
Multifunctional Zn(II)/Cd(II) metal complexes for tunable luminescence properties and highly efficient dye-sensitized solar cells†
Song Gaoa,
Rui Qing Fan*a,
Xin Ming Wanga,
Liang Sheng Qianga,
Li Guo Weia,
Ping Wanga,
Yu Lin Yang*a,
Yu Lei Wangb and
Tian Zhu Luanc
aDepartment of Chemistry, Harbin Institute of Technology, Harbin 150001, P. R. China. E-mail: fanruiqing@hit.edu.cn; ylyang@hit.edu.cn; Fax: +86-451-86413710
bNational Key Laboratory of Science and Technology on Tunable Laser, Harbin Institute of Technology, Harbin 150080, P. R. China
cThe First Affiliated Hospital of Harbin Medical University, Harbin, 150001, P. R. China
First published on 8th May 2015
Abstract
We design two novel d10 metal complexes [Zn(3-qlc)2]n (Zn1), [Cd(3-qlc)2]n (Cd1) (3-Hqlc = quinoline-3-carboxylic acid), which display similar 2D layer structures, and further form 3D 6-connected primitive cubic (pcu) network topology via π⋯π stacking interactions with the point symbol {412·63}. Complexes Zn1 and Cd1 exhibit tunable luminescence and photovoltaic properties as a new type of multifunctional materials. Zn1 and Cd1 show tunable luminescence from blue to green by varying the temperature in the solid state. What's more, both of them exhibit excellent aggregation-induced emission (AIE) properties in DMSO/water mixtures. Encouraged by the UV-visible absorption in an ethanol solution result, Zn1 and Cd1 can be considered as co-sensitizers in combination with N719 to investigate their effect on enhancing the dye-sensitized solar cells (DSSCs) performance. Zn1 and Cd1 could overcome the deficiency in the ruthenium complex N719 absorption in the ultraviolet and blue-violet region, offsetting competitive visible light absorption of I3− and reducing the charge recombination of injected electrons. After being co-sensitized with Zn1 and Cd1, the devices co-sensitized by Zn1/N719 and Cd1/N719 yield an overall efficiency of 7.75% and 7.70%, which are 44.59% and 43.66% higher than that of the device sensitized only by N719 (5.36%).
Introduction
The past two decades have witnessed a rapid development of metal coordination complexes (MCCs) due to their fantastic structures and wide potential applications.1–3 Recently, the design of MCCs with excellent luminescence and photovoltaic properties has attracted increasing attention in pursuit of multifunctional materials.4–6 The main bottleneck in improving the efficiency of photovoltaic energy conversion lies in the spectral mismatch between the energy distribution of photons in the incident solar spectrum and the band-gap of a semiconductor material.7,8 Multiple sensitizers taking advantage of MCCs as co-sensitizers would appear to be more achievable in improving the absorption ability of solar cells.9,10 As a typical representative of DSSC sensitization, N719 shows low utilization in the ultraviolet and blue-violet light region, and research about light utilization in high energy blue-violet light region has been little reported.11 Some MCCs as co-sensitizer or co-adsorbent in DSSCs not only compensate the absorption spectra but also overcome the competitive light absorption by I−/I3− and reduce charge recombination, which will enhance the DSSCs performance by co-sensitization.12,13 Consequently, the reasonable design of MCCs as co-sensitizers for high-efficiency DSSC is still a big challenge and of great importance.The key to design MCCs materials is the matching of organic ligand and central metal ion which can provide the platform to generate ultraviolet absorption and luminescence.14–17 Quinoline-3-carboxylic acid (3-Hqlc) as N-heterocyclic carboxylate ligand has attracted increasing attention in the preparation of interesting polymeric frameworks for their outstanding features of versatile coordination fashions.18 It has a relatively large π-conjugated system in the quinoline ring, which not only contribute to the desirable ultraviolet absorption and luminescence properties resulting from the interaction between 3-qlc− anions and metal ions, but also easily assemble into high dimensional supramolecular network via C–H⋯π/π⋯π stacking interactions between two adjacent aromatic rings as well as hydrogen bonding interactions, adding additional stability to these structures.19
The enormous variability of available ligand–metal combinations opens the possibility of creating luminescence “by design”. The d10 metal complexes have gained intensive attention because of relatively low-cost, intriguing structures and adequate electron transport, which are advantageous to enhance the luminescence efficiency.20,21 On the basis of the above considerations, we aim to synthesize d10 metal complexes with strong absorption in ultraviolet and blue-violet light region, in order to replace noble metal complexes (metal osmium(II), platinum(II) and ruthenium(II) MCCs doped semiconductors) as co-sensitizers.22–24
Herein, we report the design and synthesis of two new d10 metal complexes, [Zn(3-qlc)2]n (Zn1), [Cd(3-qlc)2]n (Cd1). Zn1 and Cd1 exhibit luminescence with emission maxima containing blue and green at 298 K and 77 K, respectively, in the solid state. Zn1 and Cd1 display excellent aggregation-induced emission (AIE) properties in the solid state, which show weak emission in DMSO (good solvent) but are brightly luminescent by the addition of water (poor solvent) due to aggregation. Therefore, Zn1 and Cd1 offer great opportunities for the design of luminescence solid films materials.25,26 In addition, as a result of their strong absorption in the wavelength range 300–400 nm, Zn1 and Cd1 are employed as co-sensitizers with the well-known ruthenium N719 dye based solar cell, and the co-sensitized devices exhibited enhancements of photovoltaic performance. It showed that the absorption spectra of complexes Zn1 and Cd1 could compensate that of N719 absorption in the region of ultraviolet and blue-violet, the light harvesting was enhanced. The power conversion efficiency of N719 sensitized DSSC is enhanced by 44.59% and 43.66% after co-sensitized with Zn1 and Cd1 than that for DSSCs using single N719 (5.36%), which indicating that Zn1 or Cd1 is a promising candidate as co-sensitizer for high efficiency DSSCs and does help to improve the devices performance. Hence, this strategy may provide interesting insight into the development of high-performance DSSCs devices.
Experimental section
General characterization
All materials and reagents were commercially available and used without further purification. IR spectra were obtained from KBr pellets using a Nicolet Avatar-360 infrared spectrometer in the 4000–400 cm−1 region. Elemental analyses (C, H and N) were performed on a Perkin-Elmer 240c element analyzer. The thermal analysis was performed on a ZRY-2P thermogravimetric analysis from 30 °C to 700 °C with heating rate of 10 °C min−1 under a flow of air. UV-visible spectra were obtained using a Perkin-Elmer Lambda 20 spectrometer. Powder X-ray diffraction (PXRD) patterns were PANalytical, monochromated Cu Kα radiation 40 mA, 40 kV. The solid-state and solution luminescence analyses were carried out on an Edinburgh FLS920 fluorescence spectrometer in the range of 200–800 nm. An Edinburgh Xe900 400 W xenon is lamp was used as exciting light source. The visible detector as well as the lifetime setup was red-sensitive photomultiplier (type r928). Low temperature analyses were carried out at 77 K with an Oxford Optistat DN™ cryostat (with liquid nitrogen filling). Lifetime studies were performed using photon-counting system with a microsecond pulse lamp as the excitation source. Data were analyzed through the nonlinear least squares procedure in combination with an iterative convolution method. The emission decays were analyzed by the sum of exponential functions. The decay curve is well fitted into a double exponential function: I = I0 + A1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) exp(−t/τ1) + A2exp(−t/τ2), where I and I0 are the luminescent intensities at time t = t and t = 0, respectively, whereas τ1 and τ2 are defined as the luminescent lifetimes. The average lifetime was calculated according to the following equation:
exp(−t/τ1) + A2exp(−t/τ2), where I and I0 are the luminescent intensities at time t = t and t = 0, respectively, whereas τ1 and τ2 are defined as the luminescent lifetimes. The average lifetime was calculated according to the following equation: 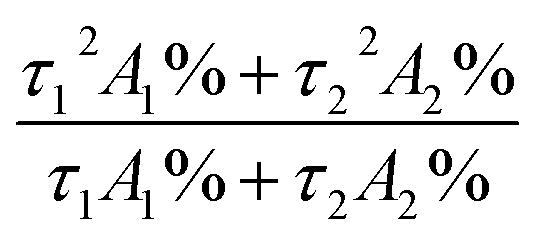 . For the solid samples, the absolute quantum yields have been measured by integrating sphere. The cyclic voltammetry (CV) were measured with a electrochemical workstation (CHI660D, Chenhua, Shanghai) using a three-electrode cell with a Pt working electrode, a Pt wire auxiliary electrode, and a saturated calomel reference electrode in saturated KCl solution. The supporting electrolyte was 0.1 M tetrabutylammonium hexafluorophosphate (TBAPF6, Fluka, electrochemical grade) in ethanol as the solvent. Photocurrent–photovoltage (I–V) curves were recorded by Keithley model 2400 digital source meter using a mask with an aperture area of 0.16 cm−2. The irradiance of AM1.5 global sunlight from a filtered 500 W xenon lamp light source was set at 100 mW cm−2 and was calibrated by a standard silicon solar cell. Based on I–V curve, the fill factor (FF) is defined as: FF = (Jmax × Vmax)/(Jsc × Voc), where Jmax and Vmax are the photocurrent density and photovoltage for maximum power output; Jsc and Voc are the short-circuit photocurrent density and open-circuit photovoltage, respectively. The overall energy conversion efficiency η is defined as: η = (FF × Jsc × Voc)/Pin where Pin is the power of the incident light. IPCE spectra were recorded with Monochromatic incident photon-to-electron conversion efficiency (IPCE) were measured on an EQE/IPCE spectral response system (Newport). Electrochemical impedance spectroscopy (EIS) was recorded by CHI660D Electrochemical Analyzer (Chenhua, China), and the measurements were taken over a frequency range of 0.1–100 kHz under standard global AM1.5 solar irradiation or in the dark by applying a forward bias equivalent to Voc.
