
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis, binding and self-assembly properties of a well-defined pillar[5]arene end functionalised polydimethylacrylamide†
N.
Laggoune
a,
F.
Delattre
b,
J.
Lyskawa
a,
F.
Stoffelbach
c,
J. M.
Guigner
d,
S.
Ruellan
b,
G.
Cooke
*e and
P.
Woisel
*a
aUniversité de Lille, ENSCL, Unité des Matériaux et Transformations, UMR CNRS 8207, Equipe Ingénierie des Systèmes Polymères (ISP), 59655 Villeneuve d'Ascq Cedex, France
bUCEIV, EA 4492, Université du Littoral Côte d'Opale, Av. M. Schuman, 59140 Dunkerque, France
cSorbonne Universités, UPMC Univ Paris 06, CNRS, Institut Parisien de Chimie Moléculaire, Equipe Chimie des Polymères, 4 place Jussieu, F-75005 Paris, France
dInstitut de Minéralogie, de Physique des Matériaux et de Cosmochimie (IMPMC), UPMC Univ Paris 06-IRD-CNRS UMR 7590-MNHN, 75005 Paris, France
eGlasgow Centre for Physical Organic Chemistry, WestCHEM, School of Chemistry, Joseph Black Building, University of Glasgow, Glasgow G12 8QQ, UK. E-mail: Graeme.Cooke@glasgow.ac.uk
First published on 3rd September 2015
Abstract
The synthesis, binding and self-assembly properties of a well-defined pillar[5]arene end-functionalised poly(dimethylacrylamide)(MePilla-PDMAC) are reported. In order to synthesise MePilla-PDMAC, a new trithiocarbonate type RAFT agent MePilla-CTA was developed incorporting a partially methylated pillar[5]arene moiety. Kinetic studies clearly indicated the propensity of MePilla-CTA to control the polymerisation of DMAC. Interestingly, as PDMAC type chains display good solubilty both in organic and aqueous media, MePilla-PDMAC was able of specifically bind electron deficient guest molecules at the α-chain-end both in chloroform and water. Complex formation was found to be reversible upon addition of chloride anions or heating in organic and aqueous media, respectively. Furthermore, cryo-TEM, VT-NMR (1H) and VT-DLS investigations also indicated the ability of MePilla-PDMAC to self-assemble into micelle-like aggregates in water showing reversible recognition properties.
Introduction
The amalgamation of polymer and supramolecular chemistry has led to the development of fascinating self-assembling polymeric materials with bespoke structures and tuneable properties.1,2 The incorporation of non-covalent recognition motifs into polymeric systems offers the unique opportunity to tune polymer structures (e.g., topology and morphology) and to impart unique additional properties, including self-healing,3 adaptability4 and responsiveness5–7 to materials. Furthermore, the non-covalent modification of polymers has allowed their solubility and correspondingly their processability to be greatly improved.8,9 The development of this field has largely gone hand-in-hand with the enormous progress that has been made in the development of well-defined polymers synthesised using controlled radical polymerisation (CRP) techniques that feature specific recognition motifs (hydrogen acceptor/donor,10,11 ionic unit,12,13 metal/ligand,14,15 host/guest16–18 molecules) attached in specific locations (end or side chain) to polymer backbones.Pillar[n]arenes (n = 5, 6, 7, 8…) have emerged as important host units for the construction of supramolecular assemblies with applications spanning materials,19,20 medicine21–23 and sensing.24,25 Their burgeoning interest as macrocyclic hosts relies mainly on their convenient synthesis,26 ability to be conveniently functionalised and their unique symmetrical and rigid electron-rich pillar architecture that enable various neutral and electron-deficient guests to be accommodated within their cavities.27–30 Due to the hydrophobic nature of these macrocycles, host–guest complexation and self-assembly of pillar[n]arene derivatives has been largely investigated in organic media. However, as many recognition events in nature occur in aqueous media, effort has also been directed towards the development of pillararene based receptors capable of recognizing guests in aqueous environments. In this context, the main strategy consists of grafting hydrophilic groups, such as carboxylate,31–33 ammonium,34,35 phosphonium,36 glycol37 and carbohydrate38 substituents onto the macrocycle.
Despite the advantages of CRP techniques including their flexibility, tolerance to a wide range of monomers, well-defined compositions, narrow molar mass distributions and sophisticated architectures, surprisingly few pillararene-based examples have been reported in the literature. In this context, the RAFT (Reversible Addition–Fragmentation Chain Transfer) has been very recently used to develop thermoresponsive pillararene-containing polymers capable of stabilizing gold nanoparticles39 and creating gel-like supramolecular networks.40
Here, we describe the synthesis of a trithiocarbonate-type RAFT agent MePilla-CTA incorporating a partially methylated pillar[5]arene moiety and its propensity to control the polymerisation of DMAC (Dimethylacrylamide) and, therefore, to produce a well-defined methylated pillar[5]arene end functionalised polydimethylacrylamide (PDMAC) (MePilla-PDMAC) (Scheme 1). An interesting characteristic of the PDMAC type chain is that it imparts solubility in both aqueous and organic solvents to the methylatedpillar[5]arene scaffold. This feature was exploited to specifically and reversibly end-modify MePilla-PDMAC (Mn,NMR = 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 g mol−1, Đ = 1.1) with electron deficient partners (G1 and G2) through host–guest complexation both in organic and aqueous media. Preliminary self-assembly properties of MePilla-PDMAC in water are also described.
500 g mol−1, Đ = 1.1) with electron deficient partners (G1 and G2) through host–guest complexation both in organic and aqueous media. Preliminary self-assembly properties of MePilla-PDMAC in water are also described.
