
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Templated polymerizations on solid supports mediated by complementary nucleoside interactions†
Margarita
Garcia
,
Kristian
Kempe
,
David M.
Haddleton
*,
Afzal
Khan
and
Andrew
Marsh
*
Department of Chemistry, University of Warwick, Coventry, CV4 7AL, UK. E-mail: d.m.haddleton@warwick.ac.uk; a.marsh@warwick.ac.uk; Tel: (+44) (0)24 7652 3256, (+44) (0)24 7652 4565
First published on 12th January 2015
Abstract
Template-directed radical polymerizations on solid supports are presented, which take advantage of complementary nucleoside interactions in non-polar solvents. The templates are prepared through copper-mediated living radical polymerization allowing control over the length and dispersity of the polymer bound to the support. We report a degree of control over subsequent polymerization of a complementary base pair monomer in the presence of the template not observed in its absence. Templates achieved from uridine, adenosine, cytidine and guanosine substituted methacrylate monomers were successfully prepared. Uridine derived templates were found to provide good fidelity of replication by incorporating mainly adenosine monomer in the final polymer, from a mixture of monomers. The latter was separated from its template and characterized demonstrating the preparation of polymers of approximately half of the template length. A model for the templating process is proposed.
Introduction
The controlled, sequence-specific replication of DNA using a template-directed mechanism is central to life.1 The apparent simplicity of this templating process has inspired synthetic2–6 and semi-synthetic polymerization systems7–14 that use hydrogen bonding as a key mediating interaction between generational strands.15,16 Indeed, studies using RNA17 and DNA18,19 have demonstrated that enzyme-free extension of primers is possible with remarkably high fidelity. In addition, small interfering- and micro-RNAs that are transcribed from DNA continue to be uncovered, together with novel mechanisms to understand and control templating and translation processes.20 Hence, oligomeric and polymeric molecules that include functionality capable of interacting in a specific way with nucleic acids21 and other cellular targets22 are of great significance and continue to present synthetic, biophysical and biological challenges.To date, various post-polymerization modification methods and polymerization techniques have been applied for the preparation of synthetic nucleic acid analogues as summarized by Inaki15 and Smith.23 More recently McHale et al. highlighted achievements in this field with a focus on poly(nucleobase)s and poly(nucleoside)s macromolecules obtained by controlled (radical) polymerization techniques.24 Amongst them, atom transfer radical polymerization (ATRP) was predominantly employed for different nucleobase-25,26 and nucleoside-based monomers27,28 and initiators.29 In addition nitroxide-mediated radical polymerization (NMP)6,30 and reversible addition-fragmentation chain transfer (RAFT) polymerization31 have been reported to yield well-defined poly(nucleobase) systems.
Furthermore, a range of monomer units and template strands ranging from DNA,32 RNA or DNA partnered with thioester peptide nucleic acids (tPNAs),33 to synthetic constructs such as ribonucleoside and nucleobase bearing acrylates28 and methacrylates25,27,34 have been used to prepare templates for polymerization studies. Lynn and co-workers demonstrated the templated polymerization of amine-containing thymidine monomeric units in solution32 and on solid supports using reductive amination to form the polymer backbone.35 For this purpose, complementary nucleobase-pairing interactions between a natural DNA template, namely (dAp)8, and