
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tuning the LSPR in copper chalcogenide nanoparticles by cation intercalation, cation exchange and metal growth†
Andreas
Wolf
,
Torben
Kodanek
and
Dirk
Dorfs
*
Institute of Physical Chemistry and Electrochemistry, Leibniz Universität Hannover, Callinstr. 3A, 30167 Hannover, Germany. E-mail: dirk.dorfs@pci.uni-hannover.de
First published on 29th October 2015
Abstract
Localized surface plasmon resonances (LSPRs) of degenerately doped copper chalcogenide nanoparticles (NPs) (Cu2−xSe berzelianite and Cu1.1S covellite) have been modified applying different methods. The comparison of the cation exchange (Cu2−xSe) and intercalation (Cu1.1S) of Ag(I) and Cu(I) has shown that Ag(I) causes a non reversible, air stable shift of the LSPR. This was compared to the influence of Au(I) cation exchange into Cu1.1S platelets under the formation of Cu1.1S–Au2S mixed nanoplatelets. Furthermore, we show the growth of Au domains on Cu2−xSe, and discuss the interaction of the two plasmonic parts of the obtained dual plasmonic Cu2−xSe–Au hybrid particles.
Introduction
Localized surface plasmon resonances (LSPRs) are typically known for nanosized metals like e.g. gold and silver. In recent years the near infrared (NIR) absorption band in several degenerately doped copper chalcogenide NPs (Cu2−xSe,1–3 Cu2−xS,4–7 Cu2−xTe8,9 and their alloys10–12) was also discovered to be of plasmonic origin. The use of LSPRs in a broad range of applications has been shown extensively.13–16 However, tuning LSPRs of metals into the NIR region is only possible with very large particles17,18 or via shape control13,19,20 (e.g. nanorods, core–shell structures, etc.). Doped semiconductors enable tuning of the plasmon band in the NIR for particles with a size as small as 3 nm.4 This enables additional applications in the second biological window (1000–1350 nm) which even has a larger penetration depth and a higher maximum permissible exposure to lasers than the first biological window (700–950 nm).21,22 The post synthesis and reversible change of the dopant concentration and hence the spectral positioning of the LSPR makes these degenerately doped quantum dots specifically interesting for nanophotonics, as this enables the possibility to switch resonances ON/OFF.23 An extensive overview of the properties of these new plasmonic materials can be found in several recent reviews to this topic.24–27In this article we analyze different approaches to modify and stabilize the LSPR of copper chalcogenide NCs in an attempt to increase their controlled flexibility for future applications: ion intercalation, ion exchange and metal growth. First, we explore the LSPR response of Cu1.1S vs. Cu2−xSe NCs to the addition of univalent ions. We compare the different behavior of the Cu2−xSe LSPR and the Cu1.1S LSPR, specifically the different shifting, damping and temporal stability of the LSPR after the integration of Cu(I) ions vs. Ag(I) ions (Scheme 1). Second, we present to the best of our knowledge for the first time growth of Au domains on pre-synthesized Cu2−xSe NPs (Scheme 1). The few previous reports on Au–Cu2−xSe28 and Au–Cu2−xS22,29–34 heterodimers have been either growing the copper chalcogenide onto a readily existing gold seed22,32–34 or by first synthesizing Au–Cu![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 29–31,35 alloy NPs, which were subsequently converted by sulfidation to Au–Cu2−xS. The here presented synthesis approach enables us to grow Au on readily existing berzelianite Cu2−xSe NPs resulting in dual-plasmonic hybrid NPs. This synthesis approach enables us to analyze the change of the chalcogenide LSPR through the interaction with differently sized metal domains on their surface. The same approach, applied to Cu1.1S NPs, results in a cation exchange to Au2S–Cu1.1S platelet shaped hybrid particles (Scheme 1).
29–31,35 alloy NPs, which were subsequently converted by sulfidation to Au–Cu2−xS. The here presented synthesis approach enables us to grow Au on readily existing berzelianite Cu2−xSe NPs resulting in dual-plasmonic hybrid NPs. This synthesis approach enables us to analyze the change of the chalcogenide LSPR through the interaction with differently sized metal domains on their surface. The same approach, applied to Cu1.1S NPs, results in a cation exchange to Au2S–Cu1.1S platelet shaped hybrid particles (Scheme 1).
 | ||
| Scheme 1 Overview of the presented approaches for tuning the LSPR in spherical Cu2−xSe NPs and Cu1.1S nanoplatelets. | ||