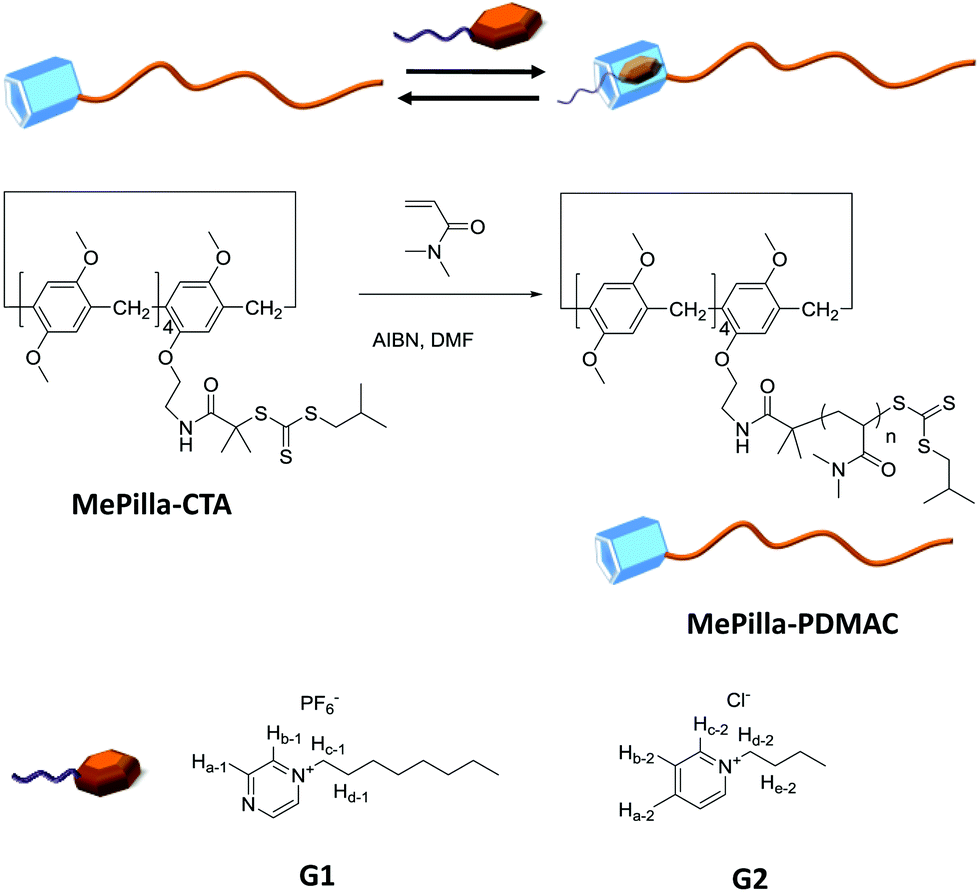 | ||
| Scheme 1 Chemical structures of MePilla-CTA, MePilla-PDMAC, G1 and G2. | ||
Results and discussion
The RAFT agent MePilla-CTA was conveniently prepared by coupling the N-hydroxysuccinimide (NHS) activated ester of the trithiocarbonate 2-(1-isobutyl)sulfanylthiocarbonylsulfanyl-2-methylpropionic acid (CTA-NHS)41 and the mono amino pillararene derivative MePilla-NH242 (Scheme 2). The analytical data for MePilla-CTA were consistent with the proposed structure. The 1H NMR spectrum of MePilla-CTA recorded in CDCl3 at 298 K shows the characteristic signals of the pillararene unit in addition to those belonging to the isobutylsulfanylthiocarbonylsulfanyl [(CH3)2CH–CH2–S–C(S)–S–] moiety (Fig. 1). The 13C NMR spectrum (Fig. S1†) also confirmed the identity of MePilla-CTA through the appearance of chemical shifts near 173 and 220 ppm ascribed to the carbonyl of the amide group and the thiocarbonyl moiety, respectively.
