
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelective ion-permeable membranes by insertion of biopores into polymersomes†
Mihai
Lomora‡
,
Fabian
Itel‡
,
Ionel Adrian
Dinu
and
Cornelia G.
Palivan
*
Department of Chemistry, University of Basel, Klingelbergstrasse 80, CH-4056 Basel, Switzerland. E-mail: cornelia.palivan@unibas.ch; Fax: +41 (0)61 267 38 55; Tel: +41 (0)61 267 38 39
First published on 25th February 2015
Abstract
In nature there are various specific reactions for which highly selective detection or support is required to preserve their bio-specificity or/and functionality. In this respect, mimics of cell membranes and bio-compartments are essential for developing tailored applications in therapeutic diagnostics. Being inspired by nature, we present here biomimetic nanocompartments with ion-selective membrane permeability engineered by insertion of ionomycin into polymersomes with sizes less than 250 nm. As a marker to assess the proper insertion and functionality of ionomycin inside the synthetic membrane, we used a Ca2+-sensitive dye encapsulated inside the polymersome cavity prior to inserting the biopore. The calcium sensitive dye, ionomycin, and Ca2+ did not influence the architecture and the size of polymersomes. Successful ionomycin functionality inside the synthetic membrane with a thickness of 10.7 nm was established by a combination of fluorescence spectroscopy and stopped-flow spectroscopy. Polymersomes equipped with ion selective membranes are ideal candidates for the development of medical applications, such as cellular ion nanosensors or nanoreactors in which ion exchange is required to support in situ reactions.
Introduction
In nature, both cells, and inner organelles as biocompartments preserve their functions and communicate by signaling with their environment via a permeable lipid membrane in which membrane proteins mediate the transport of molecules, which are vital for cell metabolism. Compartmentalization represents an extremely efficient organization of membranes and biomolecules that is needed to cope with a complex scenario of metabolic reactions in a confined space, and whose stability and functions are vital for life.1 Taking inspiration from natural biocompartments, significant efforts have been made to produce compartments that mimic cells and organelles, either in terms of their membrane properties or of the reactivity of encapsulated biomolecules.2–4 Whereas lipid based compartments (liposomes) are straightforward systems for mimicking a cell/organelle membrane, and encapsulating biomolecules to perform specific reactions,5 their mechanical instability and the presence of membrane defects limit applications that require robust compartments.6 One elegant way to avoid these drawbacks, and at the same time to add new membrane properties, such as stimuli-responsiveness or external functionalization for targeting approaches, is to use compartments generated from amphiphilic block copolymers. A particularly interesting way to generate polymer compartments with sizes in the nanometer range is to self-assemble amphiphilic copolymers with appropriate chemical and physical properties.7 Indeed, polymer vesicles (polymersomes) are spherical 3-D compartments surrounded by a synthetic membrane with greater stability than liposomes.8The architecture of polymersomes offers three different regions for combination with biomolecules: the inner cavity for encapsulating hydrophilic molecules, the membrane for insertion of hydrophobic molecules, and the external surface for attachment of molecules, by chemical modification of the hydrophilic polymer block.9,10 From a topologic point of view, polymersomes are mimics of cellular compartments, which by combination with active compounds can also be designed to act and communicate similarly to intracellular organization. As a result of simultaneously possessing hydrophilic and hydrophobic molecules in a single compartment, various applications can be developed based on polymersomes. These range from drug carriers,11–13 and containers for contrast agents,14 up to nanoreactors,15–18 and artificial organelles.19 In addition, in a biomimetic approach, multicompartmentalized polymersomes have been introduced to allow reactions between different inner compartments encapsulated in a giant compartment.20–22 A key factor for controlling in situ reactions (either in a single compartment or in multi-compartments) is the permeability of the polymersome membrane,23 which has to allow transport of molecules required for the reaction (substrates of enzymes, products). This can be achieved in various ways by: (i) using a block copolymer, which forms a porous membrane,24 (ii) chemical modification of the membrane to create pores,25,26 (iii) blending block copolymers with lipids followed by their extraction,27 (iv) insertion of biopores,28 and (v) reconstitution of channel porins.15,29–31
The permeabilization of synthetic membranes by intrinsic or induced pores supports penetration of molecules with sizes smaller than the dimension of the pores, but they are non-selective in terms of specific molecules that are allowed to pass through. A strategy to introduce selective transport of ions/molecules through membranes is to insert biopores with specific functionality, as for example ionophores. A first example of a permeabilized membrane of giant polymer vesicles (GUVs) for calcium ions was obtained by insertion of selective and non-selective ionophores, including Lasalocid A, Alamethicin (U22324), and N,N-dicyclohexyl-N′,N′-dioctadecyl-3-oxapentane-1,5-diamide (ETH5234).32 GUV membranes were permeabilized towards calcium ions by insertion of calcimycin A23187 as well, and served for development of a microreactor.33 While interesting as a microreactor model, the system is not appropriate for medical applications, due both to the size and instability of GUVs, and the use of calcimycin A23187, with a medium efficiency in ion transport.34
Introduction of selectivity of polymersome membranes should support specific reactions inside the cavities, or the development of nano-biosensors for molecules selected by their specific permeability. In particular, biosensors are in focus as diagnostic systems, because they can sense changes in biological responses, which are then converted to detectable and measurable signals.35,36 Such sensors based on nanoreactors have been developed recently, but only a few examples have been reported, either in solution37 or immobilized on surfaces.38 However, these sensors are based on the specificity of active compounds encapsulated inside nanoreactors, and are not mediated by a selective membrane, as is the case in natural biocompartments.