Results and discussion
Chalcogenide starting NPs – Cu2−xSe and Cu1.1S
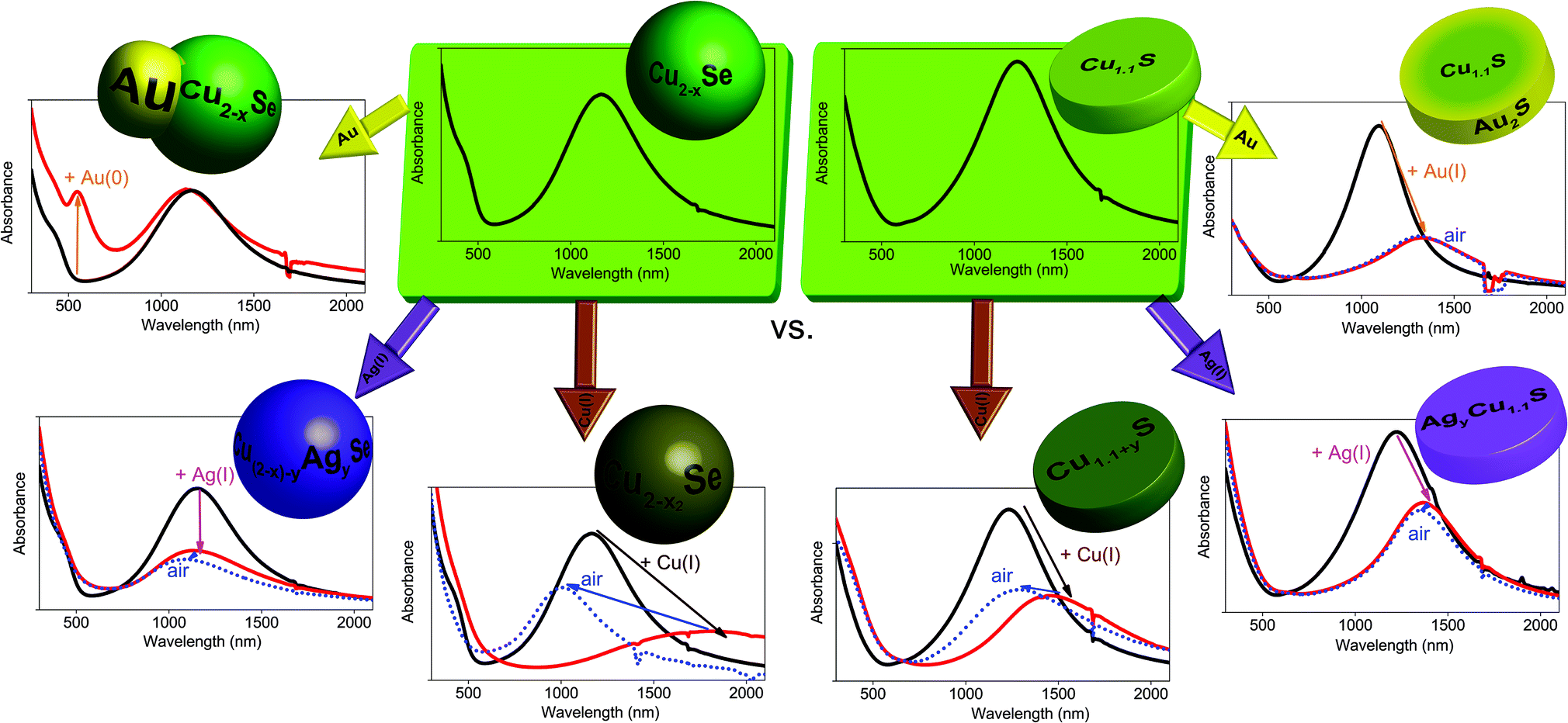
In order to study the influence of gold domains and different cations on the LSPR of copper chalcogenides, Cu2−xSe berzelianite and Cu1.1S covellite nanoparticles have been synthesized. The Cu2−xSe particles were synthesized by adapting a hot-injection synthesis previously reported resulting in 12.7 ± 1.7 nm mainly spherical nanocrystals.36 The Cu1.1S nanoparticles were synthesized by adapting the synthesis from Xie et al. resulting in nanoplatelets with the diameter of 13.2 ± 2.9 nm, and a thickness of 5.8 ± 0.9 nm.6 Cu2−xSe NPs (after exposure to air) and as-synthesized Cu1.1S show a clear LSPR with a maximum around 1155 nm and 1230 nm respectively (see Fig. 1). The LSPRs in the NIR emerge from a high density of free p-type charge carriers in the self-doped NPs.1,4,6 The origin of these are the cation vacancies in the Cu2−xSe system and intrinsic valence-band-delocalized holes in the metallic covellite system.1,6,37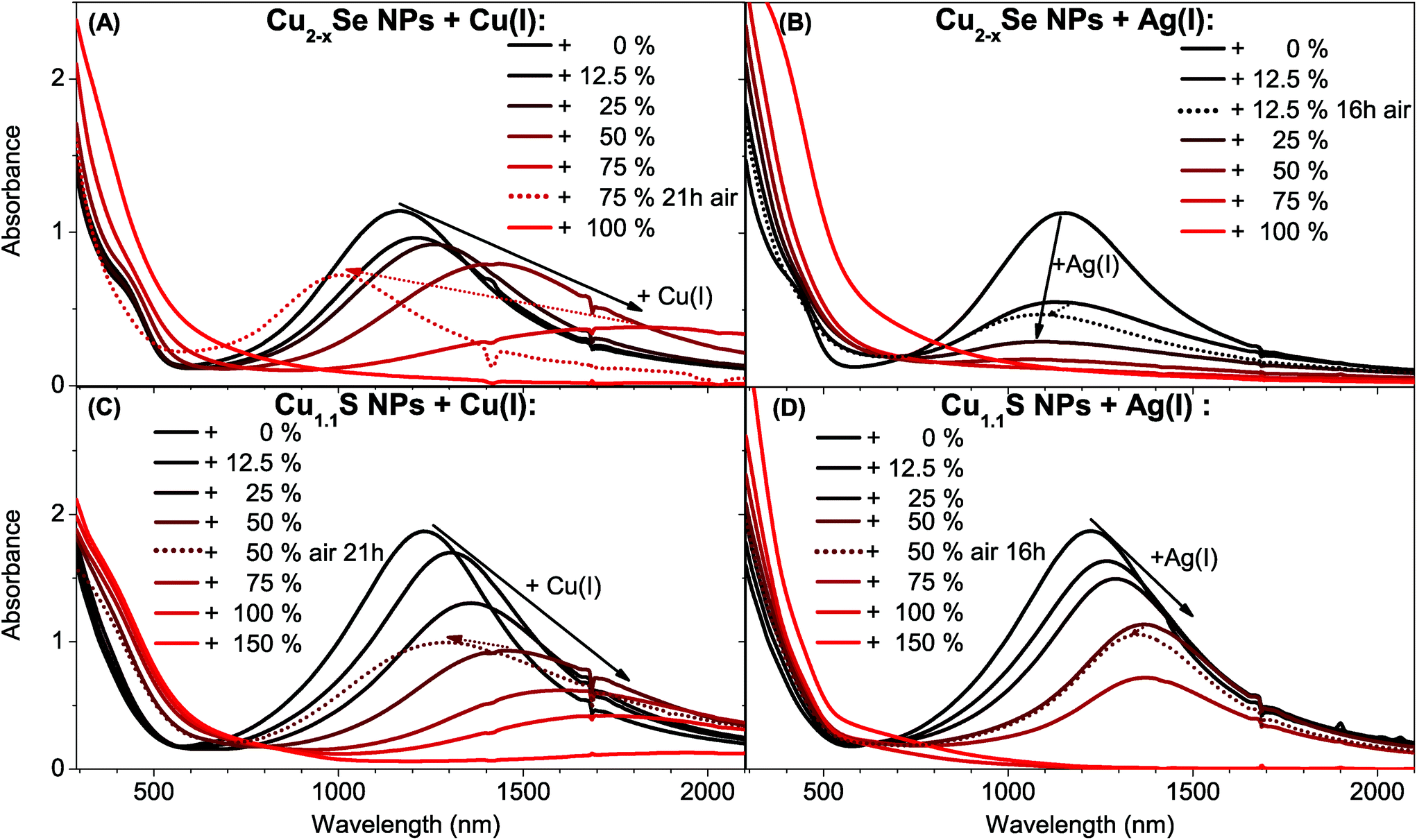 | ||
| Fig. 1 UV/Vis/NIR absorption spectra of (A, B) Cu2−xSe NPs and (C, D) Cu1.1S NPs dissolved in toluene which are treated with different amounts of a (A, C) 0.04 M Cu(I) solution or (B, D) different amounts of a 0.16 M Ag(I) solution. The spectra of the so treated particles after storing under air (dotted lines) for (A, C) 21 hours and (B, D) 16 hours are also shown. | ||
Addition of Cu(I) and Ag(I) ions to Cu2−xSe and Cu1.1S
We compare the influence on the modulation of the Cu2−xSe and Cu1.1S LSPRs through the addition of univalent ions. Specifically, we compare the different changes in the absorption spectra after the addition of different amounts of Cu(I) vs. Ag(I) ions. We discuss the spectral shifts of the LSPRs maxima and their oscillator strength as well as the long term stability of these changes under oxidizing conditions. Even though the LSPRs of the Cu2−xSe and the Cu1.1S system have a similar origin in the valence-band-delocalized holes, the above mentioned spectral changes are quite opposed for the two systems after the treatment with Cu(I) vs. Ag(I). The origin of this behavior will be discussed in the following section.Previous reports have shown that for Cu2−xSe and Cu1.1S a bathochromic shift accompanied by a decrease of the oscillator strength of the LSPR can be detected after the addition of Cu(I), that finally leads to a complete disappearance of the LSPR for higher Cu(I) amounts.1,6 This was shown to be reversible for Cu2−xSe.1 It was also shown that during the oxidation of Cu2Se to Cu2−xSe the cubic berzelianite crystal structure does not change.1 For the Cu1.1S covellite system instead, a structural change due to the Cu(I) intercalation was reported; first to a probably metastable phase and for Cu2S to either a metastable phase or a mixture of phases.6
Adding Cu(I) ions to both systems leads to the same optical response (bathochromic shift and intensity decrease of the LSPR) and ultimately to the complete disappearance of the plasmon band (Fig. 1A/C). Instead, the systems behave contrary to each other when adding an Ag(I) containing solution (Fig. 1B/D). The Cu1.1S NPs, whose LSPR have a maximum at 1230 nm, show a decrease of the oscillator strength of the LSPR, which is accompanied by a bathochromic shift until it completely disappears when adding higher Ag(I) amounts. This is similar to the addition of Cu(I). When exposed to air, however, the Ag(I) induced shift is almost irreversible (Fig. 1D) which might be interesting for applications that require specific and stable LSPRs in the NIR. This is remarkable as a Cu(I) induced shift of the LSPR maximum position is reversible (Fig. 1C) when the sample is exposed to air. Cu2−xSe NPs treated with Ag(I) show also a damping of the LSPR intensity, which is, however, not accompanied by a shift to longer wavelength. In this system we rather observe a slight hypsochromic shift of the LSPR maximum (Fig. 1B). Adding 25% Ag(I) ions in relation to the determined copper contents of the NPs leads to a LSPR shift from 1155 nm to 1130 nm after 2 h reaction time. The LSPR shifts and damping stabilizes after a few hours at around 1090 nm. Additionally this shift and the damping are non-reversible under air exposure, which is in contrast to the shift after Cu(I) addition (Fig. 1A). The bathochromic shift for Cu2−xSe after the Cu(I) addition is based on the reduction of the copper deficiencies to zero, resulting in a non plasmonic Cu2Se system.