. For the solid samples, the absolute quantum yields have been measured by integrating sphere. The cyclic voltammetry (CV) were measured with a electrochemical workstation (CHI660D, Chenhua, Shanghai) using a three-electrode cell with a Pt working electrode, a Pt wire auxiliary electrode, and a saturated calomel reference electrode in saturated KCl solution. The supporting electrolyte was 0.1 M tetrabutylammonium hexafluorophosphate (TBAPF6, Fluka, electrochemical grade) in ethanol as the solvent. Photocurrent–photovoltage (I–V) curves were recorded by Keithley model 2400 digital source meter using a mask with an aperture area of 0.16 cm−2. The irradiance of AM1.5 global sunlight from a filtered 500 W xenon lamp light source was set at 100 mW cm−2 and was calibrated by a standard silicon solar cell. Based on I–V curve, the fill factor (FF) is defined as: FF = (Jmax × Vmax)/(Jsc × Voc), where Jmax and Vmax are the photocurrent density and photovoltage for maximum power output; Jsc and Voc are the short-circuit photocurrent density and open-circuit photovoltage, respectively. The overall energy conversion efficiency η is defined as: η = (FF × Jsc × Voc)/Pin where Pin is the power of the incident light. IPCE spectra were recorded with Monochromatic incident photon-to-electron conversion efficiency (IPCE) were measured on an EQE/IPCE spectral response system (Newport). Electrochemical impedance spectroscopy (EIS) was recorded by CHI660D Electrochemical Analyzer (Chenhua, China), and the measurements were taken over a frequency range of 0.1–100 kHz under standard global AM1.5 solar irradiation or in the dark by applying a forward bias equivalent to Voc.
Fabrication of devices
The FTO conducting glass (fluorine-doped SnO2, sheet resistance 15 Ω per square, transmission 90% in the visible) was purchased from NSG, Japan, and cleaned by a standard procedure. N719 [cis-bis(isothiocyanato)bis(2,2-bipyridyl-4,4-dicarboxylato)-ruthenium(II)bis-tetrabutylammonium] was purchased from Solaronix Company, Switzerland. Dye-sensitized solar cells were fabricated using the following procedure. The TiO2 paste was cast onto the FTO substrate by the screen-printing method, followed by drying at 100 °C for 5 min and this process was repeated for six times, then followed by sintering at 500 °C for 15 min in air to obtain a transparent TiO2 photoelectrode with the thickness of ca. 10 μm. The co-adsorbent electrodes were prepared by immersing the obtained mesoporous TiO2 photoelectrode into 0.3 mM Zn1 or Cd1 solution in absolute ethanol for 2 h and washed with ethanol and dried with blower, then further immersing the electrodes in 0.3 mM N719 solution in absolute ethanol for 12 h, and then washed with ethanol and dried with blower again. The single N719 sensitized electrodes were prepared by only immersing TiO2 photoelectrode into 0.3 mM N719 solution in absolute ethanol for 14 h. The electrolyte used in this work was 0.5 M LiI + 0.05 M I2 + 0.1 M tert-butyl pyridine in a 1:1 (volume ratio) of acetonitrile–propylene carbonate. The platinum counter electrode was prepared by depositing H2PtCl6 paste onto the FTO glass substrates and then sintered at 450 °C for 30 min. The cells were assembled by sandwiching the electrolyte between the dye-sensitized photoanode and the counter electrode and assembly was held together using mini-binder clips.
Synthesis of [Zn(3-qlc)2]n (Zn1)
A mixture of Zn(NO3)2·6H2O (59.5 mg, 0.2 mmol), 3-Hqlc (17.3 mg, 0.1 mmol) were dissolved in H2O (5 mL). After stirring for 30 minutes in air, it was transferred into a 15 mL Teflon-lined stainless steel autoclave and heated at 165 °C for 6 days. Then slow cooling to room temperature, yellow block crystals of Zn1 were obtained in 72% yield (based on Zn). Anal. calcd for C20H12N2O4Zn (Mr: 409.71): C, 58.63; H, 2.95; N, 6.84%. Found: C, 58.60; H, 2.96; N, 6.85%. IR (KBr pellet, cm−1, Fig. S1†) for Zn1: 3438(br, s), 1617(vs), 1572(m), 1522(m), 1458(w), 1373(s), 1305(m), 1269(w), 1242(w), 1206(w), 1174(w), 1115(m), 1066(m), 961(m), 844(m), 781(m), 699(w), 659(m), 582(m), 532(w), 496(w).Synthesis of [Cd(3-qlc)2]n (Cd1)
Complex Cd1 was obtained by the same hydrothermal procedure as that for preparation of Zn1 except that the metal salt was replaced by Cd(NO3)2·4H2O (61.6 mg, 0.2 mmol). After the reaction mixture was cooled down to room temperature, yellow platelet crystals of Cd1 were collected with a yield of 68% (based on Cd). Anal. calcd for C20H12N2O4Cd (Mr: 456.73): C, 52.60; H, 2.65; N, 6.13%. Found: C, 52.63; H, 2.64; N, 6.12%. IR (KBr pellet, cm−1, Fig. S1†) for Cd1: 3442(br, s), 1621(vs), 1580(m), 1549(m), 1504(m), 1468(s), 1409(vs), 1386(s), 1309(s), 1278(w), 1206(m), 1151(m), 1029(w), 961(m), 898(m), 790(s), 745(m), 663(w), 596(m), 541(w), 482(w).X-ray crystal structure determination