the synthetic monomer were exploited to enable sequence- and chain-length-specific polymerization. In other studies, synthetic templates prepared by, e.g. free radical polymerization,36 NMP6 and ring-opening metathesis polymerization (ROMP) were used.37 Well-defined daughter poly(nucleobase)s were obtained with chain lengths and dispersities similar to the template polymer37 and high molar masses via segregation of propagating chains in discrete micelle cores, respectively.6 Following our initial report on templated ATRP of complementary nucleoside monomers28 it became clear that a template, which allowed the resultant polymer–polymer complex to be separated more readily would be extremely beneficial, as would a template of controlled length and dispersity. In order to address these possibilities we opted to use ATRP, which we had previously shown to be compatible with the growth of methacryloyl nucleoside polymers both in solution27 and from a solid support.38
Herein, we describe the synthesis and characterization of the more challenging cytidine and guanosine ribonucleoside-based methacrylate monomers, to extend the uridine and adenosine materials prepared in our earlier work27 (Fig. 1). Copper-mediated living radical polymerization was employed for the preparation of poly(methacryloyl nucleoside)s bound to polystyrene bead supports (Wang-type resin). The choice of this solid support allowed the cleavage of the polymer and thus its detailed characterization. Furthermore, the potential of the individual templates for templated polymerizations was investigated.‡
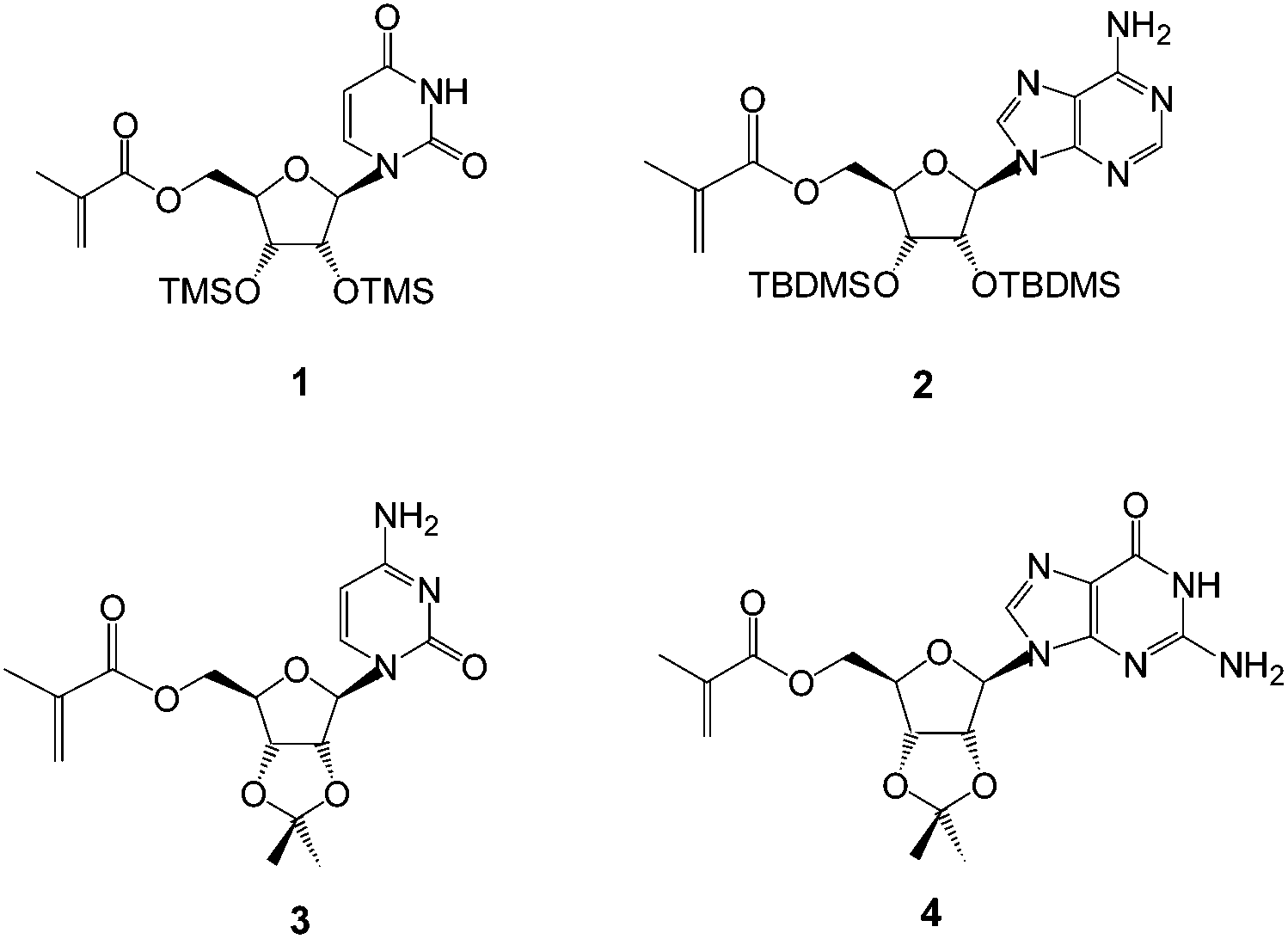 | ||
| Fig. 1 Structures of the methacryloylribonucleosides prepared, including uridine (1),27 adenosine (2),27 cytidine (3) and guanosine (4) derivatives. | ||
Results and discussion
Synthesis of monomers
The monomers chosen for this study were the 5′-methacryloylribonucleosides (Fig. 1). The preparation of uridine 1 and adenosine 2 derivatives has been described previously27 using an immobilized Candida antarctica lipase-promoted acylation reaction.39 The cytidine 3 and guanosine 4 derivatives were prepared according to Schemes 1 and 2.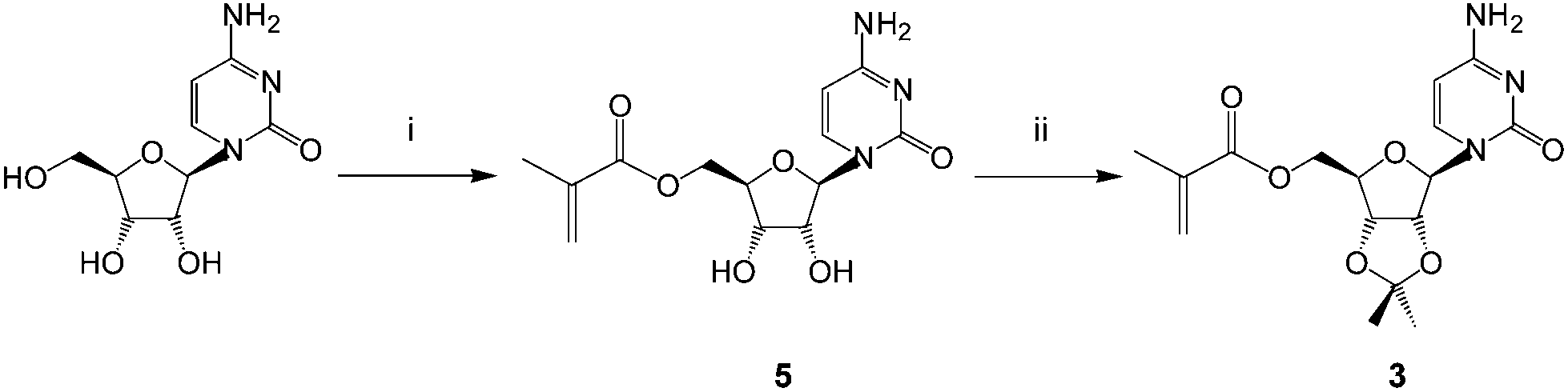 | ||
| Scheme 1 Synthesis of the cytidine derivative 3: (i) methacryloylacetone oxime, Novozyme 435®, dioxane, radical inhibitor, 60 °C, 22 h (47%); (ii) p-toluenesulfonic acid, triethylorthoformate, acetone, r.t., 43 h (55%). | ||
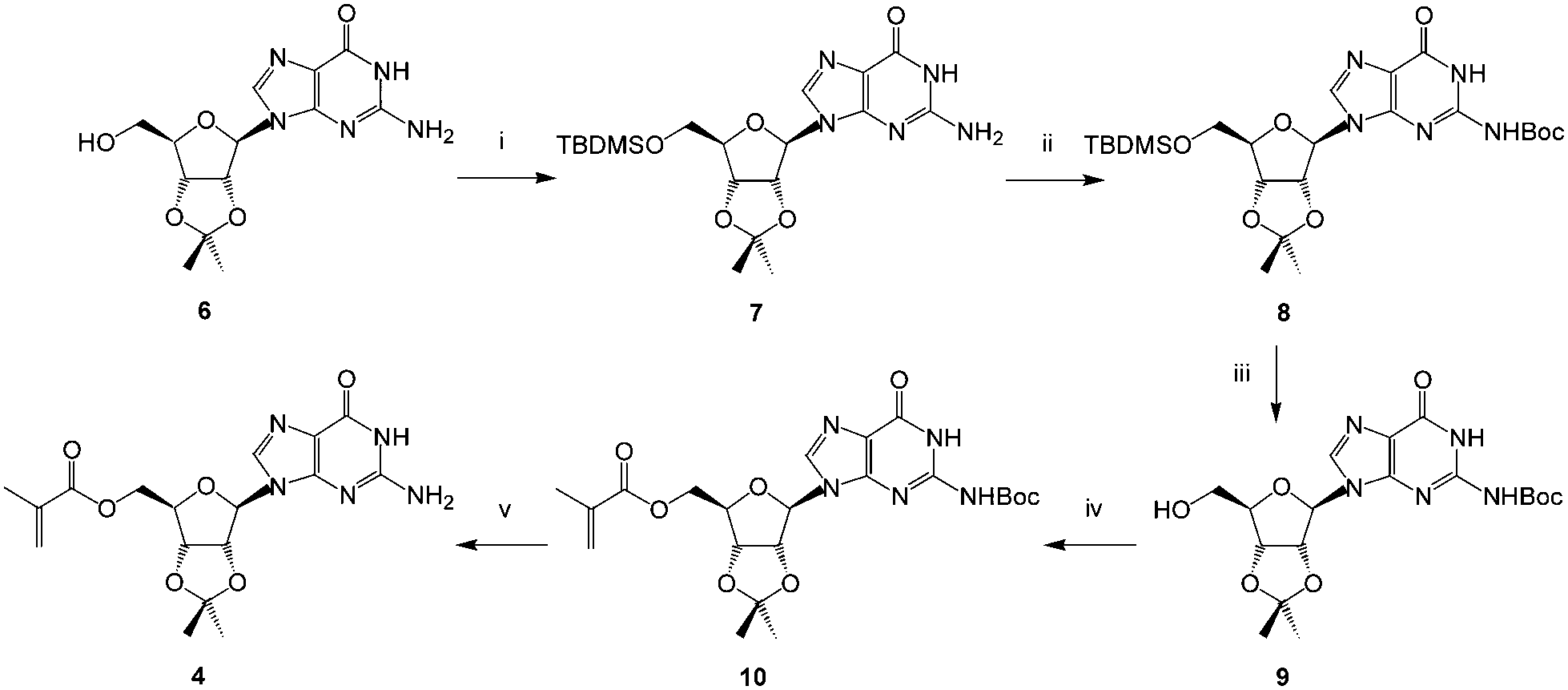 | ||
Scheme 2 Synthesis of the guanosine derivative 4: (i) TBDMSCl, Et3N, CH2Cl2, r.t., 72 h (88%); (ii) (Boc)2O, DMF, DMAP, 60 °C, 48 h (46%); (iii) TBAF 1 M in THF, r.t., 2 h (99%); (iv) methacryloylacetone oxime, Novozyme 435®, dioxane, radical inhibitor, 60 °C, 24 h (78%); (v) TFA![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :CH2Cl2 (1:1), r.t., 2 h (58%). :CH2Cl2 (1:1), r.t., 2 h (58%). | ||
Cytidine was reacted with methacryloylacetone oxime in the presence of C. antarctica lipase40 (Novozyme 435®) in dioxane and the product 5 was then protected as the 2′,3′-acetonide413 in reasonable yield (ESI†). Unlike Moris and Gotor's original report40 we found this enzyme-promoted reaction42 to allow direct 5′-O-acylation of cytidine. The acetonide protecting group was synthetically accessible and found to provide acceptable solubility in non-polar solvents in order to maximize templating hydrogen bonding and π-interactions. We found that the corresponding acrylate was not accessible by this route and required a lengthier protection sequence,43 to be reported elsewhere. Attempts to acylate guanosine in a regioselective fashion using this enzyme were also unsuccessful under a variety of conditions and again necessitated protection of the amino function as follows (Scheme 2 and ESI†).