Here, we introduce polymersomes with sizes in the nanometer range, and membranes engineered to be selectively permeable to divalent ions in order to support applications that require transport of a desired ion through the membrane, such as for specific in situ reactions or nano-sensors. We selected poly(2-methyloxazoline)-block-poly(dimethylsiloxane)-block-poly(2-methyloxazoline) (PMOXAx–PDMSy–PMOXAx) for vesicle formation, because this type of amphiphilic triblock copolymer self-assembles in polymersomes when appropriate molecular properties (block length, hydrophilic to hydrophobic ratio), and preparation conditions (method of preparation, buffer) are used.15 The membranes of such polymersomes are both stable and impermeable to ions and small molecules,39 except oxygen species.40 Various channel porins can be inserted into this type of membrane; for example outer membrane protein F (OmpF), which induces non-selective permeability for molecules ≤ 600 Da.15
As a model biopore, able to induce selective membrane permeability, we selected ionomycin, a carboxylic acid ionophore (divalent anion polyether, MW 787.08 Da), produced by Streptomyces conglobatus. Ionomycin allows specific transport of divalent ions, such as Ca2+,41–43 which we selected because it plays a pivotal role in biological systems where many cellular responses are regulated by an ON and OFF mechanism that is strongly correlated to the intracellular Ca2+ concentration.44 In nature, calcium signaling is initiated by cells using both intra- and extracellular sources of Ca2+, which are controlled by channels, pumps and exchangers to provide cellular calcium homeostasis for the cell integrity. Any perturbation in the calcium regulating gates results in channelopathies correlated with severe diseases, such as muscle or neurological disorders.45,46 The advantages of ionomycin compared to other ionophores, as for example calcimycin, are a more efficient Ca2+ transport,34 and its lack of fluorescence, allowing a non-interfering use with fluorescent molecules.34,47 A combination of light scattering (LS) and transmission electron microscopy (TEM) has been used to characterize the formation of biopore-equipped polymersomes and their stability. Both fluorescence and stopped-flow spectroscopy were used to evaluate in details whether there was functional insertion of the biopore, and thus selective permeabilization of the membrane. In a systematic manner we took into account the factors affecting the system, as for example the presence of solvent, the gradient of ionomycin or the gradient of Ca2+ concentration in order to obtain the optimum conditions for selective membrane permeabilization without affecting the architecture of polymersomes.
Our ion-selective permeable polymersome (Fig. 1) represents progress in the development of mimics of a cell membrane, and supports fine modulation of reactions inside the cavity or the functionality of biosensors based on ion specificity. The system represents an optimized candidate towards medical applications (in terms of vesicles size, efficacy of ionophore and the improved stability of the polymer membrane).
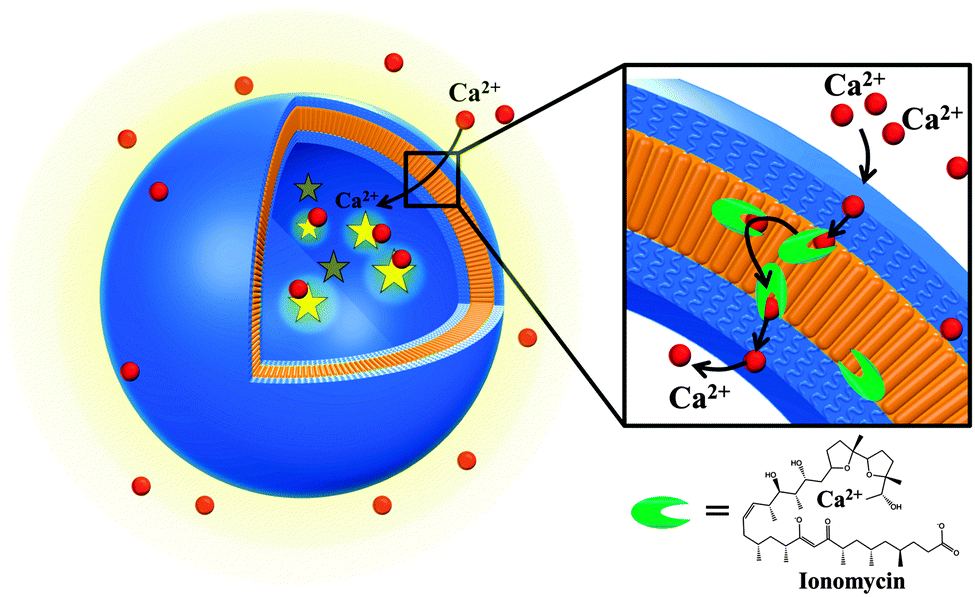 | ||
| Fig. 1 Design of Ca2+-selective permeable membranes by insertion of ionomycin into polymersomes. | ||
Experimental
Materials and methods
Asante Calcium Green (ACG) K+ salt, was purchased from TEFLabs Inc, ionomycin from Alomone Labs, and N-(2-hydroxyethyl)piperazine-N′-(2-ethanesulfonic acid) sodium salt (HEPES), sodium chloride (NaCl), calcium chloride dihydrate (CaCl2·2H2O), and ethanol (EtOH) from Sigma. All reagents and materials were used as received.Preparation of polymersomes and insertion of ionomycin
Polymersomes were prepared by the film rehydration method48 at room temperature using a PMOXA6–PDMS44–PMOXA6 triblock copolymer.49 Briefly, for each sample, 5 mg of the block copolymer was dissolved in 1 mL of ethanol and subsequently evaporated under reduced pressure (150 mbar) to render a thin film. This film was then rehydrated with: (i) 1 mL HEPES buffer (20 mM HEPES, 50 mM NaCl, pH = 7.4) (empty polymersomes), or (ii) 1 mL of 20 μM ACG solution in HEPES buffer (polymersomes with entrapped ACG). The polymer suspensions were stirred overnight at room temperature (RT) and afterwards extruded (13 times) through a 200 nm pore-size polycarbonate (PC) membrane (Merck, Germany), using an Avanti mini-extruder (Avanti Polar Lipids, USA). Extravesicular dye was separated from polymersomes that contained entrapped dye by passage through a 20 cm3 in-house prepacked size exclusion column (Sepharose 2B, Sigma Aldrich). Ionomycin was added to the polymersomes mixture from a stock solution of ionomycin in EtOH (1.17 mM). Ionomycin molecular dimensions were determined using Visual Molecular Dynamics (VMD) software.50Efficiency of the ACG entrapment process and ionomycin insertion in polymersomes
