DOI:
10.1039/C3MB70304E
(Review Article)
Mol. BioSyst., 2014,
10, 970-1003
Isothermal amplified detection of DNA and RNA
Received
24th July 2013
, Accepted 18th February 2014
First published on 18th February 2014
Abstract
This review highlights various methods that can be used for a sensitive detection of nucleic acids without using thermal cycling procedures, as is done in PCR or LCR. Topics included are nucleic acid sequence-based amplification (NASBA), strand displacement amplification (SDA), loop-mediated amplification (LAMP), Invader assay, rolling circle amplification (RCA), signal mediated amplification of RNA technology (SMART), helicase-dependent amplification (HDA), recombinase polymerase amplification (RPA), nicking endonuclease signal amplification (NESA) and nicking endonuclease assisted nanoparticle activation (NENNA), exonuclease-aided target recycling, Junction or Y-probes, split DNAZyme and deoxyribozyme amplification strategies, template-directed chemical reactions that lead to amplified signals, non-covalent DNA catalytic reactions, hybridization chain reactions (HCR) and detection via the self-assembly of DNA probes to give supramolecular structures. The majority of these isothermal amplification methods can detect DNA or RNA in complex biological matrices and have great potential for use at point-of-care.
1.0 Introduction
Sensing nucleic acid sequences is vital in modern biology and medicine and the field has grown exponentially since the 1990s, and is likely to continue to increase in importance over the next few decades. The detection of pathogenic bacteria/viruses and cancer genes in a patient's sample can be critical to decisions that impact patient's recovery. Seminal discoveries made by several groups have linked the susceptibility to diseases such as cancer or the differences in drug metabolism to single nucleotide polymorphisms (SNPs) and short insertion/deletion variations.1 Several studies have also shown that RNA plays a key role in cancer development.1–9 Human miRNA genes are often found near cancer-associated genomic regions and fragile sites and some miRNAs have lower levels of expression in cancer samples compared to normal tissues.2 Abnormal expression of miRNAs has been found in cancers including chronic lymphocytic leukaemia,3 colorectal neoplasia,4 paediatric Burkitt lymphoma,5 lung cancer,6 large cell lymphoma,7 glioblastoma,8 and B cell lymphomas.9 Profiling miRNA tools are therefore required for the understanding of the expression pattern of miRNA. The detection of nucleic acid sequences without prior purification and amplification, in cells or in biological fluids, and even directly in living organisms is highly desirable. The demand for detecting specific nucleic acid sequences of different lengths to aid clinical diagnosis and for understanding fundamental biology has spurred the development of a myriad of DNA/RNA detection technologies.10 The polymerase chain reaction (PCR) can be used to exponentially amplify a few copies of target genes to concentrations that could be readily detected via a variety of means. Due to the need for a duplex melting step during the PCR cycle, which requires heating, PCR is not appropriate for the detection of nucleic acid sequences in live cells. Isothermal nucleic acid amplification strategies, especially those that would work at cellular temperatures (30–37 °C) could aid studies that aim to understand the temporal expression of genes in live cells. For in vitro applications, the need for a thermal cycling instrument or skilled technician to conduct the PCR assay (both of which require capital investment) can sometimes be prohibitive for resource-poor settings. These aforementioned limitations of PCR-based DNA/RNA detection assays have spurred the development of various isothermal detection platforms that amplify the target, the signal or both. The aim of this review is to highlight some of the nucleic acid detection methodologies that do not require thermal cycling procedures, such as polymerase chain reactions (PCR) or ligase chain reaction (LCR). We acknowledge that this review does not cover every isothermal DNA/RNA detection technology that has been reported to date, as it would be impractical to do so in a single review, and the omission of any amplified isothermal detection strategy does not imply that the omitted strategy is deemed to be unexciting. The reader is referred to other excellent reviews on this topic, such as the review by Asiello and Baeumner, which focused on miniaturized isothermal nucleic acid amplification11, and by Wilson, which focussed on gold nanoparticles for detection.12 Also, the reviews by Dong13a and Li13b on isothermal nucleic acid detection of microRNA are complementary to this review.13
2.0 Isothermal nucleic acids detection without amplification
2.01
In situ hybridization (ISH)
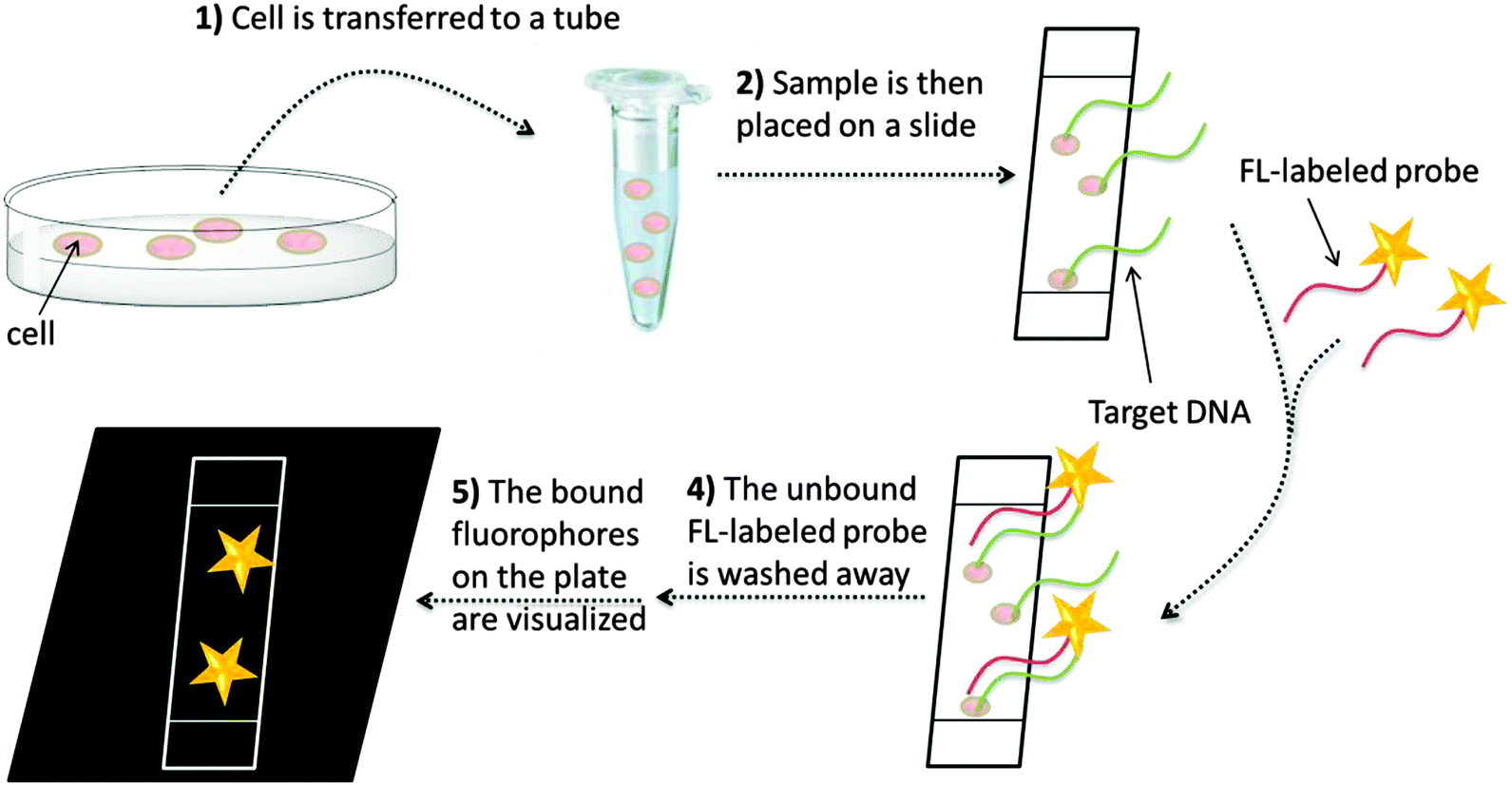
In situ hybridization (ISH) is an isothermal method that was developed in the late 1960s by Pardue and Gall.14 It was introduced into bacteriology by Giovannoni, who was the first to use radioactively labeled oligonucleotide probes for the detection of bacteria.15 Radioactive labels were gradually replaced by non-isotopic dyes with the development of fluorescent labels,15 which provide better safety and resolution (compared to the radioactive probes). Moreover, fluorescent probes can be labeled with dyes of different emission wavelengths, thus enabling detection of several target sequences during a single hybridization step (multiplexing). In 1989, fluorescently labeled oligonucleotides were used for the detection of microbial cells by DeLong.16 Since then, fluorescence in situ hybridization (FISH, see Fig. 1) has become a popular technology, especially in cytogenetics, where FISH is used to identify sequences on chromosomes.17 For sequence analysis using FISH, unhybridized probes have to be removed and this precludes the use of FISH to study nucleic acid processing in live cells. The rest of this review is mainly focused on nucleic acid detection assays that do not include tedious washing steps.
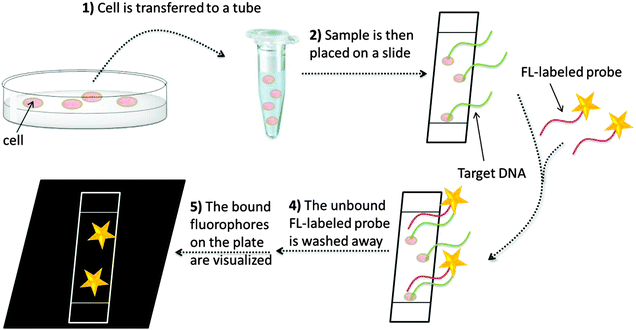 |
| | Fig. 1 A general FISH procedure. In step 1, fixation stops all biological processes in the cell. The sample is transferred to a glass plate or a surface and the DNA is then denatured so that hybridization with fluorescently labeled probe becomes possible (steps 2 and 3). Unbound fluorescently labeled probes are then washed off (step 4) and the labelled DNA can then be visualized (step 5). | |
2.02 Molecular beacon (MB)
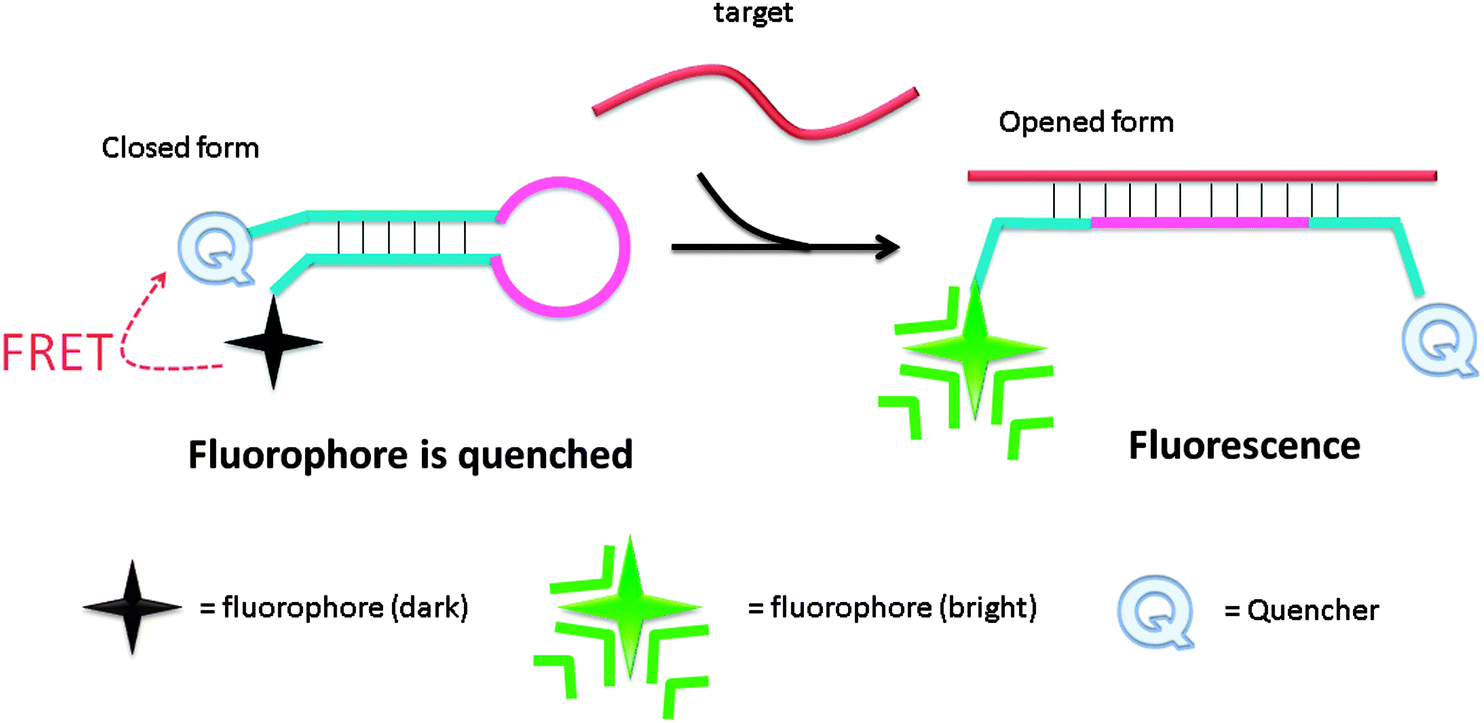
Molecular beacon (MB) was introduced in the 1990s by Tyagi and Kramer and has been successfully utilized in diverse applications.18–22 Molecular beacons are single-stranded oligonucleotide hybridization probes, which form a stem-loop structure. The sequences of the two sides of a MB probe are complementary, so a duplex stem can form by hybridization of the two arms. The loop is comprised of a sequence that is complementary to a target sequence. A fluorescent dye is labelled at the end of one arm and a quencher is labelled at the end of the other arm (see Fig. 2). The fluorophore is “dimmed” by the quencher when they are positioned close to each other in the stem-loop structure. However, when a MB binds to a complementary strand (target analyte), the stem-loop structure opens up and this results in a separation of the fluorophore and the quencher. Therefore, fluorescence is restored (Fig. 2).
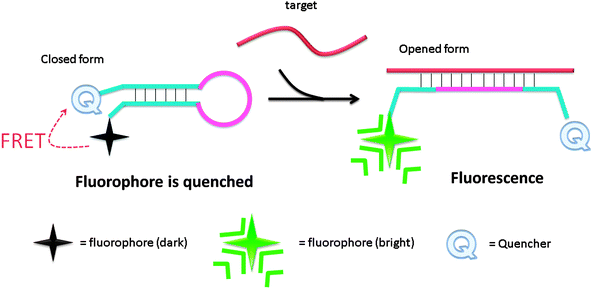 |
| | Fig. 2 Detection of analytes by molecular beacon. Initially the fluorophore is quenched due to the formation of a stem-loop, which places the quenching molecule adjacent to the fluorophore. Upon hybridization to the target sequence, the stem-loop opens and the quencher is separated from the fluorophore, causing an increase in fluorescence. | |
Multiplexing with MB, using different fluorophores has been demonstrated.20 By using several molecular beacons, each designed to recognize a different target and each labeled with a different fluorophore, multiple targets can be identified in the same solution, even if they differ from one another by as little as a single nucleotide. A major limitation of the traditional MB is low sensitivity (one target usually gives one fluorescence signal, although a few MB probes that contain multiple fluorophores per MB probe have also been described). Other limitations of MB include the interference of the MB stem with target hybridization,23 poor discrimination of single nucleotide polymorphism (SNP),24 or incomplete fluorophore quenching25,26 and although attempts have been made to resolve these aforementioned problems associated with MB, there is still room for improvement.
Other nucleic acid detection strategies that rely on the release of a quenched fluorophore have also been described.27–29 For example, toehold strand displacement has been used to detect nucleic acids (see Fig. 3 and 4). A toehold strand displacement uses a DNA duplex with a “sticky” end (usually few bases long). When a DNA/RNA analyte binds to the “sticky” end portion of the probe, the analyte exchanges with one of the strands in a displacement process. It has been shown that the length of the toehold affects the rate of strand displacement, with an enhancement factor of up to 106 being reported.30
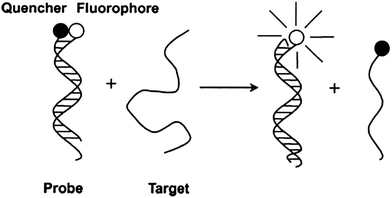 |
| | Fig. 3 Displacement of a quencher-labeled oligonucleotide, facilitated by binding to toehold. (Copied from ref. 27 with permission. Copy right 2002 Oxford University Press.) | |
 |
| | Fig. 4 The separation of fluorescently labeled probe from a gold nanoparticle quencher via initial binding of target RNA to a toehold has also been described. (Copied from ref. 29 with permission. Copyright 2007 American Chemical Society.) | |
3.0 Isothermal nucleic acids detection with amplification
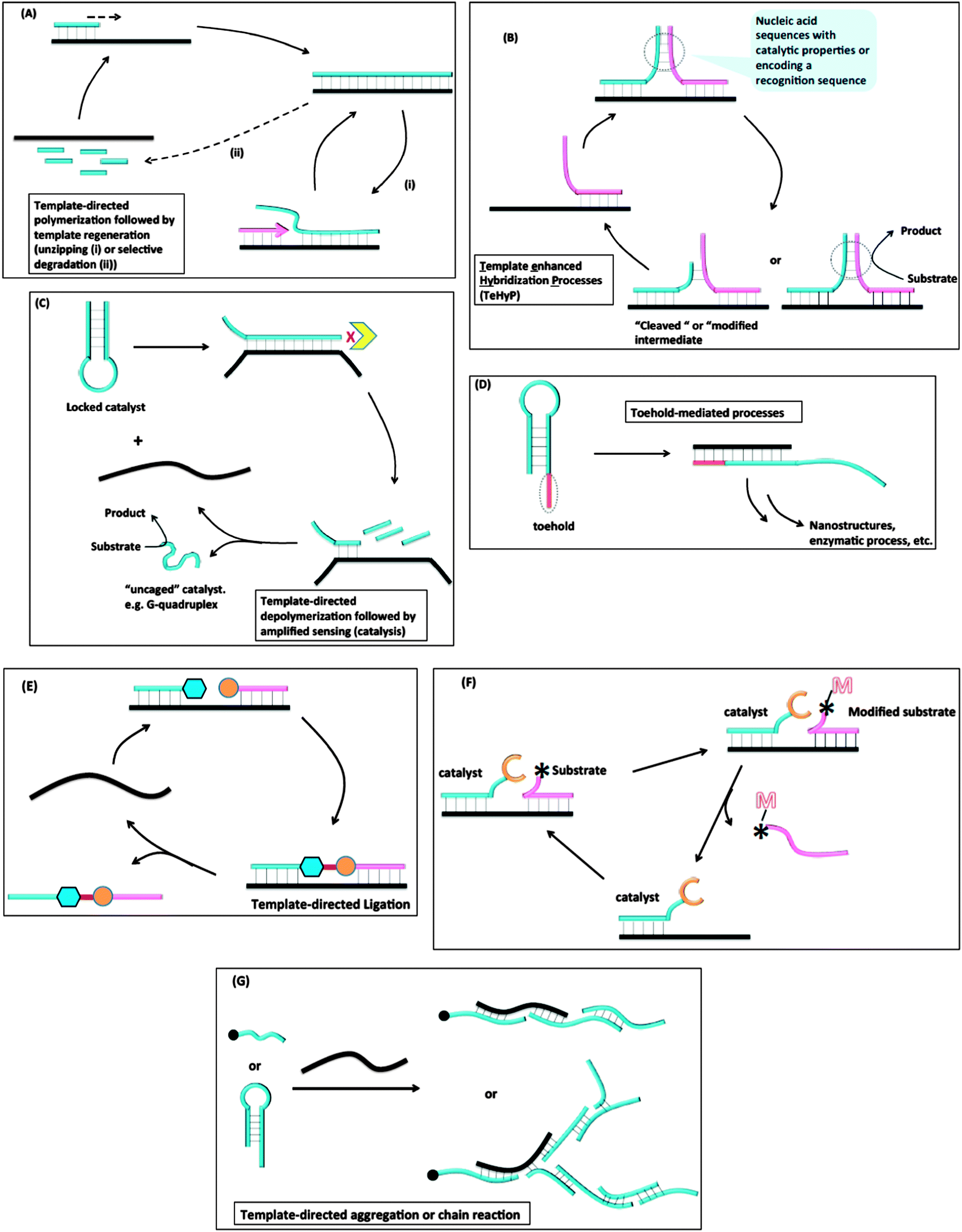
Despite the great advances that have been made in detecting nucleic acids in vitro or in vivo with MB probes or “toehold” probes, the sensitivity issue associated with non-amplifiable detection strategies has necessitated the development of other isothermal strategies that either amplify the initial target analyte or signal. The sections below highlight some of the isothermal DNA/RNA detection methods that utilize an amplification strategy (isothermal detection concepts are summarized in Fig. 5 and Table 1 summarizes the different detection platforms that are covered in this review). Fig. 5A describes template-directed polymerization followed by template regeneration. For this strategy, a polymerase converts a single stranded target analyte into a duplex and different strategies (non-thermal) are used to regenerate the single strand, which then serves as a template to start another amplification process. Fig. 5B demonstrates Template enhanced Hybridization Processes (TeHyP). For this strategy, two probes that have limited complementarity and hence do not form stable complexes at the assay temperature can be made to associate with each other in the presence of a target that binds to both probes in a juxtaposed manner. Binding of the two probes can then create functional/structural nucleic acid structures, such as an active DNAzyme or a recognition sequence for a DNA modifying enzyme, such as a nuclease. Fig. 5C depicts template-directed depolymerisation followed by amplified sensing (catalysis). For this strategy, the catalytic activity of a nucleic acid enzyme (such as a DNAzyme or RNAzyme) is inhibited via stem-loop formation. Upon binding to a target, the stem-loop inhibition is relieved and the catalyst becomes active. Incorporation of a degradation step by an exonuclease allows for target regeneration and hence amplified sensing. In toehold-mediated processes (see Fig. 5D), a stem-loop with a “sticky” end can be opened up with an analyte and the unveiled sequences, which were previously unavailable to act as templates due to participation in the stem duplex formation, can be used to trigger further stem-loop openings and formation of active enzymes or nanostructures. In the template-directed ligation process (see Fig. 5E), a template analyte is used to facilitate the ligation of two probes, which in the absence of a template would hardly react with each other. The ligation process can be coupled to a fluorogenic reaction for detection. Fig. 5F summarizes template-directed catalysis. In this process, the target analyte binds to both the catalyst and substrate to decrease the “effective Km” for the catalytic process. In template-directed aggregation or chain reaction (see Fig. 5G), a template is used to initiate a chain reaction, which results in the formation of supramolecular aggregates that are easily detected.
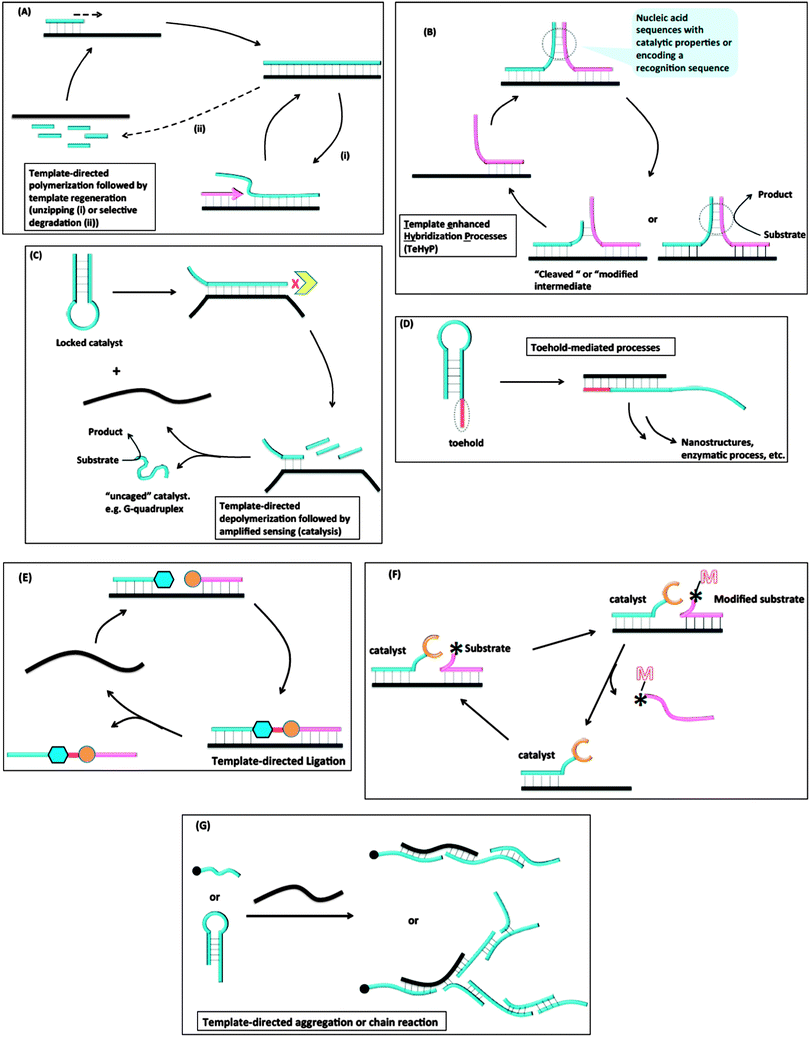 |
| | Fig. 5 Different strategies used for amplified isothermal detection of nucleic acids: (A) template-directed polymerization followed by template regeneration; (B) Template enhanced Hybridization Processes (TeHyP); (C) template-directed depolymerisation followed by amplified sensing (catalysis); (D) toehold-mediated processes; (E) template-directed ligation; (F) template-directed catalysis; (G) template-directed aggregation or chain reaction. | |
Table 1 Various isothermal detection platforms and components of the assay that are amplified
| Section |
Platform |
Amplified component |
Amplification catalyst |
| 3.01 |
NASBA |
Complementary sequence of target (RNA) |
Enzymatic |
| 3.02 |
SDA |
Probe |
Enzymatic |
| 3.03 |
LAMP |
Probe |
Enzymatic |
| 3.04 |
Invader assay |
Signal |
Enzymatic |
| 3.05 |
RCA |
Probe |
Enzymatic |
| 3.06 |
SMART |
New RNA |
Enzymatic |
| 3.07 |
HDA |
Target and the complementary sequence |
Enzymatic |
| 3.08 |
RPA |
Target and the complementary sequence |
Enzymatic |
| 3.09 |
NESA, NEANA |
Signal |
Enzymatic |
| 3.10 |
Exo III aided target recycling |
Signal |
Enzymatic |
| 3.11 |
Junction or Y probes |
Signal |
Enzymatic |
| 3.12 |
Reactivation of enzymatic activity |
Signal |
Enzymatic |
| 3.13 |
Ultrasensitive miRNA detection |
Probe and signal |
Enzymatic |
| 3.14 |
Split DNAzyme |
Signal |
Non-enzymatic |
| 3.15 |
RNA cleaving deoxyribozymes (CMB, TASC) |
Signal |
Non-enzymatic |
| 3.16 |
Template-directed chemical reaction |
Signal |
Non-enzymatic |
| 3.17 |
Non-covalent DNA catalytic reaction |
Signal |
Non-enzymatic |
| 3.18 |
HCR |
Signal |
Non-enzymatic |
| 3.19 |
Self-assembled DNA |
Signal |
Non-enzymatic |
3.01 Nucleic acid sequence-based amplification (NASBA)
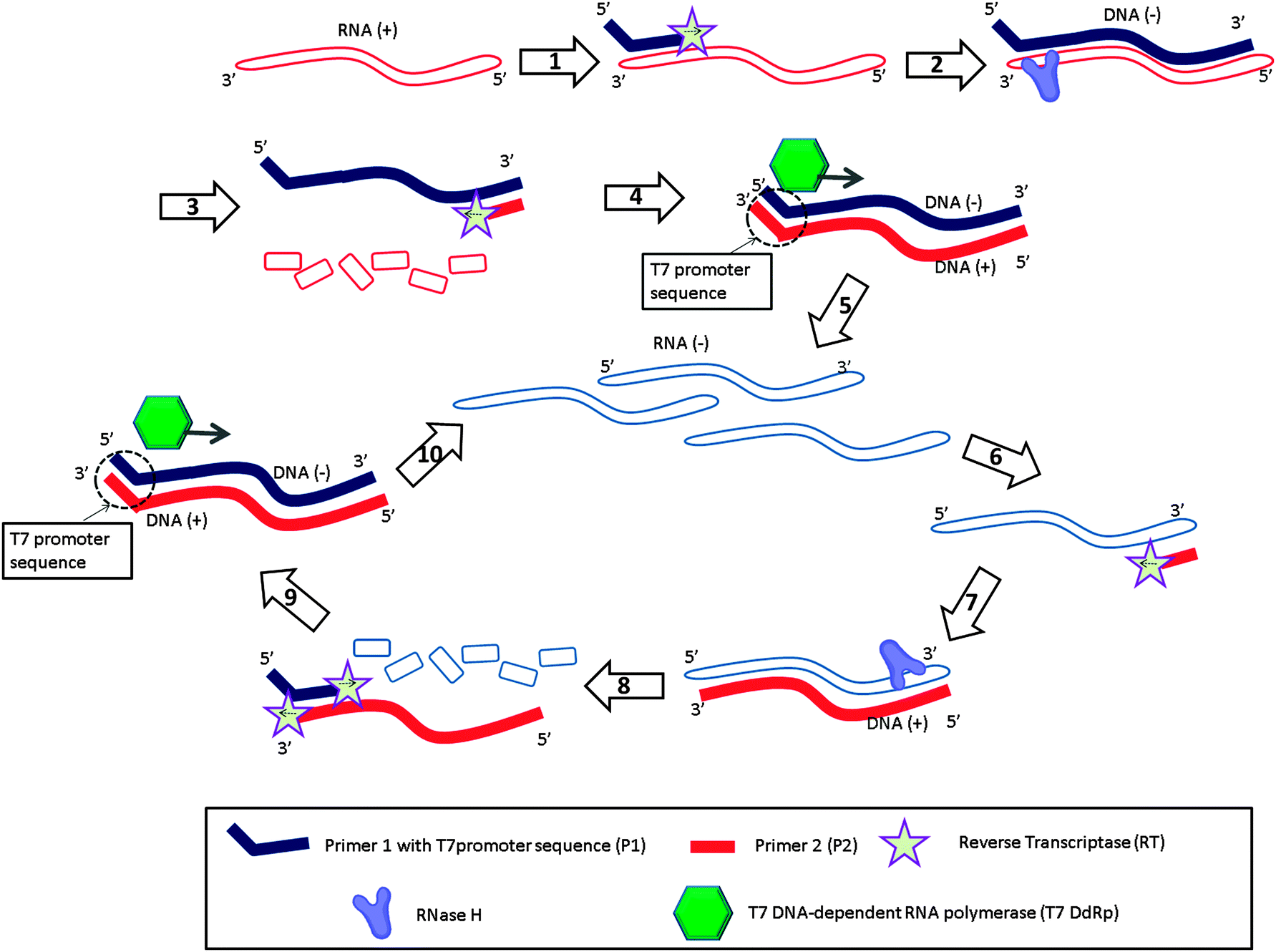
Nucleic acid sequence-based amplification (NASBA)31 is an isothermal amplification method designed to detect RNA targets (Fig. 6). With this method, the forward primer (primer 1 (P1) in Fig. 6) is composed of two parts, one of which is complementary to the 3′-end of the RNA target and the other to the T7 promoter sequence. When the P1 binds to the RNA target (RNA (+)), reverse transcriptase (RT) extends the primer into a complementary DNA (DNA (+)) of the RNA (step-1). RNase H then degrades the RNA strand of the RNA–DNA (+) hybrid (step-3). The reverse primer 2 (P2 in Fig. 6) then binds to the DNA (+) (step-3), and a reverse transcriptase (RT) produces double stranded DNA (dsDNA) (step-4), which contains a T7 promoter sequence. After this initial phase (Fig. 6, steps 1 to 5), the system enters the amplification phase (Fig. 6, steps 6 to 10). The T7 RNA polymerase generates many RNA strands (RNA (−)) based on the dsDNA, and the reverse primer (P2) binds to the newly formed RNA (−) (step-6). RT extends the reverse primer (step-7) and RNase H degrades the RNA of the RNA–cDNA duplex into ssDNA (step-8). The newly produced cDNA (DNA (+)) then becomes a template for P1 (step-9) and the cycle is repeated (step-10; amplification cycles).
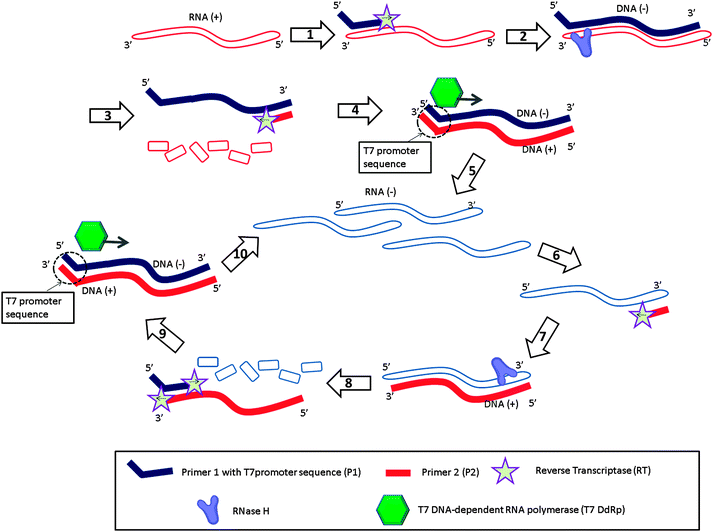 |
| | Fig. 6 Nucleic acid sequence-based amplification (NASBA). RT = Reverse Transcriptase, T7 DdRp = T7 DNA-dependent RNA polymerase. Steps 1 to 5 are the initial phase to amplify original RNA (+) target to RNA (−). Steps 6 to 10 are the amplification phase to amplify RNA. (Adapted from ref. 31 with permission. Copyright 1990, National Academy of Sciences, U.S.A.) | |
NASBA achieves 10 million fold amplification in 1–2 h.32 It has been commercialized33 and a wide range of target RNA sequences have been amplified and detected via this technique, such as HIV-1 genomic RNA,34 hepatitis C virus RNA,35 Human Cytomegalovirus mRNA,36 16S RNA in many bacterial species,37 and enterovirus genomic RNA,38etc.
