
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Continuous synthesis of diethyl carbonate from ethanol and CO2 over Ce–Zr–O catalysts
Iuliia
Prymak
,
Venkata Narayana
Kalevaru
,
Sebastian
Wohlrab
* and
Andreas
Martin
Leibniz-Institut für Katalyse e.V. an der Universität Rostock, Albert-Einstein-Str. 29a, 18059 Rostock, Germany. E-mail: Sebastian.Wohlrab@catalysis.de
First published on 29th January 2015
Abstract
CexZr1−xO2 (x = 0, 0.2, 0.5, 0.8 and 1.0) solids were prepared by a citrate method and characterized by various techniques such as N2-adsorption (BET-SA), XRD, XPS, TEM, H2-TPR, NH3- and CO2-TPD. The catalytic performance of these solids was evaluated for the direct synthesis of diethyl carbonate (DEC) from ethanol and CO2 in continuous mode using a plug-flow reactor (PFR). According to thermodynamic data, the reaction is favourable at low reaction temperatures and high reaction pressures. Thus, the catalytic experiments were carried out at reaction temperatures ranging from 80 to 180 °C and at reaction pressures from 80 to 180 bar. The CexZr1−xO2 catalysts exhibited significant differences in their performance mainly depending on (i) their Ce![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Zr ratio and (ii) the different acid–base characteristics. Among the series Ce0.8Zr0.2O2 (C80Z) and Ce0.5Zr0.5O2 (C50Z) catalysts displayed the most efficient performance. Moreover, C80Z, pretreated at 700 °C, yielded DEC at the equilibrium conversion level of YDEC ~ 0.7% at 140 °C and 140 bar at a CO2:ethanol ratio of 6:1 at a LHSV of 42 Lliq kgcat−1 h−1.
:Zr ratio and (ii) the different acid–base characteristics. Among the series Ce0.8Zr0.2O2 (C80Z) and Ce0.5Zr0.5O2 (C50Z) catalysts displayed the most efficient performance. Moreover, C80Z, pretreated at 700 °C, yielded DEC at the equilibrium conversion level of YDEC ~ 0.7% at 140 °C and 140 bar at a CO2:ethanol ratio of 6:1 at a LHSV of 42 Lliq kgcat−1 h−1.
Introduction
Carbon dioxide is a non-toxic, abundant and low-cost C1 feedstock. It is an environmentally friendly chemical reagent and can be understood as a phosgene substitute.1 Furthermore, CO2 conversion into various useful chemical products is certainly very attractive from both economic and environmental points of view. In fact, there are many methodologies to transform carbon dioxide into various commercially important products, for instance, dry reforming of CH4 with CO2 to produce syngas,2 transformation of CO2 to cyclic carbonates by cycloaddition with epoxides2a,3 or glycols3 and via oxidative carboxylation of olefins with CO2,3 synthesis of methanol from CO2 and H24etc. Among different approaches, direct synthesis of organic carbonates from alcohols and CO2,5 is gaining huge interest in recent times due to its high commercial significance. For instance, dimethyl carbonate (DMC) from methanol and CO23,6 or diethyl carbonate (DEC) from ethanol and CO27 are two challenging examples of such an approach of CO2 utilisation. DEC in particular is of special importance due to its unique commercial applications. For example, it is widely used in lubricants, cosmetics, as plasticizer, in pharmaceutical applications8 and as electrolyte in lithium ion batteries.9 Furthermore, DEC can be used as an additive to diesel fuel due to its high oxygen content (40.6%) and high octane booster power ((R + M)/2 = 105, where R is the research octane number and M stands for motor octane number)10 to improve the performance of gasoline combustion. Engine tests show that 5 wt% DEC in diesel fuel can reduce particulate emissions by up to 50%.11
There are several known conventional methods for the synthesis of DEC, such as the traditional phosgene-based route,12 oxidative carbonylation of ethanol,13 carbonylation of ethyl nitrite,14 catalytic alcoholysis of urea,15 transesterification of organic carbonates,16 and carbonylation of ethanol.17 A major drawback of these routes is the use of poisonous gases (phosgene, ethyl nitrite, carbon monoxide). On the other hand, only a few efforts are being made in recent times to develop a catalyst for direct synthesis of DEC. However, the majority of these efforts were confined to only batch processes. To the best of our knowledge, attempts to use continuous process for DEC production from ethanol are very rare. Thus, the present approach on the continuous synthesis of DEC from CO2 and ethanol is highly attractive and additionally it would also certainly allow CO2 to be used as a valuable and renewable low cost feedstock. The reaction route for the direct synthesis of DEC using CO2 is shown below in Scheme 1.
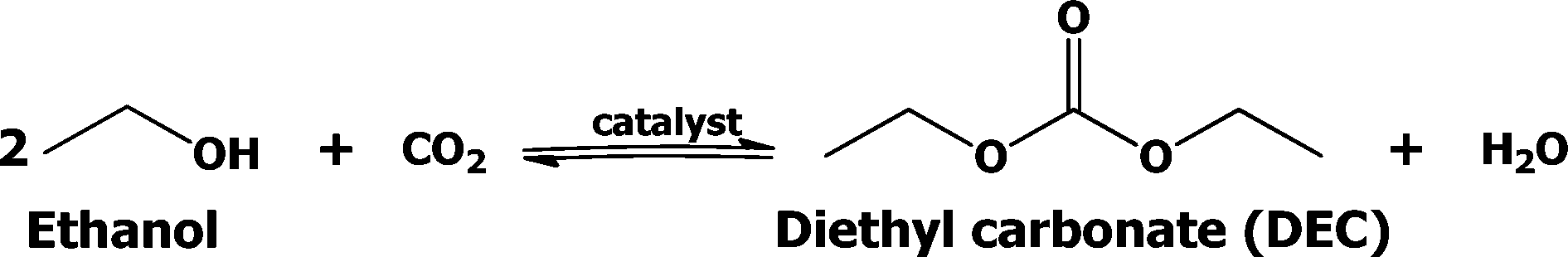 | ||
| Scheme 1 One-step synthesis of diethyl carbonate from ethanol and CO2. | ||
In spite of the obvious environmentally-friendly synthesis route, some additional difficulties of this approach also need to be taken into account. For instance, the activation of carbon dioxide is very difficult due to the fact that CO2 is highly thermodynamically stable and kinetically inert. In addition, this synthetic route also shows some thermodynamic (equilibrium) limitations and therefore the yield of DEC to be achieved is expected to be relatively low. Another problem is the formation of H2O as a by-product, which shifts the equilibrium towards the reactants side, in addition, a reverse hydrolysis of formed DEC back into ethanol and CO2 is possible. Several studies described some ways to overcome this problem, e.g. usage of certain chemical reagents or absorbents might be helpful to remove H2O from the product during the course of the reaction, for instance butylene oxide,18 benzonitrile,19 acetals or ketals,20 acetonitrile21 but also inorganic materials like zeolites.22 Alternatively, Dibenedetto et al.23 used a polymeric organic membrane PERVAP 1211 to remove the water formed during the reaction. Unfortunately, this effort was not successful due to problem that the reaction mixture cannot be directly separated as DEC passes through the membrane since it is permeable at concentrations above 0.3%. Nevertheless, in the work of Li et al.24 three types of supported membranes (mesoporous silica, polyimide silica and polyimide–titania hybrid membrane) were applied for another similar reaction, i.e. the synthesis of dimethyl carbonate (DMC) from methanol and CO2. Even though, the use of such membranes considerably improved DMC formation, however, the capability of dehydration at high pressure and temperature was reported to be very low.
Until now, wide range of catalytic systems have been studied for DMC synthesis from methanol and CO2. For instance, CeO2,7a,21,25 K2CO3,7b Ce-SBA-15,17 Nb2O5/CeO2,23 Cu–Ni/AC,7c Ce–Si-MCM-41,7a Ce–H-MCM-41,7a metal tetra-alkoxides,26 CeO2–ZrO25b,20d,27 are some of the most widely used catalyst compositions so far. Among them, literature reports indicate that Ce–Zr–O solids are somewhat more effective catalysts.27 The effectiveness of this catalyst was ascribed to the presence of acid–base sites on the surface, which consist of coordinatively unsaturated metal cations M4+ (Lewis acid-electron acceptors), oxide anions O2− (Lewis base-electron donors) and hydroxyl groups probably acting as Brønsted base centers during water formation.6a It has been proposed that dissociation of adsorbed ethanol leads to the formation of ethoxide group on the acid sites of the catalyst accompanied by a proton release, which reacts with a surface hydroxyl group to produce water. CO2 is then inserted into the M–O bond of the C2H5O–M species to produce the reaction intermediate m-C2H5OCOO–M. This process is facilitated by interactions of C and O atoms with Lewis acid–base pairs of sites (O2−–M4+–O2−). Monoethyl carbonate species react with activated ethanol on the acid sites of the catalyst to produce DEC.28 It was suggested that high selectivity of DEC formation is due to rapid conversion of the ethoxide species to ethyl carbonate species under high CO2 pressure.
In this work, we describe the application and catalytic performance of different Ce–Zr mixed oxide catalysts for the continuous synthesis of DEC under varying reaction conditions. Efforts were made to investigate the effect of varying Ce:Zr ratio on catalyst phase purity, morphologies, surface composition, reducibility, acid–base characteristics, as well as the catalytic performance.