Literature lately discusses the stoichiometric changes during reduction as an incorporation of Cu(I) combined with a reduction of the chalcogenide, while a fraction of the Cu(I) is oxidized in solution to Cu(II) in order to provide the required electrons. In fact, the Manna group showed that no Cu(II) can be detected inside copper chalcogenide NPs before or after the reduction.6,11
The LSPR frequency of both self-doped systems can be described by the Drude model.4 From eqn (S5) in the ESI† it can be seen, that at a given plasma frequency the increase of the damping factor (γ) leads to an increase of the imaginary part of the dielectric function and hence, according to eqn (S3) in the ESI† to a decrease of the extinction cross section. Following this we suggest, that the reduction of the LSPR band in Cu2−xSe through the Ag(I) addition is caused by a continuous increase of the damping in the system. This damping could be caused by Ag(I) ions that serve as charge carrier scattering centers. An increase of the silver content would hence cause an increased damping. This would cause a decrease of the extinction cross section. As the carrier density does not change significantly it is likely that Ag(I) preferentially exchanges Cu(I) instead of intercalating, as this would not significantly influence the hole density. This is supported by the significant increase of the amount of small Cu2O NPs that can be seen in the TEM (see Fig. 2A) and HAADF-STEM (see Fig. S1 in the ESI†) images in comparison to samples of the seed particles.
 | ||
| Fig. 2 TEM images and EDX mappings of Ag, Se respectively S and Cu averaged with the corresponding HAADF-STEM images, of Cu2−xSe (A) and Cu1.1S (B) NPs after addition of 25% Ag(I). | ||
Comparing γ of the Cu2−xSe system, extracted as the full width half maximum from the absorbance plotted against the energy (eV) (see Fig. S2 in the ESI†), it can be seen that γ increases from 0.43 eV (0% Ag) to 0.53 (12.5% Ag) to 0.57 eV (25% Ag). In contrast to this γ stays constant within the margin of error when Cu(I) ions are added, from 0.43 eV (0% Cu–I–) to 0.42 eV (12.5% & 25% Ag) to 0.41 eV (50% Ag). The increase in γ supports the theory that incorporated Ag(I) ions act as charge carrier scattering centers for the plasmon resonance.
STEM-EDX analysis of a Cu2−xSe sample treated with 25% Ag(I) ions relative to the copper content shows that silver is evenly distributed over a whole NP and does not form Ag2Se islands (Fig. 2A). This behavior is supported by literature that shows ternary mixed silver–copper–selenide compounds.38 Additionally we can also exclude the formation of a Ag2Se shell, as this would have led to a strong bathochromic shift of the plasmon band, due to the high frequency dielectric constant of Ag2Se (ε∞ = 11 ± 1 (ref. 39)). This finding is different to the formation of Janus-like NPs that was recently reported for the exchange with divalent cations in Cu2−xSe.40 However some EDX mappings of the same sample showed variations in the silver content between different particles (see Fig. S1 in the ESI†). Combining these findings two different mechanisms for the LSPR damping in Cu2−xSe are possible. One theory is that Ag(I) is forming charge carrier scattering centers in the NPs and hence would increase γ, which would lead to a decrease in the plasmon band intensity. The second theory is that Cu(I) is exchanged to Ag(I) preferentially in some particles and in such a high ratio that they lose their LSPR completely. Hence, in that case the weakened LSPR would be caused by Cu2−xSe NPs that do not show any Ag(I) incorporation after a total addition of 25% Ag(I).
The Cu1.1S system shows a behavior very different to the above described Cu2−xSe when Ag(I) is added (Fig. 1D). While the LSPR intensity is decreasing it additionally shifts strongly to longer wavelengths. This behavior is quite similar to the one upon addition of Cu(I) (Fig. 1C), however not reversible upon O2 exposure. This correlates with a decrease in the charge carrier density (hole density) in the NPs. γ does not increase with the addition of Ag(I) and Cu(I). Hence neither of these ions incorporation does create new charge carrier scattering centers.
Elemental mapping of the Cu1.1S NPs treated with Ag(I), does not show island or shell formation (Fig. 2B). Again the silver seems to be homogeneously distributed over each particle, and also no significant variations of the silver content between different particles can be detected.
In the above mentioned analysis it could be shown that the two copper chalcogenide systems behave optically quite opposed. The reason for this could be the different crystal systems. Cu1.1S is a hexagonal system with layers, where the reduction of the sulfur in the NCs is accompanied by the intercalation of Cu(I) between the layers of the original covellite lattice under braking of S–S bonds. At high intercalation levels a structural change to a metastable phase or a mixture between an orthorhombic and a monoclinic phase occurs.6 We suggest a similar intercalation of Ag(I) ions into the pristine covellite (00-006-0464) starting NPs. The XRD patterns show the continuous formation of a second phase due to the intercalation, the orthorhombic AgCuS (00-044-1436) (Fig. 3B). Upon further Ag(I) addition a third body-centered tetragonal Ag3CuS2 (00-012-0207) phase starts to form. This is in line with the optical observations, as this would cause a continuous shift due to an increase of the cation content in the NPs and hence a decrease of the charge carrier density. The better LSPR stability when exposed to oxygen further shows that the incorporation of the Ag(I) is not reversible under oxygen exposure, in contrast to added Cu(I) ions. This is most likely due to more oxygen stable Cu(I)–Ag(I)–S phases in comparison to the metastable Cu(I)–S phases that occur during the Cu(I) intercalation.
 | ||
| Fig. 3 XRD patterns of the evolution of crystal phases from the pristine copper chalcogenide NPs to mixed silver copper chalcogenide phases. The reference structural data shows fcc Cu2−xSe berzelianite (PDF card #: 01-072-7490), orthorhombic CuAgSe eucairite (PDF card #: 00-010-0451), orthorhombic Ag2Se naumannite (PDF card #: 01-089-2591), hexagonal Cu1.1S covellite (PDF card #: 00-006-0464), base-centered orthorhombic AgCuS stromeyerite (PDF card #: 00-044-1436), body-centered tetragonal Ag3CuS2 jalpaite (PDF card #: 00-012-0207). | ||
The XRD patterns of the cubic berzelianite Cu2−xSe (01-072-7490) NPs and 25% Ag(I) sample show the formation of a orthorhombic CuAgSe (00-010-0451) phase (Fig. 3A). If the Ag(I) content is increased further (to 100%) a full exchange can be observed, as the orthorhombic Ag2Se phase (01-089-2591) is forming. This shows a clear difference to the Cu1.1S system.
