 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Water corrosion of spent nuclear fuel: radiolysis driven dissolution at the UO2/water interface
Ross
Springell
*a,
Sophie
Rennie
a,
Leila
Costelle
a,
James
Darnbrough
a,
Camilla
Stitt
a,
Elizabeth
Cocklin
b,
Chris
Lucas
b,
Robert
Burrows
c,
Howard
Sims
d,
Didier
Wermeille†
e,
Jonathan
Rawle
f,
Chris
Nicklin
f,
William
Nuttall
g,
Thomas
Scott
a and
Gerard
Lander
h
aInterface Analysis Centre, University of Bristol, Bristol BS2 8BS, UK. E-mail: phrss@bristol.ac.uk; Tel: +44 (0)117 331 1176
bDepartment of Physics, University of Liverpool, Liverpool L69 7ZE, UK
cNational Nuclear Laboratory, 102B Stonehouse Park, Sperry Way, Stonehouse, Gloucester GL10 3UT, UK
dNational Nuclear Laboratory, Harwell Science and Innovation Campus, Oxfordshire OX11 0QT, UK
eXMaS, European Synchrotron Radiation Facility, BP220, F-38043 Grenoble Cedex 09, France
fDiamond Light Source, Harwell Science and Innovation Campus, Harwell OX11 0DE, UK
gDepartment of Engineering and Innovation, The Open University, Venables Building, Milton Keynes MK7 6AA, UK
hEuropean Commission, Joint Research Centre, Institute for Transuranium Elements, Postfach 2340, D-76125 Karlsruhe, Germany
First published on 28th January 2015
Abstract
X-ray diffraction has been used to probe the radiolytic corrosion of uranium dioxide. Single crystal thin films of UO2 were exposed to an intense X-ray beam at a synchrotron source in the presence of water, in order to simultaneously provide radiation fields required to split the water into highly oxidising radiolytic products, and to probe the crystal structure and composition of the UO2 layer, and the morphology of the UO2/water interface. By modeling the electron density, surface roughness and layer thickness, we have been able to reproduce the observed reflectivity and diffraction profiles and detect changes in oxide composition and rate of dissolution at the Ångström level, over a timescale of several minutes. A finite element calculation of the highly oxidising hydrogen peroxide product suggests that a more complex surface interaction than simple reaction with H2O2 is responsible for an enhancement in the corrosion rate directly at the interface of water and UO2, and this may impact on models of long-term storage of spent nuclear fuel.
1 Introduction
The future storage of spent nuclear fuel (SNF) poses some of the most challenging scientific and economic questions.1–3 With the growing consensus that storage of this material in a deep underground repository is the most viable long term solution, and the likely scenario of containment failure and groundwater contact,4,5 one of the central problems is to understand the reactions at the interface of the fuel with its surroundings.4,6–8 Of these, of course, water is the most important, as it can transport radioactive material away from the fuel repository and into the ecosystem.9The predominant component of this fuel is a ceramic oxide of uranium, UO2, whose solubility in water of its stoichiometric U(IV) form, is very low.10 However, post burn-up, the UO2 fuel possesses levels of activity from 1014 Bq to 1016 Bq at its surface, depending on the reactor type, and this decays by approximately four orders of magnitude over the first 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 years of proposed storage, at which point containment failure becomes significantly likely.11 The strong alpha, beta and gamma radiation fields are sufficient to radiolyse (radiolysis is the dissociation of molecules due to nuclear radiation) water in close proximity.4,12,13 The radiolysis products comprise short-lived, highly oxidising free radical species, such as ˙OH and the much longer lived hydrogen peroxide molecule, H2O2.14,15 Under these highly oxidising conditions it is possible to form UO22+ (uranyl) ions via the oxidation of U(IV) to U(VI), whose solubility in water is several orders of magnitude greater than UO2,6 leading to accelerated dissolution of the fuel matrix and potential release of radionuclides into the environment. Therefore, an understanding of the water/UO2 interface, and the ability to predict its long-term behaviour are vitally important.
000 years of proposed storage, at which point containment failure becomes significantly likely.11 The strong alpha, beta and gamma radiation fields are sufficient to radiolyse (radiolysis is the dissociation of molecules due to nuclear radiation) water in close proximity.4,12,13 The radiolysis products comprise short-lived, highly oxidising free radical species, such as ˙OH and the much longer lived hydrogen peroxide molecule, H2O2.14,15 Under these highly oxidising conditions it is possible to form UO22+ (uranyl) ions via the oxidation of U(IV) to U(VI), whose solubility in water is several orders of magnitude greater than UO2,6 leading to accelerated dissolution of the fuel matrix and potential release of radionuclides into the environment. Therefore, an understanding of the water/UO2 interface, and the ability to predict its long-term behaviour are vitally important.
A stored spent fuel pellet possesses a great deal of complexity, including defects, He bubbles, microscopic cracking and fission daughter products for example. The situation becomes even more complicated when one includes groundwater, containing various ionic species, and further still, if one includes the potential influence from the cladding material. To date, the majority of studies of this complex system have focused on the chemical composition of the dissolution and the electrochemistry of the corrosion mechanism.4,7,9,10,16 Since one of the most important factors driving oxidising conditions in the groundwater is the radiolysis of the water,6,17 some studies have gone further and have attempted to replicate the radiolytic conditions electrochemically,18,19 using external and dopant alpha sources9,20–22 and by the addition of H2O2 to the groundwater solution.6,10,23 This final method is hotly debated amongst research groups, since it is not clear precisely what the H2O2 concentration would be at the fuel surface.
