
RAFT-prepared α-difunctional poly(2-vinyl-4,4-dimethylazlactone)s and their derivatives: synthesis and effect of end-groups on aqueous inverse temperature solubility†
Jing Yang
Quek‡
a,
Xuechao
Liu§
a,
Thomas P.
Davis
bc,
Peter J.
Roth
a and
Andrew B.
Lowe
*a
aSchool of Chemical Engineering, Centre for Advanced Macromolecular Design, UNSW Australia, University of New South Wales, Kensington, Sydney, NSW 2052, Australia. E-mail: a.lowe@unsw.edu.au
bMonash Institute of Pharmaceutical Sciences, Monash University, Parkville, VIC 3052, Australia
cDepartment of Chemistry, University of Warwick, Gibbet Hill, Coventry, CV4 7AL, UK
First published on 15th September 2014
Abstract
A series of five novel R-group di-functional phenyl dithiobenzoates have been prepared and utilized in the controlled reversible addition–fragmentation chain transfer (RAFT) radical polymerization of 2-vinyl-4,4-dimethylazlactone (VDMA), yielding a series of homopolymers of similar average degrees of polymerization but variable α-end group functionality. Each of the reactive polyVDMA homopolymers was reacted with four different small molecule amines: dimethylamine, diethylamine, N,N-diethylethylenediamine and tetrahydrofurfurylamine yielding a series of novel end-functional materials. The effect of the end-groups on the inverse temperature dependent aqueous solubility of the formally hydrophilic homopolymers was then measured and compared to similar materials prepared with benzylpropyltrithiocarbonate as the RAFT agent. In virtually all instances, the introduction of the twin α-end-groups resulted in overall more hydrophobic species that exhibited cloud points spanning the range 25.1–42.7 °C. Importantly, there was a strong influence on the nature of the end groups and the associated solubility characteristics with, in some cases, cloud point behaviour only being observed in polymers with twin end groups while those derived from benzylpropyltrithiocarbonate were fully soluble.
Introduction
Reversible addition–fragmentation chain transfer (RAFT) radical polymerization1–5 is a reversible deactivation radical polymerization processes based on the degenerative transfer of thiocarbonylthio compounds, termed RAFT chain transfer agents (CTAs), between propagating (co)polymer chains. The choice of both RAFT CTA family (dithioester vs. trithiocarbonate vs. xanthate vs. dithiocarbamate) as well as specific Z and R groups within a family is crucial for achieving good control in a given (co)polymerization system.6,7 However, beyond general (co)polymerization control, the use of suitable RAFT CTAs offers the opportunity for instilling specific functional groups at the α and or ω chain ends.5 This is an important feature since polymer end-groups are well known to have an effect on physical properties, especially in low-to-medium molecular weight (co)polymers.8 For example, Li et al.9 reported the effect on the cloud point (CP) of a poly(N,N-diethylacrylamide) (PDEAM) homopolymer (![[M with combining macron]](https://www.rsc.org/images/entities/i_char_004d_0304.gif) n,SEC = 2540, ĐM = w/n = 1.10) in which the ω-thiocarbonylthio end groups were cleaved to the macromolecular thiol and then reacted with a series of small molecule isocyanates of varying hydrophobicities to give novel thiocarbamate end-functional PDEAMs. The authors reported that the measured CPs of these modified materials spanned the range 23–34 °C with the exact value being dependent on the isocyanate employed in the modification reaction. Similar observations have been made in a RAFT-prepared PDEAM homopolymer in which the ω-terminal thiocarbonylthio groups were cleaved to the thiol followed by reaction with a series of oxiranes.10 Being the most widely studied thermoresponsive polymer, the effect of end-groups on the inverse temperature solubility characteristics of poly(N-isopropylacrylamide) (PNIPAM) has been examined by a number of research groups.11–14 Studies have also been performed with other thermoresponsive polymers. For example, Huber, Hutton and Jordan reported the effect of end-group polarity on the lower critical solution temperature (LCST) of poly(2-isopropyl-2-oxazoline),15 Roth et al.16 highlighted the influence of end groups on the LCST behaviour of poly[oligo(ethylene glycol) methacrylate], and Miasnikova and Laschewsky described how end groups influence the LCST of poly(methoxy diethyleneglycol acrylate).17 In general, we find that LCSTs decrease with the introduction of hydrophobic end groups and increase with hydrophilic end groups.18,19
n,SEC = 2540, ĐM = w/n = 1.10) in which the ω-thiocarbonylthio end groups were cleaved to the macromolecular thiol and then reacted with a series of small molecule isocyanates of varying hydrophobicities to give novel thiocarbamate end-functional PDEAMs. The authors reported that the measured CPs of these modified materials spanned the range 23–34 °C with the exact value being dependent on the isocyanate employed in the modification reaction. Similar observations have been made in a RAFT-prepared PDEAM homopolymer in which the ω-terminal thiocarbonylthio groups were cleaved to the thiol followed by reaction with a series of oxiranes.10 Being the most widely studied thermoresponsive polymer, the effect of end-groups on the inverse temperature solubility characteristics of poly(N-isopropylacrylamide) (PNIPAM) has been examined by a number of research groups.11–14 Studies have also been performed with other thermoresponsive polymers. For example, Huber, Hutton and Jordan reported the effect of end-group polarity on the lower critical solution temperature (LCST) of poly(2-isopropyl-2-oxazoline),15 Roth et al.16 highlighted the influence of end groups on the LCST behaviour of poly[oligo(ethylene glycol) methacrylate], and Miasnikova and Laschewsky described how end groups influence the LCST of poly(methoxy diethyleneglycol acrylate).17 In general, we find that LCSTs decrease with the introduction of hydrophobic end groups and increase with hydrophilic end groups.18,19
In extreme cases, and if the end groups are sufficiently hydrophobic, self-assembly can be induced in otherwise hydrophilic homopolymers.20 For example, we recently reported the synthesis of a series of novel phenyl dithioester-based RAFT CTAs bearing two pyrenyl or cholesteryl groups in the R group fragment.21 As a representative example, the bis-pyrenyl CTA was employed in the homopolymerization of N,N-dimethylacrylamide (DMA) to give homopolymers with NMR measured absolute molecular weights of 5900 and 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 and low dispersities as determined by size exclusion chromatography (SEC). Importantly, the use of such CTAs instilled two large hydrophobic pyrene groups at the homopolymer α-chain end. It was reported that the impact of such groups was sufficient in water to induce self-directed assembly giving polymersomes. More recently, Roth, Davis and Lowe22 reported the synthesis of novel α,α-bischolesteryl functional poly[pentafluorophenyl (meth)acrylate]s (PPFP(M)A) with low-to-medium molecular weights and low ĐM values employing a dicholesterol functional phenyl dithioester RAFT CTA. These PPFP(M)A scaffolds were then reacted with a series of low molecular weight 1° and 2° amines including 2-hydroxypropylamine, iso-propylamine, diethylamine and N-2-glucosamine, in the presence of an appropriate Michael acceptor such as N-isopropylacrylamide or N,N-diethylacrylamide. Conversions to the corresponding (meth)acrylamido homopolymers were smooth and facile giving the modified species quantitatively. Importantly, these α-bisfunctional homopolymers were able to undergo self-directed assembly in aqueous media forming polymersomes, albeit of a rather broad size distribution.