3.02 Strand displacement amplification (SDA)
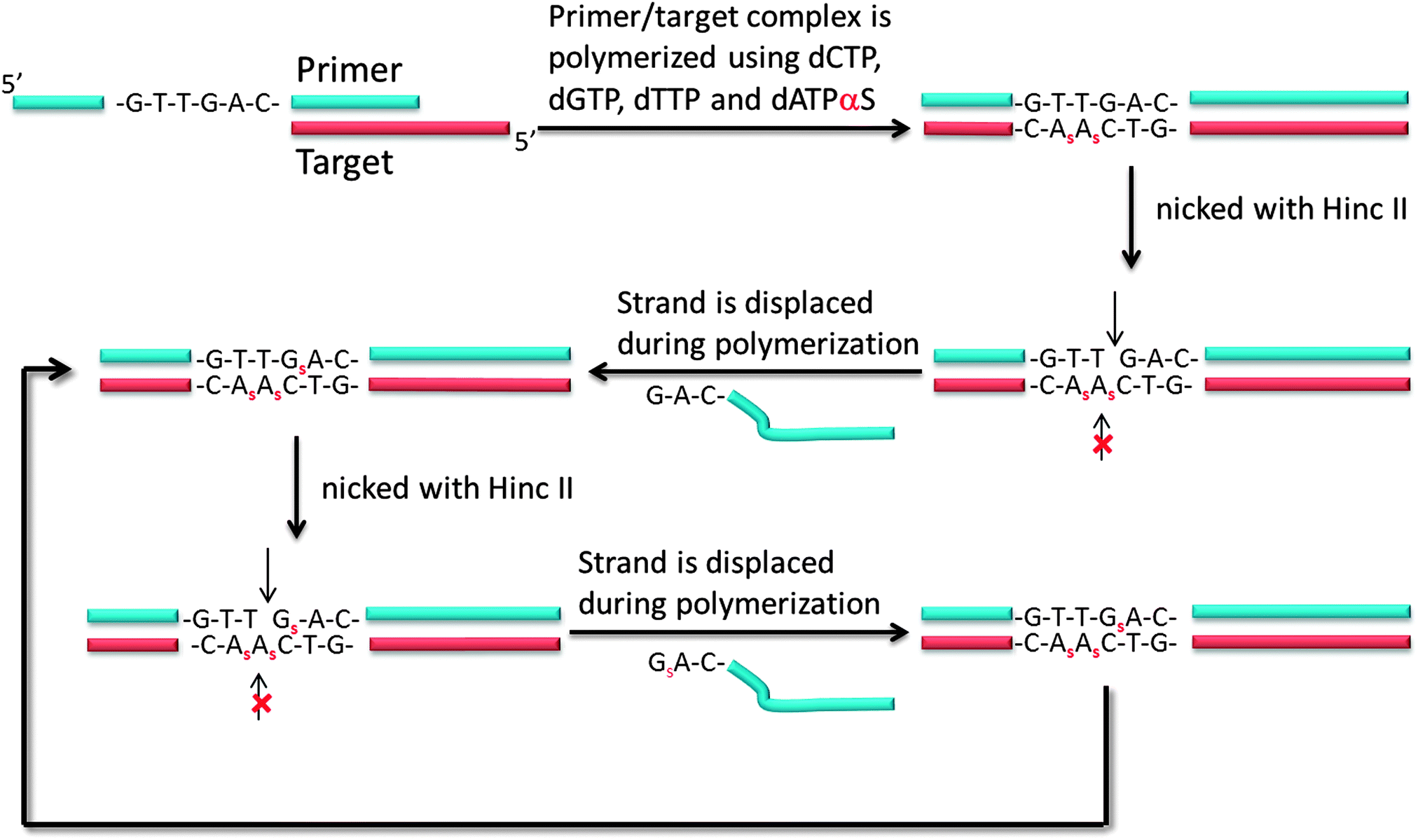
Another isothermal amplification method, which is based on DNA polymerase, is strand displacement amplification (SDA) (Fig. 7). In the original method,39 a probe contained two parts: a HincII recognition site at the 5′ end and another segment that contained sequences that are complementary to the target. DNA polymerase can extend the primer and incorporate deoxyadenosine 5′-[α-thio]triphosphate (dATP[αS]). The restriction endonuclease HincII then nicks the probe strand at the HincII recognition site because the endonuclease cannot cleave the other strand that contains the thiophosphate modification. The endonuclease cleavage reveals a 3′-OH, which is then extended by DNA polymerase. The newly generated strand still contains a nicking site for HincII. Subsequent nicking of the newly synthesized duplex, followed by DNA polymerase-mediated extension is repeated several times and this leads to an isothermal amplification cascade.
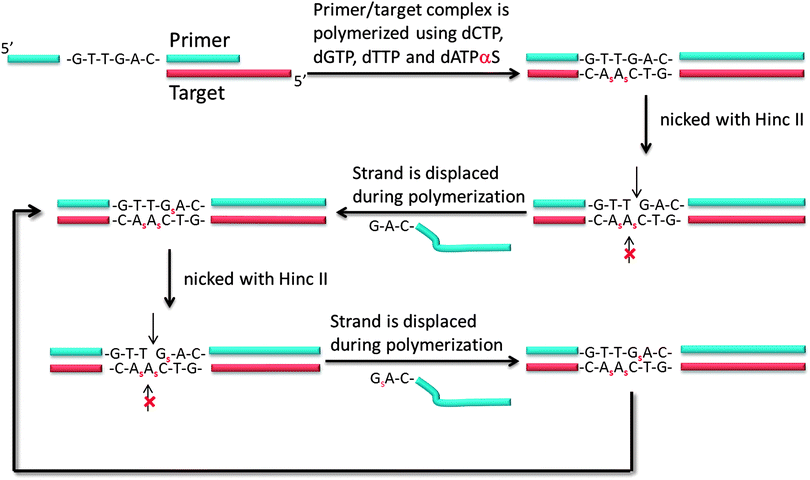 |
| | Fig. 7 First-generation strand displacement amplification (SDA). The primer has HincII recognition site (5′-GTT//GAC-3′) and target binding site. After binding of target, new DNA is synthesized by DNA polymerase. However the new DNA has thiophosphate modification because dATPαS is used instead of dATP, the original primer is cleaved by HincII but not new DNA strand. (Adapted from ref. 39 with permission. Copyright 1992, National Academy of Sciences, U.S.A.) | |
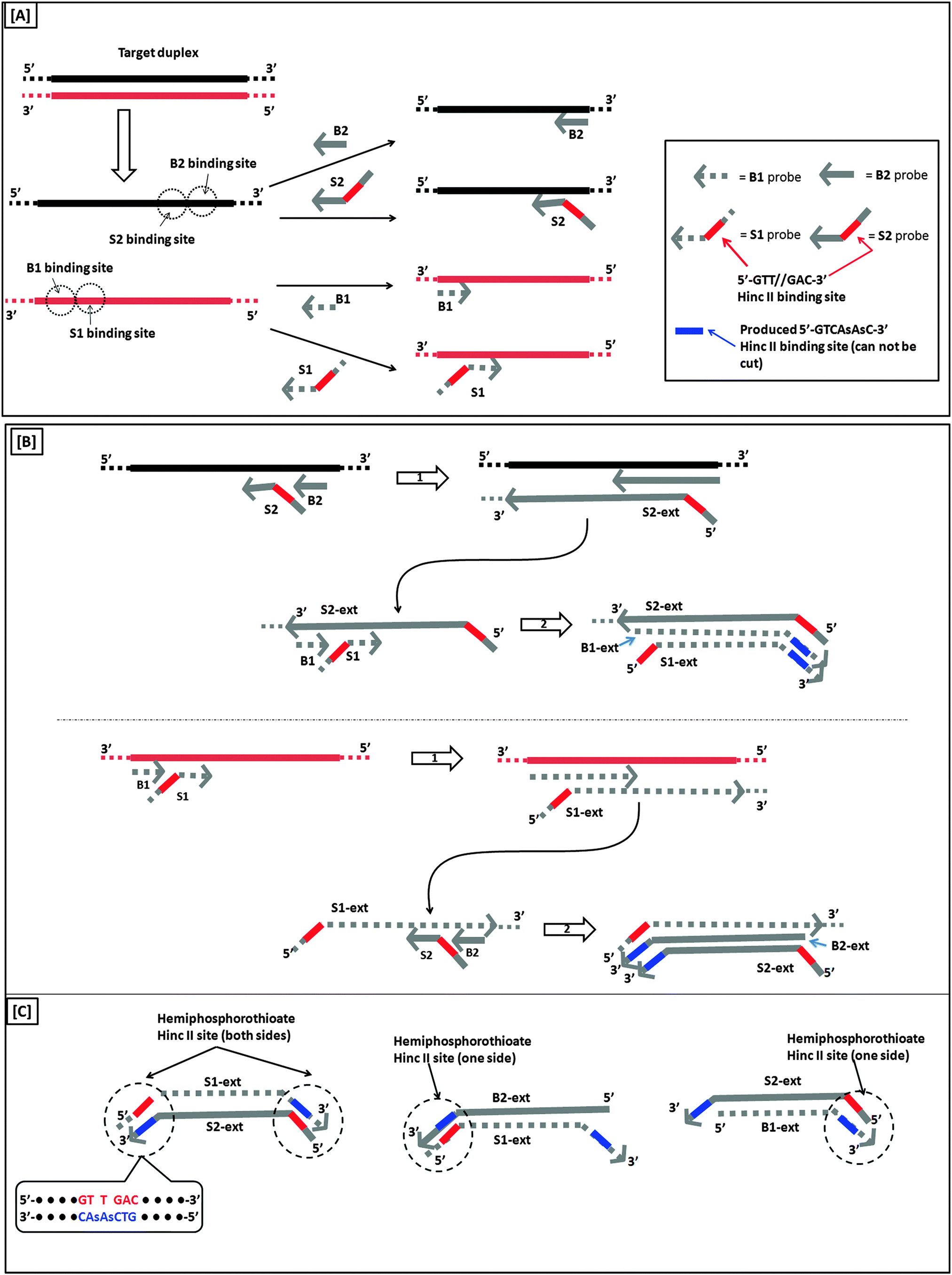
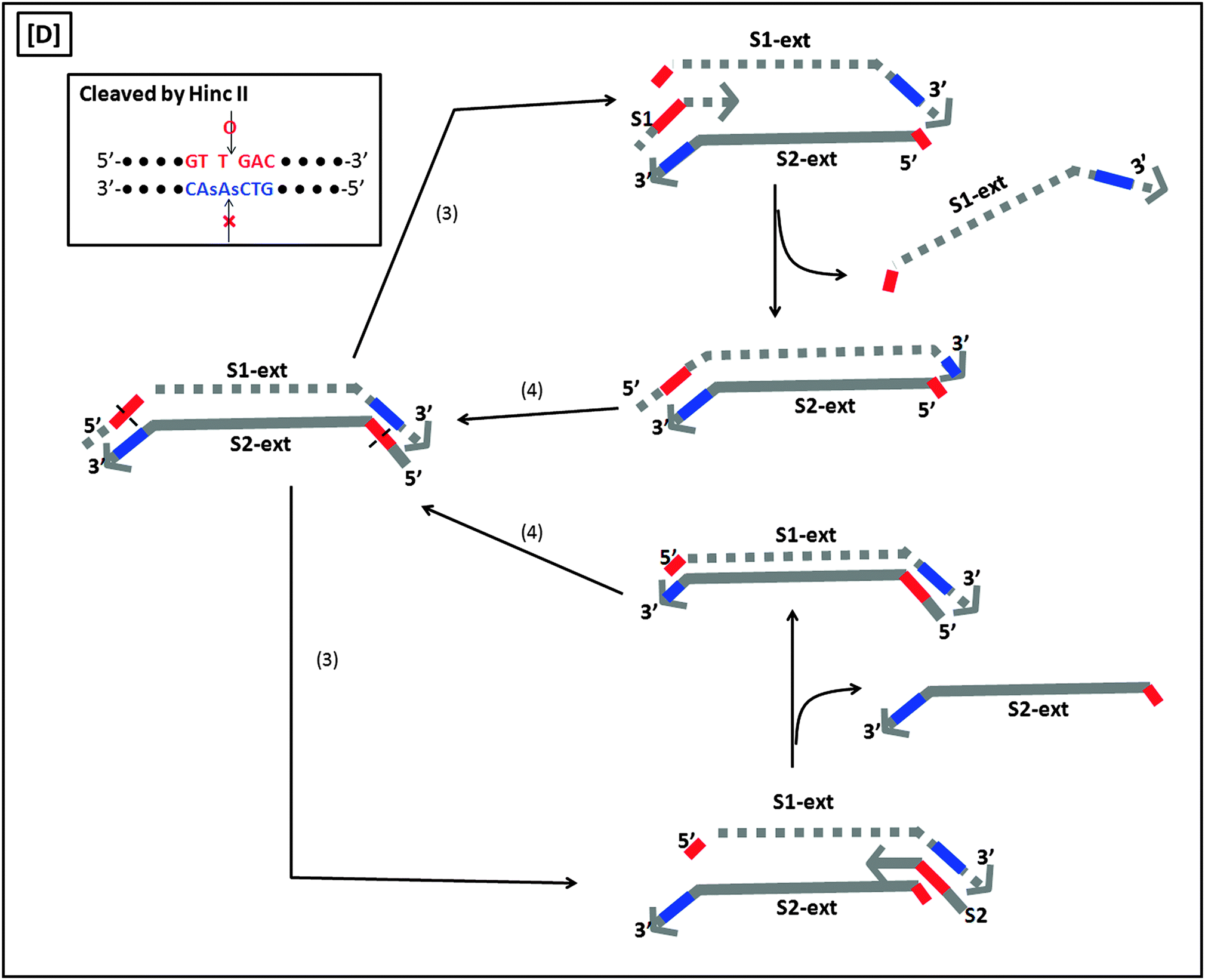
A more sensitive later generation of SDA uses four probes (B1, B2, S1 and S2), instead of two (see Fig. 8).40 In the second-generation SDA platform, probes B1 and S1 (or B2 and S2) bind to the same DNA strand (see Fig. 8). Primers S1 and S2 contain HincII recognition sequences and the four primers are simultaneously extended by DNA polymerase using dGTP, dCTP, TTP and dATPαS. Extension of B1 displaces the S1 primer extension product, S1-ext. Likewise, extension of B2 displaces S2-ext. Extension and displacement reactions on templates S1-ext and S2-ext produce two types of fragments: one has a hemiphosphorothioate HincII site at each end and the other has a hemiphosphorothioate HincII site at just one end. SDA products can be detected via a variety of means, including the use of molecular beacons41,42 and intercalating dyes,43,44 whose fluorescence gets enhanced upon binding to nucleic acids.
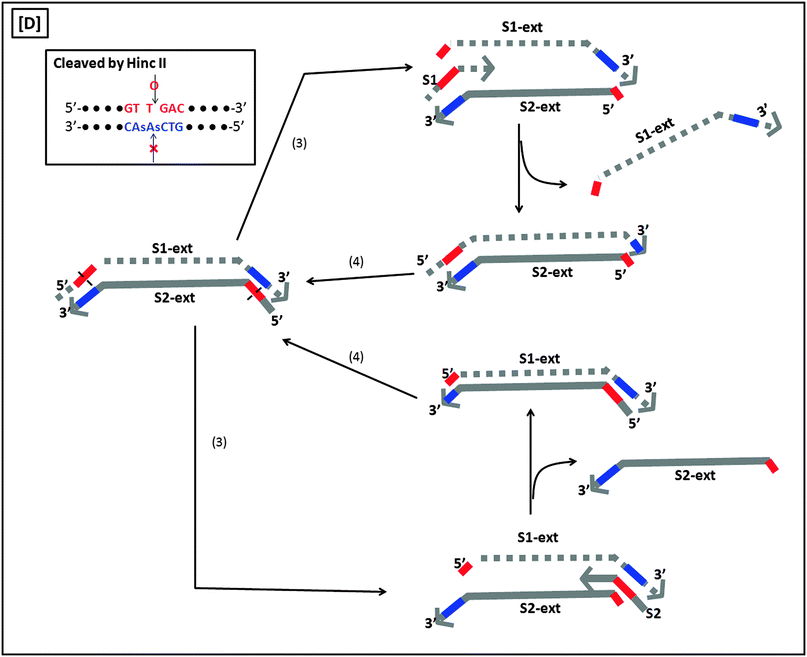 |
| | Fig. 8 Second-generation strand displacement amplification (SDA). (A) The target DNA is denatured and primers (B1, B2, S1, and S2) are allowed to bind. Primers S1 and S2 have HincII recognition sites; (B) after binding of primers, the primers are simultaneously extended by exo-klenow in the presence of dCTP, TTP, dGTP, dATPαS. Once B1 and B2 are extended they displace the S1 and S2 extension products, S1-ext and S2-ext. Remaining primers (B1, B2) bind to S1-ext and S2-ext and are again extended by exo-klenow; (C) the new extended primers contain two phosphorothioate HincII recognition sites as well as two products with just one phosphorothioate HincII recognition site; (D) the resulting amplified target sequences with phosphorothioate HincII recognition sites are nicked by HincII. Exo-klenow then extends the strand at the nick in the presence of dCTP, TTP, dGTP, and dATPαS. This can be carried out over and over again resulting in an exponential amplification (steps 3 and 4). (Adapted from ref. 40 with permission. Copyright 1992, Oxford University press.) | |
The SDA technique has been combined with a ligation reaction to detect SNP (see Fig. 9).45 For this interesting ligation-SDA technique, Wang et al. incorporated a G-quadruplex unit into their design to allow for a DNAzyme/hemin-based chemiluminescence detection.
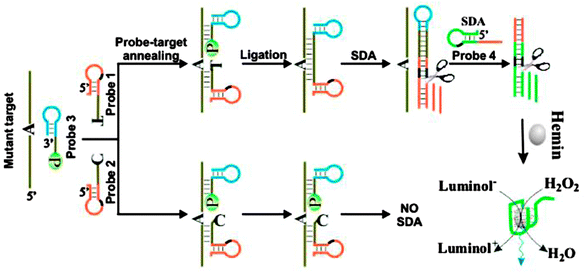 |
| | Fig. 9 SDA-Ligase-G-quadruplex peroxidase detection strategy. P = 5′-phosphate. This strategy could be used to detect single nucleotide polymorphisms (SNPs). Two discriminating probes 1 and 2 contain sequences complementary to the 5′-terminal sequence of mutant target or the wild-type target and probe 3 can hybridize with 3′-terminal sequence of both mutant target and the wild-type target. When the discriminating probe and probe 3 perfectly anneals with the target, they are ligated by a ligase, followed by two SDA reactions. The SDA products are hemin aptamer. The hemin-aptamer can catalyze the oxidation of luminol to give chemiluminescence signal. If there is a mismatch between the discriminating probe and the target, ligation does not take place and SDA reactions cannot be initiated. As a result, no chemiluminescence signal can be detected. (Copied from ref. 45 with permission. Copyright 2011, American Chemical Society.) | |
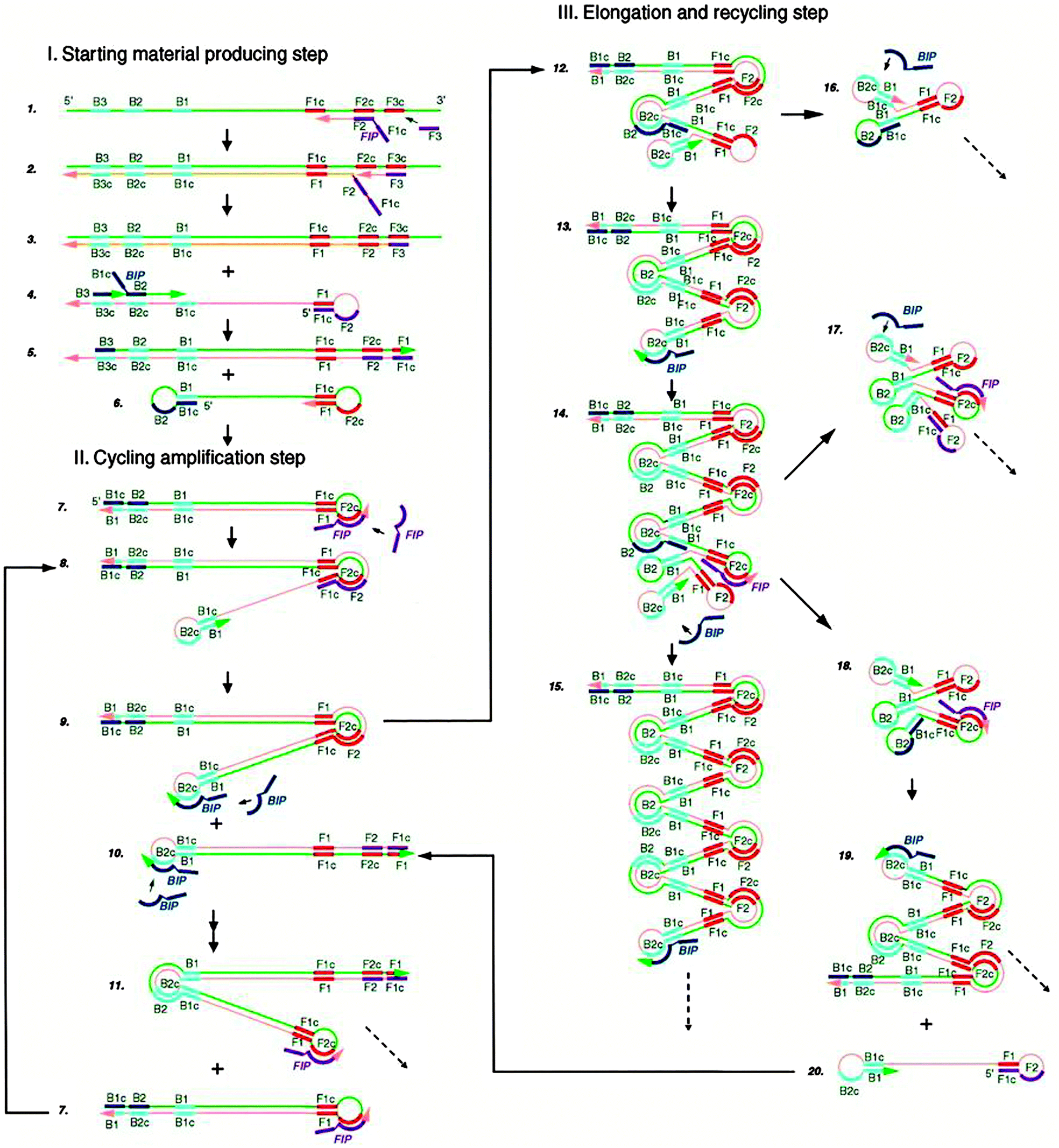
3.03 Loop-mediated amplification (LAMP)
Loop-mediated amplification (LAMP) is an isothermal method that has been shown to display amplification levels that approach that of PCR.45 LAMP also achieves high target specificity and this is due to the fact that two sets of primers spanning 6 distinct sequences of the target are used. The steps of LAMP are illustrated in Fig. 10.46 Two primers in the forward primer set are named inner (F1c-F2, c strands for “complementary”) and outer (F3) primers. At around 60 °C, the F2 region of the inner primer first hybridizes to the target, and is extended by a DNA polymerase. The outer primer F3 then binds to the same target strand at F3c, and the polymerase extends F3 to displace the newly synthesized strand. The displaced strand forms a stem-loop structure at the 5′ end due to the hybridization of F1c and F1 region. At the 3′ end, the reverse primer set can hybridize to this strand and a new strand with stem-loop structure at both ends is generated by the polymerase. The dumbbell structured DNA enters the exponential amplification cycle and strands with several inverted repeats of the target DNA can be made by repeated extension and strand displacement.
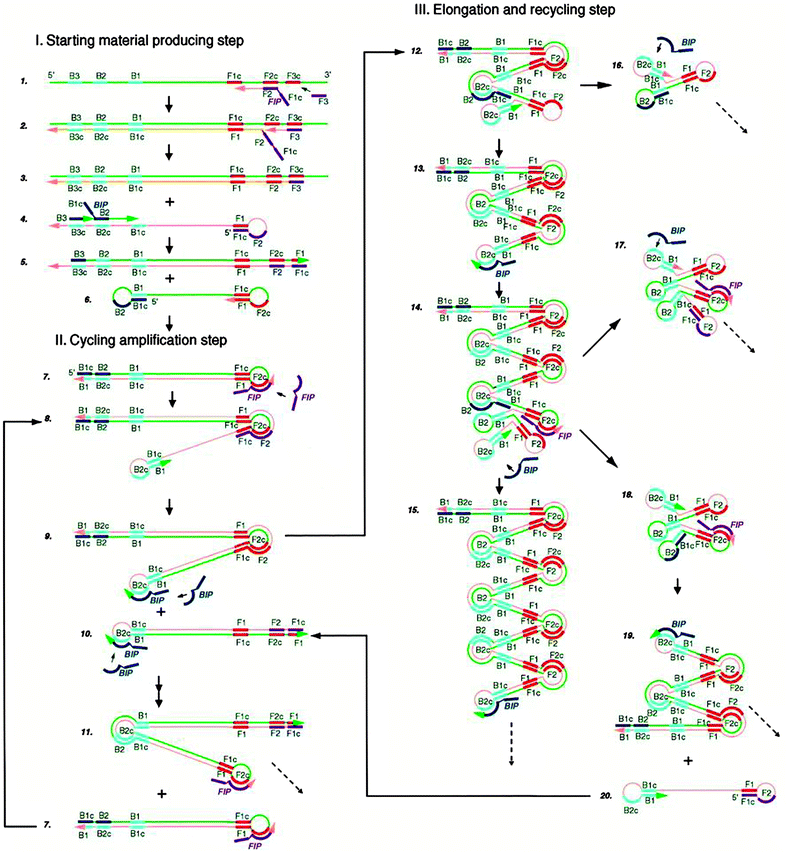 |
| | Fig. 10 Mechanism of Loop-mediated amplification (LAMP). Four probes (F1c-F2, F3, R1c-R2, R3) are used for this method. F1 is complementary with F1c (c stands for complementary sequence). (Copied from ref. 46 with permission, copyright 2000, Oxford University press.) | |
LAMP can amplify a few copies of the target to 109 in less than one hour, even when large amounts of non-target DNA are present.46 LAMP has been applied to detect a variety of viral pathogens, including dengue,47 Japanese Encephalitis,48 Chikungunya,49 West Nile,50 Severe acute respiratory syndrome (SARS),51 and highly pathogenic avian influenza (HPAI) H5N1,52etc. Recently, Fang et al. used LAMP to detect multiple genes in a microfluidic platform.52 The major disadvantage of LAMP is that the design of the primer sets can be complicated, since 6 regions of the target are covered.
3.04 Invader assay
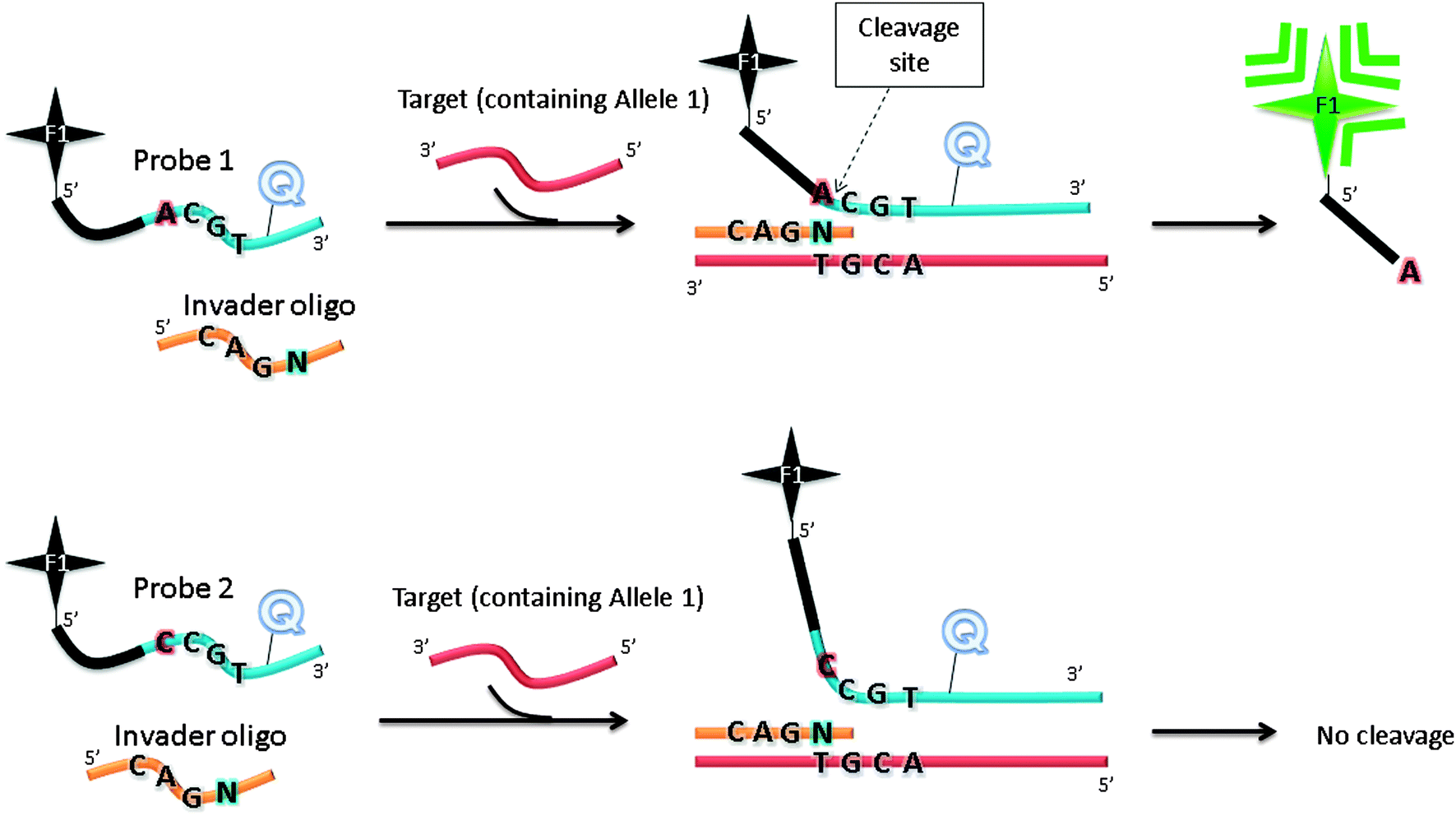
The Invader assay (Fig. 11) is a structure-specific assay that utilizes a flap endonuclease.53 The two oligonucleotide probes that are used in the invader assay bind to the target in a juxtapose manner, but upon target binding a structure-specific “flap” is created at the junction where the two probes meet (see Fig. 11). This flap can be cleaved by a cleavase (flap endonuclease) so if a fluorophore and quencher are positioned around the cleavage site, then the separation of the fluorophore and quencher would generate enhanced fluorescence. The fluorophore-labelled probe is designed to have a melting temperature close to the assay temperature to enable the probe to frequently bind and detach. When the probe binds, the flap endonuclease cleaves it, and after detachment a new intact fluorescently labelled probe anneals to the same site. It has been estimated that for one target about 3000 cleavages can be done in about 90 min.54 Although an amplification of 3000 in 90 min is good, it is not high enough for the detection of low copy number targets and so for low analyte concentrations an initial PCR amplification of the target is necessary before the invader reaction.