Here, we report a new approach; we aim to remove much of the material complexity and study the corrosion of UO2 in pure water in the presence of strong radiation fields. Using high-quality single-crystal thin films of UO2 with atomically smooth surfaces and a thin layer surface tension cell of MilliQ, pure water (nominally pH 7), we expose this model fuel/groundwater interface to an intense, monochromated beam of X-rays from a synchrotron source; an approach first employed on the XMaS beamline, BM28 at the ESRF. This source-probe method allows us to simultaneously provide strong radiation fields and probe the structure of the interface. Using a combination of X-ray reflectivity (XRR) and high angle diffraction (XRD) in a specular geometry we are able to probe changes in the interface structure, roughness, electron density, crystallinity and eventual dissolution as a function of exposure time.
It is important to stress the surface sensitivity of these techniques (XRR and XRD), and ask why this has not been observed previously with bulk UO2 samples. For typical energies and angles of incidence, the penetration of such a photon beam into UO2 is in the order several microns. Hence, there will be only a minuscule change in the Bragg reflected intensity, since >99.5% of the intensity comes from the undisturbed UO2 bulk sample. Even if a single-crystal is used, no measurable change will be observed. On the other hand, by using an epitaxial film of 50 Å, there will be enough intensity (at a synchrotron) and the changes will be very substantial, often exceeding 50%. There are also additional features in the diffraction profiles, resulting from finite thickness effects that give even more detailed information about the morphology of the interface. Such films therefore give unprecedented sensitivity to structural changes at the UO2 interface.
2 Experimental
Single-crystal thin films of UO2 were grown in a dedicated DC magnetron sputtering facility at the University of Bristol under UHV conditions. Samples were deposited in the three high symmetry directions, [001], [110] and [111], although the majority of the work presented here describes data collected from an [001]-oriented UO2 sample, deposited onto a single-crystal [001]-YSZ (yttria-stabilised zirconia) substrate of dimensions 1 cm × 1 cm × 0.5 mm.24 Reactive sputtering was used to deposit uranium in an argon pressure of ρAr = 7.2 × 10−3 mbar and an oxygen partial pressure of ρO2 = 2 × 10−5 mbar, to give a sputtering rate of 1.2 ÅUO2 s−1 in order to produce a sample of nominal thickness, tUO2 = 40 Å. Substrate heating was used to elevate the growth temperature to ∼550 °C, providing thermal energy to improve the crystalline quality, monitored using in situ reflection high-energy electron-diffraction (RHEED).Several samples were grown in order to verify the reproducibility of the experiment, and these were characterised using X-ray reflectivity (XRR) and high angle X-ray diffraction (XRD) on a Philips X'Pert Pro MRD, with a Cu Kα source (λ = 1.54 Å). This same geometry was used in the synchrotron measurements, carried out at the XMaS beamline, BM28, ESRF25 and I07, Diamond Light Source, as shown in Fig. 1.
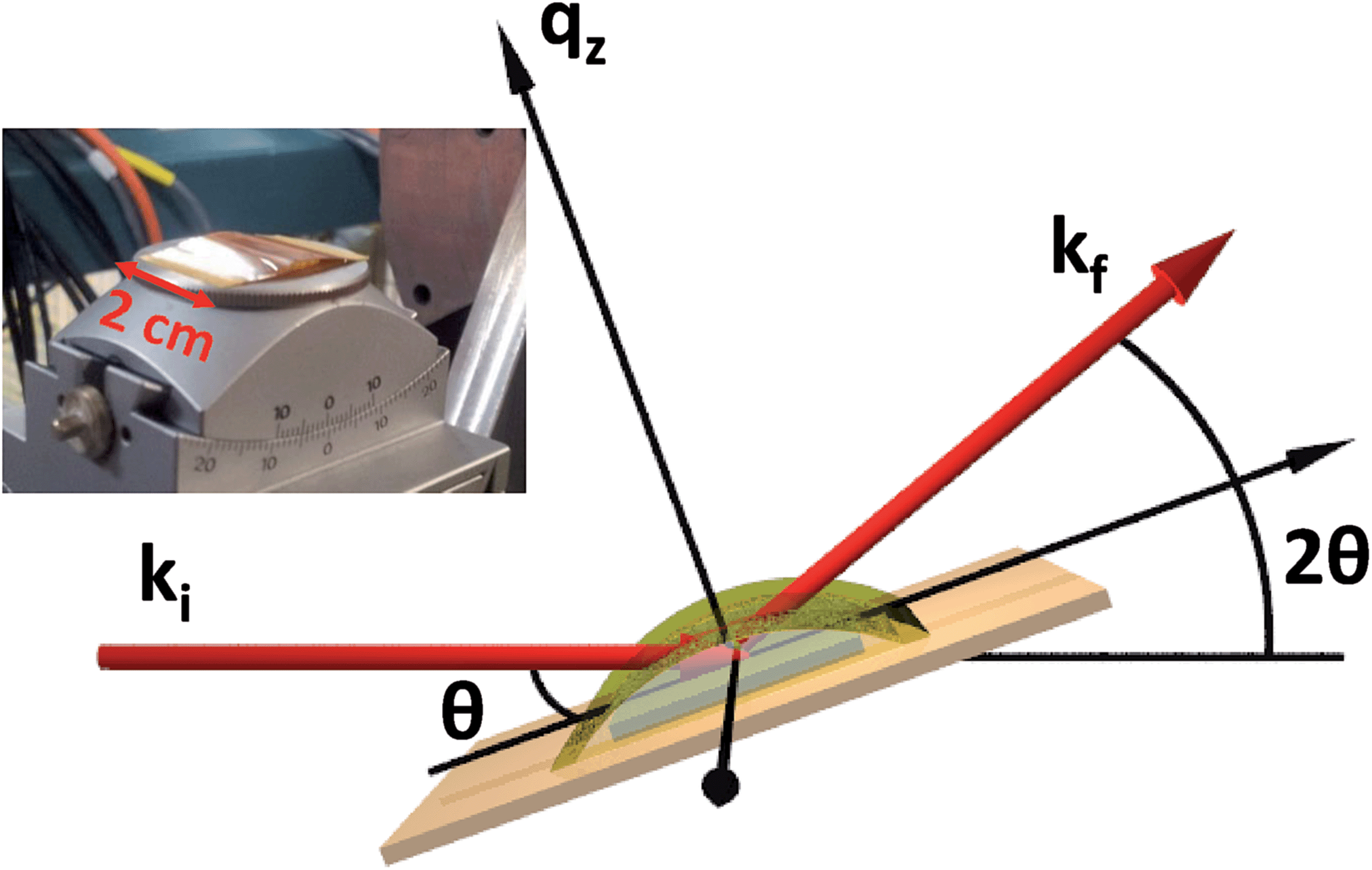 | ||
| Fig. 1 Schematic of the set-up used in the laboratory characterisation measurements and synchrotron experiments at the ESRF and Diamond Light Sources. Here, the incident and exit wavevectors (ki and kf), angle of incidence (θ), angle with respect to the detector (2θ) and the wavevector momentum transfer (qz) are labelled. The thin layer surface tension cell can be seen over the sample, which is used to hold a fixed volume of water that covers the entire sample during X-ray irradiation. The insert shows a photograph of the cell, the horizontal dimension is noted along the edge of the cell. | ||