To recap the results in this paragraph, it can be seen that due to a controlled exchange (Cu2−xSe) or intercalation (Cu1.1S) with univalent ions it is possible to damp and shift the LSPR maximum of copper chalcogenides. While both systems show the same, under O2 exposure reversible, optical trend for the addition of Cu(I), their behavior varies significantly for the addition of Ag(I). The Cu2−xSe NPs show a strong, non reversible damping after Ag(I) addition. The LSPR of the Cu1.1S however can be almost non-reversibly bathochromically shifted by the Ag(I) addition. This allows a new way to permanently tune the position of the LSPR maximum for degenerately doped chalcogenides. The use in applications that require a stable and specific LSPR under ambient conditions is hence enabled.
Growth of Au domains on Cu2−xSe
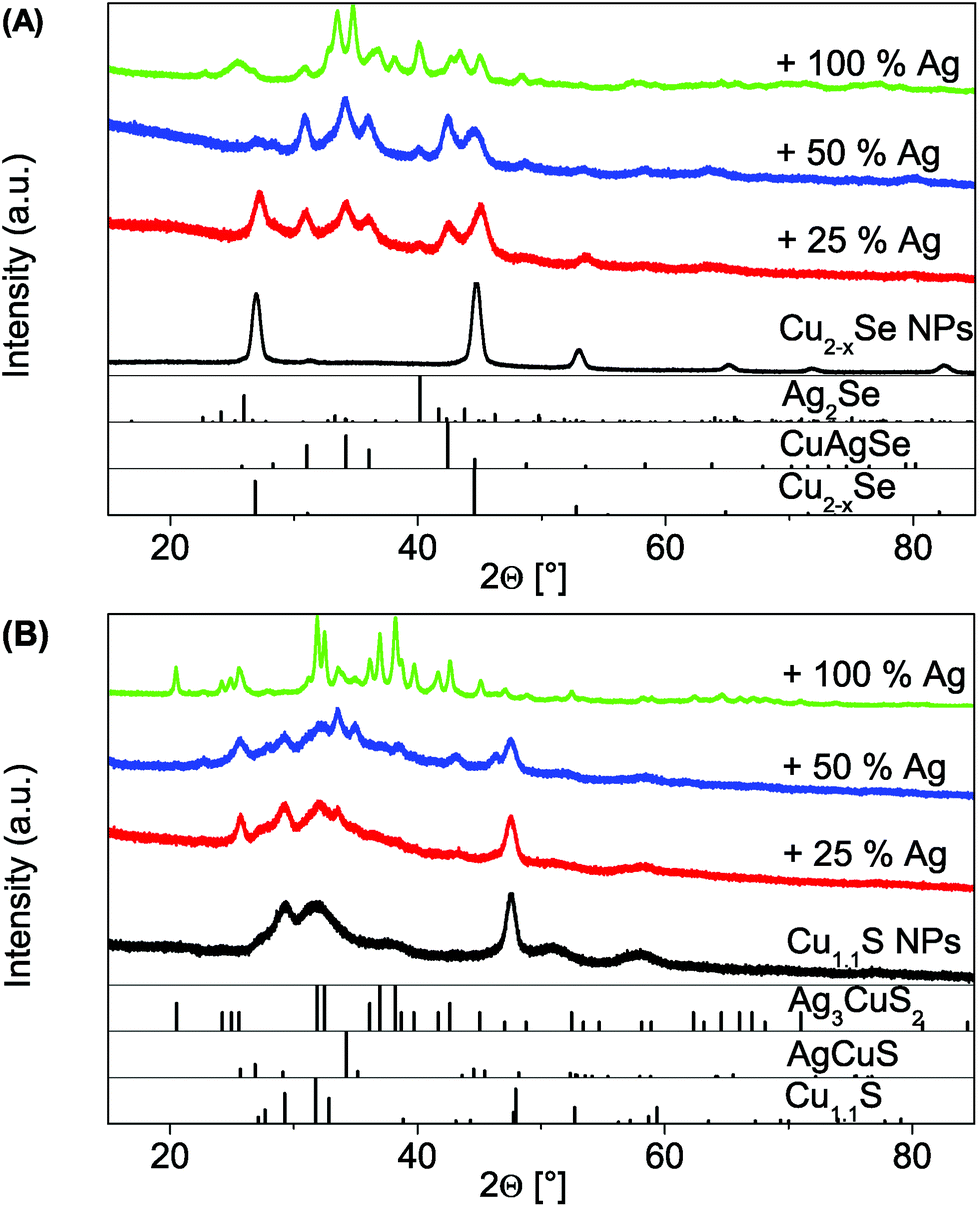
In the following paragraph we will show the synthesis of dual-plasmonic Au–Cu2−xSe hybrid NPs and the manipulation of the LSPR of the semiconductor domain due to the addition of different amounts of univalent ions. For this approach differently sized Au domains are grown on the same starting berzelianite seed particles. Furthermore the formation of Cu1.1S–Au2S hybrids is shown when the same Au growth approach is applied to covellite NPs.The growth of Au on Cu2−xSe can be controlled by changing the amount of Au-precursor added to the same amount of Cu2−xSe NP solution. Cu2Se:Au ratios between 1:0.24 and 1:1.89 have been achieved. The TEM bright field images (Fig. 4) show that high contrast Au domains have been grown on the Cu2−xSe seed NPs (lower contrast domains). For the 1:0.24 sample 76% of the Cu2−xSe seed particles have been covered with a 5.6 nm ± 1.3 nm Au domain. For the 1:0.56 sample 8.0 nm ± 1.6 nm domains have been grown on 73% of the seed particles (no separately nucleated gold particles were observed). This corresponds roughly to the calculated size of the Au domain per particle assuming 100% reaction yield, which is 6 nm for the 1:0.24 sample and 8 nm for the 1:0.56 sample. The size of the sample with the largest Au amount (1:1.89) could not be determined due to its strong tendency to agglomerate. The XRD patterns (Fig. 5) show a continuous intensity growth of the reflexes that can be assigned to fcc Au (PDF card #: 00-004-0784) while the relative intensity of the berzelianite reflexes is decreasing at the same time until they can hardly be detected for the 1:1.86 sample. The relative decrease can be explained by the higher X-ray atomic scattering factor for gold.
