
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
In-depth structure–selectivity investigations on asymmetric, copper-catalyzed oxidative biaryl coupling in the presence of 5-cis-substituted prolinamines†
Felix
Prause
a,
Benjamin
Arensmeyer
b,
Benjamin
Fröhlich
b and
Matthias
Breuning
*a
aOrganic Chemistry Laboratory, University of Bayreuth, Universitätsstraße 30, 95447 Bayreuth, Germany. E-mail: matthias.breuning@uni-bayreuth.de
bInstitute of Organic Chemistry, University of Würzburg, Am Hubland, 97074 Würzburg, Germany
First published on 19th January 2015
Abstract
Thirteen new and 25 known prolinamines carrying an additional 5-cis substituent were evaluated as the chiral ligands in asymmetric copper-catalyzed, oxidative biaryl coupling of 3-hydroxy-2-naphthoates. Comprehensive structure–selectivity investigations revealed that a phenyl group in the 5-cis position and a small substituent at the pyrrolidine nitrogen (e.g., Me) are essential for high levels of chirality transfer. The sense of the asymmetric induction depends on the steric demand of the exocyclic amino function. In the coupling of methyl 2-hydroxy-3-naphthoate, a primary amino group permitted up to 36% ee in favor of the P-enantiomer, while up to 64% ee in favor of the M-enantiomer was reached with secondary and tertiary amino functions (e.g. NMe2, (S)-NHCH(Me)Ph). A fully linear relationship between the enantiomeric excess of the prolinamine and the binaphthol was observed. A mechanism consistent with all stereochemical findings is proposed, indicating that 3-hydroxy-2-naphthoates with bulkier ester groups should permit better stereocontrol. Indeed, the enantiomeric excess was raised to good 87% when tert-butyl 3-hydroxy-2-naphthoate was used as the substrate.
Introduction
A chiral biaryl axis is the characteristic and dominating feature of a wide variety of bioactive natural products1 and privileged ligands for enantioselective synthesis.2 During the past three decades, many efficient and strategically diverse methods for the stereoselective construction of chiral biaryl bonds have been developed, from the desymmetrization of achiral, but rotationally hindered biaryls and the stereochemical fixation of configurationally labile ones through the atroposelective construction of aromatic rings to diastereo- and enantioselective biaryl coupling reactions.3,4Among the latter approaches, oxidative coupling reactions of 2-naphthols3c in the presence of chirally modified copper catalysts have received particular attention,5–7 since these reactions offer a direct access to the important class of axially chiral 1,1′-binaphthol derivatives.2 A first breakthrough in this field was achieved by Nakajima et al. in 1995.8–10 The oxidative coupling of the naphthol 1a, catalyzed by 10 mol% of a complex generated from CuCl and the prolinamine 3, provided the 1,1′-binaphthyl-2,2′-ol 2a in good 85% yield and 78% ee (Scheme 1). As for most copper–diamine catalysts developed so far, the additional ester group at C2 (or another coordinating electron-withdrawing group allowing bidentate binding) is crucial for a high level of enantioselection.11 Further seminal work was done by Kozlowski et al. introducing the C2-symmetric 1,5-diaza-cis-decalin 4.12,13 The CuI complex of 4 proved to be a highly enantioselective catalyst, providing, for example, the model biaryl 2a in good 85% yield and excellent 93% ee.12 This system was successfully applied in the total synthesis of several axially chiral biaryl natural products.13 Among all other diamines evaluated so far in the enantioselective, copper-catalyzed oxidative coupling of 1a,14 only the CuCl complex of Ha's C1-symmetric BINAM (1,1′-binaphthyl-2,2′-diamine) derivative 5 was able to provide binaphthol 2a in comparable 94% ee.15 The highest level of asymmetric induction (97% ee) was recently reported by Sekar et al., using a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of C2-symmetric BINAM (6) and CuCl in combination with the stable radical additive TEMPO ((2,2,6,6-tetramethylpiperidin-1-yl)oxyl).16
:1 ratio of C2-symmetric BINAM (6) and CuCl in combination with the stable radical additive TEMPO ((2,2,6,6-tetramethylpiperidin-1-yl)oxyl).16
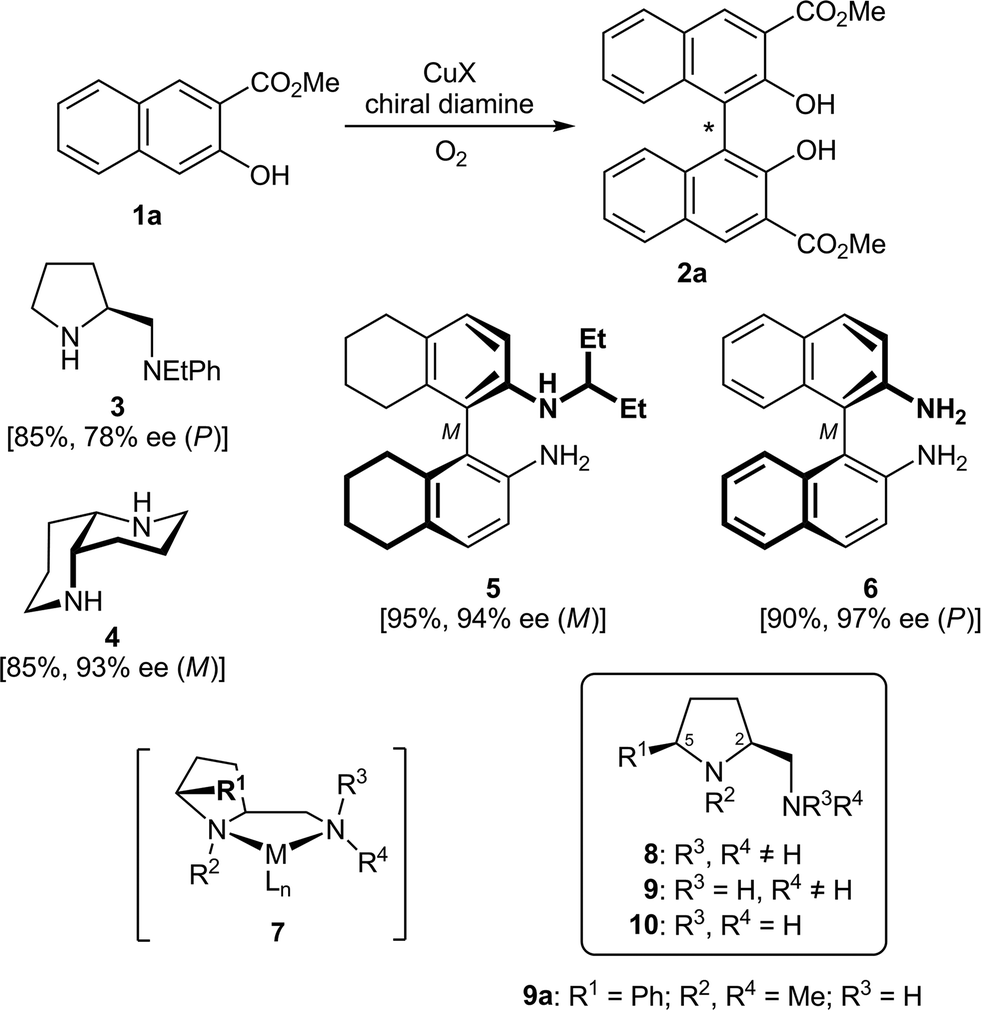 | ||
| Scheme 1 The oxidative biaryl coupling of 1a to 2a, a selection of successfully used chiral diamines (3–6), the metal complex 7 and the new 5-cis-substituted prolinamines 8–10. | ||
In the course of our investigations on conformationally rigid diamines17 we became interested in prolinamines of general types 8–10, which possess, as compared to other proline derived ligands, an additional substituent R1 in the 5-cis position. Upon chelation of a metal (see complex 7), this substituent R1 should shield the upper left face, which might result in enhanced levels of stereocontrol in asymmetric synthesis. This assumption was recently corroborated by copper-catalyzed, enantioselective Henry reactions18 of nitromethane with a series of aromatic, heteroaromatic, vinylic, and aliphatic aldehydes.19 The CuCl2 and CuBr2 complexes of the simple prolinamine 9a (R1 = Ph; R2, R4 = Me; R3 = H) provided the corresponding β-nitro alcohols with 99% ee in all cases (36 examples). This successful application and the structural similarity of 8–10 to Nakajima's diamine 3 prompted us to study the performance of 8–10 in the enantioselective, copper-catalyzed oxidative biaryl coupling of 1 to 2. With the broad variety of derivatives available, in-depth investigations on structure–selectivity relationships should be possible.