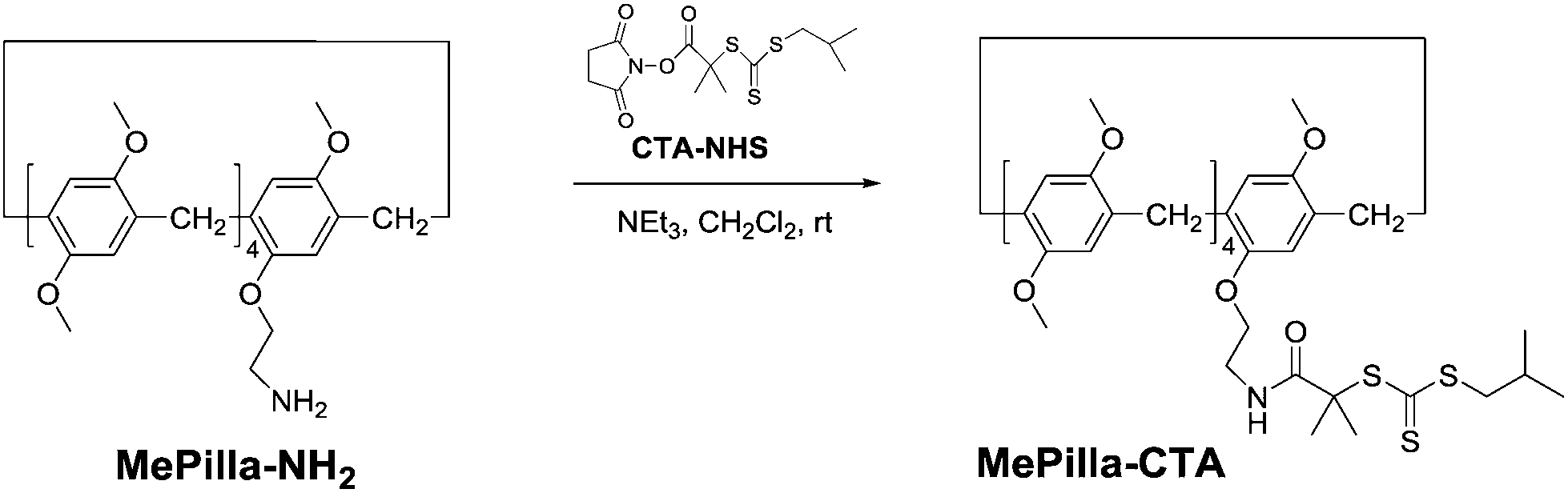 | ||
| Scheme 2 Synthesis of MePilla-CTA. | ||
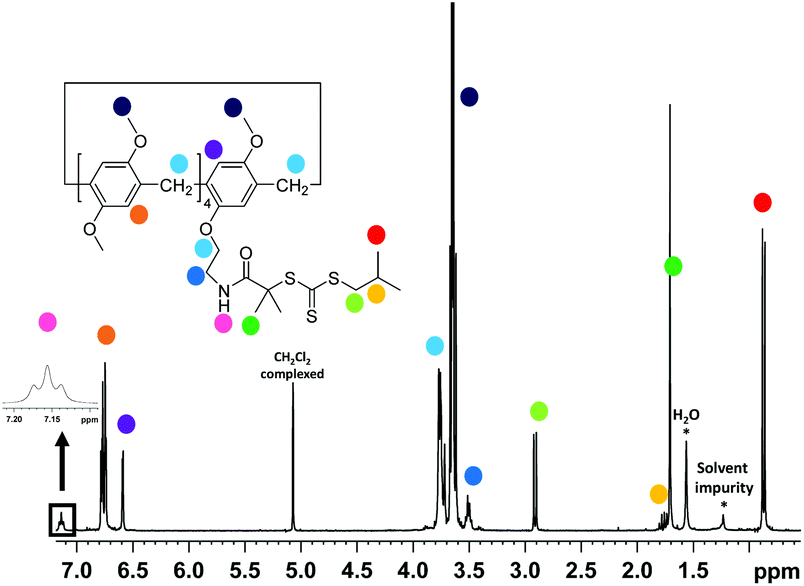 | ||
| Fig. 1 1H NMR spectrum of MePilla-CTA (300 MHz, CDCl3). | ||
Having the MePilla-CTA in hand, we next investigated its ability to control the polymerisation of DMAC via a RAFT procedure. RAFT polymerisation of DMAC was carried out by using 2,2′-azobis(2-methylpropionitrile) (AIBN) as initiator at 343 K in DMF in the presence of MePilla-CTA ([DMAC]0/[CTA]0/[AIBN]0 = 1000/5/1). As shown in Fig. 2a, the RAFT polymerisation exhibits pseudo-first order kinetics indicating a constant propagating radical concentration during the polymerisation.
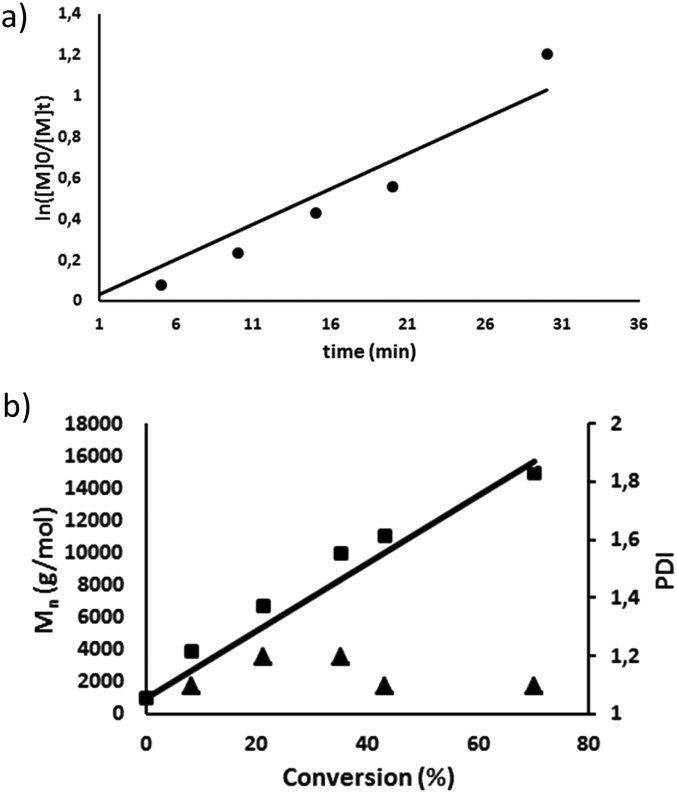 | ||
| Fig. 2 (a) First-order kinetic plot for the RAFT polymerisation of DMAC using MePilla-CTA); (b) dependence of the number-average molar mass (Mn) and dispersity (Đ) on conversion, for the RAFT polymerisation of DMAC in DMF using MePilla-CTA. | ||
Control over molecular weight was also demonstrated by the linear increase of Mnversus DMAC conversion and dispersity (Đ) values of the resulting polymers of 1.1–1.3 (Fig. 2b). SEC analysis also revealed polymers with symmetrical traces (Fig. S4†) and experimental molar masses in accordance with theoretical values, indicating that the number of chains is governed by the RAFT agent concentration which remained constant during the polymerisation. The pseudo-living character of the RAFT polymerisation was also evidenced by restarting the polymerisation of DMAC from a macromolecular RAFT agent (Mn,SEC = 22260 g mol−1) which gave rise to a well-defined polymer with a higher molar mass (Mn,SEC = 59500 g mol−1) with a narrow molecular mass distribution (Đ = 1.3) (Fig. S5†).