Experimental
Catalyst preparation
CexZr1−xO2 solids with x = 0, 0.2, 0.5, 0.8 and 1.0 were prepared by a citric acid complexation method according to Alifanti et al.29 ZrO(NO3)2·xH2O (Sigma-Aldrich, technical grade) and Ce(NO3)3·6H2O (Alfa-Aesar, 99.5%) in desired quantities were dissolved in deionized water (0.1 M). Citric acid (C6H8O7, Sigma-Aldrich, 99%) was added in 10 mol% excess for complete complexation of metal ions. The mixture was stirred for 2 hours at room temperature. The excess solvent was removed on a rotary evaporator. The obtained solid was dried overnight (16 h) under vacuum at 70 °C. This precursor was calcined at 450 °C for 3 h in air. Using this procedure, five different CexZr1−xO2 catalysts with varying Ce contents of 0, 20, 50, 80, 100 mol% denoted as Z (pure zirconia), C20Z, C50Z, C80Z and C (pure ceria), respectively, were prepared. Another batch of C80Z was calcined at 700 °C for 3 h in air.Catalysts characterization
The surface areas (SA) as determined by the BET equation and pore volumes of the samples received from BJH equation were measured using a NOVA 4200e device (Quantachrome Instruments). Prior to each nitrogen sorption measurement, the samples were evacuated for 2 h at 200 °C to remove physisorbed water.X-ray diffraction (XRD) studies were carried out on a X'Pert Pro diffractometer (Panalytical, Almelo, Netherlands) with CuKα radiation (λ = 1.5418 Å, 40 kV, 40 mA) and an X'Celerator RTMS detector. The phase composition of the samples was determined using the program suite WinXPOW by STOE & CIE with inclusion of the Powder Diffraction File PDF2 of the ICDD (International Centre of Diffraction Data). The average crystallite size (D) was calculated using Scherrer equation:30
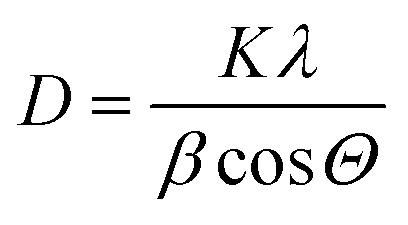
For the determination of the elemental composition, a Varian 715-ES ICP-OES (Inductively Coupled Plasma-Optical Emission Spectrometer) was used. Approximately 10 mg of the sample was mixed with 8 mL of aqua regia and 2 mL hydrofluoric acid. Digestion was performed in a microwave-assisted sample preparation system “MULTIWAVE PRO” from Anton Paar at ~200 °C and ~50 bar pressure. The data analysis was performed on the Varian 715-ES software “ICP Expert”.
X-ray photoelectron spectroscopy (XPS) was carried out using a VG ESCALAB 220iXL instrument with monochromatic AlKα radiation (E = 1486.6 eV). The samples were fixed by using a double adhesive carbon tape on a stainless steel sample holder. The peaks were fitted by Gaussian–Lorentzian curves following a Shirley background subtraction.
Transmission Electron Microscopy (TEM) investigations were carried out at 200 kV with an aberration-corrected JEM-ARM200F (JEOL, Corrector: CEOS). The aberration corrected STEM imaging (High-Angle Annular Dark Field (HAADF) and Annular Bright Field (ABF)) were done with a spot size of approximately 0.13 nm, a convergence angle of 30–36° and collection semi-angles for HAADF and ABF of 90–170 mrad and 11–22 mrad, respectively. Samples were prepared by deposing without any pre-treatment on a holey carbon supported Cu-grid (mesh 300) and transferred to the microscope.
Temperature programmed reduction (TPR) profiles were recorded in a temperature range from r.t. to 800 °C at a heating rate of 5 K min−1 on a Micromeritics AC2920 instrument. Prior to TPR measurement, all the samples were pre-treated with 5% H2/Ar at room temperature for 10 min.
The total acidity and basicity (adsorbed mmol NH3 or CO2 per gram catalyst) of the solids were determined by temperature programmed desorption of ammonia (TPD-NH3) or carbon dioxide (TPD-CO2), which was carried out in a home-made apparatus consisting of a gas flow system, a high temperature oven and a quartz reactor. For determination of acid sites, the samples (100–200 mg) were treated in nitrogen at 400 °C for 30 min to remove moisture and cooled down to 100 °C in He of high purity (6.0) prior to NH3 adsorption, which was carried out at 100 °C for 30 min in a flow of 5% NH3/He. Afterwards, the TPD-NH3 experiments were carried out from 100 to 450 °C in He flow (50 cm3 min−1) with a heating rate of 10 K min−1. Desorption of NH3 was monitored and evaluated by a thermal conductivity detector (TCD, GOW-Mac Instrument Co.). For determination of basic properties, the sample (200 mg) was treated in He (50 mL min−1) at 500 °C with a heating rate of 10 K min−1 for 15 min (for the removal of adsorbed water) and cooled down to 100 °C in He (50 mL min−1) prior to CO2 adsorption, which was carried out at 100 °C for 90 min in a flow of 1.2% CO2–He mixture. Afterwards, the TPD-CO2 experiments were carried out in He flow (50 mL min−1) at 100 °C for 30 min (for removal of physisorbed CO2). After cooling to 70 °C for 10 min, the sample was heated up to 800 °C at a rate of 10 K min−1 in a helium flow (50 mL min−1). The analysis of the effluent gases was performed by Quadrupole mass spectrometer (Balzers Omnistar).
Experimental setup and catalytic tests
The experimental setup is shown in Fig. 1. The catalytic setup mainly consisted of a high pressure reactor (inner volume = 32 mL, inner diameter = 12.7 mm, max. pressure = 200 bar), equipped with an oil heated jacket (max. temperature = 220 °C); high pressure pumps, relief valves, product collector and flow-meter. All tubings and fittings were made from stainless steel 316L (Swagelok).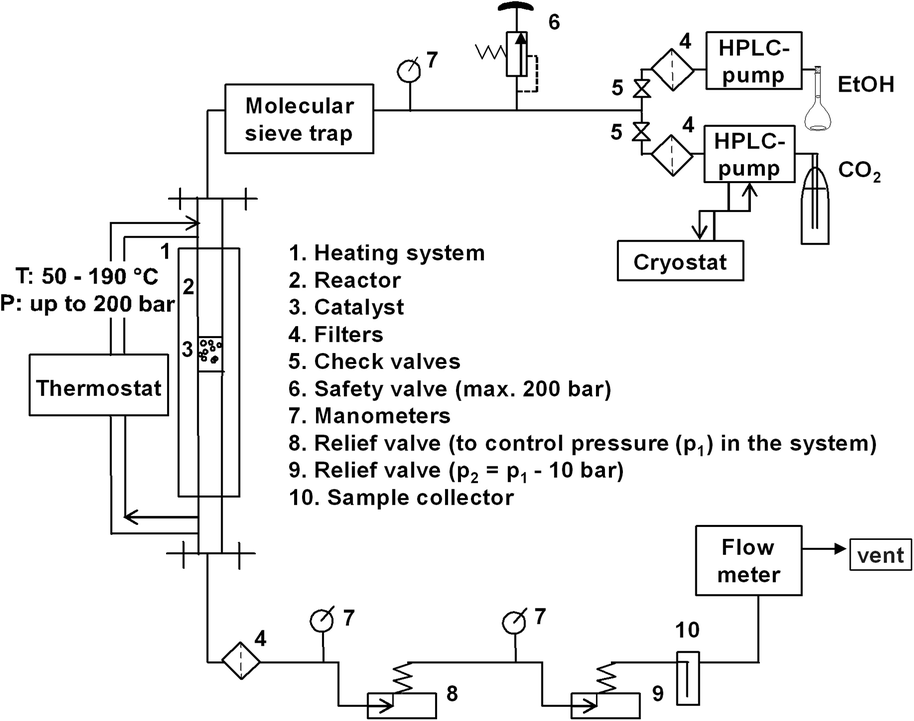 | ||
| Fig. 1 Schematic diagram of the experimental set up. | ||
In a standard procedure, the reactor was packed layer-by-layer: 27 g of corundum (size: 1 to 1.25 mm), 1 g of catalyst (size: 1 to 1.25 mm fraction), 27 g of corundum. A Gilson-pump with a thermostatic kit was employed to deliver liquid CO2 (99.9%, Air Liquide) to reactor at a flow rate of 0.6 mL min−1. A Shimadzu-HPLC pump was used in order to independently control the flow of ethanol of 0.1 mL min−1 (99.9%, H2O ≤ 0.1%, Roth). The molar ratio of EtOH:CO2 was kept constant during the mainly 1:6. Ethanol and liquid CO2 (from a dip tube cylinder) were metered through a tube that was filled with molecular sieve type 3A and placed before the entrance of the reactor to remove moisture, if any present in the used ethanol.
