
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Aqueous asymmetric aldol reactions in polymersome membranes†
Matthijs C. M.
van Oers
,
Wouter S.
Veldmate
,
Jan C. M.
van Hest
* and
Floris P. J. T.
Rutjes
*
Radboud University, Institute for Molecules and Materials, Heyendaalseweg 135, 6525 AJ Nijmegen, The Netherlands. E-mail: J.vanHest@science.ru.nl; F.Rutjes@science.ru.nl
First published on 3rd July 2015
Abstract
L-Proline catalysts have been immobilised in the hydrophobic domain of a polymersome via a copper(I)-catalysed azide–alkyne cycloaddition (CuAAC) reaction. Utilisation of these nanoreactors in the asymmetric aldol reaction of cyclohexanone with 4-nitrobenzaldehyde afforded the corresponding β-hydroxyketones in quantitative yields and with excellent enantio- and diastereoselectivities. The polymersomes were recycled up to five times without any loss in activity or selectivity.
The field of asymmetric organocatalysis has witnessed a tremendous growth in the last decade. Ever since the pioneering work of List1 and Macmillan,2 chemists have been seeking novel applications to exploit the catalytic nature of small organic molecules. One emerging trend has been the utilisation of organocatalysts in aqueous media. Despite the apparent benefits of using water as a solvent, realisation of this concept has been far from trivial since the presence of water often hampers the formation of the intermediate in enamine-mediated organocatalytic reactions. For instance, in the L-proline-catalysed asymmetric aldol reaction of 4-nitrobenzaldehyde with ketone donors, yields and selectivities significantly drop when water is added to the reaction mixture.3 To overcome these incompatibility issues, the organocatalyst has to be effectively shielded from the aqueous environment. The research groups of Barbas,4 Hayashi5 and Noto6 solved this problem by adding a hydrophobic group to the proline catalyst which sequesters the enamine intermediate away from water. Armstrong and coworkers7 used a cyclodextrin that could bind a tert-butylphenoxyproline molecule to achieve a site-isolated catalytic system in water. Other strategies involve the immobilisation of organocatalysts on polystyrene resins,8,9 acrylic beads,10–12 micelles,13,14 and stimuli-responsive polymers.15–17 Most of these heterogeneous catalysts take advantage of a confined hydrophobic microenvironment that maximises the substrate concentration around the catalyst.18 This often results in an increase in reaction rate compared to the activity of the homogeneous catalyst under non-aqueous conditions.
In a previous report19 we demonstrated the embedding of a chiral copper bis(oxazoline) catalyst inside the hydrophobic domain of a polymersome membrane via a copper(I)-catalysed azide–alkyne cycloaddition (CuAAC) reaction.20,21 By applying these polymersomes in an aqueous asymmetric cyclopropanation reaction between ethyl diazoacetate and styrene derivatives, we showed that the corresponding cyclopropane products were obtained in high yields and enantioselectivities. These results encouraged us to investigate the immobilisation of other catalysts inside the polymersome bilayer. Given the powerful nature of L-proline in catalysing asymmetric aldol reactions, we envisioned that affixation of this organocatalyst in the membrane of a polymersome would yield a catalytic system that could mimic the activity of an aldolase enzyme.22 Not only would this afford a novel catalytic nanoreactor for the synthesis of enantiopure aldol products, the presence of an inner aqueous cavity would potentially allow the encapsulation of a biocatalyst which could work in concert with the organocatalyst to enable one-pot aqueous cascade reactions. Additionally, we expected that the polymersomes could readily be separated from the products by extraction, which would allow them to be reused in subsequent aldol reactions.
To allow the immobilisation of L-proline in the polymersome membrane, it had to be functionalised with an alkyne tail. This was achieved by alkylating N-Boc-trans-4-hydroxy-L-proline with propargyl bromide, followed by a subsequent deprotection step with HCl to provide catalyst 2 in 57% overall yield (Fig. 1).
