
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencepH-responsive and switchable triplex-based DNA hydrogels†
Jiangtao
Ren‡
a,
Yuwei
Hu‡
a,
Chun-Hua
Lu
a,
Weiwei
Guo
a,
Miguel Angel
Aleman-Garcia
a,
Francesco
Ricci
bc and
Itamar
Willner
*a
aInstitute of Chemistry and the Center for Nanoscience and Nanotechnology, The Hebrew University of Jerusalem, Jerusalem 91904, Israel. E-mail: willnea@vms.huji.ac.il
bDipartimento di Scienze e Tecnologie Chimiche, University of Rome Tor Vergata, Rome 00133, Italy
cConsorzio Interuniversitario Biostrutture e Biosistemi “INBB”, Rome, Italy
First published on 13th May 2015
Abstract
New methods for the preparation of reversible pH-responsive DNA hydrogels based on Hoogsteen triplex structures are described. One system consists of a hydrogel composed of duplex DNA units that bridge acrylamide chains at pH = 7.4 and undergoes dissolution at pH = 5.0 through the reconfiguration of one of the duplex bridging units into a protonated CG·C+ triplex structure. The second system consists of a hydrogel consisting of acrylamide chains crosslinked in the presence of an auxiliary strand by Hoogsteen TA·T triplex interaction at pH = 7.0. The hydrogel transforms into a liquid phase at pH = 10.0 due to the separation of the triplex bridging units. The two hydrogel systems undergo reversible and cyclic hydrogel/solution transitions by subjecting the systems to appropriate pH values. The anti-cancer drug, coralyne, binds specifically to the TA·T triplex-crosslinked hydrogel thereby increasing its stiffness. The pH-controlled release of the coralyne from the hydrogel is demonstrated.
Introduction
Stimuli-responsive polymers undergoing reversible hydrogel/solution transitions attract substantial interest in material science.1 Different triggers such as pH,2 chemical substrates,3 redox reactions,4 supramolecular interactions,5 light6 and electrical signals7 were used to switch these phase transitions of polymers. Different applications of switchable polymers undergoing hydrogel/solution transitions were suggested, and these include the controlled drug delivery,8 tissue engineering,1a micromechanical devices such as valves or actuators,9 sensors,10 and smart coating materials.11 Within the stimuli-responsive polymers undergoing solution/hydrogel transitions, DNA-containing switchable hydrogels attract special interest, since the nucleic acid components can be encoded with appropriate base sequences that provide the mechanisms for the switchable hydrogel/solution phase transitions.12 The stimuli-responsive DNA hydrogels may be synthesized by two general approaches: by one approach, DNA units are crosslinked by stimuli-responsive nucleic acid bridging units to yield the switchable hydrogel.13 The second approach, involves the tethering of nucleic acids, that are encoded with stimuli-responsive base sequences, on polymer chains, and the crosslinking of the chains by appropriate external triggers to form the hydrogels.14 By coding programmed base sequences into the bridging units exhibiting reversible and switchable crosslinking properties, stimuli-responsive matrices undergoing reversible hydrogel/solution were designed. Different external signals were used to trigger the reversible DNA-stimulated formation and dissociation of hydrogels. These included the use of nucleic acid duplex bridging units and their separation by the strand displacement process,15 the bridging and separation of the polymer chains by K+-ion-stabilized G-quadruplexes and crown ether receptors,16 and the use of metal ions to bridge the polymer chains to hydrogels and to separate the hydrogels by ligands eliminating the metal ions.17 Also, the pH-triggered formation of i-motif structures was applied to bridge DNA units to yield all-DNA hydrogels.13b Different applications of DNA-based hydrogels were suggested, including sensing,18 removal of hazardous metal ions,19 inscription of structural information,20 and the use of the nucleic acid functionalized polymers as shape-memory matrices.21Hoogsteen base interactions allow the pH-stimulated formation and dissociation of triplex DNA structures. Specifically, protonated CG·C+ parallel domains favor the formation of triplex structures at acidic pH,22 and TA·T parallel domains stimulate triplex formation at neutral pH environments.23 The pH-triggered switchable transitions duplex/triplex structures of nucleic acids were previously implemented to develop DNA machines24 and switches.25 In the present report, we present the use of triplex DNA structures as pH triggers that stimulate switchable and reversible hydrogel/solution transitions. We demonstrate that through a rational design of the nucleic acid crosslinking units and their pH-induced triplex nanostructures, the dictated transitions of DNA matrices existing as hydrogels at neutral pH, into liquid phases proceed either under acidic or basic conditions. We further demonstrate that the anti-cancer drug, coralyne, intercalates specifically into the TA·T triplex crosslinked hydrogel. Besides the increase of the stiffness of the hydrogel through the intercalation process, the pH-controlled release of the coralyne from the hydrogel is demonstrated.