500 and low dispersities as determined by size exclusion chromatography (SEC). Importantly, the use of such CTAs instilled two large hydrophobic pyrene groups at the homopolymer α-chain end. It was reported that the impact of such groups was sufficient in water to induce self-directed assembly giving polymersomes. More recently, Roth, Davis and Lowe22 reported the synthesis of novel α,α-bischolesteryl functional poly[pentafluorophenyl (meth)acrylate]s (PPFP(M)A) with low-to-medium molecular weights and low ĐM values employing a dicholesterol functional phenyl dithioester RAFT CTA. These PPFP(M)A scaffolds were then reacted with a series of low molecular weight 1° and 2° amines including 2-hydroxypropylamine, iso-propylamine, diethylamine and N-2-glucosamine, in the presence of an appropriate Michael acceptor such as N-isopropylacrylamide or N,N-diethylacrylamide. Conversions to the corresponding (meth)acrylamido homopolymers were smooth and facile giving the modified species quantitatively. Importantly, these α-bisfunctional homopolymers were able to undergo self-directed assembly in aqueous media forming polymersomes, albeit of a rather broad size distribution.
In addition to the use of pentafluorophenylesters as reactive precursor materials there has also been recent interest in (co)polymers containing azlactones.23–30 This is due to the facile reaction of the azlactone heterocycle with various nucleophiles including 1° and 2° amines, alcohols, and thiols. For example, Sun et al.31 described the synthesis of a library of polymers derived from a parent VDMA homopolymer modified with a series of 1° amines that also contained tertiary amine functionality and subsequently evaluated them as cellular delivery vehicles for DNA. We have also been interested in reactive VDMA (co)polymers for both the preparation of novel thermoresponsive (co)polymers, as a reactive species incorporated into block copolymers amenable to facile modification for the preparation of tuneable amphiphilic AB diblock copolymers, and as a reactive building block in the preparation of new stimulus responsive organo- and hydrogels.32–34 For example, Quek et al.33 reported the trithiocarbonate-mediated RAFT synthesis of AB diblock copolymers of VDMA with DMA or NIPAM followed by nucleophilic ring-opening modification of the azlactone repeat units with N,N-dimethylethylenediamine (DMEDA), N,N-diethylethylenediamine (DEEDA) or picolylamine (PA). As an example, when a VDMA homopolymer is ring-opened with DEEDA the resulting bis-amide material exhibits inverse temperature aqueous solubility with a CP of ca. 23 °C in 0.1 M NaOH and a slightly higher value of 30 °C in DI water. Modification of the near block symmetric P(VDMA54-b-DMA46) copolymer with DEEDA gives a functional material that exists as unimers in DI water at 10 °C but forms self-assembled nanoparticles with an average hydrodynamic diameter of 79.8 nm at 50 °C. Such behaviour is completely reversible.
Building on our previous work with end functional (co)polymers and the use of VDMA as a building block in the preparation of novel stimulus-responsive (co)polymers herein we report the synthesis of novel RAFT CTAs bearing dual functionality in the R-group fragment and their use in the polymerization of VDMA. As far as we are aware we are one of only a few groups to have investigated the use of VDMA-based (co)polymers as precursors to novel thermoresponsive materials and as such this work is also motivated by the desire to investigate such bisamide derivatives in more detail. Additionally, since we have previously shown that two large bulky, bicyclic α-end groups, such as pyrene or cholesterol, can induce self-assembly in homopolymers derived from PPFP(M)A reactive precursors we additionally wanted to begin to establish the effect of ring size and structure and the ability, if any, to direct self-association with small, dual cyclic α-end-groups. Modification of the VDMA repeat units with a series of primary amines gives series of homopolymers with tuneable inverse aqueous temperature solubilities. This approach is complimentary to our previous reported studies with PFP(M)A (co)polymers with ‘common’ RAFT CTAs as well as our work detailing the direct polymerization of (meth)acrylamido monomers with dual-R group functional CTAs. Additionally, this work provides further insights into the molecular weight dependence of the cloud point on functional (co)polymers obtained from reactive VDMA scaffolds.