X-ray reflectivity is a non-destructive technique particularly well-suited to probing the fine details of surfaces or buried interfaces,26 where the X-rays probe the electron density perpendicular to the surface normal. In this instance, the diffraction is called specular (or longitudinal) elastic scattering, i.e. that the incident and exit wavevectors, ki and kf, respectively, have the same magnitude, and that the angles of incidence and exit, θi and θf, respectively, are also equivalent. The X-ray intensity is measured as a function of incidence angle, close to the critical angle, θC, for total external reflection, which is typically a combination of Fresnel reflectivity (with a |1/qz|4 dependence) and a fringe pattern (commonly Keissig fringes), due to constructive interference from scattering at the layer interfaces. Here, qz is the wavevector momentum transfer along the surface normal.
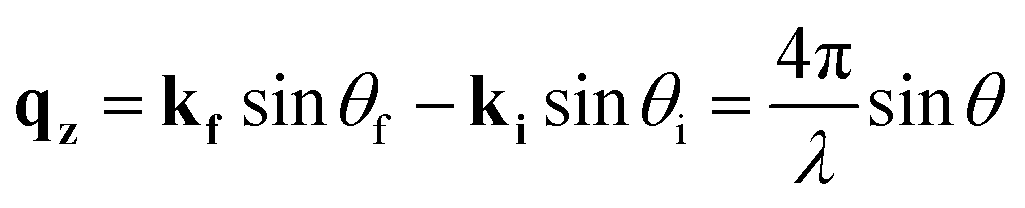 | (1) |
In this case, we have used the Parratt recursion method27 of calculating transmitted and reflected wave fields, using the GENX computer program, developed by Björck and Andersson,28 which fits calculated reflectivity profiles to experimental data, using a differential evolution algorithm that can be optimised to avoid local minima; a common problem encountered when modeling X-ray reflectivity. The variables that are used to construct the electron density profile are the densities of materials (ρsubstrate, ρUO2), the layer thickness (tUO2) and the roughness of each interface (σsubstrate, σUO2), measured as the root mean squared of the fluctuations in the height of the layer. Also included in the model is a top layer of uranium oxide that is lower in electron density than stoichiometric UO2, which we label UOX. This layer is modelled by a series of slices of varying electron density with thicknesses equivalent to a UO2 monolayer, and this attempts to interpret both the topology and hyperstoichiometric progression of uranium oxide. Later, in Table 1 an average tUOX is given, that represents the mean overall thickness of this top-layer, assuming half the electron density of UO2 (i.e. the average between the bulk value and air).
| Exposure time (s) | t UO2 | σ UO2 | t UOX | σ UOX |
|---|---|---|---|---|
| 0 | 34 | 3 | 10 | 6 |
| 30 | 27.5 | 2.5 | 16 | 9 |
| 90 | 21 | 7.6 | 20 | 11.5 |
| 120 | 13.5 | 11 | 22 | 13 |
High resolution X-ray diffraction provides a host of complementary information to describe the structural composition of thin films. In the case of a single-crystal [001]-UO2 film on YSZ, a longitudinal measurement across the (002) Bragg peak gives the average d-spacing for the thin film lattice parameter along the surface normal, and the finite thickness broadening of the Bragg peak can be used to calculate the number of scattering planes contributing to the intensity, and therefore the thickness of crystalline UO2. For smooth interfaces, fringes are also present, similar to those observed in X-ray reflectivity, which give information about the electron density profile. A rocking curve at the Bragg peak position produces an observable intensity from planes that are not perfectly parallel. This gives an indication of the crystal mosaicity and the density of dislocation defects. The rocking curve measurement is made by rotating the sample (varying θ), while keeping the detector (2θ) fixed, see Fig. 1.
