
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
On the tunneling instability of a hypercoordinated carbocation†
Sebastian
Kozuch

Chemistry Department, Ben-Gurion University of the Negev, Beer-Sheva 84105, Israel. E-mail: kozuch@bgu.ac.il
First published on 26th May 2015
Abstract
C(CH3)5+, a pentacoordinate carbocation, was recently described as a “fleeting” molecule, that is, stable only at low temperatures. Herein it is shown using theoretical methods that, owing to carbon tunneling, the molecule is not persistent even at 0 K. One possible way to lengthen its lifespan is by deuterium substitution, in spite of the fact that no bond to hydrogen is broken.
A recent communication exploring the limits of carbon bonding by McKee, Schleyer and coworkers1 described the theoretical existence of C(CH3)5+ near absolute zero as “fleeting”.2 This term was coined by Hoffmann, Schleyer and Schaeffer in their essay: “Predicting Molecules—More Realism, Please!”, indicating a molecule where the sole energetic criterion of its stability is a vibrational analysis (thus being a real minimum in the potential energy surface).
C(CH3)5+, a C3h pentacoordinate carbocation, is more a proof of concept for the feasible bonding possibilities of carbon rather than a molecule with real perspectives to be synthesized. Although the “classical” thermal barriers suggest that this carbocation might be detectable at extremely low temperatures, herein it is shown that due to quantum mechanical tunneling (QMT) the existence of C(CH3)5+ is much more ephemeral than was considered, even at absolute zero.3,4
The two decomposition pathways for C(CH3)5+ (described in detail in the original communication1) are shown in Fig. 1. The first pathway, going through a C3 symmetry transition state (TS1), involves a CH3+ loss by elongation of an axial methyl C–C bond, followed by a hydrogen abstraction and CH3 migration, generating a relatively stable tertiary carbocation. This process has a Δ(E + ZPE)‡ value of 1.65 and a ΔE‡ value of 4.35 kcal mol−1 (calculated at the CCSD(T)/CBS level, as reported in the original communication1). The second decomposition pathway, via a transition state of Cs symmetry (TS2), involves a bending mode joining an axial and an equatorial carbon to give ethane and a tert-butyl carbocation, with Δ(E + ZPE)‡ = 1.37 and ΔE‡ = 2.32 kcal mol−1. Thus, despite these extremely low degradation barriers, transition state theory in its simplest form predicts that the cation should have a fleeting,2 possibly observable existence near absolute zero.
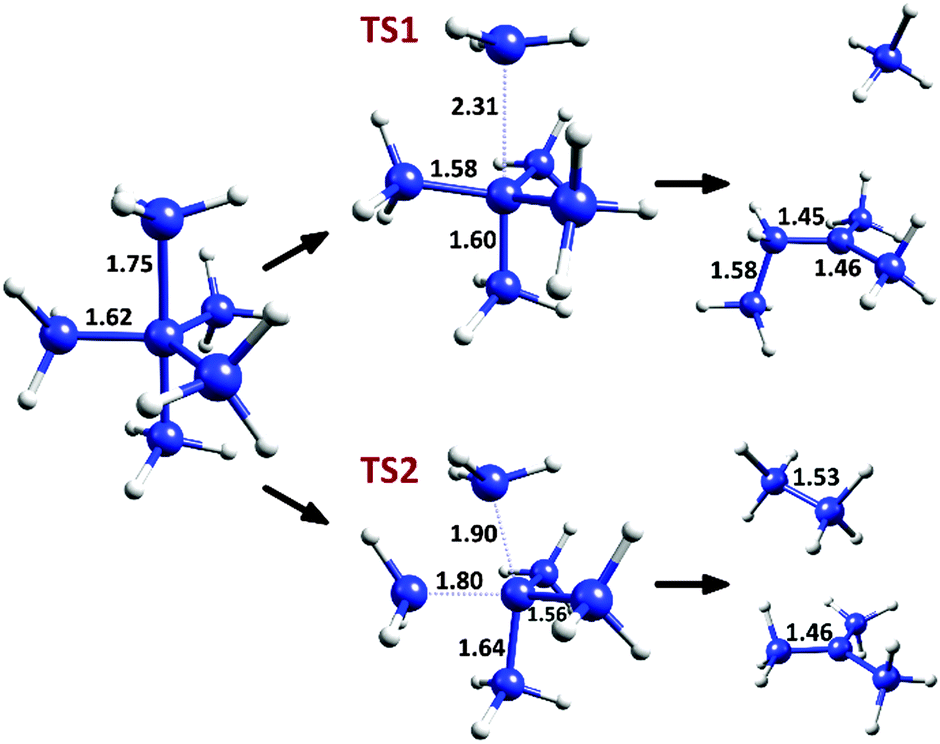 | ||