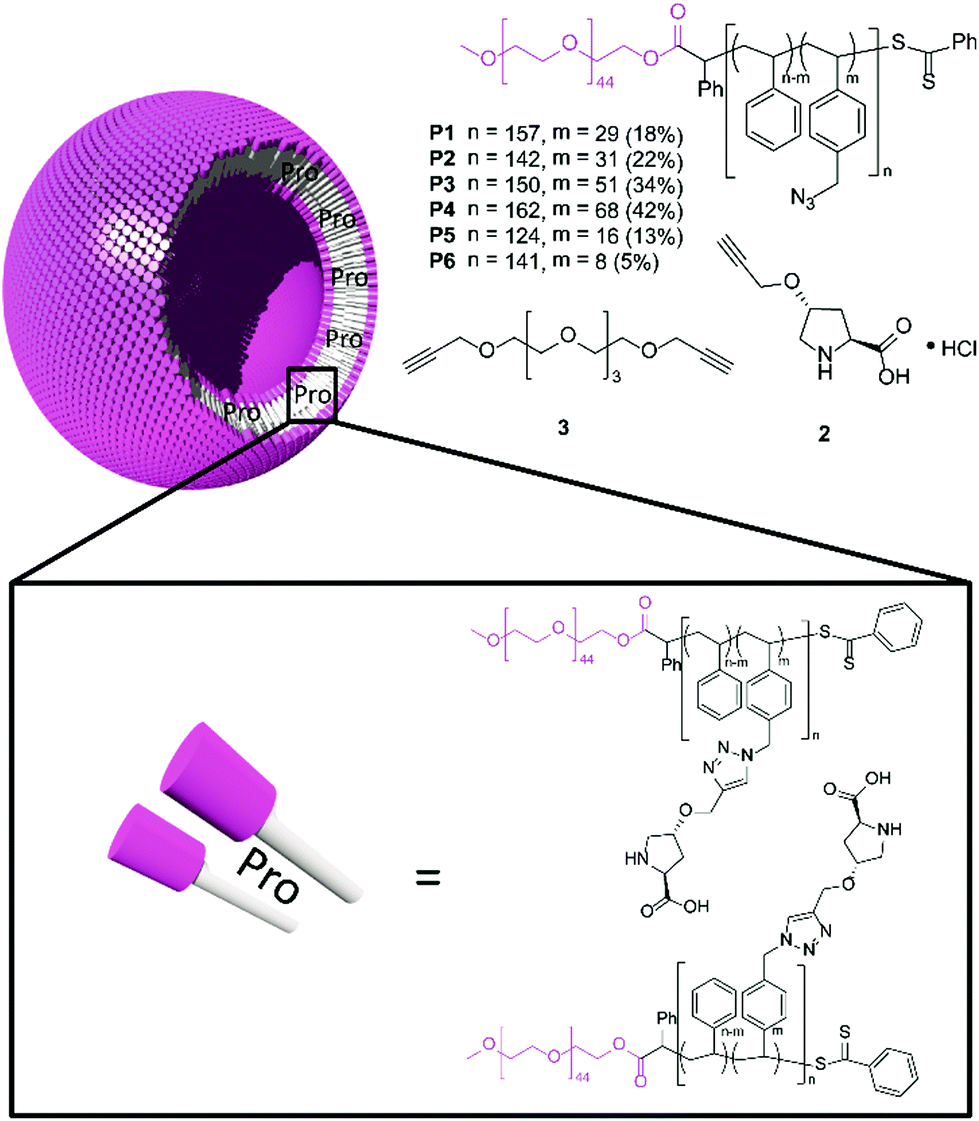 | ||
| Fig. 1 Chemical structures of azide-functionalised block copolymers, catalyst 2 and cross-linker 3 (top) and a schematic representation of a proline-loaded polymersome membrane (bottom). | ||
Next we turned our attention to the synthesis of the block copolymers. Following the described conditions for the preparation of poly(ethylene glycol)-b-poly(styrene-co-4-vinylbenzyl azide) (PEG-b-P(S-co-4-VBA)) polymers, we obtained block copolymers P1–P6 with varying azide contents, simply by changing the ratio between styrene and 4-vinylbenzyl chloride during the Reversible Addition–Fragmentation chain Transfer polymerisation (RAFT) (Table S1†).