Experimental
Instrumentation
Size exclusion chromatography (SEC) was performed on a Shimadzu system with four phenogel columns in dimethylacetamide (DMAc) (0.03% w/v LiBr, 0.05% BHT stabilizer) as eluent at a flow rate of 1.0 mL min−1 at 50 °C. A Shimadzu modular system was employed comprising a DGC-12A solvent degasser, an LC-10AT pump, a CTO 10A column oven and a RID-10A refractive index detector. The system was equipped with a Polymer Laboratories 5.0 mm bead-size guard column (50 × 7.8 mm2) followed by four 300 × 7.8 mm2 linear PL columns (105, 104, 103 and 500 Å). Chromatograms were analysed using Cirrus SEC software (version 3.0) and the system was calibrated with a series of narrow molecular weight distribution polystyrene (PS) standards with molecular weights ranging from 580–1820000 g mol−1.
Nuclear magnetic resonance (NMR) spectra were recorded on a Bruker DPX 300 spectrometer at 300 MHz for hydrogen nuclei and 75 MHz for carbon nuclei. Chemical shifts are reported in parts per million (ppm). Data is reported as follows: chemical shift [multiplicity (s = singlet, d = doublet, dd = doublet of doublets, t = triplet, m = multiplet), coupling constants in Hertz, integration]. Measurements were performed in CDCl3 with the internal solvent signal (7.26 ppm) being used as the reference signal. NMR data was evaluated using iNMR 4.0.4 Mestrelab Research software.
Turbidity measurements were performed on a Varian Cary 300 Scan spectrophotometer equipped with a Cary temperature controller and a Peltier heating element in quartz cuvettes of 10 mm path length at a wavelength of 520 nm. In all cases, a polymer concentration of 10 mg mL−1 was employed. Heating rates were 1 °C min−1 for all measurements. For all clear solutions, the baseline was corrected to zero absorbance, A. Transmittance, T = 10−A, was plotted against temperature, and cloud points were determined at 50% transmittance. The maximum temperature evaluated for all solutions was 80 °C.
Fourier transform infrared spectroscopy (FTIR) measurements were performed on a Bruker IFS 66/S instrument under attenuated total reflectance (ATR). Data was analyzed with OPUS software version 4.0.
Syntheses
4,4′-Azobis(4-cyanovaleric acid) (ACVA, Fluka, 98%), diethanolamine (Aldrich, 99%), 2-mercaptothiazoline (Aldrich, 98%), N,N′-dicyclohexylcarbodiimide (DCC, Aldrich, 99%), 4-(dimethylamino)pyridine (DMAP, Aldrich, 99%), succinic anhydride (Lancaster, 99%), 2,2-dithiodipyridine (Aldrich, 98%), mercaptoethanol (Aldrich, 99%), were used as received. 4-Dimethylaminopyridinium-4-toluene sulfonate (DPTS) was prepared according to the procedure described in the literature.35 Precursors 1–4, Scheme 1, were synthesized according to a previously published procedure.21N-(2-Hydroxyethyl)naphthalimide was synthesized according to the literature.36 2-Vinyl-4,4-dimethylazlactone (VDMA) was prepared according to a published procedure.37 Benzylpropyltrithiocarbonate (BPTC) was prepared as described elsewhere.38 2,2′-Azobis(2-methylpropionitrile) (AIBN) was recrystallized twice from MeOH and then stored at −24 °C until needed. All other chemicals were purchased at the highest purity available and used as received.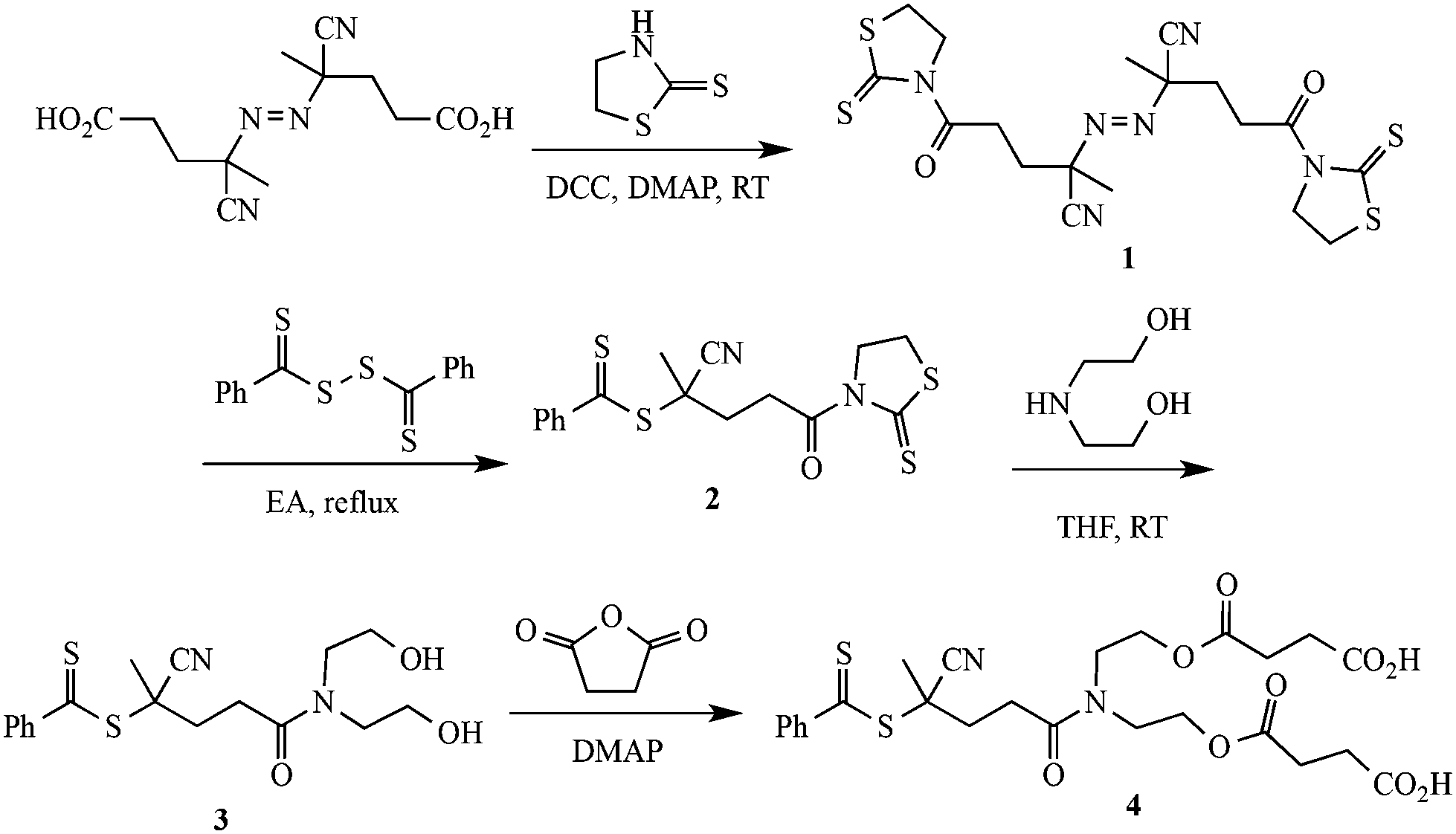 | ||
| Scheme 1 Synthetic route to a dicarboxylic functional phenyldithioester RAFT chain transfer agent, 4. | ||
:[CTA5]:[AIBN] ratio used for this instance was [215]:[1]:[0.2]. Isolated yield: 67%.
:[BPTC]:[AIBN] ratio used for this instance was [215]:[1]:[0.1]. Isolated yield: 75%.