Results and discussion
Synthesis of the prolinamines
We recently developed three tailor-made routes to prolinamines of type 8–10 that all start from cheap L-pyroglutamic acid (11), but differ in the order of introduction of the substituent at the 5-cis position (R1), at the pyrrolidine nitrogen atom (R2), and at the exocyclic aminomethyl function (R3, R4).20 The high flexibility and applicability of these approaches was demonstrated in the preparation of more than 25 derivatives with widely varying substitution patterns. Some of these compounds were used in this study. The new prolinamines 8a, b and 9b–l (Table 1) were all synthesized from the amino alcohol 12, which is available from 11 in seven steps and 49% overall yield19,20 and possesses a 5-cis-phenyl substituent and an N-methyl group at the pyrrolidine. Activation of the hydroxy function of 12 by mesylation and subsequent treatment with an excess of the respective amine HNR3R4 afforded the target prolinamines in one pot operations.21 Two bulky tertiary amines (8a, b), ten secondary amines (9b–k) with varying steric demand and, in part, additional stereogenic centers, and one aniline substituent (9l) were thus introduced in acceptable to good 47–78% yield.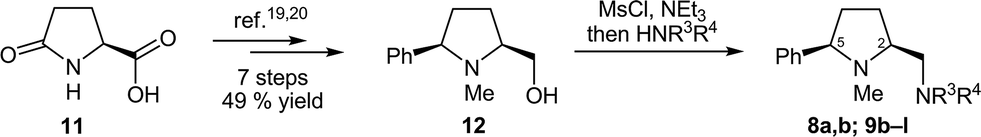
Validation of the enantiomer analysis
Initially, an accurate determination of the enantiomeric excess of the stereochemically enriched binaphthyl 2a by HPLC on chiral phase proved to be difficult.22 Just picking a small sample from the product, which was obtained as a slightly yellowish solid after column chromatography, and dissolving it in the HPLC solvent led to huge derivations in the ee measured. For example, the ee-values of a scalemic sample with 63% ee varied between 27% and 80%, depending on the position the material was taken from. Thus, the solid material of 2a is not stereochemically homogeneous, but a conglomerate of areas with different enantiopurities. Furthermore, the low solubility of 2a in typical HPLC solvents such as hexane, isopropanol, ethanol, or methanol in connection with the high tendency of 2a to form racemic (micro)crystals8b bears the risk of an exaggerated enantiomeric excess in the solution to be measured.We solved these problems by using the following procedure for sample preparation: the complete material of 2a gathered from column chromatography was dissolved in dichloromethane (ca. 1 mL per 10 mg) giving a homogeneous, clear solution. A small aliquot was taken, evaporated, and dissolved in methanol (ca. 50 μg mL−1) under ultra-sonification and warming. The resulting solution was directly injected into HPLC, providing reliably and reproducibly ee-values (Δee ≤ 1%) as checked by several control measurements.
Optimization of the reaction conditions
All copper–diamine complexes were freshly prepared prior to use by stirring a solution of the copper salt and the respective 5-cis-substituted prolinamine 8–10 in acetonitrile–dichloromethane 0.9:1 for 20 min. After evaporation, the residue was dissolved in the reaction solvent, providing a clear green solution.
The reaction conditions were optimized using the simple prolinamine 8c (R1 = Ph, R2–R4 = Me) as the chiral ligand (Table 2). Because of the close structural relationship of 8c with diamine 3,8b we initially chose Nakajima's conditions (entry 1), but added mol sieves 4 Å, which is known12a to be beneficial to the reaction rate and yield. After 72 h at 20 °C, the oxidative coupling of naphthol 1a afforded binaphthol 2a in high 91% yield and acceptable 61% ee in favor of the M-enantiomer. In agreement with the literature,8b the enantiomeric excess of 2a was easily raised by trituration with ethyl acetate, giving highly enriched (M)-2a (96% ee) in the mother liquor.
|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | Solvent | Conc. (M) | 8c (mol%) | CuX (mol%) | Temp. (°C) | Oxidant | Yielda(%) | eeb(%) |
| a Isolated yield. b Determined by HPLC on chiral phase. c After trituration with ethyl acetate. d Suspension. e Without mol sieves 4 Å. f Side products formed. g Reaction time: 18 h. h Reaction time: 7 days. | ||||||||
| 1 | CH2Cl2 | 0.1 | 10 | CuCl (10) | 20 | O2 | 91 | 61 (96)c |
| 2 | (CH2Cl)2 | 0.1 | 10 | CuCl (10) | 20 | O2 | 80 | 61 |
| 3 | CHCl3 | 0.1 | 10 | CuCl (10) | 20 | O2 | 83 | 62 |
| 4 | MeCN | 0.1 | 10 | CuCl (10) | 20 | O2 | 45 | 25 |
| 5 | Et2Od | 0.1 | 10 | CuCl (10) | 20 | O2 | 22 | 28 |
| 6e | CH2Cl2 | 0.1 | 10 | CuCl (10) | 20 | O2 | 67 | 62 |
| 7 | CH2Cl2 | 0.1 | 10 | CuCl (12) | 20 | O2 | 87 | 61 |
| 8 | CH2Cl2 | 0.1 | 10 | CuCl (9) | 20 | O2 | 94 | 62 |
| 9 | CH2Cl2 | 0.1 | 20 | CuCl (18) | 20 | O2 | 94 | 62 |
| 10 | CH2Cl2 | 0.1 | 5 | CuCl (4.5) | 20 | O2 | 59 | 61 |
| 11 | CH2Cl2 | 0.1 | 1 | CuCl (0.9) | 20 | O2 | 23 | 55 |
| 12 | CH2Cl2 | 0.1 | 10 | CuI (9) | 20 | O2 | 43f | 59 |
| 13 | MeCN | 0.1 | 10 | CuI (9) | 20 | O2 | 55f | 19 |
| 14 | CH2Cl2 | 0.1 | 10 | CuCl2·H2O (9) | 20 | O2 | 81 | 63 |
| 15 | (CH2Cl)2 | 0.1 | 10 | CuCl (9) | 40 | O2 | 78g | 53 |
| 16 | CH2Cl2 | 0.1 | 10 | CuCl (9) | 0 | O2 | 65 | 75 |
| 17 | CH2Cl2 | 0.5 | 20 | CuCl (18) | −20 | O2 | 56h | 77 |
| 18 | CH2Cl2 | 0.05 | 10 | CuCl (9) | 20 | O2 | 85 | 63 |
| 19 | CH 2 Cl 2 | 0.5 | 10 | CuCl (9) | 20 | O 2 | 91 | 64 |
| 20 | CH2Cl2 | 0.5 | 10 | CuCl (9) | 20 | Air | 84 | 63 |
| 21 | CH2Cl2 | 0.5 | 10 | CuCl (9) | 20 | tBuOOH | 12 | 55 |
| 22 | CH2Cl2 | 0.5 | 10 | CuCl (9) | 20 | AgCl | 77 | 56 |
| 23 | CH2Cl2 | 0.5 | 10 | CuCl (9) | 20 | DDQ | 0 | — |
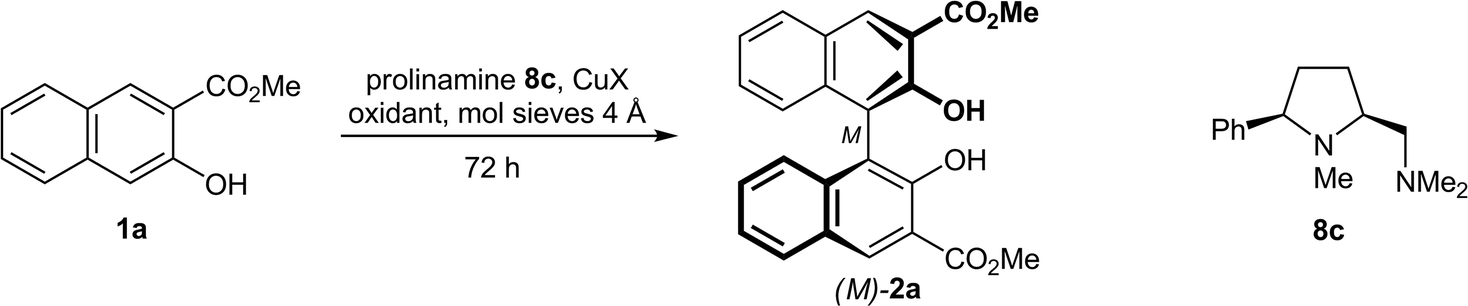
Variation of the reaction parameters showed that chlorinated hydrocarbons and mol sieves 4 Å are essential for high yields and enantioselectivities (entries 1–6). Small changes in the relative stoichiometry CuCl/8c (1.2:1–0.9:1) had no measureable effect on the chirality transfer (entries 7 and 8). Nine mol% of the catalyst was required; lower loadings resulted in significantly reduced yields (entries 9–11). CuI was not suited as the metal salt because of the formation of side products (entries 12 and 13). CuCl2·2H2O (entry 14) and CuCl gave comparable results, as expected from the redox process Cu(I) ⇄ Cu(II) in the catalytic cycle, in which both oxidation states are involved. The enantioselectivity of the oxidative biaryl coupling can be enhanced to 75% ee by lowering the temperature from 20 °C to 0 °C (entry 16), albeit at the price of a reduced yield (65% within 72 h). At −20 °C, a drastic breakdown of the reaction rate was observed (56% yield after 7 days in the presence of 18 mol% catalyst), in combination with just a poor further gain in chirality control (77% ee, entry 17). The dilution had no significant effect on the chirality transfer, although the best results at 20 °C (91% yield, 64% ee) were obtained at higher concentration (c = 0.5 M, entry 19). As the oxidant, air can be used instead of oxygen, but the reaction rate slows somewhat down (entry 20). tBuOOH and AgCl afforded lower yields and diminished enantioselectivities, while no biaryl coupling was observed with DDQ (entries 21–23).