| Fig. 1 Decomposition pathways for C(CH3)5+, with selected C–C bond distances at the M06-2X/6-31G(d) level. | ||
However, due to the low and (more critical) narrow3–6 barriers in the decomposition patterns of C(CH3)5+, the tunneling effect may be a crucial factor to take into account, even if QMT of “heavy” atoms is involved. Carbon tunneling is an unusual but well documented effect, which has been observed or calculated in reactions such as the automerization of cyclobutadiene and other antiaromatic molecules (starting from the pioneering work of Carpenter),5,7–13 or in different carbene insertion reactions and other reactive systems.6,14–17 All these systems have something in common: low and considerably narrow barriers. In the decomposition of C(CH3)5+ the carbon atoms travel over great distances, but the effective barrier for tunneling is tight (that is, from the reactant stage to the point of the same energy in the product side of the reaction coordinate), and therefore may be prone to reaction by a QMT mechanism.
It must be pointed out that QMT on the related CH5+ carbocation has been thoroughly studied (see for instance ref. 18). However, the differences between both systems are significant. For CH5+, the tunneling concerns only the hydrogens (“light atom” QMT) mostly in its fluxionality;19 for C(CH3)5+, tunneling will primarily affect the carbon structure (“heavy atom” QMT) in its chemical degradation.
To study the effect of quantum mechanical tunneling on the degradation of C(CH3)5+, we used canonical variational theory (CVT) to compute the semi-classical rate constant (without the tunneling component),20 and the small curvature tunneling approximation (SCT)21 including the quantized reactant state tunnelling (QRST) approximation22 to compute the rate constant including tunneling. The polyrate23 program was used for this effect, and Gaussian0924 was employed for the DFT calculations (with Gaussrate25 as the interface between Polyrate and Gaussian). Since high-quality, wave-function-based calculations are not practical for the demanding SCT calculations, all rate constant calculations were carried out using the M06-2X functional26 and the 6-31G(d) basis set. The combination of this functional and basis set has shown good accuracy for tunneling calculations of organic reactions,12,26 and reasonably agrees with the CCSD(T)/CBS methodology, generating values of 2.27 and 2.16 kcal mol−1 for Δ(E + ZPE)‡ of TS1 and TS2, respectively.
Table 1 shows the calculated rate constants for both decomposition pathways with (SCT) and without (CVT) tunneling effect included, and the SCT half-life at different temperatures (see the ESI† for the complete tables).