Subsequent polymersome assembly was achieved through the addition of ultrapure water to a solution of the block copolymers in THF, also known as the cosolvent method.23 In an initial attempt to produce the catalytic polymersomes, we added proline catalyst 2 together with CuSO4·5H2O, bathophenanthroline sulfonated sodium salt and ascorbic acid to a dispersion of the polymersomes in H2O![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :THF (50:50, v/v). Although Fourier Transform Infrared (FTIR) spectroscopy showed a full conversion of the azides after three days of stirring, the functionalisation was accompanied with a significant loss in turbidity of the sample. This suggested a decrease in the number of polymersomes, which can be explained by the introduction of the proline moieties inside the hydrophobic block of the membrane. Since proline is very hydrophilic, anchoring many of these catalysts to the hydrophobic styrene monomers might switch the hydrophobicity of the polystyrene block to hydrophilic and make the block copolymers soluble in the H2O:THF mixture. Adding a small amount of crosslinker 3 (Fig. 1) to the polymersome dispersion provided a solution to this problem. The CuAAC reaction could now still go to full completion (Fig. S2b†), while the integrity of the polymersomes was preserved, as confirmed by Transmission Electron Microscopy (TEM, Fig. S1†) and Dynamic Light Scattering (DLS, Fig. S2a†). The slightly smaller size of the polymersomes after functionalisation might have been the result of an increase in polarity of the hydrophobic block which is known to have a negative effect on the polymersome diameter.24 TEM analysis of the membrane revealed that the thickness was not significantly affected by the functionalisation and was estimated to be 25–35 nm (Fig. S3†).
:THF (50:50, v/v). Although Fourier Transform Infrared (FTIR) spectroscopy showed a full conversion of the azides after three days of stirring, the functionalisation was accompanied with a significant loss in turbidity of the sample. This suggested a decrease in the number of polymersomes, which can be explained by the introduction of the proline moieties inside the hydrophobic block of the membrane. Since proline is very hydrophilic, anchoring many of these catalysts to the hydrophobic styrene monomers might switch the hydrophobicity of the polystyrene block to hydrophilic and make the block copolymers soluble in the H2O:THF mixture. Adding a small amount of crosslinker 3 (Fig. 1) to the polymersome dispersion provided a solution to this problem. The CuAAC reaction could now still go to full completion (Fig. S2b†), while the integrity of the polymersomes was preserved, as confirmed by Transmission Electron Microscopy (TEM, Fig. S1†) and Dynamic Light Scattering (DLS, Fig. S2a†). The slightly smaller size of the polymersomes after functionalisation might have been the result of an increase in polarity of the hydrophobic block which is known to have a negative effect on the polymersome diameter.24 TEM analysis of the membrane revealed that the thickness was not significantly affected by the functionalisation and was estimated to be 25–35 nm (Fig. S3†).
After a dialysis step to remove THF and the CuAAC catalyst we employed the polymersomes in the benchmark reaction between 4-nitrobenzaldehyde and cyclohexanone. Starting with polymersomes constructed out of block copolymer P1, a catalyst loading of 30 mol%, and an aldehyde concentration of 83 mM, we were able to recover 9% of the desired β-hydroxyketone 4 after 22 h of stirring (Table 1, entry 1). Despite the low yield, the diastereo- and enantioselectivity of the reaction were acceptable which confirmed the viability of our concept. We argued that the low yield could be ascribed to the low concentration of water inside the hydrophobic membrane. From previous studies it is known that the addition of a small amount of water helps to speed up the proline-catalysed aldol reaction in organic solvents.8,16,25 To increase the water content in the polymersome membranes, we opted to add a small amount of THF. Due to the compatibility with both water and polystyrene, THF can act as a plasticiser that renders the membrane more accessible to the aqueous environment. Indeed, adding 10% (v/v) of THF to the reaction mixture led to an increase in conversion to 40% whilst the diastereomeric excess (de) and the enantiomeric excess (ee) rose to an excellent 90% and 95%, respectively. Nevertheless, when the THF content was further increased to 25% the yield decreased again, implying that the presence of too much water interferes with catalysis (entry 3). Another improvement in reaction rate could be established upon raising the aldehyde concentration to 120 mM, providing β-hydroxyketone 4 in 62% yield (entry 4). We refrained from using higher concentrations of substrates to avoid phase separation between cyclohexanone and water.
|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | Reaction time (h) | Conc. aldehyde (mM) | H2O:THF |
Polymer | Azide content (%) | Conversionb,c (%) | anti/sync | ee antid (%) |
|
a Reactions were carried out at room temperature using a 4:1 ratio between cyclohexanone and 4-nitrobenzaldehyde, a catalyst concentration of 30 mol% and a reaction volume of 1.0 mL.