The X-ray diffraction data taken at room temperature for complexes Zn1 and Cd1 were collected on a Rigaku R-AXIS RAPID IP diffractometer equipped with graphite-monochromated Mo Kα radiation (λ = 0.71073 Å). The structures of Zn1 and Cd1 were solved by direct methods and refined on F2 by the full-matrix least squares using the SHELXTL-97 crystallographic software.27,28 Anisotropic thermal parameters are refined to all of the non-hydrogen atoms. The hydrogen atoms were held in calculated positions on carbon atoms and nitrogen atoms.† Crystal structure data and details of the data collection and the structure refinement are listed as Table 1, selected bond lengths and bond angles of Zn1 and Cd1 are listed as Table S1.†| Identification code | Zn1 | Cd1 |
|---|---|---|
| a R1 = ∑||Fo| − |Fc||/∑|Fo.b wR2 = [∑[w(Fo2 − Fc2)2]/∑[w(Fo2)2]]1/2. | ||
| Empirical formula | C20H12N2O4Zn | C20H12N2O4Cd |
| Formula mass | 409.71 | 456.73 |
| Crystal system | Monoclinic | Monoclinic |
| Space group | C2/c | C2/c |
| a (Å) | 28.967(6) | 28.893(6) |
| b (Å) | 8.053(16) | 8.239(16) |
| c (Å) | 15.285(3) | 15.421(3) |
| α (°) | 90.00 | 90.00 |
| β (°) | 114.27(3) | 113.10(3) |
| γ (°) | 90.00 | 90.00 |
| V (Å3) | 3250.4(11) | 3376.5(12) |
| Z | 8 | 8 |
| Dc/(g cm−3) | 1.674 | 1.797 |
| μ (Mo Kα)/mm−1 | 1.542 | 1.324 |
| F(000) | 1664 | 1808 |
| θ range (°) | 3.09–27.47 | 3.07–27.48 |
| Limiting indices | −37 ≤ h ≤ 36 | −33 ≤ h ≤ 37 |
| −10 ≤ k ≤ 10 | −9 ≤ k ≤ 10 | |
| −19 ≤ l ≤ 19 | −19 ≤ l ≤ 20 | |
| Data/restraints/parameters | 3716/0/244 | 3866/0/244 |
| GOF on F2 | 1.022 | 1.194 |
| Final R indices [I > 2σ(I)] | ||
| R1a | 0.0469 | 0.0305 |
| wR2b | 0.0966 | 0.0706 |
| R indices (all data) | ||
| R1 | 0.0782 | 0.0375 |
| wR2 | 0.1082 | 0.0735 |
| CCDC | 1055702 |
1055703 |
Results and discussion
Structure description of [Zn(3-qlc)2]n (Zn1) and [Cd(3-qlc)2]n (Cd1)
The asymmetric unit consists of one Zn(II) or Cd(II) cation, two 3-qlc− anions (Fig. 1a and b) for Zn1 and Cd1. However, Zn(II) site takes the four-coordinated tetrahedral coordination geometry and the Cd(II) site takes the six-coordinated slightly distorted octahedron coordination geometry (Fig. 1c and d). Comparison of Zn(II) to Cd(II), they both have d10 full-filled electron orbit, the difference is that Zn(II) has 3d10 as outer shell while Cd(II) has 4d10 as outer shell. Accordingly, the Cd(II) ionic radius is larger than that of Zn(II) which leads to form the covalent bonds in different bond strength, bond lengths and bond angles with the same organic ligands. As a consequence, even if the complexes formed by Cd(II) and Zn(II) have the same ligands and composition, they crystallize in the different structures. X-ray analysis reveals that the hexacoordinated Cd(II) cation is connected with four oxygen atoms and two nitrogen atoms from two individual 3-qlc− anions to give a [CdN2O4] coordination geometry. The central Zn(II) cation is tetracoordinated and forms a distorted tetrahedral geometry, completed by two oxygen atoms and two nitrogen atoms from two individual 3-qlc− anions. Four oxygen atoms display the bidentate-chelate mode to connect center metal Cd(1). Two oxygen atoms adopt a monodentate fashion providing one sites to bond to center metal Zn(1). Nitrogen atoms from two quinoline rings display the ηN1 mode to connect metal center Cd(1)/Zn(1). Among them, Cd–O bond lengths range from 2.164(2) Å to 2.765(2) Å, Cd(1)–N1 and Cd(1)–N2 bond lengths are 2.332(2) Å and 2.348(2) Å. O–Cd–O bond angles range from 56.1(7)° to 158.9(9)°, N–Cd–N bond angle is 97.3(8)°, O–Cd–N bond angles are in the range of 96.5(8)–152.6(9)°. Zn(1)–O1 and Zn(1)–O3 bond lengths are 1.968(3) Å and 1.927(2) Å, Zn(1)–N1 and Zn(1)–N2 bond lengths are 2.106(3) Å and 2.097(3) Å. O–Zn–O bond angle is 138.4(1)°, N–Zn–N bond angle is 100.9(1)°, O–Zn–N bond angles are in the range of 100.3(1)–110.2(1)°. Zinc cations are connected through one 3-qlc− anion giving rise to an infinite 1D chain (Fig. 1e, as an example of Zn1). Two adjacent 1D chains are combined by other 3-qlc− anion forming 2D layer structure (Fig. 1f).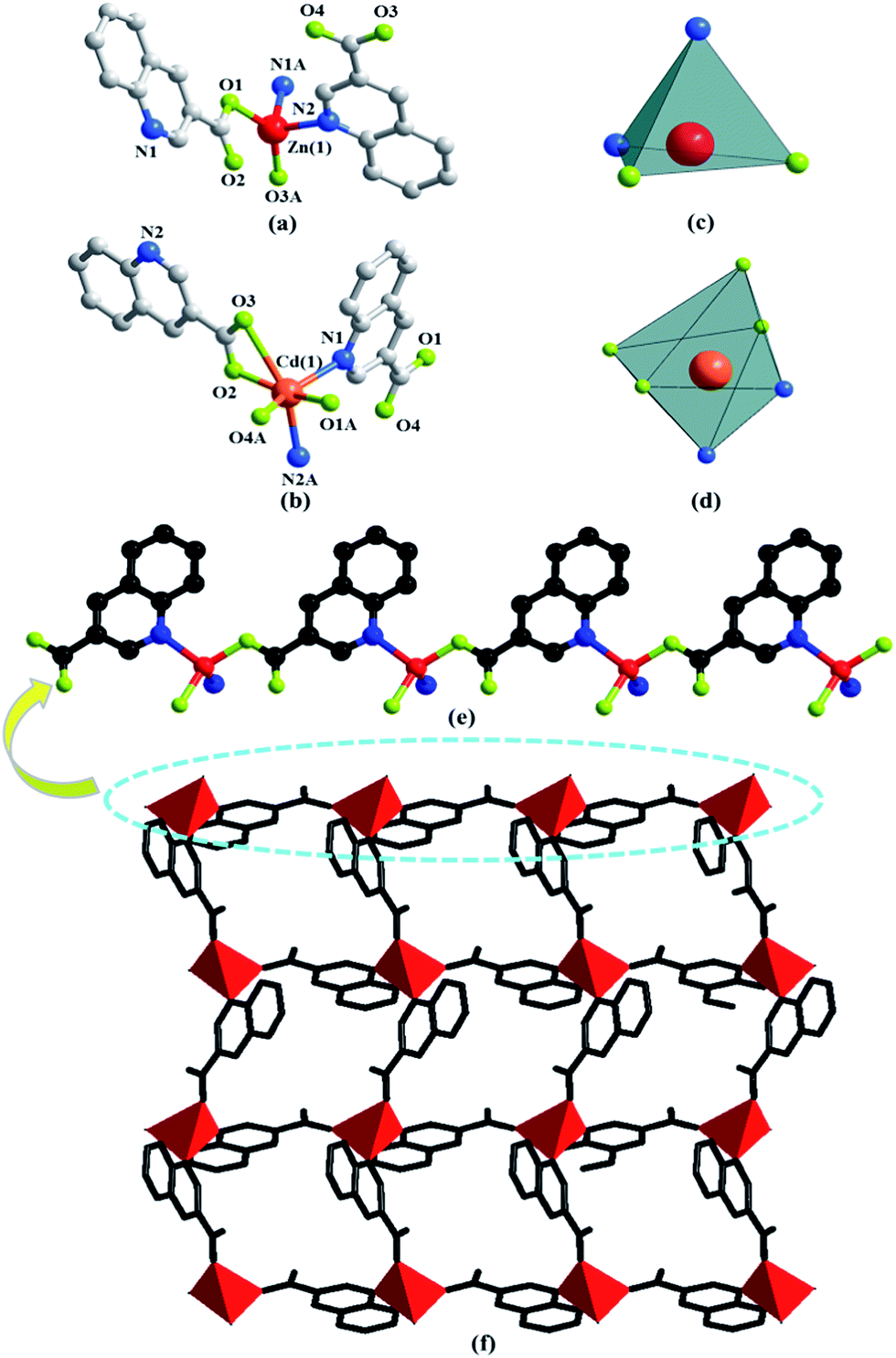 | ||
| Fig. 1 The molecular structures of complex Zn1 (a) and complex Cd1 (b). All hydrogen atoms have been omitted for clarity. (c) Polyhedral representation of the coordination sphere of the Zn(1) centre in Zn1. (d) Polyhedral representation of the coordination sphere of the Cd(1) centre in Cd1. (e) Ball-and-stick representation of the 1D chain structure in Zn1. (f) The illustration of 2D layer structure in Zn1. | ||