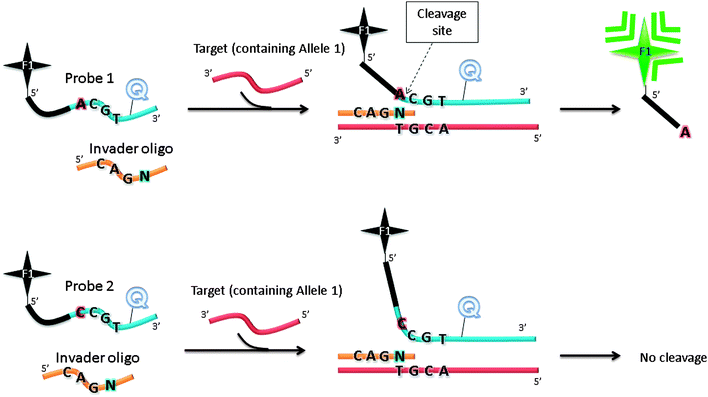 |
| | Fig. 11 Invader assay. The sequence at the overlap region is important for effective cleavage by flap endonuclease. F1 (= fluorophore), Q (= quencher). Schematic illustration of allele-specific cleavage mechanism in Invader assay, in the presence of flap endonucleases. The Invader assay contains two oligonucleotide probes, which can hybridize to target DNA. When bound to target DNA, a three-dimensional invader structure is formed, which is recognized by flap endonucleases. The cleavage reaction by flap endonucleases generates fluorescent output signal. When the probe does not match the allele present in the target DNA, there will be no overlapping invader structure and the probe cannot be cleaved. | |
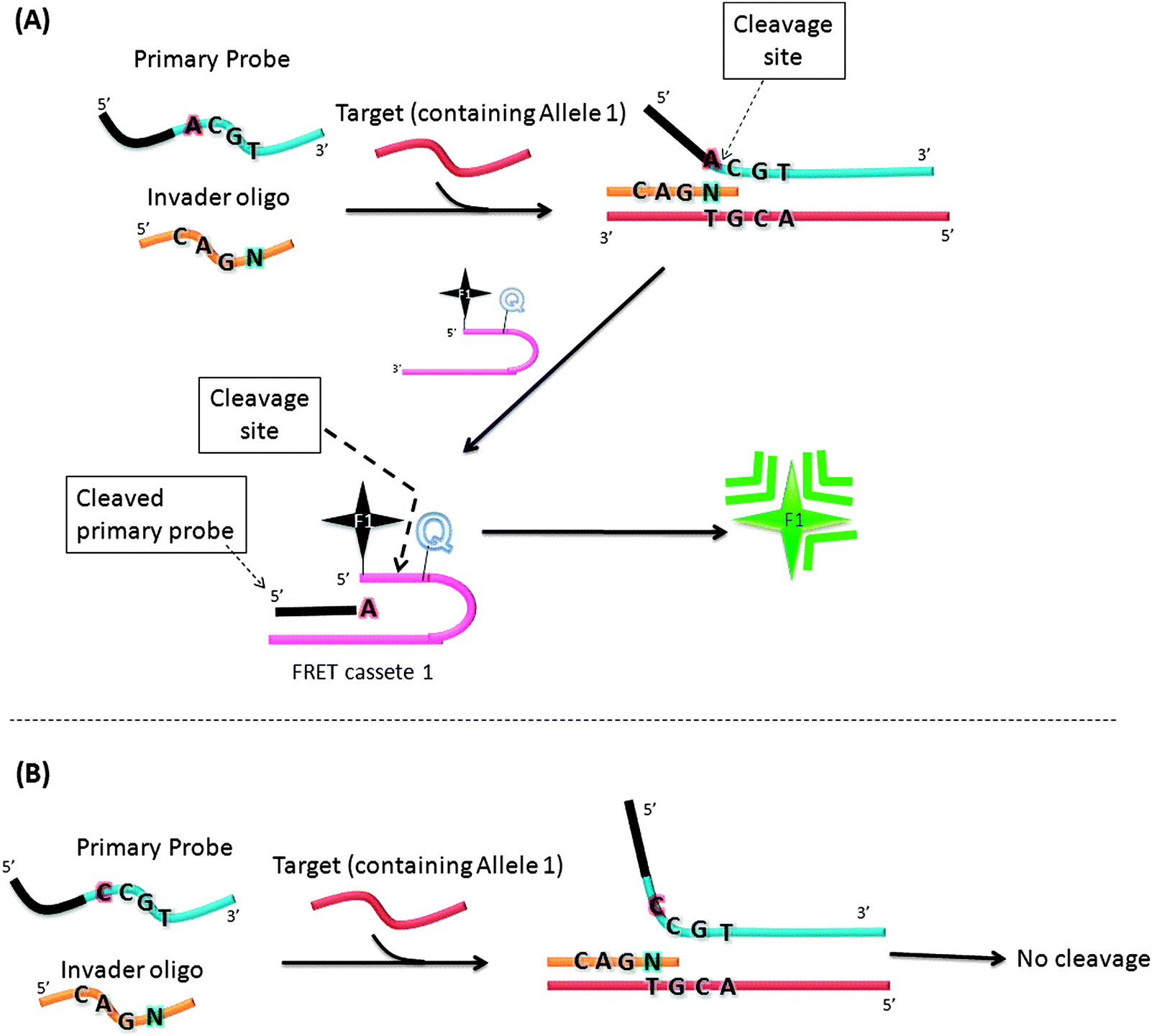
In an attempt to increase the sensitivity of the Invader assay, Hall et al. reported an interesting strategy that combined two Invader cleavage reactions into a single homogeneous assay (Fig. 12).55 The first cleavage reaction in the strategy by Hall and co-workers is similar to the original Invader assay strategy. However after the first cleavage step, the flap sequence, once released, acts as Invader oligonucleotide and anneals to another FRET cassette, which can be cleaved by flap endonuclease (see Fig. 12). Therefore a single target can lead to ∼n2 signals (where n is the signal generated by the first generation Invader assay).
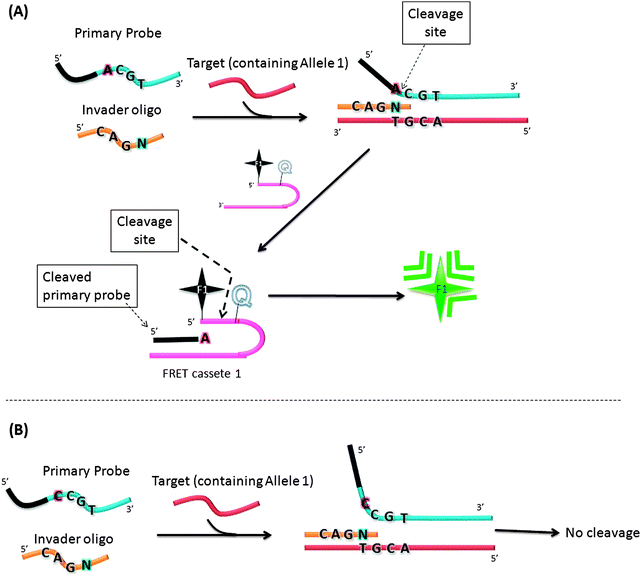 |
| | Fig. 12 A second-generation Invader assay, using a serial signal amplification strategy. (A) When complementary primary probe was used, cleavage reaction occurred. (B) When non-complementary primary probe was used, there was no cleavage. (Adapted from ref. 55 with permission. Copyright 2000, National Academy of Sciences, U.S.A.) | |
The Invader assay has been used to detect the factor V gene (named Leiden mutation, a well-known hereditary risk factor for venous thrombosis),54 genes responsible for Japanese hearing loss,56 human papillomavirus DNA,57 microRNA from HeLa and Hs578T cells,58 and herpes simplex virus types 1 and 2,59 amongst others.
3.05 Rolling circle amplification (RCA)
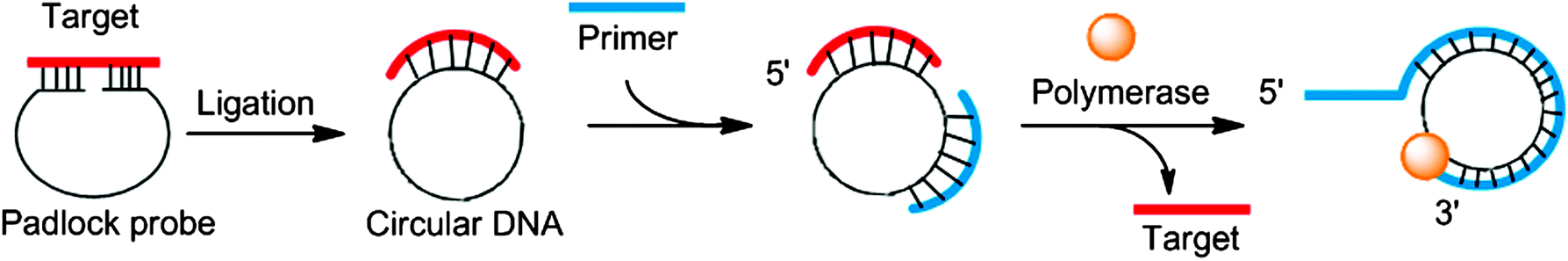
In the 1990s it was reported that DNA polymerase can utilize circular templates to make a long linear strand, containing multiple copies of a given sequence and this process was referred to as rolling circle amplification (RCA).60–62 RCA can be coupled to the ligation of a padlock probe (in the presence of a target analyte) to provide sensitive detection of nucleic acids.61,63 The padlock probe is a single probe that contains 3′- and 5′-end sequences that bind to target sequences in a juxtapose manner to aid a ligase-mediated circularization of the padlock probe if the 5′-end is phosphorylated (Fig. 13). After circularization of the padlock probe, a primer and DNA polymerase are used to produce long repeating DNA sequences, which can then be detected via a variety of methods (Fig. 13).64–68
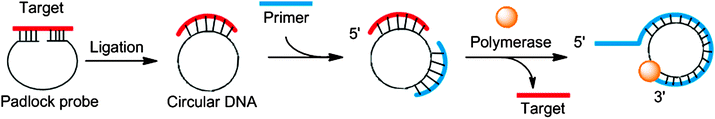 |
| | Fig. 13 Padlock probes and rolling circle amplification. | |
The padlock probe has been used to detect several disease-related genes,69 such as cystic fibrosis transmembrane conductance regulator (CFTR) G542X mutation (leading to cystic fibrosis),61 Epstein–Barr virus (EBV) in human lymphoma specimens,70 influenza A H1N1 and H3N2 mutations,71 porcine circovirus type 2,72 and Listeria monocytogenes,73 among others. In a recent interesting development, Kool and co-workers reported a ligase-free padlock-type probe assay using templated chemical ligation to make the circular probe for RCA.74
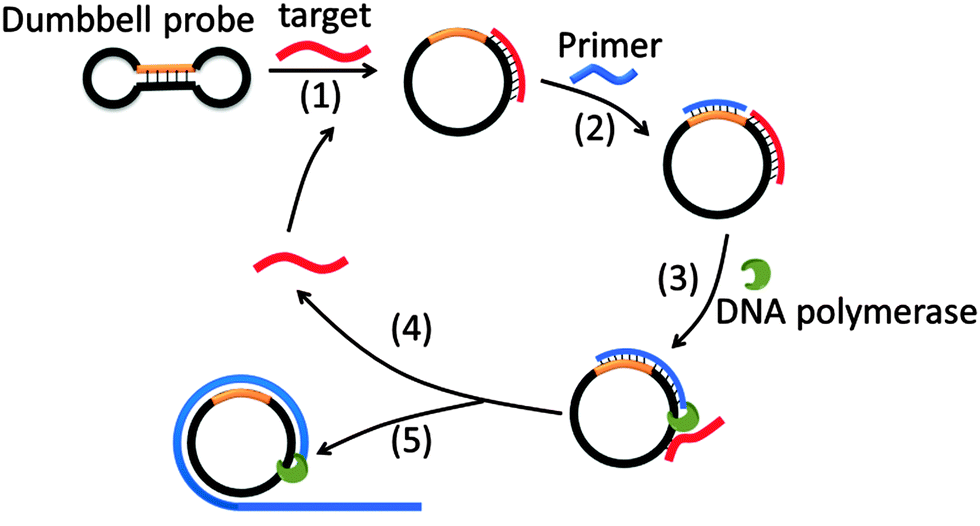
Target sequence recycled rolling circle amplification (TR-RCA) is based on RCA but does not utilize ligases as is done in padlock probes (Fig. 14).75 In the original RCA technique, the target facilitates the circularization of the probe, which is then used as a template for RCA. However, for TR-RCA, the circular DNA template that is required for the amplified sensing is already circularized but the primer-binding site is “sequestered” in the duplex region of the dumbbell. Upon target binding, the duplex region opens up to enable the primer to bind for the RCA process to begin. TR-RCA has been used to detect fM target.67,75 RCA has been used extensively to detect nucleic acids and space limitations prevents an exhaustive description of all of the applications.67,76 Of note, it has been used to detect mRNA expression levels in individual cells as well as microRNA, both in vitro and in situ.77
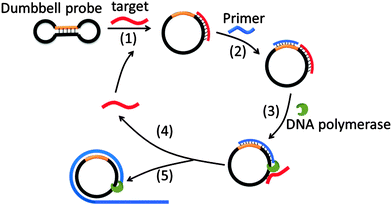 |
| | Fig. 14 Target sequence recycled rolling circle amplification (TR-RCA). Target binds to dumbbell probe to open the dumbbell structure into a circular template (step-1). The circular template can then be amplified via the regular RCA mechanism. (Copied from ref. 75 with permission. Copyright 2013, Elsevier.) | |
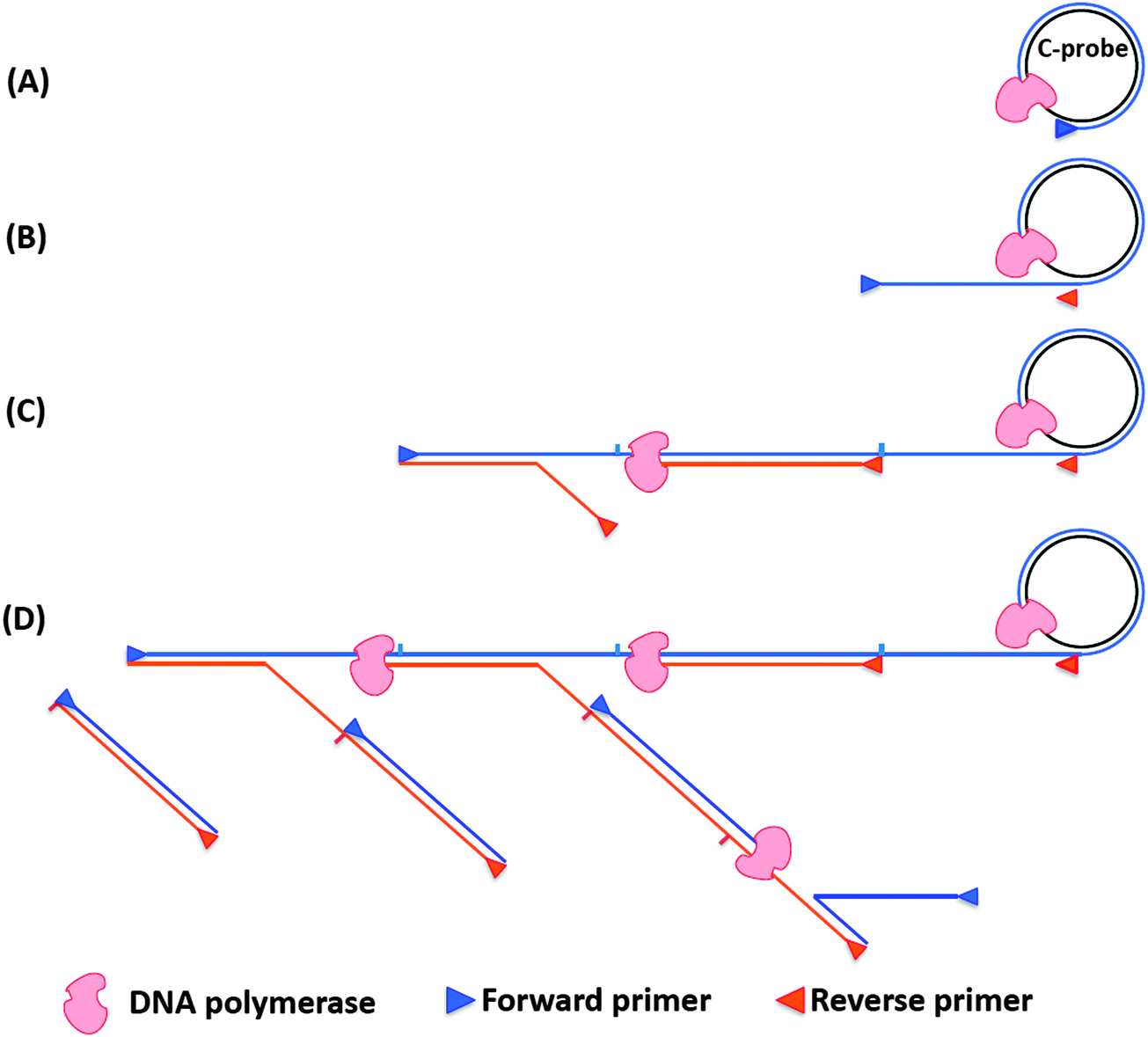
Rolling circle amplification is an efficient way to amplify RNA or DNA targets but the amplification is linear. To improve upon the sensitivity of RCA both forward and reverse primers have been used. During the normal RCA process, the forward primer produces a multimeric ssDNA, which then becomes the template for multiple reverse primers. The DNA polymerase then extends the reverse primer, and during the extension process the downstream DNA is displaced to generate branching or a ramified DNA complex. When all of the ssDNA strands have been converted into dsDNA, the process ceases. This type of amplification is called hyperbranched RCA (or ramification amplification, RAM), see Fig. 15.61,78
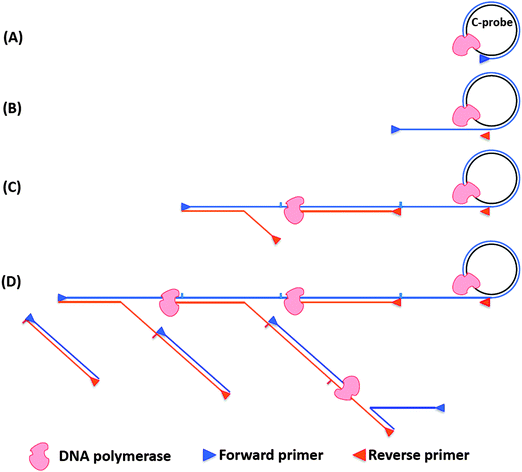 |
| | Fig. 15 Schematic representation of hyperbranched RCA. (A) Forward primer binds to ligated circular probe (C-probe) and is extended by DNA polymerase. (B) The continuous amplification generates a long ssDNA, which contains binding sites for reverse primer. (C) Reverse primers bind to the long ssDNA and are extended by DNA polymerase. (D) The nascent ssDNA generated from reverse primer extension can also serve as templates to synthesize new DNA. | |
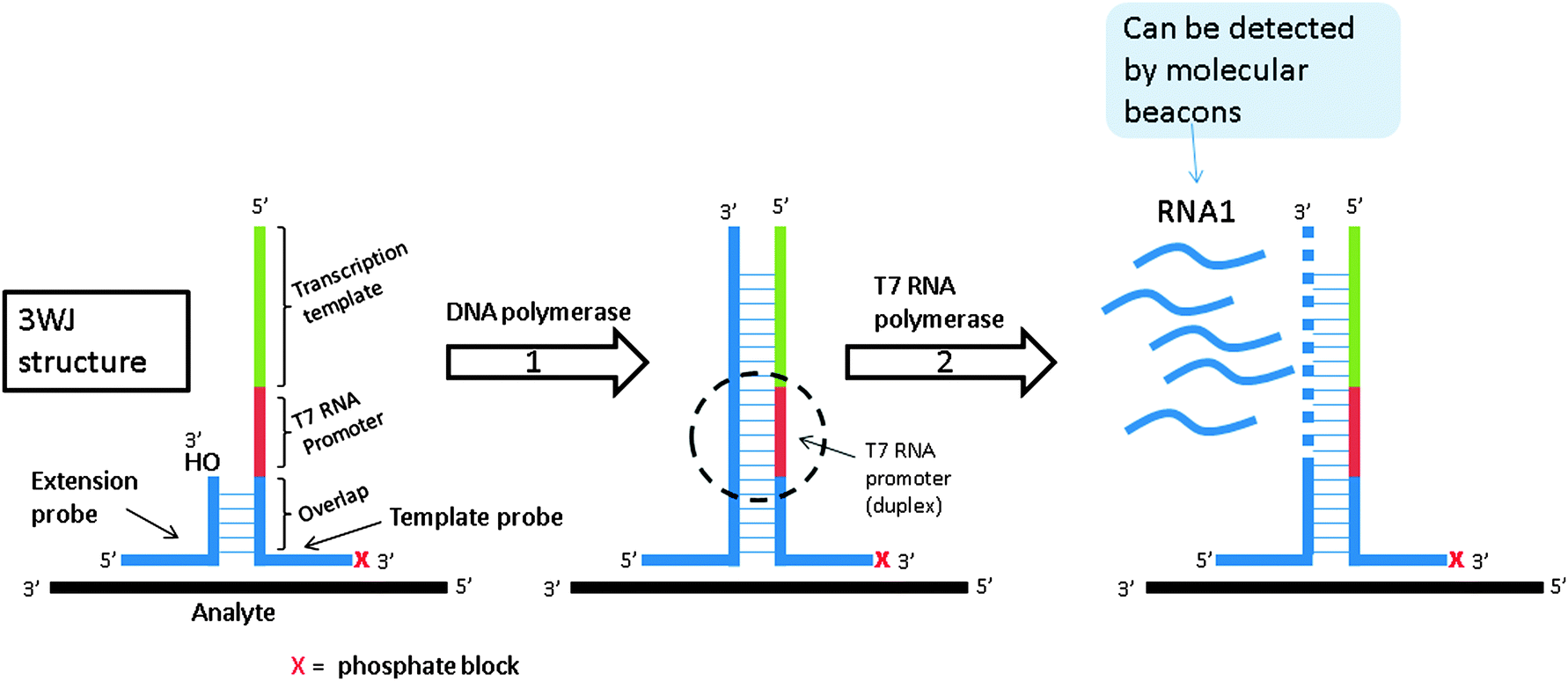
3.06 Signal mediated amplification of RNA technology (SMART)
Signal mediated amplification of RNA technology (SMART) is based on the formation of a three-way junction (3WJ) structure. As shown in Fig. 16, the amplification is initiated by hybridization of the analyte to the two probes. The two probes are also partially complementary to each other but this complementarity is not extensive enough for the two probes to associate with each other in the absence of a template under assay conditions. Upon target (or analyte) binding to the two probes a 3WJ is formed and a DNA polymerase elongates the shorter probe to produce a double stranded DNA. The longer probe contains a T7 promoter sequence so after polymerization, this promoter sequence (which is now a duplex) can facilitate the synthesis of RNA by T7 RNA polymerase (RNA1 in Fig. 16). Since a single DNA template can generate multiple RNA copies, SMART is an amplified sensing technology. The generated RNA can be detected by an enzyme linked oligosorbent assay (ELOSA) or molecular beacon for example.79
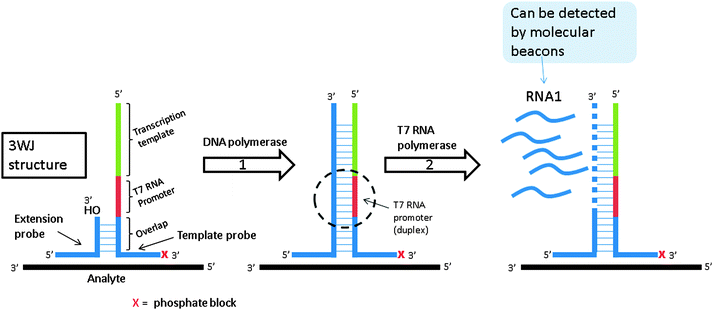 |
| | Fig. 16 Signal mediated amplification of RNA technology (SMART). Probes are designed such that they bind flanking regions of the DNA analyte and then subsequently bind one another. They do not form stable complexes in the absence of DNA analyte. Upon the formation of the three-way junction structure between probes and target, DNA polymerase extends the shorter probe. Once extended, the new double stranded DNA contains an active T7 polymerase promoter site, which can be used to make multiple copies of RNA. This RNA can then be detected via a variety of means. (Adapted from ref. 79 with permission. Copyright 2001.) | |
SMART is sensitive and has been shown to detect 50 pM analyte in a few hours.79 Genomic DNA and rRNA of E. coli has been detected with SMART, even when crude samples without sophisticated sample preparation were used79 SMART has also been applied to detect DNA and mRNA of marine cyanophage virus80 amongst others.
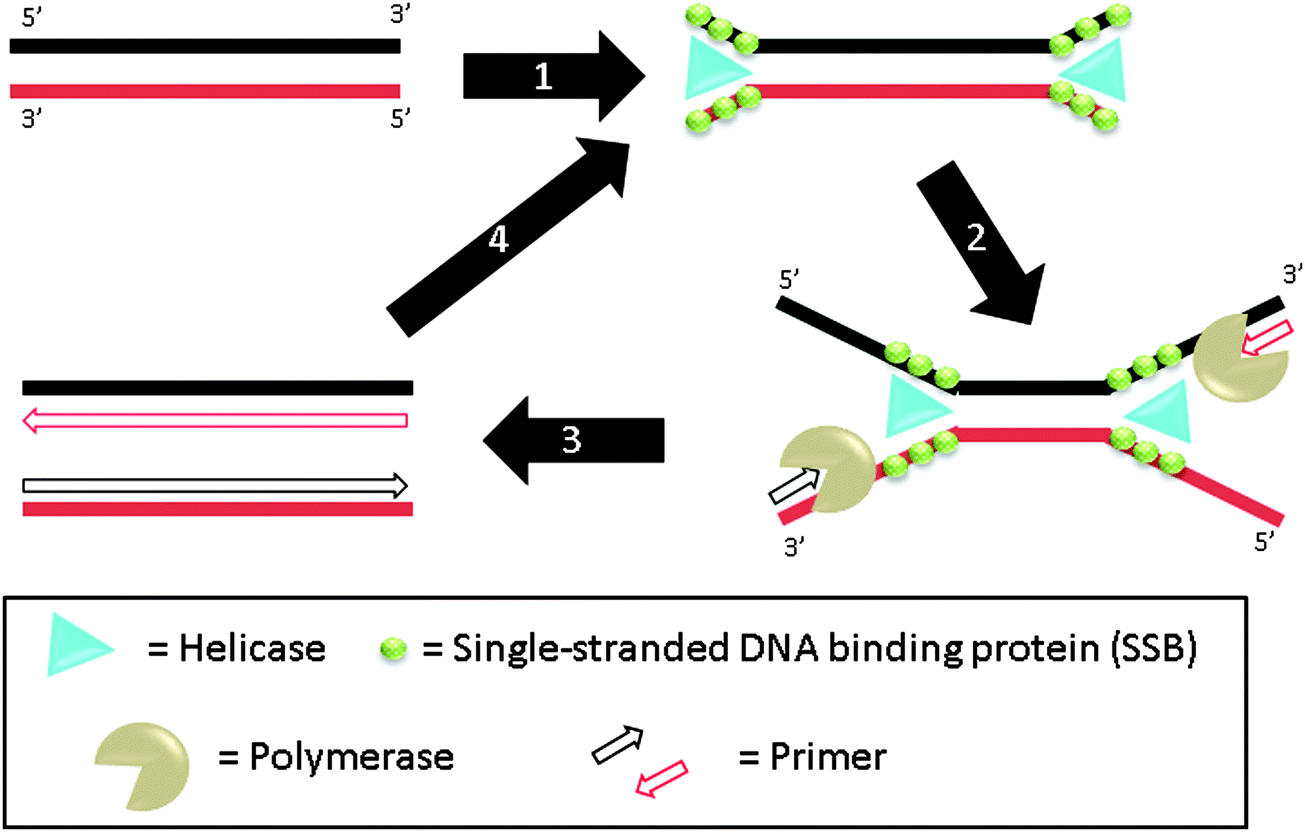
3.07 Helicase-dependent amplification (HDA)
Helicase-dependent amplification (HDA) is a polymerase-based isothermal amplification method, which uses helicase to separate double stranded DNA instead of heat.81 During the amplification, helicases unwind DNA duplexes and single-stranded DNA binding proteins (SSB) stabilize the resulting single strands (step-1), which hybridize to primers (step-2). DNA polymerase then extends the primers (step-3) and the newly formed duplexes can act as templates for the subsequent cycles (step-4) (Fig. 17). The whole process is performed at one temperature so thermal cycling is not required. By using a thermally stable helicase, the assay can be performed at 60 °C and this results in improvement in sensitivity and specificity and also eliminates the need for accessory proteins in this assay.82 A helicase–polymerase fusion complex was developed and shown to be able to amplify a 1.5 kb target. The specificity using the helicase–polymerase fusion complex was similar to that obtained using a separate DNA polymerase and helicase.83
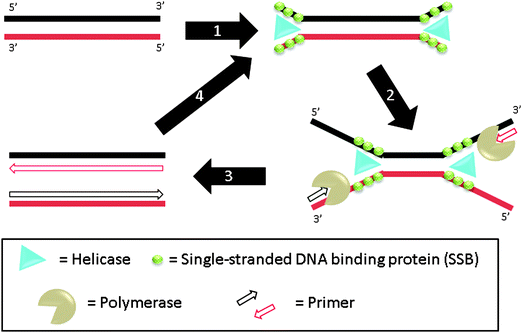 |
| | Fig. 17 Helicase-dependent amplification (HDA). During step-1, helicase unwinds DNA duplex to generate ssDNA regions. This process is aided by SSB (single-stranded DNA binding proteins) and accessory protein. The primers can then bind to the ssDNA regions, extended by polymerase (steps 2 and 3). The newly formed duplexes can then be amplified again via steps 1–3. (Adapted from ref. 81 with permission. Copyright 2004, Nature Publishing Group.) | |
HDA can be used on targets in complex biological matrices (such as crude bacterial lysates or blood) and can achieve one million-fold amplification81 and it has been used to detect multiple targets, such as the genomic fragments of Treponema denticola, and the parasite Brugia malayi.81
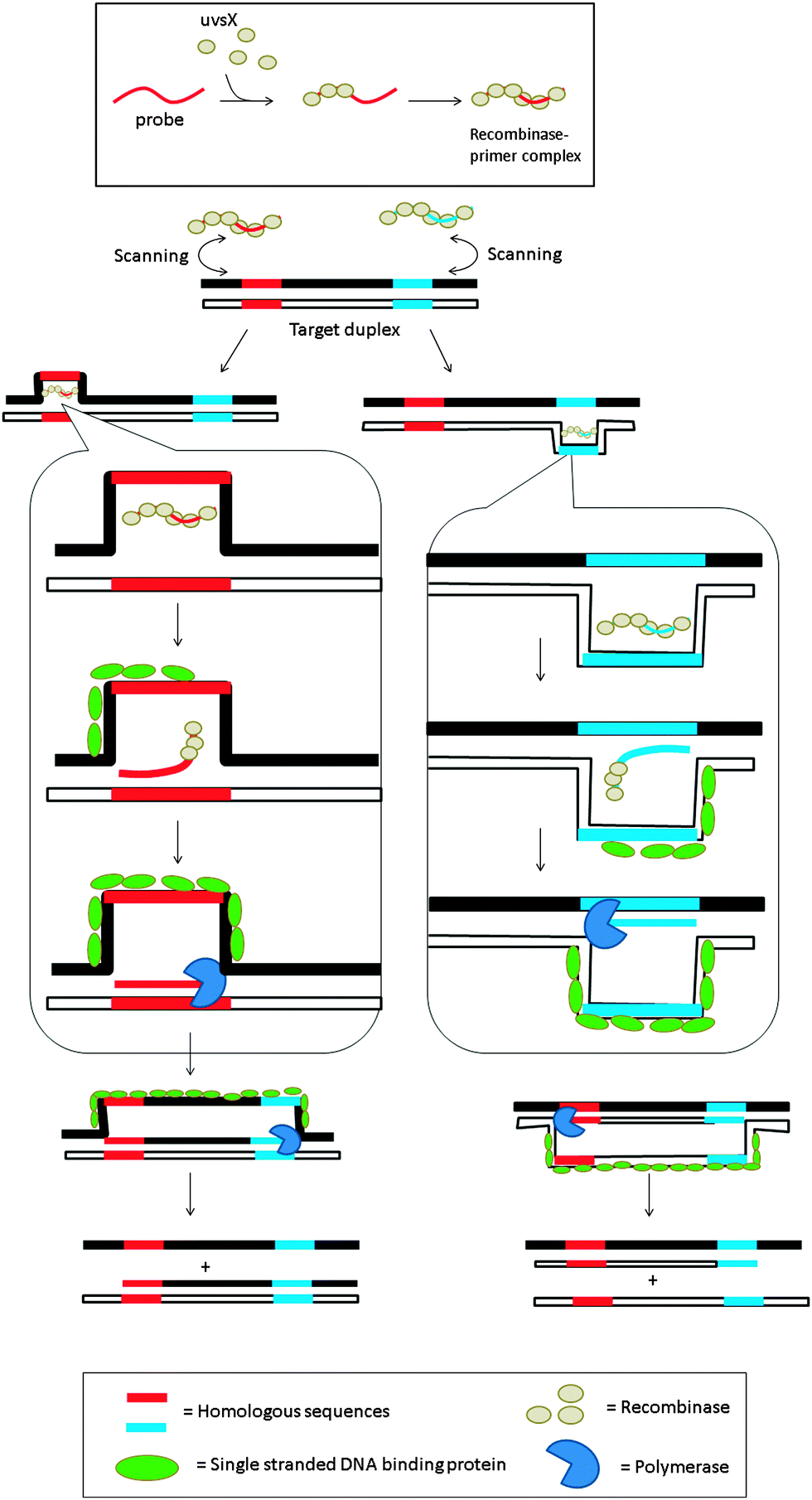
3.08 Recombinase polymerase amplification (RPA)
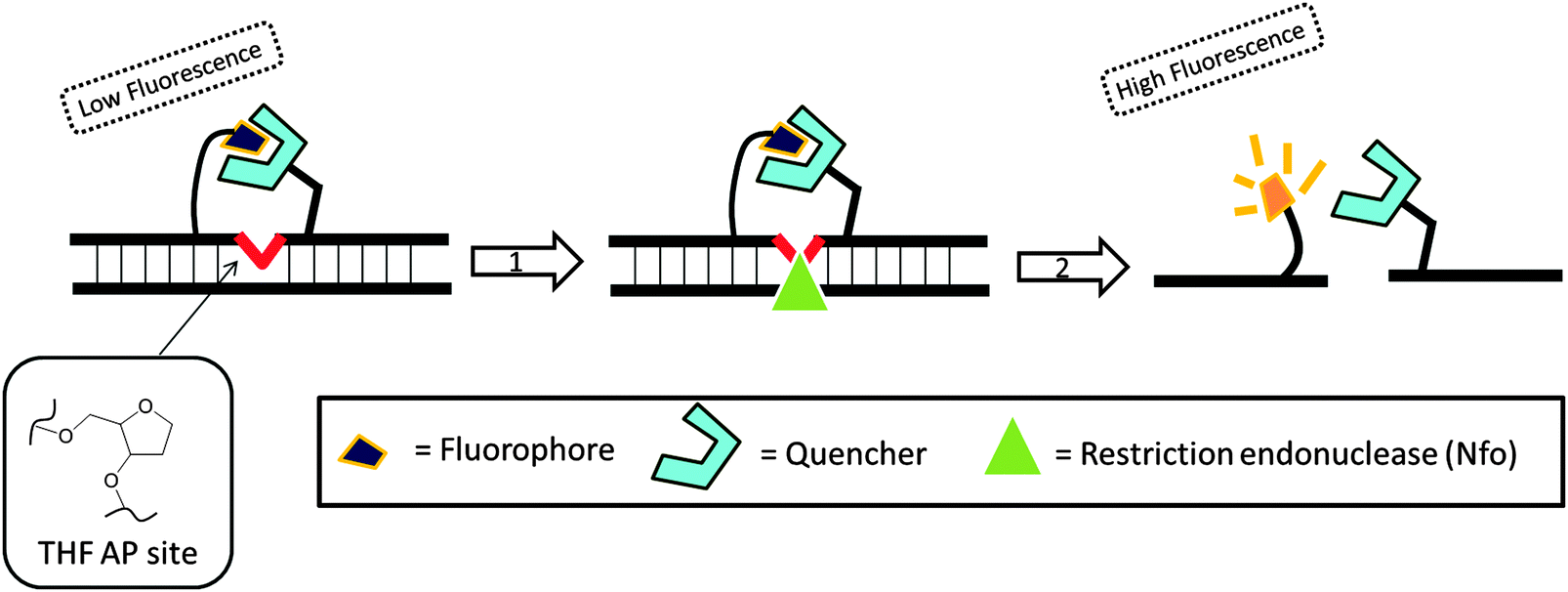
Recombinase is a protein, which catalyses the hybridization process of short oligonucleotide primers with the homologous target sequence. During a recombinase polymerase amplification (RPA), the recombinase–primer complexes initiate the amplification cycle by scanning the duplex template for homologous sequences. Once the specific site is found, the enzyme opens the double strands to allow for the hybridization of the primer and target sequence. This process is aided by ssDNA binding proteins, which bind to the “melted” DNA (see Fig. 18). The primer is then elongated with the newly synthesized strand displacing the old strand. The newly synthesized duplex can then act as a template for the next cycle (therefore exponential amplification is achieved).84 RPA is fast and sensitive, but background noise can be an issue. To eliminate background noise, a primer that contains a cleavage site for the double-strand-specific Escherichia coli endonuclease IV (Nfo), which recognizes and cleaves a tetrahydrofuran abasic site in duplexes, is used. This primer can be used for the DNA polymerase extension step only after it has been cleaved by the endonuclease to reveal a 3′-OH end. The primer can be labeled with a fluorophore and quencher so that the cleavage reaction is accompanied by enhancement of fluorescence (Fig. 19). The endonuclease cleavage step in RPA serves as an additional proofreading step to eliminate background noise.84
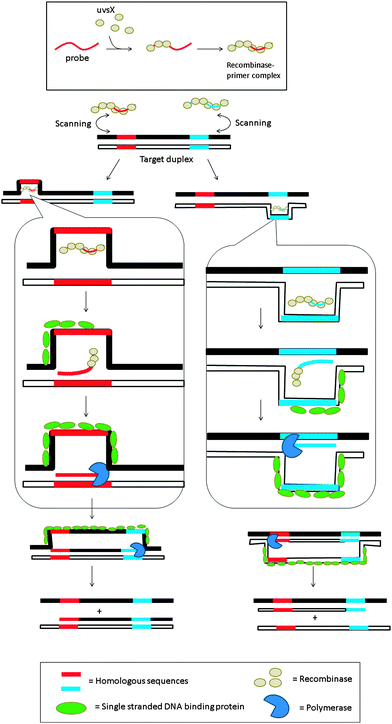 |
| | Fig. 18 Recombinase polymerase amplification (RPA) begins with the formation of the nucleoprotein complex, consisting of an oligonucleotide probe and a recombinase. The primer–recombinase complex then scans the duplex DNA until a sequence that is complementary to the primer is reached, at which point the recombinase aids the formation of a primer–DNA complex. DNA polymerase can then access the replication fork and extend the primer. (Adapted from ref. 84 with permission. Copyright 2006, Public library of science.) | |
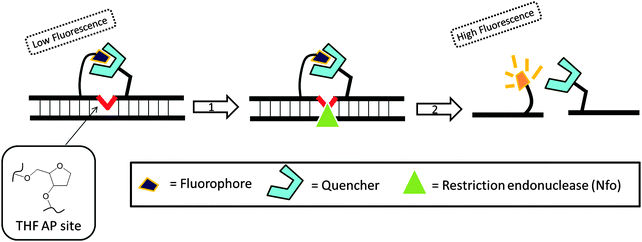 |
| | Fig. 19 Fluorescent signal is generated after the probe is cleaved by double-strand specific endonuclease, Nfo. THF AP site = tetrahydrofuran abasic site. (Copied from ref. 84 with permission. Copyright 2006, Public library of science.) | |
Using RPA, three genetic markers, apolipoprotein B, sex-determining region Y and porphobilinogen deaminase from complex human genomic DNA were amplified, and Bacillus subtilis genome (only 10 copies) could be detected.84 The reaction conditions for RPA are however stringent and detection using crude samples could be problematic.