Results and discussion
Fig. 1A depicts the synthesis and characterization of the DNA hydrogel undergoing hydrogel/liquid transitions upon triggering the system between pH = 7.4 and pH = 5.0. The acrydite nucleic acid (1) was polymerized in the presence of acrylamide to yield copolymer chains (3) composed of the (1)-tether![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :acrylamide with ratio corresponding to 1:110, average molecular weight 800000. Similarly, the acrydite nucleic acid (2) was polymerized in the presence of acrylamide to yield the copolymer chains (4) ((2)-tether:acrylamide ratio 1:98, average molecular weight 400000), for the experimental determination of the loading see Fig. S1 and S2, ESI.† The sequence domain I in (1) is complementary to (2). Mixing the two chains (3) and (4) at pH = 7.4 yields the (1)/(2)-crosslinked hydrogel. Subjecting the hydrogel to pH = 5.0 results in the separation of the crosslinking duplex units and domain I of (1) stabilizes the intramolecular triplex DNA nanostructure of (1) resulting in the separation of the hydrogel. Changing the pH of the solution to pH = 7.4 leads to the separation of the triplex units and to the re-assembly of the (3)/(4) hydrogel. By the cyclic alternation the pH of the system between values 7.4 and 5.0, reversible transitions between hydrogel and liquid phases are observed (see macroscopic images in Fig. 1B). The rheology characterization of the pH-switchable hydrogel is depicted in Fig. 1C. At pH = 5.0, the liquid phase is generated (G′ ≈ G′′ ≈ 1–2 Pa), whereas at pH = 7.4, G′ ≈ 22 Pa, and a low loss modulus G′′ ≈ 2 Pa. Fig. 1D shows the stained, freeze-dried, SEM image of the hydrogel that reveals pore sizes exhibiting a 1 μm diameter. The melting temperature (Tm) of the (3)/(4) hydrogel was followed by UV spectroscopy and it corresponds to ca. 60 °C, Fig. 2A. This phase transition was further confirmed by rheology studies, Fig. 2B. At ca. 60 °C sharp decrease in the G′ modulus is observed, consistent with the transformation of the hydrogel into a liquid phase.
:acrylamide with ratio corresponding to 1:110, average molecular weight 800000. Similarly, the acrydite nucleic acid (2) was polymerized in the presence of acrylamide to yield the copolymer chains (4) ((2)-tether:acrylamide ratio 1:98, average molecular weight 400000), for the experimental determination of the loading see Fig. S1 and S2, ESI.† The sequence domain I in (1) is complementary to (2). Mixing the two chains (3) and (4) at pH = 7.4 yields the (1)/(2)-crosslinked hydrogel. Subjecting the hydrogel to pH = 5.0 results in the separation of the crosslinking duplex units and domain I of (1) stabilizes the intramolecular triplex DNA nanostructure of (1) resulting in the separation of the hydrogel. Changing the pH of the solution to pH = 7.4 leads to the separation of the triplex units and to the re-assembly of the (3)/(4) hydrogel. By the cyclic alternation the pH of the system between values 7.4 and 5.0, reversible transitions between hydrogel and liquid phases are observed (see macroscopic images in Fig. 1B). The rheology characterization of the pH-switchable hydrogel is depicted in Fig. 1C. At pH = 5.0, the liquid phase is generated (G′ ≈ G′′ ≈ 1–2 Pa), whereas at pH = 7.4, G′ ≈ 22 Pa, and a low loss modulus G′′ ≈ 2 Pa. Fig. 1D shows the stained, freeze-dried, SEM image of the hydrogel that reveals pore sizes exhibiting a 1 μm diameter. The melting temperature (Tm) of the (3)/(4) hydrogel was followed by UV spectroscopy and it corresponds to ca. 60 °C, Fig. 2A. This phase transition was further confirmed by rheology studies, Fig. 2B. At ca. 60 °C sharp decrease in the G′ modulus is observed, consistent with the transformation of the hydrogel into a liquid phase.