Guanosine was protected as the 2′,3′-acetonide436 and then the 5′-hydroxyl protected as its TBDMS-ether 7. The heterocyclic amine was subsequently protected with a Boc group to give 8 and the silyl ether removed using tetrabutylammonium fluoride furnishing 9. Acylation of this newly exposed 5′-hydroxyl function using the methacryloylacetone oxime proceeded smoothly in the presence of Novozyme 435® giving the fully protected monomer 10.
In contrast, attempts to use non-enzymatic acylation methods including the anhydride44 or chloride led to intractable byproducts. The need to protect the exocyclic amine on the purine base is readily rationalized on the grounds that this group is more nucleophilic than the 5′-hydroxyl and may be better positioned to interact with the proposed serine-bound acylating reagent in the active site of the enzyme.45 Finally, deprotection of the amine gave the methacryloylguanosine 4 in reasonable overall yield and high purity.
Preparation of solid-support polymer templates
The method chosen for the growth of the polymer on the support was copper-mediated living radical polymerization as used previously for the preparation of silica-supported poly(methacryloyl nucleoside)s.38 In order to explore the scope of the template-directed polymerization on solid supports and exploit also the greater association constant of the G·C base pair as compared to the A·U pair, a full set of poly(methacryloyl nucleosides) bound to the resin were prepared. The supports used in the current work were polystyrene Wang-type resins that allowed acidic cleavage of the template polymer for analysis. The Wang initiator resin 11 (1.00 mmol g−1 initial loading) was prepared by analogy with a method used for derivatisation of a PEG support45 and it was possible to monitor the reaction by the appearance of the ester carbonyl stretch at 1734 cm−1 in the infrared (IR) spectrum. Polymerizations of the four different monomers (1–4) with the Wang resin solid support initiator 11 were carried out using copper(I) bromide as a catalyst and N-(n-propyl)-2-pyridylmethanimine as ligand in toluene in which the Wang resin has good swelling properties (Scheme 3).41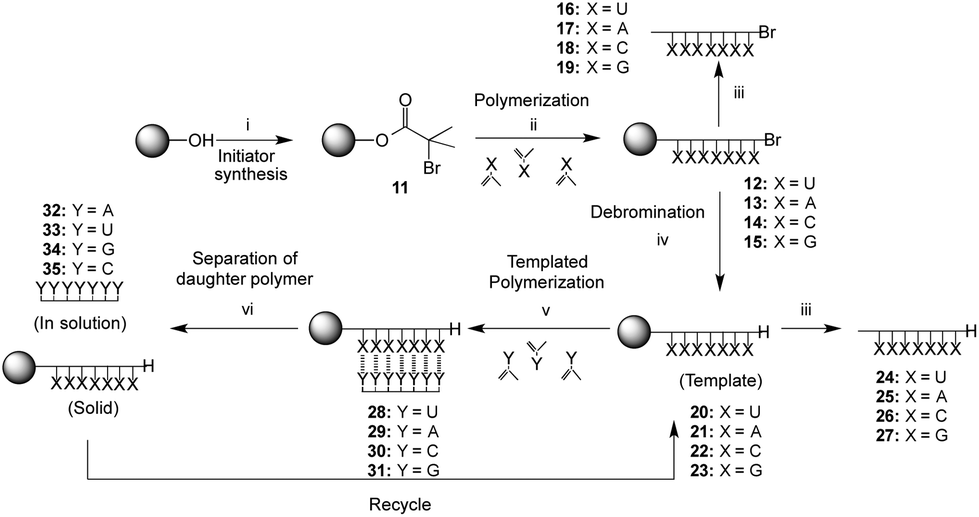 | ||
| Scheme 3 Solid support functionalization, template synthesis and templated polymerization of complementary monomer. ‘X’ and ‘Y’ represent the methacryloylribonucleosides (Fig. 1), ‘Y’ being the complementary ribonucleoside to ‘X’; Reagents and conditions: (i) bromoisobutyroylbromide, Et3N, CH2Cl2, r.t., 22 h; (ii) Y, CuBr, N-(n-propyl)-2-pyridylmethanimine, toluene, 110 °C, 64 h; (iii) TFA/CH2Cl2 (1/1), r.t., 18 h; (iv) Bu3SnH, AIBN, toluene, 100 °C, 20 h; (v) Y, AcOEt–toluene (2:1), AIBN, 60 °C, 42 h; (vi) CF3CH2OH, r.t., 1 h. | ||
In all cases, a mass increase after polymerization was observed, 180, 220, 240 and 120% for the polymerization of 1, 2, 3 and 4, respectively. The polymerizations were followed by IR spectroscopy (Fig. 2A), observing the appearance of a broad NH signal (3600–3100 cm−1) and a shift and an increase in the intensity of the carbonyl signal (1689 cm−1). Furthermore, gel phase 1H NMR provided valuable information about the successful polymerization, as exemplarily shown for poly(methacryloyl uridine) bonded to Wang resin (12) (Fig. 2).