3 Results
Using a combination of X-ray reflectivity and high resolution diffraction, it is then possible to build up a full structural picture of the thin film and the substrate/film and film/air interface. Fig. 2 shows the X-ray reflectivity (panel (a)) and high resolution diffraction (panel (b)) from a single-crystal [001]-UO2 film on YSZ, with a nominal thickness of 40 Å. The data are shown as open black circles and fitted calculations of the reflectivity and the high angle diffraction are represented by solid red lines.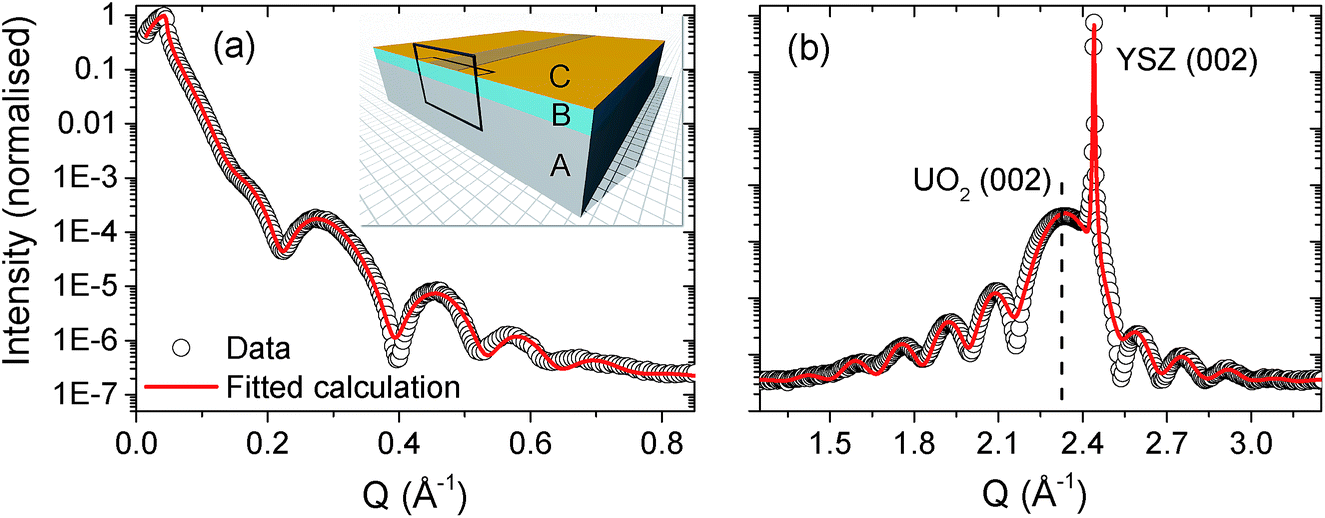 | ||
| Fig. 2 Panel (a) shows the X-ray reflectivity spectrum for a nominal 40 Å [001]-UO2 thin film, grown on YSZ. Panel (b) contains a high resolution diffraction spectrum across the UO2 (002) Bragg peak. The UO2 (002) thin film and YSZ (002) substrate Bragg peaks have been labelled for clarity. The experimental data are the open black circles and the fitted calculations are the solid red lines. The insert in panel (a) shows the model of a pristine UO2 film, labelled B, with a complex surface oxide layer, C, grown on a YSZ substrate, A. The copper coloured strip across the centre of the sample indicates the footprint of the X-ray beam at a low angle and the frame indicates the 2D profile used to indicate the corrosion front in Fig. 4. | ||
These data were taken at the I07 beamline of the Diamond Light Source. The photon flux at a synchrotron source is between 4 and 5 orders of magnitude greater than a laboratory source and so allows one to probe the reflectivity far further in qz and better resolve the fringes at higher angles. It is also ideal to study buried interfaces, which in our case will be the UO2/water interaction, since we are interested in the potential dissolution of UO2 in groundwater.
Table 1 includes the model parameters used to reproduce the calculations. It is clear that even for a pristine sample, not exposed to heavily oxidising conditions, it is necessary to model the thin film with a ∼30 Å crystalline UO2 layer and then a top layer of low electron density oxide. The insert of Fig. 2(a) shows a model of the pristine film, where the substrate layer is labelled A, the UO2, B, and the top oxide layer, C. Since we are concerned with the corrosion of UO2 in water, the next step is to repeat these measurements, using synchrotron radiation to simultaneously probe the film structure and to provide the necessary radiolytic products for oxidative dissolution.