3.09 Nicking endonuclease signal amplification (NESA)/nicking endonuclease assisted nanoparticle activation (NEANA)
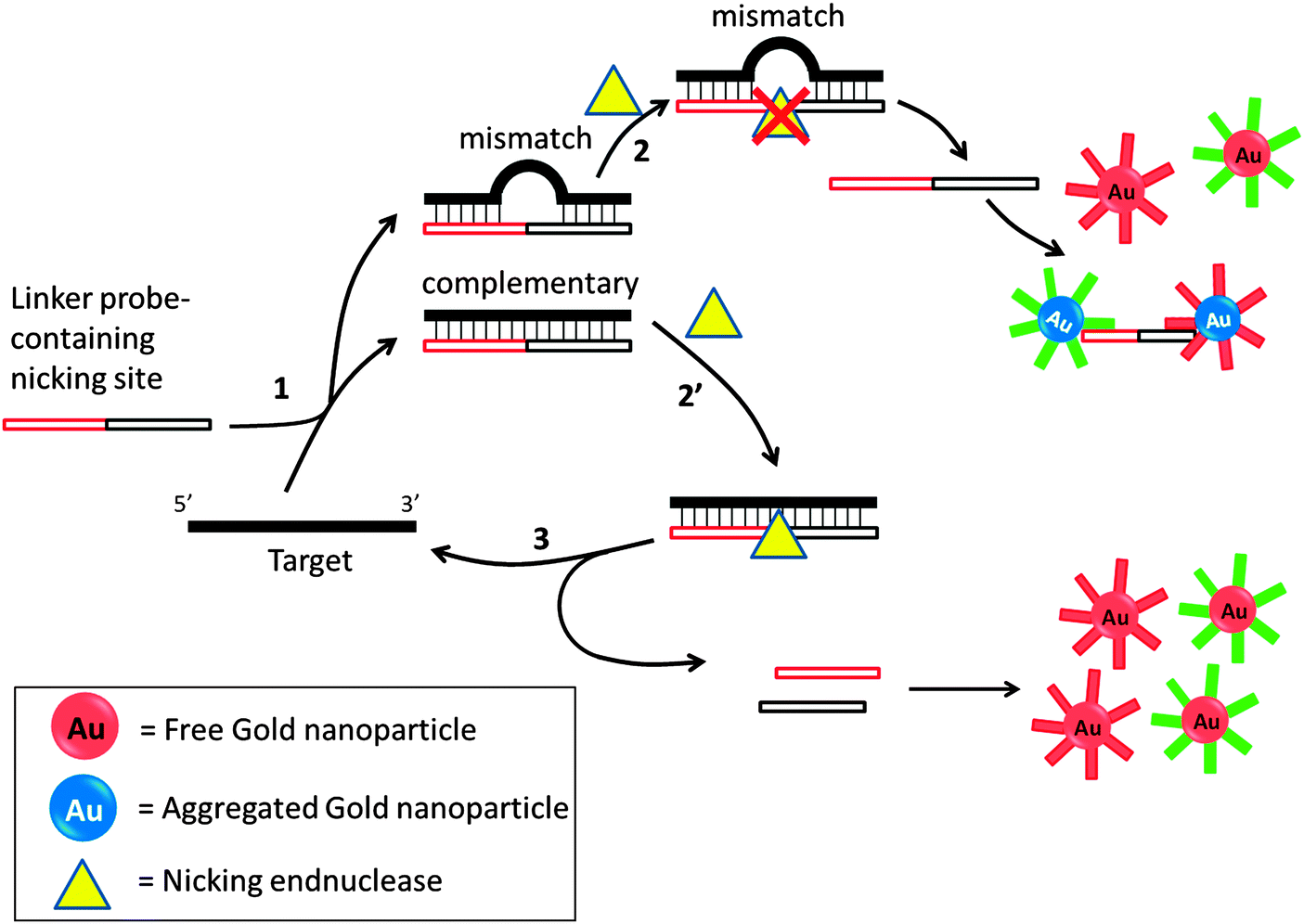
Several groups have developed amplification detection of nucleic acids detection based on nicking endonucleases.66,85–88 Nicking endonuclease usually does not cleave ssDNA, but recognize a double stranded cognate site and nick only one strand. In an interesting colorimetric assay that utilizes nicking endonuclease, developed by Liu et al., a probe can aggregate oligonucleotide-labeled gold nanoparticle to cause a color change. If a target is fully complementary to the linker probe, this linker probe would bind to it and a nicking endonuclease would cleave the probe. The cleaved probe is now unable to aggregate the gold nanoparticle so the intensity of color change would be less in the presence of the target analyte. If the target contains an SNP, then cleavage by the nicking endonuclease is inhibited so one could distinguish between SNPs using this method (Fig. 20).85 Because the target can be recycled in this assay, amplified sensing is achieved. Despite the simplicity of this assay, the requirement of a nicking endonuclease cognate site in the target sequence precludes the possibility of detecting all target sequences.
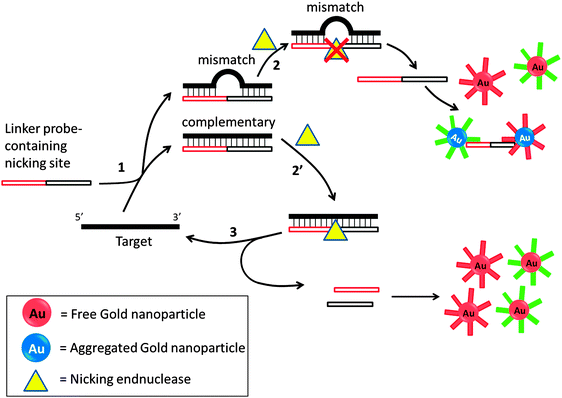 |
| | Fig. 20 Nicking endonuclease assisted nanoparticle amplification (NEANA). This method is designed to detect ultralow concentrations of single-stranded DNA sequences. A linker strand is used in the detection, which contains complementary sequences to the target DNA. If there is a mismatch, the linker strand dissociates from the target DNA at an elevated temperature. The free linker strand is able to aggregate gold nanoparticles and induce a concomitant color change. If the linker perfectly matches target DNA, then a nicking endonuclease cleaves the linker strand and no aggregation happens and no color change will be detected. (Adapted from ref. 85 with permission. Copyright 2009, John Wiley and Sons.) | |
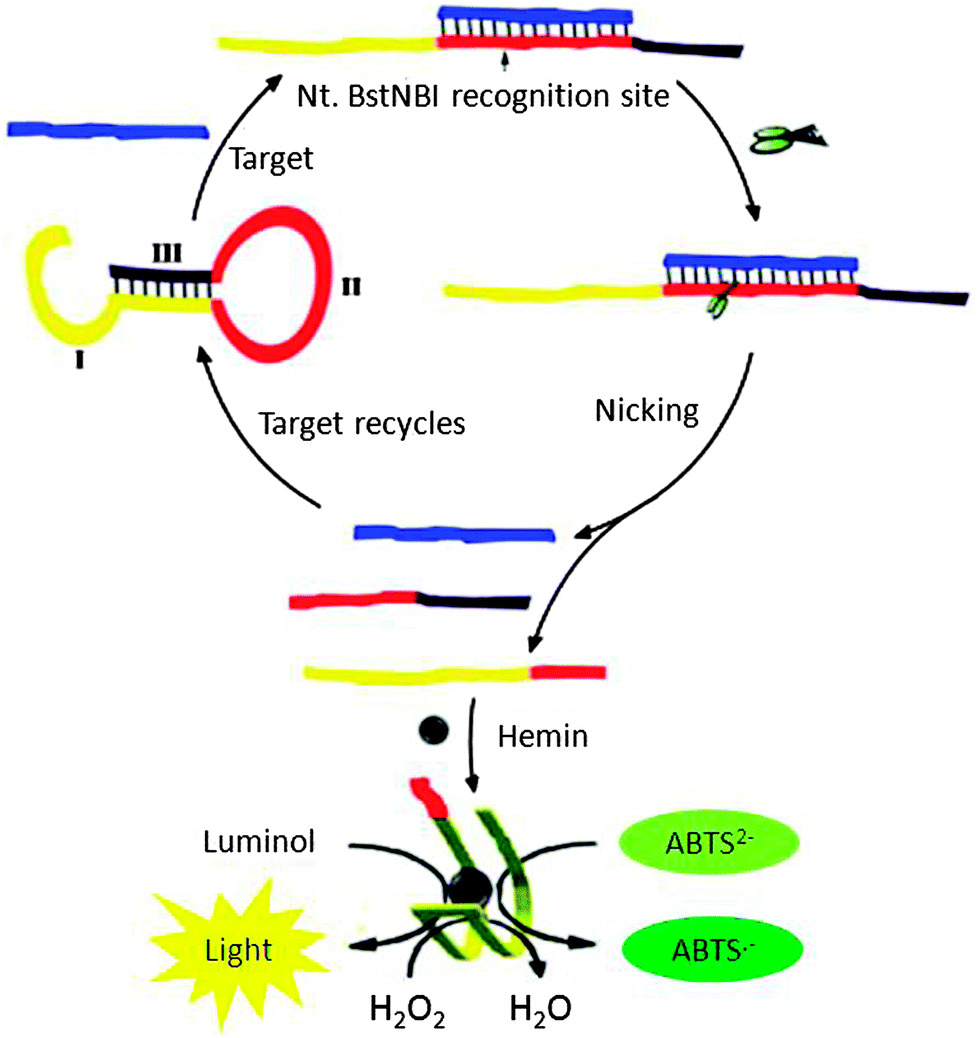
Yang et al. designed a probe, which could form a G-quadruplex upon cleavage by NEAse after binding to the target (see Fig. 21). Upon binding to a target, the NEAse enzyme cleaved the target, which could then folds into a G-quadruplex. G-quadruplexes are peroxidase mimics so this method could be used for colorimetric or chemiluminescence detection (Fig. 21).88 The detection limits using the combined NEAse/G-quadruplex strategy are 10 pM using a colorimetric reagent and 0.2 pM using a chemiluminescence reagent.
 |
| | Fig. 21 A combination of NESA and G-quadruplex peroxidises allows for a colorimetric detection or chemiluminescence detection. Region I in the hairpin DNAzyme probe is the HRP DNAzyme sequence, and it partially hybridizes with region III so the HRP DNAZyme cannot form. Region II is complementary to the target sequence, which contains a NEase recognition sequence. Upon target binding to the probe, a nicking site is unveiled and after cleavage with NEase, the HRP DNAzyme can now form the correct structure to catalyse peroxidation reactions. (Copied from ref. 88 with permission. Copyright 2011, Elsevier.) | |
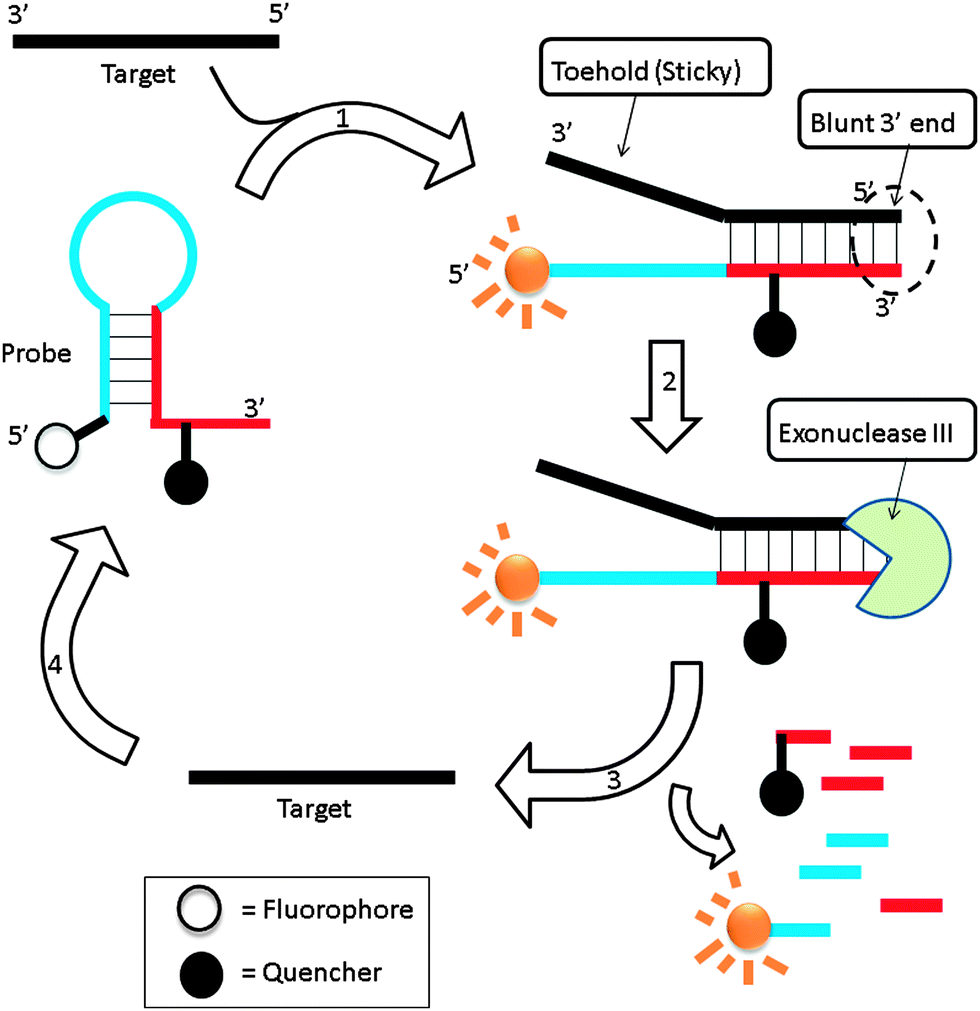
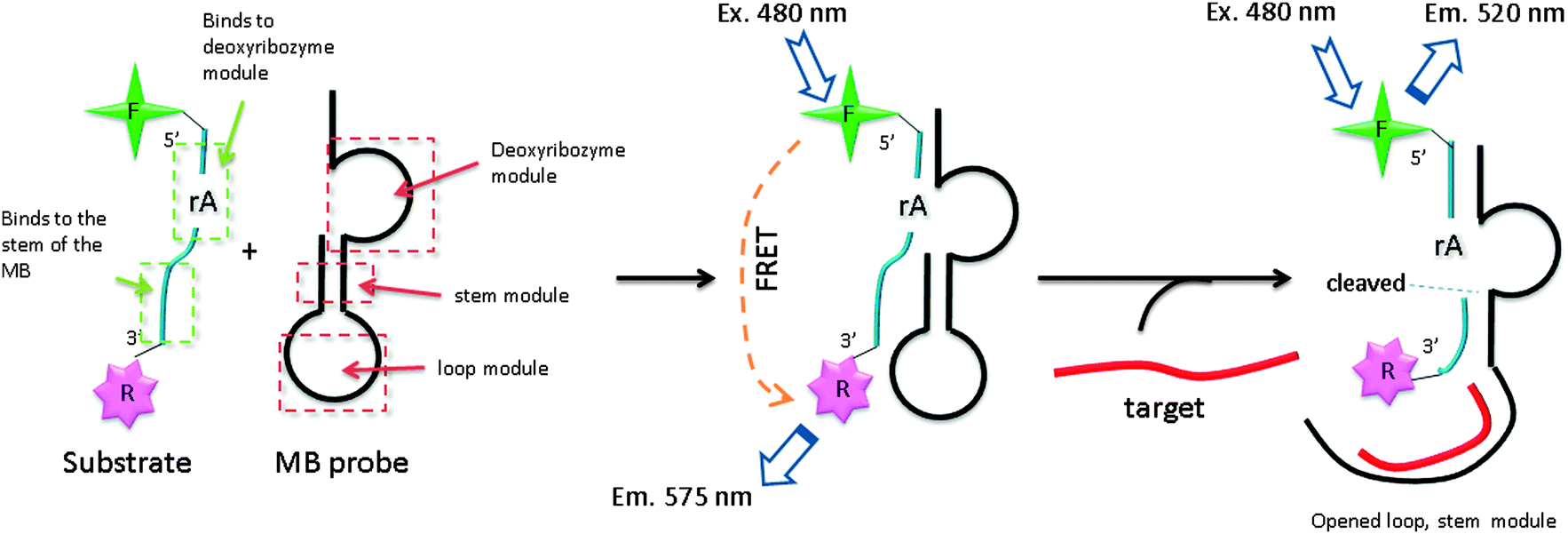
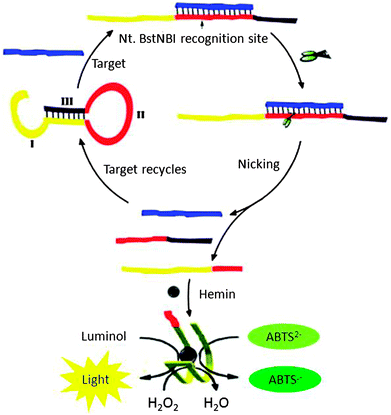
3.10 Exonuclease III-aided target recycling approach
Exonuclease III is a dsDNA specific nuclease and hardly cleaves ssDNA. It is now finding increased use in nucleic acids diagnostics.89,90 Plaxco et al. demonstrated an isothermal amplified detection of nucleic acids, using a catalytic MB approach by utilizing exonuclease III.89 Exonuclease III non-specifically digests double strands DNA, from a blunt 3′ end. The MB probe, which contains a “sticky” 3′-ssDNA end (toehold), is not cleaved by exonuclease III. Upon binding to a target DNA, a blunt 3′ end is created and this can be digested by exonuclease III (Fig. 22). Fluorescent signal enhancement can therefore be obtained if the MB probe is appropriately labelled with a fluorophore and quencher. Catalytic MB that utilizes exonuclease III provides a simple and sensitive detection method and analytes as low as 20 aM could be detected (although a long assay time of 24 hours is required for low copy number analytes).
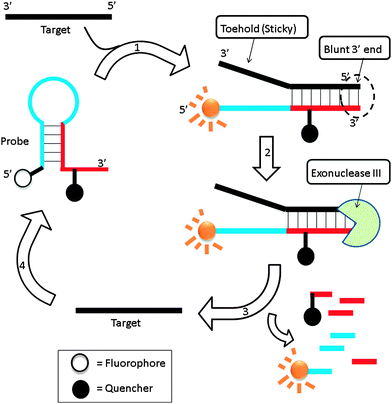 |
| | Fig. 22 Binding of probe to target facilitates the cleavage of the probe by exonuclease III. Once cleaved the target is free to bind and open another molecular beacon, which results in an amplified fluorescent signal. (Adapted from ref. 89 with permission Copyright 2010, American Chemical Society.) | |
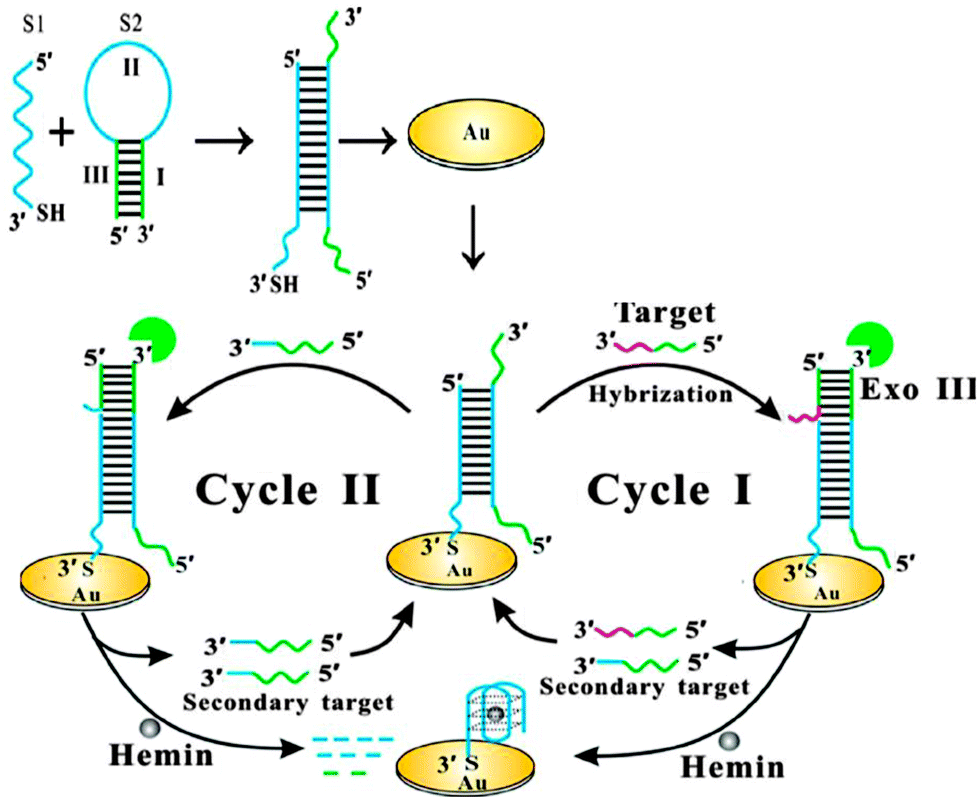
Bo Tang and co-workers reported an interesting electrochemical detection of nucleic acids using exonuclease III and probes that could form G-quadruplexes after an exonuclease III digestion (see Fig. 23).91 In this approach, a duplex probe that is attached to a gold electrode contains a single strand overhang, which can bind to a target DNA. One strand of a duplex probe is G-rich and could potentially form a G-quadruplex. After the target binds to the single strand portion of duplex probe, exonuclease III cleaves one of the probe strands and this results in the other strand folding into a G-quadruplex. The reassembled G-quadruplex then binds to hemin and the bound hemin could be detected electrochemically. A limit of detection of 10 fM was reported for this approach.