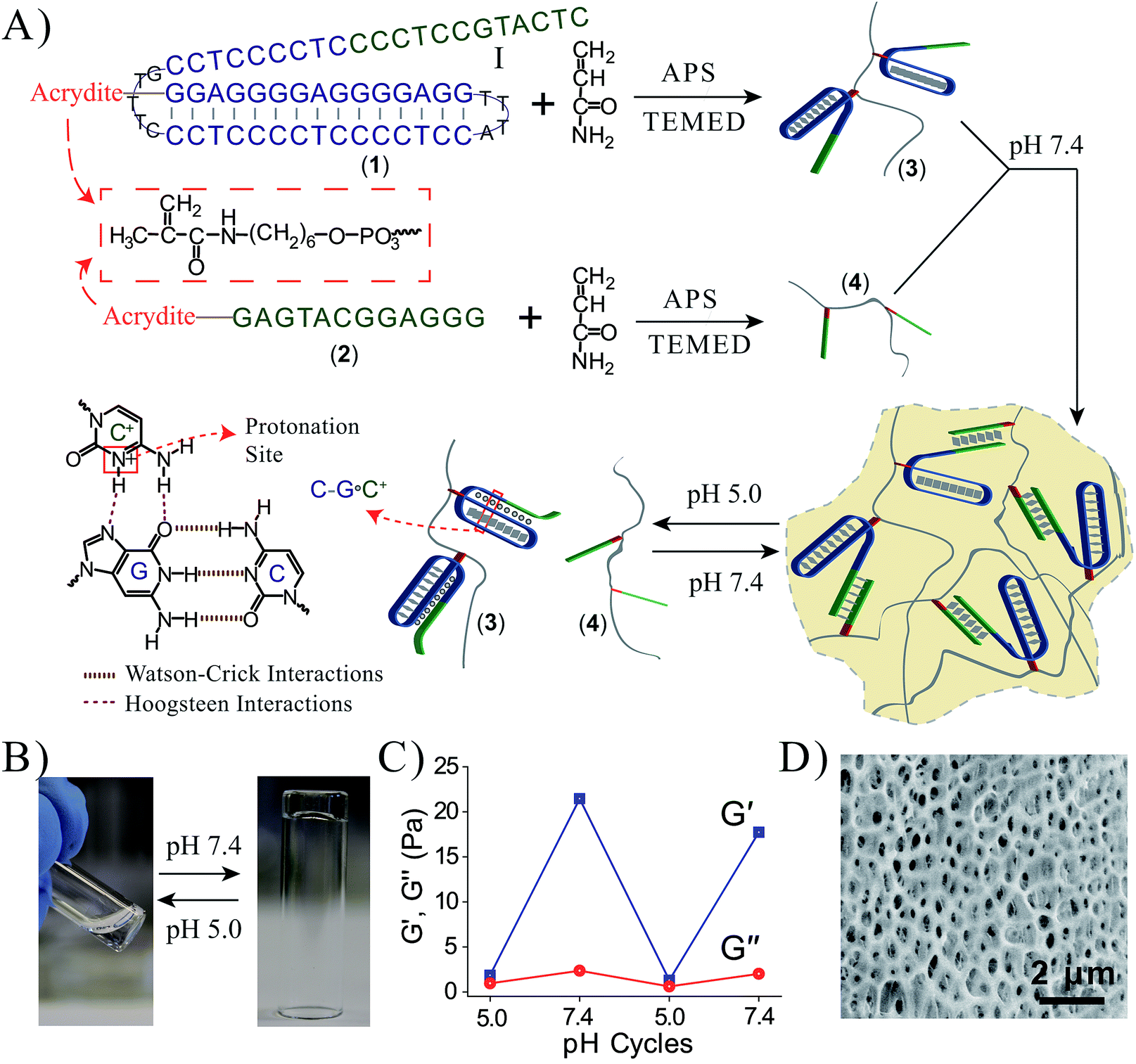 | ||
| Fig. 1 (A) Synthesis of the DNA chains undergoing pH-stimulated hydrogel formation and dissociation. Formation of the hydrogel is triggered at pH = 7.4 through the crosslinking of the polymer chains by duplex Watson–Crick interactions. The separation of the hydrogel is triggered at pH = 5.0 using Hoogsteen triplex interactions that separate the bridging units. By reversible pH triggers between the values 7.4 and 5.0, the system undergoes cyclic transitions between the hydrogel and solution phases. (B) Photographs of the solution phase of the system at pH = 5.0, and the hydrogel phase at pH = 7.4. The pH-stimulated transition of the hydrogel to the liquid phase proceeds within a time interval of <1 minute via shaking. (C) Rheology studies following the pH-switchable storage moduli, G′, and loss moduli, G′′, upon cyclic the pH of the system between the values pH = 5.0 and pH = 7.4. (D) SEM image of the Au/Pd coated (3)/(4) hydrogel. | ||
 | ||
| Fig. 2 (A) Monitoring the Tm value of the (3)/(4) hydrogel. (B) Rheology characterization of the temperature-induced transition of the (3)/(4) hydrogel into a liquid. | ||
To further demonstrate the versatility of our approach, we designed a DNA-based hydrogel that undergoes hydrogel/solution transitions under basic conditions, using Hoogsteen TA·T triplex interactions as depicted in Fig. 3A. The thymine-functionalized nucleic acid (5) was tethered to the acrylamide copolymer chains (6) by the copolymerization of the (5)-functionalized acrydite monomer with the acrylamide monomer (final ratio of (5):acrylamide is 1:79, average molecular weight 360000), for the determination of the loading see Fig. S3, ESI.† The adenine-rich nucleic acid (7) was used as “glue” to form the TA·T triplex structure. The mixture of the copolymer (6) and (7) yields at pH = 7.0 the triplex-bridged hydrogel. Subjecting the hydrogel to pH = 10.0 results in the separation of the triplex structures, due to the deprotonation of the thymine residues. The dissociation of the triplex bridges yields the polymer chains consisting of the duplex (5)/(7) tethers and free (5) tethers, leading to the transition of the hydrogel to the liquid polymer state. For the stability of the duplex (5)/(7), see the Fig. S4 in the ESI.† By the cyclic treatment of the system between the pH values 7.0 and 10.0, the system undergoes reversible transitions between the hydrogel and liquid phases, respectively. Fig. 3B depicts the images of the hydrogel and liquid states of the system at the respective pH values. Fig. 3C shows the rheology characterization of the triplex-crosslinked hydrogel at pH = 7.0. The storage modulus of the system is G′ ≈ 48 Pa whereas the loss modulus is G′′ ≈ 2 Pa, consistent with the existence of a hydrogel. The Tm values of the (6)/(7) hydrogel correspond to 32 °C (separation of the triplex) and 60 °C (separation of the duplex), see Fig. S5, ESI.† Also, we find that the quality of the triplex crosslinked hydrogel is sensitive to mutations introduced in the bridging strand (7), Fig. 3D. The use of a bridging strand (7a) that introduce two-base mismatches results in a lower storage modulus, G′ ≈ 37 Pa (as compared to G′ ≈ 48 Pa for the (6)/(7) hydrogel). The introduction of three base mismatches or four base mismatches into the bridging strand, (7b) and (7c), respectively, does not allow the formation of crosslinked hydrogel.