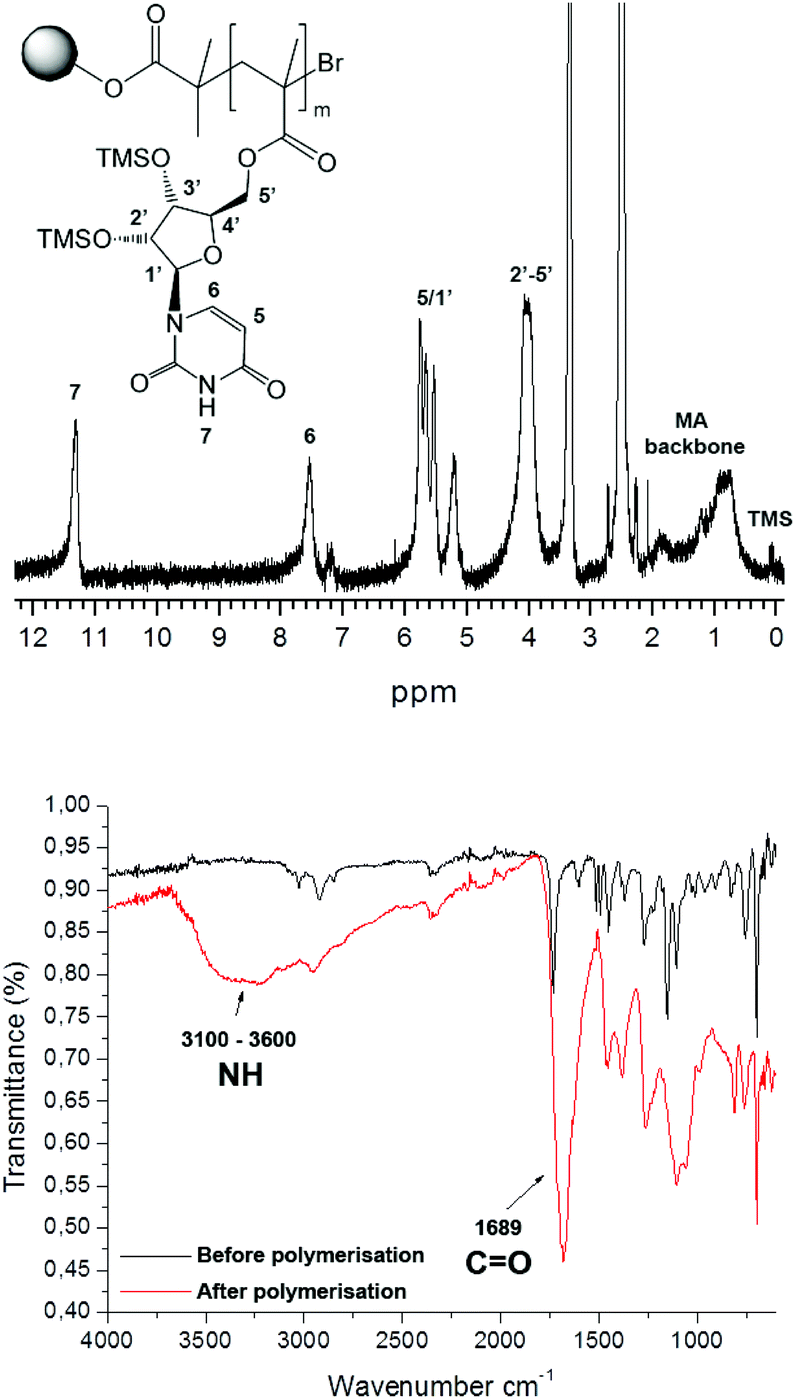 | ||
| Fig. 2 Gel phase 1H NMR (top) and IR (bottom) spectrum of poly(methacryloyl uridine) 12 bound to Wang resin. | ||
In order to characterize the poly(methacryloyl uridine) further, it was cleaved from the resin by treatment with trifluoroacetic acid. It was found that the silyl protecting groups of the uridine and adenosine units were also removed under these conditions, making the product polymers insoluble in tetrahydrofuran (THF) and also not very soluble in dimethylformamide (DMF). Full re-protection of this polymer with TMS groups was not achieved. In contrast, the isopropylidene-protecting groups of the cytidine and guanosine moieties were not affected. In order to perform templated polymerizations, debromination of the resin bound polymers with tributyltin hydride and AIBN as initiator was deemed necessary, as the terminal bromide could inhibit radical propagation. The debromination of the polymer bound to the resin was followed by IR, which indicated the loss of the C–Br bond by the disappearance of the band around 700 cm−1. Evidence for successful debromination of poly(methacryloyl cytidine) and poly(methacryloyl guanosine) was less assured since a C–Br peak was still discernible after treatment with tributyltin hydride in these cases. Prior to carrying out the template polymerization, the debrominated polymers were cleaved from the resin for full characterization. Reasonable molar masses and relatively low dispersities were found for uridine, cytidine and guanosine based polymers (Table 1), which is in accordance with a living mechanism for polymer growth.
Adenosine polymers 17 and 25 were found to be less well defined in agreement with observations made previously28 using adenosine monomers. This could be due to (i) coordination of the copper(I) catalyst by amine moieties in monomer and polymer (ii) adenine's tendency to aggregate in solution and hydrogen bond to itself, including through Hoogsteen interactions, generating a less well solvated environment in which the polymerization takes place. For comparison, it has been shown by Rotello and co-workers that triazine polymers undergo folding due to intramolecular hydrogen bonding to give highly compact micelle-like structures.46
Polymerizations in the presence of the solid-support polymer templates
The prepared poly(nucleoside) modified resins, 20 (uridine), 21 (adenosine), 22 (cytidine) and 23 (guanosine), represent excellent candidates for the templated polymerization of the complementary nucleoside monomers, allowing for a straightforward separation of the template polymer from the resin. The complementary monomers were polymerized in the presence of the respective template resin under free radical conditions using AIBN as initiator. First, the silyl protected methacryloyl adenosine 2 was polymerized with the poly(methacryloyl uridine) template 20 to generate complex 28 with a ratio of adenosine to uridine of 1:1, as determined by gel phase 1H NMR. It was possible to confirm the presence of the hydrogen bonds between these polymers by IR in the solid state, observing significant shifts in the amino N–H region [from 3223 (polymer) and 3260 (monomer) cm−1 to 3396, 3207 and 3063 cm−1] and carbonyl [from 1668 (polymer) and 1685 (monomer) to 1693 and 1634 cm−1].
The daughter polymer 32, poly(methacryloyl adenosine), was separated from the template 20 by simply washing the complex 28 with trifluoroethanol and precipitation in diethyl ether. 1H NMR and IR of both the regenerated template resin 20 and the daughter polymer 32 showed that, firstly, the template 20 can be re-used for further template polymerizations and, secondly, poly(methacryloyl adenosine) 32 was obtained. As a control experiment, adenosine homopolymer 36 was prepared in solution from methacryloyl adenosine monomer 2 under the same conditions employed for the template polymerization showing the same 1H NMR and IR spectrum as the daughter polymer 32. SEC measurements revealed molar masses and dispersities lower than those found in the corresponding parent polymer, poly(methacryloyl uridine) 24 (Table 2).47 The average DP of the poly(methacryloyl adenosine) 32 was approximately half that of the poly(methacryloyl uridine) 24, possibly caused by chain transfer during the polymerization, although the fact that this is not observed with a 1:1 mixture of methacryloyl uridine and methacryloyl adenosine monomers (Scheme 4) suggests one of several possible effects.
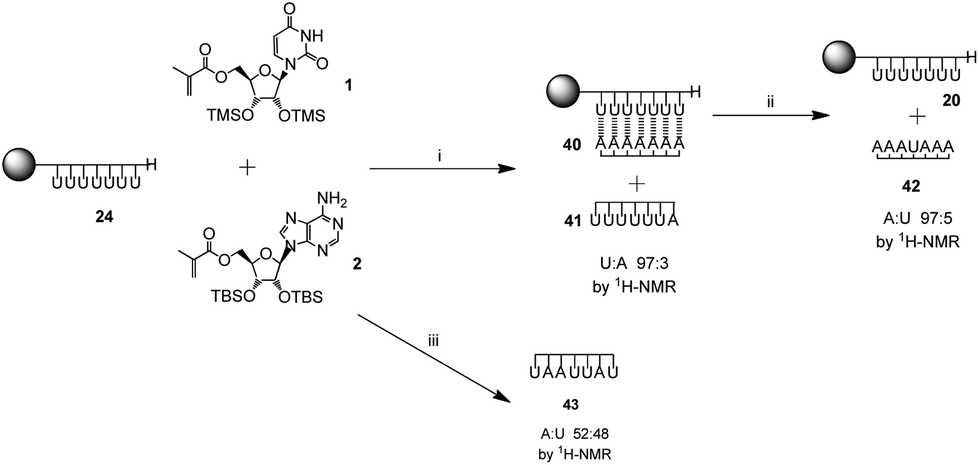 | ||
| Scheme 4 Control experiment: templated polymerization of methacryloyl adenosine 2 using template 24 in the presence methacryloyl uridine 1: (i) AIBN, toluene–ethyl acetate (1:2), 60 °C, 24 h; (ii) CF3CH2OH, r.t., 30 min. Solution polymerization of monomer 1 and 2 (iii): AIBN, toluene–ethyl acetate (1:2), 60 °C, 24 h. | ||
Firstly, a model whereby the propagating adenosine daughter polymer initially complexes to one site on the immobilized poly(methacryloyl uridine) template can be proposed. The interactions between template and growing daughter polymer are relatively weak at this stage with the most stable polymer complex formed by maximizing hydrogen bonding (Fig. 3). This use of Hoogsteen-type triplex hydrogen bonding, well-known for adenine48 strengthens the interaction whilst effectively reducing the available length of the template by a factor of two as it folds onto the propagating polymer.