The optimum conditions with respect to reaction rate, yield, and stereoselectivity, which were used in the following diamine screening, are thus as follows: diamine (10 mol%), CuCl (9 mol%), O2 (1 bar), CH2Cl2 (c = 0.5 M), mol sieves 4 Å, 20 °C, 72 h (entry 19).
Structure–selectivity studies
The facile and modular access to various prolinamines of type 8–10 permitted in-depth investigations on the structure–enantioselectivity relationship. All four substituents R1–R4 were separately varied and their influence on the chirality transfer was studied.Prolinamines 8, which are characterized by a tertiary exocyclic amino function (R3, R4 ≠ H), were screened first (Table 3). The bulkiness of the 5-cis substituent R1 was found to exert a profound influence on the chirality transfer (entries 1–8). The enantiomeric excess of 2a rose from low 4% to acceptable 62–64% by increasing the steric demand of R1 from H (8d) to 4-methoxyphenyl (8h) and phenyl (8c). Larger substituents R1 such as 3,5-(bistrifluoromethyl)phenyl in 8i and 1-naphthyl in 8j, however, resulted in a deterioration of the chirality transfer (48% and 25% ee, respectively).
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | 8 | R1 | R2 | R3 | R4 | Yielda(%) | eeb(%) |
| a Isolated yield. b Determined by HPLC on chiral phase. c See Table 2, entry 19. | |||||||
| 1 | d | H | Me | Me | Me | 95 | 4 |
| 2 | e | Me | Me | Me | Me | 95 | 7 |
| 3 | f | Bn | Me | Me | Me | 95 | 22 |
| 4 | g | iPr | Me | Me | Me | 73 | 28 |
| 5 | h | 4-MeOC6H5 | Me | Me | Me | 98 | 62 |
| 6c | c | Ph | Me | Me | Me | 91 | 64 |
| 7 | i | 3,5-(CF3)2C6H3 | Me | Me | Me | 74 | 48 |
| 8 | j | 1-Naphthyl | Me | Me | Me | 59 | 25 |
| 9 | k | Ph | H | Me | Me | 64 | 15 |
| 10 | l | Ph | Et | Me | Me | 81 | 28 |
| 11 | m | Ph | Bn | Me | Me | 93 | 19 |
| 12 | n | Ph | Me | Me | Bn | 74 | 12 |
| 13 | a | Ph | Me | Me | tBu | 46 | 10 |
| 14 | b | Ph | Me | Me | Ph | 20 | 10 |
| 15 | o | Ph | Me | –(CH2)4– | 77 | 63 | |
| 16 | p | Ph | Me | –(CH2)5– | 54 | 42 | |
| 17 | q | Ph | Me | Et | Et | 73 | 6 |

The N-methyl group at the pyrrolidine nitrogen atom is essential since all variations of R2 (8k–m, R2 = H, Et, Bn) led to lower enantioselectivities (15–28% ee, entries 9–11). A similar small substituent tolerance was observed for R3 and R4 at the exocyclic amino function (entries 12–17). Good enantioselectivities (63–64% ee) were only reached with the model diamine 8c (R3, R4 = Me) and the pyrrolidine derivative 8o. Even a slight increase in the steric demand of one or both substituents at the NR3R4 group as, for example, in 8q (NR3R4 = NEt2) and 8p (NR3R4 = piperidinyl) resulted in drastically reduced enantioselectivities (6–42% ee). Finally, it should be noted that the formation of the M-atropoenantiomer of 2a was favored in all coupling reactions in the presence of a diamine 8.
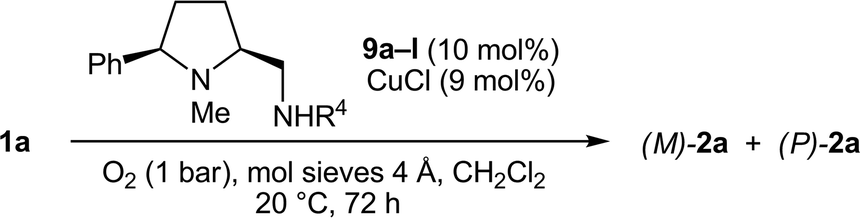
In the screening of the secondary prolinamines 9 (R3 = H), we first kept the optimized substituents R1 = Ph and R2 = Me and varied R4 at the exocyclic amino group (Table 4). To our surprise and in contrast to the results with all tertiary diamines 8 (see Table 3), the P-atropoenantiomer of 2a was preferentially formed (19% ee) in the presence of 9a, which possesses the sterically least hindered secondary aminomethyl function (R4 = Me, entry 1). Even a slight increase in the bulkiness of R4 deteriorated the P-preference and a racemic mixture was obtained with 9b (R4 = Et, entry 2). More demanding α-branched substituents R4 tilted the sense of stereoinduction in favor of the M-enantiomer. A broad maximum plateau in the range of 58–61% ee was reached for R4 = 3-pentyl, (S)-1-phenylethyl, (S)-1-phenylpropyl, and (S)-3,3-dimethylbutan-2-yl (9e–h, entries 5–8). The configuration of the stereocenter in the α-position in 9f–h was also of importance, as seen in the reaction with 9i, which carries, compared to 9f, the enantiomeric (R)-1-phenylethyl side chain, and provided (M)-2a in lower 49% ee (entry 9). A further increase in the steric demand in R4 was not favorable. With bulky α-tertiary substituents such as tBu (9j) and C(CH2OBn)3 (9k), the stereoinduction sharply dropped to 42% and 22% ee, respectively (entries 10 and 11). The reaction rates, which roughly correspond to the isolated yields after 72 h, also decreased with the rising steric demand of R4. The aniline derivative 9l failed to induce a good chirality transfer (38% ee, entry 12).
|
|
|||||
|---|---|---|---|---|---|
| Entry | 9 | R4 | Yielda(%) | eeb(%) | Config. |
| a Isolated yield. b Determined by HPLC on chiral phase. | |||||
| 1 | a | Me | 96 | 19 | P |
| 2 | b | Et | 99 | 0 | — |
| 3 | c | CH2tBu | 60 | 42 | M |
| 4 | d | iPr | 81 | 47 | M |
| 5 | e | 3-Pentyl | 64 | 58 | M |
| 6 | f | (S)-CH(Me)Ph | 72 | 61 | M |
| 7 | g | (S)-CH(Et)Ph | 73 | 61 | M |
| 8 | h | (S)-CH(Me)tBu | 77 | 61 | M |
| 9 | i | (R)-CH(Me)Ph | 68 | 49 | M |
| 10 | j | tBu | 61 | 42 | M |
| 11 | k | C(CH2OBn)3 | 38 | 22 | M |
| 12 | l | Ph | 49 | 38 | M |
Curious by the reversed stereoinduction observed with the prolinamine 9a, we wondered whether the P-preference could be raised by the appropriate choice of the substituents. Since an increase in the size of R2 at the pyrrolidine nitrogen atom had led to a loss of chirality transfer with the M-selective prolinamines 8l and 8m (see Table 3, entries 10 and 11), we anticipated that the opposite effect, an enhanced P-selectivity, should occur with the analogous derivatives of 9a. However, just slightly higher 24% ee (vs. 19% ee for 9a) was found for the N-ethyl diamine 9m, whereas the sense of stereoinduction switched back to M for 9n and 9o carrying the larger N-benzyl and N-isopropyl groups (Table 5, entries 1–3).
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Diamine | R1 | R2 | R4 | Yielda(%) | eeb(%) | Config. |
| a Isolated yield. b Determined by HPLC on chiral phase. c Naph = 1-naphthyl. | |||||||
| 1 | 9m | Ph | Et | Me | 96 | 24 | P |
| 2 | 9n | Ph | Bn | Me | 90 | 3 | M |
| 3 | 9o | Ph | iPr | Me | 74 | 15 | M |
| 4 | 10a | H | Me | H | 90 | 8 | M |
| 5 | 10b | Me | Me | H | 91 | 19 | P |
| 6 | 10c | Bn | Me | H | 89 | 29 | P |
| 7 | 10d | iPr | Me | H | 93 | 33 | P |
| 8 | 10e | Ph | Me | H | 85 | 36 | P |
| 9 | 10f | Naphc | Me | H | 82 | 19 | P |

An increase in P-selectivity might also result if the steric demand at the exocyclic NR3R4 group is minimized, as in the primary diamines 10a–f (NR3R4 = NH2, entries 4–9). Indeed, derivative 10e delivered, compared to corresponding secondary amine 9a, the binaphthol (P)-2a with an improved P-selectivity (36% ee vs. 19% ee for 9a). Increasing the size of R1 as in 10f (R1 = 1-naphthyl) as well as decreasing it as in 10b–10d (R1 = iPr, Bn, Me) resulted in diminished enantioselectivities, while the formation of the M-atropoisomer was slightly favored for the 5-cis-unsubstituted prolinamine 10a (R1 = H, 8% ee).