To a glass vial equipped with a magnetic stir bar was added PVDMA (69.6 mg, 0.5 mmol of monomer units), MMTS (5.05 μL, 10.0 equiv. with respect to polymer end-groups) and DMF (2.0 mL). To a separate vial, THFA (0.16 mL, 1.5 mmol, 3.0 equiv.) and DMF (2.0 mL) were added. After complete dissolution, the two solutions were combined and the reaction was left to stir overnight at 50 °C. The same equivalent of reagents were used in the case of reactions with DMA, DEEDA and DEA.
Results and discussion
The motivation for this work stems from our previous observations regarding the effect of large hydrophobic groups being instilled in (co)polymers at the α- and or ω-chain end, and the effect of those end groups on the aqueous solution properties. While we have previously shown that large bulky end groups, such as pyrene or cholesterol, can induce self-assembly in otherwise hydrophilic homopolymers, we wanted to explore the effect, if any, of smaller ring structures introduced as pairs at the α-chain end. To accomplish this five new phenyl dithioester-based RAFT CTAs, Fig. 1, were prepared based on the general procedure we reported previously.21 | ||
| Fig. 1 Chemical structures of phenyl dithiobenzoate-based RAFT chain transfer agents CTA1–5. | ||
CTAs2–5 were prepared from the common precursor 4, Scheme 1, while CTA1 was prepared from the reaction of 3, Scheme 1, with 3,5-dinitrobenzoyl chloride in CH2Cl2. The structures and purity of all new CTAs were confirmed via standard techniques (see Experimental and ESI†). With the new CTAs in hand they were employed in the RAFT homopolymerization of 2-vinyl-4,4-dimethylazlactone (VDMA) to give a series of reactive homopolymers amenable to modification with 1° or 2° amines. The overall synthetic strategy is shown in Scheme 2.
 | ||
| Scheme 2 RAFT homopolymerization of VDMA with CTAs1–5 and their post-polymerization modification with 1° and 2° amines. | ||
Since CTAs1–5 are new species, albeit examples of the well-known phenyl dithioester family of RAFT mediating agents, we initially conducted several straightforward VDMA homopolymerizations to confirm their general effectiveness for conferring control in RAFT polymerizations. For example, Fig. 2A shows a series of experimentally determined molecular weight distributions from the homopolymerization of VDMA with the bisbenzyl CTA, CTA2, and also lists the measured SEC-determined n's and dispersities, ĐM, while 2B shows the pseudo first order kinetic plot for the same polymerization. The molecular weight distribution shows a progressive shift to high molecular weight with, in this instance, no evidence of any high molecular weight impurity to give a species with a final ĐM of 1.15.
 | ||
| Fig. 2 (A) General scheme and a series of molecular weight distributions obtained for the homopolymerization of VDMA with the bis-benzyl RAFT CTA, CTA2, highlighting its controlled nature, and (B) the pseudo first order kinetic plot for the same polymerization demonstrating its linearity. | ||
In addition to the unimodal and narrow molecular weight distributions, the pseudo first order kinetic plot for the same polymerization, shown in Fig. 2B, exhibits linear behaviour over the course of the homopolymerization further highlighting the controlled nature of the system with this novel CTA. Simply to reaffirm the effectiveness of these new difunctional R-group CTAs, Fig. 3 shows a series of molecular weight distributions for the homopolymerization of VDMA with the decalin-based RAFT mediating species CTA4. As with CTA2 the molecular weight distributions are unimodal and narrow with final dispersities of the VDMA homopolymer of ca. 1.17.
