53, 6757–6761 CrossRef CAS PubMed.
- Although the symmetry of the spiro anion is distorted in the solid state structure (due to the asymmetric location of the sodium cation), the gas phase calculated structure of anion [5e]− (R = Me) shows C2 symmetry.
- For neutral 1,3,5,7-tetramethyl-1,3,5,7-tetraaza-4-phosphaspiro[3.3]heptane-2.6-diones axial PN bond lengths from 1.74 Å to 1.80 Å were reported in the literature: D. Schomburg, U. Wermuth and R. Schmutzler, Chem. Ber., 1987, 120, 1713–1718 CrossRef CAS Note that similar extremely long axial bonds were found for the anionic 10-P-4 species in [Li(THF)(cyclenP)]x as well (1.94 Å and 2.01 Å) where cyclen is 1,4,7,10-tetraazacyclododecan: M. Lattman, M. M. Olmstead, P. P. Power, D. W. H. Rankin and H. E. Robertson, Inorg. Chem., 1988, 27, 3012–3018 CrossRef.
- The calculated value of the J(PFA) is 124.4 Hz, while J(PFB) is 0.7 Hz at the B3LYP/6-31+G* level. In the calculation the Fermi-contact terms were calculated with orbitals obtained by uncontracting the basis and adding tight polarization functions for the core.
- This process is comparable to the observed breaking of P–O bonds in related oxygen substituted phosphoranides: D. Schomburg, W. Storzer, R. Bohlen, W. Kuhn and G. V. Röschenthaler, Chem. Ber., 1983, 116, 3301–3308 CrossRef CAS.
- K. B. Dillon, Chem. Rev., 1994, 94, 1441–1456 CrossRef CAS.
- J. N. Gibb and J. M. Goodman, Org. Biomol. Chem., 2013, 11, 90–97 CAS.
- J. S. Tang and J. G. Verkade, Angew. Chem., Int. Ed. Engl., 1993, 32, 896–898 CrossRef.
- M. Roman, B. Andrioletti, M. Lemaire, J. M. Bernard, J. Schwartz and P. Barbeau, Tetrahedron, 2011, 67, 1506–1510 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1049373, 986846, 1049374, 986881 and 986854. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5sc00963d |
| This journal is © The Royal Society of Chemistry 2015 |