At the exit of reactor, a filter with pore diameter of 0.5 μm was placed in order to avoid a discharge of catalyst with flowing reagents. The system pressure was controlled by two manually regulated relief valves in series. The first one was set to the desired reaction pressure while the second one was set to approximately 5–10 bar less. Such modification was applied to reduce a possible rapid pressure drop and to allow a more constant flow. Moreover, the second relief valve was covered by an external heating to avoid freezing of humidity. Afterwards, the liquid phase was separated from the gas phase in a cold trap placed at the exit of the reactor outlet. The samples were analysed by a gas chromatograph (GC-2014, Shimadzu) using a capillary column (CP-PoraBOND Q, 10 m × 0.53 mm × 10 μm) equipped with a FID detector. The experiments were performed at pressures of 80, 110, 140, 180 bar and at temperatures of 80, 100, 120, 140, 160, 180 °C.
Results and discussion
Nitrogen sorption analysis
The surface areas, pore volumes and average pore sizes of mixed solids catalysts are summarized in Table 1. SAs were found to depend on the content of Ce in the catalysts. Pure ZrO2 (Z) displays much higher SA compared to pure CeO2 (C). As a result, the surface areas of the Ce–Zr mixed oxides are observed to decrease with increase in Ce content due to the poor resistivity against sintering of the cerium rich solids as claimed elsewhere.31| Sample | x in CexZr1−xO2 | BET-SA, m2 g−1 | V pore, cm3 g−1 | d pore, nm | Crystal size, nm |
|---|---|---|---|---|---|
| Z | 0 | 88 | 0.069 | 2.8 | 3.8 |
| C20Z | 0.2 | 79 | 0.060 | 2.8 | 4.6 |
| C50Z | 0.5 | 57 | 0.064 | 4.2 | 4.8 |
| C80Z | 0.8 | 42 | 0.076 | 6.2 | 6.0 |
| C | 1.0 | 27 | 0.036 | 5.3 | 9.0 |
The crystallite sizes as well as the pore volumes of mixed solids are found to vary in a narrow range from 4.6 to 6.0 nm and 0.060 to 0.076 cm3 g−1, respectively, depending upon the content of Ce in the system. Pure CeO2 (C) exhibits the largest crystallite size (9 nm) and lowest pore volume, while pure ZrO2 (Z) possesses the smallest crystallite size and pore volume in the range of the mixed oxides. The combination of zirconium and cerium enhances the stability of mixed oxides and suppresses the crystal growth during catalyst preparation. Additionally, it can be seen that mesopores are present in all CexZr1−xO2 samples.
XRD analysis
Fig. 2 shows XRD patterns of different compositions of fresh CexZr1−xO2 catalysts. Characteristic peaks for sample C can be attributed to a cubic CeO2 phase.7a,32 With increase in zirconium loading in the solid composition, the main peaks shift to higher diffraction angles leading to changes of unit cell parameters and lattice deformation.33 This can be associated with a progressive substitution of Ce4+ (ionic radius 0.097 nm) with the smaller Zr4+ (ionic radius 0.084 nm).34 The results, obtained by Yashima et al.35 revealed that phase transitions occurring in CexZr1−xO2 depend on their composition. The tetragonal CexZr1−xO2 phase appears with Ce < 50 mol% (C20Z), whereas above 50 mol%, a cubic CexZr1−xO2 (C50Z and C80Z) phase is formed.33c,34b,36 There is no evidence for phase segregation in wide range of composition. For pure ZrO2 (Z) the diffraction peaks can be assigned to the tetragonal ZrO2 structure.32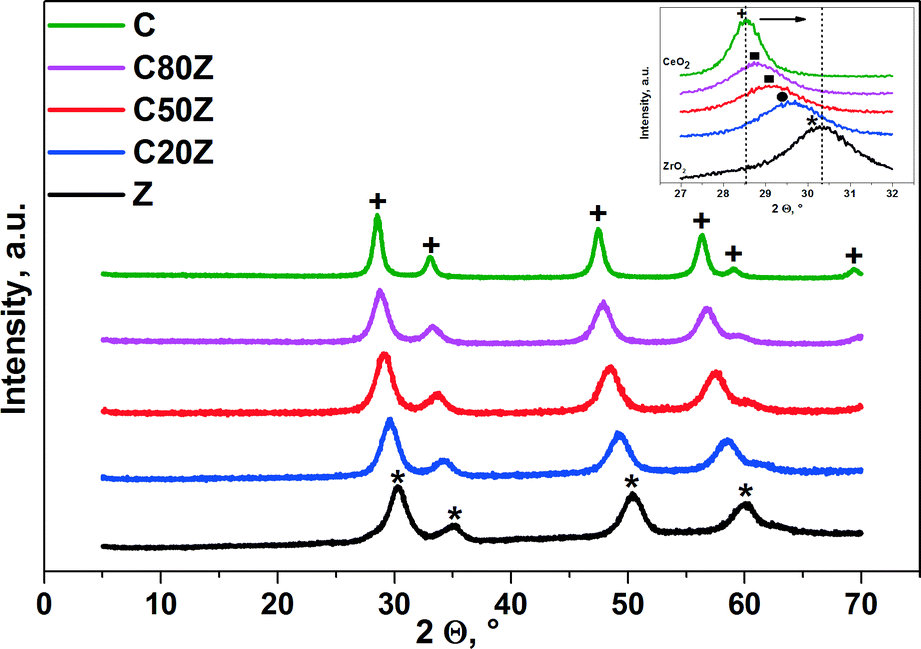 | ||
| Fig. 2 XRD patterns for CexZr1−xO2 catalysts. Phase composition: + cubic CeO2 (JCPDS 65-5923), * tetragonal ZrO2 (JCPDS 79-1769). Inset: ■ c-Ce–Zr–O mixed phase, ● t-Ce–Zr–O mixed phase. | ||
Near-surface characterization by XPS
XPS was used to investigate the near-surface composition of the samples. Fig. 3 compares the relative Ce, Zr and O concentrations measured by XPS with that of bulk composition obtained by ICP. It can be seen that Zr is significantly enriched in the near-surface-region of all mixed oxides. The oxygen content is also above its stoichiometric value except for C20Z. In case of near-surface concentration of Ce, there is no general tendency. For instance Ce is either enriched (C80Z) or remained more or less at the same concentration (C50Z) compared to the bulk. But in the case of pure CeO2 and low Ce content catalyst (C20Z), the surface concentration of Ce is slightly decreased compared to its bulk.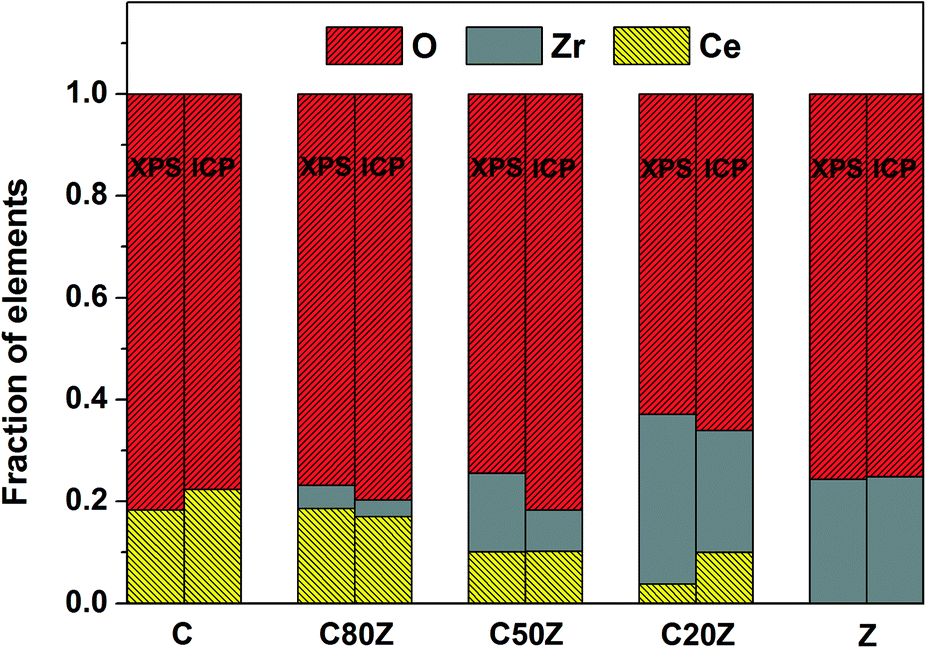 | ||
| Fig. 3 Comparison of the relative Ce, Zr and O concentrations in the near-surface-region as measured by XPS with bulk composition (ICP) for CexZr1−xO2 (x = 0, 0.2, 0.5, 0.8, 1). | ||
TEM analysis
TEM (Transmission Electron Microscopy) studies were performed on the pure oxides C and Z as well on C80Z. Representative images are displayed in Fig. 4 revealing nanometer sized particles. The particle size of CeO2 is about 8 nm, which is quite close to the value calculated from Scherrer equation (9 nm). In the case of C80Z particle sizes of about 5 nm are obtained but these particles are not uniform in shape. TEM-HAADF images (Fig. 4 (1b, 2b and 3b)) confirm the mesoporous nature of the samples. In some cases, larger pores up to 40 nm in diameter can also be detected. | ||
| Fig. 4 ABF-STEM (a) and HAADF-STEM (b) images of (1) C, (2) C80Z, (3) Z. | ||
H2-TPR analysis