 | ||
| Fig. 3 General scheme and a series of molecular weight distributions obtained for the homopolymerization of VDMA with the bis-decalin RAFT CTA, CTA4, demonstrating the systematic shift to higher molecular weight with increasing polymerization time and the narrow, unimodal nature of the distributions. | ||
Having confirmed the new functional CTAs to be generally effective mediating agents for VDMA homopolymerization we prepared a series of homopolymers with low-to-medium targeted molecular weights with CTAs1–5 as well as the monofunctional trithiocarbonate benzylpropyl trithiocarbonate (BPTC). The latter was prepared to give VDMA homopolymers with single functional α and ω end-groups facilitating a direct comparison with doubly end functional analogues. Table 1 gives a summary of the VDMA homopolymers prepared, their SEC and NMR determined number average molecular weights, dispersities and yields.
| CTA |
n,theory
|
n,NMR
|
n,SEC
|
Đ M | Yield (%) |
|---|---|---|---|---|---|
| a As determined by size exclusion chromatography on a system calibrated with narrow molecular weight distribution polystyrene standards. | |||||
| 1 | 9000 | 6500 | 25700 |
1.10 | 49 |
| 2 | 10000 |
6500 | 17300 |
1.02 | 63 |
| 3 | 8000 | 6000 | 5800 | 1.27 | 45 |
| 4 | 15000 |
13000 |
15500 |
1.23 | 65 |
| 5 | 8000 | 6000 | 6600 | 1.12 | 68 |
| 5 | 30000 |
28800 |
68900 |
1.16 | 67 |
| BPTC1 | 8000 | 5000 | 22500 |
1.38 | 69 |
| BPTC2 | 30000 |
20500 |
83100 |
1.44 | 75 |
In all instances, VDMA homopolymers with low ĐM's and NMR-measured molecular weights close to those expected based on the conversion were obtained. For the lower molecular weight species the final, absolute molecular weights were kept as close as possible to facilitate meaningful comparisons while examples of higher molecular weight samples were also prepared to allow for a preliminary evaluation on the effect of homopolymer molecular weight, on the resulting aqueous solution properties.
With a range of reactive, functional precursors available we next converted them to a series of new end functional poly(acrylamido isobutyramide)s via reaction with the four small molecule amines shown in Scheme 2, namely dimethylamine (DMA), diethylamine (DEA), tetrahydrofurfurylamine (THFA) and N,N-diethylethylenediamine (DEEDA). All nucleophilic ring-opening reactions were performed in the presence of methyl methanethiosulfonate (MMTS)38–41 since reaction with these amines will also cleave the ω-thiocarbonylthio end groups giving the corresponding macromolecular thiols which, if not trapped, are prone to oxidative coupling.42,43 Successful trapping was confirmed via SEC with the modified homopolymers all retaining the narrow, unimodal, molecular weight distributions observed for the precursor VDMA homopolymers. Successful conversion of the parent VDMA homopolymers to the various functional poly(acrylamido isobutyramide)s was quantitatively confirmed by 1H NMR spectroscopy and qualitatively by FTIR spectroscopy. As an example, Fig. 4 shows the 1H NMR spectra of the PVDMA-diNm homopolymer (entry 2, Table 1) and the product obtained after reaction with DEEDA.
 | ||
| Fig. 4 (A) 1H NMR spectra, recorded in CDCl3, of PVDMA-diNm, and (B) the same homopolymer after reaction of the VDMA repeat units with N,N-diethylethylenediamine (DEEDA), also recorded in CDCl3. | ||
In the case of determining the absolute molecular weights this was most readily accomplished using the signals labelled f, in Fig. 4A, associated with the R group fragment of CTA3, with the resonance labelled c associated with the polymer backbone. In the case of modified polymers the absolute molecular weights and degree of modification was calculated via comparison of the integration of the resonances associated with the two methyl groups of the 2-methylalanyl spacer with those assigned to the newly introduced side groups. With respect to FTIR analysis, the key diagnostic absorptions include the total disappearance of the band centred at ca. 1818 cm−1 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O of azlactone ring), the disappearance of the band at ca. 1670 cm−1 (the imine CN absorption band of the VDMA precursor), the appearance of new CO bands at ca. 1640 cm−1 (amide absorption) in the products and the clear appearance of N–H stretching bands at ca. 3280 cm−1. Table 2 gives a summary of the functionalized VDMA homopolymers, their SEC-measured molecular weights, dispersities and cloud points.
O of azlactone ring), the disappearance of the band at ca. 1670 cm−1 (the imine CN absorption band of the VDMA precursor), the appearance of new CO bands at ca. 1640 cm−1 (amide absorption) in the products and the clear appearance of N–H stretching bands at ca. 3280 cm−1. Table 2 gives a summary of the functionalized VDMA homopolymers, their SEC-measured molecular weights, dispersities and cloud points.