In order to probe the change in the uranium oxide film in detail as a function of exposure time, first we set the detector position to the centre of the UO2 (002) Bragg peak and measured the intensity as a function of time. The following data were taken on the I07 beamline at the Diamond Light Source, UK. The beam energy was monochromated to 17.116 keV (50 eV below the U L3 absorption edge), which was then focused in the vertical and horizontal directions to give an approximate beam size at the sample position of 200 μm × 200 μm. The incident beam slits were set to 100 μm × 100 μm for all measurements and the scattered photons were detected, using a Pilatus 100K detector.
In Fig. 3(a) and (b) we present data at three points in time, 30 s, 90 s and 120 s. The thin layer surface tension cell was filled with Milli-Q ultra pure type 1 water and the incident slits were set to a 200 micron square. The surface was exposed at an incident angle of ∼0.5°, such that the footprint was approx. 2 cm, more than covering the whole length of the sample. The water was then removed, the slits were closed down to 100 μm × 100 μm and the diffraction spectra were recorded. The water removal was carried out using a pipette, and an N2 gas flow jet was positioned close to the sample surface during measurements. Two simple tests were carried out to confirm the necessary conditions of a surface/water interface and radiation strong enough to drive radiolysis: the first involved exposing the UO2 surface to water for two hours and then measuring the X-ray reflectivity and diffraction profiles; no changes were detected. The second test involved exposing a sample to an intense X-ray beam for an hour with no water present; again, no changes to the scattered intensity were observed.
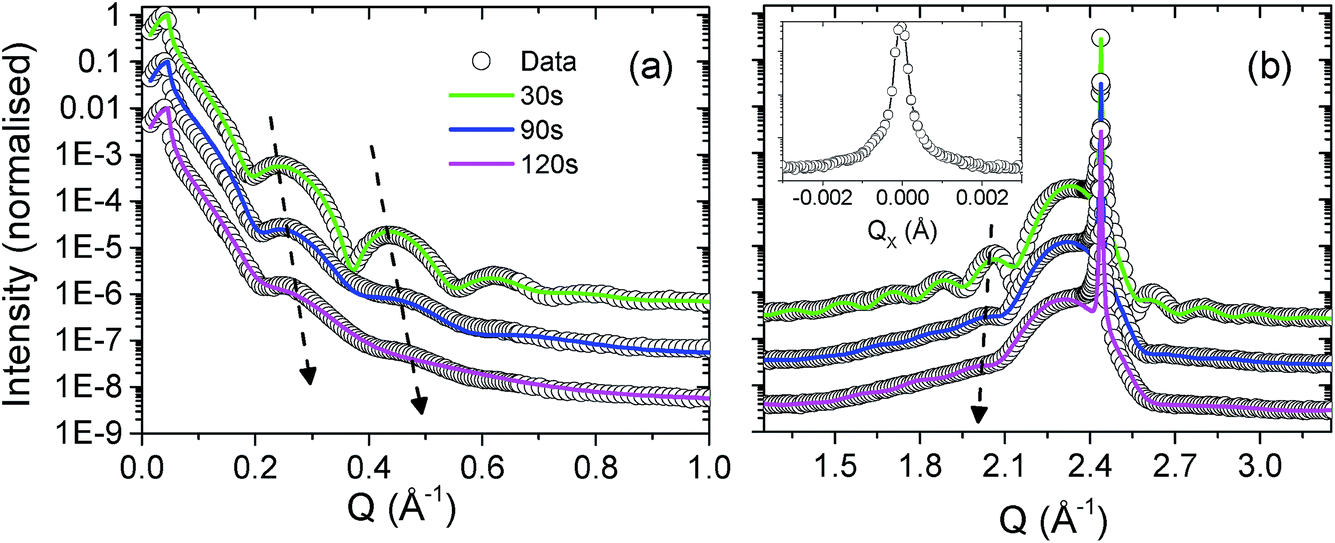 | ||
| Fig. 3 Panel (a) shows X-ray reflectivity data and panel (b) shows high angle diffraction data, measured at exposure times of 30 s, 90 s and 120 s; the experimental data are represented by the open black circles and the fitted calculations by the solid green, blue and magenta lines, respectively. The insert of panel (b) shows the rocking curve of the (002) Bragg peak for the 30 s exposure. The dashed black arrows indicate an increase in fringe separation as a function of exposure time, which suggests a concomitant loss of material. | ||
A calibrated Si photodiode was placed in the beam at the sample position in order to accurately calculate the number of photons per second per unit area incident for a number of slit settings. Together with the beam energy, these values could then be used to calculate the likely number of oxidising species present in the X-ray beam path in the water, specifically, to calculate the number of long lived, oxidising H2O2 molecules. Fig. 3 shows fitted calculations to the experimental data, based on a structural model of the UO2 film that consists of a layer of crystalline UO2 with the standard bulk density and a surface layer of reduced electron density, labeled UOX. Roughnesses for each of the substrate/UO2 (∼2 Å in each case), UO2/UOX and UOX/water interfaces were also computed. Table 1 summarises all of the parameters and Fig. 4 shows a pictorial representation of the corrosion region at each of the 30 s, 90 s and 120 s exposures.