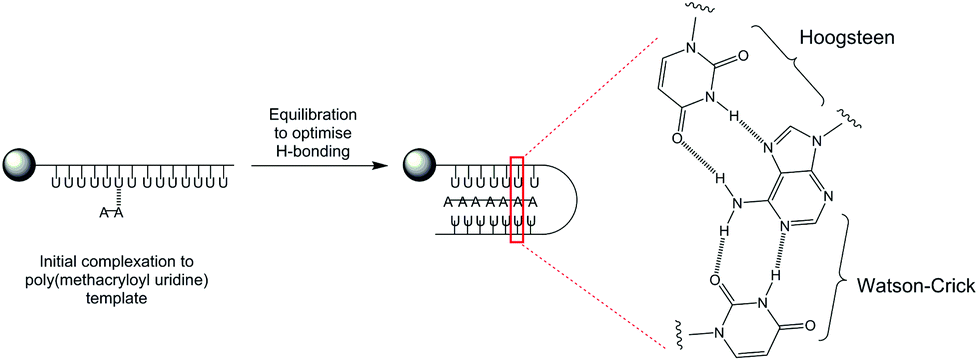 | ||
| Fig. 3 Proposed templating mechanism with hydrogen-bonding interactions between poly(methacryloyl uridine) template and growing poly(methacryloyl adenosine) using Watson-Crick and Hoogsteen interactions. | ||
Secondly, another possible explanation might be that only the shorter uridine polymers unfold or are solvated sufficiently on the resin to act as a template, leading to the observed shorter polyadenosine product in this case, although uridine does not significantly self-associate.
The templating process provided more control over the polymerization of methacryloyl adenosine 2 than the homopolymerization in absence of the template. This is evident when comparing the SEC analyses obtained for the daughter polymer 32 with the homopolymer 37 (Table 2). However, the inconsistency of these results with those obtained by gel phase 1H NMR from complex 28 (a ratio of adenosine to uridine of 1:1) suggests, thirdly, that some polymer products may be retained on the support, despite the effective solvation provided by trifluoroethanol.
To further explore the limits of the templating effect of the immobilized poly(methacryloyl uridine) 20, the polymerization was carried out both in the presence of the complementary monomer methacryloyl adenosine 2 and the monomer present in the template, i.e. the non-complementary methacryloyl uridine 1 (Scheme 4). Both monomers 1 and 2 were present in the same ratio and in equal amount to the accessible sites of the template 20. An insoluble complex 40 was obtained which showed a ratio of uridine to adenosine of 1:1 by gel phase 1H NMR. This was similar to the complex 28 obtained for the template polymerization of pure methacryloyl adenine 2 and resembles the results obtained in solution for acryloyl monomers.28 A soluble polymer 41, which mostly contained uridine repeating units, was also obtained (uridine to adenosine 97:3 by 1H NMR). Moreover, separation of the strands in complex 40 with trifluoroethanol showed a polymer 42 mostly composed of adenosine repeating units (adenosine to uridine 95:5 by 1H NMR). The template 20 was also recovered. These results indicate that, as expected, poly(methacryloyl uridine) is a good template for the polymerization of methacryloyl adenosine with high selectivity.
When a statistical copolymerization was carried out in solution under the same conditions as those used for the template polymerization (monomers in equimolar amount in the presence of 3% AIBN with toluene as solvent), a copolymer 43 with ratio of uridine and adenosine of 52:48 by 1H NMR was obtained (data not shown). Hence the observation that a greater proportion of adenosine is incorporated in the daughter polymer under templated conditions is good evidence that solid-supported polymerizations exert control through specific complementary ribonucleoside interactions over the polymerization process. This experiment is consistent with templating through a combination of both Watson-Crick and Hoogsteen interactions, depicted in Fig. 3 since incorporation of uridine would lack these stabilizing interactions.
Polymerization using immobilized poly(methacryloyl adenosine) 21 as template was performed with the complementary monomer, methacryloyl uridine 1. Based on the increase in weight, similar yields were obtained as for the polymerization of methacryloyl adenosine 2 using immobilized poly(methacryloyl uridine) 20. Furthermore, hydrogen bonds were observed between the polymers by IR. 1H NMR and IR spectra of the poly(methacryloyl uridine) daughter polymer 33 were similar to those obtained of the cleaved immobilized poly(methacryloyl uridine) 24. SEC measurements of polymer 33 showed lower molar masses (Mn = 2100 g mol−1; Đ = 1.7) than those of its parent polymer 25. This is in agreement with what was found for the acryloyl monomers28 and it has been shown previously that the poly(acryloyl adenosine) shows a less good performance as template than poly(acryloyl uridine). Moreover, it appears that the complementary uridine by itself did not provoke sufficient unfolding of the adenosine polymer 21. Inaki and co-workers have shown that polymers containing the adenine base are prone to intramolecular hydrogen bonding interactions.49 When the poly(methacryloyl uridine) 36 was prepared by a non-templated polymerization process, SEC analysis of the corresponding homopolymer 36 showed a dispersity similar to that of polymer 33. However, the molecular weight was higher (Mn = 14400; Đ = 1.51). This confirms that the presence of the poly(methacryloyl adenosine) as template inhibits the synthesis of poly(methacryloyl uridine).
In a similar manner poly(methacryloyl cytidine) 22 and poly(methacryloyl guanosine) 23 modified resins were employed for the template polymerization of their complementary monomers, methacryloyl guanosine 4 and methacryloyl cytidine 3, respectively. IR measurements of the complexes demonstrated the presence of hydrogen bonds. Separation of the putative daughter polymers from the complexes afforded only unchanged monomers with no evidence of polymer. In both cases, template polymerization apparently did not take place under the conditions chosen and it seems the resins have inhibited the radical polymerization observed in the absence of the supported template. This effect seems not to be due to the presence of a bromine end-group on the resin bound polymer, nor specific to Wang resin since the radical polymerizations were carried out on resins treated with tribuyltin hydride. It is entirely possible that self-association in both the cases of cytidine, where (C·C)n i-motif tetraplexes50 are well recognized, especially in longer pyrimidine tracts, and guanosine, which is undoubtedly forming a variety of robust ribbon51 and quartet structures52 potentially inhibiting template effects. Other explanations include the isopropylidene protecting group adversely altering sugar or polymer backbone conformation.