The concentration of encapsulated ACG in polymersomes was determined by UV-Vis spectroscopy at λ = 517 nm. For this, a value of 24![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 310 M−1 cm−1 for the extinction coefficient of ACG in HEPES buffer was obtained based on a calibration curve for the dye. The entrapment efficiency of the process (EEP) was determined as a percentage of the ratio between the concentration of ACG in polymersomes after encapsulation and purification (cep) and the initial concentration of ACG used for the film rehydration (ci):48
310 M−1 cm−1 for the extinction coefficient of ACG in HEPES buffer was obtained based on a calibration curve for the dye. The entrapment efficiency of the process (EEP) was determined as a percentage of the ratio between the concentration of ACG in polymersomes after encapsulation and purification (cep) and the initial concentration of ACG used for the film rehydration (ci):48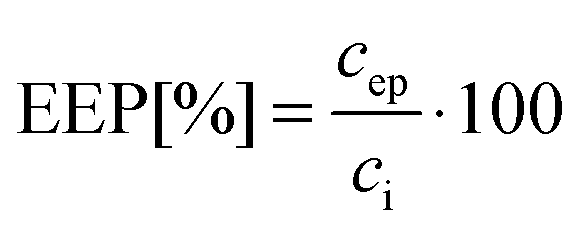 | (1) |
Preparation of giant unilamellar vesicles (GUVs)
For the generation of giant unilamellar polymeric vesicles (GUVs), 30 μL of the polymer dissolved in EtOH (4 mg mL−1) was spread on an ITO-coated glass plate. The polymer solution was dried in high vacuum for 2 h in order to form a smooth polymer film on the glass surface. A chamber was formed with an O-ring and filled with 100 mM sucrose solution containing 20 μM of ACG. The chamber was closed with a second ITO-coated glass plate facing the ITO side to the solution, and then exposed to an AC current (2.5 V, 3.0 Hz) overnight at 20 °C.Characterization techniques
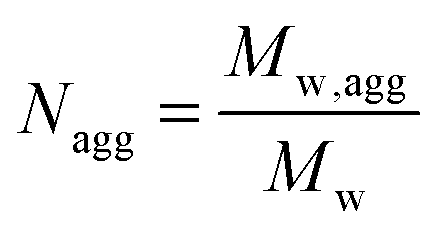 | (2) |
| A(t) = ΔA1·exp(k1·t) + ΔA2·exp(k2·t) + A∞ | (3) |
For FCS measurements, the diameter of the pinhole was set to 74 μm, and the diffusion time for ACG (25 nM) in Hepes buffer was fixed in the fitting procedure. Empty polymersomes were incubated overnight with 20 μM ACG at RT, then 10 μL of these solutions were placed on microscopy glass slides, and FCS measured at an excitation transmission of 10% over 20 × 10 s. Autocorrelation functions were fitted with a two-component model including triplet state.51 From the two-component fitting model, the percentages of fast-diffusing (free ACG) and slow-diffusing fractions (ACG-containing polymersomes) were obtained.
Z-scan FCS. GUV membranes were stained with Bodipy 630/650 NHS ester (Thermo Fisher Scientific Inc; Waltham, MA, USA) by the addition of 5 μL of a 25 nM Bodipy stock solution in buffer to the immobilized GUVs on the glass surface. Z-scans were recorded as previously described,49 except that a He–Ne laser was used (λ = 633 nm, 1% transmission at 15 mW).
Results and discussion
The process of generation of polymersomes with selective permeable membranes (Polsp) by insertion of biopores has to preserve both the architecture of the polymer vesicles and the functionality of the biopores which, depending on their functionality, should span or cross the entire membrane. The membrane permeabilization upon insertion of ionophores results from a selective transport of ions, which is mediated by the ionophore through the membrane. In this respect, we selected PMOXA6–PDMS44–PMOXA6 copolymers with a membrane thickness of 10.7 nm (almost twice as thick as a lipid bilayer)49 and ionomycin, a model ionophore for efficient membrane permeabilization for Ca2+ ions.Architecture of polymersomes before and after ionomycin insertion
We combined light scattering (LS) and transmission electron microscopy (TEM) to evaluate the formation of polymersomes using specific conditions aimed at not affecting ionomycin functionality. We were interested in establishing whether the engineering of Polsp is influenced in terms of polymersomes size, shape and stability by the components of the system: i.e. whether the selected calcium sensitive dye (ACG), ionomycin used for polymer membranes permeabilization, or the presence of calcium ions, influences the self-assembly process.First, 3-D supramolecular assemblies were formed in the presence of ACG. TEM indicated the formation of spherical structures upon encapsulation of ACG, with radii around 100 nm (Fig. 2A), and no changes were observed in either shape or size of the 3-D supramolecular assemblies upon addition of ionomycin (Fig. S1, ESI†), or ionomycin and CaCl2 (Fig. 2B).
 | ||
| Fig. 2 TEM micrograph of A6B44A6 polymersomes with encapsulated ACG (A), and in the presence of 3.74 μM ionomycin and 830 μM CaCl2 (B). | ||
To obtain insight into the size, shape and stability of the 3-D supramolecular assemblies that resulted from encapsulation of ACG, in the presence of ionomycin, or/and Ca2+ ions, we also used light scattering measurements. Their hydrodynamic radius (Rh) was determined by dynamic light scattering, whilst the radius of gyration (Rg) was measured by static light scattering (Table 1).