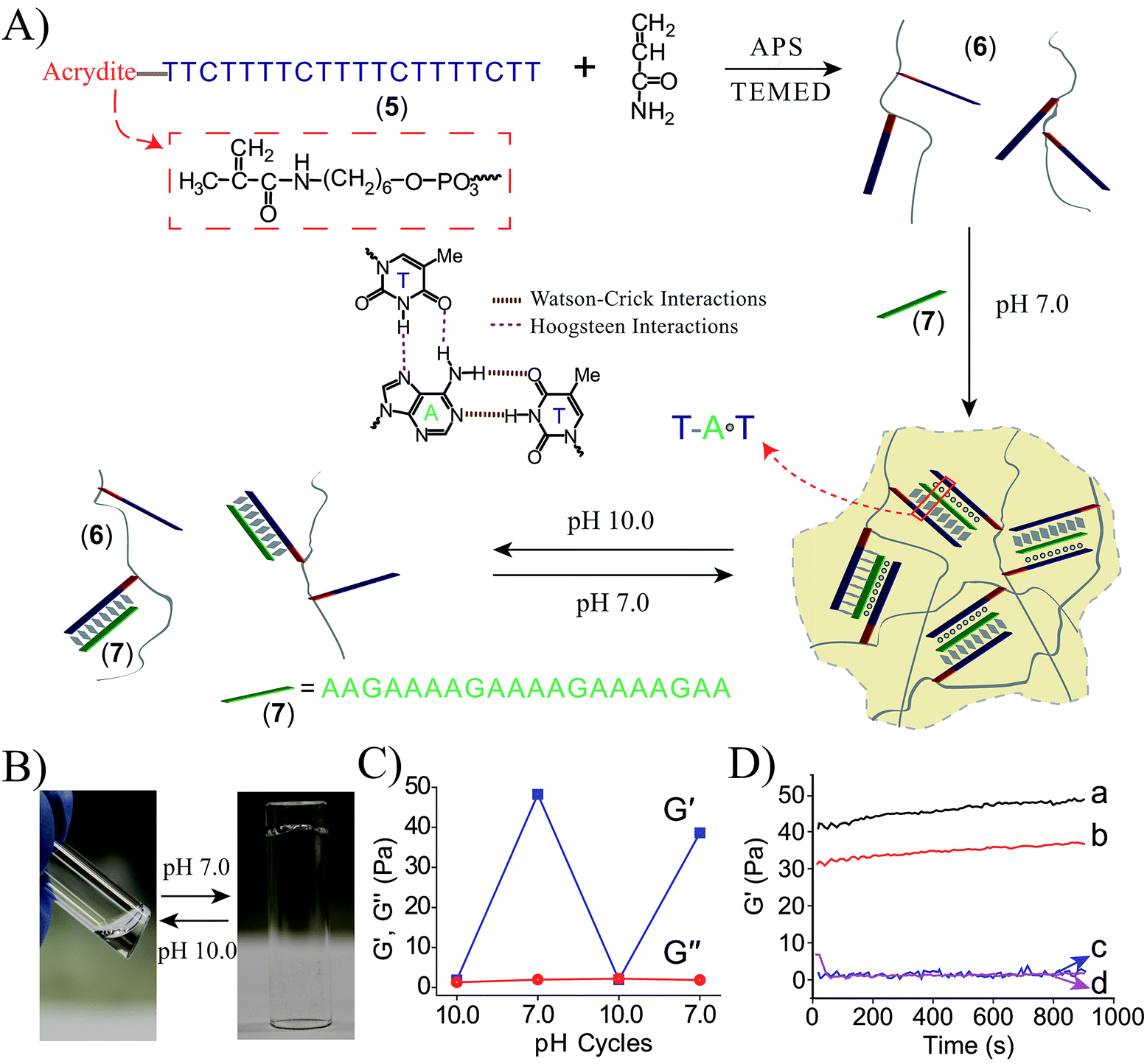 | ||
| Fig. 3 (A) Synthesis of the DNA chains undergoing pH-induced hydrogel formation and dissociation. Formation of the hydrogel proceeds at pH = 7.0 using the strand (7) as activator for the formation of Hoogsteen triplex bridges between the (5)-modified chains. At pH = 10.0 the triplex bridging units dissociate to form the separated polymer chains. (B) Images of the polymer hydrogel formed at pH = 7.0 and the polymer solution generated at pH = 10.0. (C) Rheology studies following the pH-switchable storage moduli, G′, and loss moduli, G′′, upon switching the TA·T triplex-crosslinked system between the pH values 10.0 and 7.0. (D) Time-dependent feature of the G′ modulus corresponding to systems composed of: (a) the (6)/(7) triplex crosslinked hydrogel. (b) The polymer chains (6) treated with (7a) (two base mismatches). (c) The polymer chains (6) treated with (7b) (three base mismatches). (d) The polymer chains (6) treated with (7c) (four base mismatches). | ||
Coralyne is an anti-cancer drug26 and it preferentially binds to TA·T triplex structures.27 Accordingly, coralyne was loaded in the TA·T triplex crosslinked hydrogel, and its release upon the dissolution of the hydrogel was examined, Fig. 4A. The comparison of the rheology features of the coralyne-loaded hydrogel to the unloaded triplex-crosslinked hydrogel is depicted in Fig. 4B. The storage modulus of the coralyne-loaded hydrogel corresponds to G′ ≈ 64 Pa (as compared to G′ ≈ 48 Pa of the unloaded hydrogel), implying that the binding of coralyne to the TA·T triplex units stabilizes the hydrogel and increases its stiffness. Fig. 4C depicts the time-dependent release of coralyne upon treatment of the coralyne-loaded (6)/(7) hydrogel at different pH conditions. While at pH = 7.0, the release of coralyne from the hydrogel is slow and inefficient, as the pH of the hydrogel increases the release of coralyne is enhanced. At pH = 10.0, effective release of coralyne from the matrix is observed, consistent with the dissociation of the hydrogel. From the fluorescence intensities generated by the release of coralyne, and we estimate that at pH = 7.0, 19% of the loaded coralyne are released after a time-interval of 400 minutes, whereas 56% of coralyne are released after the same time-interval from the dissociated hydrogel at pH = 10.0. The incomplete release of coralyne is attributed to the partial electrostatic association of coralyne to the DNA-modified polymer chains separated from the delivery coralyne reservoir.