Conclusions
The potential of immobilized poly(methacryloyl nucleosides) as templates for the polymerization of their complementary nucleoside monomers is presented for the first time. Methacryloyl cytidine and guanosine ribonucleoside monomers were prepared to add to uridine and adenosine, monomers available from our previous work and used in copper-mediated living radical polymerizations initiated by functional Wang resins. The poly(methacryloyl nucleoside) modified resins were characterized by IR and 1H NMR spectroscopy. The choice of the resin enabled the acidic cleavage of the polymer and thus more complete characterization using solution NMR and SEC. All four immobilized polymers were applied in templated polymerizations of their respective complementary nucleoside monomer exploiting complementary base pair interactions of uridine and adenosine as well as cytidine and guanosine. Poly(methacryloyl uridine) templates were found to be suitable candidates for the templated polymerization of the complementary methacryloyl adenosine monomer. This result reflected what was observed in solution.28 The great advantage of this approach, however, is the covalent linkage of the parent polymer to the solid support, which allows for a straightforward separation of the templated daughter polymer. The latter appeared to differ from the parent polymer in length and dispersity as measured by SEC, which was attributed to a dual hydrogen bonding complexation of the growing daughter polymer to the template through Watson-Crick, and possibly Hoogsteen-type, hydrogen bonding interactions. Competition experiments with non-complementary monomer and in the absence of the immobilized template, respectively, demonstrate the superiority of the template process. This particular templated polymerization shows that solid-supported polymerizations can provide good control over the polymerization process through specific hydrogen bonding interactions.Our results contrast both with cellular replication in the presence of polymerases, transcription factors and other proteins which help maintain replicating oligonucleotide strands in their functional state53 and with enzyme-free primer extensions.14,18,19 In summary, we demonstrate the utility of a polymer-supported template, and our experiments also highlight the potential for tuning solvent,54 and chaperone molecules to create abiotic replicating systems.55,56
Experimental
Materials and instrumentation
All reactions were carried out under an atmosphere of nitrogen using standard Schlenk techniques. The synthesis of 5′-methacryloyluridine 1,27 5′-methacryloyladenosine 2,27 5′-methacryloylcytidine 5,27 methacryloylacetone oxime,27 Wang resin initiator 1157 and the ligand N-(n-pentyl)-2-pyridylmethanimine58 were synthesized as previously reported and stored under anhydrous conditions prior to use. Copper(I) bromide (Aldrich, 98%) was purified according to the method of Keller and Wrcoff.59 All other reagents were purchased from Aldrich or Lancaster Synthesis at the highest purity available and used without further purification unless otherwise stated. Solvents were of the highest grade available and were used as supplied. DSC was carried out on a Perkin Elmer Pyris 1 differential scanning calorimeter. TGA were recorded on a Perkin Elmer TGA 7 apparatus. Temperatures reported are Tonset. IR spectra were recorded on an Avatar 320 FT-IR fitted with a “Golden gate” attenuated total reflection (ATR) cell attachment. NMR spectra were recorded with a Bruker DPX 300. Chemical shifts (δ) are quoted in ppm using residual non-deuterated solvents as internal standard [d6-DMSO (1H, δ: 2.50 ppm; 13C, δ: 39.5 ppm)]. Coupling constants (J) are quoted in Hz. Molar mass distributions with PMMA standards in the range (200 to 6.85 × 105 g mol−1) used for specific calibration were measured using size exclusion chromatography (SEC) at ambient temperature, on a system equipped with a guard column and on 2× mixed-C columns (Agilent) with differential refractive index detection using DMF as eluent, at a flow rate of 0.5 ml min−1. Sample solutions were prepared by adding 2.0 ml of solvent to 4.0 mg of sample; leaving 72 h at 60 °C to dissolve. Molar mass distributions with PEO/PEG standards were carried out at RAPRA Polymer Laboratories. They were measured using size exclusion chromatography (SEC) at 80 °C, on a system equipped with a PLgel 2× mixed bed-B 30 cm and 10 μm columns with differential refractive index detection using DMF with 1% of lithium bromide as eluent, at a flow rate of 1.0 ml min−1. Sample solutions were prepared by adding 10 ml of solvent to 20 mg of sample, leaving overnight and warming, if necessary, at 80 °C for 20 minutes to dissolve. After thorough mixing, the solutions were filtered through a 0.2 μm PTFE membrane prior to chromatography. Nomenclature of the compounds refers to a mixture of systematic and trivial nomenclature, which is in accordance with that used by other authors. The letters U, A, C and G during the text corresponds to methacryloyl uridine, methacryloyl adenosine, methacryloyl cytidine and methacryloyl guanosine, respectively. The letters U and A in the NMR refers to protons of uridine and adenosine, correspondingly. Yields of the polymerizations were determined gravimetrically.
Resin initiated polymerization of 1 (general procedure)
A mixture of the Wang initiator-resin 11 (300.0 mg, 0.30 mmol), 5′-O-methacryloyl-2′,3′-O-trimethylsilyluridine 1 (1.37 g, 3.00 mmol) and copper(I) bromide (43.0 mg, 0.30 mmol) was placed in a Schlenk and de-aerated. The ligand N-(n-pentyl)-2-pyridylmethanimine (105.8 mg, 0.60 mmol) was added in de-aerated toluene (5.00 ml) and the reaction mixture was de-aerated four times using a freeze–pump–thaw cycle and then heated at 110 °C for 64 h. The solid was filtered, washed with water, methanol and acetone and dried in vacuo to afford the poly(methacryloyl uridine) substituted Wang resin 12 (849.3 mg, 120% weight increased) as a light brown solid; TGA 310.3, 355.8, 354.2, 461.0 and 540.0 °C; Tonset 244.5 °C; νmax/cm−1 (solid): 3430 (N–H st), 2970 (C–H st), 1737 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O st), 1668 (CO st), 1149 and 1103 (C–O as st), 1059 (C–O sym st), 698 (C–Br st); δH (d6-DMSO, gel phase, 300 MHz): 11.27 (1H, bs, NH), 7.58 [1H, bs, H(6)], 5.78–5.38 [2H, m, H(1′) and H(5)], 4.33–3.78 [5H, m, H(2′–5′)], 1.00–0.60 (5H, m, CH3 and CH2), 0.05 [9H, bs, Si(CH3)3], −0.05 [9H, bs, Si(CH3)3] ppm.
O st), 1668 (CO st), 1149 and 1103 (C–O as st), 1059 (C–O sym st), 698 (C–Br st); δH (d6-DMSO, gel phase, 300 MHz): 11.27 (1H, bs, NH), 7.58 [1H, bs, H(6)], 5.78–5.38 [2H, m, H(1′) and H(5)], 4.33–3.78 [5H, m, H(2′–5′)], 1.00–0.60 (5H, m, CH3 and CH2), 0.05 [9H, bs, Si(CH3)3], −0.05 [9H, bs, Si(CH3)3] ppm.
Cleavage of poly(methacryloyl uridine) from resin (general procedure)
Poly(methacryloyl uridine) substituted Wang resin 12 (95.1 mg) was swollen in a mixture of trifluoroacetic acid–dichloromethane (1:1, 4.0 ml). The solution was stirred at ambient temperature for 18 h, and then the resin was filtered and washed with more trifluoroacetic acid–dichloromethane (1:1, 3.0 ml). The filtrates were combined and cold diethyl ether was added (30 ml). The mixture was kept in the fridge overnight to precipitate the product 16 which was filtered and washed with cold diethyl ether to afford the unprotected poly-5′-O-methacryloyluridine 16 (76. mg, 80%)60 as a pale brown solid; TGA 98.0, 312.1 and 542.5 °C; Tonset 270.6 °C; Mn = 7700; Đ = 1.19 (SEC-DMF, PMMA calibrated); νmax/cm−1 (solid): 3350 (N–H st), 3072 (C–H st), 1773 (CO st), 1690 (CO st), 1620 (CC st), 1423 (N–H δ ip), 1338 (C–N st), 1139 and 1077 (C–O as st), 995 (C–O–C st); δH (d6-DMSO, 300 MHz): 11.39 (1H, bs, NH), 7.54 [1H, bs, H(6)], 5.76–5.65 [2H, m, H(1′) and H(5)], 4.00–3.55 [5H, m, H(2′–5′)], 1.13–0.76 (5H, m, CH3 and CH2) ppm.
Debromination of the poly(methacryloyl uridine) substituted Wang resin (general procedure)
A mixture of the poly-5′-O-methacryloyl-2′,3′-O-trimethylsilyluridine substituted Wang resin 12 (600.0 mg, 388.1 mg polymer, 0.84 mmol polymer), tri-n-butyltin hydride (307.3 mg, 1.27 mmol), AIBN (18.0 mg) and de-aerated toluene (6.0 ml) was de-aerated in a Schlenk by four freeze–pump–thaw cycles. It was heated at 100 °C for 20 h. The resin was filtered, washed with water, methanol and acetone and dried in vacuo to give the debrominated poly-5′-O-methacryloyl-2′,3′-O-trimethylsilyl-uridine substituted Wang resin 20 (546 mg) as a dark brown solid; TGA 313.7 and 408.1 °C; Tonset 230.1 °C; νmax/cm−1 (solid): 1750 (CO st), 1693 (CO st), 1454 (N–H δ ip), 1252 (C–N st), 1152 and 1079 (C–O as st), 839 (C–O–C st); δH (d6-DMSO, gel phase, 300 MHz): 11.79 (1H, bs, NH), 7.85 [1H, bs, H(6)], 5.91–5.68 [2H, m, H(1′) and H(5)], 4.03–3.58 [5H, m, H(2′–5′)], 0.99–(−0.98) (23 H, m, 2Si(CH3)3), CH3 and CH2) ppm.