| Size and stability of calcium sensing nanodevices | DLS/SLS | ||
|---|---|---|---|
| R g [nm] | R h [nm] | ρ = Rg/Rh | |
| A6B44A6 | 115 | 128 | 0.89 |
| A6B44A6 + ionomycin | 132 | 145 | 0.91 |
| A6B44A6 + ionomycin + CaCl2 | 117 | 122 | 0.96 |
The 3-D supramolecular assemblies resulting from encapsulation of ACG, and in the presence of ionomycin or/and Ca2+ ions, have radii of gyration around 100 nm, in agreement with the values obtained from TEM micrographs (Table 1 and Fig. 2). The observation of a monomodal distribution peak with a narrow size distribution indicates that only one population of 3-D supramolecular structures is present in solution (Table S1, ESI,† PDI values). For the 3-D supramolecular assemblies, the structural parameter ρ (ρ = Rg/Rh) has values of 0.89–0.96, which are close to the value of 1.0 for hollow spheres. Therefore, the 3-D supramolecular assemblies are polymersomes after encapsulation of ACG, and there is no influence on their architecture when ionomycin is inserted into their membranes and/or Ca2+ ions are present in their surroundings. Moreover, within the investigated concentration range, there was no significant variation in their radii, indicating that the measurements were in a concentration regime without interactions between polymersomes. The increase in Mw,agg and Nagg by addition of ionomycin confirms the presence of ion-carriers in the self-assembled structures, in agreement with reports indicating that Rh and Nagg values of nano-objects are highly dependent on the length and content of the constituent molecules52,53 (Table S1, ESI†). By changing the hydrophobic to hydrophilic ratio of the membrane, a nonlinear increase of Nagg has been observed, whilst the size of the vesicles only slightly increased,53 which is also the case for our polymersomes upon insertion of ionomycin (Table 1 and Table S1, ESI†). In addition, the insertion of the charged molecules of ionomycin in the hydrophobic domain of the membrane decreased the ζ-potential of polymersomes, and contributed to the increase in the Nagg value (Tables S1 and S2, ESI†). The stability of supramolecular assemblies in the absence and in the presence of ionomycin, as well as upon addition of CaCl2, is supported by the very small and slightly negative values of the second virial coefficients (A2 ∼ 10−8). Both the low PDI values and the very low values of the second virial coefficient A2 indicate no interaction of polymersomes, which would result in a merge of vesicles. Therefore, the change in Mw,agg and Nagg values is attributed only to the insertion of the ionophore into the vesicle membrane.
By dividing the fraction of encapsulated ACG by the initial amount used for encapsulation, we calculated the entrapment efficiency process of ACG as 7.5%.
Polymersomes with inserted and functional ionomycin
Although ionomycin was successfully inserted and functional in lipid membranes,54 it is interesting to study whether it is still able to mediate Ca2+ transport when inserted in polymer membranes with double the thickness of lipid membranes. Ionomycin has a diameter of 1.5 nm, as calculated from the crystal structure of the calcium complex55 (Fig. S2, ESI†).First, we evaluated the changes in fluorescence intensity of the free ACG, induced by the presence of a CaCl2 solution under similar conditions to those used for assessing ionomycin functionality in ACG-loaded polymersomes. The fluorescence intensity of ACG was evaluated in the presence of ionomycin, and EtOH was used for ionomycin solubilization. Neither ionomycin nor EtOH had any influence on ACG fluorescence intensity (Fig. S3, ESI†). Secondly, the change in fluorescence intensity of encapsulated dye was evaluated, both without and in the presence of ionomycin, to provide evidence of eventual defects in the polymer membrane (in the absence of ionomycin), and on successful insertion of ionomycin. If ionomycin is inserted into the membrane, Ca2+ ions from the environment of the vesicles will penetrate the membrane, resulting in an increase in fluorescence intensity of the encapsulated dye.
ACG-loaded polymersomes without ionomycin had low fluorescence intensity in the presence of 830 μM CaCl2 (Fig. 3, black curve), and did not change in the presence of EtOH (Fig. 3, red curve). The slight increase in fluorescence intensity upon addition of CaCl2 may be due to a small fraction of ACG that was incompletely removed during purification of the ACG-containing polymersomes.
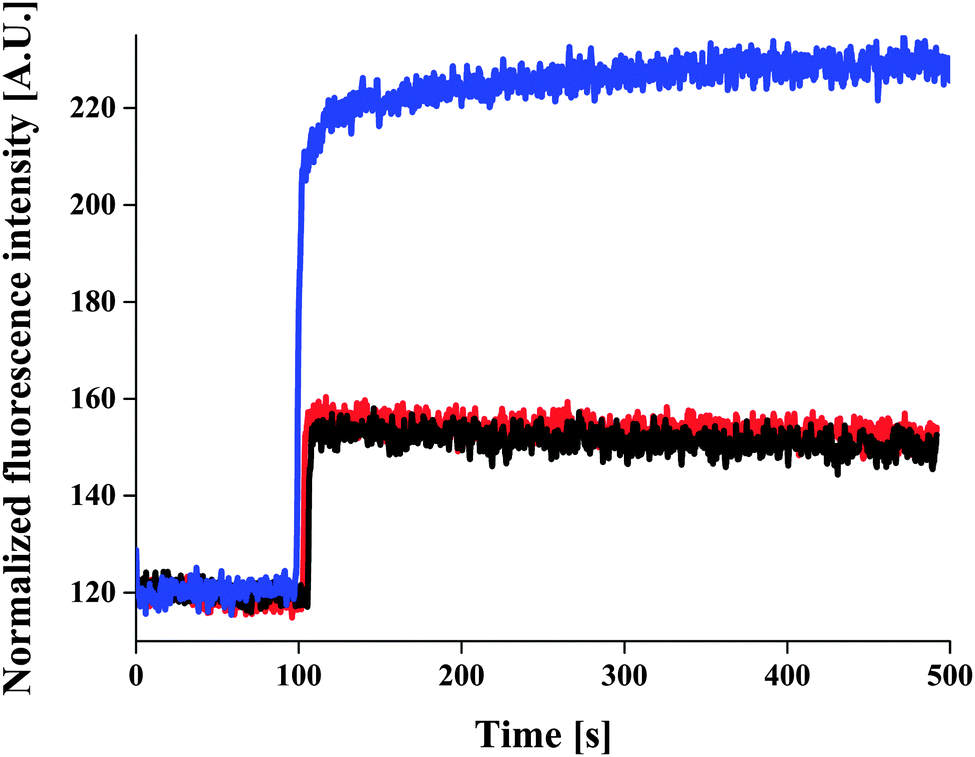 | ||
| Fig. 3 Ca2+ influx through A6B44A6 polymer membrane. ACG-loaded A6B44A6 polymersomes (black curve), with EtOH (red curve), at the optimum concentration of 3.74 μM ionomycin (blue curve), in the presence of 830 μM CaCl2. | ||
It was only when ionomycin was added that the fluorescence intensity increased significantly. This indicates a calcium influx through the ionomycin (Fig. 3, blue curve), and thus demonstrates their proper insertion and functionality. The fluorescence intensity of ACG-loaded polymersomes increased with increasing ionomycin concentration in a range of 117–3740 nM (in the presence of 830 μM CaCl2), at which maximum of fluorescence intensity was reached (Fig. S4, ESI†). For low initial amounts of ionomycin, an insertion efficiency close to 100% was obtained, which slightly decreased upon increasing the ionomycin amount (for 3.74 μM, the insertion efficiency was of 93%). To calculate the number of ionomycin molecules/polymersome we used a simple model based on uniform distribution of ionomycin molecules in polymersomes, and neglected the statistical character of the self-assembly process of polymersome formation. This results in aprox. 9.14 × 103 ionomycin molecules/polymersome for the initial amount of 3.74 μM ionomycin, and 2.28 × 1011 polymersomes (obtained from light scattering data).