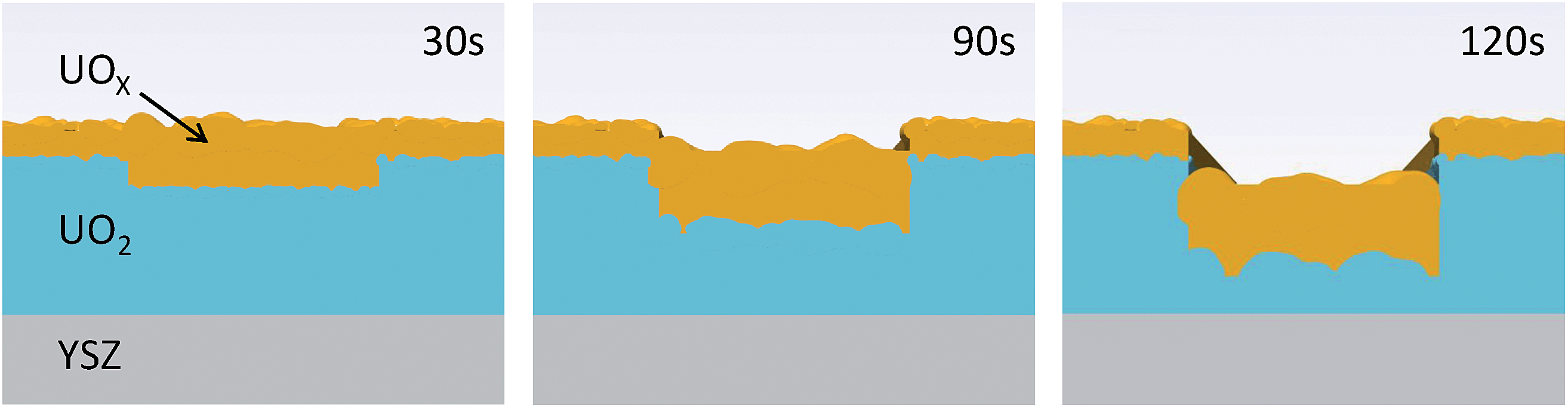 | ||
| Fig. 4 Pictorial representation of the increase in roughness and UOX thickness, and the amount of dissolution as the surface undergoes corrosion. | ||
Initially, the intensity of the UO2 (002) Bragg peak was measured as a function of the exposure time. This is not precisely a measure of the dissolution, since a decrease in intensity will also result from a surface roughening and oxidation, but it does provide a good estimate for the rate of change of the interface structure. This measurement was carried out for thin film samples of [001] (polar surface), [110] and [111] orientations (non-polar surfaces), which due to their different surface energies and water adsorption energies are expected to have significant impact on the rate of dissolution. However, contrary to this assumption, within the errors of this experiment, we saw no evidence to suggest that this may play a significant role. This confirmed earlier measurements carried out at an incident energy of 15 keV at the XMaS beamline, where also the experiment was repeated under alkaline (pH ∼ 11) and acidic (pH ∼ 2) conditions. As expected, in the case of increased acidity, the rate of corrosion dramatically increased, whereas under heavily alkaline conditions the rate of change in the UO2 Bragg peak intensity all but stopped, i.e. dissolution was halted.
Furthermore, since we were also interested in any potential surface enhancement of the corrosion by the photocatalytic process, we measured the rate of change in the Bragg peak intensity before (17.116 keV), at (17.166 keV) and after (17.216 keV) the uranium L3 absorption edge. There is a huge resonant enhancement in the number of electrons excited to the continuum at such an absorption edge, particularly for the U L3, so this ought to have a pronounced effect on any possible photocatalytic process, however, we did not observe any difference in the rate of the decrease in the Bragg peak intensity, within experimental errors.
4 Discussion
So far we have seen experimentally the effect of radiolytically driven oxidation and dissolution of the UO2 surface, but it is also possible to calculate the quantity of radiolysis products from first principles, based on the energy and flux of the X-ray source incident on a known water thickness. In the first instance, we are making the same assumption as the vast majority of the literature, that the longest lived and dominant oxidising product is hydrogen peroxide.6,23 Here, we have ∼1 × 1012 photons per s of 17.116 keV X-rays incident on a 0.5 mm thick water layer on the UO2 film surface. The G-value, the number of molecules of reactant consumed or product formed (in our case) per unit of incident energy absorbed, is 0.6 molecules of H2O2/100 eV.For this thickness of water at this photon energy, the transmission of photons to the surface is ∼59%, which results in ∼7 × 10−11 moles H2O2 produced. Since the volume of the water exposed to the beam is approx. 200 μm wide, 0.5 mm high and 1 cm long, i.e. 0.001 cm3, the H2O2 concentration increases by ∼7 × 10−5 M per second. The equilibrium concentration is in the region of 1 × 10−4 M, which means that it is reached almost instantly, relative to the timescales of this experiment. The question then arises – is H2O2 alone enough to drive the changes that we are observing in our experiment? This is the general assumption purported by the literature.6,23
Fig. 5 may provide a clue. Panel (a) is a scanning electron microscopy image, obtained using a Zeiss Sigma FEG-SEM; utilising secondary electron detection with electron gun settings of 10 kV and 30 μA. The image shows the area of the sample that has been exposed to the beam. This area is heavily corroded and so is not as conductive as the surrounding UO2 film. What is observed here is the resultant charging of the corroded region. There is one particularly remarkable feature and that is that the width of the corrosion track is 100 μm, which is precisely the slit settings used in the experiment. During the duration of the experiment, one might imagine that due to diffusion of the H2O2 species there would be a far wider area of corroded material. Panels (b) and (c) of Fig. 5 represent a finite element model, including (i) a short-lived species, which is confined to the radiolysis volume within the beam path, or one produced only at the sample surface and (ii) a long-lived species (such as H2O2) subject to diffusive transport through the water layer in order to determine the likely footprint of corrosion.