The structure–enantioselectivity relationships found show that there is a complex interplay between the relative and absolute steric bulk of the substituents R1–R4. Significant findings are: (i) the catalyst system is highly sensitive to steric overcrowding. In particular at the positions R2 and R3, only the small substituents (R2 = Me and R3 = Me, H) are tolerated. (ii) The stereoselection rises with an enhanced steric demand of R1. This accounts for the M-selective prolinamines as well as for the P-selective ones. (iii) In the prolinamine series with R1 = Ph and R2 = Me (8a–c, n–q, 9a–l and 10e), the sense of stereoinduction can be steered by the size and degree of substitution of the exocyclic NR3R4 group. Tertiary diamines of type 8 generally provide the M-enantiomer of 2a, but good levels of enantioselection require small substituents as in 8c and 8o (NR3R4 = NMe2, pyrrolidinyl). Roughly the same chirality transfer is achieved with the secondary diamines 9e–h possessing a sterically more demanding, α-branched alkyl substituent R4. (iv) P-configured 2a is preferentially formed, albeit with lower stereocontrol, if an NHMe group as in 9a and 9m or a primary NH2 group as in 10b–f is present. (v) A hydrogen bridging between the catalyst and the naphthols to be coupled can be excluded for the M-selective prolinamines since the best ligand, 8c, does not possess an acidic proton; for the P-selective ligands 9a, m and 10b–f (NR3R4 = NH2), however, such an additional prefixation might be possible.
Furthermore, the screening revealed that there are significant differences between our prolinamines and the known diamines 38 and 4.12 For example, both latter ligands require at least one secondary amino function for high levels of asymmetric induction, while 8c has just tertiary ones. In addition, the optimum reaction conditions elaborated for diamine 4 (CuI, solvent MeCN) gave only unsatisfying results with our prolinamine 8c (see Table 2, entries 4, 12, and 13).
Mechanistic and stereochemical considerations
Kozlowski et al.12 did extensive studies on the mechanism of enantioselective, oxidative biaryl coupling23 in the presence of their catalyst CuI·4, finding first order dependences on the oxygen and catalyst concentrations.12d The rate determining step of the catalytic cycle is the reoxidation of the catalyst by O2,12d which presumably involves several oxygenated dimeric or oligomeric species.24,25 The stereochemically decisive formation of the biaryl axis is proposed to proceed in two consecutive steps, a face-selective coupling of two naphthyl radicals,26 of which at least one is chelated to a tetrahedral diamine–Cu-complex, followed by a central-to-axial chirality transfer upon rearomatization.12c,27 Since the counter ion has no effect on the stereoselection, it is likely that the reaction takes place at a cationic metal complex.12b Finally, a positive nonlinear effect28 was observed, hinting at dimeric or oligomeric catalyst species in solution.12b This assumption was furthermore corroborated by VPO measurements.12bOur mechanistic studies started with the proof that the 64% ee in the product (M)-2a, as achieved with the catalyst CuCl·8c, is based on a stereodifferentiating coupling step, and not, as observed for coupling with stoichiometric amounts of chirally modified Cu complexes,9 on a non-stereoselective coupling followed by resolution or deracemization of the primarily formed, racemic biaryl. A mere diastereoselective crystallization of CuCl·8c·(M)-2 can safely be excluded because the amount of (M)-2 isolated was by far larger than the amount of the catalyst used. A subsequent deracemization by atropodiastereomerization of configurationally unstable copper complexes, namely CuCl·8c·(M/P)-2 to CuCl·8c·(M)-2, can be ruled out since there was no change in the optical purity if scalemic or racemic 2a were treated with the catalyst for several days.
A fully linear relationship between the enantiomeric excess of the prolinamine 8c and the product 2a was found (Fig. 1). The absence of a nonlinear effect28 makes the existence of dimeric or oligomeric catalyst species as well as a participation of two molecules of the catalyst in the stereochemically decisive biaryl coupling step unlikely (although both possibilities cannot fully be ruled out).
 | ||
| Fig. 1 Fully linear relationship between the enantiomeric excess of the prolinamine 8c and the binaphthol 2a. | ||
Based on the aforementioned mechanistic investigations and our studies we propose the following mechanism for the oxidative biaryl coupling of 1a in the presence of our prolinamine-derived copper catalysts (Scheme 2). For simplification of the discussion, a phenyl group in the 5-cis position (R1 = Ph) and an N-methyl group at the pyrrolidine nitrogen atom (R2 = Me), which are both essential for good levels of stereoselection, and a secondary or primary exocyclic amino function are set. The ligands 9e–h (NR3R4 = NHR4; R4 = secondary alkyl, 58–61% ee in favor of M) and 10e (NR3R4 = NH2, 36% ee in favor of P) fulfill these premises. Taking the relative and absolute steric demands of the substituents R1–R4 into account, this mechanism can be extended to all prolinamines that provided significant levels of asymmetric induction.
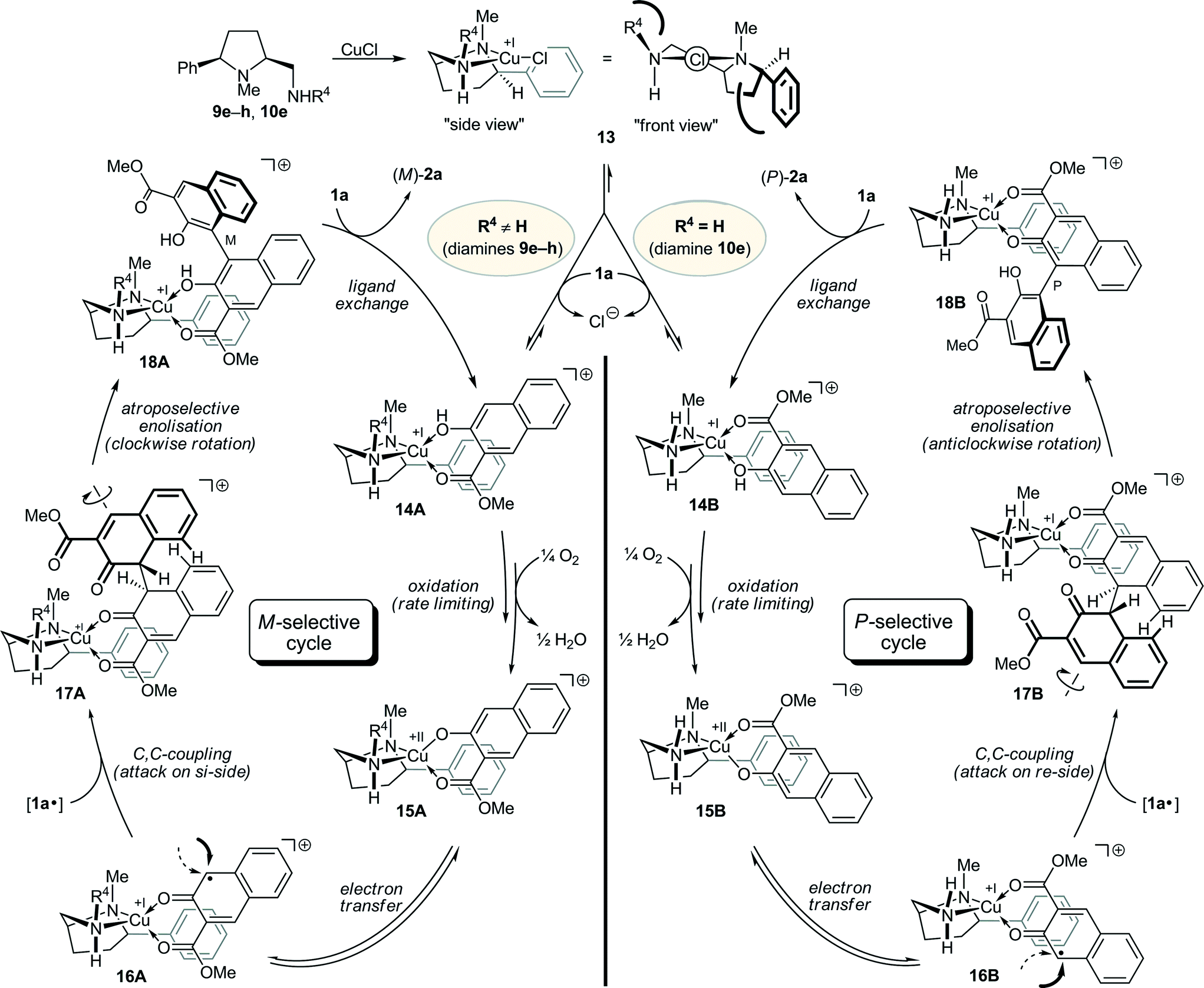 | ||
| Scheme 2 Proposed mechanism for the M- and P-selective oxidative biaryl coupling of 1a in the presence of the prolinamines 9e–h and 10e. | ||
Initial chelation of the chiral prolinamine to CuCl provides the bicyclic, C1-symmetric complex 13, to which the naphthol 1a principally can bind in two different orientations, as illustrated in the tetrahedral29 complexes 14A and 14B.30 The preference for one or the other is controlled by steric factors. On the side of the naphthol 1a, the methoxy group of the ester function is more demanding than the two carbon atoms C-4 and C-4a of the aryl ring. On the side of the chiral catalyst, it is reasonable that, in the energetically most favored conformation, the larger substituents at the two nitrogen atoms (R4 and the annelated pyrrolidine) occupy opposite positions with respect to the central copper heterocycle and align pseudo-equatorially, as the phenyl group does. As a consequence of this arrangement depicted in 13, the lower right quadrant is shielded by the phenyl group and the upper left one by R4 (see “front view”). In the case of 10e with R4 = H, only the repulsion by the phenyl group exists, thus favoring the formation of 14B, while in the cases of 9e–h (R4 = secondary alkyl), the higher steric demand of R4 dominates, thus favoring the orientation shown in 14A.