Further increases in ionomycin concentration up to 10.40 μM resulted in lower increases in the fluorescence intensity of the encapsulated ACG. It indicates that a high amount of ionomycin influence the transport of Ca2+ ions through the polymersome membrane by unspecific attachment of ionomycin molecules on its external surface or by formation of 1:2 Ca–ionomycin complexes (Fig. S4, ESI†). Only 80% of the initial ionomycin amount (10.4 μM) was inserted in polymersome membrane (Fig. S5, ESI†). At higher initial concentration of ionomycin (15 μM) a significant fraction of polymersomes did not preserve their architecture (Fig. S6B, ESI†), resulting in leakage of the entrapped dye, as observed by the very high fluorescence intensity (Fig. S4, ESI†).
To obtain insights into the efficiency of calcium influx through the ionomycin ion-carrier in the first short period of time (≤15 s), we used stopped-flow spectroscopy (Fig. S7, ESI†). An increase in the fluorescence intensity of the ACG in polymersomes upon insertion of 3.74 μM ionomycin was observed, in agreement with the behavior of Polsp, as established by fluorescence spectroscopy. Ca2+ ions are transported at a fast rate by the functional inserted ionomycin, resulting in a instant Ca2+-selective and permeable polymer membrane.
The mechanism of membrane permeabilization for Ca2+ ions upon insertion of ionomycin was further investigated by evaluating diffusion within a very similar triblock copolymer membrane (A7B49A7) of the fluorescent dye molecule Bodipy, which is similar in size to ionomycin (Fig. S8 and Table S3, ESI†). The analysis of the diffusion by Z-scan FCS shows that Bodipy has a free-diffusion character (t0 = 0, Fig. S8B, ESI†) as already observed for fluid phase of lipid membranes.49,56 This indicates that PMOXA–PDMS–PMOXA triblock copolymer membranes provides a fluid homogenous hydrophobic medium, in which the ionophores are able to freely move. We have recently showed for a library of polymer membranes that the fluidity of such polymersome membranes based on PMOXA–PDMS amphiphilic block copolymers is similar to the fluidity of lipid bilayers.49
Studies of ionomycin in model liposome membranes revealed a Ca2+ permeabilisation of the membrane by formation of Ca–ionomycin complexes, and transport through the membrane.41,54 Interestingly, calcium transport through polymersome membranes, which are twice thicker than lipid membranes (10.7 nm thickness), was successful upon insertion of ionomycin, resulting from the appropriate fluid environment of the membrane.49
Sensitivity of the calcium selective membrane of polymersomes: nano-sensor functionality
Intracellular calcium concentrations vary from 100 nM, considered as the concentration when cells are at rest, up to 1 μM when processes inside cells are activated. In the extracellular space, the calcium amount can be as much as 104-fold higher than the intracellular concentrations.57 As in a biomimetic approach, since our Ca2+ selective polymersomes were intended to sense calcium amounts in various biological conditions, we were interested in determining the lowest concentration of Ca2+ that can be sensed by our polymersomes containing ionomycin. In order to do this, we varied the concentration of Ca2+ in the environment of Polsp using the optimum concentration of 3.74 μM ionomycin (Fig. 4). Polsps respond to the presence of 100 μM CaCl2, resulting in an increase of the fluorescence intensity of encapsulated ACG. Increased concentrations of CaCl2 induced a higher fluorescence intensity of Polsp, up to 830 μM CaCl2. Significant increases in the initial concentration of CaCl2 in the environment of polymersomes resulted in a plateau and slight decrease of the fluorescence intensity from encapsulated ACG (a saturation effect). Similar behavior has been reported for the initial rate of calcium transport through liposome membranes as function of Ca2+ concentration.54 The saturation effect for high Ca2+ concentrations was explained by a favored formation of 1:2 Ca–ionomycin complexes, which decrease the efficiency of Ca transport through the membrane.41,54
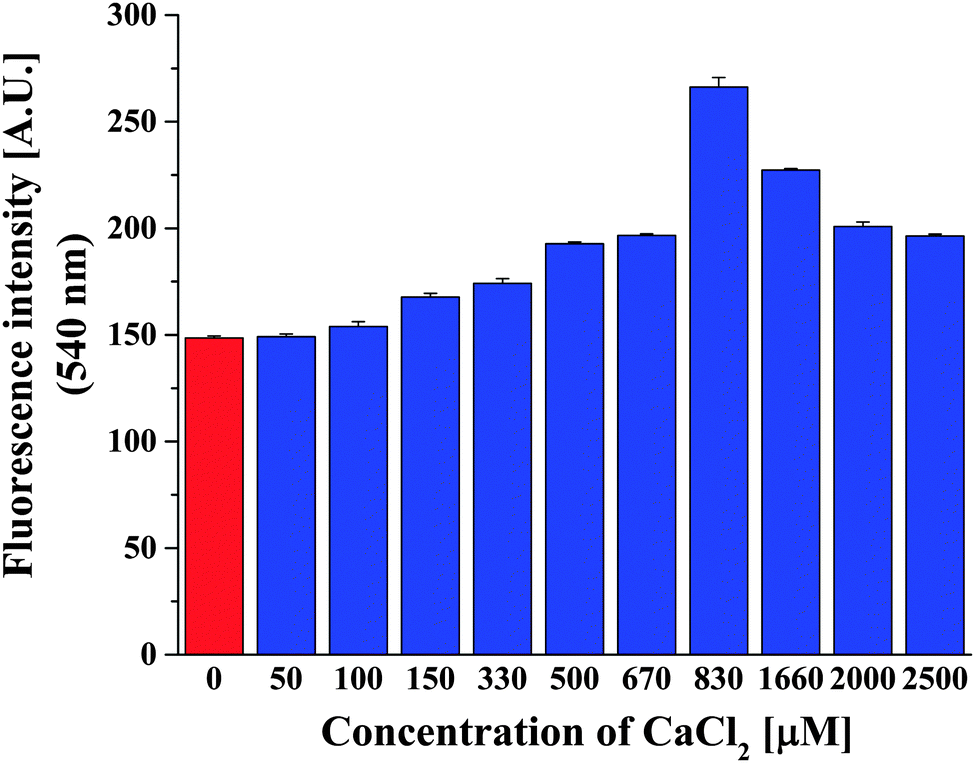 | ||