Moreover, inter-layer π⋯π stacking interaction with the distance of 3.479(2) Å and 3.506(1) Å between the centers of two quinoline rings, links the adjacent coordination layers to form a 3D network (Fig. 2a). Topological analysis has been applied for better understanding the connectivity of 3D framework in Zn1. When we consider the Zn(II) cation as the 6-connected node, the 3-qlc− anions and two kinds of inter-layer π⋯π stacking interactions as the linkers, the simplified topological representation of Zn1 can be described in Fig. 2b, which exhibits a 3D 6-connected primitive cubic (pcu) topological type with point symbol (412·63) and vertex symbol (4·4·4·4·4·4·4·4·4·4·4·4·*·*·*).
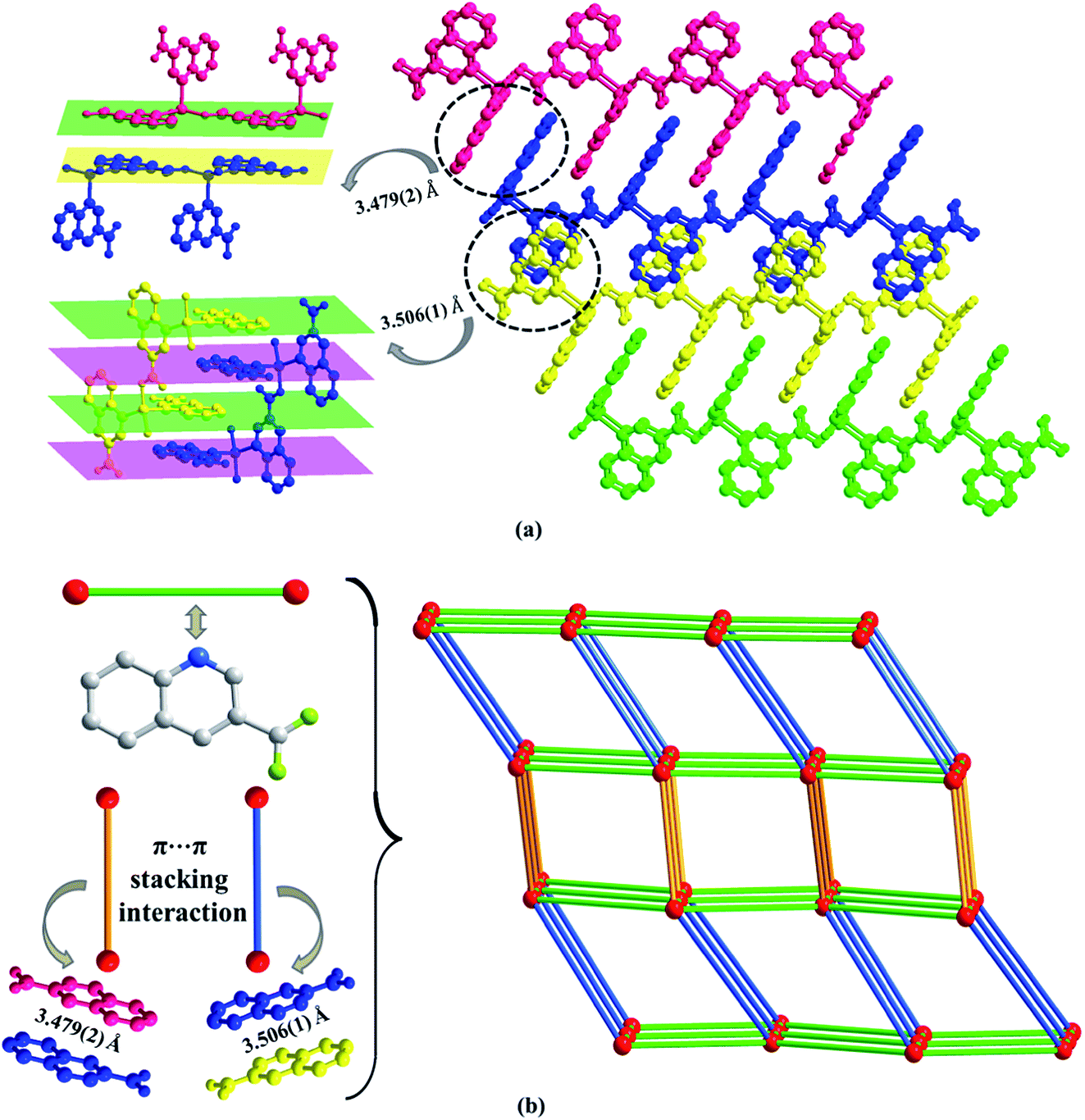 | ||
| Fig. 2 (a) 3D supramolecular network constructed by inter-layer π⋯π interaction in Zn1. (b) The 3D pcu topology in its most symmetrical form distinguished by different colors (color code: Zn(II) cation, red ball; 3-qlc− anions, green line; inter-layer π⋯π stacking interaction with the distance of 3.479(2) Å, golden line; inter-layer π⋯π stacking interaction with the distance of 3.506(1) Å, blue line). | ||
Thermal gravimetric analysis and powder X-ray diffraction patterns
To characterize the complexes in terms of thermal stability, thermal gravimetric analysis (TGA) of Zn1 and Cd1 were carried out from room temperature to 700 °C under a flow of air (Fig. S2†). Zn1 shows the mass loss of 81.76% in the range 376–488 °C, corresponding to the release of two 3-Hqlc ligands (calcd 84.04%), and the main framework has collapse. The remaining residue of 18.24% is consistent with the formation of ZnO (calcd 19.86%). In Cd1, the weight loss of 72.81% from 369 °C to 461 °C corresponds to the loss of two 3-Hqlc ligands (calcd 75.39%), and the host framework has collapse. The remaining residue of 27.19% (calcd 28.12%) is consistent with the formation of CdO.29,30 Powder X-ray diffraction (PXRD) was used to check the purity of complexes Zn1 and Cd1 (Fig. S3†). All the peaks displayed in the measured patterns are similar to those in the simulated patterns generated from single-crystal diffraction data, indicating single phases of Zn1 and Cd1 are formed. The differences in intensity may be owing to the preferred orientation of the powder samples.31Luminescence properties
The d10 metal ions have drawn much attention in the construction of luminescent complexes not only for their variable surroundings but also for their changeable luminescent features by adopting different coordination modes, connection or interaction with functional ligands and the change of temperature.32,33 The emission spectral data along with the lifetimes for complexes Zn1 and Cd1, both in solution and in the solid state, are summarized in Table S2.† The luminescent spectra of Zn1, Cd1 were measured at 298 K and 77 K in the solid state (Fig. 3a). The maximum emission band for Zn1 is located at 468 nm (450 nm for Cd1), exhibiting blue luminescent emissions at 298 K. These luminescent emissions may be attributed to the ligand-centered π* → π transition (Fig. S4†).34 The difference in luminescent details between Zn1 and Cd1 may be assigned to the different coordinating surroundings of the metal atoms and their distinct structure features. At 77 K, Zn1 has higher-energy emission of one shoulder-peaks appearing around 483sh. The maximum emission band for Zn1 is located at 510 nm and the lower-energy emission bond centered at 571sh nm (449sh, 485, 542sh nm for Cd1), which are in the green region. The luminescence quantum yields (Φ) of complexes Zn1 and Cd1 were determined by means of an integrating sphere at 298 K and 77 K under the excitation wavelengths. The solid state measurements gave luminescence quantum yields of 11.68% for Zn1 and 11.03% for Cd1 at 298 K. The corresponding Φ value of Cd1 is smaller than that of Zn1, because of the heavy-atom effect. When measurement temperature is reduced to 77 K, the quantum yield mounts up to 16.46% for Zn1 and 15.71% for Cd1. Meanwhile, the lifetimes of Zn1 and Cd1 increase conspicuously compared to those at 298 K (298 K: τ = 7.76 μs for Zn1, τ = 7.50 μs for Cd1; 77 K: τ = 10.76 μs for Zn1, τ = 9.91 μs for Cd1) (Fig. 3b). This is most likely due to the fact that cold conditions should be favorable for the rigidity of ligands with reducing the non-radiation decay and collisional quenching. Thereby the increase of quantum yields and the lifetimes could be observed eventually at lowering the temperature.35,36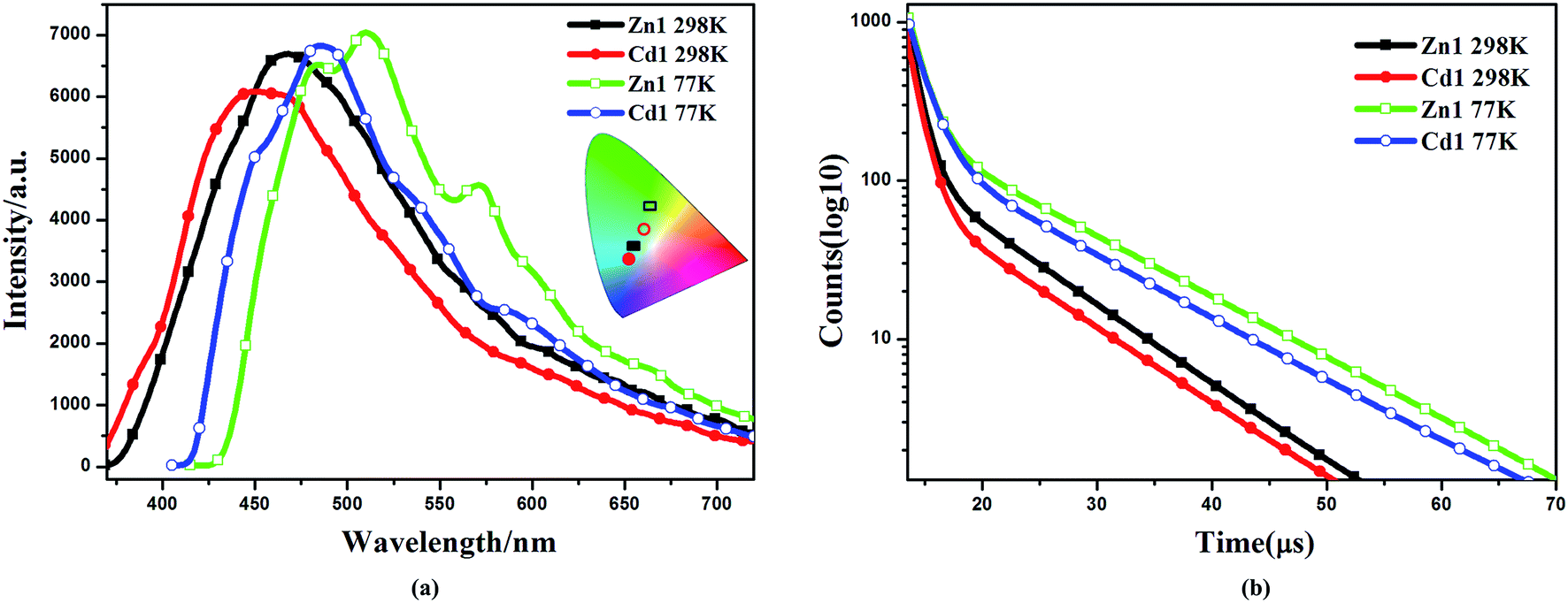 | ||
| Fig. 3 (a) Emission spectra of complexes Zn1 and Cd1 at 298 K, 77 K in solid state and the corresponding color coordinate diagram of emission. (b) The decay curves of complexes Zn1 and Cd1 at 298 K and 77 K in solid state. | ||
The obtained Zn1 and Cd1 readily dissolve in common organic solvents, such as dimethyl sulfoxide (DMSO), methanol (CH3OH), chloroform (CHCl3) and tetrahydrofuran (THF). The maximum emission wavelengths of the Zn1 and Cd1 are red-shifted by ∼10 nm (409 nm in THF, 412 nm in CHCl3 and 418 nm in CH3OH for Zn1; 405 nm in THF, 407 nm in CHCl3 and 412 nm in CH3OH for Cd1) in the corresponding solvents, and all the studied moieties generated blue emission with CIE coordinates of 0.18, 0.14–0.20, 0.21 (Fig. 4). As solvent polarity increases (polarity: THF < CHCl3 < CH3OH < DMSO), the emission intensity of the complexes gradually diminishes. In particular, the emission spectrum of Zn1 and Cd1 becomes broader and weaker in the DMSO solvent (424 nm and 418 nm), with CIE coordinates of 0.19, 0.20 and 0.19, 0.16.