| T (K) | TS1 | TS2 | ||||
|---|---|---|---|---|---|---|
| CVT | SCT | t 1/2 | CVT | SCT | t 1/2 | |
| a Due to the change in the symmetry number of TS2 (going from a C3h reactant to a Cs transition state), the rate constants for this pathway were tripled and t1/2 were divided by 3.27 | ||||||
| 10 | 1.2 × 10−39 | 1.2 × 105 | 6 × 10−6 | 6.8 × 10−36 | 1.5 × 104 | 4 × 10−5 |
| 20 | 3.2 × 10−14 | 1.2 × 105 | 6 × 10−6 | 4.2 × 10−12 | 1.6 × 104 | 4 × 10−5 |
| 50 | 1.0 × 102 | 2.2 × 105 | 3 ×10−6 | 1.5 × 103 | 5.6 × 105 | 1 × 10−6 |
| 100 | 2.8 × 107 | 8.7 × 107 | 8 × 10−9 | 2.2 × 108 | 7.0 × 108 | 1 × 10−9 |
| 150 | 2.5 × 109 | 4.2 × 109 | 2 × 10−10 | 1.5 × 1010 | 2.2 × 1010 | 3 × 10−11 |
| 200 | 2.6 × 1010 | 3.6 × 1010 | 2 × 10−11 | 1.5 × 1011 | 1.6 × 1011 | 4 × 10−12 |
| 300 | 3.1 × 1011 | 3.6 × 1011 | 2 × 10−12 | 1.6 × 1012 | 1.6 × 1012 | 5 × 10−13 |
It is clear that the stability of C(CH3)5+ is strongly influenced by carbon tunneling, with extremely high SCT rate constants, even at cryogenic temperatures. Half-lives (t1/2 = ln![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 2/k) of only 6 and 40 μs for the decomposition of C(CH3)5+ were computed for pathways 1 and 2, respectively, below 20 K. Without the inclusion of QMT, the reaction is predicted to take several millennia to occur even at 20 K, thus confirming the fleeting characteristics of the system based on transition state theory alone (i.e. without considering QMT).
2/k) of only 6 and 40 μs for the decomposition of C(CH3)5+ were computed for pathways 1 and 2, respectively, below 20 K. Without the inclusion of QMT, the reaction is predicted to take several millennia to occur even at 20 K, thus confirming the fleeting characteristics of the system based on transition state theory alone (i.e. without considering QMT).
Although the whole QMT picture is more complex than the analysis of the motion of individual particles (since the reduced mass of the whole system in the least action pathway must be considered28,29), the influence of each atom can still be analyzed by studying the carbon (12C/13C) and hydrogen (H/D) kinetic isotope effects (KIE) on the SCT rate constant.
Fig. 2 shows the KIE of each inequivalent atom on the TS1 pathway at cryogenic temperatures. The carbon of the departing methyl group is the most crucial carbon atom for the QMT process with a KIE of 1.72, but the central carbon also exerts a significant effect (KIE of 1.20). Surprisingly, the lower axial carbon provides a negative secondary KIE of 0.94. The KIE after substituting all the carbons by 13C is 2.03, virtually identical to the product of all the individual substitutions (1.72 × 1.20 × 0.94 × 1.023 = 2.05). Thus, there is a largely independent contribution of each carbon to the tunneling probability.
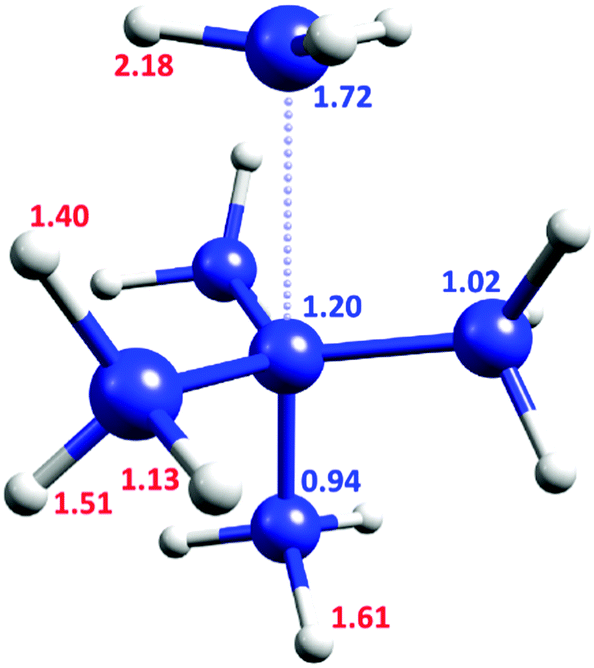 | ||
| Fig. 2 TS1 geometry, with the KIE values for inequivalent atoms in C3 symmetry. Blue values are for 12C/13C KIE, red for H/D KIE. | ||
However, the contribution of each H/D substitution is not totally independent. Replacing all of the hydrogen atoms with deuterium results in a KIE of 613. Since no C–H bond is broken, this large value is remarkable. Moreover, it is higher than the product of individual contributions (2.183 × 1.613 × 1.513 × 1.403 × 1.133 = 592, see Fig. 2), suggesting a small synergistic hydrogen contribution to the tunneling probability.