Cleavage of debrominated poly(methacryloyl uridine) from Wang resin
Following the general procedure with the debrominated poly(methacryloyl uridine) substituted Wang resin 20 (342.6 mg), and stirring in a mixture of trifluoroacetic acid–dichloromethane (1:1, 10.0 ml) at ambient temperature for 22 h to give the title product24 (180 mg, 53%)60 as a pale brown solid; TGA 189.9, 276.8, 304.7 and 411.5 °C; Tonset 247.5 °C; Mn = 7800, Đ = 1.23 (SEC-DMF, PMMA calibrated); νmax/cm−1 (solid): 3223 (N–H and O–H st), 2970 (C–H st), 1668 (CO st), 1464 (N–H δ ip), 1387 (C–N st), 1268, 1106 and 1077 (C–O as st); δH (d6-DMSO, 300 MHz): 11.39 (1H, bs, NH), 7.61 [1H, bs, H(6)], 5.93–5.66 [3H, m, H(1′), H(5) and OH(2′)], 5.25 [1H, bs, OH(3′)], 4.14–3.88 [5H, m, H(2′–5′)], 1.78–0.74 (5H, m, CH3 and CH2) ppm.
Polymerization of 2 using an immobilized template of poly(methacryloyl uridine) (general procedure)
A solution of AIBN (0.3 ml of a 30.0 mg in 3.0 ml methanol solution) was placed in a Schlenk tube and the solvent removed in vacuo. 2′,3′-O-tert-Butyldimethylsilyl-5′-O-methacryloyl adenosine 2 (159.7 mg, 0.28 mmol), the immobilized template of poly(methacryloyl uridine) 20 (200.0 mg, 129.7 mg polymer, 0.28 mmol polymer), ethyl acetate (4.0 ml) and de-aerated toluene (2.0 ml) were added. The reaction mixture was de-aerated four times using the freeze–pump–thaw cycle and heated at 60 °C for 42.5 h. It was then filtered and washed with toluene and dried in vacuo to give the complex 28 (212 mg, 59%)61 as a cream colored solid. 1H NMR (d6-DMSO, gel phase) of this complex 28 showed a 1:1 ratio of uridine to adenosine; TGA 336.5, 406.9 and 656.0 °C; Tonset 276.9 °C; νmax/cm−1 (solid): 3396, 3207 and 3063 (N–H hydrogen bonded), 2928 and 2857 (C–H st), 1693 (CO st), 1634 (CO st), 1453 (N–H δ ip), 1373 (N–H st), 1249 (C–O–C as st), 1152 and 1077 (C–O as st), 836 (Si–CH3), 751 (Si–C st); δH (d6-DMSO, gel phase, 300 MHz): 11.53 (1H, bs, NH), 8.34 [1H, bs, H(2A)], 8.08 [1H, bs, H(8A)], 7.44–7.16 [3H, m, NH2 and H(6U)], 5.98–5.87 [1H, m, H(5U)], 5.59–5.32 [2H, m, 2H(1′)], 4.50–3.70 [10H, m, 2H(2′–5′)], 1.70–(−0.80) (58, m, 2Si(CH3)3, 2Si(CH3)2C(CH3)3, 2CH3 and 2CH2) ppm.
Separation of the daughter polymer poly(methacryloyl adenosine) 32 from its parent polymer the immobilised poly(methacryloyl uridine) 24 (general procedure)
The previous complex 28 (172.6 mg) was stirred in trifluoroethanol (5.0 ml) at ambient temperature for 1 h. It was then filtered and washed with more trifluoroethanol. The trifluoroethanol washings were removed under reduced pressure to give the poly-2′,3′-O-tert-butyldimethylsilyl-5′-O-methacryloyladenosine 32 (48 mg, 30%)62 as a white solid. Analytical data for the solid supported parent polymer was identical to the parent polymer 24 prepared above. Analytical data of the daughter polymer 32 is as follows: TGA 349 °C; Tonset 275.7 °C; Mn = 4300, Đ = 1.60 (SEC-THF, PMMA calibrated); Mn = 3400, Đ = 1.15 (SEC-DMF, PMMA calibrated); νmax/cm−1 (solid): 3330 (NH2st), 2931 (C–H st), 1694 (CO st), 1636 (CN st), 1472 (N–CO sym st and NH amide II), 1251 (C–C–N bending), 1155 and 1078 (C–O as st), 834 (Si–CH3), 776 (C–Si st); δH (d6-DMSO, 300 MHz): 8.38 [1H, bs, H(2)], 8.08 [1H, bs, H(8)], 7.29 (2H, bs, NH), 5.92–5.89 [1H, m, H(1′)], 4.65–3.75 [5H, m, H(2′–5′)], 1.30–(−0.6) (35H, m, 2Si(CH3)2C(CH3)3, CH3 and CH2) ppm.
Acknowledgements
We thank Dr Andrew Steward (University of Warwick) for help with polymer analyses. We thank EPSRC for a Fast Stream studentship (GR/L71933) held by MG and the University of Warwick for postdoctoral fellowships (AK, KK). MG thanks the Warwick-Wisconsin Exchange Fellowship for an award and we thank Professor S. H. Gellman (University of Wisconsin-Madison) in whose laboratory some of this work was carried out for additional support and guidance. DMH is a Royal Society Wolfson fellow.Notes and references
- J. D. Watson and F. H. C. Crick, Nature, 1953, 171, 737–738 CrossRef CAS.
- G. Challa and Y. Y. Tan, Pure Appl. Chem., 1981, 53, 627–641 CrossRef CAS.
- K. Takemoto and Y. Inaki, Adv. Polym. Sci., 1981, 41, 1–51 CrossRef CAS.
- A. W. Schwartz and L. E. Orgel, Science, 1985, 228, 585–587 CAS.
- Y. Y. Tan, Prog. Polym. Sci., 1994, 19, 561–588 CrossRef CAS.
- R. McHale, J. P. Patterson, P. B. Zetterlund and R. K. O'Reilly, Nat. Chem., 2012, 4, 491–497 CrossRef CAS PubMed.
- L. E. Orgel, Nature, 1992, 358, 203–209 CrossRef CAS PubMed.
- I. A. Kozlov, S. Pitsch and L. E. Orgel, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 13448–13452 CrossRef CAS.
- X. Y. Li and D. R. Liu, Angew. Chem., Int. Ed., 2004, 43, 4848–4870 CrossRef CAS PubMed.
- C. T. Calderone and D. R. Liu, Curr. Opin. Chem. Biol., 2004, 8, 645–653 CrossRef CAS PubMed.
- R. E. Kleiner, Y. Brudno, M. E. Birnbaum and D. R. Liu, J. Am. Chem. Soc., 2008, 130, 4646–4659 CrossRef CAS PubMed.
- Y. Brudno and D. R. Liu, Chem. Biol., 2009, 16, 265–276 CrossRef CAS PubMed.
- Y. Brudno, M. E. Birnbaum, R. E. Kleiner and D. R. Liu, Nat. Chem. Biol., 2010, 6, 148–155 CrossRef CAS PubMed.