The following steps are identical for both catalytic cycles. Multistage24 and rate-limiting12d,25 oxidation of the copper(I) atom in 14 by O2 affords the copper(II) complexes 15, which can undergo electron transfer from the naphthol to the copper atom to give the naphthyl radicals 16. In both complexes, the backside of the naphthyl radical is efficiently shielded by the phenyl group, possibly supported by some π-stacking, which directs the attack of a second naphthyl radical [1a˙] to the front side, thus leading to 17. It should be noted that the true nature of [1a˙] is still unclear,26 although the observed absence of a nonlinear effect (see Fig. 1) makes the complexation of [1a˙] to a second, chirally modified copper atom and, thus, a coupling between two molecules of 16, unlikely.
During the rearomatization process via twofold ketoenol tautomerism, the two (pre)aromatic moieties have to rotate in order to reach the orthogonal alignment in biaryls. This rotation follows the pathway of the least steric hindrance, which means that the carbonyl groups pass each other and not the aromatic rings (repulsion of the peri-H).12c,27 Consequently, the rotation is clockwise in 17A, leading to M-configuration at the newly created biaryl axis in 18A, whereas an anticlockwise rotation takes place in 17B, creating the P-configured biaryl axis in 18B. Final exchange of the binaphthol 2a against naphthol 1a completes the catalytic cycle. For the orientation of 1a upon complexation, the very same steric arguments apply as in the chelation of 1a to 13 giving 14A and 14B.
Since most of the prolinamines 8–10 used in this study favored the formation of (M)-2a, the top faces of the respective copper complexes must be more strongly shielded than the bottom faces, which forces the incoming naphthol 1a to bind in a fashion shown in 14A. This also means that the steric demand of the annelated, R1-substituted pyrrolidine cannot be high, probably due to its pseudo-equatorial orientation with respect to the central copper heterocycle. The size of R1 at this ring, however, exerts a drastic effect on the level of stereoselection. Since the same trends – decreasing the bulkiness of R1 led to a reduced chirality transfer (see 8d–j, Table 3, and 10a–f, Table 5) – were observed for the M- and the P-directing prolinamines, this substituent cannot play an important role in the binding of 1a, but must be decisive in the shielding of the backside of the complexed naphthyl radical in 16A and 16B (see Scheme 2), which is in good agreement with the mechanism proposed.
Consequences of the mechanism
Under the assumption that the backside-shielding in 16 and the central-to-axial chirality transfer occur with high selectivity, the resulting enantiomeric excess in the binaphthol 2a is thus determined by the orientation of 1a during binding to 18 (and 13 in the starting sequence). As a consequence, the enantioselection of the M-selective catalysts should increase if the steric differentiation in the naphthol substrate is more pronounced, which can be achieved by raising the steric bulk of the ester group at C-2. In order to consolidate this theory, we synthesized the naphthol esters 1b–d and subjected these compounds to the coupling procedures (Table 6). Indeed, the levels of stereoselection significantly increased by using the sterically more hindered esters. The binaphthol 2d (R = tBu) was produced in good 78% and 75% ee with the prolinamines 8c and 9f as the chiral ligands (entries 4 and 8). This trend is in sharp contrast to the observations made in other oxidative biaryl coupling reactions in the presence of diamine–copper catalysts, in which the chirality transfer dropped when the size of the ester group was increased.8b,12c,14a,16 By lowering the reaction temperature to 0 °C, the enantiomeric excess in the CuCl·8c catalyzed coupling of 1d to 2d was further improved to 87%, without any noticeable loss in yield (96%, entry 9). The latter result is the best asymmetric induction so far reached with the naphthyl ester 1d.|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | Biaryl 1, 2 | R | Diamine | Temp. (°C) | t (day) | Yielda(%) | eeb(%) | Config. |
| a Isolated yield. b Determined by HPLC on chiral phase. c See Table 2, entry 19. d See Table 4, entry 6. e See Table 5, entry 8. | ||||||||
| 1c | 1a | Me | 8c | 20 | 3 | 91 | 64 | M |
| 2 | 1b | iPr | 8c | 20 | 3 | 94 | 69 | M |
| 3 | 1c | Bn | 8c | 20 | 3 | 93 | 73 | M |
| 4 | 1d | tBu | 8c | 20 | 6 | 99 | 78 | M |
| 5d | 2a | Me | 9f | 20 | 3 | 72 | 61 | M |
| 6 | 2b | iPr | 9f | 20 | 5 | 94 | 75 | M |
| 7 | 2c | Bn | 9f | 20 | 5 | 99 | 72 | M |
| 8 | 2d | tBu | 9f | 20 | 7 | 94 | 75 | M |
| 9 | 1d | tBu | 8c | 0 | 8 | 96 | 87 | M |
| 10e | 1a | Me | 10e | 20 | 3 | 85 | 36 | P |
| 11 | 1d | tBu | 10e | 20 | 5 | 99 | 36 | P |
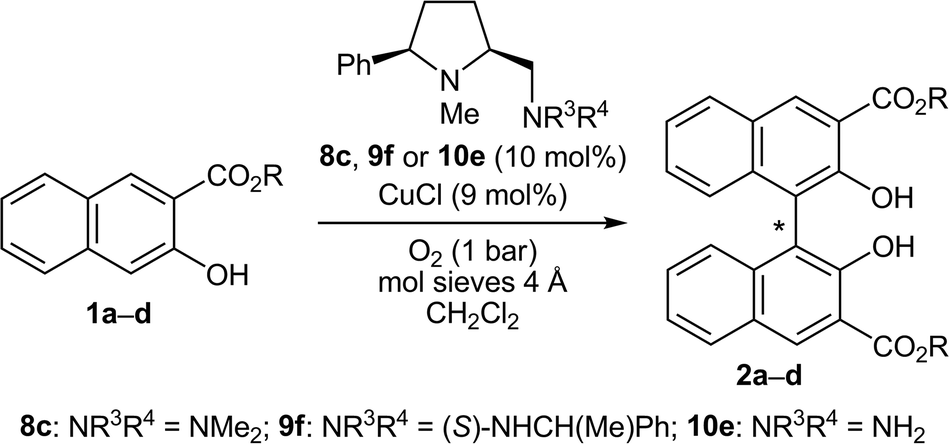
For the P-selective catalyst CuCl·10e, the enantioselection achieved with the methyl ester 1a and the t-butyl ester 1d was virtually identical (entries 10 and 11). This result is also in good agreement with the proposed catalytic cycle, since the interaction between the ester group and the chiral backbone is just weak (see 14B, Scheme 2).
Conclusions
A series of 38 prolinamines 8–10, which differ in the substituents R1 at the 5-cis position, R2 at the pyrrolidine nitrogen atom, and R3R4 at the exocyclic amino function, were evaluated in their performance as chiral ligands in the copper-catalyzed oxidative biaryl coupling of the naphthol 1a. Essential for good enantioselectivities were a 5-cis-phenyl (R1) and an N-methyl group (R2). With these two substituents given, the level and sense of the asymmetric induction can be steered by the NR3R4 group. Good 58–64% ee in favor of the M-enantiomer of 2a were reached with the tertiary amine 8c (NR3R4 = NMe2) and the secondary amines 9e–h (NR3R4 = NHR4, with R4 = secondary alkyl), while the P-enantiomer of 2a was preferentially formed with the primary amine 10e (36% ee, NR3R4 = NH2). A mechanism, in which the steric demand of the NR3R4 group of the chiral ligand determines the orientation of 1a upon complexation to the copper atom and, thus, the sense of the chirality transfer, was proposed. The 5-cis-phenyl group (R1) plays the decisive role in the face-selective C,C-coupling step by shielding one side of the complexed naphthyl radical. As a consequence of the structure–enantioselectivity investigations, we concluded that naphthols with bulkier ester groups should permit better stereocontrol. Indeed, the enantiomeric excess of the oxidative coupling of 1d (CO2R = CO2tBu) was improved to up to 87% ee by using CuCl·8c as the chiral catalyst.Experimental
All reactions with moisture-sensitive reagents were carried out under an argon atmosphere in anhydrous solvents, prepared using standard procedures.31 Commercially available reagents (highest quality available) were used as received. Reactions were monitored by thin layer chromatography on precoated silica gel (Macherey-Nagel, Alugram SIL G/UV254). Spots were visualized by UV light (254 nm) or by staining with aqueous KMnO4, vanillin, or ceric ammonium molybdate. Silica gel (Macherey-Nagel, particle size 40–63 μm) was used for column chromatography. Melting points were measured by a Stuart SMP10 digital or a Thermo Scientific 9300 melting point apparatus and are uncorrected. Optical rotations were recorded on a Jasco P-1020 polarimeter (10 cm cell). NMR spectra were taken on a Bruker Avance 300, a Bruker Avance 400, or a Bruker Avance III HD 500 instrument and calibrated using the residual undeuterated solvent as an internal reference. The peak assignments in the 1H and 13C NMR data were performed on the basis of 2D NMR methods (COSY, HSQC, HMBC). Infrared spectra were recorded on a Jasco FT-IR-410 or a PerkinElmer Spectrum 100 FT-IR spectrometer, high resolution mass spectra on a Bruker Daltonics micrOTOF focus mass spectrometer using ESI (electronspray ionization). The enantiomeric excess of the binaphthols 2a–d was determined by HPLC analysis (Waters Alliance HPLC; Waters 2695 Separation Module, Waters 2487 Dual λ Absorbance Detector) on chiral phase (Daicel Chiralpak AD-H).The synthesis of 9f and the general procedure for the oxidative biaryl coupling under optimized conditions are described here exemplary. For the preparation of all other new compounds, see the ESI.†
(2R,5S)-1-Methyl-2-phenyl-5-((((S)-1-phenylethyl)amino)methyl)pyrrolidine (9f)
NEt3 (197 μL, 143 mg, 1.41 mmol) and MsCl (87.4 μL, 129 mg, 1.13 mmol) were added at 0 °C to a solution of the alcohol 1219 (180 mg, 941 μmol) in anhydrous CH2Cl2 (8 mL). After 1 day at r.t., the solution was treated with (S)-1-phenylethylamine (2.40 mL, 2.28 g, 18.8 mmol) and stirring was continued for 5 days. The solvent was removed under reduced pressure and the crude material was directly subjected to column chromatography (1. silica gel, CH2Cl2/MeOH, 100:0–97:3, 2. silica gel, EtOAc). Filtration through a pad of basic alumina (activity I, CH2Cl2/MeOH, 9:1) delivered the prolinamine 9f (205 mg, 697 μmol, 74%) as a yellowish oil.