b Conversion of 4-nitrobenzaldehyde into β-hydroxyketone 4.
c Determined by 1H-NMR spectroscopy of crude product.
d Determined by chiral HPLC.
e
L-Proline was used as catalyst.
|
||||||||
| 1 | 22 | 83 | 100:0 |
P1 | 18 | 9 | 87/13 | 88 |
| 2 | 22 | 83 | 90:10 |
P1 | 18 | 40 | 95/5 | 95 |
| 3 | 22 | 83 | 72:25 |
P1 | 18 | 29 | 95/5 | 96 |
| 4 | 22 | 120 | 90:10 |
P1 | 18 | 62 | 95/5 | 95 |
| 5 | 22 | 120 | 90:10 |
P2 | 22 | 60 | 96/4 | 98 |
| 6 | 22 | 120 | 90:10 |
P3 | 34 | 17 | 92/8 | 95 |
| 7 | 22 | 120 | 90:10 |
P4 | 42 | 21 | 95/5 | 94 |
| 8 | 22 | 120 | 90:10 |
P5 | 13 | 31 | 94/6 | 92 |
| 9 | 22 | 120 | 90:10 |
P6 | 5 | 16 | 89/11 | 89 |
| 10e | 22 | 120 | 90:10 |
— | — | 4 | 71/29 | 1 |
| 11 | 72 | 120 | 90:10 |
P2 | 22 | 99 | 96/4 | 98 |
An interesting observation was made when we varied the catalyst content in the polymersomes. This was achieved by using block copolymers P2–P6 for the assembly of the polymersomes, all of which contained a different degree of azide functionalisation. It is worth mentioning that for fair comparison the catalyst concentration was kept at 30 mol% in all experiments. When the catalyst loading per polymersome was increased to 22% the reaction rate remained more or less constant (entry 5). Substantially lower yields were obtained, however, when polymersomes were used in which 34% and 42% of the styrene monomers were functionalized with a proline catalyst (entries 6–7). On the other hand, when the catalyst loading was lowered to 13% or 5% the reaction rate declined as well (entries 8–9). Although the exact reason for this trend remains unknown, we speculate that access of the substrates to the catalyst plays an important role. At low catalyst loadings the hydrophobic volume is relatively large due to the higher number of polymersomes that are required to attain a 30 mol% catalyst concentration. This leads to a reduced substrate concentration per membrane volume and therefore to a lower reactant availability at the catalytic site. In contrast, there is a considerably smaller substrate capacity in the membranes when using polymers with a high degree of functionalisation. As these membranes contain more catalysts, there is less room available to accommodate the substrates. This also results in a lower overall concentration of reactants accessible to the catalyst. It could well be that at an 18–22% functionalisation level the substrate concentration surrounding the catalyst is at a maximum which causes the reaction rate to peak. A similar effect was observed by Lu et al. when they investigated the aqueous asymmetric aldol reaction in PMMA nanogels.11 At identical catalyst loadings the conversion reached an optimum when the nanogel was functionalised for 2 wt% with L-proline monomers.
Applying nanogels with higher or lower degrees of functionalisation led to a significant drop in reaction rate. The sequestering effect of the polymersome membrane was demonstrated when we employed L-proline as a catalyst in the aqueous asymmetric aldol reaction. Under identical circumstances we could now only recover 4% of product 4. More compelling evidence was obtained when we measured the de and ee of the reaction. With an anti/syn ratio of 71/29 and an almost racemic mixture of enantiomers the control reaction could be considered as non-asymmetric. In a final attempt to improve the yield of the aldol reaction we extended the reaction time to 72 h (entry 11). Much to our satisfaction, β-hydroxyketone 4 was isolated nearly at full conversion while the diastereo- and enantioselectivity were preserved to an excellent degree.
With the optimised conditions established, we investigated the recyclability of the catalytic polymersomes. To ensure that all product after the first cycle was removed we decided to spin the polymersomes down after completion of the reaction. Next, we separated the aqueous supernatant, redispersed the polymersomes in ethyl acetate and centrifuged them again. After the organic phase was removed and combined with the aqueous supernatant, the polymersomes were redispersed in a H2O:THF mixture and new substrates were added to initiate the next reaction cycle.