The redox properties of the CexZr1−xO2 solids were evaluated by temperature-programmed reduction (TPR) with 5% H2/Ar (50 mL min−1). Fig. 5 depicts the TPR profiles of the CexZr1−xO2 catalysts, where the peak maximum indicates the temperature that corresponds to the maximum rate of reduction. It is known, that cerium can exist in CexZr1−xO2 solid solutions as Ce3+ and Ce4+ ions while zirconium exists as Zr4+ only.37 Pure CeO2 (C) has a high oxygen storage capacity and possess a larger number of oxygen vacancies.38 These redox properties can strongly be enhanced when Zr4+ cations are introduced into the CeO2 lattice by higher oxygen ion mobility and increased vacancy sites inside the modified lattice.37,39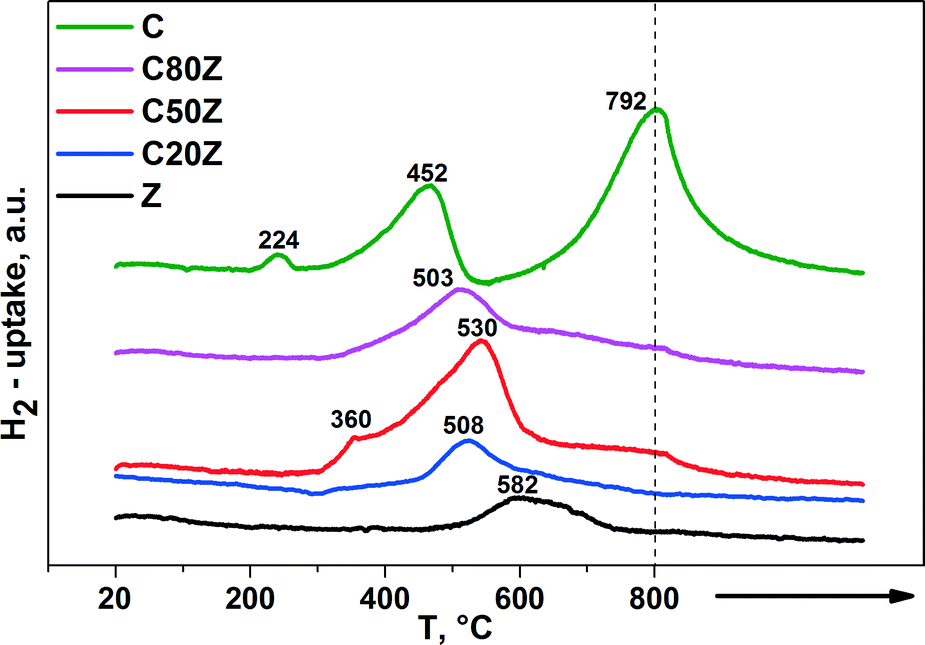 | ||
| Fig. 5 H2-TPR profiles of different CexZr1−xO2 solids. (→: indicates stationary treatment of sample at 800 °C for 2 h). | ||
The reduction process of pure CeO2 (C) involves two main steps. The first region is located between 350 and 600 °C with Tmax around 452 °C and second region starts from 600 °C with Tmax around 792 °C. The low-temperature peak is due to the most easily removable surface capping oxygen of CeO2, while the high-temperature signal at 792 °C is caused by the removal of bulk oxygen.40 The TPR profiles of the mixed CexZr1−xO2 oxides show a main broad reduction in the region between 500–530 °C with different Tmax values. As reported by de Rivas et al. this fact suggests that the addition of Zr to CeO2 sample remarkably causes reduction of surface and bulk in one step at medium temperatures.41 Besides, some H2 consumption can be noticed at higher temperatures for C80Z and C50Z, but to a lower extent than for CeO2. Furthermore, the position of surface reduction peak shifted from 452 to 508 °C with increasing of zirconium content. It was noted that the extent of the reduction seems to be the highest for C50Z sample. Moreover, the weak peak at 360 °C can be attributed to subsurface Ce4+ in different chemical environment.42 The H2 uptake of pure CeO2 (C) at 224 °C might be traced back to an adsorptive process as reported by Fierro et al.43
The relative hydrogen consumption, expressed as mmol of H2 per gram of catalyst and peak maxima are shown in Table 2 as a direct measure of the amount of water evolved from the sample under flowing 5% H2/Ar. The increase in H2 uptake from 1.15 mmol g−1 for pure ceria (C) to 1.793 mmol g−1 for C50Z provides hints to enhanced reducibility by the addition of Zr to CeO2 solid.41a Further increase in zirconium content beyond 50 mol% progressively reduces the H2 consumption. Moreover, for pure ZrO2 (Z) the H2 consumption was only 0.192 mmol g−1. This denotes the Zr4+ cations are hardly being reduced under the conditions applied. Our findings are in a good agreement with those described by Trovarelli et al.40 where the reducibility of CexZr1−xO2 solid solutions is also strongly dependent on the crystal structure.
| Sample | H2 uptake, mmol g−1 | Peak max, °C |
|---|---|---|
| Z | 0.192 | 582 |
| C20Z | 0.704 | 508 |
| C50Z | 1.793 | 530 |
| C80Z | 1.482 | 503 |
| C | 0.015, 0.271, 0.869 | 224, 452, 792 |
NH3- and CO2-TPD
The concentration and strength of the acid sites was evaluated by NH3-TPD (i.e. expressed as an amount of NH3 desorbed per gram of catalyst) and presented in Table 3. The acidity characteristics of the solids is strongly affected by the addition of zirconium into ceria lattice.44 It is evident from Table 3 that pure ZrO2 (Z) is more acidic (i.e. 0.185 mmol NH3 g−1) than pure CeO2 (C). It can be seen that the total acidity was the lowest for solid C (0.034 mmol g−1), which is however considerably improved by the addition of zirconium.| Sample | NH3 desorbed, mmol g−1 | CO2 desorbed, mmol g−1 |
|---|---|---|
| Z | 0.185 | 0.112 |
| C20Z | 0.130 | 0.235 |
| C50Z | 0.118 | 0.117 |
| C80Z | 0.094 | 0.104 |
| C | 0.034 | 0.052 |
The NH3-TPD profiles of the CexZr1−xO2 solids are shown in Fig. 6. The results demonstrate the presence of acid sites of different strength in these solids. The desorption peaks of TPD profiles located at 100–200 °C, 200–400 °C and 400–450 °C can be assigned to weak, moderate and strong acid sites, respectively.45 Both the weak and moderate acid sites were observed for all catalysts. However, the strong acid sites with characteristic desorption temperature of about 450 °C were observed in Z, C20Z and C50Z solids only.
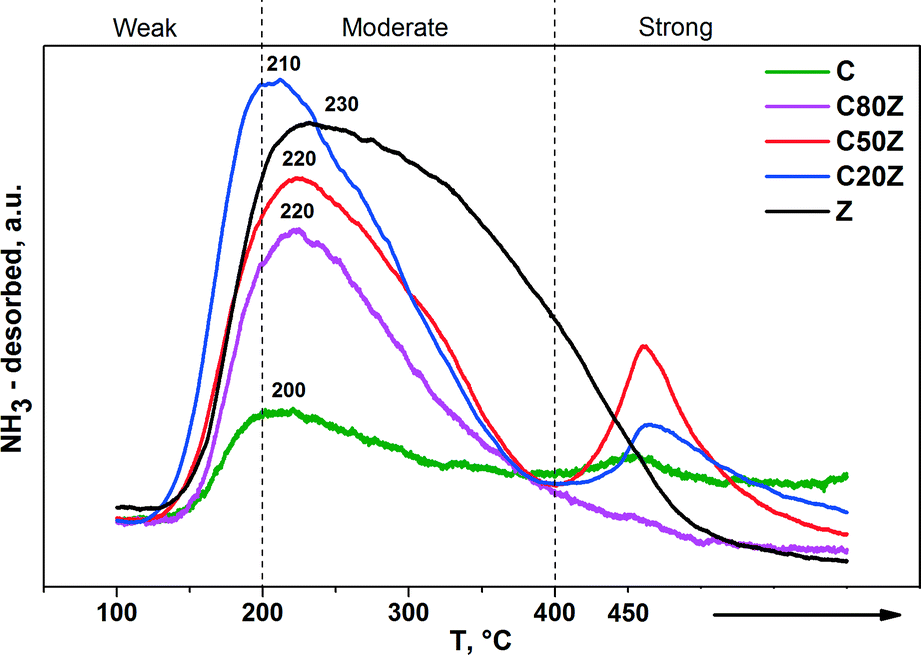 | ||
| Fig. 6 NH3-TPD profiles of CexZr1−xO2 solids. | ||
To investigate the effect of Ce content on basic properties, CO2-TPD experiments were also carried out. Fig. 7 illustrates the CO2-TPD profiles and Table 3 lists the amount of CO2 desorbed during temperature programmed desorption measurements of different CexZr1−xO2 solids. All Ce-based catalysts exhibit two broad desorption peaks at varying temperatures indicating that different types of basic sites are present with weak (100–230 °C), moderate (230–500 °C) and strong (above 500 °C) basic strengths.18a,46 It can be seen, that the total concentration of CO2 desorption from sample C is very low. Among all prepared mixed oxides, C20Z exhibited the highest concentration of basic sites (0.235 mmol g−1).