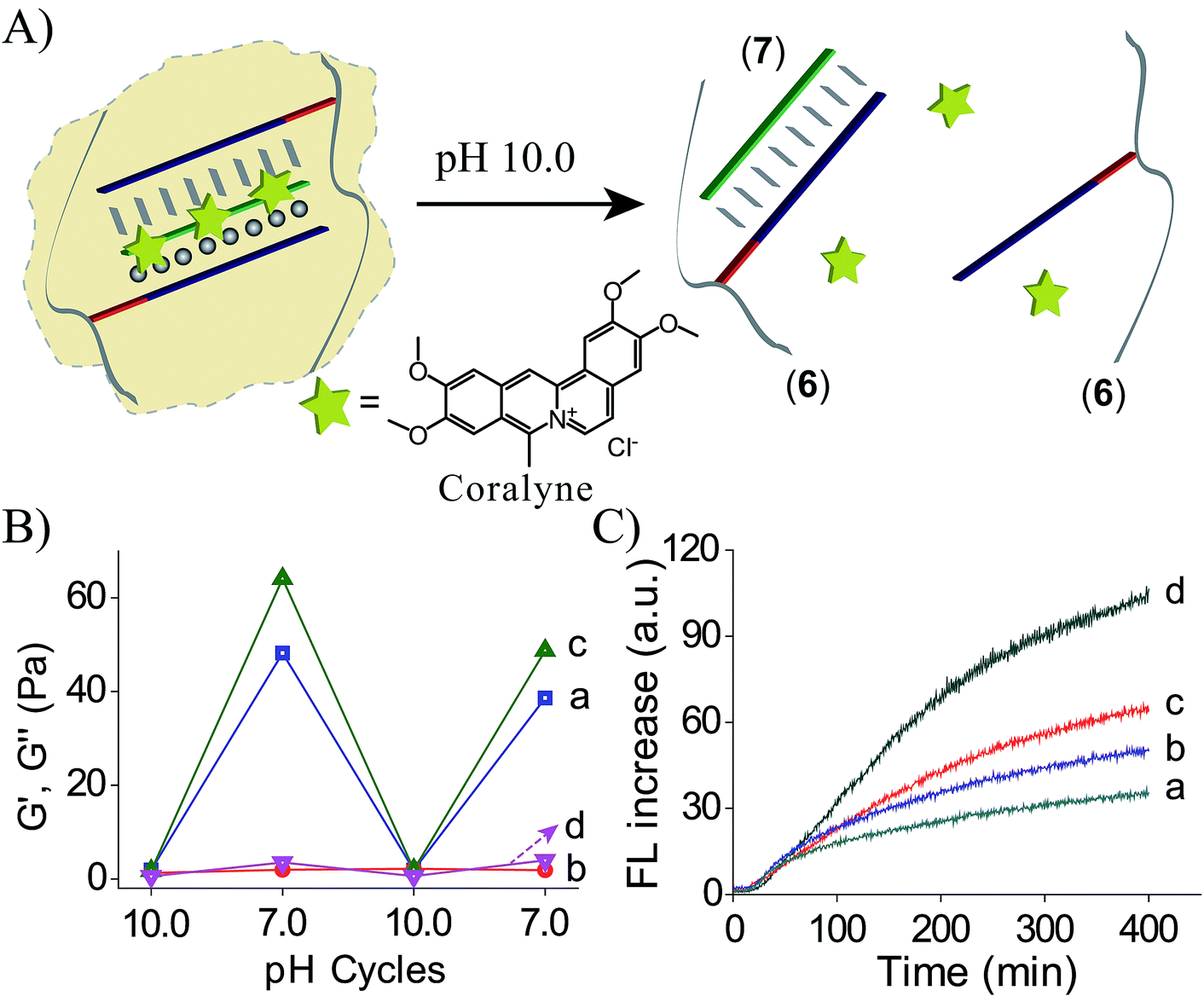 | ||
| Fig. 4 (A) Schematic pH-stimulated release of coralyne from the coralyne-loaded triplex-stabilized hydrogel upon subjecting the hydrogel to pH = 10.0. (B) Rheology studies comparing the effect of pH on the storage moduli, G′, and the loss moduli, G′′, of the (6)/(7) hydrogel, curves (a) and (b), respectively, to the effect of pH on the storage moduli, G′, and loss moduli, G′′, of the coralyne-loaded hydrogel, curves (c) and (d). (C) Time-dependent fluorescence changes upon subjecting the coralyne-loaded (6)/(7) hydrogel to variable pH values, resulting in the release of coralyne: (a) pH = 7.0 (b) pH = 8.0 (c) pH = 9.0 (d) pH = 10.0. | ||
Conclusions
To conclude, the present study has introduced new methods to synthesize stimuli-responsive DNA hydrogels based on triplex DNA structures. By the appropriate design of the triplex DNA units hydrogels existing at neutral pH could be dissociated either under acid conditions, pH = 5.0, or under basic conditions (pH = 10.0). We have further demonstrated that the specific interaction of coralyne into the TA·T triplex-bridged hydrogel increases the stiffness of the resulting hydrogel. The dissociation of the triplex, at pH = 10.0, leads then to the separation of hydrogel and to the release of coralyne that lacks affinity toward single-strand or duplex DNA structures.28Experimental section
Materials
The oligonucleotides used in the study were purchased from Integrated DNA Technologies (Coralville, IA, USA). The sequences used in the study are summarized in Table 1. For electrospray ionization (ESI) mass spectrometry confirmed purity of the different oligonucleotides see Fig. S9–S15.†| (1) | 5′-Acrydite-GGAGGGGAGGGGAGGTTTACCTCC CCTCC CCTCCCTTTGCCTCCCCTCCCCTCCGT ACTC-3′ (18426.9; 18427.1) |
| (2) | 5′-Acrydite-GAGTACGGAGGG-3′ (4022.7; 4023.1) |
| (5) | 5′-Acrydite-TTCTTTTCTTTTCTTTTCTT-3′ (6209.1; 6209.0) |
| (7) | 5′-AAGAAAAGAAAAGAAAAGAA-3′ (6266.2; 6266.7) |
| (7a) | 5′-AAGAAAAGTAATGAAAAGAA-3′ (6248.2; 6248.2) |
| (7b) | 5′-AAGATAAGATAAGATAAGAA-3′ (6239.2; 6239.0) |
| (7b) | 5′-AAGATAAGTAATGAATAGAA-3′ (6230.2; 6230.5) |
Synthesis of the acrydite-modified oligo/acrylamide copolymers