The 1H NMR spectrum recorded in CDCl3 of MePilla-PDMAC (Fig. 3) clearly shows the presence of the pillararene unit connected to polymer chains with the presence of characteristic resonances of H1,2 (δ = 6.7 ppm) and H3 (δ = 3.7 ppm) belonging to the host unit and PDMAC chains (0.8 < δ < 3.2 ppm). This was further confirmed by performing 2D diffusion-ordered spectroscopy (DOSY) 1H NMR experiments that showed a single diffusion coefficient value of 0.6 × 10−10 m2 s−1 at 298 K for the aforementioned protons in CDCl3.
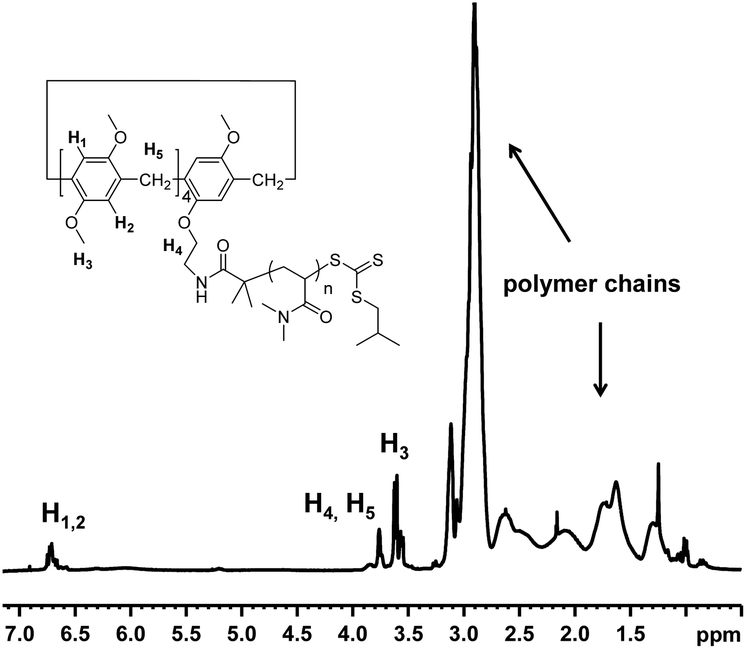 | ||
| Fig. 3
1H NMR spectrum of MePilla-PDMAC (Mn = 14500 g mol−1) recorded in CDCl3 at 298 K. | ||
An interesting characteristic of the PDMAC type chain is that it imparts solubility in water and organic solvents to the methylatedpillar[5]arene scaffold, thereby allowing binding properties of MePilla-PDMAC to be investigated in both aqueous and organic media. The complexation properties of MePilla-PDMAC were first investigated by 1H NMR spectroscopy in CDCl3 (Fig. 4A) and D2O (Fig. S6†) in the presence of G1 and G2 as guest molecules, respectively. In both cases, a broadening and a shielding effect for the resonances of protons of the guest and host partners were observed. This is particularly noteworthy for Ha-1, Hb-1, Hc-1 of G1 that show upfield shifts of 0.3, 2.2, 3.6 ppm upon complexation in CDCl3, respectively. A slight downfield shift (+0.05 ppm) was also observed for H1,2 protons of MePilla-PDMAC. A similar trend was evidenced for Hc-2 and Hd-2 of G2 that shift upfield by 0.3 and 3.2 ppm in D2O, respectively, upon the addition of an equimolar solution of MePilla-PDMAC (Fig. S6†).