The high H/D KIE value goes beyond the effect of the higher mass of the particles on the tunneling probability. As pointed out by a reviewer, more than being exclusively a carbon tunneling process, the process can be considered as a methyl group tunneling, since the hydrogens connected to the dissociating carbon move as well. In that sense, the mass change of one H/D substitution on the departing CH3 group should be as important for the tunnelling probability as a 13C/12C substitution on the same group. However, the H/D replacement produces a significantly higher KIE than the carbon replacement (2.18 vs. 1.72). Moreover, all the other hydrogens have a strong influence on the rate, while other carbons do not. This is despite the fact that these hydrogens are not directly “involved” in the C–C breaking process (at least not in the reaction section important to QMT). More than the direct effect of the mass, what is strongly influencing the tunneling rate is a factor that is traditionally connected to a change in the semi-classical KIE, the zero point energy. The ZPE change is also a mass effect, but the extra mass indirectly affects the QMT probability through a higher and broader barrier.
The additive lowering of the ZPE of the reactant by each H/D substitution increases the Δ(E + ZPE)‡ value and reduces the tunneling probability. One H/D axial methyl group substitution raises the Δ(E + ZPE)‡ value from 2.27 to 2.39 kcal mol−1, while the replacement of all 15 H's by D's raises the Δ(E + ZPE)‡ value to 2.99 kcal mol−1 for decomposition viaTS1. Considering that this ZPE difference is a major component relative to the activation energy, the lower ZPE of the reactant in deuterated systems significantly affects the tunneling-based KIE30 of the degradation of C(CH3)5+ by raising and broadening the barrier, in addition to the increase of the methyl mass. Contrary to this, the 12C/13C substitution does not change the ZPE appreciably, and therefore its KIE only has a direct mass effect on the tunneling probability.
Notably, this change in the ZPE greatly extends the lifetime of C(CD3)5+. Its computed half-life of 4 ms is three orders of magnitude longer than that of the C(CH3)5+ isotopologue. Replacing all the hydrogen atoms with tritium would lead to a Δ(E + ZPE)‡ value of 3.31 kcal mol−1 and a still brief, but drastically longer, half-life of 0.2 seconds. The Δ(E + ZPE)‡ values for decomposition through TS2 are less sensitive to an isotopic replacement, with values of 2.16, 2.41 and 2.53 kcal mol−1 for C(CH3)5+, C(CD3)5+ and C(CT3)5+, respectively. Nevertheless, due to the wider barriers, the half-lives of these species considering only decomposition through TS2 are longer than those viaTS1: 40 μs, 60 ms, and 10 s. This technique of prolonging the lifetime and stabilizing a molecule prone to degradation by QMT with H/D substitutions is, to the best of our knowledge, unheard of in reactions not based on hydrogen migration.