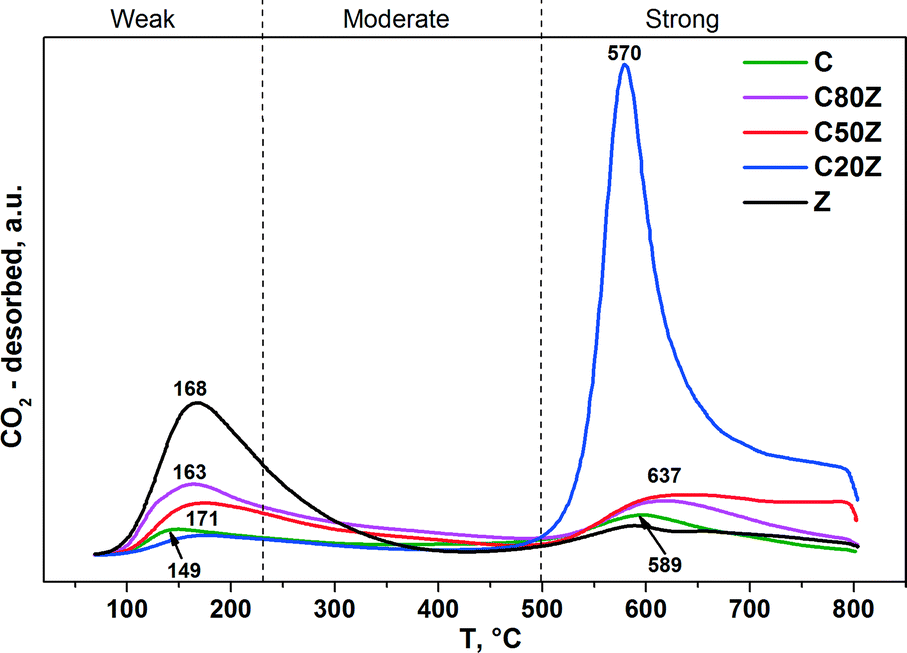 | ||
| Fig. 7 CO2-TPD profiles of CexZr1−xO2 solids. | ||
A main conclusion from these studies is that surfaces of CexZr1−xO2 catalysts possess both acidic and basic sites. Interestingly, the acid-to-base site ratio was about 1 for C80Z and C50Z catalysts, which seemed to be optimum for improved catalytic properties.
Influence of the reaction conditions on catalytic performance
The critical points (pc, Tc) for the C2H5OH–CO2 binary system were reported by various researchers in the past, for instance by Baker,47 Takishima,48 Lim,49 Yeo,50 and Galicia-Luna et al.51 The supercritical region for C2H5OH–CO2 binary system is reached maximum near 160 bar and temperature in a range of 120–160 °C with mixtures whose initial CO2 molar fraction are between 0.7 and 0.9. Moreover, the larger the ethanol concentration in the feed mixture is, the higher is the required temperature to reach the critical point. Based on such reports, we suppose that these trends are still valid for quaternary system, due to the low DEC amount (predicted x ~ 0.004 at reaction equilibrium) and H2O (predicted x ~ 0.004 at reaction equilibrium) productions. One can also expect that above 160 bar the reaction mixture might be in the liquid or supercritical state, depending upon the system temperature. According to literature,23,52 the use of supercritical conditions allow to reach a higher ethanol conversion with respect to using liquid ethanol pressurized by CO2. Such conclusion was made based on fact that under supercritical conditions, EtOH and CO2 are in a single phase and the effect of solubility-dependent concentration of CO2 in the liquid phase is cancelled out. Cai et al.53 and Leino et al.5b assumed in a theoretical study for DMC synthesis that the high performance had been partly attributed to the fact that reaction becomes thermodynamically favourable as the system pressure increases.The present work was focused on transferring the knowledge from batch approaches to a continuous process, thus the optimal flow rate of reagents is a very important parameter to achieve a highest DEC yield and space-time-yield as well. From preliminary tests performed at 140 °C and 140 bar, a maximum possible flow rate (characterized by no significant YDEC loss) of 42 Lliq kgcat−1 h−1 (τ = 68.6 s) was identified and selected for the study. The influence of EtOH:CO2 ratios on ethanol conversion into DEC was already described by Dibenedetto et al.23 They found a correlation between DEC formation and EtOH:CO2 ratio, i.e. increasing the ratio leads to decreased EtOH conversion. Therefore, the ratio of EtOH:CO2 = 1:6 was selected for our experiments.
The influence of the total reaction pressure and temperature on DEC formation was studied over C, C80Z, C50Z, C20Z and Z catalysts in continuous running plug flow reactor (see Fig. 8). The areas in the three-dimensional figures reflect the catalytic behaviour of the CexZr1−xO2 catalysts depending on their composition. We observed an increase of DEC yield with rise of pressure for all CexZr1−xO2 catalysts. The influence of reaction temperature is more complex. For C, C80Z and C50Z, where the yield of DEC gradually increased with elevating reaction temperature it reaches a maximum at 140 °C and then drops to lower values at higher temperatures. It is well-known from literature18a,27 that carboxylation of alcohols is an exothermic reaction and from thermodynamic point of view the high reaction temperature is unfavourable for organic carbonates formation. The activity of pure ZrO2 (Z) and C20Z slowly increased with rise in temperature in the pressure region of 140–160 bar and reached the highest value at 180 °C. Highest DEC yields were obtained for C50Z and C80Z possessing maxima at a reaction pressure of 160 and 140 bar, respectively.
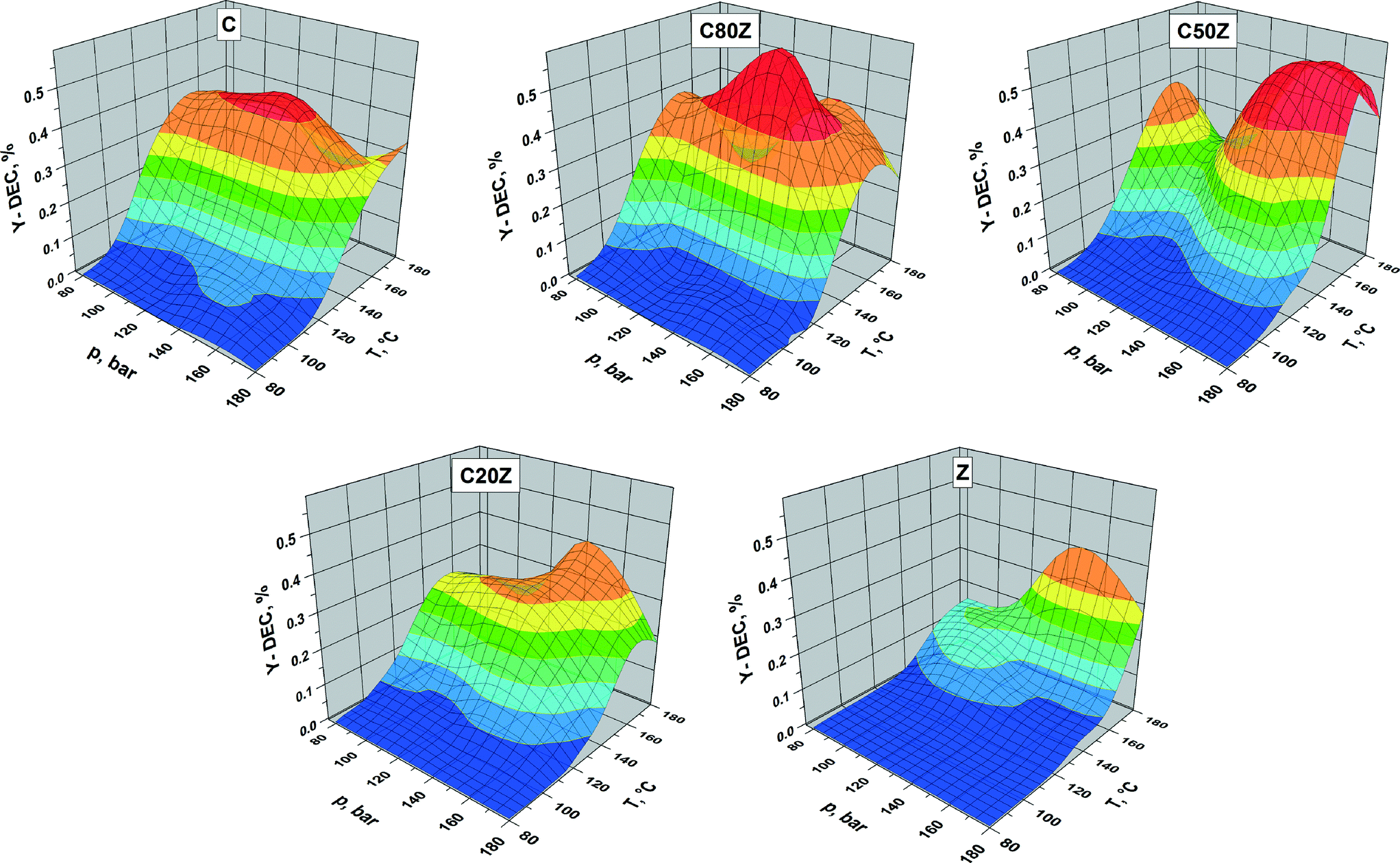 | ||
| Fig. 8 Effect of reaction pressure and temperature on the yield of DEC over different CexZr1−xO2 catalysts. Reaction conditions: catalyst weight 1 g, EtOH:CO2 = 1:6, 1 h time-on-stream, LHSV = 42 Lliq kgcat−1 h−1, τ = 68.6 s. | ||
Materials properties affecting the catalyst activity
According to literature the activity of pure CeO2 and CexZr1−xO2 catalysts towards DMC and DEC formation had been related to its specific surface areas.20d,25b In our contribution, the BET-SA of the CexZr1−xO2 solids increase with higher Zr content and show a maximum value for pure ZrO2 (Z). However, CexZr1−xO2 catalyst with x = 0.8 and 0.5 exhibited better DEC yields. Thus, a relation between DEC formation and BET-SA cannot be derived. Possible reasons for more efficient performance of C80Z and C50Z solids compared to other Ce:Zr ratios could be the differences in their crystal structure, surface composition and acid–base properties. The cubic structure was formed for Ce-rich samples (e.g. Ce ≥ 50 mol%), whereas a tetragonal phase was found to be predominant in the solids with lesser Ce contents. Based on this, we can conclude that the most active phase for DEC formation is cubic CexZr1−xO2 (C, C80Z, C50Z). This result is well consistent with previous works where pure CeO227 and mixed oxides such as Ce0.6Zr0.4O26b and Ce0.8Zr0.2O254 existed in cubic phase were the most active catalysts in direct DEC/DMC batch syntheses. In contrast, Zhang52 and Wang27 have shown that the tetragonal Ce0.5Zr0.5O2 and Ce0.07Zr0.93O2 phases exhibited the highest DMC/DEC formations among other Ce–Zr mixed oxides. We assume that the high performance of tetragonal CexZr1−xO2 phase claimed by Zhang et al. can be related to different synthesis protocols applied. The