| Fig. 4 Optimum concentration of CaCl2. ACG-loaded A6B44A6 polymersomes without ionomycin, in the presence of EtOH and 830 μM CaCl2 (red bar), compared with ACG-loaded A6B44A6 polymersomes equipped with 3.74 μM ionomycin and increasing concentrations of CaCl2 (blue bars). Error bars are shown as standard deviations of three individual measurements. | ||
Interaction of ACG with the polymer membrane
We used fluorescence correlation spectroscopy (FCS)19,58 to investigate why a slight increase in ACG fluorescence intensity appears in ACG-containing polymersomes without ionomycin after purification by size exclusion chromatography (Fig. S9, ESI†). FCS provides information on the sizes of particles by determining their diffusion time through the confocal volume of the laser beam. The short diffusion time of free-diffusing ACG in HEPES buffer (τd,ACG = 40 μs) is typical for small molecular mass fluorescent dyes. Interaction of ACG with the polymersome membrane was assessed by adding ACG (20 μM) to empty polymersomes, which initially showed no fluorescence signal. After overnight incubation, the autocorrelation function of the fluorescence signal could be fitted with a bimodal model, which indicates the presence of two different populations of diffusing particles in the solution: a fraction of free ACG, and a second population with a significantly slower diffusion time (τd,vesicle ≈ 7000 μs). As the second population has a diffusion time characteristic of polymersomes with hydrodynamic radii of 50–100 nm,58 this fraction represents polymersomes to which ACG is attached by nonspecific binding. To gain more details about this nonspecific interaction of ACG with the polymersomes membrane, we formed giant unilamellar vesicles (GUVs) with entrapped ACG, and visualized them by confocal laser scanning microscopy (CLSM). CLSM micrographs (Fig. S10, left, ESI†) revealed GUVs containing free ACG molecules in the intravesicular volume. Upon increasing the laser intensity, the encapsulated dye was bleached, and the fluorescence intensity of the GUV membrane clearly indicates that the dye is interacting with the membrane (Fig. S10, right, ESI†). This behaviour was unexpected, due to the hydrophilic nature of ACG, and explains the slight increase of the fluorescence intensity of ACG-containing polymersomes upon addition of CaCl2 (Fig. 3, black curve).To take into account both fractions of ACG (encapsulated and attached to the membrane), we analysed the stopped-flow data by a double-exponential equation. The change over time in the intensity of the fluorescence signal upon mixing the ACG-containing polymersomes with CaCl2 solution indicates that there are two kinetic processes taking place. The kinetic parameter of the first exponential term (k1 ≈ 4 s−1) obtained from a sudden increase in fluorescence intensity had similar values for ACG-containing polymersomes, ACG-containing polymersomes upon addition of EtOH, and Polsp (Table S3, ESI†). Therefore this parameter describes the interaction of Ca2+ ions with ACG molecules attached on the outer surface of the polymersome membrane. The kinetic time constant of the second exponential term (k2) is ∼50 times slower than for ACG-containing polymersomes without ionomycin (k2 ≈ 0.8 s−1) in the presence of CaCl2. When ACG-containing polymersomes contained ionomycin, k2 was three times higher (k2 = 0.27 s−1) in the presence of CaCl2, indicating that transport of Ca2+ ions through the membrane occurs as a result of functional insertion of ionomycin. In addition, both the amplitude of the second time rate (ΔA2), related to the change in the signal intensity of the encapsulated dye, and the amplitude (ΔA1), related to the dye attached to the membrane, are almost double for the Polsp compared to the ACG-containing polymersomes without ionomycin (Table S3, in ESI†). The higher value of ΔA2 in the case of Polsp is related to the freely diffusing dye molecules inside the polymersome cavity, which react with Ca2+ ions that diffuse through the ionomycin. The difference in ΔA1 values between Polsp and ACG-containing polymersomes, indicates that ACG is also attached to the inner surface of the membrane, as expected due to the symmetric character of membrane formation by the self-assembly process.
Overall, the stopped-flow parameters indicate a selective permeabilization of the membrane by appropriate and functional insertion of ionomycin, and give an insight into the interaction of the selected dye with the polymer membrane.