| Entry | Amine | Parent VDMA |
n,SEC
|
Đ M | Cloud pointb (°C) |
|---|---|---|---|---|---|
| a As measured by size exclusion chromatography in DMAc. Molecular weights are reported as polystyrene equivalents. b All measurements for the turbidity studies were conducted at a polymer concentration of 5.0 g L−1 in distilled water. c S = soluble. | |||||
| P1 | DMA | BPTC1 | 116700 |
1.35 | Sc |
| P2 | DEEDA | BPTC1 | 49000 |
1.45 | Sc |
| P3 | DEA | BPTC1 | 136000 |
1.28 | Sc |
| P4 | THFA | BPTC1 | 64600 |
1.45 | 29.1 |
| P5 | DMA | diBn | 41900 |
1.06 | Sc |
| P6 | DEEDA | diBn | 20800 |
1.05 | 32.2 |
| P7 | DEA | diBn | 57200 |
1.09 | 34.3 |
| P8 | THFA | diBn | 24800 |
1.05 | 26.5 |
| P9 | DMA | diDn | 42800 |
1.14 | Sc |
| P10 | DEEDA | diDn | 18800 |
1.16 | 30.8 |
| P11 | DEA | diDn | 57800 |
1.06 | 32.1 |
| P12 | THFA | diDn | 23200 |
1.19 | 25.1 |
| P13 | DMA | diNm | 14800 |
1.15 | Sc |
| P14 | DEEDA | diNm | 7600 | 1.27 | 39.6 |
| P15 | DEA | diNm | 18200 |
1.12 | 28.5 |
| P16 | THFA | diNm | 8600 | 1.31 | 32.6 |
| P17 | DMA | diNt | 20100 |
1.10 | Sc |
| P18 | DEEDA | diNt | 12200 |
1.02 | 32.9 |
| P19 | DEA | diNt | 27100 |
1.06 | 30.2 |
| P20 | THFA | diNt | 11600 |
1.02 | 28.3 |
| P21 | DMA | diNBz | 24900 |
1.10 | Sc |
| P22 | DEEDA | diNBz | 13200 |
1.02 | 36.0 |
| P23 | DEA | diNBz | 37800 |
1.08 | 33.7 |
| P24 | THFA | diNBz | 15300 |
1.02 | 42.7 |
| P25 | DEEDA | BPTC2 | 73000 |
1.84 | 28.1 |
| P26 | THFA | BPTC2 | 113300 |
1.60 | 26.1 |
| P27 | DEEDA | diNt | 88600 |
1.07 | 35.8 |
| P28 | THFA | diNt | 115100 |
1.06 | 27.1 |
SEC analysis indicated that in addition to retaining the well-defined features of the VDMA precursor polymers in terms of retaining a unimodal, essentially symmetric distribution, the use of MMTS as a trapping agent for the intermediated polymeric thiols was also successful given the absence of any detectable coupled polymeric species. This is also evident in the relatively low measured dispersities for the modified homopolymers as listed in Table 2.
With a library of modified homopolymers in hand giving series of materials with identical average degrees of polymerization (![[X with combining macron]](https://www.rsc.org/images/entities/i_char_0058_0304.gif) n) and α and ω end-groups, but differing only in the nature of the pendent groups, their aqueous solution solubility behaviour was examined with an emphasis on inverse temperature solubility characteristics. To start, it is worth highlighting the properties exhibited by functional derivatives prepared from the VDMA homopolymer with α-benzyl and ω-propyltrithiocarbonate groups (BPTC1, Table 1), P1–P4Table 2.
n) and α and ω end-groups, but differing only in the nature of the pendent groups, their aqueous solution solubility behaviour was examined with an emphasis on inverse temperature solubility characteristics. To start, it is worth highlighting the properties exhibited by functional derivatives prepared from the VDMA homopolymer with α-benzyl and ω-propyltrithiocarbonate groups (BPTC1, Table 1), P1–P4Table 2.
Most importantly, with the exception of the THFA derivative the polymers remained soluble over the temperature range evaluated (ambient up to 80 °C). While the measured CP of 29.1 °C for the THFA derivative agrees well our recently reported value of ∼ 31.0 °C for a structurally similar homopolymer, albeit of differing molecular weight and ω-end group functionality, the behaviour of the remaining derivatives, P1–P3, is different.32 However, we note a significant difference in the molecular weights of the samples reported here with those previously highlighted and this alone could account for the observed difference in thermal behaviour.12,44–46 This seems to be, at least partially, the case since the THFA derivative of BPTC2 (entry P26, Table 2), prepared from a higher molecular weight PVDMA homopolymer, shows a reduced CP of 26.1 °C, while the DEEDA derivative, P25, also exhibits inverse temperature dependent solubility with a measured CP of 28.1 °C, a value almost identical to that we reported previously.32,33
With regards to functional homopolymers prepared from the new RAFT CTAs, as a representative example, Fig. 5 shows the heating and cooling curves for the derivatives of the PVDMA-diNBz homopolymer (VDAM prepared with CTA1), P21–P24Table 2. Since these derivatives were prepared from the same parent VDMA homopolymer with identical α and ω end groups any observed difference in the measured CP can be attributed to the nature of the newly introduced functionality. Within this particular series, the most hydrophilic material is the DMA derivative (Fig. 5A). This polymer did not exhibit a CP over the heating range examined (up to 80 °C). Indeed, every DMA derivative (P1, P5, P9, P13, P17 and P21) exhibited identical behaviour regardless of the chemical nature of the end groups, remaining completely soluble during the heating/cooling cycles. The next most hydrophilic species in this series was the derivative P24 with the tetrahydrofurfuryl side chain, Fig. 5D, that exhibited a sharp transition with a corresponding measured CP of 42.7 °C. This value is some 10 °C higher than similar homopolymers, albeit with slightly different end groups, that we reported recently,32 is in stark contrast to other tetrahydrofurfurylamine derivatives reported herein, vide infra. The cooling curve exhibits some hysteresis but redissolution is also rapid and occurs just below 40 °C. Such hysteresis is not uncommon and is due to the difference in kinetics and the physical processes associated with phase separation and solvation/dissolution. The DEEDA derivative, P22Fig. 5B, has a measured CP of 36.0 °C and also exhibits a reasonably sharp transition and small degree of hysteresis. This value is also slightly higher than that noted above for the BPTC2 derivative and our previously reported value. The DEA derivative, P23, has the lowest measured CP of the series derived from PVDMA-diNBz 33.7 °C and also the broadest phase transition, spanning nearly 20 °C.
 | ||
| Fig. 5 Heating/cooling turbidity curves for the functional derivatives of a VDMA homopolymer prepared with the bis-nitrobenzoyl functional RAFT chain transfer agent. Measurements made at a concentration of 5.0 g L−1 in distilled water. | ||
The overall CP values for the remaining samples, and those noted above, are shown graphically below, Fig. 6.
 | ||
| Fig. 6 Graphical representation of the measured CP values for the DEEDA, DEA and THFA derivatives of VDMA homopolymers prepared with BPTC, and the new RAFT CTAs CTA1–5. | ||
As a general observation, the introduction of the relatively hydrophobic groups at the α-chain ends (associated with the R groups of the RAFT CTAs) is sufficient to induce inverse aqueous temperature solubility characteristics for all derivatives (compared to those prepared from the VDMA homopolymer and labelled BPTC1), except for DMA modified polymers which, as noted above, remain totally soluble over the temperature range evaluated.