incorporation of Zr into the cubic CeO2 lattice remarkably affects the amount of oxygen vacancies and the basic properties of the materials therewith.41a Besides, XPS revealed that there is a clear enrichment of Zr in the near-surface-region of all samples. Interestingly, such enrichment is much more pronounced in the case of C80Z and C50Z samples, which seemed to be one of the major reasons for their improved performance. In addition, the concentration of acid sites in CexZr1−xO2 catalysts also depends on Zr content, which in turn led to the different catalytic behaviour. However, a large amount of strong acid and base sites has a negative effect on the DEC formation. As a result, the pure ZrO2 (Z) and C20Z having stronger acid–base properties showed rather poor performance. However, Wang et al. showed the absence of strong basicity and acidity on the surface of the CexZr1−xO2 solid with the high Zr content (0.93) whereas samples with low Zr (0.2) concentration demonstrated the strong strength of acid–base sites. In contrast, our C80Z catalyst that exhibits weak to medium strength of acidic sites seems to be a good balance between the acidity and catalytic activity. As stated above, it is also known from the literature27,55 that the catalytic activity of CexZr1−xO2 is reduced to some extent by the presence of strong acid–base sites on the surface of the catalysts. This might be another reason for the requirement of higher reaction pressure (supercritical CO2) and higher temperature for C50Z sample compared to C80Z to display maximum YDEC. Moreover, previous reports20d,56 have shown that the presence of weak to medium acidity on Ce–Zr surfaces is the key factor in a selective DEC/DMC syntheses. On strong acid sites the formation of diethyl ether/dimethyl ether (DEE/DME) is favoured. Furthermore, DEE/DME together with H2O suppresses the formation of DEC/DMC. In addition, Tomishige and co-authors20d found that during the reaction of EtOH with CO2 ethylene as by-product also being formed. Zhang et al.52 investigated the influence of the Ce/Zr ratio on the formation of by-products in the direct synthesis of DMC from methanol and CO2. The catalyst with a Ce:Zr molar ratio of 1 showed the highest activity without forming by-products except H2O. In addition to this, in all our continuous mode experiments no by-products were detected revealing the advantage of the continuous process.
As mentioned above, the catalytic activity was primarily related to the strength of acid–base sites located on the Ce–Zr surface. Interestingly, both C80Z and C50Z have shown nearly an equal amount of acid–base sites and the highest catalytic performance as well. This is also in accordance to Tomishige et al. who claimed that an equal number of neighbouring acid–base sites is required for optimal catalyst performance, whereas they found such an effect in the direct synthesis of DMC over ZrO2 catalysts.28a
Besides acid–base strength and the ratio of such sites, carbon dioxide activation, which is the most difficult part of the reaction, needs to be evaluated in a systemic manner. According to the reaction mechanism proposed by Wada et al.57 for the formation of DMC over Cu–CeO2 catalyst, the carbon dioxide adsorption was related to oxidation state of surface cerium. It was speculated that oxygen vacancies are defect sites, which can adsorb CO2. The population of O vacancies might increase by the reduction in H2 and/or by the presence of Cu sites in catalysts. In case of CexZr1−xO2 solid solutions, the presence of Zr4+ in the CeO2 lattice causes distortion in the ceria lattice resulting in an increase of oxygen mobility and also an increase of the number of anion vacancies on the oxide surface.41a These oxygen vacancies can also act as CO2 adsorption sites, which were however confirmed by CO2-TPD analysis of the present study. Due to the significant increased adsorption of CO2, an improved catalytic performance could be achieved on C50Z and C80Z solids compared to pure CeO2. In view of the highest YDEC obtained on C80Z at possibly lowest temperatures and pressures, this solid was further used to check the long-term stability of the catalyst.
In order to explore the variation of DEC formation with time, the reaction was performed at 140 °C, 140 bar for 20 hours over C80Z catalyst. The experimental results revealed that the formation of DEC slightly increased with reaction time and levelled off after 6 hours (YDEC = 0.55%). Beyond, an influence of calcination temperature of CexZr1−xO2 catalysts was already reported by Tomishige et al. for the cyclic carbonate synthesis from glycol and carbon dioxide.46 Accordingly, an even higher catalytic performance could be achieved with C80Z which was pretreated at 700 °C and tested at 140 °C and 140 bar at different EtOH:CO2 ratios (Fig. 9a) as well as different flow rates (Fig. 9b).
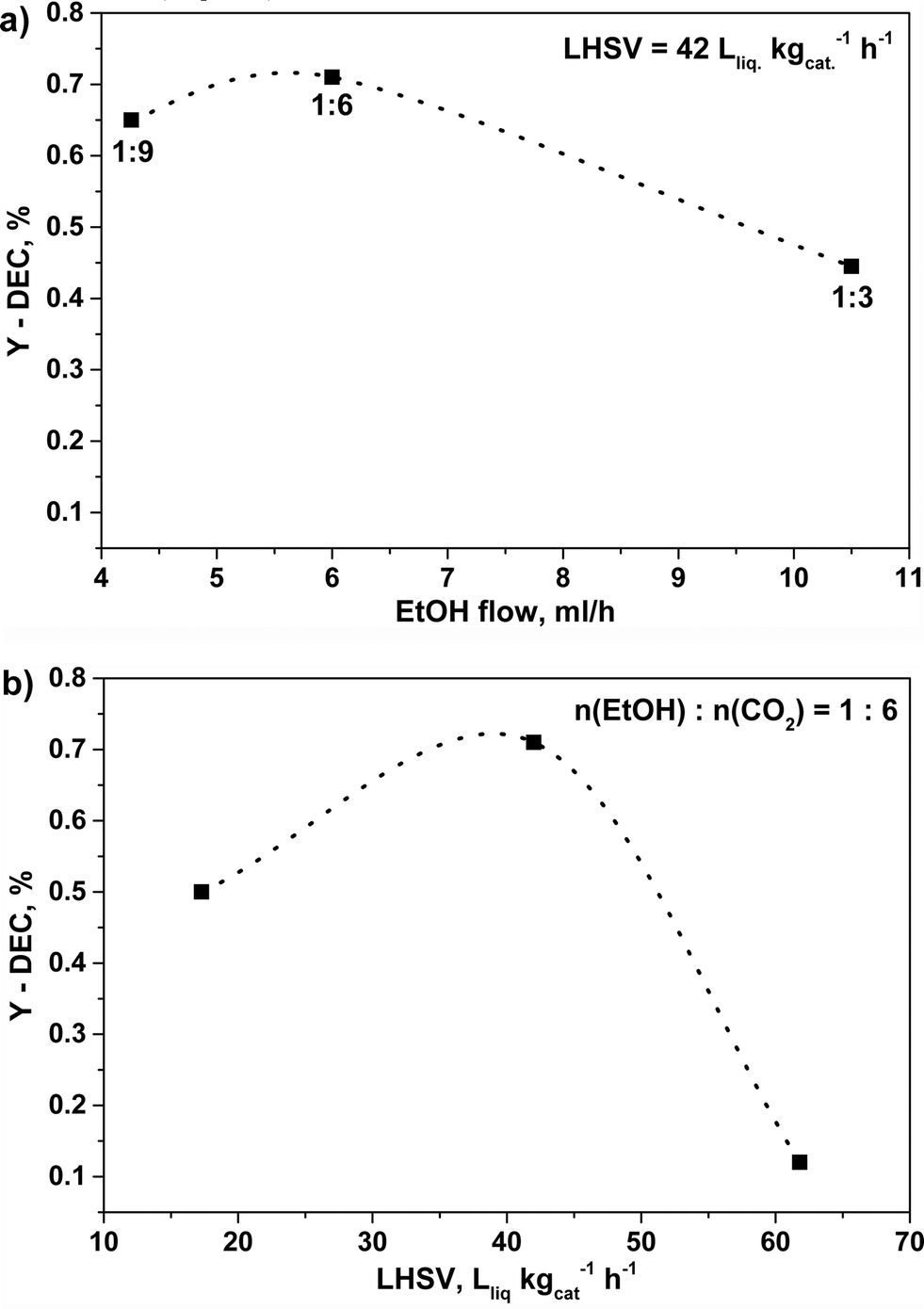 | ||
| Fig. 9 Effect of different a) EtOH:CO2 ratios and b) LHSV's on DEC yield over C80Z calcined at 700 °C. Reaction conditions: catalyst weight 1 g, 1 h time-on-stream, T = 140 °C, p = 140 bar. | ||
Here, the EtOH:CO2 ratio was also at its optimum at 1:6 in accordance to Dibenedetto et al.23 Furthermore, up to a LHSV of 42 Lliq kgcat−1 h−1 (τ = 68.6 s) an increase of the DEC-yield up to YDEC ~ 0.7%. was observed. However, a further increase in LHSV, e.g. 62 Lliq kgcat−1 h−1 (τ = 46.5 s), caused a dramatic drop of the DEC yield (YDEC ~ 0.1%). The change of total CO2–EtOH flow greatly affects the contact time, which however was found to be unexpectedly very low for this continuous reaction mode.