Conclusions
We have introduced synthetic compartments with sizes in the nanometer range (polymersomes) equipped with ionomycin in order to induce selective membrane permeability for supporting applications in which it is necessary to provide an influx of ions to the inner cavity for in situ reactions, or to sense low changes in the concentrations of ions in the environment. Polymersomes with ionomycin inserted in their membrane were produced by self-assembly in dilute aqueous solutions, as a straightforward mild method. PMOXAx–PDMSy–PMOXAx amphiphilic copolymers were used to generate polymersomes, with a membrane thickness of 10.7 nm, in order to see whether functional insertion of ionomycin (1.5 nm size) is possible in such thick membranes. Ionomycin was successfully inserted into membranes up to 3.74 μM, without affecting the stability and architecture of the polymersomes, and allowed a rapid influx of Ca2+ ions through the synthetic membrane, as established by a combination of fluorescence spectroscopy and stopped-flow spectroscopy. Polymersomes equipped with ionomycin allowed a rapid sensing of Ca2+ ions in the concentration range 100–860 μM, and the rate of Ca2+ transfer was evaluated for the first time in a synthetic membrane. This concentration range covers both intracellular and extracellular conditions, and thus supports possible biological applications as sensitive cellular ion sensors, or for the development of nanoreactors in which an influx of specific ions is necessary for reactions inside the inner cavity. By a systematic study of the factors affecting the insertion and functionality of the ionophore in polymersome membranes we were able to successfully obtain a selective nanosized polymersome membrane as an optimized candidate towards medical applications (in terms of vesicles size, efficacy of ionophore and the improved stability of the polymer membrane). In addition, our selective ion polymersomes can support applications in environmental science, food technology or catalysis, because they are able both to shield the encapsulated molecules from harsh external conditions, and to sense a large ionic range with rapid and selective efficacy.Funding sources
We gratefully acknowledge the financial support provided by the Swiss National Science Foundation, the National Center of Competence in Research Nanoscale Science, and the National Center of Competence in Molecular Systems Engineering.Acknowledgements
We thank Prof. W. Meier from the University of Basel for providing the amphiphilic block copolymers, G. Persy and Adrian Najer (University of Basel) for the TEM-measurements. ML thanks Dr Mariana Spulber, Ina Andrea Ontiveros Casas, and Serena Rigo (University of Basel) for preliminary experiments on ionomycin insertion and to Samuel Lörcher (University of Basel) for the synthesis of A7B49A7 triblock copolymer. ML and FI acknowledge Dr Reinhard Kissner (Laboratory of Inorganic Chemistry, ETH Zurich) for providing support and useful discussions related to stopped-flow measurements. Dr B. A. Goodman is thanked for editing the manuscript.References
- A. H. Chen and P. A. Silver, Trends Cell Biol., 2012, 22, 662–670 CrossRef CAS PubMed.
- J. W. Szostak, D. P. Bartel and P. L. Luisi, Nature, 2001, 409, 387–390 CrossRef CAS PubMed.
- P. Tanner, O. Onaca, V. Balasubramanian, W. Meier and C. G. Palivan, Chem. – Eur. J., 2011, 17, 4552–4560 CrossRef CAS PubMed.
- C. G. Palivan, O. Fischer-Onaca, M. Delcea, F. Itel and W. Meier, Chem. Soc. Rev., 2012, 41, 2800–2823 RSC.
- C. Chiarabelli, P. Stano and P. L. Luisi, Front. Microbiol., 2013, 4, 285 Search PubMed.
- S. Nath, J. Dancourt, V. Shteyn, G. Puente, W. M. Fong, S. Nag, J. Bewersdorf, A. Yamamoto, B. Antonny and T. J. Melia, Nat. Cell Biol., 2014, 16, 415–424 CrossRef CAS PubMed.
- M. Massignani, H. Lomas and G. Battaglia, Adv. Polym. Sci., 2010, 229, 115–154 CrossRef CAS.
- B. M. Discher, Y.-Y. Won, D. S. Ege, J. C. M. Lee, F. S. Bates, D. E. Discher and D. A. Hammer, Science, 1999, 284, 1143–1146 CrossRef CAS PubMed.
- M. Antonietti and S. Förster, Adv. Mater., 2003, 15, 1323–1333 CrossRef CAS.
- L. Messager, J. Gaitzsch, L. Chierico and G. Battaglia, Curr. Opin. Pharmacol., 2014, 18C, 104–111 CrossRef PubMed.
- F. Ahmed, R. I. Pakunlu, A. Brannan, F. Bates, T. Minko and D. E. Discher, J. Controlled Release, 2006, 116, 150–158 CrossRef CAS PubMed.
- W. Chen and J. Du, Sci. Rep., 2013, 3, 2162 Search PubMed.
- J. P. Jain, W. Y. Ayen and N. Kumar, Curr. Pharm. Des., 2011, 17, 65–79 CrossRef CAS PubMed.
- P. Broz, N. Ben-Haim, F. Santini, S. Marsch, K. Scheffler, W. Meier and P. Hunziker, Eur. J. Nanomed., 2009, 2, 43–48 Search PubMed.
- C. Nardin, S. Thoeni, J. Widmer, M. Winterhalter and W. Meier, Chem. Commun., 2000, 1433–1434 RSC.
- D. Dobrunz, A. C. Toma, P. Tanner, T. Pfohl and C. G. Palivan, Langmuir, 2012, 28, 15889–15899 CrossRef CAS PubMed.
- S. F. van Dongen, M. Nallani, J. J. Cornelissen, R. J. Nolte and J. C. van Hest, Chem. – Eur. J., 2009, 15, 1107–1114 CrossRef CAS PubMed.
- D. M. Vriezema, P. M. Garcia, N. Sancho Oltra, N. S. Hatzakis, S. M. Kuiper, R. J. Nolte, A. E. Rowan and J. C. van Hest, Angew. Chem., Int. Ed., 2007, 46, 7378–7382 CrossRef CAS PubMed.
- P. Tanner, V. Balasubramanian and C. G. Palivan, Nano Lett., 2013, 13, 2875–2883 CrossRef CAS PubMed.
- Z. Fu, M. A. Ochsner, H. P. de Hoog, N. Tomczak and M. Nallani, Chem. Commun., 2011, 47, 2862–2864 RSC.
- M. Marguet, O. Sandre and S. Lecommandoux, Langmuir, 2012, 28, 2035–2043 CrossRef CAS PubMed.
- R. J. Peters, M. Marguet, S. Marais, M. W. Fraaije, J. C. van Hest and S. Lecommandoux, Angew. Chem., Int. Ed., 2014, 53, 146–150 CrossRef CAS PubMed.
- R. Rodríguez-García, M. Mell, I. López-Montero, J. Netzel, T. Hellweg and F. Monroy, Soft Matter, 2011, 7, 1532 RSC.