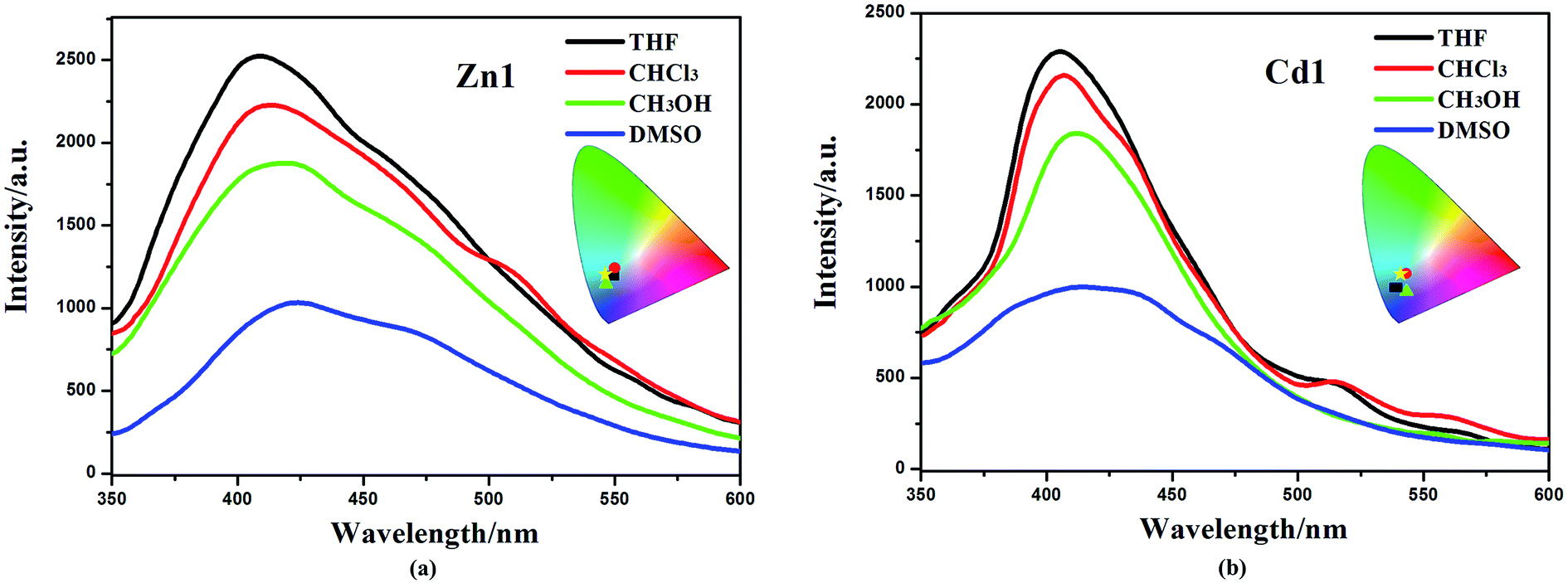 | ||
| Fig. 4 Emission spectra of Zn1 (a) and Cd1 (b) in different solvents (excited at 330 nm) and the corresponding color coordinate diagram of emission (rectangle: THF; roundness: CHCl3; triangle: CH3OH; pentacle: DMSO). | ||
In addition, Zn1 and Cd1 are insoluble in water (H2O). The aggregated particles should be precipitated as the fraction of water is increased. The emission spectra of Zn1 and Cd1 in DMSO/water mixtures with different water content (0–99%) were obtained to verify if the Zn1 and Cd1 displayed AIE properties. This is done by adding different amount of water to the pure DMSO solutions to obtain water fraction (fw) of 0–99% and monitored the changes of luminescence. The concentration was maintained at 10−5 M. At every increment of water fraction (fw), the emission spectrum is collected. All f values are volume/volume percentages. As shown in Fig. 5, there is a clear trend that Zn1 and Cd1 are weak emissive in DMSO solution, but when they aggregates due to the addition of water, as a poor solvent, the emission dramatically increase after a large amount of water have been added. The luminescence enhancement from the solution state to the aggregated state is defined by the AIE factor (αAIE): αAIE,I = Iaggr/Isoln, where Iaggr and Isoln are the luminescence intensity of the aggregates and solution, respectively. The luminescence intensity of Zn1 and Cd1 in the DMSO/water mixture with 99 vol% H2O is 6.06-fold and 5.43-fold higher than that in DMSO solution, respectively.37 This is most likely due to the fact, when the molecules are aggregated; such free rotations are hindered because of the short distances among the crowded molecules. Thus, the nonradiative energy consuming pathway is blocked and strong emission is vitalized. For Zn1 and Cd1: the building blocks of the crystals are not the monomers, but rather the dimers, based on the intermolecular π⋯π stacking interactions between adjacent carboxylic acid groups and quinoline rings, this structure further increases the rigidity of the molecules to generate a more extended planar skeleton, which restricts the intermolecular rotations, and may extend the conjugation system by incorporating the carboxylate oxygen and nitrogen atoms to the 3-Hqlc ligand, and thus might influence the luminescent. Zn1 and Cd1 as new AIE materials show advantages of high sensitivity, and may shed light on the development of new probes for luminescent applications.38
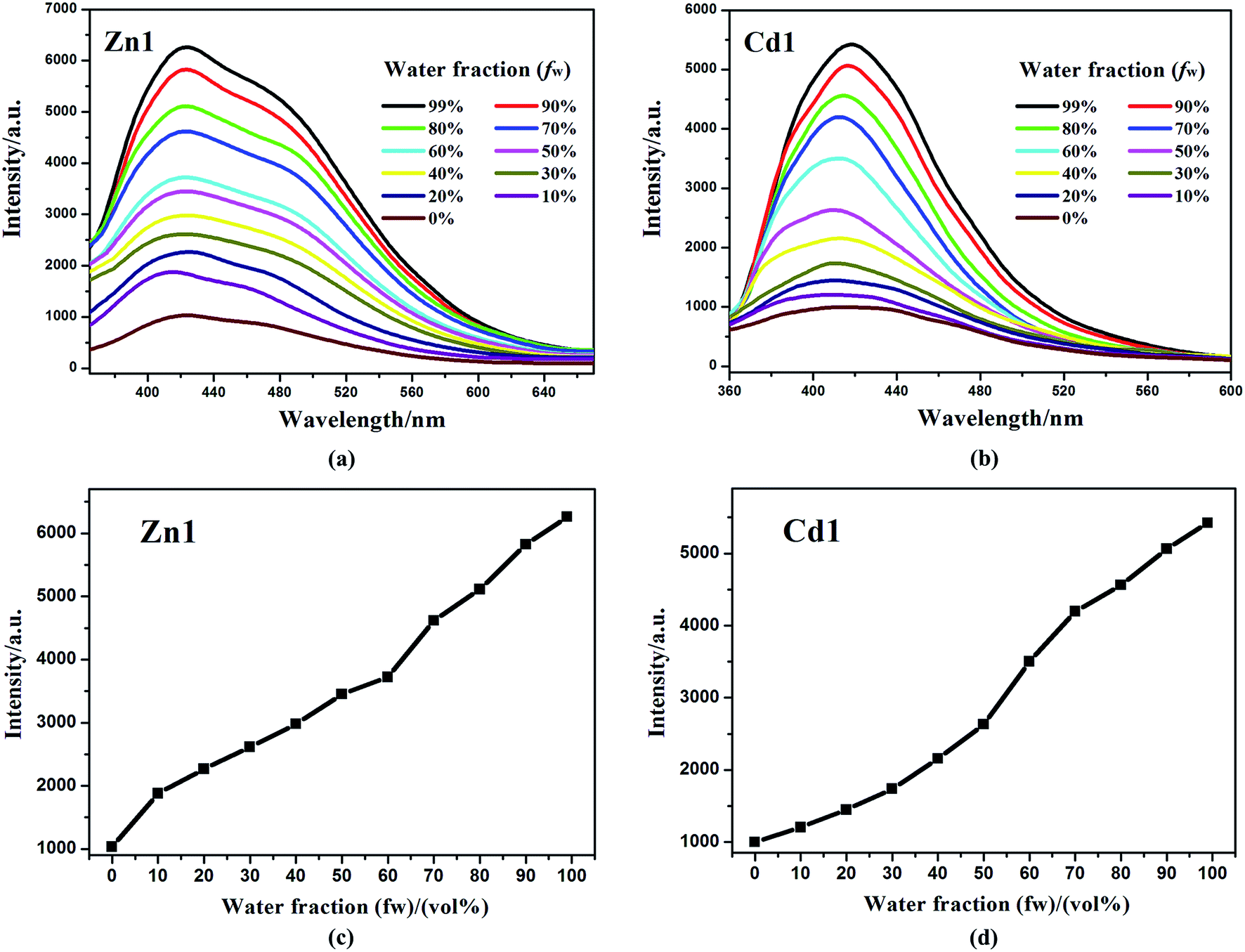 | ||
| Fig. 5 Emission spectra of Zn1 (a) and Cd1 (b) in DMSO/water mixtures with different water fractions (fw) at 298 K under excitation at 330 nm. (c) The changes in the luminescence peak intensity with different water fractions of Zn1. (d) The changes in the luminescence peak intensity with different water fractions of Cd1. | ||
Application in DSSCs
The UV-visible absorption spectra of Zn1, Cd1 and N719 in ethanol solution are displayed in Fig. 6, and the corresponding data are listed in Table 2. Two main absorption bands for Zn1 and Cd1 can be observed. The two prepared complexes display weak absorption bands at around 325 nm with strong absorption peaks at ca. 386 nm. Encouraged by the UV-visible absorption result, Zn1 and Cd1 can be considered as co-sensitizers used in N719 sensitized DSSCs. The molar extinction coefficients in the blue-violet region are 51079 M−1 cm−1 for Zn1 and 48693 M−1 cm−1 for Cd1. All of these are much higher than that of the ruthenium complex N719.39 A higher molar extinction coefficient indicates that the Zn1 or Cd1 possesses a higher light harvesting ability in this wavelength region compared with N719 and I3− (25000 M−1 cm−1).40 Hence it can be predicted that the photon lost due to the light absorption by I3− will be suppressed by the use of Zn1 or Cd1 as a co-sensitizer due to the competition between Zn1 or Cd1 and I3− to absorb light.
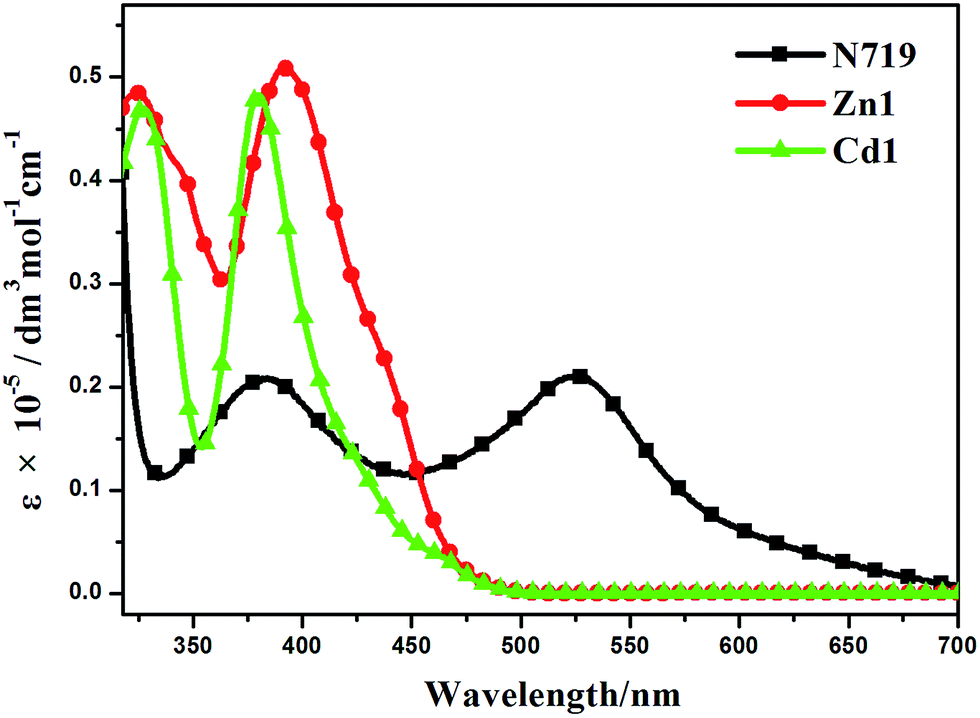 | ||
| Fig. 6 UV-visible absorption spectra of Zn1, Cd1 and N719 in ethanol. | ||
| Dyes | λabsa (nm) | εa (M−1 cm−1) | λema,b (nm) | E0–0c (eV) | Eox/V vs. SCEd | EHOMOe (eV) | ELUMOe (eV) |
|---|---|---|---|---|---|---|---|
| a Absorption and emission spectra were recorded in ethanol solution (10−5 M) at room temperature.b Complexes were excited at their absorption maximum value.c Optical band gap calculated from intersection between the absorption and emission spectra.d The first oxidation potentials of complexes were obtained by CV measurement.e The values of EHOMO and ELUMO were calculated with the following formula: HOMO (eV) = −e(Eoxonset V + 4.4 V); LUMO (eV) = EHOMO + E0–0 where E0–0 is the intersection of absorption and emission of the complexes Zn1 and Cd1. | |||||||