150 μL of buffer solution (10 mM HEPES, 10 mM MgCl2 and pH = 7.0) containing 1 mM acrydite-modified DNA (1, 2 or 5) and 2% acrylamide was bubbled with nitrogen gas for 3 minutes. 9 μL of initiator mixture (20 mg ammonium persulfate, APS, and 10 μL N,N,N′,N′-tetramethylenediamine, TEMED, in 0.2 mL H2O) was added into the solution. Nitrogen gas was bubbled into the solution for another 5 minutes. Then it was incubated at 4 °C overnight. The resulting copolymer was purified with a Microcon (Millipore) spin filter unit (MWCO 10 kD) to remove the unreacted monomers and other impurities. Finally, the copolymer solution was dried under a gentle flow of nitrogen gas and redispersed in buffer. Based on the calibration curves shown in Fig. S1–S3,† DNA loading (the ratio of the acrylamide/acrydite-oligo) of the different polymers was determined spectroscopically. All the UV-vis absorption data were recorded using a UV-2450 spectrophotometer (Shimadzu). For the determination of average molecular weights of the different polymers see Fig. S6.† It should be noted that the DNA loading of the polymer chains to yield the hydrogels represent optimized value. For optimization of the loading, polymer chains exhibiting variable loading of the DNA were prepared, and the resulting hydrogels were characterized by rheology, Fig. S7A and B.†Preparation of the hydrogels and their switchable hydrogel/liquid transitions
To prepare the (3)/(4) hydrogel, copolymer (3) and (4) were mixed in a buffer solution (10 mM HEPES, 10 mM MgCl2 and pH = 7.4) with the final concentrations of nucleic acid (1) and (2) to be 0.43 mM. The gelation was completed within 5 minutes. To dissociate the hydrogel, a minute volume of acetic acid (20%) was used to adjust pH to 5.0. The dissociation was completed within 1 minute via shaking. Ammonium hydroxide solution (10%) was used to change the solution pH to 7.4.To prepare the (6)/(7) hydrogel, the nucleic acid (7) was mixed with copolymer (6) in the HEPES buffer (10 mM HEPES, 10 mM MgCl2 and pH = 7.0). The final concentrations of (5) and (7) were 1.2 mM and 0.6 mM, respectively. Ammonium hydroxide solution (25%) and acetic acid (40%) were employed to change the solution pH between 10.0 and 7.0. The gelation was completed within 5 minutes and dissociation was completed within 1 minute via shaking.