To get a better assessment of catalytic activity of C80Z, the predictive Soave–Redlich–Kwong equation58 was used to calculate the equilibrium DEC yield of ~0.7% under these selected reaction conditions (T = 140 °C, p = 140 bar). The comparison shows that a continuous process for the direct formation of DEC from carbon dioxide and ethanol can be operated at the reaction equilibrium level.
Conclusions
CexZr1−xO2 catalysts have been tested in the direct and continuous synthesis of DEC from EtOH and CO2. The Ce:Zr ratio displayed a considerable effect on the catalytic performance. Results revealed that activation of Zr-rich catalysts require higher temperature regimes than Zr-lean catalysts. CexZr1−xO2 solid solutions with x ≥ 0.5 showed relatively good catalytic performance. In addition, the reaction pressure and temperature are also crucial parameters for improving the catalytic performance. A reaction pressure of 140 bar and a temperature of 140 °C are effective for continuous and direct synthesis of diethyl carbonate.
It was found that total concentration of acid–base sites is the lowest for pure ceria, but markedly increased with the addition of Zr due to its higher acidity. Consequently, the highest value of NH3 consumption was noticed on pure zirconia. Moreover, the introduction of Zr into the CeO2 lattice remarkably enhances the amount of oxygen vacancies due to the formation of Ce3+ species. These additional adsorption sites lead to a significant increase in YDEC by cubic CexZr1−xO2 solid solutions instead of pure CeO2.
In conclusion, it can be stated that the Ce:Zr ratios play an important role on the catalytic performance. They can be used to tune the acid–base properties, reducibility, surface vacancies, phase composition of the solids and hence the catalytic properties as well. Among various catalysts tested C80Z and C50Z samples with balanced concentration of acid and base sites (1:1) exhibited reasonably good DEC yields. An outstanding performance (YDEC ~ 0.7%) could be achieved with C80Z which was pretreated at 700 °C and tested at 140 °C and 140 bar at a CO2:ethanol ratio of 6:1 at a LHSV of 42 Lliq kgcat−1 h−1 (τ = 68.6 s). Hence, it is possible to run the reaction continuously at the equilibrium level at very low contact times.
Acknowledgements
The authors like to thank the Leibniz Association for funding (Project SAW-2011-LIKAT-2). Thanks are also due to Mr. R. Eckelt for BET-SA, Dr. M. Schneider for XRD measurements, Ms. A. Simmula for ICP-OES, Dr. J. Radnik for XPS measurements, Dr. M.-M. Pohl for TEM results, Dr. H. Atia for H2-TPR, NH3- and CO2-TPD.Notes and references
- (a) M. Aresta, A. Dibenedetto and I. Tommasi, Energy Fuels, 2001, 15, 269 CrossRef CAS; (b) M. Aresta and M. Galatola, J. Cleaner Prod., 1999, 7, 181 CrossRef.
- (a) J. Ma, N. Sun, X. Zhang, N. Zhao, F. Xiao, W. Wei and Y. Sun, Catal. Today, 2009, 148, 221 CrossRef CAS; (b) S. Therdthianwong, A. Therdthianwong, C. Siangchin and S. Yongprapat, Int. J. Hydrogen Energy, 2008, 33, 991 CrossRef CAS; (c) N. A. S. Amin and T. C. Yaw, Int. J. Hydrogen Energy, 2007, 32, 1789 CrossRef CAS; (d) M. Rezaei, S. M. Alavi, S. Sahebdelfar and Z.-F. Yan, J. Nat. Gas Chem., 2006, 15, 327 CrossRef CAS.
- W.-L. Dai, S.-L. Luo, S.-F. Yin and C.-T. Au, Appl. Catal., A, 2009, 366, 2 CrossRef CAS.
- (a) R. Raudaskoski, M. Niemelä and R. Keiski, Top. Catal., 2007, 45, 57 CrossRef CAS; (b) X. Dong, H.-B. Zhang, G.-D. Lin, Y.-Z. Yuan and K. R. Tsai, Catal. Lett., 2003, 85, 237 CrossRef CAS; (c) K. Ushikoshi, K. Mori, T. Kubota, T. Watanabe and M. Saito, Appl. Organomet. Chem., 2000, 14, 819 CrossRef CAS.
- (a) T. Sakakura and K. Kohno, Chem. Commun., 2009, 1312 RSC; (b) E. Leino, P. Mäki-Arvela, V. Eta, D. Y. Murzin, T. Salmi and J. P. Mikkola, Appl. Catal., A, 2010, 383, 1 CrossRef CAS.
- (a) H. J. Hofmann, A. Brandner and P. Claus, Chem. Ing. Tech., 2011, 83, 1711 CrossRef CAS; (b) H. Lee, S. Park, I. Song and J. Jung, Catal. Lett., 2011, 141, 531 CrossRef CAS.
- (a) N. Kumar, E. Leino, P. Mäki-Arvela, A. Aho, M. Käldström, M. Tuominen, P. Laukkanen, K. Eränen, J.-P. Mikkola, T. Salmi and D. Y. Murzin, Microporous Mesoporous Mater., 2012, 152, 71 CrossRef CAS; (b) F. Gasc, S. Thiebaud-Roux and Z. Mouloungui, J. Supercrit. Fluids, 2009, 50, 46 CrossRef CAS; (c) O. Arbeláez, A. Orrego, F. Bustamante and A. Villa, Top. Catal., 2012, 55, 668 CrossRef.
- (a) A.-A. G. Shaikh and S. Sivaram, Chem. Rev., 1996, 96, 951 CrossRef CAS PubMed; (b) M. Berridge, D. Comar and C. Crouzel, Int. J. Appl. Radiat. Isot., 1983, 34, 1657 CrossRef CAS.
- S.-K. Jeong, M. Inaba, Y. Iriyama, T. Abe and Z. Ogumi, J. Power Sources, 2003, 119–121, 555 CrossRef CAS.
- N. Keller, G. Rebmann and V. Keller, J. Mol. Catal. A: Chem., 2010, 317, 1 CrossRef CAS.
- B. C. Dunn, C. Guenneau, S. A. Hilton, J. Pahnke, E. M. Eyring, J. Dworzanski, H. L. C. Meuzelaar, J. Z. Hu, M. S. Solum and R. J. Pugmire, Energy Fuels, 2002, 16, 177 CrossRef CAS.
- I. E. Muskat and F. Strain, Preparation of carbonic acid esters, US Pat., 2 379 250, 1941 Search PubMed.
- U. Romano, R. Tessi, G. Ciprianni and L. Micucci, Method for the preparation of Esters of Carbonic acids, US Pat., 4 218 391, 1980 Search PubMed.
- J. X. Zhen, S.-Y. Hua and C.-S. Hua, Catal. Lett., 2000, 69, 153 CrossRef CAS.
- D. Wang, B. Yang, X. Zhai and L. Zhou, Fuel Process. Technol., 2007, 88, 807 CrossRef CAS.
- (a) C. Murugan and H. C. Bajaj, Fuel Process. Technol., 2011, 92, 77 CrossRef CAS; (b) H.-P. Luo and W.-D. Xiao, Chem. Eng. Sci., 2001, 56, 403 CrossRef CAS.
- E. Leino, P. Mäki-Arvela, V. Eta, N. Kumar, F. Demoisson, A. Samikannu, A.-R. Leino, A. Shchukarev, D. Y. Murzin and J.-P. Mikkola, Catal. Today, 2013, 210, 47 CrossRef CAS.