Rf 0.65 (EtOAc). [α]D21 8.2 (c 0.50 in MeOH). IR (ATR) νmax/cm−1 2968w, 2784w, 1491w, 1450s, 1122w, 1041w, 1027w, 757s, 697vs; 1H NMR δH (500 MHz; CDCl3) 1.43 (3 H, d, J = 6.7 Hz, CHCH3), 1.67 (1 H, m, 3-HH), 1.83 (1 H, m, 4-HH), 1.98 (1 H, m, 4-HH), 2.05 (1 H, m, 3-HH), 2.06 (3 H, s, 1-CH3), 2.52 (1 H, dd, J = 11.1, 6.4 Hz, 5-CHH), 2.58 (1 H, m, 5-H), 2.75 (1 H, dd, J = 11.1, 3.1 Hz, 5-CHH), 3.27 (1 H, dd, J = 9.7, 6.7 Hz, 2-H), 3.82 (1 H, q, J = 6.7 Hz, CHCH3), 7.26 (2 H, m, Ar–H), 7.33 (8 H, m, Ar–H) ppm. 13C NMR δC (125 MHz, CDCl3) 24.4 (CHCH3), 28.1 (C-4), 34.3 (C-3), 39.3 (1-CH3), 50.7 (5-CH2), 58.8 (CHCH3), 65.9 (C-5), 72.6 (C-2), 126.7, 127.0, 127.1, 127.4, 128.4, 128.6 (CH–Ar), 143.9, 145.9 (Cq-Ar) ppm. HRMS (ESI, pos.) m/z calcd for C20H27N2 [M + H]+ 295.2169, found 295.2169.
General procedure for the oxidative biaryl coupling under optimized conditions
:1–2:1; for 2b–d: silica gel, petroleum ether/CH2Cl2, 3:1–1:9), delivering the product 2a–d8b as a yellowish solid.
:2, 1.0 mL min−1, 254 nm: tR (P-enantiomer) = 8.8 min; tR (M-enantiomer) = 14.3 min;8b2b: n-hexane/iPrOH 98:02, 1.0 mL min−1, 254 nm: tR (P-enantiomer) = 7.9 min; tR (M-enantiomer) = 9.5 min; the absolute configuration of 2b was determined after transesterification of 2b into 2a; 2c: n-hexane/iPrOH 9:1, 1.0 mL min−1, 254 nm: tR (P-enantiomer) = 14.6 min; tR (M-enantiomer) = 23.1 min;8b2d: n-hexane/iPrOH 98:02, 1.0 mL min−1, 254 nm: tR (M-enantiomer) = 6.8 min; tR (P-enantiomer) = 7.6 min.8b
Acknowledgements
The financial support of the German Research Foundation (DFG) is gratefully acknowledged.Notes and references
- Reviews: (a) U. Weiss, L. Merlini and G. Nasini, Prog. Chem. Org. Nat. Prod., 1987, 52, 1 CAS; (b) G. Bringmann, C. Günther, M. Ochse, O. Schupp and S. Tasler, Prog. Chem. Org. Nat. Prod., 2001, 82, 1 CAS; (c) O. Baudoin and F. Guéritte, Stud. Nat. Prod. Chem., 2003, 29(Part J), 355 CrossRef CAS; (d) H. Aldemir, R. Richarz and T. A. M. Gulder, Angew. Chem., Int. Ed., 2014, 53, 8286 CrossRef CAS PubMed.
- Reviews: (a) L. Pu, Chem. Rev., 1998, 98, 2405 CrossRef CAS PubMed; (b) R. Noyori, Asymmetric Catalysis in Organic Synthesis, Wiley, New York, 1994 Search PubMed; (c) C. Rosini, L. Franzini, A. Raffaelli and P. Salvadori, Synthesis, 1992, 503 CrossRef CAS; (d) T. Hayashi, Acc. Chem. Res., 2000, 33, 354 CrossRef CAS PubMed; (e) J. M. Brunel, Chem. Rev., 2005, 105, 857 CrossRef CAS PubMed; (f) M. Berthod, G. Mignani, G. Woodward and M. Lemaire, Chem. Rev., 2005, 105, 1801 CrossRef CAS PubMed.
- For reviews about the atroposelective synthesis of axially chiral biaryls (methodology and application), see: (a) G. Bringmann, A. J. Price Mortimer, P. A. Keller, M. J. Gresser, J. Garner and M. Breuning, Angew. Chem., Int. Ed., 2005, 44, 5384 CrossRef CAS PubMed; (b) O. Baudoin, Eur. J. Org. Chem., 2005, 4223 CrossRef CAS; (c) M. C. Kozlowski, B. J. Morgan and E. C. Linton, Chem. Soc. Rev., 2009, 38, 3193 RSC; (d) G. Bringmann, T. Gulder, T. A. M. Gulder and M. Breuning, Chem. Rev., 2011, 111, 563 CrossRef CAS PubMed; (e) S. E. Allen, R. R. Walvoord, R. Padilla-Salinas and M. C. Kozlowski, Chem. Rev., 2013, 113, 6234 CrossRef CAS PubMed.
- For selected recent developments in the atroposelective synthesis of biaryls, see: (a) X. Shen, G. O. Jones, D. A. Watson, B. Bhayana and S. L. Buchwald, J. Am. Chem. Soc., 2010, 132, 11278 CrossRef CAS PubMed; (b) J. L. Gustafson, D. Lim and S. J. Miller, Science, 2010, 328, 1251 CrossRef CAS PubMed; (c) F. Guo, L. C. Konkol and R. J. Thomson, J. Am. Chem. Soc., 2011, 133, 18 CrossRef CAS PubMed; (d) N. Asakura, S. Fujimoto, N. Michihata, K. Nishii, H. Imagawa and H. Yamada, J. Org. Chem., 2011, 76, 9711 CrossRef CAS PubMed; (e) P. Wessig, C. Pick and U. Schilde, Tetrahedron Lett., 2011, 52, 4221 CrossRef CAS; (f) S. Ogaki, Y. Shibata, K. Noguchi and K. Tanaka, J. Org. Chem., 2011, 76, 1926 CrossRef CAS PubMed; (g) M. Leibeling and D. B. Werz, Chem. – Eur. J., 2012, 18, 6138 CrossRef CAS PubMed; (h) A. Ros, B. Estepa, A. Bermejo, E. Álvarez, R. Fernández and J. M. Lassaletta, J. Org. Chem., 2012, 77, 4740 CrossRef CAS PubMed; (i) Y. Kasai, N. Michihata, H. Nishimura, T. Hirokane and H. Yamada, Angew. Chem., Int. Ed., 2012, 51, 8026 CrossRef CAS PubMed; (j) Z.-H. Shan, J. Liu, L.-M. Xu, Y.-F. Tang, J.-H. Chen and Z. Yang, Org. Lett., 2012, 14, 3712 CrossRef CAS PubMed; (k) M. Groh, D. Meidlinger, G. Bringmann and A. Speicher, Org. Lett., 2012, 14, 4548 CrossRef CAS PubMed; (l) F. R. Leroux, A. Berthelot, L. Bonnafoux, A. Panossian and F. Colobert, Chem. – Eur. J., 2012, 18, 14232 CrossRef CAS PubMed; (m) S. Postikova, M. Sabbah, D. Wightman, I. T. Nguyen, M. Sanselme, T. Besson, J.-F. Brière, S. Oudeyer and V. Levacher, J. Org. Chem., 2013, 78, 8191 CrossRef CAS PubMed; (n) G.-Q. Li, H. Gao, C. Keene, M. Devonas, D. H. Ess and L. Kürti, J. Am. Chem. Soc., 2013, 135, 7414 CrossRef CAS PubMed; (o) N. Kadoya, M. Murai, M. Ishiguro, J. Uenishi and M. Uemura, Tetrahedron Lett., 2013, 54, 512 CrossRef CAS; (p) A. Link and C. Sparr, Angew. Chem., Int. Ed., 2014, 53, 5458 CrossRef CAS PubMed; (q) G. Xu, W. Fu, G. Liu, C. H. Senanayake and W. Tang, J. Am. Chem. Soc., 2014, 136, 570 CrossRef CAS PubMed; (r) Y. Zhou, X. Zhang, H. Liang, Z. Cao, X. Zhao, Y. He, S. Wang, J. Pang, Z. Zhou, Z. Ke and L. Qiu, ACS Catal., 2014, 4, 1390 CrossRef CAS; (s) C. K. Hazra, Q. Dherbassy, J. Wencel-Delord and F. Colobert, Angew. Chem., Int. Ed., 2014, 53, 13871 CrossRef CAS PubMed.