 | ||
| Fig. 4 UV/Vis/NIR absorption spectra (measured with an integrating sphere) and TEM images of Au growth on Cu2−xSe NCs with different amounts of Au precursor (Cu2−xSe:Au = 1:0.24, 1:0.56 and 1:1.89). As synthesized (black lines) and tuning of the LSPR by addition of Cu(I). Each step from black to red corresponds to an addition of 5 μl of a 0.4 M Cu(I) solution. *The sharp peaks (around 1700 nm) in the absorption are due to solvent absorption. | ||
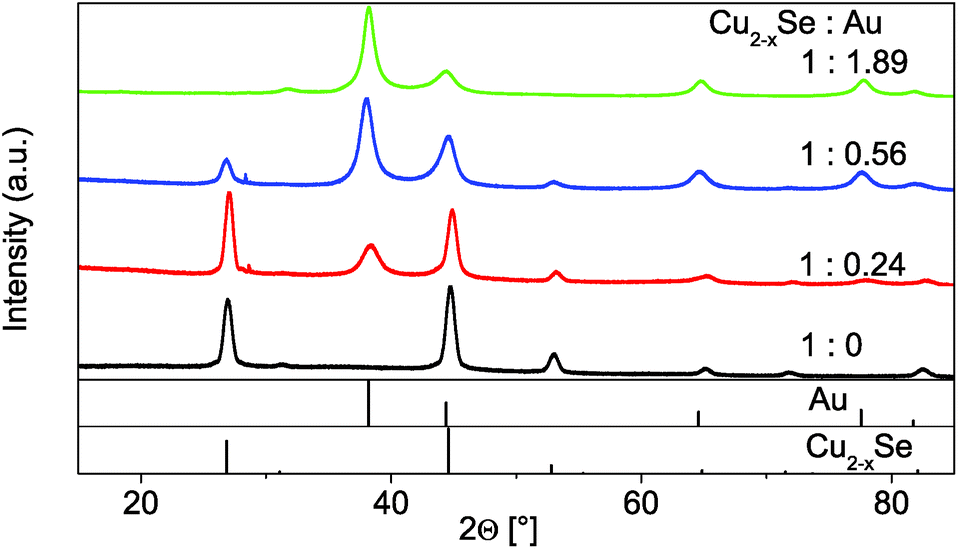 | ||
| Fig. 5 XRD patterns of the original Cu2−xSe seed particles (1:0) and of the Cu2−xSe–Au hybrid structures with different Cu2−xSe:Au ratios (1:0.24, 1:0.56 and 1:1.89). The reference structural data shows fcc Cu2−xSe berzelianite (PDF card #: 01-072-7490) and fcc Au (PDF card #: 00-004-0784). | ||
With increasing Au content, the absorption spectra (measured with an integrating sphere) of the as-prepared samples show a more pronounced absorption peak centered at 550–600 nm which originates from an LSPR in the Au domains of the hybrid NPs (Fig. 4). The intensity of the absorption band of the Cu2−xSe LSPR on the other hand decreases with an increasing amount of Au and shows a small hypsochromic shift of about 100 nm from the 1:0.24 to 1:0.56 sample. A further increase of the Au amount (1:1.89 sample) leads to a broad absorption band where no distinct Cu2−xSe LSPR absorption can be distinguished. The broad absorption band rather shows a maximum around 600 nm, however with an absorption onset above 2000 nm. It cannot be excluded that the difference in absorption behavior in comparison to the other samples originates in the strong agglomeration that occurred for this high Au contents.
The as-prepared samples show a LSPR in the NIR region due to the copper deficiency in the Cu2−xSe part. By the addition of Cu(I) ions to this hybrid system, additional Cu(I) is built into the Cu2−xSe, and partially in solution oxidized to Cu(II) (in order to reduce the selenium in the NPs). Hence, the free charge carrier concentration (holes) in the semiconductor domain is reduced. This, as has been discussed in literature1,4,6 and previously in this publication, leads to a bathochromic shift and decreased intensity of the copper chalcogenide LSPR as can be seen in Fig. 4.
In contrast to the two distinct and separate LSPR maxima that can be observed for our 1:0.24 and 1:0.56 sample (Fig. 4), Liu et al. described for Cu2−xSe grown on Au seed particles a broad LSPR across visible and NIR wavelengths. They qualitatively analyzed this using a so called quantum model.28 As Cu2−xSe NPs are strongly p-doped, the Fermi level is close to the valence band. In order to achieve thermal equilibrium at the interface, electrons of the metal will diffuse into the semiconductor. The consequence will be an electron density reduction in the Au part, and hence a bathochromic shift of its LSPR. The semiconductor LSPR should show a broadening and reduced intensity, due to a lower charge carrier density.28 Following this model a decrease of p-doping would lead to less electrons being able to diffuse from the metal to the semiconductor part, as to the decrease of p-doping. This, however, would be expected to result in a hypsochromic shift for the Au LSPR for higher Cu(I) amounts being added. No such shift can be observed (Fig. 4). In fact the metal LSPR shifts bathochromically for about 20–30 nm for the 1:0.24 and the 1:0.56 sample and around 50–60 nm for the 1:0.89 sample. One explanation for this shift is the bathochromic shift of the Cu2−xSe band gap absorption, due to its reduction according to the reverse Burstein–Moss effect.
It is worth to note that the position of the plasmon maximum above 550 nm would require a pure Au NP to have a size well above 50 nm. As no such Au particles could be found in any sample the LSPR maximum must hence be explained differently. Another reason for the absorption maximum at relatively long wavelengths is the high frequency dielectric constant of Cu2−xSe (ε∞ = 11.0–11.6)41 compared to toluene. This approach also explains why no hypsochromic shift of the Au related LSPR is observed during the addition of Cu(I).
Adding Ag(I) ions to the Au–Cu2−xSe hybrid NPs the semiconductor LSPR can be non reversible, bathochromically shifted as we showed for the pristine Cu2−xSe particles (see Fig. S3 in the ESI†).
We showed here by Au growth experiments that with our synthesis strategy we received dual-plasmonic hybrid Au–Cu2−xSe NPs which exhibit two surprisingly little interacting LSPRs. This is, to the best of our knowledge, the first report of weakly coupled LSPRs in Au–copper chalcogenide hybrid systems. The difference to previous reported hybrid systems might be the reverse growth strategy that allows the coupling of the two domains at room temperature.