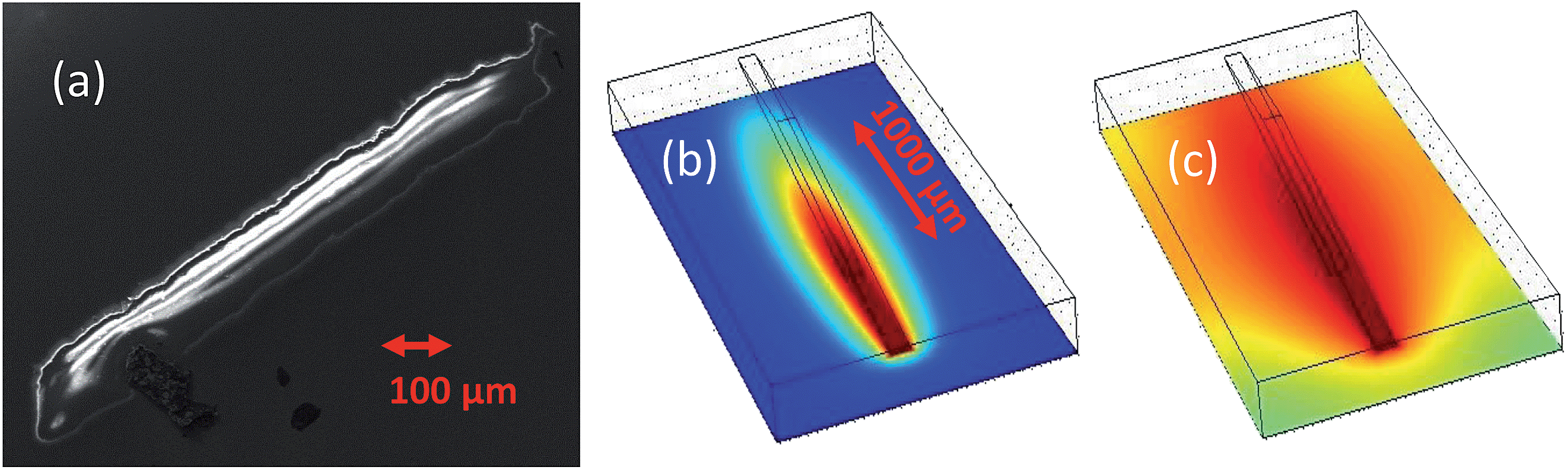 | ||
| Fig. 5 Panel (a) shows an SEM image of a corroded UO2 single-crystal thin film, measured at the UO2 Bragg peak position for 500 s (an incident angle of about 7°) with 100 μm vertical and horizontal slit settings. Panels (b) and (c) are images representing radiolysis product concentrations close to the beam footprint after 500 s, as calculated using finite element modeling, based on diffusion from the beam volume into the bulk liquid for short- and long-lived species, respectively, with the latter being the representative of H2O2. | ||
It is clear that a long-lived species, subject to a bulk diffusion, cannot be solely responsible, so this suggests that the corrosion, which is restricted to the beam footprint, is driven by interactions at the surface. There are several candidate propositions that can be explored, for example, it might be due to a photocatalytic effect driven by the high photon flux. As UO2 has a band gap in the region of 2 eV, the high flux of X-rays may result in the production of electron–hole pairs, that can further enhance the formation of reactive oxygen species. To test this theory we measured the rate of corrosion at a range of X-ray energies spanning the uranium L3 absorption edge. Crossing through this edge enhances the number of electrons in the valence band, and thus would increase the oxidant species produced via photocatalysis. However, on passing through the U L3 edge, no statistically significant increase in the corrosion rate was observed, indicating that in this case a photocatalytic process is not responsible. While it is unclear why we observe this surface enhanced corrosion, other possibilities may include a significant concentration of short-lived oxidising species, such as OH radicals. Due to the extremely short-lived nature of such species, diffusion outside of the beam footprint would prove unlikely.
5 Conclusions
In these experiments we have demonstrated that we can induce significant oxidation and further, dissolution of a UO2 surface, by using an intense beam of X-rays, mimicking the radiation fields found at the surface of spent nuclear fuel. Both the X-rays and the water interface are essential ingredients for these changes. We have been able to measure variations in the electron density, surface roughness and rate of dissolution of a radiolysis driven corrosion front in a nuclear fuel material at the Ångström length-scale.There still remain some open questions regarding the precise mechanism for the observed corrosion, which seems unlikely to be due to hydrogen peroxide alone and may include a more complex surface effect. This could have significant consequences for previous research that has predominantly relied on this assumption to simulate the conditions driven by radiation fields in real SNF.6,23
This technique, using thin (<100 Å) epitaxial films of UO2 gives unprecedented (sub-nanometer) surface sensitivity, and in the future, paves the way for a new set of experiments, using synchrotron X-rays in a series of source-probe measurements, as complexities in fuel structure, cladding and groundwater composition can be incorporated.