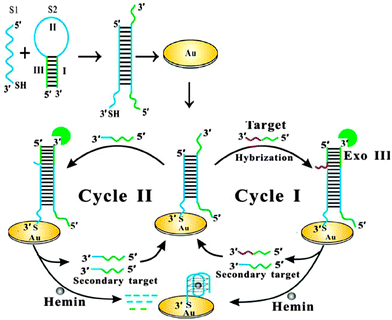 |
| | Fig. 23 Electrochemical detection of target after G-quadruplex assembly, after exonuclease III degradation. (Copied from ref. 91 with permission. Copyright 2013, American Chemical Society.) | |
3.11 Junction or Y probes
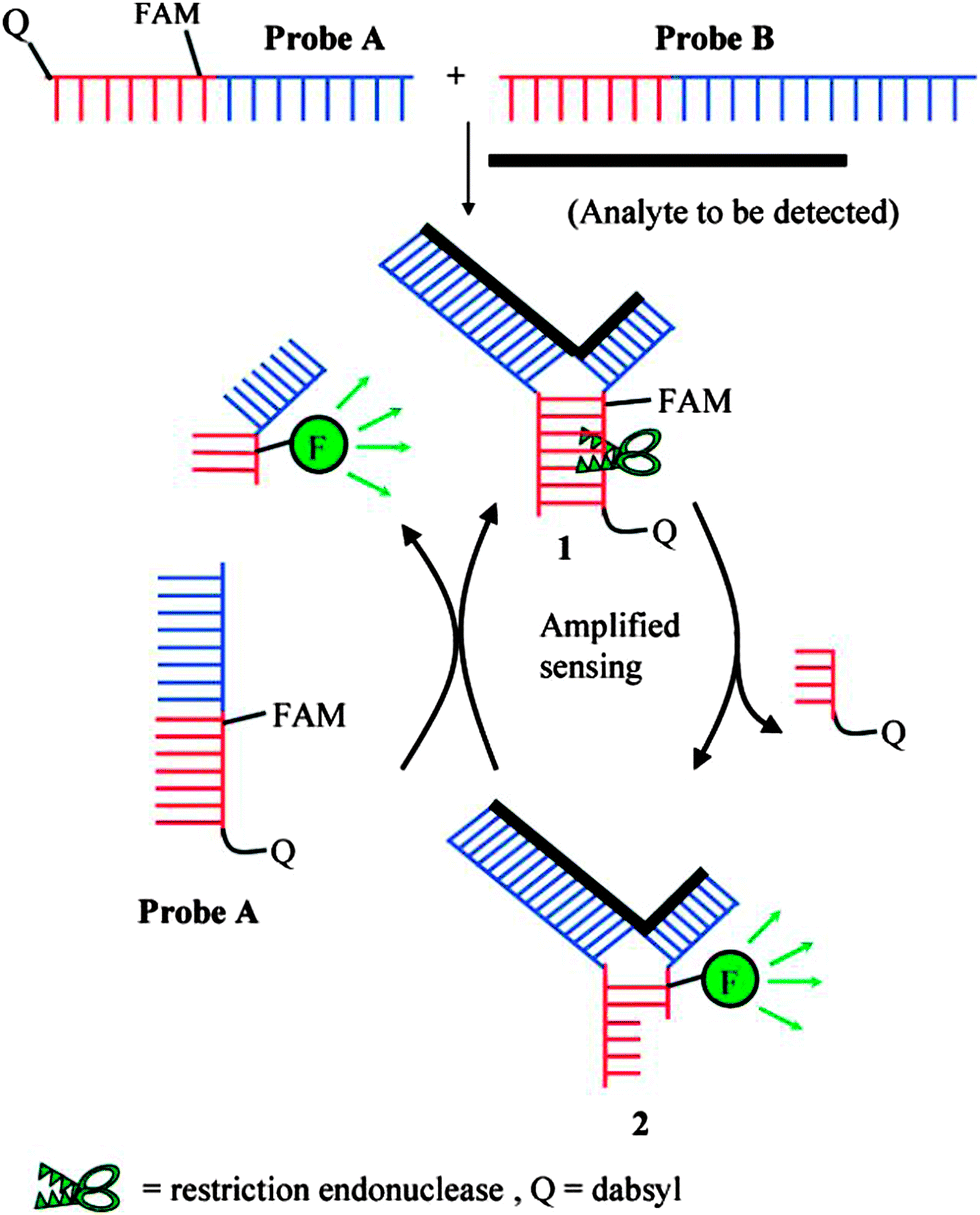
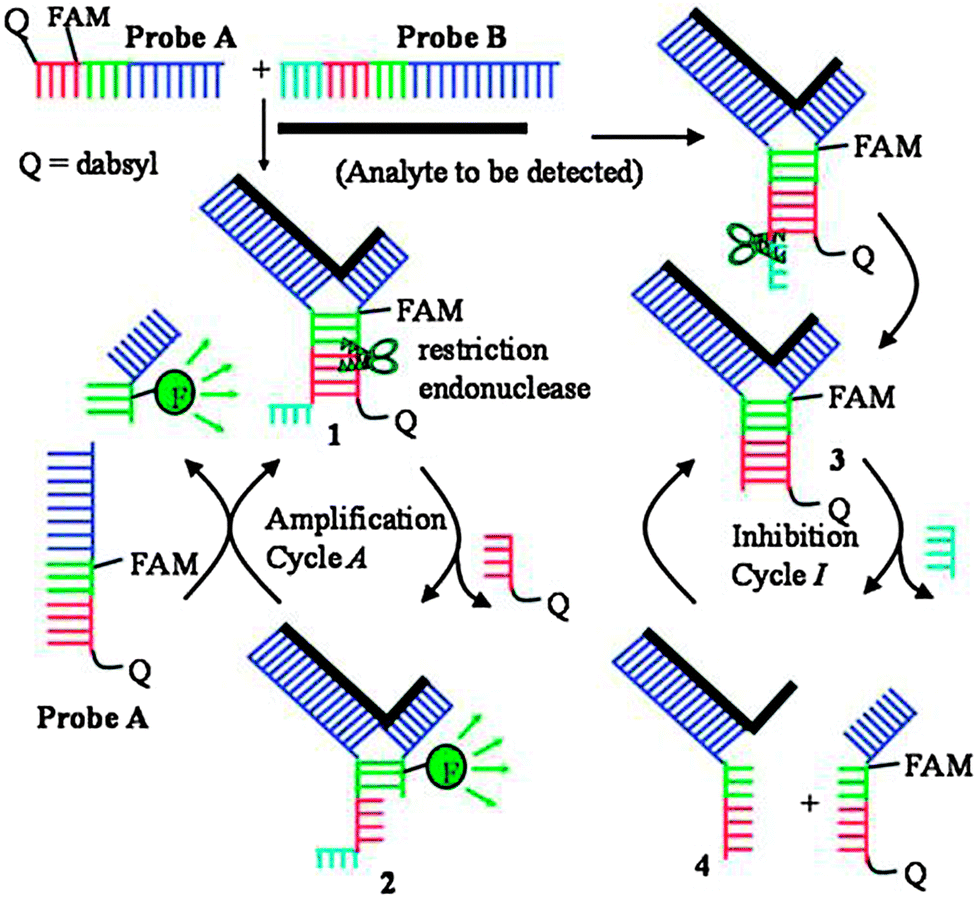
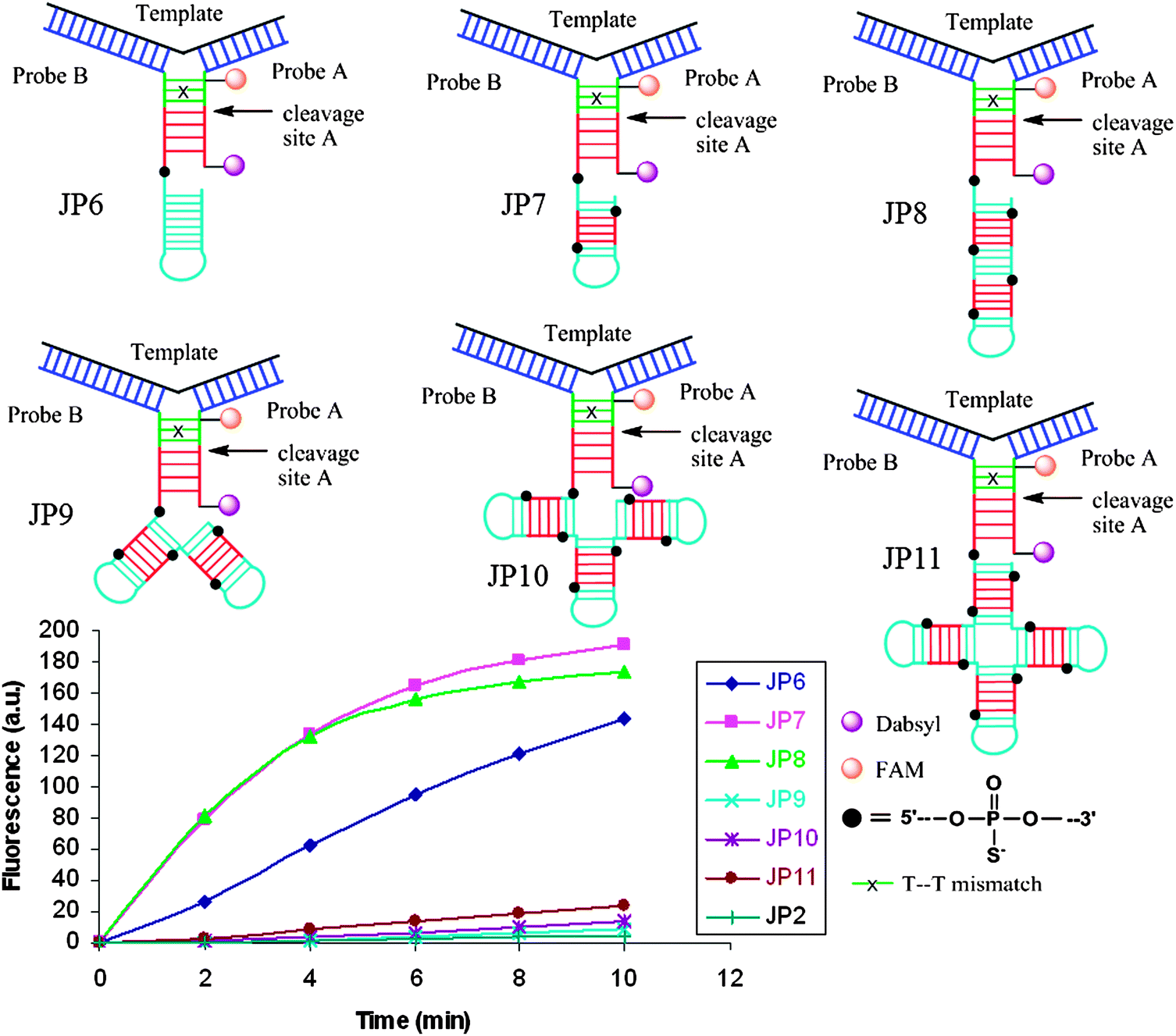
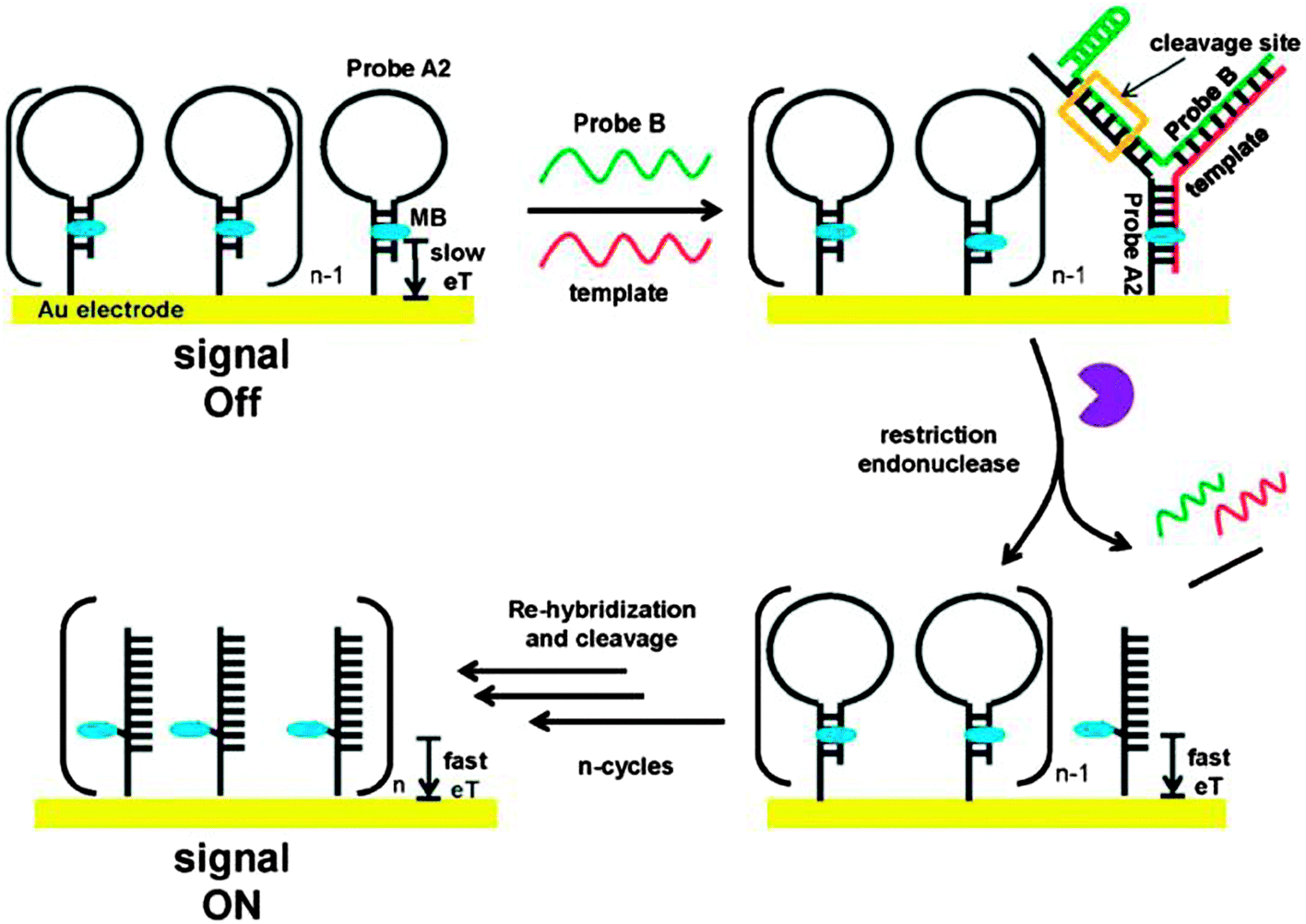
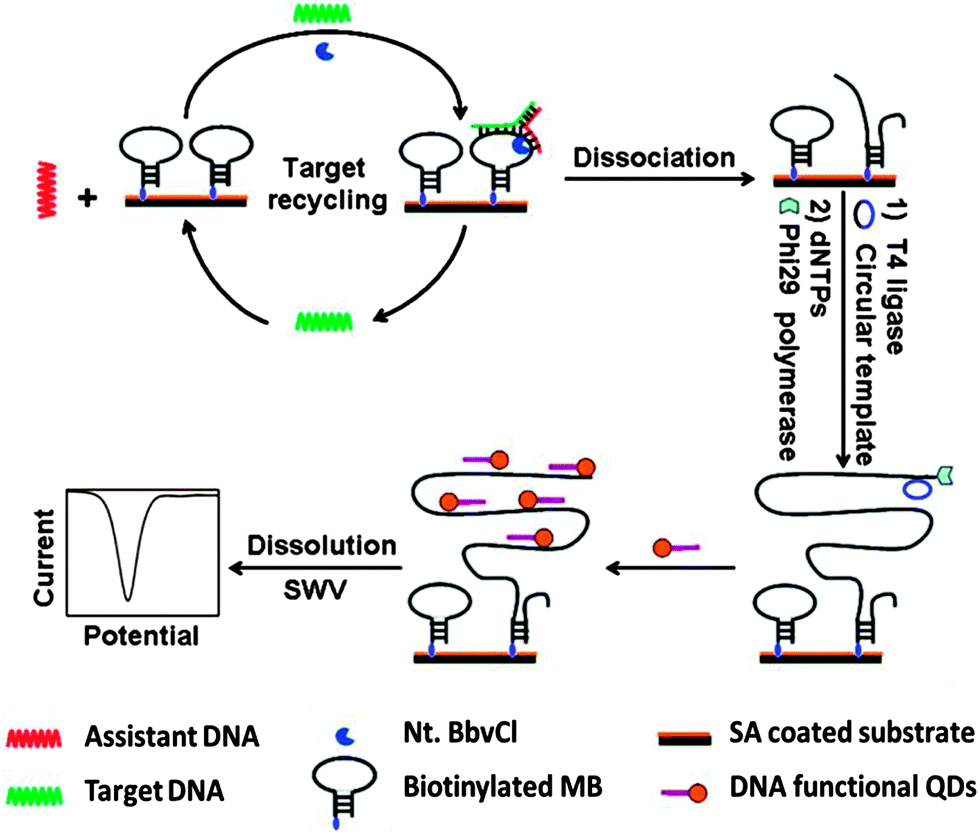
Restriction endonucleases (REAses) cleave duplex DNA to form either a blunt end or a sticky end. These enzymes are finding increasing use in nucleic acid diagnostics. There are over 3500 characterized REAses so there are many opportunities to use this class of enzyme for varied diagnostic purposes. One of the disadvantages of using REAses for nucleic acid diagnostics is the need to have a specific recognition site in the target analyte. It has been demonstrated that this requirement to have the recognition site in the target sequence can be obviated when probes that form Y or junction structures are used (see Fig. 24).92 In the junction probe strategy, two probes that have limited complementarity (usually 4–6 base pairs) do not bind to each other to an appreciable extent but when an analyte that binds to the two probes in a juxtapose manner is present, a Y-structure is formed (a Template-enhanced Hybridization Process, TeHyP). If the segment of complementarity between the two probes contains a restriction endonuclease recognition site, then the formation of the Y-structure would lead to REAse cleavage and the process can be repeated, leading to amplified sensing under isothermal conditions. In the last few years, various modifications to the junction probe (JP) platform have been reported.93–97 For example, it has been shown that the incorporation of a phosphorothioate moiety in one of the probes (called the helper probe, and not cleaved by the REAse) results in a nicking scenario and a more sensitive detection.93 It has been suggested that the incorporation of a phosphorothioate into one of the probes of the JP system prevents an inhibition cycle (see Fig. 25). Sintim and co-workers have shown that the architecture of the probes (they refer to these as probe overhangs) can have dramatic influences on the rate of Y-structures by REAses (see Fig. 26).93,95 Others have also reported a nicking-based junction probe strategy (using nicking endonucleases)98 but the nicking endonucleases used required 7 base pairs for recognition98 (compared to only 4 required by REAses, such as BfuCI). It has been shown that increasing the hybridization number between the two JP probes can lead to significant background noise93 so REAses, with phosphorothioate protection, might be better suited for a more sensitive detection than nicking endonucleases. Electrochemical JP has also been described, using methylene blue-labeled probe that is affixed to a gold electrode for a turn-off97 and turn-on94 detection (see Fig. 27). In a recent interesting development, Ju and co-workers combined junction probes, immobilized on a surface, with rolling circle amplification to achieve an impressive detection limit of 11 aM (see Fig. 28).99
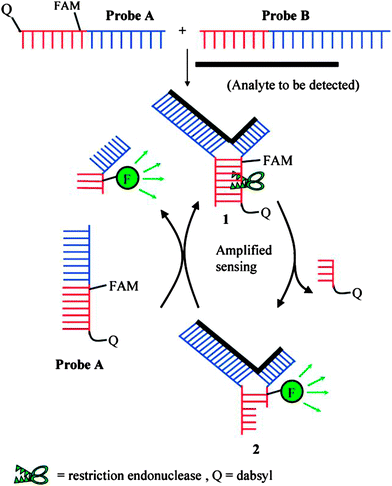 |
| | Fig. 24 Junction probe via Template Enhanced Hybridization Processes (TeHyP). Due to limited complementarity between the two probes, the complex formed between probes A and B is not stable. When an analyte is present, each probe will hybridize to a portion of the analyte, and subsequently hybridize to the other probe to generate a double stranded region that contains a cleavage site for REAse. Cleavage by REAse results in a complex that is less stable than the uncleaved Y-structure and therefore new probes can associate with the analyte, giving rise to an amplification cycle. (Copied from ref. 92 with permission. Copyright 2008, American Chemical Society.) | |
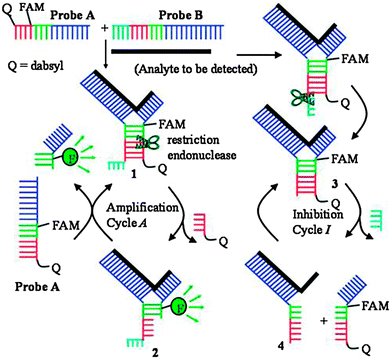 |
| | Fig. 25 An inhibition cycle reduces the sensitivity of first-generation junction probe. The incorporation of phosphorothioates eliminates this inhibition cycle. (Copied from ref. 93 with permission. Copyright 2010, Royal Society of Chemistry.) | |
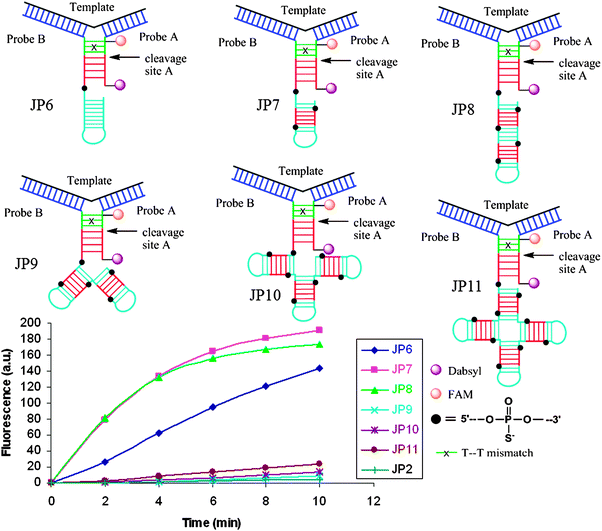 |
| | Fig. 26 The architecture of Probe B (Helper probe) can have a dramatic influence on the rate of JP cleavage. Addition of a second and third recognition site the overhang region of probe B (JP7 and JP8) dramatically increased the cleavage rate. Non-canonical overhang architectures JP9-JP11 decreased cleavage rates, likely due to the steric sensitivity of the enzyme. (Copied from ref. 93 with permission. Copyright 2010, Royal Society of Chemistry.) | |
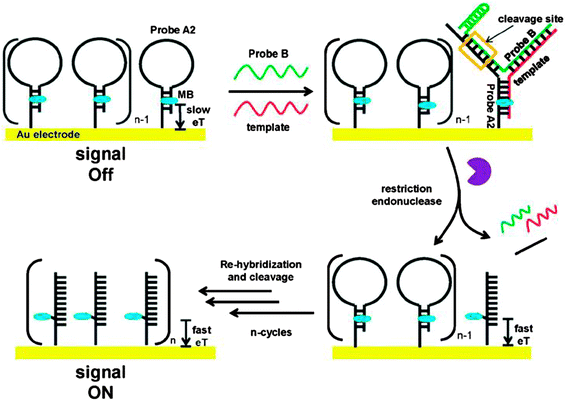 |
| | Fig. 27 A turn-on electrochemical junction probe. The methylene blue (MB), which is attached to the immobilized probe, intercalates into the duplex region of the stem. Upon formation of the junction probe and subsequent REAse cleavage, the stem region of the immobilized probe is lost and MB is now attached to a single strand. Since the single strand is more flexible than the duplex, the MB on a single strand can now approach the Au surface. Therefore the electron transfer between the MB moiety and the Au surface is enhanced upon the loss of the stem region. (Copied from ref. 94 with permission. Copyright 2012, Royal Society of Chemistry.) | |
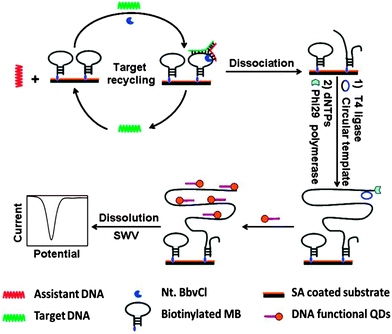 |
| | Fig. 28 Combining junction probe and rolling cycle amplification to achieve ultrasensitive detection of nucleic acids. In this strategy, the cleaved immobilised junction probe is used for RCA amplification. The amplified strand is then bound to multiple oligonucleotides, which are conjugated to quantum dots (QDs). As these oligo-QD probes are brought closer to the electrode, facile electron transfer can occur, leading to detectable electrochemical signal. T4 ligase is used to make circular template in situ. (Copied from ref. 99 with permission. Copyright 2012, American Chemical Society.) | |
Komiyama and co-workers have also described an interesting three-way junction probe primer-generating rolling circle amplification to sensitively detect RNA and achieved 15.9 zmol synthetic RNA and 143 zmol in vitro transcribed human CD4 mRNA.101 Other Y structure-based strategies to detect nucleic acids, which do not utilize endonucleases have also been described. For example Dervan showed that Y-forming probes could be used to detect duplex DNA as far back as 1991 (see Fig. 29).100,102 Fu and co-workers have also described an electrochemical detection of sub-picomolar DNA using an electrochemical junction probe that utilizes [Ru(NH3)6]3+ (RuHex) for the electrochemical process.103
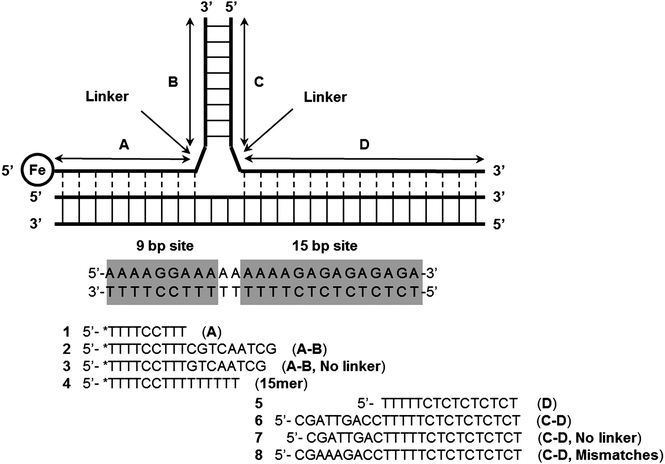 |
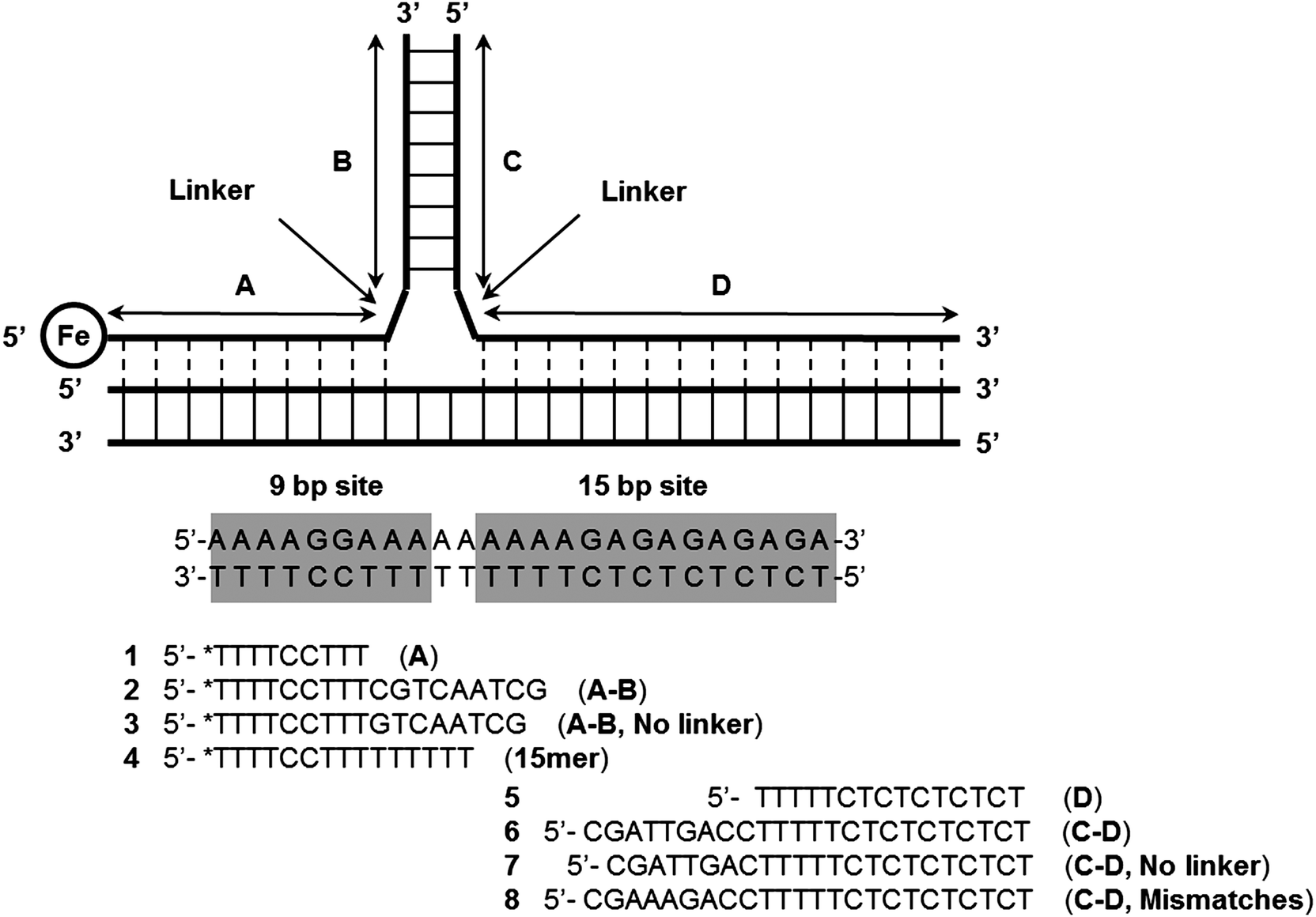
| | Fig. 29 Formation of Y-structures via triplex formation. After triplex formation the modified thymine, (*T, an EDTA-Fe modified Thymidine) at the 5′-end of the 9 bp site can cleave the DNA upon addition of dithiothreitol and O2. (Copied from ref. 100(a) with permission. Copyright 1991, American Chemical Society.) | |
3.12 Catalytic beacons based on reactivation of enzymatic activity
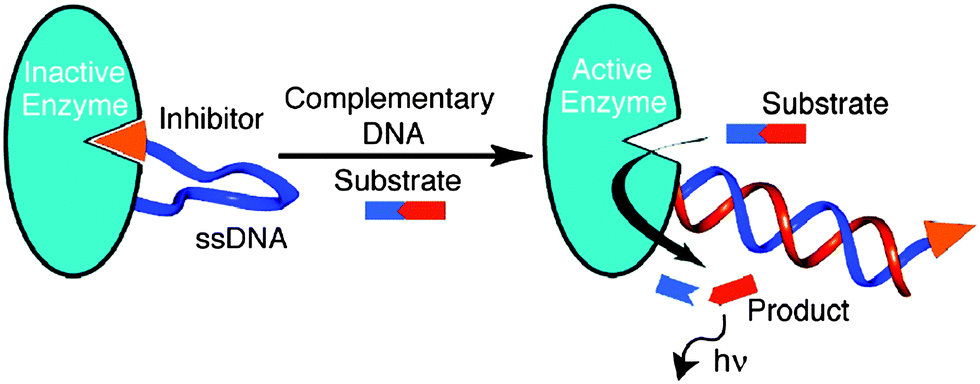
Ghadiri and co-workers demonstrated amplified detection of nucleic acids using a ssDNA sensor that was labeled at one end with a protease from Bacillus cereus and its inhibitor (phosphoramidon)104 at the other end to form an inactive protease. Upon target binding to the ssDNA portion of the sensor, the protease inhibition was relieved and the active enzyme could then cleave a Fluorogenic substrate to generate a signal in an amplified fashion (see Fig. 30).105
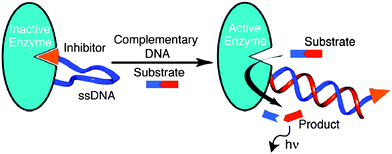 |
| | Fig. 30 Protease–oligonucleotide conjugate used for nucleic acid sensing. When ssDNA binds to the target (with complementary sequence), the enzyme becomes active, which cleaves the fluorogenic substrate to generate optical output signal. (Copied from ref. 105 with permission, Copyright 2003, American Chemical Society). | |
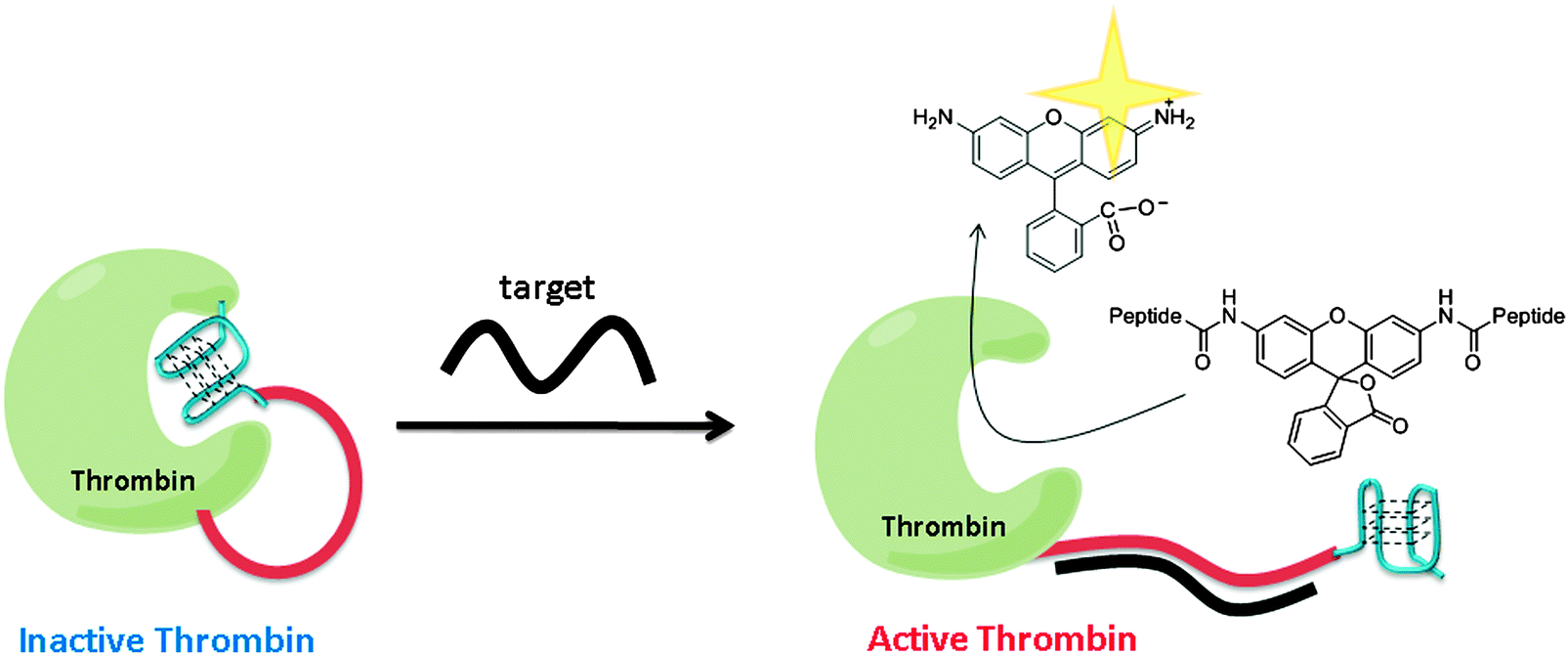
Using a similar strategy to Ghadiri's, Willner and co-workers utilized a G-quadruplex to inhibit thrombin. Upon target hybridization to the ssDNA part of the thrombin-ssDNA G-quadruplex conjugate, the inhibition of thrombin was relieved to allow for the cleavage of a fluorogenic substrate to give amplified fluorescent signals (see Fig. 31).106
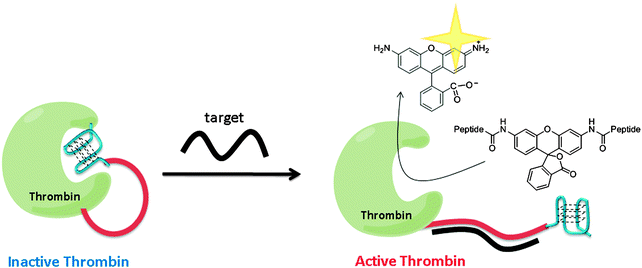 |
| | Fig. 31 Thrombin activity switching by G-quadruplex and target gene binding. The inactive thrombin-ssDNA G-quadruplex conjugate contains three parts: thrombin (green), a sensing ssDNA linker (red) and G-quadruplex (blue), which inhibits the thrombin. When target DNA hybridizes with the target ssDNA to form double strand, the G-quadruplex is unable to inhibit thrombin and hence the active thrombin can cleave the fluorogenic peptide substrate. (Adapted from ref. 106 with permission, Copyright 2005, American Chemical Society.) | |
3.13 Ultrasensitive detection using concurrent isothermal detection strategies
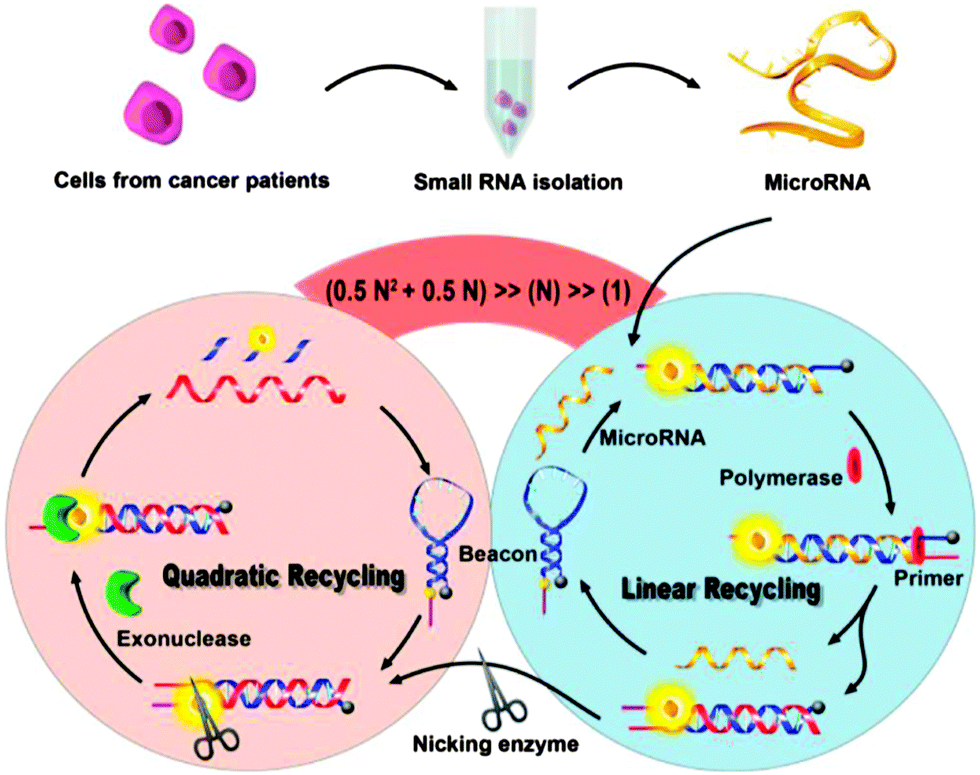
The various isothermal detection strategies that have been developed over the years are beginning to be put into use for the detection of nucleic acids from biological samples. Recently Xia and co-workers demonstrated an ultrasensitive detection of microRNA (Limit of Detection (LOD) of 1 aM at 4 °C) using concurrent amplification cycles involving polymerase extension, nicking endonuclease cleavage and exonuclease-mediated target recycling (see Fig. 32).107 Encouragingly, this detection assay could be used to detect microRNAs in crude extracts of cancer cells, MCF-7 and PC3.