| Zn1 | 392 | 51079 |
409 | 3.11 | 0.84 | −5.24 | −2.13 |
| Cd1 | 380 | 48693 |
406 | 3.17 | 0.93 | −5.33 | −2.16 |
Energy matching is crucial in selecting sensitizers for DSSCs. Therefore, in order to evaluate if there are favorable energy offsets of the prepared complexes with respect to the TiO2 film and the electrolyte, the HOMO and LUMO energy levels of Zn1 and Cd1 were investigated by cyclic voltammetry (CV) measurement in a three-electrode cell and an electrochemistry workstation. The experimental data are summarized in Table 2. As estimated from the intersection of the absorption and emission spectra, the excitation transition energy (E0–0) of Zn1 and Cd1 are 3.11 and 3.17 eV, respectively. Consequently, the HOMO values of Zn1 and Cd1 are calculated based on their first redox potentials as −5.24 and −5.33 eV, respectively, and the LUMO levels of Zn1 and Cd1 calculated from EHOMO + E0–0, are −2.13 and −2.16 eV, respectively.41 The HOMO and LUMO energy levels of Zn1 and Cd1 are shown in Scheme 1. It shows that the energy levels of the prepared complexes are appropriate for the DSSCs system containing TiO2. The LUMO levels lied above the conduction band of the TiO2 semiconductor (−4.40 eV vs. vacuum), indicating efficient electron injection, and the HOMO energy levels lied below the I−/I3 redox electrolyte (−4.60 eV vs. vacuum) which is further improved negatively about 0.3 V by adding additives such as 4-tert-butyl pyridine (TBP) to the I−/I3− redox electrolyte, providing sufficient driving force for dye regeneration.42
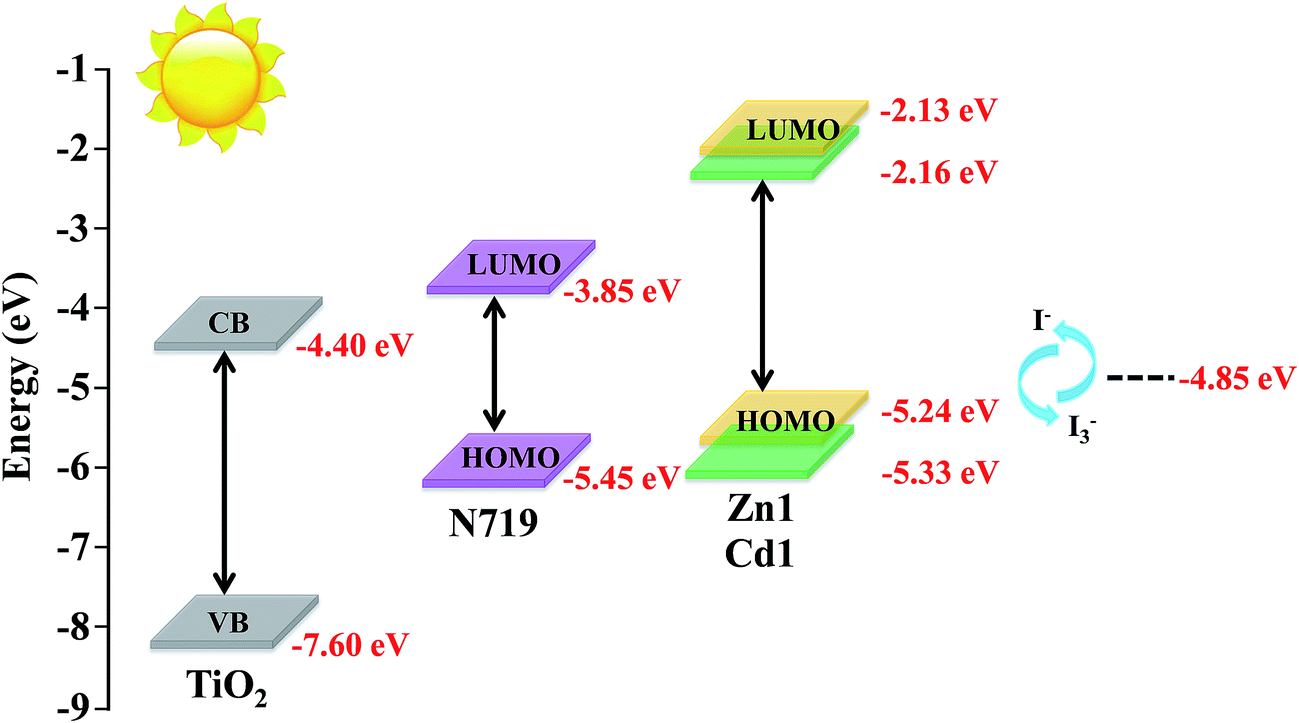 | ||
| Scheme 1 Schematic energy diagram of HOMO and LUMO for dyes compared to the energy levels calculated for TiO2. | ||
Fig. 7 presents the performances of the co-sensitized DSSCs in terms of Jsc and Voc, and the results of all DSSCs are list in Table 3. The result indicates that the photochemical performance of co-sensitized photoelectrodes has been enhanced. The present work indicated that d10 metal coordination complexes could affect the photochemical performance of DSSCs due to proton number. Fig. 7a and Table 3 indicate that the performances of cells co-sensitized with Zn1 or Cd1 and N719 are improved in the order of Zn1/N719 > Cd1/N719 > N719. As shows in Table 3, the co-sensitized solar cell based on a Zn1/N719/TiO2 and Cd1/N719/TiO2 electrode yield a remarkably high photocurrent density (Jsc) of 18.61 mA cm−2 and 18.36 mA cm−2 under standard global AM1.5 solar irradiation conditions without an expense of lower open circuit voltage (Voc). The cell shows a better photo-electricity conversion efficiency of 7.75% and 7.70%, which is higher than that for DSSCs using single N719 (5.36%). In this way the cell efficiency was improved by more than 44.59% and 43.66% compared with that for single N719. This result suggests that co-sensitization of TiO2 photoelectrode with the Zn1 or Cd1 and N719 is an effective way to improve the efficiency of DSSCs.
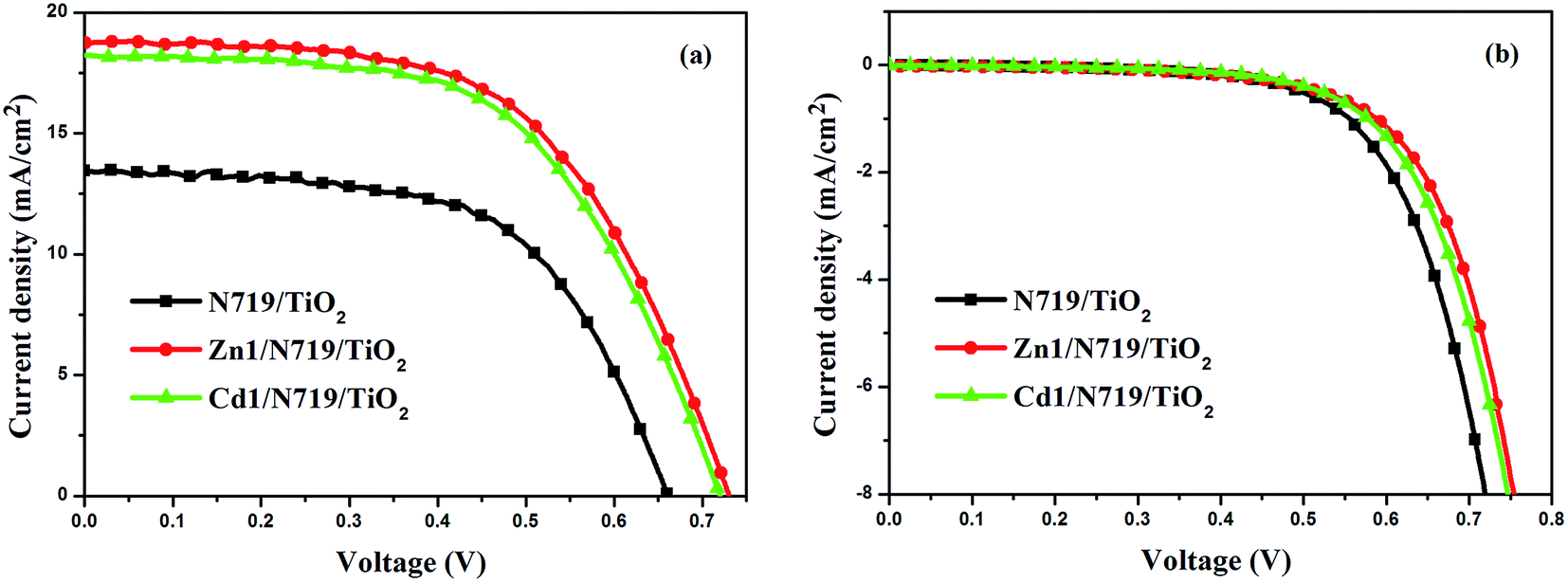 | ||
| Fig. 7 J–V curves for DSSCs based on co-sensitized photoelectrodes and N719 sensitized photoelectrode under irradiation (a) and in dark (b). | ||
| Photoelectrode | Jsc/mA cm2 | Voc/V | FF | η/% |
|---|---|---|---|---|
| N719/TiO2 | 13.42 | 0.66 | 0.60 | 5.36 |
| Zn1/N719/TiO2 | 18.61 | 0.73 | 0.57 | 7.75 |
| Cd1/N719/TiO2 | 18.36 | 0.72 | 0.58 | 7.70 |
Dark-current measurement of DSSCs has been considered as a qualitative technique to describe the extent of the back electron transfer. A comparison of dark current between the investigated cells can provide useful information regarding the back electron transfer process. Fig. 7b shows the dark current–voltage characteristics of the DSSCs based on different photoelectrodes with the applied bias from 0 to +0.80 V. The onset of the dark current for individual N719-sensitized DSSC occurs at a bias of about +0.48 V, with a subsequent dramatic increase of dark current with the increase of potential. In contrast, for the co-sensitized DSSCs, the onset potential shifted to about +0.54 V for Zn1/N719 and Cd1/N719; furthermore, the dark current of the co-sensitized DSSCs increased much more slowly than that of N719-sensitized DSSC when the potential was greater than +0.54 V. Under the same potential bias, when the potential was ≥0.48 V, the dark current for the co-sensitized DSSCs was smaller than that for the N719-sensitized DSSC. The increase of the onset potential and the reduction of the dark current indicated that Zn1 and Cd1 successfully suppress the electron back reaction with I3− in the electrolyte.