Cation exchange of Au(I) into Cu1.1S
In contrary to the Cu2−xSe–Au hybrid structures, a Cu1.1S–Au hybrid structure cannot be observed following our synthetic conditions (Fig. 6). Instead a continuous cation exchange from Cu1.1S to Au2S starting from the edges of the Cu1.1S discs resulting in a Cu1.1S–Au2S structure takes place. This is accompanied with a bathochromic shift and an intensity decrease of the LSPR, and was reported so far only for quasi-spherical Cu2−xS NPs.42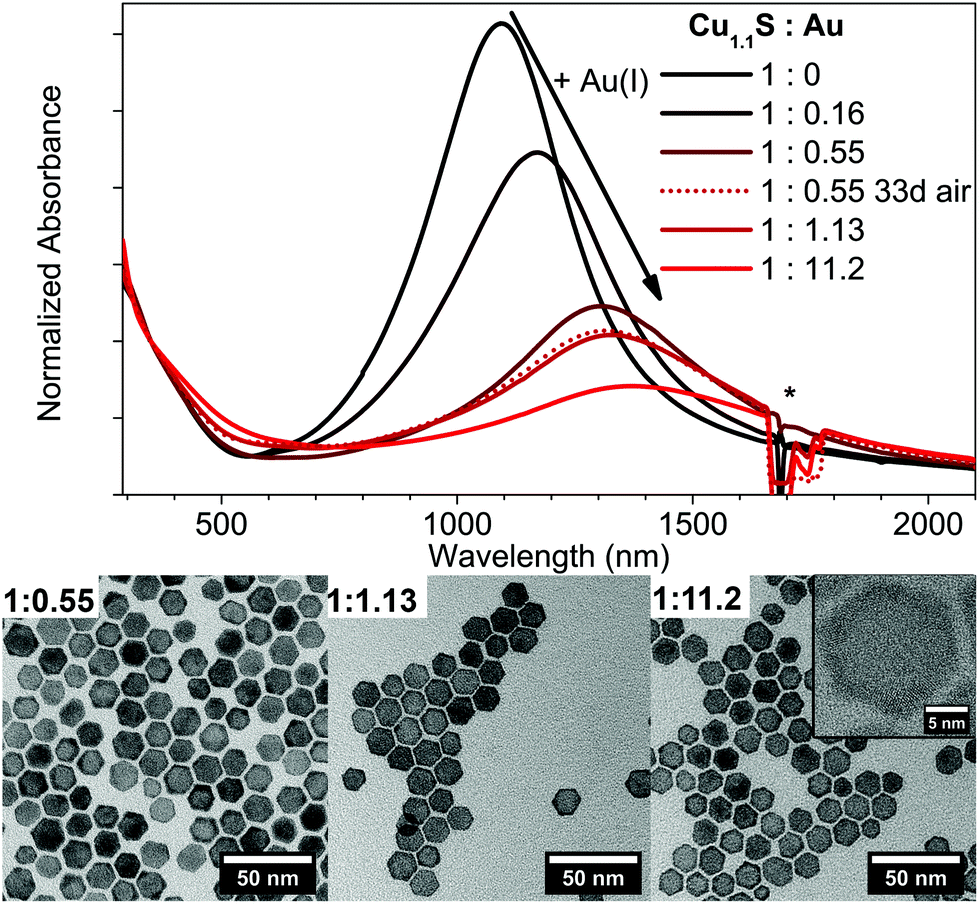 | ||
| Fig. 6 Normalized absorption spectra and TEM images of covellite Cu1.1S NPs treated with different AuCl3 amounts (Cu1.1S:Au = 1:0.16, 1:055, 1:1.13, 1:11.2). The spectra of the 1:0.55 sample stored under air for 33 d (dotted line) is also shown. *The peaks around 1700 nm are due to solvent absorption. | ||
The absorbance spectra show a LSPR maximum shift from 1100 nm to 1365 nm from no Au added to a Cu1.1S:Au ratio of 1:11.2 (Fig. 6). The shift of the LSPR is highly stable under air exposure over several days and only shows slight variations in the oscillator strength. The LSPR shift behaves very similar to the one observed for the addition of Ag(I), however due to the slower reaction kinetics an even better control of the exact maximum position can be achieved. The high stability of the LSPR shift under oxygen exposure can be explained due to the formation of the stable Au2S phase. The increasing formation of Au2S can be clearly seen in the provided TEM images in Fig. 6. In comparison to pure covellite NPs (see Fig. S4 in the ESI†) these samples show a high contrast crown, due to the higher contrast of elements with a higher atomic number in bright field imaging, which is increasing with higher Au amounts added. By controlling the amount of Au added this technique gives an additional method to shift and stabilize the LSPR maximum at a desired wavelength.
A possible reason for the different behavior of the two systems are their different redox potentials. These result in the reduction of the Au(III) precursor to Au(0) for the Cu2−xSe system but only to Au(I) for the Cu1.1S system.
In summary the growth of Au domains on Cu2−xSe with size control can be achieved with the here presented method. This leads to Au–Cu2−xSe dual-plasmonic hybrid particles with two LSPRs originating from the two domains of the particles. It was shown for the first time that the LSPRs of such Au–copper chalcogenide hybrids influence each other little. Applying the same strategy to grow Au on the Cu1.1S NPs does not result in the growth of Au domains but rather in a cation exchange and the formation of Au2S–Cu1.1S NPs (see Fig. 6). As with the presented method the exchange is not complete, the LSPR can still be detected; even though it is strongly bathochromically shifted. Additionally the shift is stable also under air exposure for several days.
Conclusions
We have presented different methods to tune the LSPR of degenerately doped Cu2−xSe berzelianite and Cu1.1S covellite NPs. We showed and compared different strategies to change in a controlled manner the LSPR maximum position and its oscillator strength intensity. Specifically we compared the temporal stability of these changes for the ion exchange respectively intercalation of Ag(I) in comparison to the changes with Cu(I). We could show that the intercalation of Ag(I) and the ion exchange of Au(I) into covellite Cu1.1S NPs causes a LSPR bathochromic shift that is more stable also under exposure to air. This reduced air sensitivity enables us to stabilize the LSPR band for possible applications in different wavelength regions than those accessible by the pristine NPs. In comparison the Ag(I) ion exchange in the berzelianite system leads to a damping of the LSPR without a significant shift. We assigned this observation to an increased damping due to the additional Ag(I) in the NPs. Furthermore, we presented a new strategy to synthesize Au–Cu2−xSe dual-plasmonic hybrid nanoparticles. We were able to show with Au growth experiments that the two LSPRs, originating from the different domains of the hybrid NPs, interact surprisingly little with each other.Overall, the post-synthetic treatment of plasmonic copper chalcogenides gives, despite of its complexity, a manifold way to tune the LSPR of these particles and especially to stabilize the LSPR position also under ambient conditions.
Experimental section
Materials
Copper(I) chloride (CuCl, 99.99% extra pure), ethanol (EtOH, 99.5%, extra dry) and silver nitrate (99.85%) were purchased from Acros Organics. Selenium (Se, 99.999%, 200 mesh), methanol (MeOH, 99.9%, anhydrous), toluene (99.8%, anhydrous packed under argon) and n-butanol (99.9% packed under argon) were purchased from Alfa Aesar. Sulfur (S, 99.98%), tetrakis (acetonitrile)copper(I) hexafluorophosphate ([Cu(CH3CN)4]PF6, 97%), dodecylamine (DDA, 98%), didodecyl-dimethylammonium bromide (DDAB, 98%), octadecene (ODE, 90%) and oleylamine (OLA, >70%) were purchased from Sigma Aldrich. Gold(III) chloride (99%) was purchased from ABCR. Hydrochloric acid (>37%, for trace analysis) and nitric acid (>69%, for trace analysis) were purchased from Fluka.Pre-degassed OLA and ODE were prepared by placing each solvent in a separate flask and heating them under reflux (≤1 × 10−3 mbar, 115 °C) for 6 h, while purging them multiple times with argon. Afterwards they are transferred and stored inside the glove box under inert gas.
All other chemicals were used without further purification.
If not otherwise mentioned all experiments have been done under argon using standard Schlenk line techniques or were done inside a nitrogen filled glove box.
Synthesis of Cu2−xSe nanoparticles
The Cu2−xSe spherical nanoparticles have been synthesized adapting a procedure from Deka et al.36 15 ml ODE and 15 ml OLA were degassed for 3 h under vacuum (≤1 × 10−3 mbar) at 115 °C in a 100 ml 3-neck flask heated by a heating mantle. After cooling the mixture to room temperature CuCl (297 mg, 3 mmol) is added under a strong argon flow. The mixture was again set under vacuum and heated to 115 °C for an additional 15 min. After setting the flask under a constant argon flow the mixture was rapidly heated in 5–6 min to 300 °C. During that heating the color changed from first yellowish hazy to clear yellow to black. The selenium precursor solution was prepared by mixing Se (117 mg, 1.5 mmol) with pre-degassed 9 ml OLA and evacuating it for 30 min at 115 °C. After setting the flask under a constant argon flow the temperature was left at 190–200 °C. When all Se was dissolved the temperature was raised to 230 °C for 20 min before it was cooled down to 150 °C. At this temperature the solution was transferred into a glass syringe and rapidly injected into the copper precursor solution. Following this, the reaction mixture's temperature dropped to 270 °C and was allowed to recover to 290 °C. 15 min after the injection the solution was rapidly cooled down to room temperature at 150 °C 20 ml toluene were injected into the flask to prevent agglomeration. The particles have been precipitated by addition of 20 ml EtOH and 10 ml MeOH and centrifuged at 3700 g for 20 min. The precipitate was redispersed in 20 ml toluene by ultrasonication for 5 min. After leaving the particles for 12 h they were again centrifuged for 20 min at 3700 g and the supernatant used for all further experiments.A typical Cu2−xSe NP solution in toluene contains 0.053 mmol per ml Cu2−xSe, this corresponds to roughly 2.5 × 10−6 mmol NPs per ml.