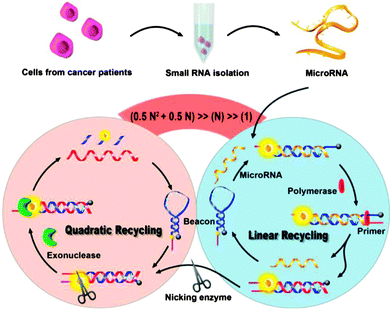 |
| | Fig. 32 Combining a polymerase, nicking endonuclease and exonuclease to achieve ultrasensitive detection of microRNA. (Copied from ref. 106 with permission Copyright 2013, American Chemical Society.) | |
Ellington and co-workers have also demonstrated that catalytic hairpin assembly (CHA) can be combined with several isothermal amplification methods, including LAMP,108 SDA109 and RCA,109 to provide a more sensitive detection of nucleic acids. The authors pointed out that CHA is adaptable to different temperatures, buffers, enzymes and detection modalities and therefore holds promise for improving upon many of the isothermal detection strategies developed to date.109
3.14 Split peroxidase-like DNA enzyme probes
A hemin binding G-quadruplex DNA peroxidase110 has been incorporated in the design of split peroxidase-like DNAzyme probes (Fig. 33).111–113 In this strategy, the two probes contain two regions: the analyte binding arms and the split peroxidase parts. The splitting pattern can be even ((A) in Fig. 33)111 or uneven ((B) in Fig. 33).112,113 When the two probes are separate, they show no or very low peroxidase activity. When the target hybridizes to both of the two probes, the G-quadruplex peroxidase-like DNAzyme is assembled. The hemin–G-quadruplex complex can oxidize ABTS (2,2′-azino-bis(3-ethylbenz-thiazoline-6-sulfonic acid)) with H2O2 and the color change associated with the reaction can be visually monitored. It has been reported that the introduction of a stem-loop or loop-stem-loop motif connecting the G3-tracts in a G-quadruplex structure can effectively reduce the background noise (signal generated in the absence of target gene) and sub-nanomolar target could be detected (Fig. 33(C)).113
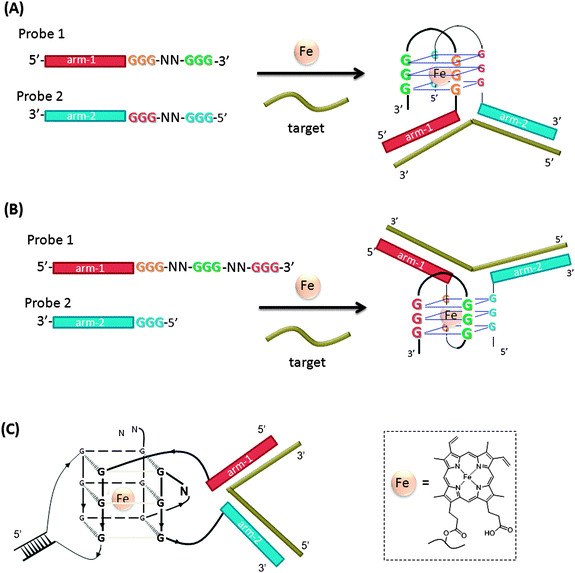 |
| | Fig. 33 Split peroxidase-like DNA enzyme probes. (A) Evenly split DNAzyme probes. (B) Unevenly split DNAzyme probes. (C) Split DNAzyme probes that contain duplex forming regions. | |
Split peroxidase-like DNA enzyme probes assay was applied for the detection of PCR amplified DNA from Salmonella and mycobacterium.114 The assays are fast (reactions complete in minutes) and convenient (visually detectable signals are produced), but it is limited by the detection of nanomolar analyte.
Several molecular beacon-based peroxidase-mimicking DNAzymes for the detection of DNA and RNA under isothermal conditions have also been reported.115 In addition, these DNAzyme peroxidises have also been combined with several DNA modifying enzymes, such as polymerases (including RCA)116 and nicking endonucleases86,88,117 for amplified sensing of nucleic acids.
3.15 RNA cleaving deoxyribozymes
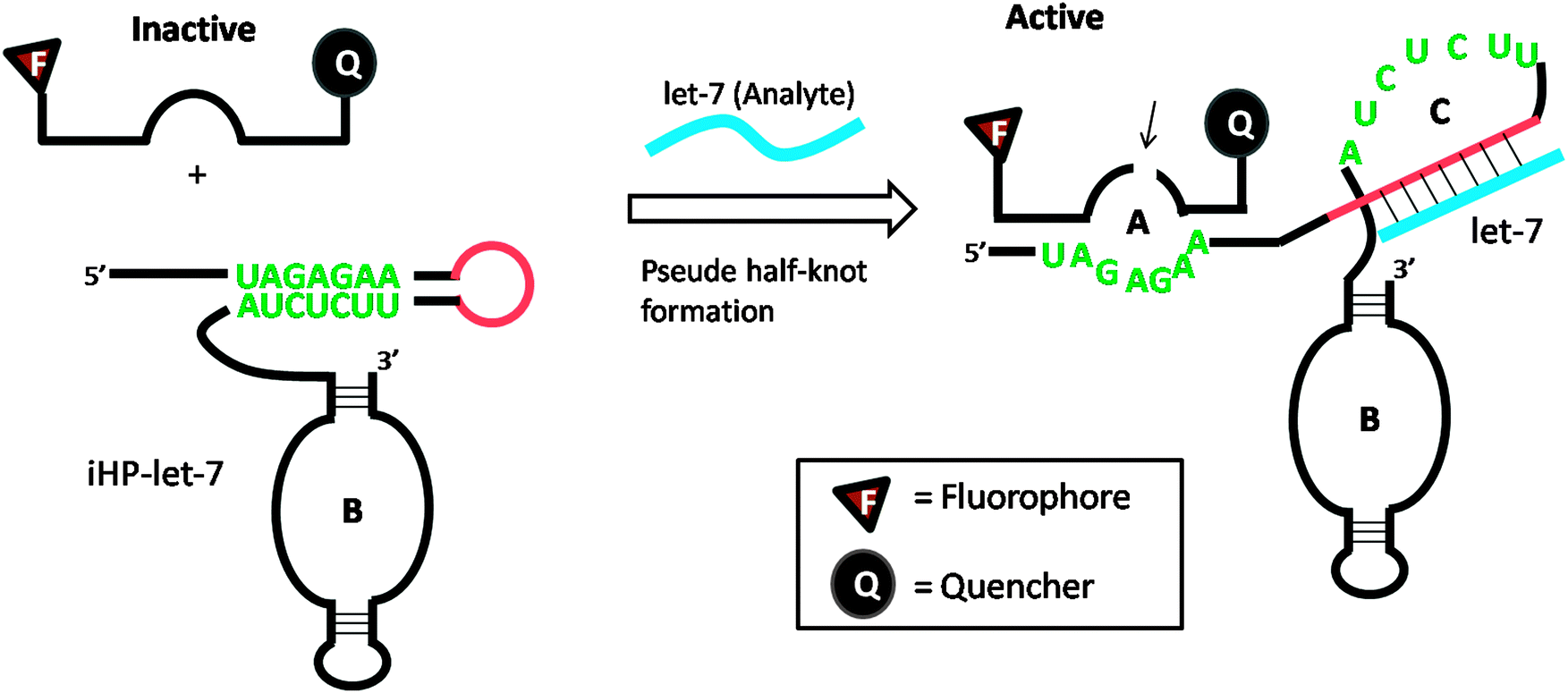
Several DNA or RNA detection strategies that use either ribozymes or RNA cleaving DNAzymes have been described. Stojanovic and co-workers demonstrated an interesting concept of catalytic molecular beacons, using a target to open an “inhibited” DNAzyme (which was inactivated in a stem-loop format) to reveal a proficient DNAzyme (see Fig. 34).118 A few years after the report by Stojanovic et al. Famulok and co-workers reported the detection of microRNA using hairpin ribozyme119 (see Fig. 35).
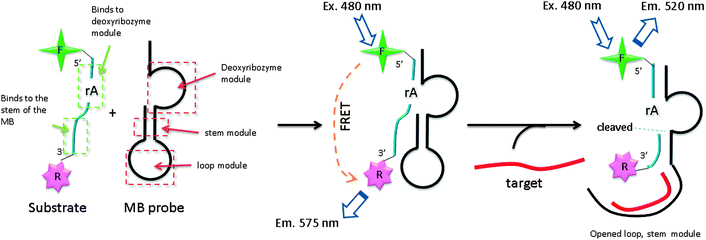 |
| | Fig. 34 Catalytic molecular beacon strategy (CMB). F (fluorescein) and R (tetramethylrhodamine) By binding of target gene, inactivated stem-loop molecular beacon (MB) probe is opened and fluorescent-labelled substrate is cleaved, resulting in a change in the fluorescence profile (520 nm). (Adapted from ref. 118 with permission. Copyright 2001, John Wiley and Sons.) | |
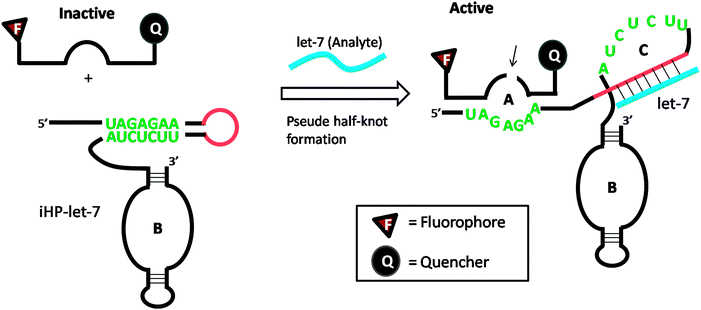 |
| | Fig. 35 Detection of microRNA using hairpin ribozyme. Domain-C is the binding site for target microRNA (let-7). In the presence of let-7, the hairpin ribozyme (iHP-let-7) is opened up and the probe substrate is cleaved. (Copied from ref. 119 with permission copyright 2004, American Chemical Society.) | |
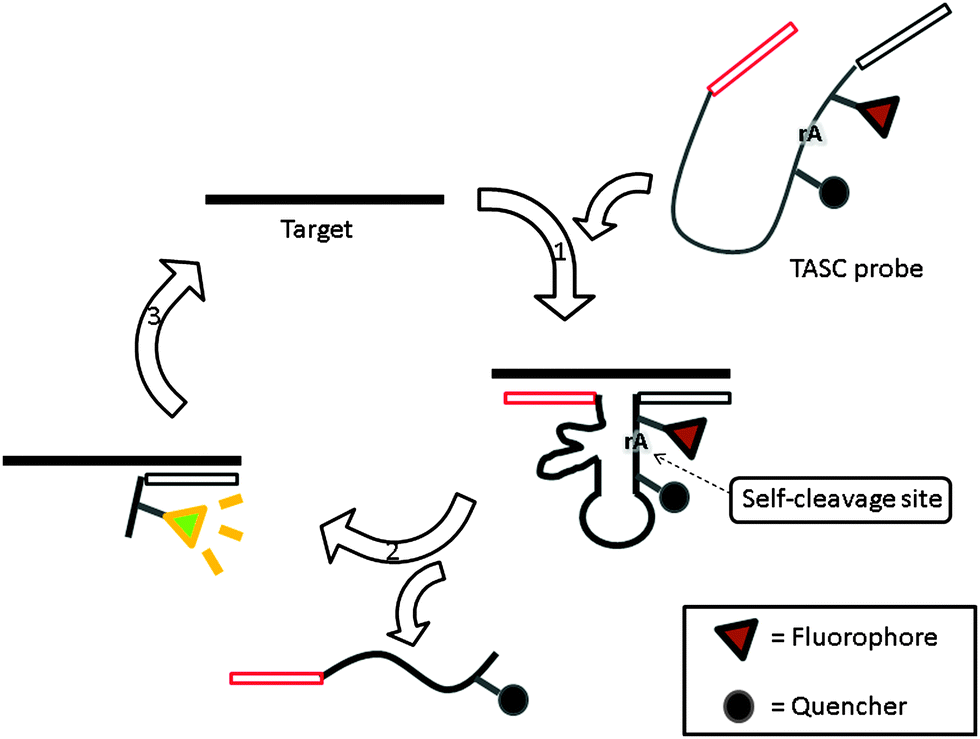
Target-assisted self-cleavage (TASC) probe contains a deoxyribozymes (DNA enzymes, DNAzyme) moiety, which can catalytically cleave RNA.120,121 The probe contains target-binding arms and is labeled with a fluorophore (FAM) and a quencher (Dabcyl), separated by a short nucleic acids sequence embedding a ribonucleotide phosphodiester bond. When the probe hybridizes with the target, an active DNAzyme is enabled, leading to self-cleavage, thus separating the fluorophore–quencher pair originally placed on the two sides of the ribonucleotide moiety of the DNAzyme. The two fragments then come off from the target allowing another probe to bind again to drive a catalytic cycle (Fig. 36).120
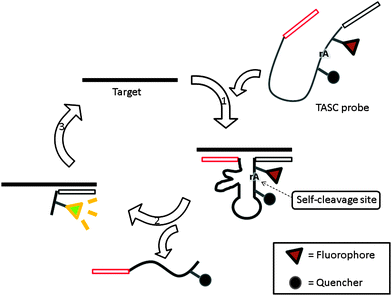 |
| | Fig. 36 Target-assisted self-cleavage (TASC) probe. After binding to a target gene, a self-cleaving DNAzyme is reconstituted and self-cleaves at the ribonucleotide, rA, site. The cleavage results in the separation of the fluorophore and quencher, giving rise to an enhanced fluorescence signal and the release of target DNA, which is then reused in subsequent catalysis. (Adapted from ref. 120 with permission Copyright 2003, American Chemical Society.) | |
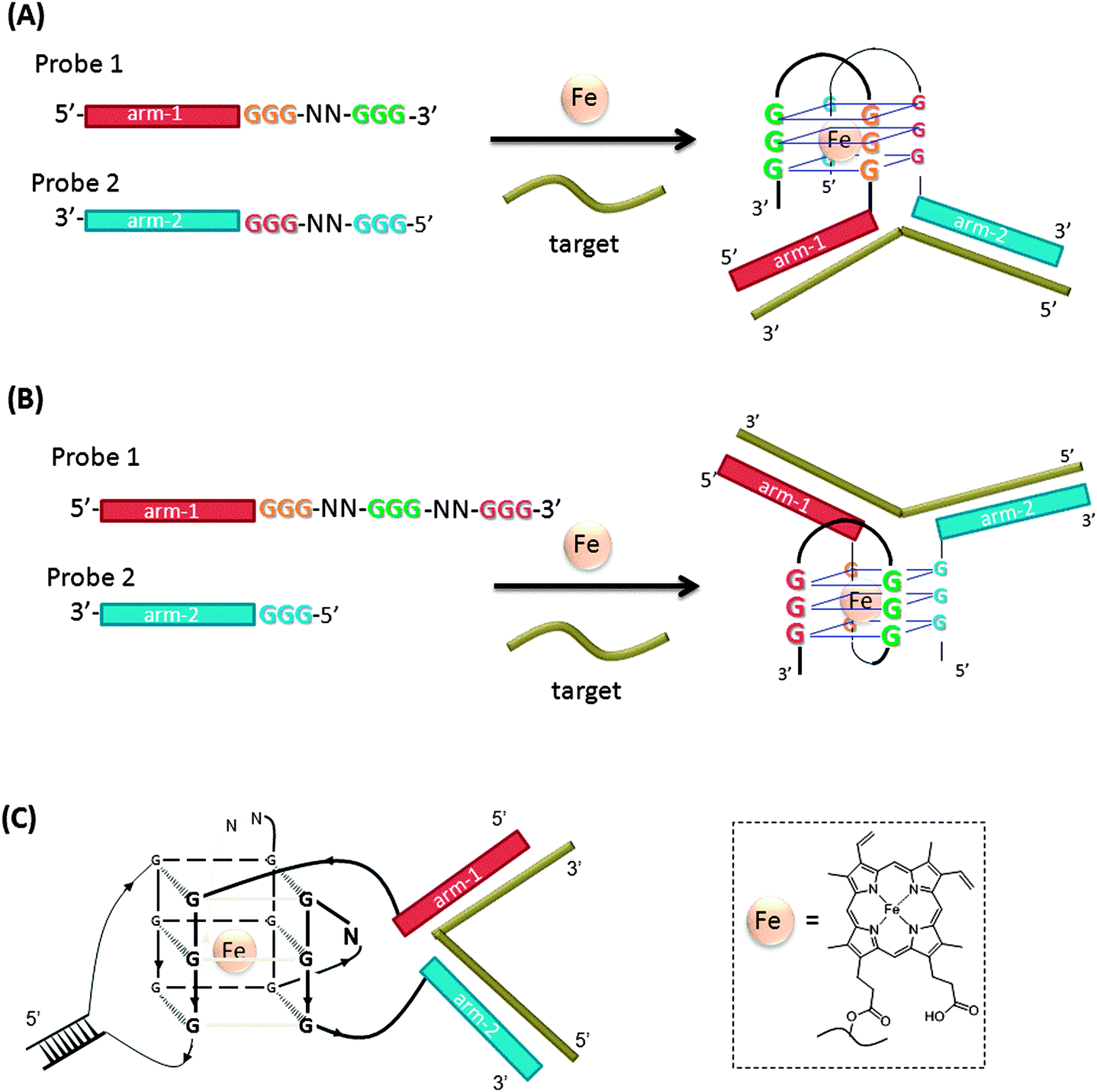
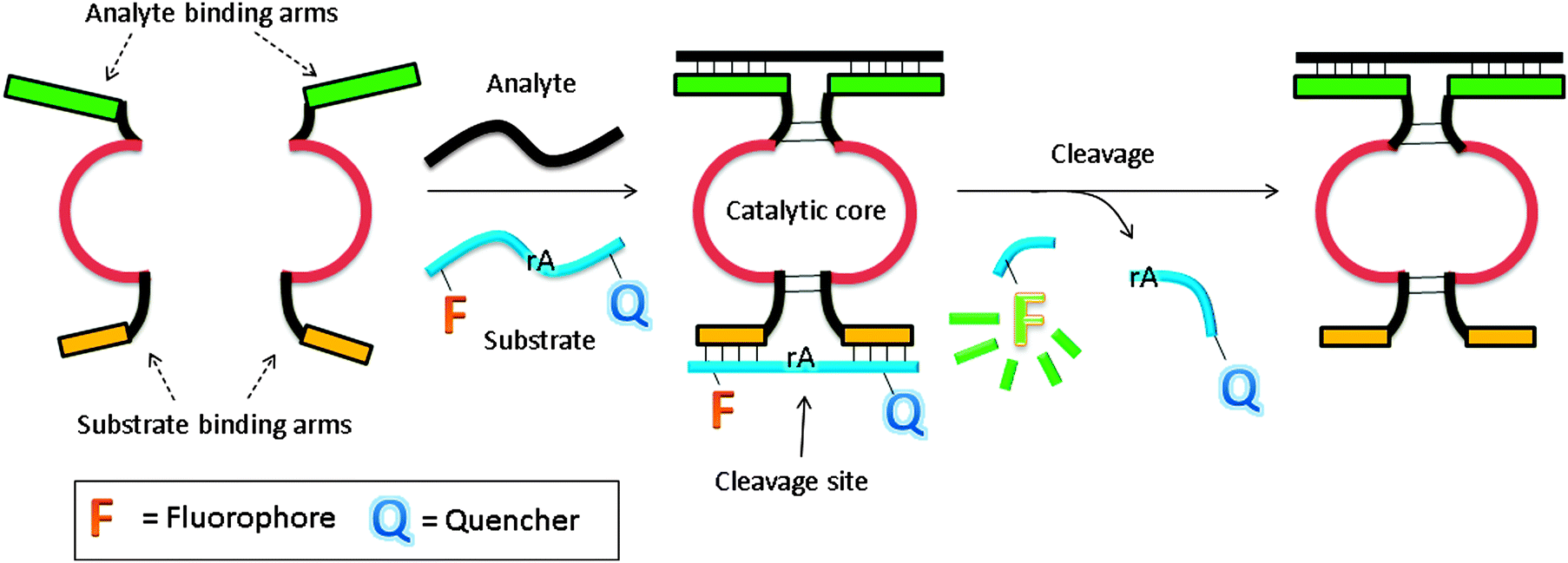
In another design, based on RNA cleaving DNAzyme, the catalytic core is split into two inactive parts, and analyte binding arms are linked to each part (MNAzymes, which means multicomponent nucleic acid enzymes).122–126 The analyte hybridizes to the two probes, and active DNAzyme is assembled. A fluorescent substrate is cleaved and the fluorescence signal is enhanced (Fig. 37).123 In a similar design, based on a more efficient DNAzyme, ∼0.2 nM analyte was detected after a 3 h incubation.124 Coupling with other non-linear amplification platforms may further lower the limit of detection.125,126
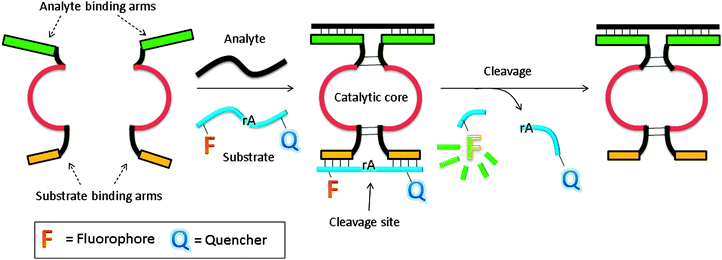 |
| | Fig. 37 Binary deoxyribozyme probes, using a TeHyP strategy. This strategy uses two probes, which have analyte binding arms and substrate binding arms. In the presence of analyte, the two probes bind to the analyte to form a tripartite structure, which is an active DNAzyme. The DNAzyme can then cleave a fluorogenic oligonucleotide, containing rA cleavage site. | |
3.16 Template directed chemical reactions
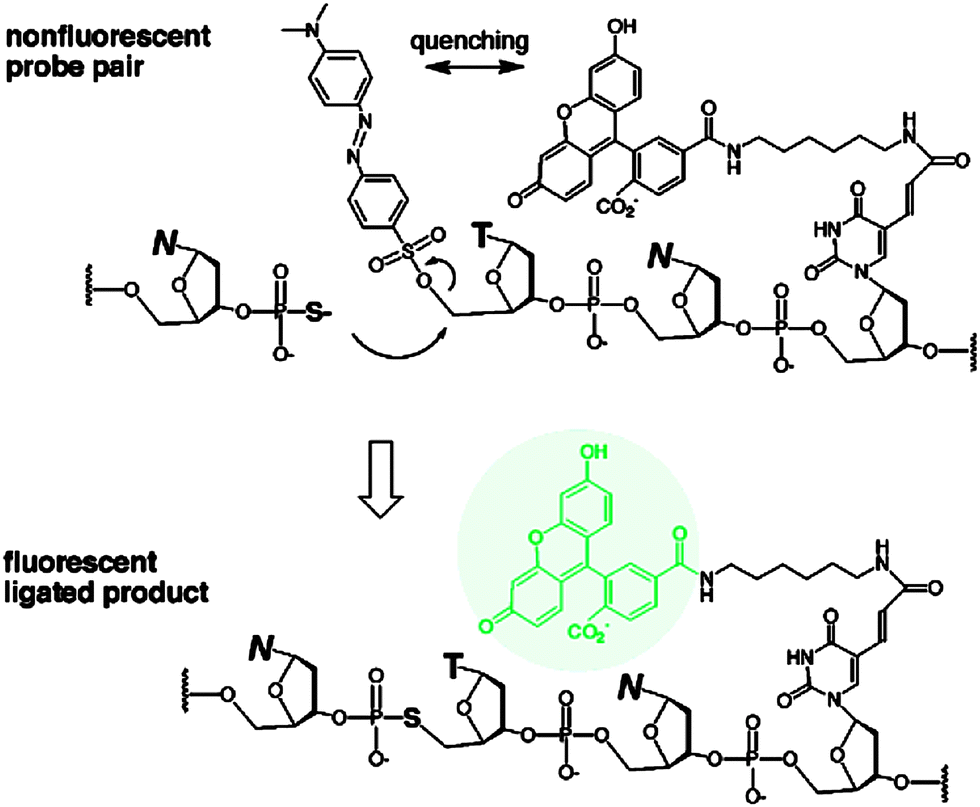
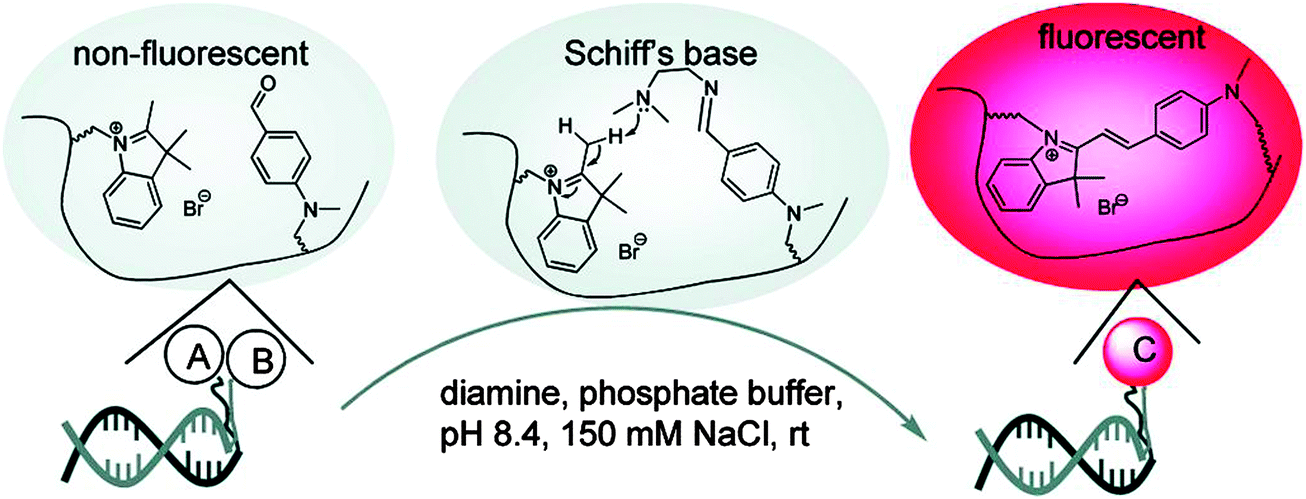
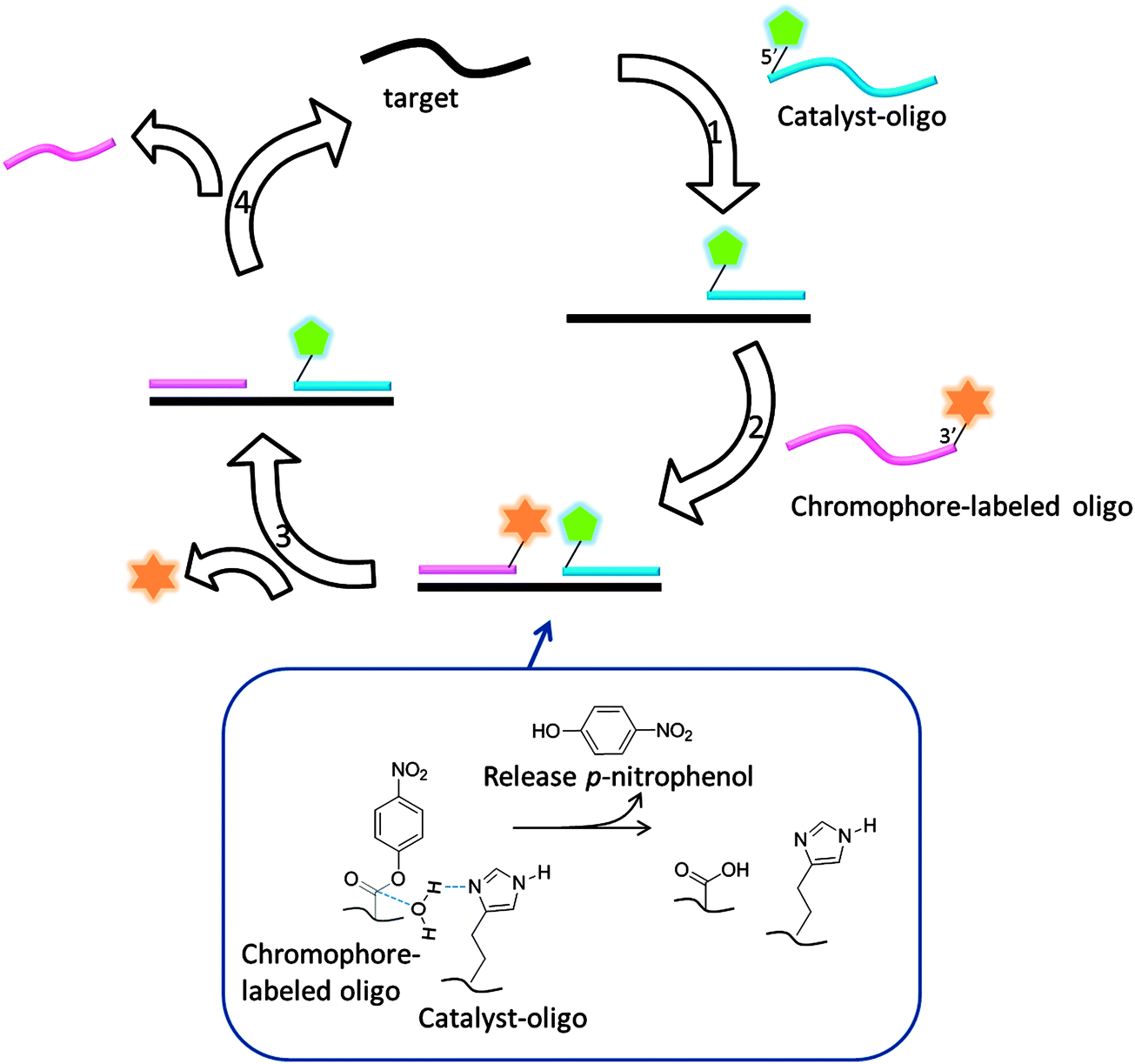
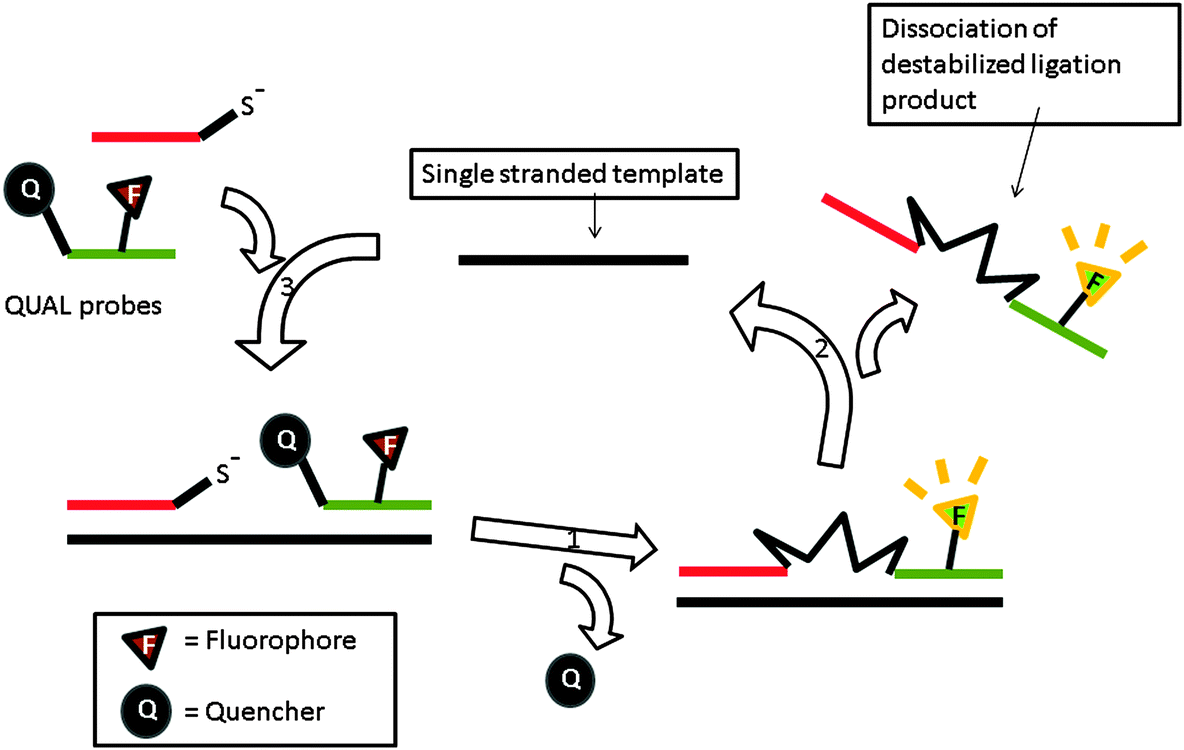
The preceding section has highlighted enzyme-mediated isothermal detection of DNA or RNA. One of the limitations of enzyme-based methods is that sample quality and the presence of other biomolecules can affect the sensitivity of the method. Secondly, the adaptation of these strategies for detecting nucleic acids in live cells is non-trivial. Therefore there have been efforts aimed at developing non-enzymatic DNA/RNA-templated reactions to be used in genetic diagnostics or for studying nucleic acid processes in vivo.127 Numerous types of DNA template-catalyzed reactions for detection purposes have emerged in the last twenty years.128 These approaches are divided into two types: chemical ligation-based method129 and ligation-free detection.130–132 Quenched autoligation-fluorescence energy transfer (QUAL-FRET, see Fig. 38)133,134 and fluorophore reconstitution (Fig. 39)135 are two classic examples of the detection of nucleic acids using a ligation-based method whereas template-assisted fluorophore/chromophore generation (Fig. 40)130–132,136,137 and reporter transfer reaction (Fig. 41)138 represent ligation-free methods.