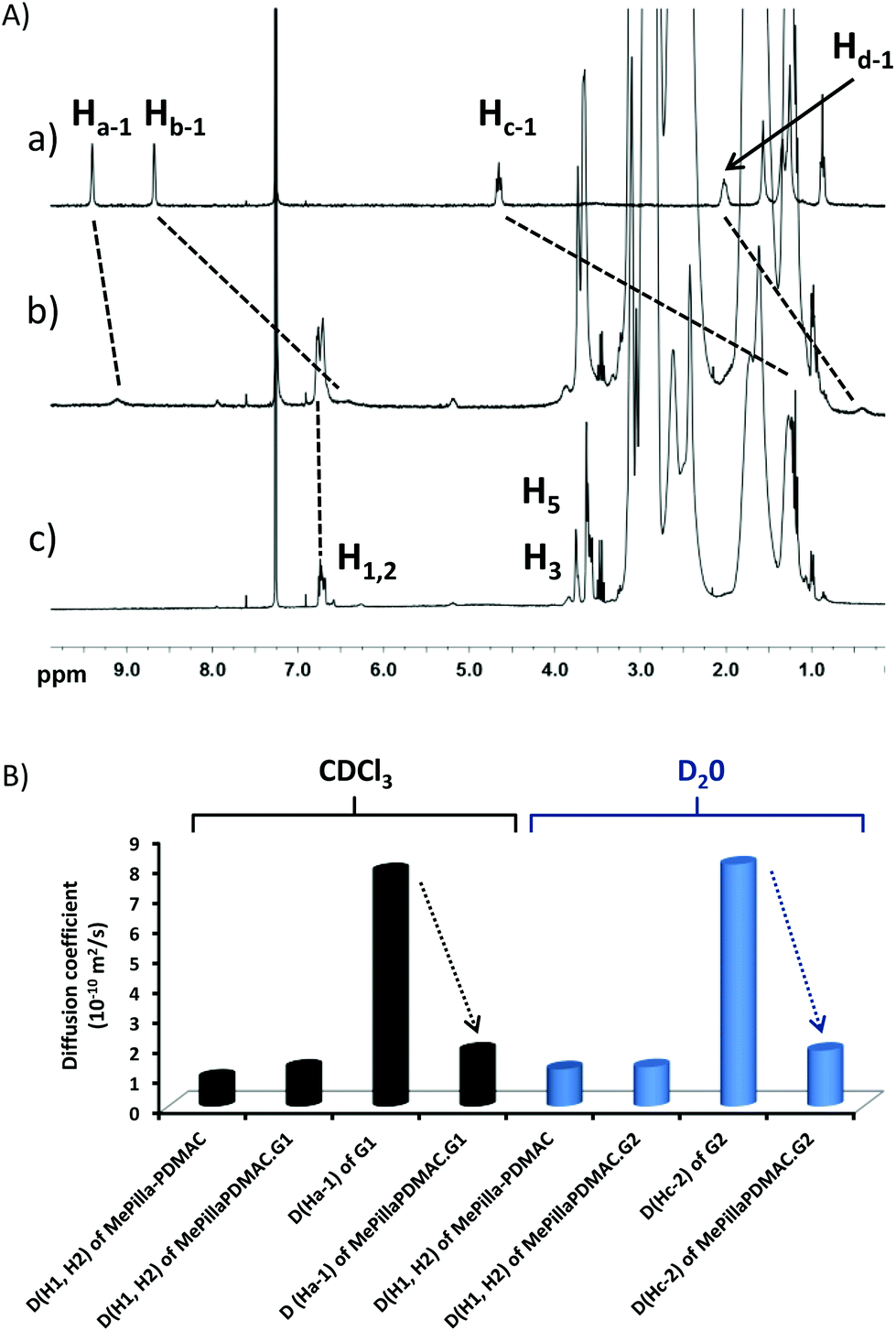 | ||
| Fig. 4 (A) 1H NMR spectra (300 MHz, CDCl3, 298 K) of (a) G1 (4.5 mM), (b) a 1:1 mixture of G1 and MePilla-PDMAC and (c) MePilla-PDMAC (4.5 mM), (B) diffusion coefficients of Ha-1, Hc-2 and H1,2 after and before complexation in CDCl3 or D2O. | ||
To further prove the ability of MePilla-PDMAC to form pseudorotaxane-like complexes with G1 and G2, 2D-NOESY (Fig. S7†) and 2D-DOSY (Fig. 4B) experiments were undertaken. As expected, correlations between most aromatic and alkyl protons of guest molecules G1/G2 and protons located on the host unit were observed, suggesting the formation of pseudorotaxane architectures between MePilla-PDMAC and G1 and G2. 2D-DOSY experiments showed that H1,2 protons of MePilla-PDMAC were not impacted in terms of diffusion coefficient by the presence of the significantly smaller guest molecules G1 and G2. On contrary, the diffusion coefficient values of Ha-1 and Hc-2 of G1 and G2, respectively, were dramatically affected upon complexation and moved to much lower and almost identical values to those obtained for MePilla-PDMAC in the different media, thereby further demonstrating the formation of complexes. Finally, association constants for MePilla-PDMAC complexing with G1 and G2 were estimated by 1H NMR (in CDCl3) and fluorescence (in H2O) titration experiments and were found to be 0.9 (±0.1) × 102 (Fig. S8†) and 1.5 (±0.1) × 103 M−1 (Fig. S9†), respectively, by employing a non-linear fitting method using one-site binding models. In addition, the 1:1 binding mode was further evidenced through the continuous variation method (Job plot method) using 1H NMR spectroscopy (Fig. S10†).
With complexation confirmed for MePilla-PDMAC·G1 and MePilla-PDMAC·G2 in chloroform and water, respectively, we next turned our attention to whether complex formation of this type could be controlled by applying an external stimulus. First, we investigated the opportunity to switch off the complexation between MePilla-PDMAC and G1 in chloroform by adding chloride anions. Indeed, Huang43 and Wang24 have recently demonstrated that pillar[5]arene based complexes could be dissociated in chloroform by adding tetrabutylammonium chloride (TBACl), leading to the exchange of the soft counter ion PF6− of the guests by a hard counter ion Cl− and the formation of an intimate ion pair between ammonium cations and Cl−.
Fig. 5A shows the evolution of chemical shifts corresponding to the MePilla-PDMAC·G1 complex in the presence of an excess of TBACl. As expected, a disassembly process between G1 and MePilla-PDMAC can be observed through a downfield and upfield shift of Ha-1 and H1,2 protons, respectively, that move toward the resonances observed for the uncomplexed G1 and MePilla-PDMAC.