Rheology measurements
The formation, dissociation and mechanical properties of the hydrogels were characterized by a HAAKE MARS III rheometer (Thermo Scientific). Oscillatory frequency sweep test and the strain test of the (3)/(4) hydrogel were performed, Fig. S8A and B,† respectively. The linear viscoelastic region was found to be in the range of 1% strain and 1 Hz frequency. Time-sweep oscillatory tests were performed using a 20 mm parallel-plate geometry and with 135 μL of a fresh solution (resulting in a gap size of 0.30 mm). For the (3)/(4) hydrogel, final concentrations of (1) and (2) were both 0.20 mM. For the (6)/(7) hydrogel, the concentrations of (5), (7) and coralyne were 0.5 mM, 0.25 mM and 0.72 mM, respectively.Release of coralyne from the (6)/(7) hydrogel
The release of coralyne from the triplex-crosslinked hydrogel was also recorded by its fluorescence. 3.2 μL of a mixture solution of copolymer (6) (1.34 mM (5)) and coralyne (1.62 mM) was placed in the corner of a cuvette, and then mixed with 0.38 μL of (7) to yield the hydrogel. 2 mL HEPES buffers of different pHs (pH = 7.0, 8.0, 9.0 and 10.0) were injected into four cuvettes containing hydrogels, and the fluorescence of supernatant was recorded immediately (Ex: 422 nm, Em: 480 nm, slits: 2.5/5 nm).Scanning electron microscope (SEM)
SEM images were collected by an Extra High Resolution Scanning Electron Microscope Magellan (TM) 400L (2 kV, 6.3 pA). The silicon slides (Virginia semiconductor, Inc.) were cleaned by washing with distilled water, ethanol and acetone in succession, and processing by UV/zone in a T1O × 10/OES/E UV/ozone chamber from UVOCS, Inc. (USA). According to the procedures, the hydrogels were prepared, placed on the cleaned silicon slides and freeze-dried, and then coated with an Au/Pd film.Acknowledgements
This research is supported by the EU FET Open MICREAGENTS Project.Notes and references
- (a) K. Y. Lee and D. J. Mooney, Chem. Rev., 2001, 101, 1869 CrossRef CAS PubMed; (b) N. A. Peppas, J. Z. Hilt, A. Khademhosseini and R. Langer, Adv. Mater., 2006, 18, 1345 CrossRef CAS PubMed; (c) F. Liu and M. W. Urban, Prog. Polym. Sci., 2010, 35, 3 CrossRef CAS PubMed; (d) M. A. C. Stuart, W. T. S. Huck, J. Genzer, M. Müller, C. Ober, M. Stamm, G. B. Sukhorukov, I. Szleifer, V. V. Tsukruk, M. Urban, F. Winnik, S. Zauscher, I. Luzinov and S. Minko, Nat. Mater., 2010, 9, 101 CrossRef PubMed.
- (a) S. Dai, P. Ravi and K. C. Tam, Soft Matter, 2008, 4, 435 RSC; (b) S. R. Haines and R. G. Harrison, Chem. Commun., 2002, 2846 RSC.
- Y. Lvov, A. A. Antipov, A. Mamedov, H. Möhwald and G. B. Sukhorukov, Nano Lett., 2001, 1, 125 CrossRef CAS.
- A. Akhoury, L. Bromberg and T. A. Hatton, ACS Appl. Mater. Interfaces, 2011, 3, 1167 CAS.
- (a) J. B. Beck and S. J. Rowan, J. Am. Chem. Soc., 2003, 125, 13922 CrossRef CAS PubMed; (b) J. Yuan, D. Wen, N. Gaponik and A. Eychmüller, Angew. Chem., Int. Ed., 2013, 52, 976 CrossRef CAS PubMed.
- Y. Zhao, J. Mater. Chem., 2009, 19, 4887 RSC.
- Q. Yan, J. Yuan, Z. Cai, Y. Xin, Y. Kang and Y. Yin, J. Am. Chem. Soc., 2010, 132, 9268 CrossRef CAS PubMed.
- (a) M. E. Byrne, K. Park and N. A. Peppas, Adv. Drug Delivery Rev., 2002, 54, 149 CrossRef CAS; (b) J. Z. Hilt and M. E. Byrne, Adv. Drug Delivery Rev., 2004, 56, 1599 CrossRef CAS PubMed; (c) A. S. Hoffman, J. Controlled Release, 2008, 132, 153 CrossRef CAS PubMed.
