
Self organization of main-chain/side-chain liquid crystalline polymer based on “jacketing” effect with different lengths of spacer: from smectic to hierarchically ordered structure†
Abstract
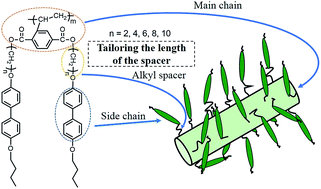
A series of combined main-chain/side-chain liquid crystalline polymers (MCSCLCP) based on “jacketing” effect, poly{(2,5-bis[n-(4-butoxy-4′-oxybiphenyl)n-alkyl]oxycarbonyl}styrene) with different lengths of alkyl spacers (denoted as Pn, n represents the number of carbon atoms in the alkyl spacers, n = 2–10) have been successfully synthesized via atom transfer radical polymerization (ATRP). The chemical structures of Pns and the corresponding monomers were characterized using combined techniques with satisfactory analysis data. The phase structures and transitions of Pn were investigated using differential scanning calorimetry (DSC), polarized optical microscope (POM), and one- and two-dimensional wide-angle X-ray diffraction (1D and 2D WAXD). It has been identified that P2 and P4 with short alkyl spacers form typical smectic phase. For n ≥ 6, Pns exhibit similar hierarchical ordered structure at low temperatures, bearing double orderings on the nanometer and sub-nanometer scales. In the hierarchical structure, the main-chains based on “jacketing” effect form a 2D centered rectangular scaffold, and the side-chain biphenyl mesogens within the scaffold pack into a smectic E-like structure. The a dimension of rectangular lattice enlarges with n. When the temperature is increased, different from P6, P8 and P10 present the same phase behavior, forming smectic B-like packing of side chains and maintaining their main-chain scaffold until isotropization.
 Please wait while we load your content...
Please wait while we load your content...