Synthesis of Cu1.1S nanoplatelets
The Cu1.1S nanoplatelets have been synthesized by up-scaling a method previously reported by Y. Xie et al.:6 A mixture of 40 ml OLA, 40 ml ODE and sulfur (256.5 mg; 8 mmol) were degassed for 30 min under vacuum (≤1 × 10−3 mbar) at 120 °C in a 100 ml 3-neck flask. Under argon the temperature was raised to 130 °C and held at this temperature for 5 min. The solution was cooled to room temperature and CuCl (400 mg; 4 mmol) was added to the solution. At addition the solution turns black starting from a clear yellow-orange solution. The flask was again set to vacuum for 60 min at room temperature. The temperature was raised with a ramp of 8 °C min−1 to 200 °C and kept there for 30 min. The reaction solution was rapidly cooled to room temperature. After the addition of 40 ml EtOH the particles have been centrifuged for 20 min at 3700g. The precipitate was redissolved in 28 ml toluene. The resulting dark green particle solution was centrifuged at 100g for 5 min to remove larger side products. The supernatant was used for all further experiments. A typical Cu1.1S NP solution in toluene contains 0.049 mmol per ml Cu1.1S, this corresponds to roughly 1.6 × 10−6 mmol NPs per ml.Cu(I) cation exchange
[Cu(CH3CN)4]PF6 (74.5 mg; 0.200 mmol) was dissolved in 5 ml MeOH to receive a 0.04 M Cu(I) solution. A diluted solution of copper chalcogenide particles (oxidized Cu2−xSe respectively Cu1.1S) was prepared. 3 ml of that solution were given inside a cuvette together with the calculated amount of Cu(I) solution and shaken vigorously. The samples were measured 1 h after preparation.Ag(I) cation exchange
AgNO3·6H2O (25 mg; 0.147 mmol) was dissolved in 0.9 ml MeOH to receive a 0.164 M Ag(I) solution. A diluted solution of copper chalcogenide particles (Cu1.1S respectively oxidized Cu2−xSe) was prepared. 3 ml of that solution were given inside a cuvette together with the calculated amount of Ag(I) solution and shaken vigorously. The samples were measured 2 h after preparation.Au growth on Cu2−xSe and Au cation exchange in Cu1.1S NPs
The growth of Au on chalcogenide NPs was adapted from the Banin synthesis of Au on CdSe particles.43An Au-precursor stock solution was freshly prepared with an Au:DDAB:DDA ratio of 1:5.5:22 and an Au concentration of 0.006 mmol per ml toluene. For that AuCl3 (72.9 mg; 0.24 mmol), DDAB (611.7 mg; 1.32 mmol) and DDA (980.3 mg; 5.29 mmol) were dissolved in 40 ml toluene. 1.7 ml of a diluted Cu2−xSe solution (cCu2−xSe = 0.033 mmol ml−1) and 25 μl OLA are placed in a sample vial and under stirring calculated amounts of the Au-precursor solution are added slowly. The solution is left stirring for 30 min. The particles are separated by adding MeOH and centrifugation at 3700g. The NPs are redispersed in 4 ml toluene.
For Cu1.1S 2 ml of a diluted solution (cCu1.1S = 0.009 mmol ml−1) were placed in a sample vial. All further steps have been done like for the Au growth on Cu2−xSe.
UV/Vis/NIR absorption spectroscopy
All spectra were measured on an Agilent Cary 5000 UV/Vis/NIR spectrophotometer. Absorption spectra for Cu2−xSe–Au hybrid NPs were recorded with the spectrophotometer being equipped with an Agilent DRA-2500 Ulbricht sphere to account for slight light scattering of the sample. Samples were prepared by dispersing the NPs in 3 ml of toluene in a 10 mm path length quartz glass cuvette.Elemental analysis
Quantitative copper elemental analysis was done by atomic absorption spectroscopy (AAS) using a Varian AA 140 spectrometer. The samples were dissolved in aqua regia (1 part HNO3 and 3 parts HCL) and diluted after several hours with deionized water. The measurements were carried out at a wavelength of 324.8 nm and an acetylene air flame.Electron microscopy
Conventional TEM and HRTEM measurements were carried out using a FEI Tecnai G2 F20, equipped with a field emission gun operated at 200 kV. EDX measurements were conducted in scanning TEM mode using a JEOL JEM-2100F, operated at 200 kV and equipped with a field emission gun. Sample preparation was done by placing a QUANTIFOIL carbon coated copper grid on a filter paper and dropping 10 μl of a diluted particle solution on it.Powder X-ray diffraction analysis
XRD analysis was carried out using a Bruker D8 Advance in reflection mode with a Cu source operating at 40 kV and 40 mA. Samples have been prepared by drying a concentrated NP solution on a single crystalline silicon sample holder.Acknowledgements
D.D. wants to thank the German research foundation (DFG, research grants DO1580/2-1 and DO1580/3-1) and the Volkswagen foundation (lower Saxony/Israel cooperation, Grant ZN2916) for funding. The authors are grateful to N. C. Bigall for helpful discussions. The authors thank the Laboratory of Nano and Quantum Engineering of the Leibniz Universität Hannover for support. T. K. is grateful to the Hannover School for Nanotechnology (HSN) for funding.References
- D. Dorfs, T. Hartling, K. Miszta, N. C. Bigall, M. R. Kim, A. Genovese, A. Falqui, M. Povia and L. Manna, J. Am. Chem. Soc., 2011, 133, 11175–11180 CrossRef CAS PubMed
.