Conclusions
Considering the short lifetime of the hypothetical C(CH3)5+ pentacoordinate carbocation, the definition of a “fleeting system”2 as one that can be detected at extremely low temperatures may require revision. Molecules that have a low but definite decomposition barrier (with all positive frequencies in a vibrational analysis) might be expected to be persistent close to 0 K, but QMT calculations may show otherwise. Therefore, is C(CH3)5+ “fleeting” or not? This semantic problem may be solved by adding a new term, “tunneling stability” (similar to the concept of “tunneling control”31) to the already established “kinetic persistence” and “thermodynamic stability” definitions. Indeed, other molecules predicted to be fleeting32,33 have been computed to be unstable due to tunneling,34 indicating that QMT should be a factor to take into account in stability analyses.In this sense, the decomposition of C(CH3)5+ through QMT by no means refutes the hypothetical existence of the five-fold C–C bonding motif of C(CH3)5+ as predicted by Schleyer et al.1 However, we now know that it will probably be impossible to detect this cation experimentally due to its exceptionally short life expectancy.
Interestingly, deuterium or tritium substitution is predicted to extend the lifespan of C(CH3)5+ much more than 13C substitution, in spite of the fact that we are breaking C–C and not C–H bonds. The added mass is only one factor for this extra longevity, which is mainly caused by the effect of hydrogen isotopes on the ZPE.
One can only wonder how many other fleeting systems have been theoretically predicted but will not pass the tunneling test. Or, quoting the title of the essay of Hoffmann, Schleyer and Schaeffer,2 we may say: “Predicting Molecules—More Realism, Please!”.
Acknowledgements
This research project begun at the University of North Texas, in the Department of Chemistry and Center for Advanced Scientific Computing and Modeling (CASCaM), and finished at Ben-Gurion University. I thank Jay Agarwal for helpful discussions and for aiding in the writing of this manuscript, Professor Weston T. Borden for his chemical insight, and Dr David A. Hrovat for technical and professional help. This communication is dedicated to the memory of Professor Paul von Ragué Schleyer, who actively encouraged this project and the study of chemical stability in all their dimensions.Notes and references
- W. C. McKee, J. Agarwal, H. F. Schaefer and P. v. R. Schleyer, Angew. Chem., Int. Ed., 2014, 53, 7875–7878 CrossRef CAS PubMed.
- R. Hoffmann, P. v. R. Schleyer and H. F. Schaefer, Angew. Chem., Int. Ed., 2008, 47, 7164–7167 CrossRef PubMed.
- R. P. Bell, The tunnel effect in chemistry, Chapman and Hall, London, New York, 1980 Search PubMed.
- M. Razavy, Quantum theory of tunneling, World Scientific, River Edge, NJ, 2003 Search PubMed.
- B. K. Carpenter, J. Am. Chem. Soc., 1983, 105, 1700–1701 CrossRef CAS.
- S. Kozuch, Phys. Chem. Chem. Phys., 2014, 16, 7718–7727 RSC.
- M. J. Huang and M. Wolfsberg, J. Am. Chem. Soc., 1984, 106, 4039–4040 CrossRef CAS.
- M. J. S. Dewar, K. M. Merz and J. J. P. Stewart, J. Am. Chem. Soc., 1984, 106, 4040–4041 CrossRef CAS.
- P. Čársky, R. J. Bartlett, G. Fitzgerald, J. Noga and V. Špirko, J. Chem. Phys., 1988, 89, 3008–3015 CrossRef PubMed.
- R. Lefebvre and N. Moiseyev, J. Am. Chem. Soc., 1990, 112, 5052–5054 CrossRef CAS.
- Kinetics and spectroscopy of carbenes and biradicals, ed. M. Platz, Plenum Press, New York, 1990 Search PubMed.
- S. Kozuch, RSC Adv., 2014, 4, 21650–21656 RSC.
- S. Kozuch, DOI: 10.1021/acs.jctc.5b00321.
- P. S. Zuev, R. S. Sheridan, T. V. Albu, D. G. Truhlar, D. A. Hrovat and W. T. Borden, Science, 2003, 299, 867–870 CrossRef CAS PubMed.