- For selected examples of oxidative biaryl couplings catalyzed by chiral vanadium complexes, see: (a) S.-W. Hon, C.-H. Li, J.-H. Kuo, N. B. Barhate, Y.-H. Liu, Y. Wang and C.-T. Chen, Org. Lett., 2001, 3, 869 CrossRef CAS PubMed; (b) C.-Y. Chu, D.-R. Hwang, S.-K. Wang and B.-J. Uang, Chem. Commun., 2001, 980 RSC; (c) Z. Luo, Q. Liu, L. Gong, X. Cui, A. Mi and Y. Jiang, Chem. Commun., 2002, 914 RSC; (d) N. B. Barhate and C.-T. Chen, Org. Lett., 2002, 4, 2529 CrossRef CAS PubMed; (e) C.-Y. Chu and B.-J. Uang, Tetrahedron: Asymmetry, 2003, 14, 53 CrossRef CAS; (f) H. Somei, Y. Asano, T. Yoshida, S. Takizawa, H. Yamataka and H. Sasai, Tetrahedron Lett., 2004, 45, 1841 CrossRef CAS; (g) M. Tada, T. Taniike, L. M. Kantam and Y. Iwasawa, Chem. Commun., 2004, 2542 RSC; (h) M. Tada, N. Kojima, Y. Izumi, T. Taniike and Y. Iwasawa, J. Phys. Chem. B, 2005, 109, 9905 CrossRef CAS PubMed; (i) Q.-X. Guo, Z.-J. Wu, Z.-B. Luo, Q.-Z. Liu, J.-L. Ye, S.-W. Luo, L.-F. Cun and L.-Z. Gong, J. Am. Chem. Soc., 2007, 129, 13927 CrossRef CAS PubMed; (j) S. Takizawa, T. Katayama, H. Somei, Y. Asano, T. Yoshida, C. Kameyama, D. Rajesh, K. Onitsuka, T. Suzuki, M. Mikami, H. Yamataka, D. Jayaprakash and H. Sasai, Tetrahedron, 2008, 64, 3361 CrossRef CAS; (k) S. Takizawa, J. Kodera, Y. Yoshida, M. Sako, S. Breukers, D. Enders and H. Sasai, Tetrahedron, 2014, 70, 1786 CrossRef CAS.
- For oxidative biaryl couplings catalyzed by other chiral transition metal complexes, see: Ru: (a) T. Hamada, H. Ishida, S. Usui, Y. Watanabe, K. Tsumura and K. Ohkubo, J. Chem. Soc., Chem. Commun., 1993, 909 RSC; (b) R. Irie, K. Masutani and T. Katsuki, Synlett, 2000, 1433 CAS ; Fe; (c) H. Egami and T. Katsuki, J. Am. Chem. Soc., 2009, 131, 6082 CrossRef CAS PubMed; (d) H. Egami, K. Matsumoto, T. Oguma, T. Kunisu and T. Katsuki, J. Am. Chem. Soc., 2010, 132, 13633 CrossRef CAS PubMed.
- For other types of enantioselective oxidative biaryl couplings, see: Electrochemically: (a) T. Osa, Y. Kashiwagi, Y. Yanagisawa and J. M. Bobbitt, J. Chem. Soc., Chem. Commun., 1994, 2535 RSC ; enzymatic; (b) M. Takemoto, Y. Suzuki and K. Tanaka, Tetrahedron Lett., 2002, 43, 8499 CrossRef CAS; (c) F. Trotta, G. Cravotto and S. Rossignoli, J. Inclusion Phenom. Macrocyclic Chem., 2002, 44, 293 CrossRef CAS; (d) F. Trotta, G. Cravotto and G. Casile, Environ. Sci. Pollut. Res., 2003, 10, 144 CrossRef CAS.
- (a) M. Nakajima, K. Kanayama, I. Miyoshi and S.-I. Hashimoto, Tetrahedron Lett., 1995, 36, 9519 CrossRef CAS; (b) M. Nakajima, I. Miyoshi, K. Kanayama, S.-I. Hashimoto, M. Noji and K. Koga, J. Org. Chem., 1999, 64, 2264 CrossRef CAS.
- The atroposelectivities observed in earlier biaryl couplings of 2-naphthols mediated by an (over)stoichiometric amount of a copper–diamine complex are not the result of an asymmetric coupling step, but of a succeeding dynamic kinetic resolution or a diastereoselective crystallization of the initially formed biaryl–copper–diamine complexes: (a) J. Brussee and A. C. A. Jansen, Tetrahedron Lett., 1983, 24, 3261 CrossRef CAS; (b) J. Brussee, J. L. G. Groenendijk, J. M. te Koppele and A. C. A. Jansen, Tetrahedron, 1985, 41, 3313 CrossRef CAS; (c) M. Smrčina, M. Lorenc, V. Hanuš, P. Sedmera and P. Kočovský, J. Org. Chem., 1992, 57, 1917 CrossRef; (d) M. Smrčina, J. Poláková, Š. Vyskočil and P. Kočovský, J. Org. Chem., 1993, 58, 4534 CrossRef.
- The first truly9 asymmetric oxidative biaryl coupling was reported in 1993 by Smrčina and Kočovský et al.9d In the presence of 10 mol% of a sparteine-CuCl2 complex and with AgCl as the stoichiometric oxidant, the product biaryl was obtained in low 32% ee.
- For a copper-catalyzed, enantioselective oxidative biaryl coupling of 1-naphthol, see: J. Gao, J. H. Reibenspies and A. E. Martell, Angew. Chem., Int. Ed., 2003, 42, 6008 CrossRef CAS PubMed.
- (a) X. Li, J. Yang and M. C. Kozlowski, Org. Lett., 2001, 3, 1137 CrossRef CAS PubMed; (b) M. C. Kozlowski, X. Li, P. J. Carroll and Z. Xu, Organometallics, 2002, 21, 4513 CrossRef CAS; (c) X. Li, J. B. Hewgley, C. A. Mulrooney, J. Yang and M. C. Kozlowski, J. Org. Chem., 2003, 68, 5500 CrossRef CAS PubMed; (d) J. B. Hewgley, S. S. Stahl and M. C. Kozlowski, J. Am. Chem. Soc., 2008, 130, 12232 CrossRef CAS PubMed.
- Applications of diamine 4 in the atroposelective synthesis of biaryl natural products: (a) C. A. Mulrooney, X. Li, E. S. DiVirgilio and M. C. Kozlowski, J. Am. Chem. Soc., 2003, 125, 6856 CrossRef CAS PubMed; (b) M. C. Kozlowski, E. C. Dugan, E. S. DiVirgilio, K. Maksimenka and G. Bringmann, Adv. Synth. Catal., 2007, 349, 583 CrossRef CAS; (c) E. S. DiVirgilio, E. C. Dugan, C. A. Mulrooney and M. C. Kozlowski, Org. Lett., 2007, 9, 385 CrossRef CAS PubMed; (d) E. M. O'Brien, B. J. Morgan and M. C. Kozlowski, Angew. Chem., Int. Ed., 2008, 47, 6877 CrossRef PubMed; (e) B. J. Morgan, S. Dey, S. W. Johnson and M. C. Kozlowski, J. Am. Chem. Soc., 2009, 131, 9413 CrossRef CAS PubMed; (f) C. A. Mulrooney, B. J. Morgan, X. Li and M. C. Kozlowski, J. Org. Chem., 2010, 75, 16 CrossRef CAS PubMed; (g) B. J. Morgan, C. A. Mulrooney, E. M. O'Brien and M. C. Kozlowski, J. Org. Chem., 2010, 75, 30 CrossRef CAS PubMed; (h) B. J. Morgan, C. A. Mulrooney and M. C. Kozlowski, J. Org. Chem., 2010, 75, 44 CrossRef CAS PubMed; (i) E. M. O'Brien, B. J. Morgan, C. A. Mulrooney, P. J. Carroll and M. C. Kozlowski, J. Org. Chem., 2010, 75, 57 CrossRef PubMed; (j) E. E. Podlesny and M. C. Kozlowski, Org. Lett., 2012, 14, 1408 CrossRef CAS PubMed; (k) E. E. Podlesny and M. C. Kozlowski, J. Org. Chem., 2013, 78, 466 CrossRef CAS PubMed.