- O. A. Balitskii, M. Sytnyk, J. Stangl, D. Primetzhofer, H. Groiss and W. Heiss, ACS Appl. Mater. Interfaces, 2014, 6, 17770–17775 CAS
- Y. Wang, M. Zhukovskyi, P. Tongying, Y. Tian and M. Kuno, J. Phys. Chem. Lett., 2014, 5, 3608–3613 CrossRef CAS PubMed
- J. M. Luther, P. K. Jain, T. Ewers and A. P. Alivisatos, Nat. Mater., 2011, 10, 361–366 CrossRef CAS PubMed
- Y. Zhao, H. Pan, Y. Lou, X. Qiu, J. Zhu and C. Burda, J. Am. Chem. Soc., 2009, 131, 4253–4261 CrossRef CAS PubMed
- Y. Xie, A. Riedinger, M. Prato, A. Casu, A. Genovese, P. Guardia, S. Sottini, C. Sangregorio, K. Miszta, S. Ghosh, T. Pellegrino and L. Manna, J. Am. Chem. Soc., 2013, 135, 17630–17637 CrossRef CAS PubMed
- M. Kruszynska, H. Borchert, A. Bachmatiuk, M. H. Ruemmeli, B. Buechner, J. Parisi and J. Kolny-Olesiak, ACS Nano, 2012, 6, 5889–5896 CrossRef CAS PubMed
- I. Kriegel, J. Rodríguez-Fernández, A. Wisnet, H. Zhang, C. Waurisch, A. Eychmüller, A. Dubavik, A. O. Govorov and J. Feldmann, ACS Nano, 2013, 7, 4367–4377 CrossRef CAS PubMed
- W. Li, R. Zamani, G. Rivera Pilar, B. Pelaz, M. Ibanez, D. Cadavid, A. Shavel, R. A. Alvarez-Puebla, W. J. Parak, J. Arbiol and A. Cabot, J. Am. Chem. Soc., 2013, 135, 7098–7101 CrossRef CAS PubMed
- E. Dilena, D. Dorfs, C. George, K. Miszta, M. Povia, A. Genovese, A. Casu, M. Prato and L. Manna, J. Mater. Chem., 2012, 22, 13023–13031 RSC
- P. L. Saldanha, R. Brescia, M. Prato, H. Li, M. Povia, L. Manna and V. Lesnyak, Chem. Mater., 2014, 26, 1442–1449 CrossRef CAS
- X. Liu, X. Wang and M. T. Swihart, Chem. Mater., 2013, 25, 4402–4408 CrossRef CAS
- T. K. Sau, A. L. Rogach, F. Jaeckel, T. A. Klar and J. Feldmann, Adv. Mater., 2010, 22, 1805–1825 CrossRef CAS PubMed
- P. K. Jain, X. Huang, I. El-Sayed and M. El-Sayed, Acc. Chem. Res., 2008, 41, 1578–1586 CrossRef CAS PubMed
- N. J. Halas, S. Lal, W. S. Chang, S. Link and P. Nordlander, Chem. Rev., 2011, 111, 3913–3961 CrossRef CAS PubMed
- B. Sepulveda, P. C. Angelome, L. M. Lechuga and L. Liz-Marzan, Nano Today, 2009, 4, 244–251 CrossRef CAS
- S. Link and M. El-Sayed, J. Phys. Chem. B, 1999, 103, 4212–4217 CrossRef CAS
- C. Ziegler and A. Eychmüller, J. Phys. Chem. C, 2011, 115, 4502–4506 CAS
- S. J. Oldenburg, R. D. Averitt, S. L. Westcott and N. J. Halas, Chem. Phys. Lett., 1998, 288, 243–247 CrossRef CAS
- C. J. Murphy, T. K. Sau, A. M. Gole, C. J. Orendorff, J. Gao, L. Gou, S. E. Hunyadi and T. Li, J. Phys. Chem. B, 2005, 109, 13857–13870 CrossRef CAS PubMed
- A. N. Bashkatov, E. A. Genina, V. I. Kochubey and V. V. Tuchin, J. Phys. D: Appl. Phys., 2005, 38, 2543–2555 CrossRef CAS
- X. Ding, C. H. Liow, M. Zhang, R. Huang, C. Li, H. Shen, M. Liu, Y. Zou, N. Gao, Z. Zhang, Y. Li, Q. Wang, S. Li and J. Jiang, J. Am. Chem. Soc., 2014, 136, 15684–15693 CrossRef CAS PubMed
- G. Garcia, R. Buonsanti, E. L. Runnerstrom, R. J. Mendelsberg, A. Llordes, A. Anders, T. J. Richardson and D. J. Milliron, Nano Lett., 2011, 11, 4415–4420 CrossRef CAS PubMed
- Y. Zhao and C. Burda, Energy Environ. Sci., 2012, 5, 5564–5576 CAS
- J. A. Faucheaux, A. L. D. Stanton and P. K. Jain, J. Phys. Chem. Lett., 2014, 5, 976–985 CrossRef CAS PubMed
- X. Liu and M. T. Swihart, Chem. Soc. Rev., 2014, 43, 3908–3920 RSC
- A. Comin and L. Manna, Chem. Soc. Rev., 2014, 43, 3957–3975 RSC
- X. Liu, C. Lee, W. Law, D. Zhu, M. Liu, M. Jeon, J. Kim, P. N. Prasad, C. Kim and M. T. Swihart, Nano Lett., 2013, 13, 4333–4339 CrossRef CAS PubMed
- X. Ding, Y. Zou and J. Jiang, J. Mater. Chem., 2012, 22, 23169–23174 RSC
- N. E. Motl, J. F. Bondi and R. E. Schaak, Chem. Mater., 2012, 24, 1552–1554 CrossRef CAS
- S. Shen, Z. Tang, Q. Liu and X. Wang, Inorg. Chem., 2010, 49, 7799–7807 CrossRef CAS PubMed
- Y. Kim, K. Y. Park, D. M. Jang, Y. M. Song, H. S. Kim, Y. J. Cho, Y. Myung and J. Park, J. Phys. Chem. C, 2010, 114, 22141–22146 CAS
- S. Khanal, G. Casillas, N. Bhattarai, J. Velazquez-Salazar, U. Santiago, A. Ponce, S. Mejia-Rosales and M. Jose-Yacaman, Langmuir, 2013, 29, 9231–9239 CrossRef CAS PubMed
- Z. Sun, Z. Yang, J. Zhou, M. H. Yeung, W. Ni, H. Wu and J. Wang, Angew. Chem., Int. Ed., 2009, 48, 2881–2885 CrossRef CAS PubMed
- W. Sang, T. Zheng, Y. Wang, X. Li, X. Zhao, J. Zeng and J. G. Hou, Nano Lett., 2014, 14, 6666–6671 CrossRef CAS PubMed
- S. Deka, A. Genovese, Y. Zhang, K. Miszta, G. Bertoni, R. Krahne, C. Giannini and L. Manna, J. Am. Chem. Soc., 2010, 132, 8912–8914 CrossRef CAS PubMed
- Y. Xie, L. Carbone, C. Nobile, V. Grillo, S. D'Agostino, F. Della Sala, C. Giannini, D. Altamura, C. Oelsner, C. Kryschi and P. D. Cozzoli, ACS Nano, 2013, 7, 7352–7369 CrossRef CAS PubMed
-
D. M. Trots, Structure and Lattice Dynamics of Copper- and Silver-based Superionic Conducting Chalcogenides, TU Darmstadt, Darmstadt, 2007 Search PubMed
- V. V. Gorbachev and I. M. Putilin, Phys. Status Solidi B, 1975, 69, K153–K156 CrossRef CAS
- V. Lesnyak, R. Brescia, G. C. Messina and L. Manna, J. Am. Chem. Soc., 2015, 137, 9315–9323 CrossRef CAS PubMed
- V. V. Gorbachev and I. M. Putilin, Phys. Status Solidi A, 1973, 16, 553–559 CrossRef CAS
- X. Wang, X. Liu, D. Zhu and M. T. Swihart, Nanoscale, 2014, 6, 8852–8857 RSC
- T. Mokari, E. Rothenberg, I. Popov, R. Costi and U. Banin, Science, 2004, 304, 1787–1790 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Further HAADF-STEM images and EDX mappings, further absorbance spectra and the theoretical dependence of the extinction cross section against γ. See DOI: 10.1039/c5nr05425g |
| This journal is © The Royal Society of Chemistry 2015 |