The higher η value of co-sensitized solar cell compared with the individually N719 sensitized devices, is attributed to the enhanced photovoltaic parameters Jsc and Voc. Particularly, the enhanced Jsc value is ascribed to the enhanced IPCE response of the cell, since they are related by the equation:
| Jsc = ∫eϕph.AM1.5 G(λ)dλ |
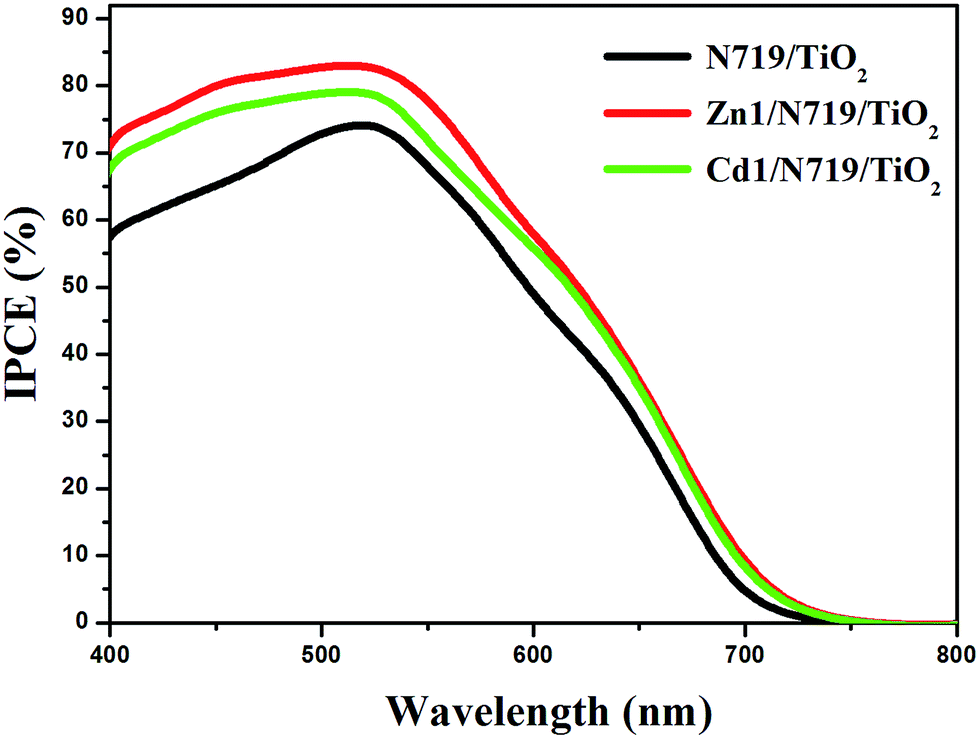 | ||
| Fig. 8 The incident photon-to-current conversion efficiency spectra of devices based on single N719 sensitized and co-sensitized photoanodes. | ||
The electrochemical impedance spectroscopy (EIS) is a steady-state method for examining the interfacial resistances and electron transport properties within an electrochemical system.46 The EIS investigation of the DSSC device provides valuable information for the understanding of photovoltaic parameters (Jsc, Voc, FF, and η). Herein, we utilize EIS to analyze charge carrier dynamics in the interfacial regions of solid–liquid layers. Impedance spectra of DSSC devices with Zn1/N719 and Cd1/N719 co-sensitized photoanode were measured under standard global AM1.5 solar irradiation or in the dark by applying a forward bias equivalent to open circuit potential.47 As indicated in Fig. 9, a typical EIS spectrum exhibits three semicircles in the Nyquist plot. In the order of increasing frequency the features are attributed to the Nernst diffusion in the electrolyte, electron transfer at the TiO2/electrolyte interface, and redox charge transfer at the counter electrode, respectively.48
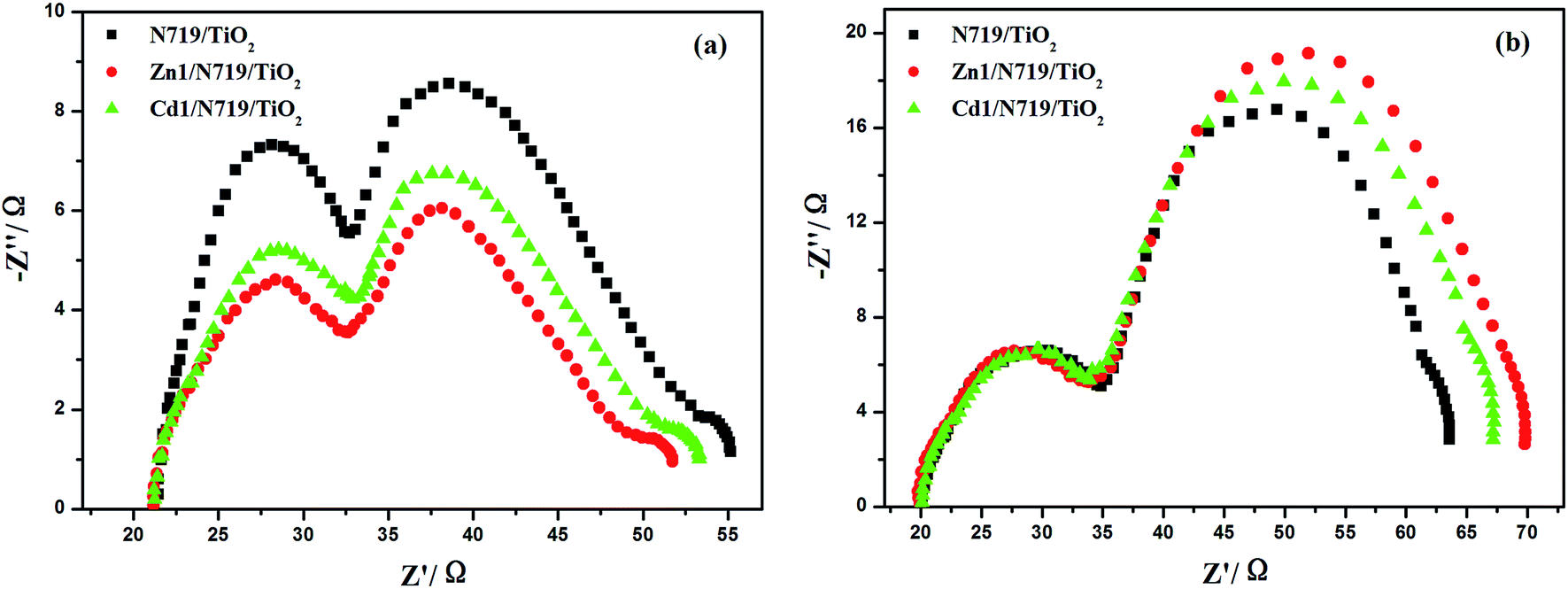 | ||
| Fig. 9 Nyquist plots of EIS for DSSCs based on different photoelectrodes measured under standard AM1.5 G solar irradiation (a) and in dark (b) at forward bias −0.75 V. | ||
Under light illumination, the second semicircle radius in the Nyquist plot decreased after co-sensitized with Zn1 or Cd1, and the values are in the order of Zn1/N719 < Cd1/N719 < N719, which indicates a decrease of the electron transfer impedance and an increase of charge transfer rate at this interface (Fig. 9a); while in the dark, this semicircle radius in the Nyquist plot increased after co-sensitized with Zn1 or Cd1, and the values are in the order of Zn1/N719 > Cd1/N719 > N719, which indicates an increase of electron recombination impedance and a reduction of the interfacial charge recombination rate (Fig. 9b).
In an effort to understand the enhancement of the Voc value in the dye-sensitized solar cells, the electron lifetime (τe) in different devices were calculated by fitting the Bode plots of the EIS spectra of different solar cells in dark (Fig. 10), according to the relationship: τe = 1/(2πfmax), where fmax is the frequency at the maximum of the curve in the intermediate frequency region in Bode phase plot.49 The electron lifetimes (τe) for the devices co-sensitized with prepared complexes (16.42 ms for Zn1 and 15.77 ms for Cd1) were found to be longer than individually N719 sensitized device (8.59 ms). This difference might be expected, since the adsorption of prepared complexes may form a better dye coverage to help to passivate the TiO2 surface or form an insulating molecular layer composed of prepared complexes and N719 molecules. It will reduce the recombination due to electron back-transfer between TiO2 and I3−. This retardation of the charge recombination between injected electron and I3− ions in the electrolyte, leads to a consequent increase of Voc. This appears to be consistent with the larger Voc values sequence which is in the order of Zn1/N719 > Cd1/N719 > N719.
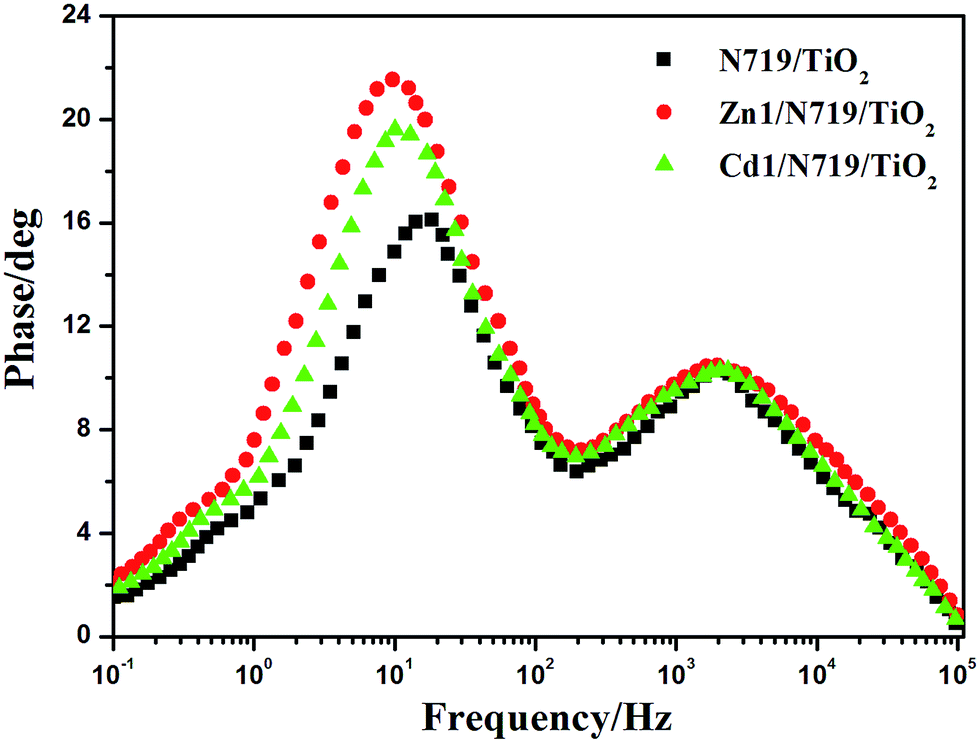 | ||
| Fig. 10 Bode plots of EIS for DSSCs based on different photoelectrodes measured in dark conditions. | ||
When the geometric and electronic structures of the complexes are taken into account, it is found that there is a relationship between crystal structure and DSSCs performance. For complexes Zn1 and Cd1, they adsorbed on Nano TiO2 thin film surface through carboxyl which have various coordination modes with metal ions. Although carboxyl has coordinated to the Cd2+/Zn2+ ion, Ti4+ ion may also be coordinated with carboxyl due to the existence of lone pair electrons in carboxylate oxygen atom which could combine with Ti4+ ion to form Ti–O coordination bond, and further adsorbed on the TiO2 film. Furthermore, the complexes Zn1 and Cd1 extend as 3D networks through π–π stacking interactions, which make the electron-donating ability of Zn1 and Cd1 better when they are used in DSSCs as co-sensitizers. All of these structural features of Zn1 and Cd1 result in obtaining higher performance of co-sensitized cells than that of cells sensitized by single N719. Meanwhile, for Cd1, the larger atom radius of Cd likely increases the Cd–O and Cd–N average bond lengths, which are 2.403 Å and 2.340 Å (the average Zn–O and Zn–N bond lengths are 1.948 Å and 2.102 Å), indicating that Zn–O and Zn–N bond are more stable than Cd–O and Cd–N bond (Table S1†). Apparently, the increased distance is not good for electron transfer. Therefore, the performance of the co-sensitized cell is in the order of Zn1/N719 > Cd1/N719.
Conclusion
Two new 2D d10 metal complexes with rigid 3-Hqlc ligand have been prepared and structurally characterized. The crystallographic data of Zn1 and Cd1 demonstrate that the formation of π⋯π stacking interactions can act as primary tool for constructing fascinating 3D network architectures. Complexes Zn1 and Cd1 show tunable luminescence (from blue to green) and photovoltaic properties. Both of them can display obvious AIE properties when they are dissolved in DMSO/water mixtures. The results reveal the potential of molecular design in functional luminescent complexes. Beyond that, we systematically investigated Zn1 and Cd1 as co-sensitizers in a ruthenium dye N719 based solar cell. The co-sensitization could enhance the spectral response of the N719 dye sensitized TiO2 film, suppress the electron recombination and decrease the total resistance of DSSCs. The improvement in efficiency is attributed to the fact that Zn1 and Cd1 overcome the deficiency of N719 absorption in the region of ultraviolet and blue-violet. We believe that such complexes will further facilitate the exploration of new type of multifunctional material with interesting properties, including the combination of luminescence and dye-sensitized solar cells.Acknowledgements
This work was supported by National Natural Science Foundation of China (Grant 21371040 and 21171044), the National key Basic Research Program of China (973 Program, no. 2013CB632900), supported by the Fundamental Research Funds for the Central Universities (Grant no. HIT. IBRSEM. A. 201409), and Program for Innovation Research of Science in Harbin Institute of Technology (PIRS of HIT no. A201416 and B201414).References
- H. K. Chae, J. Kim, O. D. Friedrichs, M. O'Keeffe and O. M. Yaghi, Angew. Chem., Int. Ed., 2003, 42, 3907–3909 CrossRef CAS PubMed
.