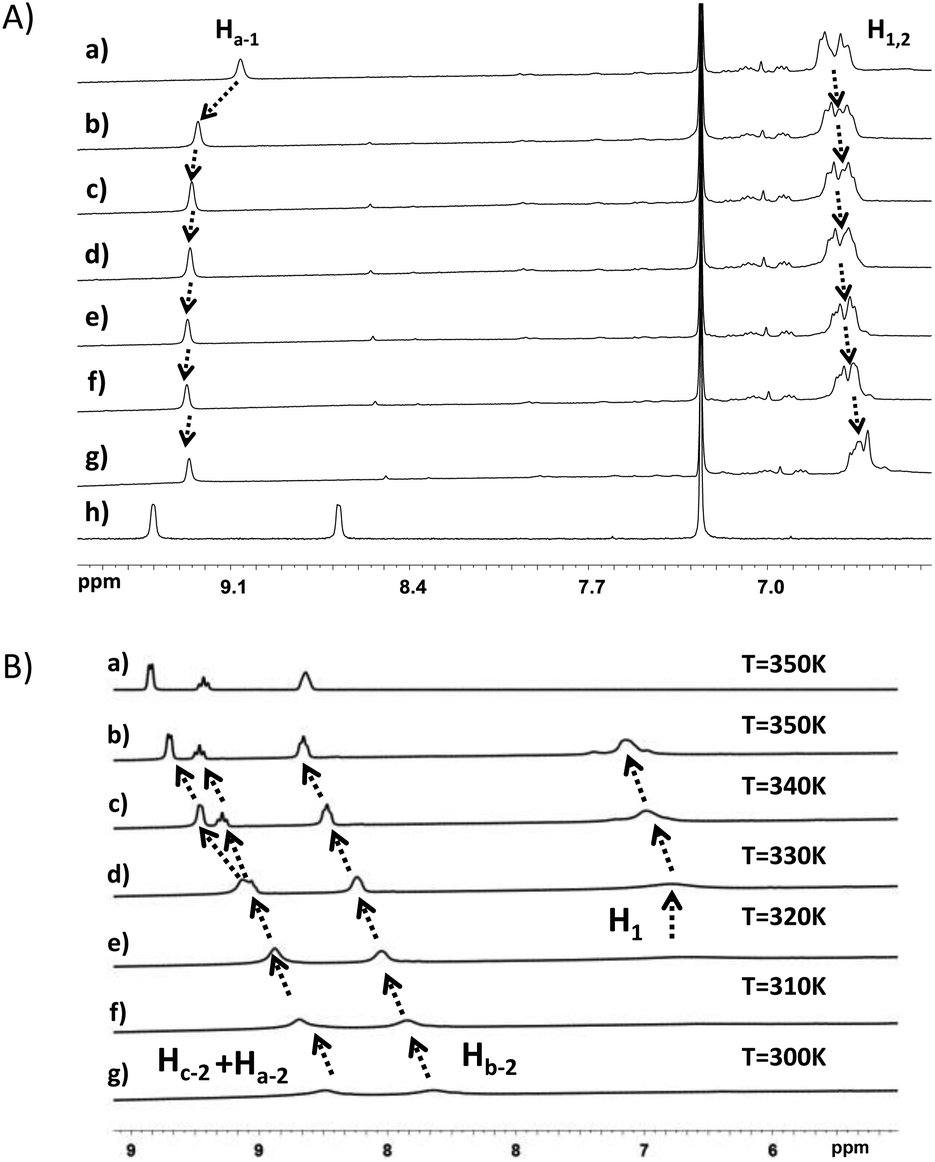 | ||
| Fig. 5 (A) Partial 1H NMR spectra (300 K, CDCl3) of MePilla-PDMAC·G1 (4.5 mM) upon addition of (a) 0.0 mM, (b) 2 mM, (c) 4 mM, (d) 8 mM, (e) 12 mM, (f)16 mM, (g) 20 mM of TBACl. (h) Partial 1H NMR spectrum (300 K, CDCl3) of G1 (4.5 mM); (B) (a) partial 1H NMR spectra of G2 (4.5 mM) in D2O at 350 K and partial 1H NMR spectra (300 K, D2O) of G2·MePilla-PDMAC (6.4 mM) complex (b) 350 K, (c) 340 K, (d) 330 K, (e) 320 K, (f) 310 K, (g) 300 K. | ||
Next, we have explored the effect of heating the MePilla-PDMAC·G2 complex using variable-temperature 1H NMR (Fig. 5B) and 2D-DOSY experiments (Fig. S11†). As shown in Fig. 5B, a clear gradual downfield shift in the resonances of Ha-2, Hb-2, Hc-2 of G2 with a concomitant sharpening of NMR signals for both guest and host moieties was observed. This is particularly noticeable for Ha-2 and Hc-2 which spilt into two distinct and sharp signals at 340 K, which is similar to the resonances observed in the NMR spectrum of the uncomplexed G2. Moreover, a similar behaviour was evidenced by estimating diffusion coefficients for Hc-2 and H1,2versus temperature. Indeed, while D(H1,2) remained almost identical with temperature, D(Hc-2) increased progressively from 300 to 340 K and then rose sharply above this temperature, thereby suggesting a disassembly of the complex (Fig. S11†). To confirm this dissociation with temperature, VT-fluorescence studies were carried out on the uncomplexed MePilla-PDMAC and the MePilla-PDMAC·G2 complex. While the complexed MePilla-PDMAC shows a much lower relative fluorescence intensity than the free MePilla-PDMAC at 300 K, above 340 K both species display a nearly identical relative fluorescence intensity to their non-complexed states, thereby suggesting the dissociation of the MePilla-PDMAC·G2 upon heating (Fig. S12†).