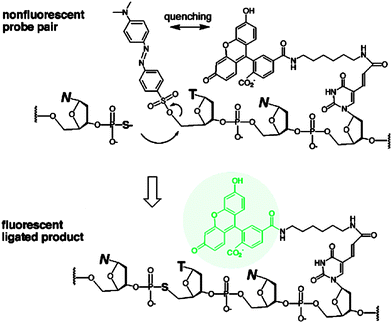 |
| | Fig. 38 Quenched autoligation-fluorescence energy transfer (QUAL-FRET). In the above strategy, the fluorophore on one of the probes is quenched by a dabsyl group, which can act as a leaving group. After a template-catalyzed ligation of two probes (via SN2 displacement), the dabsyl quencher becomes separated from the FAM fluorophore, leading to enhancement of fluorescence. (Adapted from ref. 133 with permission. Copyright 2002, American Chemical Society.) | |
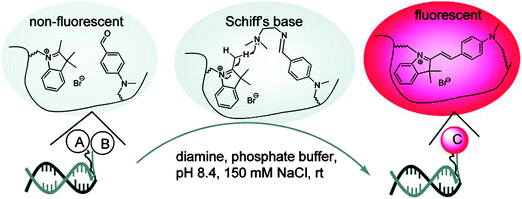 |
| | Fig. 39 A fluorophore reconstitution method reported by Huang and Coull. The non-fluorescent indolinium- and aldehyde-labeled complementary DNAs react to form a hemicyanine, catalyzed by a diamine. The ligated reaction product is fluorescent. (Copied from ref. 135 with permission. Copyright 2008, American Chemical Society.) | |
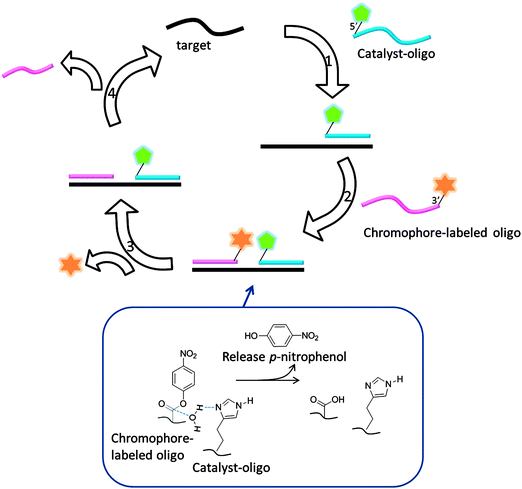 |
| | Fig. 40 Template enhanced ester hydrolysis to generate a chromophore. (Adapted from ref. 131 with permission. Copyright 2000, National Academy of Sciences, U.S.A.) | |
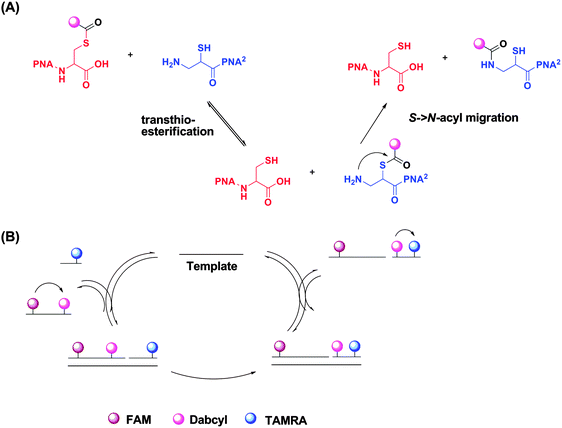 |
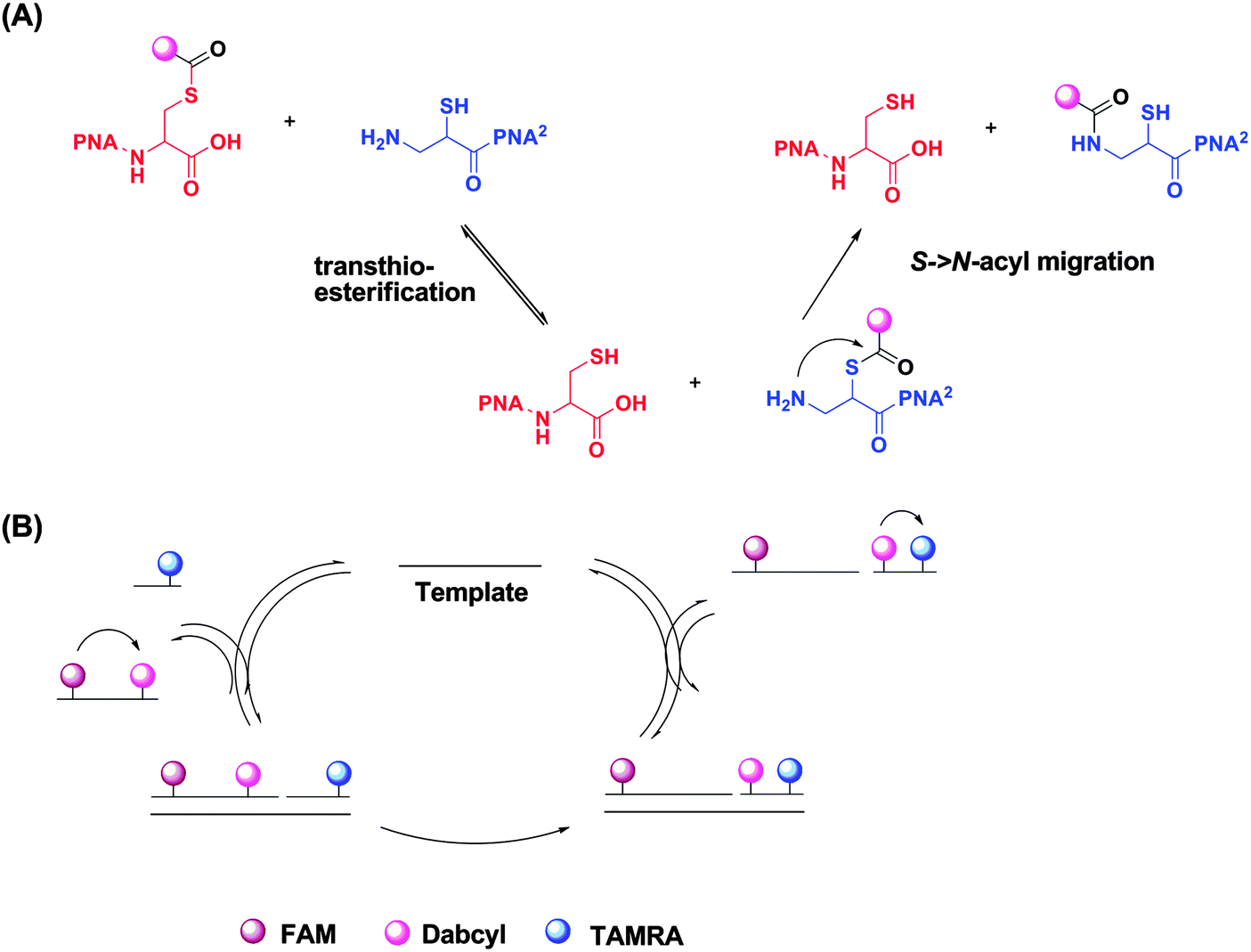
| | Fig. 41 Reporter transfer reaction. (a) Quencher transfer reaction (transthio-esterification and S->N-acyl migration). (b) Catalytic cycle of template-catalyzed quencher transfer. (Adapted from ref. 138 with permission. Copyright 2006, American Chemical Society.) | |
QUAL probes can react with each other to generate a signal but the reaction is significantly enhanced when the two probes bind to the same target analyte (effective molarity argument). Typically one of the probes contains a nucleophilic moiety, e.g. phosphorothioate, whereas the other probe contains a leaving group. Autoligation of the probes (SN2 reaction) ligates the two probes together. If the probe that contains the leaving group contains a quenched fluorophore and if the SN2 reaction results in the quencher leaving, then fluorescence enhancement will be obtained (Fig. 38).133
Reporter transfer reaction, using peptide nucleic acid (PNA) probes, has been used to detect DNA.138 For this strategy, the PNA probes are both labeled with fluorophores. The donating probe has a fluorophore (FAM) and nearby quencher (Dabcyl), while the accepting probe has a different fluorophore (TAMRA). When the two probes are positioned in proximity to each other by the analyte, a trans-thioesterification transfers the dabcyl moiety to the probe originally labelled with only TAMRA. The subsequent S→N-acyl shift stabilizes the product, making the reaction irreversible. This dabcyl migration from the donating probe to the accepting probe enhances the fluorescence of FAM, while decreasing the fluorescence of TAMRA (Fig. 41).138 Therefore the ratio of the fluorescence intensities of FAM and TAMRA (fFAM/fTAMRA, where superscript f means fluorescence intensity) can be used to quantify the target concentration. Since the reaction is not a ligation, the affinity of the final products should be similar to (and not more than) the original probes so product inhibition is not an issue with this approach. It has been demonstrated that a 20 pM synthetic DNA target could be detected with a signal/background ratio of ∼1.5 using the reporter transfer strategy.139 Also this strategy has been combined with an enzyme-linked immunosorbent assay (ELISA) to detect HIV genomic RNA.140
Assays that employ template-directed chemical reactions are enzyme-free and multiple analytes can be identified by employing probes that bear different fluorophores.141 Similar to enzyme-catalyzed ligation reactions, chemical ligations also suffer from product inhibition. Kool demonstrated a clever solution to this problem by using destabilizing universal linkers (which are hydrocarbon linkers) to decrease the stability of ligated product–target complex (Fig. 42).134 Chemical ligations that utilize leaving groups could also suffer from background noise due to the abundance of several nucleophiles, such as amino acids, thiols, and water. Kool has demonstrated that the background problem could be reduced using a FRET approach.142 In this approach, the nucleophile-modified probe is also labeled with a fluorophore-a FRET acceptor such as Cy5 whereas the probe with the leaving group also contains a FRET donor, such as FAM. Chemical ligation brings the FRET donor and acceptor together for sensing. Since the target-independent hydrolysis of the leaving group would not bring the FRET pair together, undesired hydrolysis does not lead to a FRET signal, although non-specific hydrolysis would erode the concentration of useful probes. The incorporation of a phosphotrithioate into ligation-based methods has been shown to speed up the template-enhanced ligation so that the kinetics of template-directed ligation could compete with the non-specific hydrolysis.143 Using template-directed reactions, RNAs in cells have been detected.142,144
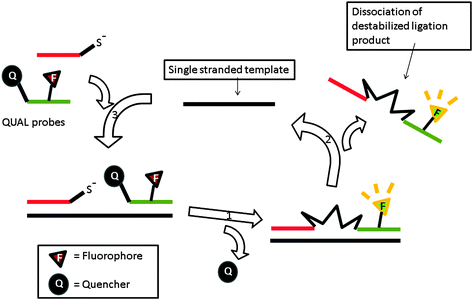 |
| | Fig. 42 Schematic illustration of destabilizing linkers for signal amplification in autoligating probes. The linker is designed to have a dabsyl group as leaving group during ligation with nucleophilic DNA probes. The hydrophobic linker, which forms after ligation, destabilizes the duplex structure, leading to strand disassociation and the release of the template for further catalysis. (Copied from ref. 134 with permission. Copyright 2006, National Academy of Sciences, U.S.A.) | |
Several other non-enzymatic template-directed chemical reactions that have the potential to be used for diagnostics, using crude samples have also been described; however the sensitivities of methods that rely on chemical reactions have not matched those that employ DNA modifying enzymes, such as polymerases.130,136,145 Non-enzymatic detection of nucleic acids, however, has the potential to work in harsher biological environments or matrices, where the activities of enzymes are usually compromised due to enzyme inhibition. We anticipate that ongoing efforts to develop fast template-directed chemical reactions for sensing applications will continue.
3.17 Non-covalent DNA catalytic reactions
Non-covalent DNA catalytic reaction, which uses an input signal (target) to catalyze the release of oligonucleotide that can be used to catalyze the next reaction (see Fig. 43)146 has been used to detect nucleic acids.146,147 In most cases the strand-displacement begins after the target nucleic acid analyte has bound to the toehold segment of the initiation probe. Yin et al. designed an exponential detection strategy that utilized numerous catalytic non-covalent DNA reactions to detect as low as 5 fM DNA.146 In an interesting development, Willner and co-workers have combined toehold-mediated strand displacement and a Zn2+-dependent DNAzyme, which ligates two strands, to detect 10 pM Tay-Sachs gene mutant.148
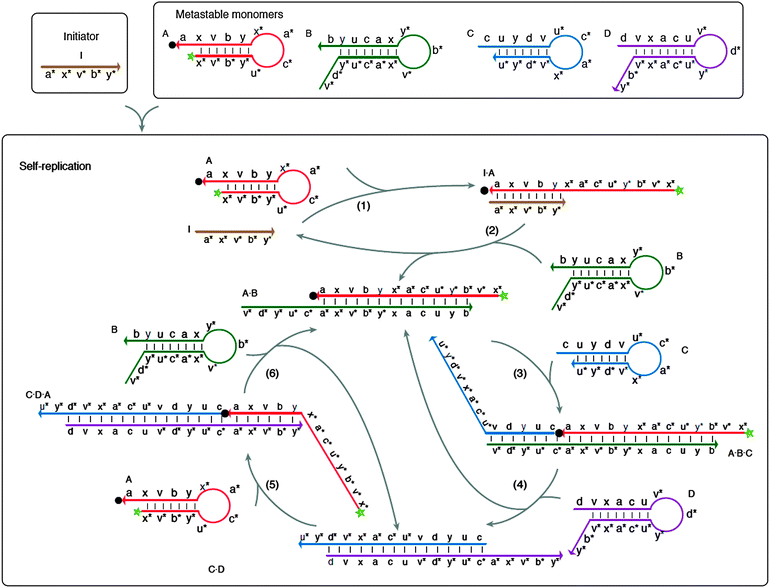 |
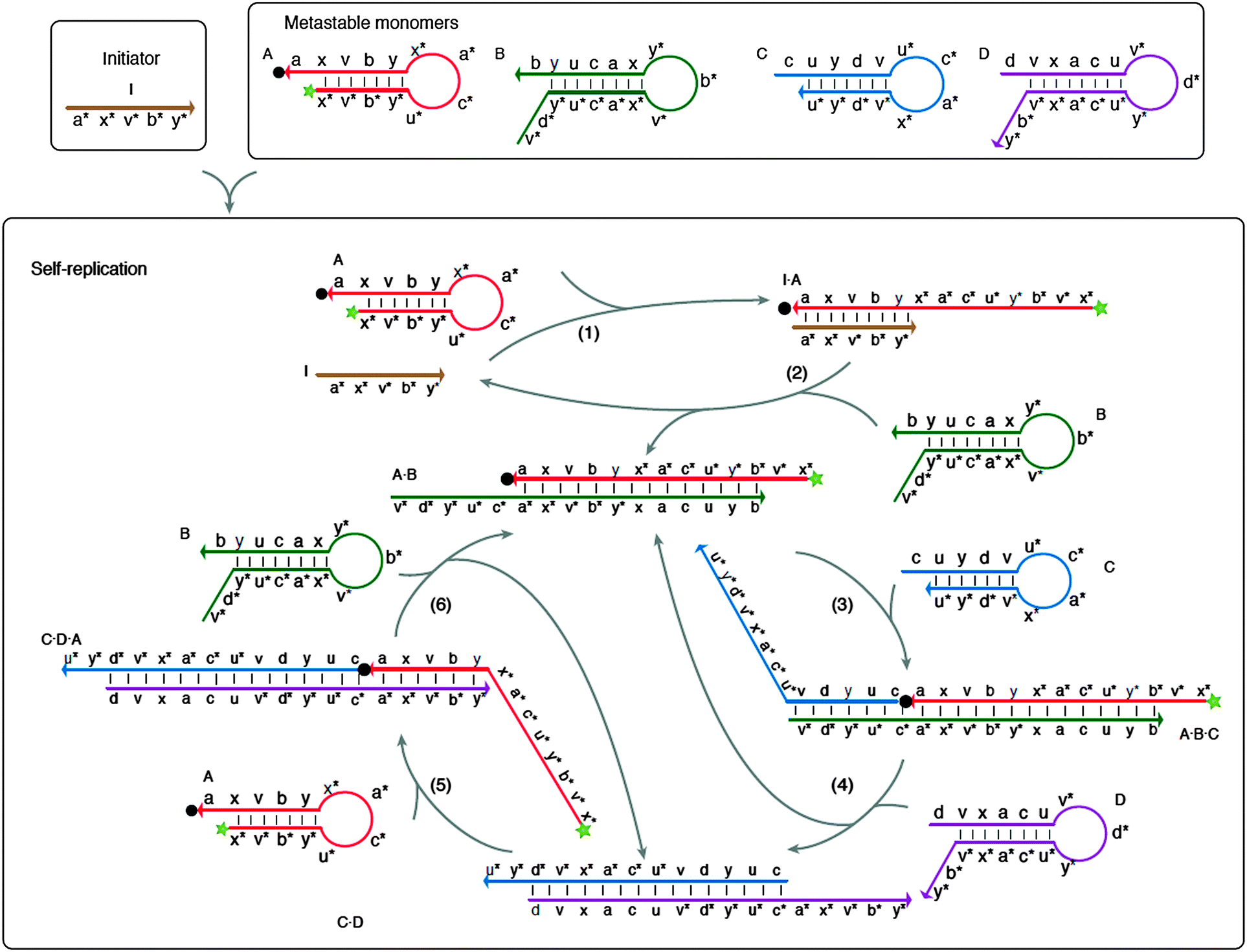
| | Fig. 43 Multiple strand displacement via non-covalent DNA catalysis. Green star indicates fluorophore and black dot indicates quencher. Four metastable probes (stem-loops), A, B, C and D are used. Probe A can form a stable complex with probe B but there is a kinetic barrier to this complex formation. A target (or an initiator) can bind to probe A to open the stem-loop and this facilitates the binding of probe A to probe B. Probe A–B complex has an overhang, which can bind to probe C to open up the stem-loop structure in probe C. The probe A–B–C complex now contains an overhang that can bind to probe D to open the stem-loop structure of probe D to form a transient probe A–B–C–D complex. Because probes D and B compete for binding to the same site on probe C, the binding of probe D to the probe A–B–C complex result in the dissociation of probe A–B complex, which can initiate another round of probe C/D opening. Probe D is designed to also contain a region of complementarity to probe A, so that the probe C–D complex can also open the stem-loop structure of probe A. These series of cascade reactions lead to exponential amplification. (Copied from ref. 146 with permission. Copyright 2008, Nature Publishing Group.) | |
3.18 Hybridization chain reactions (HCR)
Pierce and co-workers described hybridization chain reaction (HCR) as a process whereby a target DNA or RNA can initiate the assembly of long dsDNA structures, which contain nicked regions (see Fig. 44).149,150 Probes that are used for HCR are usually in a hairpin form so they do not associate with each other. However, in the presence of a target analyte, one of the stem-loop probes is opened up and this initiates a cascade process, which opens up other hairpin probes to form a long alternating dsDNA structure. By incorporating fluorescent turn-on probes149,151 or latent DNAzymes152 into the hairpin probes, the HCR process can lead to amplified sensing.
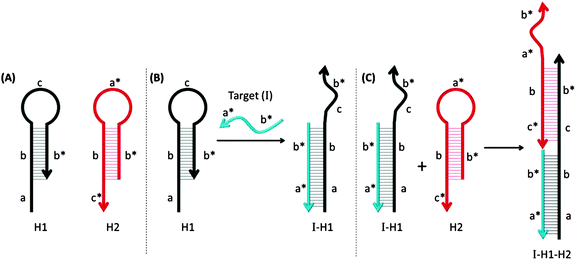 |
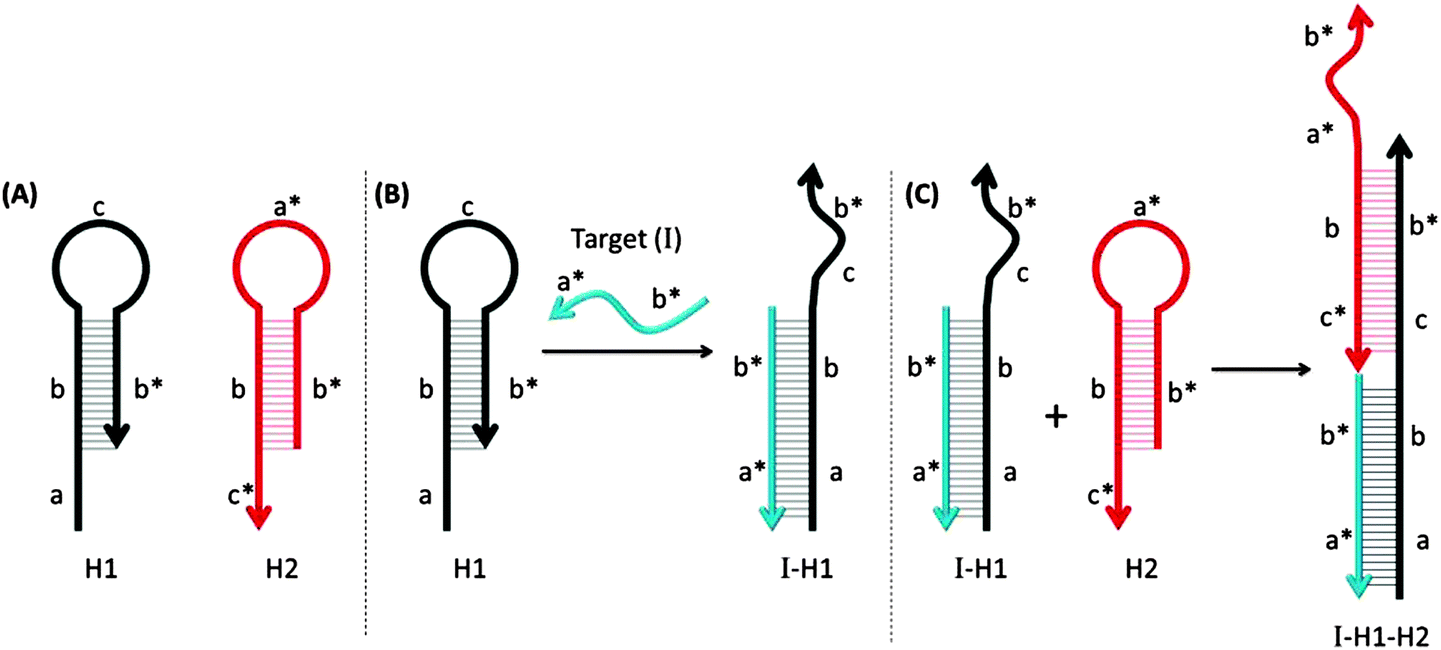
| | Fig. 44 Hybridization chain reactions (HCR). The two MB probes, shown in (A) can hybridize with each other only after the hairpin structure of one of the probes (colored black) is opened upon target binding. (B) The target gene (I) binds to region-a in the probe-H1 to open up the stem-loop structure (initiation step). (C) The complex I-H1 binds region-c* in probe-H2 and probe-H2 is opened up. Complex I-H1–H2 binds region-a in the probe-H1 and continue this hybridization chain reaction. (Adapted from ref. 149 with permission. Copyright 2004, National Academy of Sciences, U.S.A.) | |
Ellington and co-workers have demonstrated that the combination of CHA and HCR allows for the detection of nucleic acid hybridization defects and holds promise for the detection of single nucleotide polymorphism.153
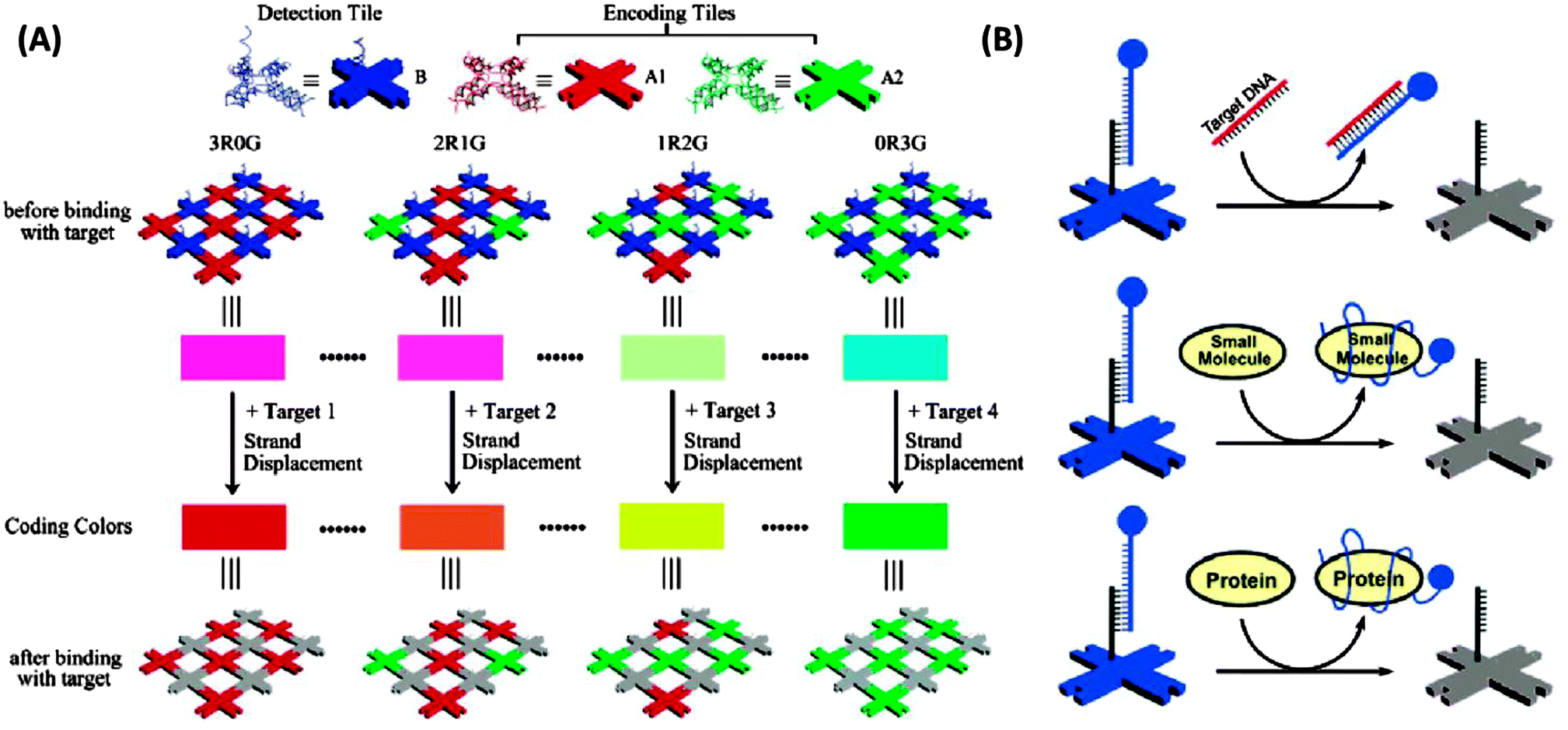
3.19 Detection via self-assembled DNA nanostructures
DNA nanostructures have also emerged as excellent platforms to detect DNA or RNA.154–156 For example Li et al. have described interesting dendrimer DNA nanobarcodes that could be used to detect attomole pathogenic DNA, using a microscope.156 Yan and co-workers also used a microscope to detect color changes on a DNA tile when a target analyte displaced a fluorescent probe on the DNA tile (see Fig. 45).154
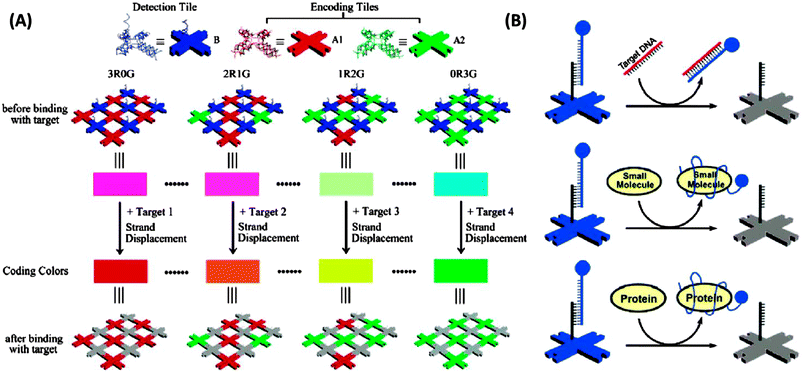 |
| | Fig. 45 Detection via self-assembled DNA nanostructures. (Copied from ref. 154 with permission. Copyright 2007, American Chemical Society.) | |
Table 2 Summary of isothermal amplification/detection the platforms
| Platform |
Advantage |
Disadvantage |
Components |
Application |
|
These methods are relatively new; hence application to clinical targets has not been demonstrated yet.
|
| NASBA |
107 fold amplification in 2 h, commercial kits available |
Not ideal for DNA analyte |
2 primers, reverse transcriptase, RNase H, RNA polymerase, dNTP, rNTP |
HIV-1 genomic RNA,34 hepatitis C virus RNA,35 human cytomegalovirus mRNA,36 16S RNA in many bacterial species,37 and enterovirus genomic RNA,38etc. |
|
|
| SDA |
105 fold amplification in 2 h |
Less efficient on long targets, sample preparation is required |
4 primers, DNA polymerase, REase HincII, dGTP, dCTP, dTTP, dATPαS |
Mycobacterium tuberculosis genomic DNA81 |
|
|
| LAMP |
∼108 amplification in less than 1 h in the presence of interfering sequences |
Complicated primer design, not suitable for short analytes |
4 primers, DNA polymerase, dNTP |
Viral pathogens, including dengue,47 Japanese encephalitis,48 Chikungunya,49 West Nile,50Severe acute respiratory syndrome (SARS),51 and highly pathogenic avian influenza (HPAI) H5N1,52etc. |
|
|
| Invader assay |
Commercial kits available, ideal for SNP genotyping |
Initial PCR required |
2 or 3 probes, flap endonuclease |
Factor V gene,54 genes responsible for Japanese hearing loss,56 human papillomavirus DNA,57 microRNA from HeLa and Hs578T cells,58 and herpes simplex virus types 1 and 2,59etc. |
|
|
| RCA |
ssDNA product makes this compatible with other isothermal amplification strategies |
Ligation using RNA analyte could be problematic |
1 probe, ligase, polymerase and dNTP |
Cystic fibrosis gene, Epstein–Barr virus in,70 influenza A H1N1 and H3N2 mutations,71 porcine circovirus type 2,72 and Listeria monocytogenes,73etc. |
|
|
| SMART |
50 pM analyte detected in ∼4 h, tolerates crude sample |
Two-step process |
2 probes, DNA polymerase, RNA polymerase, dNTP, rNTP |
DNA and rRNA of E. coli,79 DNA and mRNA of marine cyanophage virus80 |
|
|
| HDA |
106 fold amplification in 3 h, commercial kits available |
Complicated buffer optimization |
2 primers, helicase, SSB, DNA polymerase, dNTP |
Treponema denticola gene,81Brugia malayigene81 |
|
|
| RPA |
100 copies of analyte were detected in 1 h, selective |
Stringent reaction condition |
2 primers, recombinase, SSB, DNA polymerase, REAse Nfo, dNTP |
Bacillus subtilis genome84 |
|
|
| NEANA |
Endonucleases are robust enzymes and tolerate crude samples |
Requirement of certain recognition sites in analytes |
1 probe, nicking endonuclease, gold nanoparticle |
N/Aa |
|
|
| Exonuclease III aided target recycling approach |
20 aM analyte can be detected |
Not suitable for RNA |
1 probe, exonuclease III |
N/Aa |
|
|
| Junction or Y probes |
Works with crude samples |
Needs to be combined with other amplification strategies for sensitive detection |
Two probes, restriction enzyme |
DNA version of hsa-miR-16 |
|
|
| Reactivation of enzymatic activity |
∼10 fM of analyte detected |
The formation of DNA protein conjugate is non-trivial and could make the probes expensive |
Enzyme (protease, thrombin etc). DNA binding arm |
N/Aa |
|
|
| Concurrent use of polymerase, nicking endonuclease and exonuclease |
miRNA detection with 10 fM detection limit at 37 °C and 1 aM at 4 °C. Crude cellular lysate can be used |
Several enzymes (polymerase, nicking endonuclease and exonuclease) are needed |
Bst polymerase, lambda exonuclease, nicking endonuclease, one MB probe, one primer |
N/Aa |
|
|
| Split peroxidase-like DNA enzyme probes |
Simple, no protein enzyme needed, fast, colorimetric |
Not sensitive (detection of biologically-relevant samples demonstrated only after initial PCR) |
2 probes, hemin |
Salmonella and mycobacterium114 |
|
|
| RNA cleaving deoxy-ribozymes |
No protein enzyme needed, ∼200 pM was detected after 3 h of incubation124 |
Probes containing ribonucleotides are expensive and not as stable as deoxynucleotide-based probes |
1 to 3 probes |
N/Aa |
|
|
| Template-directed chemical reactions |
Simple, no enzyme needed |
Turnover is significantly lower than enzyme-based methods |
2 primers |
16S RNA of Escherichia coli,133 HIV genomic RNA140 |
|
|
| Non-covalent DNA catalytic reactions |
5 fM DNA could be detected. Method can be combined with other detection methods |
For low analyte copy numbers, assay time can be long |
Four probes (one is labelled with fluorophore and quencher) |
Tay-Sachs gene mutant148 |
|
|
| HCR |
Method can be combined with other detection methods |
|
Two probes |
N/Aa |
|
|
| Self-assembled DNA nanostructure |
5 nM of four different DNA targets could be detected in 15–20 min |
Preparation and assembly of tiles is not straightforward |
Detection tiles |
SARS virus DNA,154 HIV viral DNA154 |
4.0 Conclusions
In the last few years, multitudes of nucleic acid detection methods that operate under isothermal conditions have been described (see Table 2). Some of these methods have been shown to detect few copy numbers of nucleic acids and hold promise for point-of-care diagnostics. Detection strategies that utilize tandem/concurrent amplification strategies could allow for ultrasensitive detection of analytes under isothermal conditions but these strategies utilize many enzymes and reagents, such as nucleotides, and could prove to be expensive. Additionally, the use of many enzymes in a detection assay brings into play other inconveniences such as enzyme storage and the need to find assay conditions that are permissive for all of the enzymes being used. Most DNA modifying enzymes require divalent cations for efficient catalysis but the optimal concentrations of cations are not necessarily the same for all enzymes, so it is almost impossible to use specific cation concentrations that are optimal for all of the enzymes in concurrent catalysis. In some instances, enzymes being used in concurrent reactions might require different additives or temperatures for optimal performance and in some cases an additive that enhances the catalysis of one enzyme might inhibit another enzyme, making the optimization of concurrent catalysis non-trivial. In this regard, templated chemical reactions and nucleic acid enzymes, such as G-quadruplexes, could become popular in point-of-care diagnostics if such systems could be engineered to detect analytes in the femtomolar and atomolar ranges. Additionally, the incorporation of microbes, whose growth could be triggered by a DNA analyte157 and visualized using cheap instruments could revolutionalize nucleic acid diagnostics, especially in poor areas.
Notes and references
- R. Sachidanandam, D. Weissman, S. C. Schmidt, J. M. Kakol, L. D. Stein, G. Marth, S. Sherry, J. C. Mullikin, B. J. Mortimore, D. L. Willey, S. E. Hunt, C. G. Cole, P. C. Coggill, C. M. Rice, Z. M. Ning, J. Rogers, D. R. Bentley, P. Y. Kwok, E. R. Mardis, R. T. Yeh, B. Schultz, L. Cook, R. Davenport, M. Dante, L. Fulton, L. Hillier, R. H. Waterston, J. D. McPherson, B. Gilman, S. Schaffner, W. J. Van Etten, D. Reich, J. Higgins, M. J. Daly, B. Blumenstiel, J. Baldwin, N. S. Stange-Thomann, M. C. Zody, L. Linton, E. S. Lander, D. Altshuler and Int. S. N. P. M. W. G., Nature, 2001, 409, 928–933 CrossRef CAS PubMed.