Acknowledgements
The authors wish to thank Paul Thompson and Laura Glaubes for their assistance in the first experiments on this project at the XMaS beamline BM28, ESRF. Ross Springell would like to thank the Royal Commission for the Exhibition of 1851 for the receipt of a research fellowship. The authors would also like to thank the EPSRC for funding the DISTINCTIVE consortium grant on nuclear waste.References
- A. Corner, D. Venables, A. Spence, W. Poortinga, C. Demski and N. F. Pidgeon, Energy Policy, 2011, 39, 4823–4833 CrossRef PubMed.
- C. Pescatore and A. Vari, J. Risk Res., 2006, 9, 13–40 CrossRef PubMed.
- U. Strandberg and M. Andren, J. Risk Res., 2009, 12, 879–895 CrossRef PubMed.
- D. W. Shoesmith, Nuclear Waste Management Organisation, 2007, Report no. NWMO TR-2007-03 Search PubMed.
- T. E. Eriksen, D. W. Shoesmith and M. Jonsson, J. Nucl. Mater., 2012, 420, 409–423 CrossRef CAS PubMed.
- D. W. Shoesmith, J. Nucl. Mater., 2000, 282, 1–31 CrossRef CAS.
- H. He, M. Broczkowski, K. O'Neil, D. Ofori, O. Semenikhin and D. Shoesmith, Nuclear Waste Management Organisation, 2012, NWMO TR-2012-09 Search PubMed.
- F. Garisto, D. Barber, E. Chen, A. Inglot and C. Morrison, Nuclear Waste Management Organisation, 2009, NWMO TR-2009-27 Search PubMed.
- S. Sunder, G. D. Boyer and N. H. Miller, J. Nucl. Mater., 2003, 322, 163–169 Search PubMed.
- D. W. Shoesmith and S. Sunder, SKB Technical Report, 1991, SKB-TR–91–63 Search PubMed.
- M. I. Ojovan and W. E. Lee, An Introduction to Nuclear Waste Immobilisation, Elsevier, 2005, vol. 1 Search PubMed.
- E. Ekeroth, O. Roth and M. Jonsson, J. Nucl. Mater., 2006, 355, 38–46 CrossRef CAS PubMed.
- C. Corbel, G. Sattonnay, S. Guilbert, F. Garrido, M.-F. Barthe and C. Jegou, J. Nucl. Mater., 2006, 348, 117 CrossRef PubMed.
- J. Spinks and R. Woods, An Introduction to Radiation Chemistry, John Wiley and Sons, Inc., New York, 3rd edn, 1990 Search PubMed.
- S. LeCaër, Water, 2011, 3, 235–253 CrossRef PubMed.
- N. Rauff-Nisthar, C. Boxall, I. Farnan, Z. Hiezl, W. Lee, C. Perkins and R. Wilbraham, Corrosion in Nuclear Energy Systems: From Cradle to Grave, ECS Trans., 2013, 53, 95–104 CrossRef CAS PubMed.
- V. Čuba, V. Múčka and M. Pospíšil, in Radiation Induced Corrosion of Nuclear Fuel, ed. D. S. T. Revankar, 2012, pp. 27–52 Search PubMed.
- F. Miserque, T. Gouder, D. Wegen and P. Bottomley, J. Nucl. Mater., 2001, 298, 280–290 CrossRef CAS.
- A. Seibert, D. Wegen, T. Gouder, J. Ramer, T. Wiss and J. P. Glatz, J. Nucl. Mater., 2011, 419, 112–121 CrossRef CAS PubMed.
- B. Muzeau, C. Jagou, F. Delaunay, V. Broudic, A. Brevet, H. Catalette, E. Simoni and C. Corbel, J. Alloys Compd., 2009, 467, 578–589 CrossRef CAS PubMed.
- M. G. Bailey, L. H. Johnson and D. W. Shoesmith, Corros. Sci., 1985, 25, 233–238 CrossRef CAS.
- S. Sunder, G. D. Boyer and N. H. Miller, J. Nucl. Mater., 1997, 244, 66–74 CrossRef CAS.
- S. Sunder, N. H. Miller and D. W. Shoesmith, Corros. Sci., 2004, 46, 1095–1111 CrossRef CAS PubMed.
- M. M. Strehle, B. J. Heuser, M. S. Elbakhshwan, X. Han, D. J. Gennardo, H. K. Pappas and H. Ju, Thin Solid Films, 2012, 520, 5616–5626 CrossRef CAS PubMed.
- S. D. Brown, L. Bouchenoire, D. Bowyer, J. Kervin, D. Laundy, M. J. Longfield, D. Mannix, D. F. Paul, A. Stunault, P. Thompson, M. J. Cooper, C. A. Lucas and W. G. Stirling, J. Synchrotron Radiat., 2001, 8, 1172–1181 CrossRef CAS PubMed.
- P. F. Fewster, Rep. Prog. Phys., 1996, 59, 1339 CrossRef CAS.
- L. G. Parratt, Phys. Rev., 1954, 95, 359 CrossRef.
- M. Björck and G. Andersson, J. Appl. Crystallogr., 2007, 40, 1174 CrossRef.
Footnote |
| † Alternative address: Department of Physics, University of Liverpool, Liverpool L69 7ZE, UK. |
| This journal is © The Royal Society of Chemistry 2015 |