Having demonstrated that the methylatedpillar[5]arene moiety was located at one end of PDMAC chain, and that specific non-covalent modification with electron deficient guests was possible, we next investigated the impact of the presence of the hydrophobic pillarane unit on PDMAC chains and host–guest complexation has on the self-assembly properties of MePilla-PDMAC in water. DLS (Fig. 6A) and cryo-TEM (Fig. 6B) experiments were carried out on MePilla-PDMAC which revealed the existence of nanosized aggregates with an average hydrodynamic diameter (<Dh>) of 24 nm (poly: 0.02) corresponding to micelle-like aggregates (according the cryo-TEM). Hence, these results clearly indicate that the hydrophobic behaviour of methylatedpillar[5]arene moiety can be exploited to create self-assembled architectures when connected to hydrophilic polymers. The critical aggregation concentration (CAC) in water was estimated to be 0.5 mg mL−1 based on the dependence of the fluorescence intensity of MePilla-PDMACversus its concentration (Fig. S13†).
 | ||
| Fig. 6 (A) DLS data for MePilla-PDMAC (4.5 mM) in water recorded at 298 K, (B) cryo-TEM image of nano-aggregates obtained for MePilla-PDMAC. (C) Cartoon representing the proposed dethreading of G2 from micelle-like MePilla-PDMAC·G2 upon heating. | ||
Interestingly, VT-DLS experiments performed on MePilla-PDMAC and its complex MePilla-PDMAC·G2 indicated that both complex formation and temperature had no effect on the size distribution of micelles (Fig. S14†). Therefore, taking into account that above 340 K, VT-NMR (1H) experiments performed (at the same concentration as the VT-DLS measurements) on MePilla-PDMAC·G2 (Fig. 5B) indicated the dissociation of the complex, and that VT-DLS investigations demonstrated the persistence of micelles upon heating, the micelle-like architectures prepared from MePilla-PDMAC appear to have the propensity to bind, and then to release on demand upon heating, the electron deficient guest G2 (Fig. 6C).
Conclusions
In conclusion, we have described the synthesis of a trithiocarbonate type RAFT agent incorporating a partially methylated pillar[5]arene moiety. By using this specifically designed RAFT agent, the polymerisation of DMAC was found to be controlled, affording a well-defined dimethypillarene end-decorated PDMAC. This polymer has the ability to recognize, in a reversible manner, electron deficient guests both in organic and aqueous media. Furthermore, the hydrophobic nature of the pillararane unit has permitted the self-assembly of MePilla-PDMAC in water into micelle-like aggregates displaying reversible recognition properties. Since the RAFT procedure may be applied to the synthesis of a range well-defined macromolecular architectures, this study paves the way for the development of other pillararene-polymer conjugates with new applications.Acknowledgements
P. W. gratefully acknowledges financial support from the CNRS and the Agence Nationale de la Recherche (STRAPA project, ANR-12-BS08-0005). GC thanks the EPSRC (EP/E036244/1) for funding.Notes and references
- A. Harada, Supramolecular Polymer Chemistry, WILEY-VCH Weinheim, 2011 Search PubMed.
- Supramolecular Chemistry: From Molecules to Nanomaterials, ed. P. A. Gale and J. W. Steed, John Wiley & Sons, 2012 Search PubMed.
- F. Herbst, D. Döhler, P. Michael and W. H. Binder, Macromol. Rapid Commun., 2013, 34, 203–220 CrossRef CAS PubMed.
- P. Wei, X. Yan and F. Huang, Chem. Soc. Rev., 2015, 44, 815–832 RSC.
- J. Bigot, B. Charleux, G. Cooke, D. Fournier and P. Woisel, J. Am. Chem. Soc., 2010, 132, 10796–10801 CrossRef CAS PubMed.
- J. Lyskawa, D. Fournier, L. Bouteiller, B. Charleux, G. Cooke and P. Woisel, Macromolecules, 2011, 44, 6532–6538 CrossRef.
- G. Cooke, J. F. Garety, S. G. Hewage, B. J. Jordan, G. Rabani, V. M. Rotello and P. Woisel, Org. Lett., 2007, 9, 481–484 CrossRef CAS PubMed.
- V. R. de la Rosa, W. M. Nau and R. Hoogenboom, Org. Biomol. Chem., 2015, 13, 3048–3057 CAS.
- L. Sambe, V. R. de La Rosa, K. Belal, F. Stoffelbach, J. Lyskawa, F. Delattre, M. Bria, G. Cooke, R. Hoogenboom and P. Woisel, Angew. Chem., Int. Ed., 2014, 53, 5044–5048 CAS.
- S. Chen, A. Bertrand, X. Chang, P. Alcouffe, C. Ladavière, J. F. Gérard, F. Lortie and J. Bernard, Macromolecules, 2010, 43, 5981–5988 CrossRef CAS.
- K. E. Feldman, M. J. Kade, T. F. a De Greef, E. W. Meijer, E. J. Kramer and C. J. Hawker, Macromolecules, 2008, 41, 4694–4700 CrossRef CAS.
- M. A. Ayoubi, K. Zhu, B. Nystrom, K. Almdal, U. Olsson and L. Piculell, Soft Matter, 2013, 9, 1540–1555 RSC.
- M. A. Ayoubi, K. Almdal, K. Zhu, B. Nyström, U. Olsson and L. Piculell, RSC Adv., 2015, 5, 31091–31103 RSC.
- Y. Yan, N. A. M. Besseling, A. De Keizer, A. T. M. Marcelis, M. Drechsler and M. A. Cohen Stuart, Angew. Chem., Int. Ed., 2007, 46, 1807–1809 CrossRef CAS PubMed.
- S. Piogé, C. A. Fustin and J. F. Gohy, Macromol. Rapid Commun., 2012, 33, 534–539 CrossRef PubMed.