- D. J. Beebe, J. S. Moore, J. M. Bauer, Q. Yu, R. H. Liu, C. Devadoss and B.-H. Jo, Nature, 2000, 404, 588 CrossRef CAS PubMed.
- (a) J. N. Anker, W. P. Hall, O. Lyandres, N. C. Shah, J. Zhao and R. P. Van Duyne, Nat. Mater., 2008, 7, 442 CrossRef CAS PubMed; (b) I. Tokarev and S. Minko, Soft Matter, 2009, 5, 511 RSC.
- (a) P. M. Mendes, Chem. Soc. Rev., 2008, 37, 2512 RSC; (b) I. Luzinov, S. Minko and V. V. Tsukruk, Soft Matter, 2008, 4, 714 RSC.
- (a) Y. H. Roh, R. C. H. Ruiz, S. Peng, J. B. Lee and D. Luo, Chem. Soc. Rev., 2011, 40, 5730 RSC; (b) J. Liu, Soft Matter, 2011, 7, 6757 RSC.
- (a) S. H. Um, J. B. Lee, N. Park, S. Y. Kwon, C. C. Umbach and D. Luo, Nat. Mater., 2006, 5, 797 CrossRef CAS PubMed; (b) E. Cheng, Y. Xing, P. Chen, Y. Yang, Y. Sun, D. Zhou, L. Xu, Q. Fan and D. Liu, Angew. Chem., Int. Ed., 2009, 48, 7660 CrossRef CAS PubMed.
- B. Wei, I. Cheng, K. Q. Luo and Y. Mi, Angew. Chem., Int. Ed., 2008, 47, 331 CrossRef CAS PubMed.
- T. Liedl, H. Dietz, B. Yurke and F. Simmel, Small, 2007, 3, 1688 CrossRef CAS PubMed.
- C.-H. Lu, X.-J. Qi, R. Orbach, H.-H. Yang, I. Mironi-Harpaz, D. Seliktar and I. Willner, Nano Lett., 2013, 13, 1298 CrossRef CAS PubMed.
- (a) W. Guo, X. Qi, R. Orbach, C.-H. Lu, L. Freage, I. Mironi-Harpaz, D. Seliktar, H. H. Yang and I. Willner, Chem. Commun., 2014, 50, 4065 RSC; (b) W. Guo, C.-H. Lu, X.-J. Qi, R. Orbach, M. Fadeev, H.-H. Yang and I. Willner, Angew. Chem., Int. Ed., 2014, 53, 10134 CrossRef CAS PubMed.
- K. A. Joseph, N. Dave and J. Liu, ACS Appl. Mater. Interfaces, 2011, 3, 733 CAS.
- N. Dave, M. Y. Chan, P.-J. J. Huang, B. D. Smith and J. Liu, J. Am. Chem. Soc., 2010, 132, 12668 CrossRef CAS PubMed.
- J. B. Lee, S. Peng, D. Yang, Y. H. Roh, H. Funabashi, N. Park, E. J. Rice, L. Chen, R. Long, M. Wu and D. Luo, Nat. Nanotechnol., 2012, 7, 816 CrossRef CAS PubMed.
- W. Guo, C.-H. Lu, R. Orbach, F. Wang, X.-J. Qi, A. Cecconello, D. Seliktar and I. Willner, Adv. Mater., 2015, 27, 73 CrossRef CAS PubMed.
- (a) D. Leitner, W. Schröder and K. Weisz, Biochemistry, 2000, 39, 5886 CrossRef CAS PubMed; (b) J. L. Asensio, A. N. Lane, J. Dhesi, S. Bergqvist and T. Brown, J. Mol. Biol., 1998, 275, 811 CrossRef CAS PubMed; (c) A. M. Soto, J. Loo and L. A. Marky, J. Am. Chem. Soc., 2002, 124, 14355 CrossRef CAS PubMed.
- W. Saenger, Principles of Nucleic Acid Structure, ed. C. R. Cantor, Springer–Verlag, New York, 1984, 143–149 Search PubMed.
- (a) A. Idili, K. W. Plaxco, A. Vallée-Bélisle and F. Ricci, ACS Nano, 2013, 7, 10863 CrossRef CAS PubMed; (b) Y. Chen, S.-H. Lee and C. Mao, Angew. Chem., Int. Ed., 2004, 43, 5335 CrossRef CAS PubMed.
- A. Idili, A. Vallée-Bélisle and F. Ricci, J. Am. Chem. Soc., 2014, 136, 5836 CrossRef CAS PubMed.
- W. D. Wilson, A. N. Gough, J. J. Doyle and M. W. Davidson, J. Med. Chem., 1976, 19, 1261 CrossRef CAS.
- A. A. Moraru-Allen, S. Cassidy, J.-L. A. Alvarez, K. R. Fox, T. Brown and A. N. Lane, Nucleic Acids Res., 1997, 25, 1890 CrossRef CAS PubMed.
- J. Ren and J. B. Chaires, Biochemistry, 1999, 38, 16067 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5sc00594a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2015 |