- J. C. Blain, A. Ricardo and J. W. Szostak, J. Am. Chem. Soc., 2014, 136, 2033–2039 CrossRef CAS PubMed.
- Y. Inaki, Prog. Polym. Sci., 1992, 17, 515–570 CrossRef CAS.
- J. G. Schmidt, L. Christensen, P. E. Nielsen and L. E. Orgel, Nucleic Acids Res., 1997, 25, 4792–4796 CrossRef CAS PubMed.
- T. Inoue and L. E. Orgel, Science, 1983, 219, 859–862 CAS.
- E. Kervio, A. Hochgesand, U. E. Steiner and C. Richert, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 12074–12079 CrossRef CAS PubMed.
- E. Kervio, B. Claasen, U. E. Steiner and C. Richert, Nucleic Acids Res., 2014, 42, 7409–7420 CrossRef CAS PubMed.
- J. Kurreck, Angew. Chem., Int. Ed., 2009, 48, 1378–1398 CrossRef CAS PubMed.
- J. Niu, R. Hili and D. R. Liu, Nat. Chem., 2013, 5, 282–292 CrossRef PubMed.
- L. Zhang, W. Liu, L. Lin, D. Chen and M. H. Stenzel, Biomacromolecules, 2008, 9, 3321–3331 CrossRef CAS PubMed.
- W. T. Smith, Prog. Polym. Sci., 1996, 21, 209–253 CrossRef CAS.
- R. McHale and R. K. O'Reilly, Macromolecules, 2012, 45, 7665–7675 CrossRef CAS.
- H. J. Spijker, F. L. van Delft and J. C. M. van Hest, Macromolecules, 2007, 40, 12–18 CrossRef CAS.
- H. J. Spijker, A. J. Dirks and J. C. M. Van Hest, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 4242–4250 CrossRef CAS.
- A. Marsh, A. Khan, D. M. Haddleton and M. J. Hannon, Macromolecules, 1999, 32, 8725–8731 CrossRef CAS.
- A. Khan, D. M. Haddleton, M. J. Hannon, D. Kukulj and A. Marsh, Macromolecules, 1999, 32, 6560–6564 CrossRef CAS.
- A. Likhitsup, S. Yu, Y.-H. Ng, C. L. L. Chai and E. K. W. Tam, Chem. Commun., 2009, 4070–4072 RSC.
- B. D. Mather, M. B. Baker, F. L. Beyer, M. A. G. Berg, M. D. Green and T. E. Long, Macromolecules, 2007, 40, 6834–6845 CrossRef CAS.
- Y. Kang, A. Lu, A. Ellington, M. C. Jewett and R. K. O'Reilly, ACS Macro Lett., 2013, 2, 581–586 CrossRef CAS.
- Z. Y. Li, Z. Y. J. Zhang, R. Knipe and D. G. Lynn, J. Am. Chem. Soc., 2002, 124, 746–747 CrossRef PubMed.
- Y. Ura, J. M. Beierle, L. J. Leman, L. E. Orgel and M. R. Ghadiri, Science, 2009, 325, 73–77 CrossRef CAS PubMed.
- H. J. Spijker, A. J. Dirks and J. C. M. van Hest, Polymer, 2005, 46, 8528–8535 CrossRef CAS PubMed.
- X. Li and D. G. Lynn, Angew. Chem., Int. Ed., 2002, 41, 4567–4569 CrossRef CAS.
- M. Hattori, H. Nakagawa and M. Kinoshita, Makromol. Chem., 1980, 181, 2325–2333 CrossRef CAS.
- P. K. Lo and H. F. Sleiman, J. Am. Chem. Soc., 2009, 131, 4182–4183 CrossRef CAS PubMed.
- A. Marsh, A. Khan, M. Garcia and D. M. Haddleton, Chem. Commun., 2000, 2083–2084 RSC.
- M. Ferrero and V. Gotor, Chem. Rev., 2000, 100, 4319–4348 CrossRef CAS PubMed.
- F. Moris and V. Gotor, J. Org. Chem., 1993, 58, 653–660 CrossRef CAS.
- R. W. Chambers, P. Shapiro and V. Kurkov, J. Am. Chem. Soc., 1960, 82, 970–975 CrossRef CAS.
- I. Lavandera, S. Fernandez, J. Magdalena, M. Ferrero, R. J. Kazlauskas and V. Gotor, ChemBioChem, 2005, 6, 1381–1390 CrossRef CAS PubMed.
- R. W. Chambers, J. G. Moffatt and H. G. Khorana, J. Am. Chem. Soc., 1957, 79, 3747–3752 CrossRef CAS.
- M. J. Cooper, R. S. Goody, A. S. Jones, J. R. Tittensor and R. T. Walker, J. Chem. Soc. C, 1971, 3183–3187 RSC.
- K. Jankova, X. Y. Chen, J. Kops and W. Batsberg, Macromolecules, 1998, 31, 538–541 CrossRef CAS.
- R. Deans, F. Ilhan and V. M. Rotello, Macromolecules, 1999, 32, 4956–4960 CrossRef CAS.
- The poly(methacryloyl adenosine) 32 obtained from the template polymerization was found to be very soluble in THF, so it was possible to perform full SEC analysis in both THF and DMF (PMMA as the standard).
- R. K. Castellano, V. Gramlich and F. Diederich, Chem. – Eur. J., 2002, 8, 118–129 CrossRef CAS.
- Y. Inaki, K. Ebisutani and K. Takemoto, J. Polym. Sci., Part A: Polym. Chem., 1986, 24, 3249–3262 CrossRef CAS.
- C. H. Kang, I. Berger, C. Lockshin, R. Ratliff, R. Moyzis and A. Rich, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 11636–11640 CrossRef CAS.
- A. L. Webber, S. Masiero, S. Pieraccini, J. C. Burley, A. S. Tatton, D. Iuga, T. N. Pham, G. P. Spada and S. P. Brown, J. Am. Chem. Soc., 2011, 133, 19777–19795 CrossRef CAS PubMed.
- J. T. Davis and G. P. Spada, Chem. Soc. Rev., 2007, 36, 296–313 RSC.
- T. H. Dickey, S. E. Altschuler and D. S. Wuttke, Structure, 2013, 21, 1074–1084 CrossRef CAS PubMed.
- P. A. Korevaar, C. Schaefer, T. F. de Greef and E. W. Meijer, J. Am. Chem. Soc., 2012, 134, 13482–13491 CrossRef CAS PubMed.
- A. E. Engelhart and N. V. Hud, Cold Spring Harbor Perspect. Biol., 2010, 2, a002196 CAS.
- S. I. Walker, M. A. Grover and N. V. Hud, PLoS One, 2012, 7, e34166 CAS.
- S. Angot, N. Ayres, S. A. F. Bon and D. M. Haddleton, Macromolecules, 2001, 34, 768–774 CrossRef CAS.
- D. M. Haddleton, M. C. Crossman, B. H. Dana, D. J. Duncalf, A. M. Heming, D. Kukulj and A. J. Shooter, Macromolecules, 1999, 32, 2110–2119 CrossRef CAS.
- R. N. Keller, H. D. Wrcoff and L. E. Marchi, in Inorganic Syntheses, John Wiley & Sons, Inc., 2007, pp. 1–4 Search PubMed.
- Based on the amount recovered from the resin.
- Based on the amount of complex obtained and the amount of template and complementary monomer used.
- Based on the amount of daughter polymer recovered and the amount of daughter monomer used for the templating process.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental data of monomer synthesis, preparation of adenosine, cytidine and guanosine templates and templated polymerization. See DOI: 10.1039/c4py01783h |
| ‡ Preliminary communication: M. Garcia, Ph.D. Thesis, University of Warwick, 2003. |
| This journal is © The Royal Society of Chemistry 2015 |