Following this procedure we were able to reuse our catalytic polymersomes five consecutive times without noticeable deterioration of the catalyst or the interior structure of the polymersome membrane. More strikingly, both the yield and the de/ee did not show any decline over the course of the five cycles (Chart 1). This proves the robustness of our system and illustrates the potential of these polymersomes as green recyclable catalysts in aqueous asymmetric reactions.
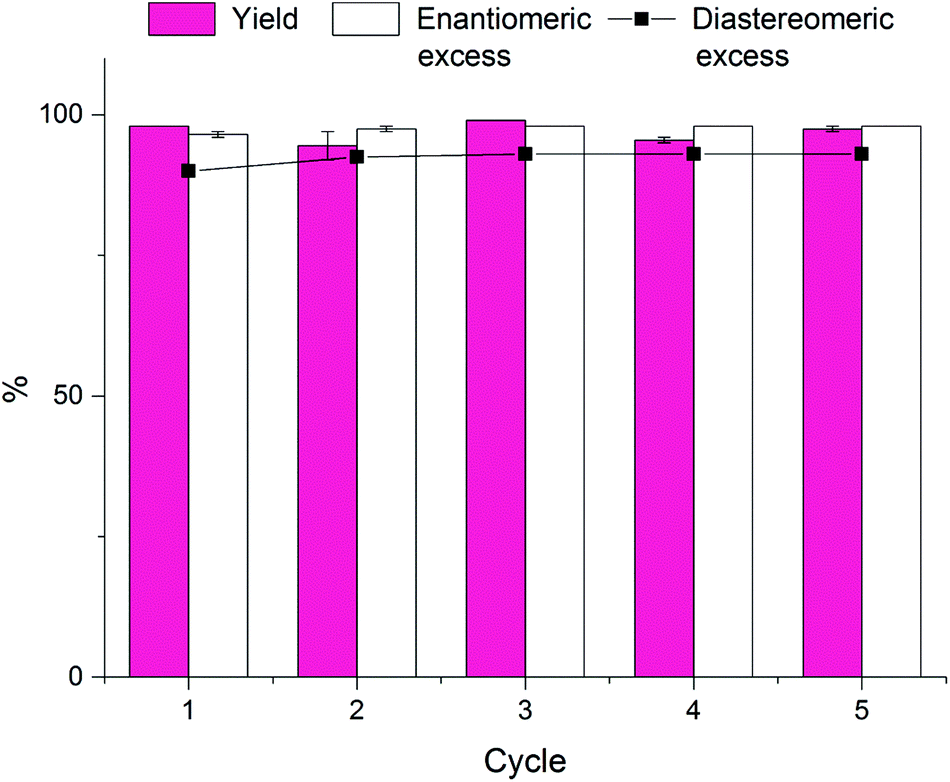 | ||
| Chart 1 Diagram of the isolated yield, enantiomeric excess, and diastereomeric excess of the asymmetric aldol reaction of 4-nitrobenzaldehyde with cyclohexanone repeated over five cycles with L-proline-functionalised polymersomes. Reactions were carried out in duplo in a 1.0 mL MilliQ:THF (90:10) mixture at room temperature using 4-nitrobenzaldehyde (120 mM), cyclohexanone (480 mM) and a 30 mol% catalyst loading. | ||
Conclusions
In summary, we have designed a polymersome nanoreactor capable of catalysing asymmetric aldol reactions in water with excellent yields, diastereoselectivities, and enantioselectivities. The successful immobilisation of an L-proline catalyst in the polymersome membrane was achieved via a CuAAC reaction which provided a protective hydrophobic environment for the enamine intermediate. The rate of the aldol reaction turned out to be highly dependent on the substrate concentration, the catalyst loading per polymersome, and the amount of plasticiser present in the aqueous solution. Optimising these conditions allowed us to produce β-hydroxyketone 4 in almost quantitative yield and with high enantio- and diasteromeric purity. Furthermore, we managed to recycle the polymeric vesicles up to five times while preserving the excellent catalytic properties of the polymersomes. With the possibility to expand the catalytic system with a second site-isolated (bio)catalyst and driven by the increasing demand for green reusable catalysts, we foresee a promising future for these polymeric nanoreactors as versatile platforms for multistep asymmetric reactions in aqueous solutions.Acknowledgements
We thank the Netherlands Research School Combination Catalysis (NRSCC) and the Ministry of Education, Culture and Science (Gravitation programme 024.001.035) for funding.Notes and references
- B. List, R. A. Lerner and C. F. Barbas, J. Am. Chem. Soc., 2000, 122, 2395 CrossRef CAS.