- For the oxidative biaryl coupling of 1 catalyzed by other chiral copper–diamine complexes, see: (a) A. Caselli, G. B. Giovenzana, G. Palmisano, M. Sisti and T. Pilati, Tetrahedron: Asymmetry, 2003, 14, 1451 CrossRef CAS; (b) J. Illesinghe, R. Ebeling, B. Ferguson, J. Patel, E. M. Campi, W. R. Jackson and A. J. Robinson, Aust. J. Chem., 2004, 57, 167 CrossRef CAS; (c) S. Arai, S. Takita and A. Nishida, Eur. J. Org. Chem., 2005, 5262 CrossRef CAS; (d) L. Chen, J.-b. Lan and R.-g. Xie, Hecheng Huaxue, 2006, 14, 581 CAS; (e) P. Yan, Y. Sugiyama, Y. Takahashi, H. Kinemuchi, T. Temma and S. Habaue, Tetrahedron, 2008, 64, 4325 CrossRef CAS; (f) Y. Yusa, I. Kaito, K. Akiyama and K. Mikami, Chirality, 2010, 22, 224 CAS; (g) S. Sabarinathan, G. Vasuki and P. S. Rao, Eur. J. Chem., 2010, 1, 360 CrossRef CAS; (h) G. Grach, G. Pieters, A. Dinut, V. Terrasson, R. Medimagh, A. Bridoux, V. Razafimahaleo, A. Gaucher, S. Marque, J. Marrot, D. Prim, R. Gil, J. G. Planas, C. Viñas, I. Thomas, J.-P. Roblin and Y. Troin, Organometallics, 2011, 30, 4074 CrossRef CAS.
- K. H. Kim, D.-W. Lee, Y.-S. Lee, D.-H. Ko and D.-C. Ha, Tetrahedron, 2004, 60, 9037 CrossRef CAS.
- S. K. Alamsetti, E. Poonguzhali, D. Ganapathy and G. Sekar, Adv. Synth. Catal., 2013, 355, 2803 CrossRef CAS.
- (a) M. Breuning, D. Hein, M. Steiner, V. H. Gessner and C. Strohmann, Chem. – Eur. J., 2009, 15, 12764 CrossRef CAS PubMed; (b) M. Breuning, M. Steiner, C. Mehler, A. Paasche and D. Hein, J. Org. Chem., 2009, 74, 1407 CrossRef CAS PubMed; (c) M. Breuning, T. Häuser, C. Mehler, C. Däschlein, C. Strohmann, A. Oechsner and H. Braunschweig, Beilstein J. Org. Chem., 2009, 5(81) CAS; (d) M. Breuning, A. Paasche, M. Steiner, S. Dilsky, V. H. Gessner, C. Strohmann and B. Engels, J. Mol. Struct., 2011, 1005, 178 CrossRef CAS; (e) M. Breuning and D. Hein, Eur. J. Org. Chem., 2013, 7575 CrossRef CAS.
- Reviews: (a) J. Boruwa, N. Gogoi, P. P. Saikia and N. C. Barua, Tetrahedron: Asymmetry, 2006, 17, 3315 CrossRef CAS; (b) C. Palomo, M. Oiarbide and A. Laso, Eur. J. Org. Chem., 2007, 2561 CrossRef CAS; (c) G. Blay, V. Hernández-Olmos and J. R. Pedro, Synlett, 2011, 1195 CrossRef CAS; (d) G. Chelucci, Coord. Chem. Rev., 2013, 257, 1887 CrossRef CAS; (e) N. Ananthi and S. Velmathi, Indian J. Chem., Sect. B: Org. Chem. Incl. Med. Chem., 2013, 52, 87 Search PubMed.
- D. Scharnagel, F. Prause, J. Kaldun, R. G. Haase and M. Breuning, Chem. Commun., 2014, 50, 6623 RSC.
- F. Prause, J. Kaldun, B. Arensmeyer, B. Wennemann, B. Fröhlich, D. Scharnagel and M. Breuning, Synthesis, 2015 DOI:10.1055/s-0034-1379457.
- The structures of the products 8 and 9 prepared by hydroxy–amine exchange were unambiguously confirmed by 2D NMR experiments. Rearrangements of 12 to β-amino piperidines via the sequence of mesylation–aziridinium formation–nucleophilic attack at C-2 under ring enlargement were not observed. It had been shown that such rearrangements do not occur under mild mesylation conditions and with amines as the nucleophiles, see: (a) D. G. Pargo and J. Cossy, Chem. – Eur. J., 2014, 20, 4516 CrossRef PubMedSee also: (b) R. H. Reitsema, J. Am. Chem. Soc., 1949, 71, 2041 CrossRef CAS; (c) J. H. Biel, W. K. Hoya and H. A. Leiser, J. Am. Chem. Soc., 1959, 81, 2527 CrossRef CAS; (d) C. F. Hammer, S. R. Heller and J. H. Craig, Tetrahedron, 1972, 28, 239 CrossRef CAS.
- To our surprise, this problem has not yet been mentioned in any other publication8,12,14a–d,f–h,15,16 dealing with the enantiomer analysis of 2.
- For proposed transition states in the presence of other chiral diamines, see ref. 8b,12a–c,14g and 15.
- (a) Ref. 12b, and 14f ; (b) L. M. Mirica, X. Ottenwaelder and T. D. P. Stack, Chem. Rev., 2004, 104, 1013 CrossRef CAS PubMed; (c) V. Mahadevan, Z. Hou, A. P. Cole, D. E. Root, T. K. Lal, E. I. Solomon and T. D. P. Stack, J. Am. Chem. Soc., 1997, 119, 11996 CrossRef CAS; (d) S. Mahapatra, J. A. Halfen and W. B. Tolman, J. Am. Chem. Soc., 1996, 118, 11575 CrossRef CAS; (e) V. Mahadevan, M. J. Henson, E. I. Solomon and T. D. P. Stack, J. Am. Chem. Soc., 2000, 122, 10249 CrossRef CAS; (f) S. Dong, J. Zhu and J. A. Porco Jr., J. Am. Chem. Soc., 2008, 130, 2738 CrossRef CAS PubMed; (g) P. Kang, E. Bobyr, J. Dustman, K. O. Hodgson, B. Hedman, E. I. Solomon and T. D. P. Stack, Inorg. Chem., 2010, 49, 11030 CrossRef CAS PubMed; (h) J. Jover, P. Spuhler, L. Zhao, C. McArdle and F. Maseras, Catal. Sci. Technol., 2014, 4, 4200 RSC.
- Kozlowski et al. also observed a fast uptake of O2 and 1a in the beginning of the reaction, but without any product formation.12d On this basis they proposed that the catalytically active Cu-species contains an additional, oxidized naphthyl ligand, presumably an ortho-quinonic species, which functions like a cofactor in enzymes. We do not think that this is the only possible explanation for the observation made. We suppose that there is a fast ligand exchange at and oxygenation of the initially applied complex CuI·4 (“burst phase”), leading to a catalytically inactive, oxygenated copper complex of type [CuOx·4·1a]n (resting state; for examples of oxygenated di- and oligomeric Cu-complexes, see ref. 24), which only slowly breaks down to the oxygen-free, catalytically active CuII-species. This pathway is also fully consistent with the experimental results. We therefore did not include an oxidized naphthol ligand in our mechanistic model.
- The product distribution in competition experiments is most consistent with a radical–radical coupling.12a,c In contrast, a radical–anion coupling seems to occur in the presence of a stoichiometric amount of a copper–diamine complex, see: (a) M. Hovorka, J. Günterová and J. Závada, Tetrahedron Lett., 1990, 31, 413 CrossRef CAS; (b) M. Smrčina, Š. Vyskočil, B. Máca, M. Polášek, T. A. Claxton, A. P. Abbott and P. Kočovský, J. Org. Chem., 1994, 59, 2156 CrossRef.
- (a) A. I. Meyers and K. A. Lutomski, J. Am. Chem. Soc., 1982, 104, 879 CrossRef CAS; (b) A. I. Meyers and D. G. Wettlaufer, J. Am. Chem. Soc., 1984, 106, 1135 CrossRef CAS.
- (a) C. Girard and H. B. Kagan, Angew. Chem., Int. Ed., 1998, 37, 2922 CrossRef; (b) D. Heller, H.-J. Drexler, C. Fischer, H. Buschmann, W. Baumann and B. Heller, Angew. Chem., Int. Ed., 2000, 39, 495 CrossRef CAS; (c) D. G. Blackmond, Acc. Chem. Res., 2000, 33, 402 CrossRef CAS PubMed; (d) H. B. Kagan, Synlett, 2001, 888 CrossRef CAS.
- A catalytic cycle via octahedral copper species cannot be fully excluded, but is very unlikely. For a short discussion, see footnote 58 in ref. 12c.
- An oxidation of the copper(I) atom in 13 prior to the chelation of 1a is also possible (and stereochemically not of relevance). This sequence, however, seems to be less likely since the oxidation is slow (rate-limiting step in the catalytic cycle).12d.
- W. L. F. Armarego and D. D. Perrin, Purification of Laboratory Chemicals, Butterworth-Heinemann, Oxford, 4th edn, 2000 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, HPLC and NMR spectra. See DOI: 10.1039/c4cy01676a |
| This journal is © The Royal Society of Chemistry 2015 |