As a representative example, Fig. 7 shows the measured turbidity curves for the DEEDA modified polymers containing different α-functional groups. Within this series of homopolymers, the largest ‘hydrophobic’ effect is seen with the diDn-modified species (obtained from CTA4. Note however that the absolute molecular weight of the parent VDMA homopolymer is approximately double that of the remaining derivatives and this may be partly responsible for this observation), exhibits a sharp CP at 30.8 °C. The diBn (P6) and diNt (P18) derivatives, with essentially identical average degrees of polymerization, exhibit similar solubility characteristics with measured CP's of 32.2 and 32.9 °C respectively. A broader, phase transition is observed in the case of P22 with a measured CP of 33.7 °C. Perhaps somewhat surprisingly, the P14, with the twin methylnaphthyl end groups, appears to be the most ‘hydrophilic’ in this series undergoing a phase transition at 39.6 °C.
 | ||
| Fig. 7 Turbidity curves, measured at a concentration of 5.0 g L−1, of the DEEDA-modified VDMA homopolymers prepared with CTAs1–5. | ||
Surprisingly, beyond the general induction of inverse temperature dependent solubility behaviour in water, there does not appear to be a clear trend with respect to CTA structure While the dual functional species clearly possess different properties, when compared to the more common benzylpropyltrithiocarbonate RAFT CTA, it may be that the new RAFT CTAs are too structurally similar to impart distinct and unique properties with aqueous solution behaviour still, perhaps, largely dominated by the nature of the homopolymer repeat units.
Conclusions
Herein we have reported the synthesis of five new phenyl dithioester-based RAFT chain transfer agents with dual functionality in the R-group fragment. Having demonstrated their general suitability as mediating agents in RAFT homopolymerizations, they were subsequently employed in the preparation of 2-vinyl-4,4-dimethylazlactone (VDMA) homopolymers to give a small library of α-difunctional materials. Nucleophilic ring opening of the azlactone heterocycles with four different amines was accomplished in a quantitative fashion in the presence of a thiol-trapping agent yielding a new series of end functional polymers. A comparison of the temperature dependent aqueous solubility of these new materials with similar species prepared with benzylpropyltrithiocarbonate indicated that the introduction of two α-end groups, regardless of their structure, was generally sufficient to impart reversible solubility characteristics. In most instances the effect was substantial. While it is common to compare hydrophobic with hydrophilic end-groups here we have compared single with twin hydrophobic end-groups. The dramatic change in the observed solubility characteristics for some polymers can now be attributed specifically to the special design of the novel twin-functional R-group RAFT CTAs. However, while phase behavior effects are often dramatic there is no clear and obvious trend, which is somewhat unexpected, and suggests the structural features governing the inverse temperature dependent solubility are not yet fully understood.Acknowledgements
ABL would like to thank the Australian Research Council (ARC) for funding this work under the Future Fellowship program (FT110100046).References
- J. Chiefari, Y. K. Chong, F. Ercole, J. Krstina, J. Jeffery, T. P. T. Le, R. T. A. Mayadunne, G. F. Meijs, C. L. Moad, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 1998, 31, 5559–5562 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2005, 58, 379–410 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2006, 59, 669–692 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2009, 62, 1402–1472 CrossRef CAS.
- G. Moad, Y. K. Chong, A. Postma, E. Rizzardo and S. H. Thang, Polymer, 2005, 46, 8458–8468 CrossRef CAS PubMed.
- Y. K. Chong, J. Krstina, T. P. T. Le, G. Moad, A. Postma, E. Rizzardo and S. H. Thang, Macromolecules, 2003, 36, 2256–2272 CrossRef CAS.
- J. Chiefari, R. T. A. Mayadunne, C. L. Moad, G. Moad, E. Rizzardo, A. Postma, M. A. Skidmore and S. H. Thang, Macromolecules, 2003, 36, 2273–2283 CrossRef CAS.
- F. Segui, X.-P. Qiu and F. M. Winnik, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 314–326 CrossRef CAS.
- H. Li, B. Yu, H. Matsushima, C. E. Hoyle and A. B. Lowe, Macromolecules, 2009, 42, 6537–6542 CrossRef CAS.
- M. A. Harvison, T. P. Davis and A. B. Lowe, Polym. Chem., 2011, 2, 1347–1354 RSC.
- J. E. Chung, M. Yokoyama, T. Aoyagi, Y. Sakurai and T. Okano, J. Controlled Release., 1998, 53, 119 CrossRef CAS.
- S. Furyk, Y. Zhang, D. Ortiz-Acosta, P. S. Cremer and D. E. Bergbreiter, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 1492–1501 CrossRef CAS.
- Y. Xia, N. A. D. Burke and H. D. H. Stöver, Macromolecules, 2006, 39, 2275 CrossRef CAS.
- R. Pamies, K. Zhu, A.-L. Kjøniksen and B. Nyström, Polym. Bull., 2009, 62, 487 CrossRef CAS PubMed.