- S. M. Kuiper, M. Nallani, D. M. Vriezema, J. J. Cornelissen, J. C. van Hest, R. J. Nolte and A. E. Rowan, Org. Biomol. Chem., 2008, 6, 4315–4318 CAS.
- M. Spulber, A. Najer, K. Winkelbach, O. Glaied, M. Waser, U. Pieles, W. Meier and N. Bruns, J. Am. Chem. Soc., 2013, 135, 9204–9212 CrossRef CAS PubMed.
- X. Wang, G. Liu, J. Hu, G. Zhang and S. Liu, Angew. Chem., Int. Ed., 2014, 53, 3138–3142 CrossRef CAS PubMed.
- Z. Cheng and A. Tsourkas, Langmuir, 2008, 24, 8169–8173 CrossRef CAS PubMed.
- A. J. Kim, M. S. Kaucher, K. P. Davis, M. Peterca, M. R. Imam, N. A. Christian, D. H. Levine, F. S. Bates, V. Percec and D. A. Hammer, Adv. Funct. Mater., 2009, 19, 2930–2936 CrossRef CAS.
- M. Nallani, S. Benito, O. Onaca, A. Graff, M. Lindemann, M. Winterhalter, W. Meier and U. Schwaneberg, J. Biotechnol., 2006, 123, 50–59 CrossRef CAS PubMed.
- A. Graff, M. Sauer, P. Van Gelder and W. Meier, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 5064–5068 CrossRef CAS PubMed.
- A. Graff, C. Fraysse-Ailhas, C. G. Palivan, M. Grzelakowski, T. Friedrich, C. Vebert, G. Gescheidt and W. Meier, Macromol. Chem. Phys., 2010, 211, 229–238 CrossRef CAS.
- M. Sauer, T. Haefele, A. Graff, C. Nardin and W. Meier, Chem. Commun., 2001, 2452–2453 RSC.
- A. Picker, H. Nuss, P. Guenoun and C. Chevallard, Langmuir, 2011, 27, 3213–3218 CrossRef CAS PubMed.
- C. Liu and T. E. Hermann, J. Biol. Chem., 1978, 17, 5892–5894 Search PubMed.
- J. P. Chambers, B. P. Arulanandam, L. L. Matta, A. Weis and J. J. Valdes, Curr. Issues Mol. Biol., 2008, 10, 1–12 CAS.
- B. Bohunicky and S. A. Mousa, Nanotechnol., Sci. Appl., 2010, 4, 1–10 Search PubMed.
- A. Napoli, M. J. Boerakker, N. Tirelli, R. J. M. Nolte, N. A. Sommerdijk and J. A. Hubbell, Langmuir, 2004, 20, 3487–3491 CrossRef CAS PubMed.
- M. Grzelakowski, O. Onaca, P. Rigler, M. Kumar and W. Meier, Small, 2009, 5, 2545–2548 CrossRef CAS PubMed.
- M. Kumar, M. Grzelakowski, J. Zilles, M. Clark and W. Meier, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 20719–20724 CrossRef CAS PubMed.
- F. Axthelm, O. Casse, W. H. Koppenol, T. Nauser, W. Meier and C. G. Palivan, J. Phys. Chem. B, 2008, 112, 8211–8217 CrossRef CAS PubMed.
- C. Fasolato and T. Pozzan, J. Biol. Chem., 1989, 264, 19630–19636 CAS.
- T. J. Beeler, I. Jona and A. Martonosi, J. Biol. Chem., 1979, 254, 6229–6231 CAS.
- M. Deleers, E. Couturier and W. J. Malaisse, Cell Calcium, 1981, 2, 159–171 CrossRef CAS.
- M. J. Berridge, P. Lipp and M. D. Bootman, Nat. Rev. Mol. Cell Biol., 2000, 1, 11–21 CrossRef CAS PubMed.
- J. Krebs and M. Michalak, Calcium: A Matter of Life or Death, Elsevier, 2007 Search PubMed.
- N. M. Lorenzon and K. G. Beam, Kidney Int., 2000, 57, 794–802 CrossRef CAS PubMed.
- W.-C. Liu, D. S. Slusarchyk, G. Astle, W. H. Trejo, W. E. Brown and E. Meyers, J. Antibiot., 1978, 31, 815–819 CrossRef CAS PubMed.
- P. Baumann, M. Spulber, I. A. Dinu and C. G. Palivan, J. Phys. Chem. B, 2014, 118, 9361–9370 CrossRef CAS PubMed.
- F. Itel, M. Chami, A. Najer, S. Lörcher, D. Wu, I. A. Dinu and W. Meier, Macromolecules, 2014, 47, 7588–7596 CrossRef CAS.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed.
- A. Najer, D. Wu, A. Bieri, F. Brand, C. G. Palivan, H.-P. Beck and W. Meier, ACS Nano, 2014, 12, 12560–12571 CrossRef PubMed.
- H. Gharibi, Z. Moosavi-Movahedi, S. Javadian, K. Nazari and A. A. Moosavi-Movahedi, J. Phys. Chem. B, 2011, 115, 4671–4679 CrossRef CAS PubMed.
- C. Feng, C. L. Ren and Y. Q. Ma, Soft Matter, 2014, 10, 5523–5531 RSC.
- W. L. Erdahl, C. J. Chapman, R. W. Taylor and D. R. Pfeiffer, Biophys. J., 1994, 66, 1678–1693 CrossRef CAS PubMed.
- Z. Gao, Y. Li, J. P. Cooksey, T. N. Snaddon, S. Schunk, E. M. Viseux, S. M. McAteer and P. J. Kocienski, Angew. Chem., Int. Ed., 2009, 48, 5022–5025 CrossRef CAS PubMed.
- F. Heinemann and P. Schwille, ChemPhysChem, 2011, 12, 2568–2571 CrossRef CAS PubMed.
- M. D. Bootman and P. Lipp, Curr. Biol., 1999, 9, R876–R878 CrossRef CAS PubMed.
- P. Rigler and W. Meier, J. Am. Chem. Soc., 2006, 128, 367–373 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4cp05879h |
| ‡ Authors contributed equally to this study. |
| This journal is © the Owner Societies 2015 |