- L. Sambe, K. Belal, F. Stoffelbach, J. Lyskawa, F. Delattre, M. Bria, F. X. Sauvage, M. Sliwa, V. Humblot, B. Charleux, G. Cooke and P. Woisel, Polym. Chem., 2014, 5, 1031–1036 RSC.
- U. Rauwald and O. A. Scherman, Angew. Chem., Int. Ed., 2008, 47, 3950–3953 CrossRef CAS PubMed.
- A. F. Hirschbiel, B. V. K. J. Schmidt, P. Krolla-Sidenstein, J. P. Blinco and C. Barner-Kowollik, Macromolecules, 2015, 48, 4410–4420 CrossRef CAS.
- Z. Zhang, Q. Zhao, J. Yuan, M. Antonietti and F. Huang, Chem. Commun., 2014, 50, 2595–2597 RSC.
- Y.-L. Sun, Y.-W. Yang, D.-X. Chen, G. Wang, Y. Zhou, C.-Y. Wang and J. F. Stoddart, Small, 2013, 9, 3224–3229 CAS.
- L.-L. Tan, H. Li, Y.-C. Qiu, D.-X. Chen, X. Wang, R.-Y. Pan, Y. Wang, X.-A. Zhang, S. B. Wang and Y.-W. Yang, Chem. Sci., 2015, 6, 1640–1644 RSC.
- X.-Y. Hu, K. Jia, Y. Cao, Y. Li, S. Qin, F. Zhou, C. Lin, D. Zhang and L. Wang, Chem. – Eur. J., 2015, 21, 1208–1220 CrossRef CAS PubMed.
- Y. Cao, Y. Li, X.-Y. Hu, X. Zou, S. Xiong, C. Lin and L. Wang, Chem. Mater., 2015, 27, 1110–1119 CrossRef CAS.
- S. Sun, X.-Y. Hu, D. Chen, J. Shi, Y. Dong, C. Lin, Y. Pan and L. Wang, Polym. Chem., 2013, 4, 2224–2229 RSC.
- N. L. Strutt, R. S. Forgan, J. M. Spruell, Y. Y. Botros and J. F. Stoddart, J. Am. Chem. Soc., 2011, 133, 5668–5671 CrossRef CAS PubMed.
- T. Ogoshi, T. Aoki, K. Kitajima, S. Fujinami, T. Yamagishi and Y. Nakamoto, J. Org. Chem., 2011, 76, 328–331 CrossRef CAS PubMed.
- T. Ogoshi and T. Yamagishi, Chem. Commun., 2014, 50, 4776–4787 RSC.
- G. Yu, C. Han, Z. Zhang, J. Chen, X. Yan, B. Zheng, S. Liu and F. Huang, J. Am. Chem. Soc., 2012, 134, 8711–8717 CrossRef CAS PubMed.
- C. Li, Q. Xu, J. Li, F. Yao and X. Jia, Org. Biomol. Chem., 2010, 8, 1568–1576 CAS.
- J. Yang, Z. Li, Y. Zhou and G. Yu, Polym. Chem., 2014, 5, 6645–6650 RSC.
- C. J. Li, J. Ma, L. Zhao, Y. Zhang, Y. Yu, X. Shu, J. Li and X. Jia, Chem. Commun., 2013, 49, 1924–1926 RSC.
- Z. Li, J. Yang, G. Yu, J. He, Z. Abliz and F. Huang, Chem. Commun., 2014, 50, 2841–2843 RSC.
- T. Ogoshi, M. Hashizume, T. Yamagishi and Y. Nakamoto, Chem. Commun., 2010, 46, 3708–3710 RSC.
- Y. Ma, X. Ji, F. Xiang, X. Chi, C. Han, J. He, Z. Abliz, W. Chen and F. Huang, Chem. Commun., 2011, 47, 12340–12342 RSC.
- Y. Yao, M. Xue, X. Chi, Y. Ma, J. He, Z. Abliz and F. Huang, Chem. Commun., 2012, 48, 6505–6507 RSC.
- T. Ogoshi, N. Ueshima and T. Yamagishi, Org. Lett., 2013, 15, 3742–3745 CrossRef CAS PubMed.
- T. Ogoshi, R. Shiga and T. Yamagishi, J. Am. Chem. Soc., 2012, 134, 4577–4580 CrossRef CAS PubMed.
- H. Li, Q. Chen, C. Schönbeck and B.-H. Han, RSC Adv., 2015, 5, 19041–19047 RSC.
- X. Liao, L. Guo, J. Chang, S. Liu, M. Xie and G. Chen, Macromol. Rapid Commun., 2015, 36, 1492–1497 CrossRef CAS PubMed.
- W. Xia, M. Ni, C. Yao, X. Wang, D. Chen, C. Lin, X.-Y. Hu and L. Wang, Macromolecules, 2015, 48, 4403–4409 CrossRef CAS.
- C. Zobrist, J. Sobocinski, J. Lyskawa, D. Fournier, V. Miri, M. Traisnel, M. Jimenez and P. Woisel, Macromolecules, 2011, 44, 5883–5892 CrossRef CAS.
- Y. Guan, M. Ni, X. Hu, T. Xiao, S. Xiong, C. Lin and L. Wang, Chem. Commun., 2012, 48, 8529–8531 RSC.
- C. Han, G. Yu, B. Zheng and F. H. Huang, Org. Lett., 2012, 14, 1712–1715 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5py01186h |
| This journal is © The Royal Society of Chemistry 2015 |