- J. Seo, C. Bonneau, R. Matsuda, M. Takata and S. Kitagawa, J. Am. Chem. Soc., 2011, 133, 9005–9013 CrossRef CAS PubMed
- Z. H. Yan, W. Wang, L. L. Zhang, X. W. Zhang, L. Wang, R. M. Wang and D. F. Sun, RSC Adv., 2015, 5, 16190–16198 RSC
- Z. C. Hu, B. J. Deibert and J. Li, Chem. Soc. Rev., 2014, 43, 5815–5840 RSC
- R. R. Hu, N. L. C. Leung and B. Z. Tang, Chem. Soc. Rev., 2014, 43, 4494–4562 RSC
- Y. X. Guo, X. Feng, T. Y. Han, S. Wang, Z. G. Lin, Y. P. Dong and B. Wang, J. Am. Chem. Soc., 2014, 136, 15485–15488 CrossRef CAS PubMed
- X. Y. Huang, S. Y. Han, W. Huang and X. G. Liu, Chem. Soc. Rev., 2013, 42, 173–201 RSC
- J. Z. Zhang, J. Zhang, H. B. Li, Y. Wu, H. L. Xu, M. Zhang, Y. Geng and Z. M. Su, J. Power Sources, 2014, 267, 300–308 CrossRef CAS PubMed
- G. De Filpo, F. P. Nicoletta, L. Ciliberti, P. Formoso and G. Chidichimo, J. Power Sources, 2015, 274, 274–279 CrossRef CAS PubMed
- D. D. Wang, Y. Wu, H. Dong, Z. X. Qin, D. Zhao, Y. Yu, G. J. Zhou, B. Jiao, Z. X. Wu, M. Gao and G. Wang, Org. Electron., 2013, 14, 3297–3305 CrossRef CAS PubMed
- L. G. Wei, Y. L. Yang, R. Q. Fan, Y. Na, P. Wang, Y. W. Dong, B. Yang and W. W. Cao, Dalton Trans., 2014, 43, 11361–11370 RSC
- G. D. Sharma, D. Daphnomili, K. S. V. Gupta, T. Gayathri, S. P. Singh, P. A. Angaridis, T. N. Kitsopoulos, D. Tasis and A. G. Coutsolelos, RSC Adv., 2013, 3, 22412–22420 RSC
- X. Wang, Y. L. Yang, P. Wang, L. Li, R. Q. Fan, W. W. Cao, B. Yang, H. Wang and J. Y. Liu, Dalton Trans., 2012, 41, 10619–10625 RSC
- X. X. Li, H. Y. Xu, F. Z. Kong and R. H. Wang, Angew. Chem., Int. Ed., 2013, 52, 13769–13773 CrossRef CAS PubMed
- A. Schneemann, V. Bon, I. Schwedler, I. Senkovska, S. Kaskel and R. A. Fischer, Chem. Soc. Rev., 2014, 43, 6062–6096 RSC
- F. A. A. Paz, J. Klinowski, S. M. F. Vilela, J. P. C. Tomé, J. A. S. Cavaleiro and J. Rocha, Chem. Soc. Rev., 2012, 41, 1088–1110 RSC
- A. Báez-Castro, J. Baldenebro-López, A. Cruz-Enríquez, H. Höpfl, D. Glossman-Mitnik, M.-S. Valentín, M. Parra-Hake and J. J. Campos-Gaxiola, RSC Adv., 2014, 4, 42624–42631 RSC
- C. B. Liu, Q. Li, X. Wang, G. B. Che and X. J. Zhang, Inorg. Chem. Commun., 2014, 39, 56–60 CrossRef CAS PubMed
- Y. Gong, M. M. Zhang, J. B. Qin, J. Li, J. P. Meng and J. H. Lin, Dalton Trans., 2014, 43, 8454–8460 RSC
- L. Croitor, E. B. Coropceanu, A. E. Masunov, H. J. Rivera-Jacquez, A. V. Siminel and M. S. Fonari, J. Phys. Chem. C, 2014, 118, 9217–9227 CAS
- L. Croitor, E. B. Coropceanu, A. E. Masunov, H. J. Rivera-Jacquez, A. V. Siminel, V. I. Zelentsov, T. Y. Datsko and M. S. Fonari, Cryst. Growth Des., 2014, 14, 3935–3948 CAS
- S. Altobello, R. Argazzi, S. Caramori, C. Contado, S. Da Fré, P. Rubino, C. Choné, G. Larramona and C. A. Bignozzi, J. Am. Chem. Soc., 2005, 127, 15342–15343 CrossRef CAS PubMed
- S. Archer and J. A. Weinstein, Coord. Chem. Rev., 2012, 256, 2530–2561 CrossRef CAS PubMed
- T. Swetha, S. Niveditha, K. Bhanuprakash and S. P. Singh, Electrochim. Acta, 2015, 153, 343–351 CrossRef CAS PubMed
- Y. Z. Xie, G. G. Shan, P. Li, Z. Y. Zhou and Z. M. Su, Dyes Pigm., 2013, 96, 467–474 CrossRef CAS PubMed
- L. Chen, Y. B. Jiang, H. Nie, R. R. Hu, H. S. Kwok, F. Huang, A. J. Qin, Z. J. Zhao and B. Z. Tang, ACS Appl. Mater. Interfaces, 2014, 6, 17215–17225 CAS
- G. M. Sheldrick, SHELXL 97 Program for Crystal Structure Refinement, University of GÖttingen, GÖttingen, Germany, 1997 Search PubMed
- G. M. Sheldrick, SHELXL 97 Program for Crystal Structure Solution, University of GÖttingen, GÖttingen, Germany, 1997 Search PubMed
- D. M. Chen, X. P. Zhang, W. Shi and P. Cheng, Cryst. Growth Des., 2014, 14, 6261–6268 CAS
- L. M. Fan, W. L. Fan, B. Li, X. Z. Liu, X. Zhao and X. T. Zhang, RSC Adv., 2015, 5, 14897–14905 RSC
- J. Yang, J. F. Ma, Y. Y. Liu and S. R. Batten, CrystEngComm, 2009, 11, 151–159 RSC
- Y. Kim, J. H. Song, W. R. Lee, W. J. Phang, K. S. Lim and C. S. Hong, Cryst. Growth Des., 2014, 14, 1933–1937 CAS
- P. Chakraborty, J. Adhikary, S. Samanta, D. Escudero, A. C. Castro, M. Swart, S. Ghosh, A. Bauzá, A. Frontera, E. Zangrando and D. Das, Cryst. Growth Des., 2014, 14, 4111–4123 CAS
- R. Q. Fan, Y. L. Yang, Y. B. Yin, W. Hasi and Y. Mu, Inorg. Chem., 2009, 48, 6034–6043 CrossRef CAS PubMed
- R. R. Hu, C. F. A. Gómez-Durán, J. W. Y. Lam, J. L. Belmonte-Vázquez, C. M. Deng, S. J. Chen, R. Q. Ye, E. Peña-Cabrera, Y. C. Zhong, K. S. Wong and B. Z. Tang, Chem. Commun., 2012, 48, 10099–10101 RSC
- Q. L. Zhu, C. J. Shen, C. H. Tan, T. L. Sheng, S. M. Hu and X. T. Wu, Chem. Commun., 2012, 48, 531–533 RSC
- P. Y. Gu, C. J. Lu, Z. J. Hu, N. J. Li, T. T. Zhao, Q. F. Xu, Q. H. Xu, J. D. Zhang and J. M. Lu, J. Mater. Chem. C, 2013, 1, 2599–2606 RSC
- M. Zhang, G. X. Feng, Z. G. Song, Y. P. Zhou, H. Y. Chao, D. Q. Yuan, T. T. Y. Tan, Z. G. Guo, Z. G. Hu, B. Z. Tang, B. Liu and D. Zhao, J. Am. Chem. Soc., 2014, 136, 7241–7244 CrossRef CAS PubMed
- D. B. Kuang, S. Ito, B. Wenger, C. Klein, J. E. Moser, R. Humphry-Baker, S. M. Zakeeruddin and M. Grätzel, J. Am. Chem. Soc., 2006, 128, 4146–4154 CrossRef CAS PubMed
- G. D. Sharma, S. P. Singh, R. Kurchania and R. J. Ball, RSC Adv., 2013, 3, 6036–6043 RSC
- L. G. Wei, Y. Na, Y. L. Yang, R. Q. Fan, P. Wang and L. Li, Phys. Chem. Chem. Phys., 2015, 17, 1273–1280 RSC
- K. R. J. Thomas, Y. C. Hsu, J. T. Lin, K. M. Lee, K. C. Ho, C. H. Lai, Y. M. Cheng and P. T. Chou, Chem. Mater., 2008, 20, 1830–1840 CrossRef CAS
- J. A. Mikroyannidis, M. S. Roy and G. D. Sharma, J. Power Sources, 2010, 195, 5391–5398 CrossRef CAS PubMed
- G. H. Wu, F. T. Kong, J. Z. Li, X. Q. Fang, Y. Li, S. Y. Dai, Q. Q. Chen and X. X. Zhang, J. Power Sources, 2013, 243, 131–137 CrossRef CAS PubMed
- J. Cui, J. F. Lu, X. B. Xu, K. Cao, Z. Wang, G. Alemu, H. L. Yuang, Y. Shen, J. Xu, Y. B. Cheng and M. K. Wang, J. Phys. Chem. C, 2014, 118, 16433–16440 CAS
- F. Malara, A. Cannavale, S. Carallo and G. Gigli, ACS Appl. Mater. Interfaces, 2014, 6, 9290–9297 CAS
- J. Bisquert, A. Zaban, M. Greenshtein and I. Mora-Sero, J. Am. Chem. Soc., 2004, 126, 13550–13559 CrossRef CAS PubMed
- D. B. Kuang, S. Uchida, R. Humphry-Baker, S. M. Zakeeruddin and M. Grätzel, Angew. Chem., Int. Ed., 2008, 47, 1923–1927 CrossRef CAS PubMed
- S. P. Singh, K. S. V. Gupta, M. Chandrasekharam, A. Islam, L. Y. Han, S. Yoshikawa, M. Haga, M. S. Roy and G. D. Sharma, ACS Appl. Mater. Interfaces, 2013, 5, 11623–11630 CAS
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1055702 and 1055703. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ra07160g |
| This journal is © The Royal Society of Chemistry 2015 |