- K. A. Ahrendt, C. J. Borths and D. W. C. MacMillan, J. Am. Chem. Soc., 2000, 122, 4243 CrossRef CAS.
- A. Cordova, W. Notz and C. F. Barbas, Chem. Commun., 2002, 3024 CAS.
- N. Mase, Y. Nakai, N. Ohara, H. Yoda, K. Takabe, F. Tanaka and C. F. Barbas, J. Am. Chem. Soc., 2006, 128, 734 CrossRef CAS PubMed.
- Y. Hayashi, S. Aratake, T. Okano, J. Takahashi, T. Sumiya and M. Shoji, Angew. Chem., Int. Ed., 2006, 45, 5527 CrossRef CAS PubMed.
- F. Giacalone, M. Gruttadauria, P. Agrigento, P. Lo Meo and R. Noto, Eur. J. Org. Chem., 2010, 5696 CrossRef CAS PubMed.
- J. Huang, X. Zhang and D. W. Armstrong, Angew. Chem., Int. Ed., 2007, 46, 9073 CrossRef CAS PubMed.
- M. Gruttadauria, F. Giacalone, A. M. Marculescu, P. Lo Meo, S. Riela and R. Noto, Eur. J. Org. Chem., 2007, 4688 CrossRef CAS PubMed.
- D. Font, C. Jimeno and M. A. Pericas, Org. Lett., 2006, 8, 4653 CrossRef CAS PubMed.
- T. E. Kristensen, K. Vestli, K. A. Fredriksen, F. K. Hansen and T. Hansen, Org. Lett., 2009, 11, 2968 CrossRef CAS PubMed.
- A. Lu, D. Moatsou, D. A. Longbottom and R. K. O'Reilly, Chem. Sci., 2013, 4, 965 RSC.
- A. Lu, D. Moatsou, I. Hands-Portman, D. A. Longbottom and R. K. O'Reilly, ACS Macro Lett., 2014, 3, 1235 CrossRef CAS.
- B. H. Lipshutz and S. Ghorai, Org. Lett., 2012, 14, 422 CrossRef CAS PubMed.
- A. Lu, P. Cotanda, J. P. Patterson, D. A. Longbottom and R. K. O'Reilly, Chem. Commun., 2012, 48, 9699 RSC.
- E. Huerta, P. J. M. Stals, E. W. Meijer and A. R. A. Palmans, Angew. Chem., Int. Ed., 2013, 52, 2906 CrossRef CAS PubMed.
- A. Lu, T. P. Smart, T. H. Epps, D. A. Longbottom and R. K. O'Reilly, Macromolecules, 2011, 44, 7233 CrossRef CAS PubMed.
- L. Qin, L. Zhang, Q. X. Jin, J. L. Zhang, B. X. Han and M. H. Liu, Angew. Chem., Int. Ed., 2013, 52, 7761 CrossRef CAS PubMed.
- J. L. Zhang, M. X. Zhang, K. J. Tang, F. Verpoort and T. L. Sun, Small, 2014, 10, 32 CrossRef CAS PubMed.
- M. C. M. van Oers, L. K. E. A. Abdelmohsen, F. P. J. T. Rutjes and J. C. M. van Hest, Chem. Commun., 2014, 50, 4040 RSC.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004 CrossRef CAS.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596 CrossRef CAS.
- A. Heine, G. DeSantis, J. G. Luz, M. Mitchell, C. H. Wong and I. A. Wilson, Science, 2001, 294, 369 CrossRef CAS PubMed.
- P. L. Soo and A. Eisenberg, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 923 CrossRef CAS PubMed.
- T. Azzam and A. Eisenberg, Angew. Chem., Int. Ed., 2006, 45, 7443 CrossRef PubMed.
- N. Zotova, A. Franzke, A. Armstrong and D. G. Blackmond, J. Am. Chem. Soc., 2007, 129, 15100 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5py00872g |
| This journal is © The Royal Society of Chemistry 2015 |