- (a) E. Leino, N. Kumar, P. Mäki-Arvela, A. Aho, K. Kordás, A.-R. Leino, A. Shchukarev, D. Y. Murzin and J.-P. Mikkola, Mater. Chem. Phys., 2013, 143, 65 CrossRef CAS; (b) V. Eta, P. I. Mäki-Arvela, A.-R. Leino, K. N. Kordás, T. Salmi, D. Y. Murzin and J.-P. Mikkola, Ind. Eng. Chem. Res., 2010, 49, 9609 CrossRef CAS.
- M. Honda, S. Kuno, S. Sonehara, K.-I. Fujimoto, K. Suzuki, Y. Nakagawa and K. Tomishige, ChemCatChem, 2011, 3, 365 CrossRef CAS.
- (a) J.-C. Choi, K. Kohno, Y. Ohshima, H. Yasuda and T. Sakakura, Catal. Commun., 2008, 9, 1630 CrossRef CAS; (b) S. Hong, H. Park, J. Lim, Y.-W. Lee, M. Anpo and J.-D. Kim, Res. Chem. Intermed., 2006, 32, 737 CrossRef CAS; (c) K. Kohno, J.-C. Choi, Y. Ohshima, H. Yasuda and T. Sakakura, ChemSusChem, 2008, 1, 186 CrossRef CAS PubMed; (d) K. Tomishige and K. Kunimori, Appl. Catal., A, 2002, 237, 103 CrossRef CAS; (e) B. Fan, H. Li, W. Fan, J. Zhang and R. Li, Appl. Catal., A, 2010, 372, 94 CrossRef CAS.
- M. Honda, S. Kuno, N. Begum, K.-I. Fujimoto, K. Suzuki, Y. Nakagawa and K. Tomishige, Appl. Catal., A, 2010, 384, 165 CrossRef CAS.
- J.-C. Choi, L.-N. He, H. Yasuda and T. Sakakura, Green Chem., 2002, 4, 230 RSC.
- A. Dibenedetto, M. Aresta, A. Angelini, J. Ethiraj and B. M. Aresta, Chem. – Eur. J., 2012, 18, 10324 CrossRef CAS PubMed.
- C.-F. Li and S.-H. Zhong, Catal. Today, 2003, 82, 83 CrossRef CAS.
- (a) E. Leino, P. Mäki-Arvela, K. Eränen, M. Tenho, D. Y. Murzin, T. Salmi and J.-P. Mikkola, Chem. Eng. J., 2011, 176–177, 124 CrossRef CAS; (b) Y. Yoshida, Y. Arai, S. Kado, K. Kunimori and K. Tomishige, Catal. Today, 2006, 115, 95 CrossRef CAS.
- A. Dibenedetto, C. Pastore and M. Aresta, Catal. Today, 2006, 115, 88 CrossRef CAS.
- W. Wang, S. Wang, X. Ma and J. Gong, Catal. Today, 2009, 148, 323 CrossRef CAS.
- (a) K. Tomishige, Y. Ikeda, T. Sakaihori and K. Fujimoto, J. Catal., 2000, 192, 355 CrossRef CAS; (b) S. Xie and A. Bell, Catal. Lett., 2000, 70, 137 CrossRef CAS; (c) K. T. Jung and A. T. Bell, J. Catal., 2001, 204, 339 CrossRef CAS.
- M. Alifanti, B. Baps, N. Blangenois, J. Naud, P. Grange and B. Delmon, Chem. Mater., 2003, 15, 395 CrossRef CAS.
- P. Scherrer, Nachrichten von der Gesellschaft der Wissenschaften zu Göttingen, Mathematisch-Physikalische Klasse, 1918, 2, 98 Search PubMed.
- E. L. Crepaldi, G. J. de A. A. Soler-Illia, A. Bouchara, D. Grosso, D. Durand and C. Sanchez, Angew. Chem., Int. Ed., 2003, 42, 347 CrossRef CAS PubMed.
- W. Huang, J. Yang, C. Wang, B. Zou, X. Meng, Y. Wang, X. Cao and Z. Wang, Mater. Res. Bull., 2012, 47, 2349 CrossRef CAS.
- (a) L. Xu, H. Song and L. Chou, Int. J. Hydrogen Energy, 2012, 37, 18001 CrossRef CAS; (b) M. Yashima, K. Morimoto, N. Ishizawa and M. Yoshimura, J. Am. Ceram. Soc., 1993, 76, 2865 CrossRef CAS; (c) M. Yashima, K. Morimoto, N. Ishizawa and M. Yoshimura, J. Am. Ceram. Soc., 1993, 76, 1745 CrossRef CAS.
- (a) J. I. Gutiérrez-Ortiz, B. de Rivas, R. López-Fonseca and J. R. González-Velasco, Appl. Catal., A, 2004, 269, 147 CrossRef; (b) A. Kambolis, H. Matralis, A. Trovarelli and C. Papadopoulou, Appl. Catal., A, 2010, 377, 16 CrossRef CAS; (c) S. Abdollahzadeh-Ghom, C. Zamani, T. Andreu, M. Epifani and J. R. Morante, Appl. Catal., B, 2011, 108–109, 32 CrossRef CAS.
- M. Yashima, H. Arashi, M. Kakihana and M. Yoshimura, J. Am. Ceram. Soc., 1994, 77, 1067 CrossRef CAS.
- E. Aneggi, C. de Leitenburg, G. Dolcetti and A. Trovarelli, Catal. Today, 2006, 114, 40 CrossRef CAS.
- P. Fornasiero, R. Dimonte, G. R. Rao, J. Kaspar, S. Meriani, A. Trovarelli and M. Graziani, J. Catal., 1995, 151, 168 CrossRef CAS.
- D. Srinivas, C. V. V. Satyanarayana, H. S. Potdar and P. Ratnasamy, Appl. Catal., A, 2003, 246, 323 CrossRef CAS.
- T. Murota, T. Hasegawa, S. Aozasa, H. Matsui and M. Motoyama, J. Alloys Compd., 1993, 193, 298 CrossRef CAS.
- A. Trovarelli, Catal. Rev.: Sci. Eng., 1996, 38, 439 CAS.
- (a) B. de Rivas, R. López-Fonseca, C. Sampedro and J. I. Gutiérrez-Ortiz, Appl. Catal., B, 2009, 90, 545 CrossRef CAS; (b) B. de Rivas, C. Sampedro, M. García-Real, R. López-Fonseca and J. I. Gutiérrez-Ortiz, Appl. Catal., B, 2013, 129, 225 CrossRef CAS.
- C. Pojanavaraphan, A. Luengnaruemitchai and E. Gulari, Int. J. Hydrogen Energy, 2013, 38, 1348 CrossRef CAS.
- J. L. G. Fierro, J. Soria, J. Sanz and J. M. Rojo, J. Solid State Chem., 1987, 66, 154 CrossRef CAS.
- W. Khaodee, B. Jongsomjit, S. Assabumrungrat, P. Praserthdam and S. Goto, Catal. Commun., 2007, 8, 548 CrossRef CAS.
- S. Wang, L. Zhao, W. Wang, Y. Zhao, G. Zhang, X. Ma and J. Gong, Nanoscale, 2013, 5, 5582 RSC.
- K. Tomishige, H. Yasuda, Y. Yoshida, M. Nurunnabi, B. Li and K. Kunimori, Green Chem., 2004, 6, 206 RSC.
- L. C. W. Baker and T. F. Anderson, J. Am. Chem. Soc., 1957, 79, 2071 CrossRef CAS.
- S. Takishima, K. Saiki, K. Arai and S. Saito, J. Chem. Eng. Jpn., 1986, 19, 48 CrossRef CAS.
- J. S. Lim, Y. Y. Lee and H. S. Chun, J. Supercrit. Fluids, 1994, 7, 219 CrossRef CAS.
- S.-D. Yeo, S.-J. Park, J.-W. Kim and J.-C. Kim, J. Chem. Eng. Data, 2000, 45, 932 CrossRef CAS.
- (a) L. A. Galicia-Luna, A. Ortega-Rodriguez and D. Richon, J. Chem. Eng. Data, 2000, 45, 265 CrossRef CAS; (b) C. Secuianu, V. Feroiu and D. Geană, J. Supercrit. Fluids, 2008, 47, 109 CrossRef CAS.
- Z.-F. Zhang, Z.-W. Liu, J. Lu and Z.-T. Liu, Ind. Eng. Chem. Res., 2011, 50, 1981 CrossRef CAS.
- Q. Cai, B. Lu, L. Guo and Y. Shan, Catal. Commun., 2009, 10, 605 CrossRef CAS.
- H. J. Hofmann, A. Brandner and P. Claus, Chem. Eng. Technol., 2012, 35, 2140 CrossRef CAS.
- Y. Ikeda, M. Asadullah, K. Fujimoto and K. Tomishige, J. Phys. Chem. B, 2001, 105, 10653 CrossRef CAS.
- I. Yoshiki, F. Yutaka, T. Keiichi and F. Kaoru, in CO2 Conversion and Utilization, American Chemical Society, 2002, vol. 809, p. 71 Search PubMed.
- S. Wada, K. Oka, K. Watanabe and Y. Izumi, Front. Chem., 2013, 1, 1 CAS.
- G. Soave, Chem. Eng. Sci., 1972, 27, 1197 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2015 |