- S. Huber, N. Hutter and R. Jordan, Colloid Polym. Sci., 2008, 286, 1653 CAS.
- P. J. Roth, F. D. Jochum, F. R. Forst, R. Zentel and P. Theato, Macromolecules, 2010, 43, 4638 CrossRef CAS.
- A. Miasnikova and A. Laschewsky, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 3313 CrossRef CAS.
- M. J. Summers, D. J. Philips and M. I. Gibson, Chem. Commun., 2013, 49, 4223 RSC.
- G. Ru and J. Feng, J. Polym. Sci., Part B: Polym. Phys., 2011, 49, 749 CrossRef CAS.
- J. Du, H. Willcock, J. P. Patterson, I. Portman and R. K. O'Reilly, Small, 2011, 7, 2070–2080 CrossRef CAS PubMed.
- J. Xu, L. Tao, C. Boyer, A. B. Lowe and T. P. Davis, Macromolecules, 2011, 44, 299–312 CrossRef CAS.
- P. J. Roth, T. P. Davis and A. B. Lowe, Macromol. Rapid Commun., 2014, 35, 391–404 CrossRef PubMed.
- C. M. Gardner, C. E. Brown and H. D. H. Stöver, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 4674–4685 CrossRef CAS.
- S. M. Heilmann, J. K. Rasmussen and L. R. Krepski, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 3655–3677 CrossRef CAS.
- H. T. Ho, M. E. Levere, S. Pascual, V. Montembault, N. Casse, A. Caruso and L. Fontaine, Polym. Chem., 2013, 4, 675–685 RSC.
- B. S. Lokitz, J. M. Messman, J. P. Hinestrosa, J. Alonzo, R. Verduzco, R. H. Brown, M. Osa, J. F. Ankner and S. M. Kilbey, Macromolecules, 2009, 42, 9018–9026 CrossRef CAS.
- J. M. Messman, B. S. Lokitz, J. M. Pickel and S. M. Kilbey, Macromolecules, 2009, 42, 3933–3941 CrossRef CAS.
- B. S. Lokitz, J. Wei, J. P. Hinestrosa, I. Ivanov, J. F. Browning, J. F. Ankner, S. M. Kilbey and J. M. Messman, Macromolecules, 2012, 45, 6438–6449 CrossRef CAS.
- M. E. Buck and D. M. Lynn, Polym. Chem., 2012, 3, 66–80 RSC.
- H. T. Ho, M. E. Levere, D. Fournier, V. Montembault, S. Pascual and L. Fontaine, Aust. J. Chem., 2012, 65, 970–977 CrossRef CAS.
- B. Sun, X. Liu, M. E. Buck and D. M. Lynn, Chem. Commun., 2010, 46, 2016–2018 RSC.
- Y. Zhu, J. Y. Quek, A. B. Lowe and P. J. Roth, Macromolecules, 2013, 46, 6475–6484 CrossRef CAS.
- J. Y. Quek, Y. Zhu, P. J. Roth, T. P. Davis and A. B. Lowe, Macromolecules, 2013, 46, 7290–7302 CrossRef CAS.
- Y. Pei, O. R. Sugita, J. Y. Quek, P. J. Roth and A. B. Lowe, Eur. Polym. J., 2014 Search PubMed , submitted.
- J. S. Moore and S. I. Stupp, Macromolecules, 1990, 23, 65 CrossRef CAS.
- A. H. Shelton, I. V. Sazanovich, J. A. Weinstein and M. D. Ward, Chem. Commun., 2012, 48, 2749 RSC.
- M. E. Levere, H. T. Ho, S. Pascual and L. Fontaine, Polym. Chem., 2011, 2, 2878–2887 RSC.
- J. Y. Quek, P. J. Roth, R. A. Evans, T. P. Davis and A. B. Lowe, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 394–404 CrossRef CAS.
- G. B. H. Chua, P. J. Roth, H. T. T. Duong, T. P. Davis and A. B. Lowe, Macromolecules, 2012, 45, 1362–1374 CrossRef CAS.
- P. J. Roth, D. Kessler, R. Zentel and P. Theato, Macromolecules, 2008, 41, 8316–8319 CrossRef CAS.
- P. J. Roth, D. Kessler, R. Zentel and P. Theato, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 3118–3130 CrossRef CAS.
- H. Willcock and R. K. O'Reilly, Polym. Chem., 2010, 1, 149 RSC.
- M. A. Harvison, P. J. Roth, T. P. Davis and A. B. Lowe, Aust. J. Chem., 2011, 64, 992 CrossRef CAS.
- N. S. Ieong, M. Hasan, D. J. Phillips, Y. Saake, R. K. O'Reilly and M. I. Gibson, Polym. Chem., 2012, 3, 794–799 RSC.
- R. Hoogenboom, H. Thijs, M. J. H. C. Jochems, B. M. van Lankvelt, M. W. M. Fijten and U. S. Schubert, Chem. Commun., 2008, 5758–5760 RSC.
- D. G. Lessard, M. Ousalem and X. X. Zhu, Can. J. Chem., 2001, 79, 1870–1874 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4py01108b |
| ‡ Current address: Engineering Product Development, Singapore University of Technology and Design, 20 Dover Drive, Singapore 138682. E-mail: ray_quek@sutd.edu.sg |
| § Current address: Biological & Organometallic Catalysis Laboratories Building 3, Office 4221, 4700 King Abdullah University of Science and Technology, KAUST, Thuwal 23955-6900, Kingdom of Saudi Arabia. E-mail: xuechao.liu@kaust.edu.sa |
| This journal is © The Royal Society of Chemistry 2015 |