- R. A. Moss, R. R. Sauers, R. S. Sheridan, J. Tian and P. S. Zuev, J. Am. Chem. Soc., 2004, 126, 10196–10197 CrossRef CAS PubMed.
- S. Kozuch, X. Zhang, D. A. Hrovat and W. T. Borden, J. Am. Chem. Soc., 2013, 135, 17274–17277 CrossRef CAS PubMed.
- J. Meisner, J. B. Rommel and J. Kästner, J. Comput. Chem., 2011, 32, 3456–3463 CrossRef CAS PubMed.
- D. Marx and M. Parrinello, Science, 1999, 284, 59–61 CrossRef CAS.
- P. R. Schreiner, S.-J. Kim, H. F. Schaefer and P. v R. Schleyer, J. Chem. Phys., 1993, 99, 3716–3720 CrossRef CAS PubMed.
- D. G. Truhlar and B. C. Garrett, Annu. Rev. Phys. Chem., 1984, 35, 159–189 CrossRef CAS.
- A. Fernandez-Ramos, B. A. Ellingson, B. C. Garrett and D. G. Truhlar, in Reviews in Computational Chemistry, ed. K. B. Lipkowitz and T. R. Cundari, John Wiley & Sons, Inc., 2007, vol. 3, pp. 125–232 Search PubMed.
- J. G. Lauderdale and D. G. Truhlar, Surf. Sci., 1985, 164, 558–588 CrossRef CAS.
- J. Zheng, S. Zhang, B. J. Lynch, J. C. Corchado, Y.-Y. Chuang, P. L. Fast, W.-P. Hu, Y.-P. Liu, G. C. Lynch, K. A. Nguyen, C. F. Jackels, A. Fernandez Ramos, B. A. Ellingson, V. S. Melissas, J. Villà, I. Rossi, E. L. Coitiño, J. Pu, T. V. Albu, R. Steckler, B. C. Garrett, A. D. Isaacson and D. G. Truhlar, POLYRATE 2010-A: Computer Program for the Calculation of Chemical Reaction Rates for Polyatomics.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision B.1, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- J. Zheng, S. Zhang, J. C. Corchado, Y.-Y. Chuang, B. A. Ellingson, E. L. Coitiño and D. G. Truhlar, GAUSSRATE 2009-A, 2000 Search PubMed.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 CrossRef CAS.
- A. Fernández-Ramos, B. A. Ellingson, R. Meana-Pañeda, J. M. C. Marques and D. G. Truhlar, Theor. Chem. Acc, 2007, 118, 813–826 CrossRef PubMed.
- B. C. Garrett and D. G. Truhlar, J. Chem. Phys., 1983, 79, 4931 CrossRef CAS PubMed.
- R. Meana-Pañeda, D. G. Truhlar and A. Fernández-Ramos, J. Chem. Theory Comput., 2010, 6, 6–17 CrossRef.
- X. Zhang, A. Datta, D. A. Hrovat and W. T. Borden, J. Am. Chem. Soc., 2009, 131, 16002–16003 CrossRef CAS PubMed.
- D. Ley, D. Gerbig and P. R. Schreiner, Org. Biomol. Chem., 2012, 10, 3781 CAS.
- G. Martínez-Guajardo, K. J. Donald, B. K. Wittmaack, M. A. Vazquez and G. Merino, Org. Lett., 2010, 12, 4058–4061 CrossRef PubMed.
- G. Markopoulos and J. Grunenberg, Angew. Chem., Int. Ed., 2013, 52, 10648–10651 CrossRef CAS PubMed.
- S. Kozuch, Org. Lett., 2014, 16, 4102–4105 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Complete CVT/SCT rate constant tables, KIE tables, geometries, absolute energies, an example of a Polyrate input, and Vmep and VaG for the root reaction of the TS1 pathway. See DOI: 10.1039/c5cp02080h |
| This journal is © the Owner Societies 2015 |