- G. A. Calin, C. Sevignani, C. Dan Dumitru, T. Hyslop, E. Noch, S. Yendamuri, M. Shimizu, S. Rattan, F. Bullrich, M. Negrini and C. M. Croce, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 2999–3004 CrossRef CAS PubMed.
- G. A. Calin, C. D. Dumitru, M. Shimizu, R. Bichi, S. Zupo, E. Noch, H. Aldler, S. Rattan, M. Keating, K. Rai, L. Rassenti, T. Kipps, M. Negrini, F. Bullrich and C. M. Croce, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 15524–15529 CrossRef CAS PubMed.
- M. Z. Michael, S. M. O'Connor, N. G. V. Pellekaan, G. P. Young and R. J. James, Mol. Cancer Res., 2003, 1, 882–891 CAS.
- M. Metzler, M. Wilda, K. Busch, S. Viehmann and A. Borkhardt, Genes, Chromosomes Cancer, 2004, 39, 167–169 CrossRef CAS PubMed.
- J. Takamizawa, H. Konishi, K. Yanagisawa, S. Tomida, H. Osada, H. Endoh, T. Harano, Y. Yatabe, M. Nagino, Y. Nimura, T. Mitsudomi and T. Takahashi, Cancer Res., 2004, 64, 3753–3756 CrossRef CAS PubMed.
- P. S. Eis, W. Tam, L. P. Sun, A. Chadburn, Z. D. Li, M. F. Gomez, E. Lund and J. E. Dahlberg, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 3627–3632 CAS.
- J. A. Chan, A. M. Krichevsky and K. S. Kosik, Cancer Res., 2005, 65, 6029–6033 CAS.
- J. Kluiver, S. Poppema, D. de Jong, T. Blokzijl, G. Harms, S. Jacobs, B. J. Kroesen and A. van den Berg, J. Pathol., 2005, 207, 243–249 CAS.
- D. M. Kolpashchikov, Chem. Rev., 2010, 110, 4709–4723 CrossRef CAS PubMed.
- P. J. Asiello and A. J. Baeumner, Lab Chip, 2011, 11, 1420–1430 RSC.
- R. Wilson, Chem. Soc. Rev., 2008, 37, 2028–2045 RSC.
-
(a) H. Dong, J. Lei, L. Ding, Y. Wen, H. Ju and X. Zhang, Chem. Rev., 2013, 113, 6207–6233 CrossRef CAS PubMed;
(b) J. Li, S. Tan, R. Kooger, C. Zhang and Y. Zhang, Chem. Soc. Rev., 2014, 43, 506–517 RSC.
- M. L. Pardue and J. G. Gall, Proc. Natl. Acad. Sci. U. S. A., 1969, 64, 600–604 CrossRef CAS.
-
(a) J. E. Landegent, N. J. I. Dewal, R. A. Baan, J. H. J. Hoeijmakers and M. Van der ploeg, Exp. Cell Res., 1984, 153, 61–72 CrossRef CAS;
(b) D. Pinkel, T. Straume and J. W. Gray, Proc. Natl. Acad. Sci. U. S. A., 1986, 83, 2934–2938 CrossRef CAS;
(c) D. Pinkel, J. Landegent, C. Collins, J. Fuscoe, R. Segraves, J. Lucas and J. Gray, Proc. Natl. Acad. Sci. U. S. A., 1988, 85, 9138–9142 CrossRef CAS.
- E. F. DeLong, G. S. Wickham and N. R. Pace, Science, 1989, 243, 1360–1363 CAS.
- R. I. Amann, L. Krumholz and D. A. Stahl, J. Bacteriol., 1990, 172, 762–770 CAS.
-
(a) S. Tyagi and F. R. Kramer, Nat. Biotechnol., 1996, 14, 303–308 CrossRef CAS PubMed;
(b) K. Wang, Z. Tang, C. J. Yang, Y. Kim, X. Fang, W. Li, Y. Wu, C. D. Medley, Z. Cao, J. Li, P. Colon, H. Lin and W. Tan, Angew. Chem., Int. Ed., 2009, 48, 856–870 CrossRef CAS PubMed;
(c) Y. Li, X. Zhou and D. Ye, Biochem. Biophys. Res. Commun., 2008, 373, 457–461 CrossRef CAS PubMed;
(d) R. Monroy-Contreras and L. Vaca, J. Nucleic Acids, 2011, 2011, 741723 Search PubMed;
(e) N. E. Broude, Mol. Microbiol., 2011, 80, 1137–1147 CrossRef CAS PubMed.
- K. Huang and A. A. Marti, Anal. Bioanal. Chem., 2012, 402, 3091–3102 CrossRef CAS PubMed.
- S. Tyagi, D. P. Bratu and F. R. Kramer, Nat. Biotechnol., 1998, 16, 49–53 CrossRef CAS PubMed.
- S. Tyagi, S. A. E. Marras and F. R. Kramer, Nat. Biotechnol., 2000, 18, 1191–1196 CrossRef CAS PubMed.
- S. A. E. Marras, F. R. Kramer and S. Tyagi, Nucleic Acids Res., 2002, 30, e122 CrossRef PubMed.
-
(a) K. A. Browne, J. Am. Chem. Soc., 2005, 127, 1989–1994 CrossRef CAS PubMed;
(b) A. T. Krueger and E. T. Kool, J. Am. Chem. Soc., 2008, 130, 3989–3999 CrossRef CAS PubMed;
(c) P. Sheng, Z. Yang, Y. Kim, Y. Wu, W. Tan and S. A. Benner, Chem. Commun., 2008, 5128–5130 RSC;
(d) Y. Kim, C. J. Yang and W. Tan, Nucleic Acids Res., 2007, 35, 7279–7287 CrossRef CAS PubMed;
(e) C. Crey-Desbiolles, D. R. Ahn and C. J. Leumann, Nucleic Acids Res., 2005, 33, e77 CrossRef PubMed;
(f) Y. Ueno, A. Kawamura, K. Takasu, S. Komatsuzaki, T. Kato, S. Kuboe, Y. Kitamura and Y. Kitade, Org. Biomol. Chem., 2009, 7, 2761–2769 RSC.
-
(a) D. M. Kolpashchikov, J. Am. Chem. Soc., 2006, 128, 10625–10628 CrossRef CAS PubMed;
(b) Y. Xiao, K. J. I. Plakos, X. Lou, R. J. White, J. Qian, K. W. Plaxco and H. T. Soh, Angew. Chem., Int. Ed., 2009, 48, 4354–4358 CrossRef CAS PubMed;
(c) C. Lv, L. Yu, J. Wang and X. Tang, Bioorg. Med. Chem. Lett., 2010, 20, 6547–6550 CrossRef CAS PubMed.
-
(a) C.-H. Lu, J. Li, J.-J. Liu, H.-H. Yang, X. Chen and G.-N. Chen, Chem.–Eur. J., 2010, 16, 4889–4894 CrossRef CAS PubMed;
(b) H. Asanuma, T. Osawa, H. Kashida, T. Fujii, X. Liang, K. Niwa, Y. Yoshida, N. Shimadad and A. Maruyama, Chem. Commun., 2012, 48, 1760–1762 RSC;
(c) H. Kashida, T. Takatsu, T. Fujii, K. Sekiguchi, X. Liang, K. Niwa, T. Takase, Y. Yoshida and H. Asanuma, Angew. Chem., Int. Ed., 2009, 48, 7044–7047 CrossRef CAS PubMed;
(d) C. Y. J. Yang, H. Lin and W. H. Tan, J. Am. Chem. Soc., 2005, 127, 12772–12773 CrossRef CAS PubMed;
(e) B. Dubertret, M. Calame and A. J. Libchaber, Nat. Biotechnol., 2001, 19, 365–370 CrossRef CAS PubMed;
(f) T. N. Grossmann, L. Roglin and O. Seitz, Angew. Chem., Int. Ed., 2007, 46, 5223–5225 CrossRef CAS PubMed;
(g) Y. Hara, T. Fujii, H. Kashida, K. Sekiguchi, X. Liang, K. Niwa, T. Takase, Y. Yoshida and H. Asanuma, Angew. Chem., Int. Ed., 2010, 49, 5502–5506 CrossRef CAS PubMed.
-
(a) Y. Sato, S. Nishizawa and N. Teramae, Chem.–Eur. J., 2011, 17, 11650–11656 CrossRef CAS PubMed;
(b) A. Okamoto, K. Tainaka, Y. Ochi, K. Kanatani and I. Saito, Mol. Biosyst., 2006, 2, 122–127 RSC;
(c) H. Kashida, K. Yamaguchi, Y. Hara and H. Asanuma, Bioorg. Med. Chem., 2012, 20, 4310–4315 CrossRef CAS PubMed;
(d) T. Heinlein, J. P. Knemeyer, O. Piestert and M. Sauer, J. Phys. Chem. B, 2003, 107, 7957–7964 CrossRef CAS;
(e) P. Hou, Y. Long, J. Zhao, J. Wang and F. Zhou, Spectrochim. Acta, Part A, 2012, 86, 76–79 CrossRef CAS PubMed.
- Q. Q. Li, G. Y. Luan, Q. P. Guo and J. X. Liang, Nucleic Acids Res., 2002, 30, e5 CrossRef PubMed.
-
(a) J. M. Picuri, B. M. Frezza and M. R. Ghadiri, J. Am. Chem. Soc., 2009, 131, 9368–9377 CrossRef CAS PubMed;
(b) M. Zhou, Y. Liu, Y. Tu, G. Tao and J. Yan, Biosens. Bioelectron., 2012, 35, 489–492 CrossRef CAS PubMed;
(c) F. He, F. Feng, X. Duan, S. Wang, Y. Li and D. Zhu, Anal. Chem., 2008, 80, 2239–2243 CrossRef CAS PubMed;
(d) D. Y. Duose, R. M. Schweller, J. Zimak, A. R. Rogers, W. N. Hittelman and M. R. Diehl, Nucleic Acids Res., 2012, 40, 3289–3298 CrossRef CAS PubMed;
(e) A. E. Prigodich, P. S. Randeria, W. E. Briley, N. J. Kim, W. L. Daniel, D. A. Giljohann and C. A. Mirkin, Anal. Chem., 2012, 84, 2062–2066 CrossRef CAS PubMed;
(f) J. Hemphill and A. Deiters, J. Am. Chem. Soc., 2013, 135, 10512–10518 CrossRef CAS PubMed.
- D. S. Seferos, D. A. Giljohann, H. D. Hill, A. E. Prigodich and C. A. Mirkin, J. Am. Chem. Soc., 2007, 129, 15477–15479 CrossRef CAS PubMed.
- D. Y. Zhang and E. Winfree, J. Am. Chem. Soc., 2009, 131, 17303–17314 CrossRef CAS PubMed.
- J. C. Guatelli, K. M. Whitfield, D. Y. Kwoh, K. J. Barringer, D. D. Richman and T. R. Gingeras, Proc. Natl. Acad. Sci. U. S. A., 1990, 87, 1874–1878 CrossRef CAS.
- J. Compton, Nature, 1991, 350, 91–92 CrossRef CAS PubMed.
-
(a) K. Loens, M. Ieven, D. Ursi, H. Foolen, P. Sillekens and H. Goossens, J. Microbiol. Methods, 2003, 54, 127–130 CrossRef CAS;
(b) D. Jeantet, F. Schwarzmann, J. Tromp, W. J. G. Melchers, A. A. M. van der Wurff, T. Oosterlaken, M. Jacobs and A. Troesch, J. Clin. Virol., 2009, 45, S29–S37 CrossRef CAS.
- D. G. Murphy, L. Cote, M. Fauvel, P. Rene and J. Vincelette, J. Clin. Microbiol., 2000, 38, 4034–4041 CAS.
- M. Damen, P. Sillekens, H. T. M. Cuypers, I. Frantzen and R. Melsert, J. Virol. Methods, 1999, 82, 45–54 CrossRef CAS.
- F. Zhang, S. Tetali, X. P. Wang, M. H. Kaplan, F. V. Cromme and C. C. Ginocchio, J. Clin. Microbiol., 2000, 38, 1920–1925 CAS.
-
(a) S. A. Morre, P. T. G. Sillekens, M. V. Jacobs, S. de Blok, J. M. Ossewaarde, P. van Aarle, B. van Gemen, J. M. M. Walboomers, C. Meijer and A. C. van den Brule, J. Clin. Pathol.: Clin. Mol. Pathol., 1998, 51, 149–154 CAS;
(b) G. M. E. van der Vliet, S. N. Cho, K. Kampirapap, J. vanLeeuwen, R. A. F. Schukkink, B. vanGemen, P. K. Das, W. R. Faber, G. P. Walsh and P. R. Klatser, Int. J. Lepr. Other Mycobact. Dis., 1996, 64, 396–403 CAS;
(c) C. Ovyn, D. van Strijp, M. Leven, D. Ursi, B. van Gemen and H. Goossens, Mol. Cell. Probes, 1996, 10, 319–324 CrossRef CAS;
(d) M. Uyttendaele, R. Schukkink, B. Vangemen and J. Debevere, Int. J. Food Microbiol., 1995, 27, 77–89 CrossRef CAS;
(e) X. Song, B. K. Coombes and J. B. Mahony, Comb. Chem. High Throughput Screening, 2000, 3, 303–313 CrossRef CAS.
- J. D. Fox, S. Han, A. Samuelson, Y. D. Zhang, M. L. Neale and D. Westmoreland, J. Clin. Virol., 2002, 24, 117–130 CrossRef CAS.
- G. T. Walker, M. C. Little, J. G. Nadeau and D. D. Shank, Proc. Natl. Acad. Sci. U. S. A., 1992, 89, 392–396 CrossRef CAS.
- G. T. Walker, M. S. Fraiser, J. L. Schram, M. C. Little, J. G. Nadeau and D. P. Malinowski, Nucleic Acids Res., 1992, 20, 1691–1696 CrossRef CAS PubMed.
-
(a) M. C. Little, J. Andrews, R. Moore, S. Bustos, L. Jones, C. Embres, G. Durmowicz, J. Harris, D. Berger, K. Yanson, C. Rostkowski, D. Yursis, J. Price, T. Fort, A. Walters, M. Collis, O. Llorin, J. Wood, F. Failing, C. O'Keefe, B. Scrivens, B. Pope, T. Hansen, K. Marino, K. Williams and M. Boenisch, Clin. Chem., 1999, 45, 777–784 CAS;
(b) E. Tan, J. Wong, D. Nguyen, Y. Zhang, B. Erwin, L. K. Van Ness, S. M. Baker, D. J. Galas and A. Niemz, Anal. Chem., 2005, 77, 7984–7992 CrossRef CAS PubMed;
(c) H. Dong, J. Zhang, H. Ju, H. Lu, S. Wang, S. Jin, K. Hao, H. Du and X. Zhang, Anal. Chem., 2012, 84, 4587–4593 CrossRef CAS PubMed;
(d) A. Ogawa, Bioorg. Med. Chem. Lett., 2010, 20, 6056–6060 CrossRef CAS PubMed;
(e) C. Ding, X. Li, Y. Ge and S. Zhang, Anal. Chem., 2010, 82, 2850–2855 CrossRef CAS PubMed;
(f) A. R. Connolly and M. Trau, Angew. Chem., Int. Ed., 2010, 49, 2720–2723 CrossRef CAS PubMed;
(g) C. Shi, C. Zhao, Q. Guo and C. Ma, Chem. Commun., 2011, 47, 2895–2897 RSC;
(h) A. R. Connolly and M. Trau, Nat. Protoc., 2011, 6, 772–778 CrossRef CAS PubMed.
- Q. Guo, X. Yang, K. Wang, W. Tan, W. Li, H. Tang and H. Li, Nucleic Acids Res., 2009, 37, e20 CrossRef PubMed.
-
(a) J. Van Ness, L. K. Van Ness and D. J. Galas, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 4504–4509 CrossRef CAS PubMed;
(b) H. Jia, Z. Li, C. Liu and Y. Cheng, Angew. Chem., Int. Ed., 2010, 49, 5498–5501 CrossRef CAS PubMed.
- Z.-z. Zhang and C.-y. Zhang, Anal. Chem., 2012, 84, 1623–1629 CrossRef CAS PubMed.
- H.-Q. Wang, W.-Y. Liu, Z. Wu, L.-J. Tang, X.-M. Xu, R.-Q. Yu and J.-H. Jiang, Anal. Chem., 2011, 83, 1883–1889 CrossRef CAS PubMed.
- T. Notomi, H. Okayama, H. Masubuchi, T. Yonekawa, K. Watanabe, N. Amino and T. Hase, Nucleic Acids Res., 2000, 28, e63 CrossRef CAS PubMed.
- M. Parida, K. Horioke, H. Ishida, P. K. Dash, P. Saxena, A. M. Jana, M. A. Islam, S. Inoue, N. Hosaka and K. Morita, J. Clin. Microbiol., 2005, 43, 2895–2903 CrossRef CAS PubMed.
- M. M. Parida, S. R. Santhosh, P. K. Dash, N. K. Tripathi, P. Saxena, S. Ambuj, A. K. Sahni, P. V. L. Rao and K. Morita, J. Clin. Microbiol., 2006, 44, 4172–4178 CrossRef CAS PubMed.
- M. M. Parida, S. R. Santhosh, P. K. Dash, N. K. Tripathi, V. Lakshmi, N. Mamidi, A. Shrivastva, N. Gupta, P. Saxena, J. P. Babu, P. V. L. Rao and K. Morita, J. Clin. Microbiol., 2007, 45, 351–357 CrossRef CAS PubMed.
- M. Parida, G. Posadas, S. Inoue, F. Hasebe and K. Morita, J. Clin. Microbiol., 2004, 42, 257–263 CrossRef CAS.
- T. C. T. Hong, Q. L. Mai, D. V. Cuong, M. Parida, H. Minekawa, T. Notomi, F. Hasebe and K. Morita, J. Clin. Microbiol., 2004, 42, 1956–1961 CrossRef PubMed.
- M. Imai, A. Ninomiya, H. Minekawa, T. Notomi, T. Ishizaki, P. V. Tu, N. T. K. Tien, M. Tashiro and T. Odagiri, J. Virol. Methods, 2007, 141, 173–180 CrossRef CAS PubMed.
- M. Olivier, Mutat. Res., Fundam. Mol. Mech. Mutagen., 2005, 573, 103–110 CrossRef CAS PubMed.
- V. Lyamichev, A. L. Mast, J. G. Hall, J. R. Prudent, M. W. Kaiser, T. Takova, R. W. Kwiatkowski, T. J. Sander, M. de Arruda, D. A. Arco, B. P. Neri and M. A. D. Brow, Nat. Biotechnol., 1999, 17, 292–296 CrossRef CAS PubMed.
- J. G. Hall, P. S. Eis, S. M. Law, L. P. Reynaldo, J. R. Prudent, D. J. Marshall, H. T. Allawi, A. L. Mast, J. E. Dahlberg, R. W. Kwiatkowski, M. de Arruda, B. P. Neri and V. I. Lyamichev, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 8272–8277 CrossRef CAS PubMed.
- S.-I. Usami, M. Wagatsuma, H. Fukuoka, H. Suzuki, K. Tsukada, S. Nishio, Y. Takumi and S. Abe, Acta Otolaryngol., 2008, 128, 446–454 CrossRef CAS PubMed.
- M. J. Stillman, S. P. Day and T. E. Schutzbank, J. Clin. Virol., 2009, 45, S73–S77 CrossRef CAS.
- H. T. Allawi, J. E. Dahlberg, S. Olson, E. Lund, M. Olson, W. P. Ma, T. Takova, B. P. Neri and V. I. Lyamichev, RNA, 2004, 10, 1153–1161 CrossRef CAS PubMed.
- H. T. Allawi, H. Li, T. Sander, A. Aslanukov, V. I. Lyamichev, A. Blackman, S. Elagin and Y.-W. Tang, J. Clin. Microbiol., 2006, 44, 3443–3447 CrossRef CAS PubMed.
- A. Fire and S. Q. Xu, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 4641–4645 CrossRef CAS.
- P. M. Lizardi, X. H. Huang, Z. R. Zhu, P. Bray-Ward, D. C. Thomas and D. C. Ward, Nat. Genet., 1998, 19, 225–232 CrossRef CAS PubMed.
- D. Y. Liu, S. L. Daubendiek, M. A. Zillman, K. Ryan and E. T. Kool, J. Am. Chem. Soc., 1996, 118, 1587–1594 CrossRef CAS PubMed.
- J. Baner, M. Nilsson, M. Mendel-Hartvig and U. Landegren, Nucleic Acids Res., 1998, 26, 5073–5078 CrossRef CAS PubMed.
- E. J. Cho, L. T. Yang, M. Levy and A. D. Ellington, J. Am. Chem. Soc., 2005, 127, 2022–2023 CrossRef CAS PubMed.
- V. V. Demidov, Expert Rev. Mol. Diagn., 2002, 2, 542–548 CrossRef CAS PubMed.
- W. Xu, X. Xie, D. Li, Z. Yang, T. Li and X. Liu, Small, 2012, 8, 1846–1850 CrossRef CAS PubMed.
- Y. Zhou, Q. Huang, J. Gao, J. Lu, X. Shen and C. Fan, Nucleic Acids Res., 2010, 38, e156 CrossRef PubMed.
- M. Nilsson, M. Gullberg, F. Dahl, K. Szuhai and A. K. Raap, Nucleic Acids Res., 2002, 30, e66 CrossRef PubMed.
- G. A. Blab, T. Schmidt and M. Nilsson, Anal. Chem., 2004, 76, 495–498 CrossRef CAS PubMed.
- D. Y. Zhang, W. D. Zhang, X. P. Li and Y. Konomi, Gene, 2001, 274, 209–216 CrossRef CAS.
- M. C. Steain, D. E. Dwyer, A. C. Hurt, C. Kol, N. K. Saksena, A. L. Cunningham and B. Wang, Antiviral Res., 2009, 84, 242–248 CrossRef CAS PubMed.
- S. Henriksson, A.-L. Blomstrom, L. Fuxler, C. Fossum, M. Berg and M. Nilsson, Virol. J., 2011, 8, 37 CrossRef CAS PubMed.
- Y. Long, X. Zhou and D. Xing, Biosens. Bioelectron., 2011, 26, 2897–2904 CrossRef CAS PubMed.
- E. M. Harcourt and E. T. Kool, Nucleic Acids Res., 2012, 40, e65 CrossRef CAS PubMed.
- Y. Long, X. Zhou and D. Xing, Biosens. Bioelectron., 2013, 46, 102–107 CrossRef CAS PubMed.
-
(a) T. Murakami, J. Sumaoka and M. Komiyama, Nucleic Acids Res., 2009, 37, e19 CrossRef PubMed;
(b) F. Dahl, J. Baner, M. Gullberg, M. Mendel-Hartvig, U. Landegren and M. Nilsson, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 4548–4553 CrossRef CAS PubMed.
-
(a) A. T. Christian, M. S. Pattee, C. M. Attix, B. E. Reed, K. J. Sorensen and J. D. Tucker, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 14238–14243 CrossRef CAS PubMed;
(b) E. Merkiene, E. Gaidamaviciute, L. Riauba, A. Janulaitis and A. Lagunavicius, RNA, 2010, 16, 1508–1515 CrossRef CAS PubMed;
(c) C. Larsson, J. Koch, A. Nygren, G. Janssen, A. K. Raap, U. Landegren and M. Nilsson, Nat. Methods, 2004, 1, 227–232 CrossRef CAS PubMed;
(d) M. Stougaard, J. S. Lohmann, M. Zajac, S. Hamilton-Dutoit and J. Koch, BMC Biotechnol., 2007, 7, 69 CrossRef PubMed;
(e) Y. Cheng, X. Zhang, Z. Li, X. Jiao, Y. Wang and Y. Zhang, Angew. Chem., Int. Ed., 2009, 48, 3268–3272 CrossRef CAS PubMed;
(f) N. Li, C. Jablonowski, H. Jin and W. Zhong, Anal. Chem., 2009, 81, 4906–4913 CrossRef CAS PubMed;
(g) S. P. Jonstrup, J. Koch and J. Kjems, RNA, 2006, 12, 1747–1752 CrossRef CAS PubMed.
- D. Y. Zhang, M. Brandwein, T. Hsuih and H. B. Li, Mol. Diagn., 2001, 6, 141–150 CrossRef CAS PubMed.
- D. Wharam, P. Marsh, J. S. Lloyd, T. D. Ray, G. A. Mock, R. Assenberg, J. E. McPhee, P. Brown, A. Weston and D. L. Cardy, Nucleic Acids Res., 2001, 29, e54 CrossRef PubMed.
-
(a) M. J. Hall, S. D. Wharam, A. Weston, D. L. N. Cardy and W. H. Wilson, Biotechniques, 2002, 32, 604–606 CAS;
(b) S. D. Wharam, M. J. Hall and W. H. Wilson, Virol. J., 2007, 4, 52 CrossRef PubMed.
- M. Vincent, Y. Xu and H. M. Kong, EMBO Rep., 2004, 5, 795–800 CrossRef CAS PubMed.
- L. X. An, W. Tang, T. A. Ranalli, H. J. Kim, J. Wytiaz and H. M. Kong, J. Biol. Chem., 2005, 280, 28952–28958 CrossRef CAS PubMed.
- A. Motre, Y. Li and H. Kong, Gene, 2008, 420, 17–22 CrossRef CAS PubMed.
- O. Piepenburg, C. H. Williams, D. L. Stemple and N. A. Armes, PLoS Biol., 2006, 4, e204 Search PubMed ,1115–1121.
- W. Xu, X. Xue, T. Li, H. Zeng and X. Liu, Angew. Chem., Int. Ed., 2009, 48, 6849–6852 CrossRef CAS PubMed.
- J. J. Li, Y. Chu, B. Y.-H. Lee and X. S. Xie, Nucleic Acids Res., 2008, 36, e36 CrossRef PubMed.
-
(a) S. Bi, J. Zhang and S. Zhang, Chem. Commun., 2010, 46, 5509–5511 RSC;
(b) T. Ando, J. Takagi, T. Kosawa and Y. Ikeda, J. Biochem., 1969, 66, 1–10 CAS;
(c) Y. Weizmann, M. K. Beissenhirtz, Z. Cheglakov, R. Nowarski, M. Kotler and I. Willner, Angew. Chem., Int. Ed., 2006, 45, 7384–7388 CrossRef CAS PubMed;
(d) B. Zou, Y. Ma, H. Wu and G. Zhou, Angew. Chem., Int. Ed., 2011, 50, 7395–7398 CrossRef CAS PubMed;
(e) J. Song, Z. Li, Y. Cheng and C. Liu, Chem. Commun., 2010, 46, 5548–5550 RSC;
(f) Z. Lin, W. Yang, G. Zhang, Q. Liu, B. Qiu, Z. Cai and G. Chen, Chem. Commun., 2011, 47, 9069–9071 RSC;
(g) R. D. Morgan, C. Calvet, M. Demeter, R. Agra and H. M. Kong, Biol. Chem., 2000, 381, 1123–1125 CrossRef CAS PubMed;
(h) T. Kiesling, K. Cox, E. A. Davidson, K. Dretchen, G. Grater, S. Hibbard, R. S. Lasken, J. Leshin, E. Skowronski and M. Danielsen, Nucleic Acids Res., 2007, 35, e117 CrossRef PubMed;
(i) L. A. Zheleznaya, G. S. Kachalova, R. I. Artyukh, A. K. Yunusova, T. A. Perevyazova and N. I. Matvienko, Biochemistry, 2009, 74, 1457–1466 CAS;
(j) L. Xu, Y. Zhu, W. Ma, H. Kuang, L. Liu, L. Wang and C. Xu, J. Phys. Chem. C, 2011, 115, 16315–16321 CrossRef CAS.
- J. Li, Q.-H. Yao, H.-E. Fu, X.-L. Zhang and H.-H. Yang, Talanta, 2011, 85, 91–96 CrossRef CAS PubMed.
- X. Zuo, F. Xia, Y. Xiao and K. W. Plaxco, J. Am. Chem. Soc., 2010, 132, 1816–1818 CrossRef CAS PubMed.
-
(a) B. Z. Ren, J. M. Zhou and M. Komiyama, Nucleic Acids Res., 2004, 32, e42 CrossRef PubMed;
(b) H. J. Lee, Y. Li, A. W. Wark and R. M. Corn, Anal. Chem., 2005, 77, 5096–5100 CrossRef CAS PubMed;
(c) F. Xuan, X. Luo and I. M. Hsing, Anal. Chem., 2012, 84, 5216–5220 CrossRef CAS PubMed;
(d) L. Cui, G. Ke, C. Wang and C. J. Yang, Analyst, 2010, 135, 2069–2073 RSC.
- S. Liu, C. Wang, C. Zhang, Y. Wang and B. Tang, Anal. Chem., 2013, 85, 2282–2288 CrossRef CAS PubMed.
- S. Nakayama, L. Yan and H. O. Sintim, J. Am. Chem. Soc., 2008, 130, 12560–12561 CrossRef CAS PubMed.
- L. Yan, S. Nakayama, S. Yitbarek, I. Greenfield and H. O. Sintim, Chem. Commun., 2011, 47, 200–202 RSC.
- Z. Shen, S. Nakayama, S. Semancik and H. O. Sintim, Chem. Commun., 2012, 48, 7580–7582 RSC.
- L. Yan, S. Nakayama and H. O. Sintim, Bioorg. Med. Chem., 2013, 21, 6181–6185 CrossRef CAS PubMed.
-
(a) Z. Zhu, F. Gao, J. Lei, H. Dong and H. Ju, Chem.–Eur. J., 2012, 18, 13871–13876 CrossRef CAS PubMed;
(b) J. Li, X.-J. Qi, Y.-Y. Du, H.-E. Fu, G.-N. Chen and H.-H. Yang, Biosens. Bioelectron., 2012, 36, 142–146 CrossRef CAS PubMed.
- Q. Wang, L. Yang, X. Yang, K. Wang, L. He, J. Zhu and T. Su, Chem. Commun., 2012, 48, 2982–2984 RSC.
- R. M. Kong, X. B. Zhang, L. L. Zhang, Y. Huang, D. Q. Lu, W. H. Tan, G. L. Shen and R. Q. Yu, Anal. Chem., 2011, 83, 14–17 CrossRef CAS PubMed.
- H. X. Ji, F. Yan, J. P. Lei and H. X. Ju, Anal. Chem., 2012, 84, 7166–7171 CrossRef CAS PubMed.
-
(a) M. D. Distefano, J. A. Shin and P. B. Dervan, J. Am. Chem. Soc., 1991, 113, 5901–5902 CrossRef CAS;
(b) G. B. Dreyer and P. B. Dervan, Proc. Natl. Acad. Sci. U. S. A., 1985, 82, 968–972 CrossRef CAS.
- T. Murakami, J. Sumaoka and M. Komiyama, Nucleic Acids Res., 2012, 40, e22 CrossRef CAS PubMed.
-
(a) M. D. Distefano and P. B. Dervan, J. Am. Chem. Soc., 1992, 114, 11006–11007 CrossRef CAS;
(b) M. D. Distefano and P. B. Dervan, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 1179–1183 CrossRef CAS.
-
(a) J. Zhang, J. H. Chen, R. C. Chen, G. N. Chen and F. F. Fu, Biosens. Bioelectron., 2009, 25, 815–819 CrossRef CAS PubMed;
(b) J. Zhang, X. Wu, P. Chen, N. Lin, J. Chen, G. Chen and F. Fu, Chem. Commun., 2010, 46, 6986–6988 RSC.
- B. W. Matthews, Acc. Chem. Res., 1988, 21, 333–340 CrossRef CAS.
- A. Saghatelian, K. M. Guckian, D. A. Thayer and M. R. Ghadiri, J. Am. Chem. Soc., 2003, 125, 344–345 CrossRef CAS PubMed.
- V. Pavlov, B. Shlyahovsky and I. Willner, J. Am. Chem. Soc., 2005, 127, 6522–6523 CrossRef CAS PubMed.
- R. Duan, X. Zuo, S. Wang, X. Quan, D. Chen, Z. Chen, L. Jiang, C. Fan and F. Xia, J. Am. Chem. Soc., 2013, 135, 4604–4607 CrossRef CAS PubMed.
- B. Li, X. Chen and A. D. Ellington, Anal. Chem., 2012, 84, 8371–8377 CrossRef CAS PubMed.
- Y. Jiang, B. Li, J. N. Milligan, S. Bhadra and A. D. Ellington, J. Am. Chem. Soc., 2013, 135, 7430–7433 CrossRef CAS PubMed.
-
(a) Y. F. Li, C. R. Geyer and D. Sen, Biochemistry, 1996, 35, 6911–6922 CrossRef CAS PubMed;
(b) P. Travascio, Y. F. Li and D. Sen, Chem. Biol., 1998, 5, 505–517 CrossRef CAS;
(c) P. Travascio, A. J. Bennet, D. Y. Wang and D. Sen, Chem. Biol., 1999, 6, 779–787 CrossRef CAS.
- D. M. Kolpashchikov, J. Am. Chem. Soc., 2008, 130, 2934–2935 CrossRef CAS PubMed.
- M. Deng, D. Zhang, Y. Zhou and X. Zhou, J. Am. Chem. Soc., 2008, 130, 13095–13102 CrossRef CAS PubMed.
- S. Nakayama and H. O. Sintim, J. Am. Chem. Soc., 2009, 131, 10320–10333 CrossRef CAS PubMed.
- A. K. L. Darius, N. J. Ling and U. Mahesh, Mol. Biosyst., 2010, 6, 792–794 RSC.
-
(a) Y. Xiao, V. Pavlov, T. Niazov, A. Dishon, M. Kotler and I. Willner, J. Am. Chem. Soc., 2004, 126, 7430–7431 CrossRef CAS PubMed;
(b) L. Zhang, J. Zhu, T. Li and E. Wang, Anal. Chem., 2011, 83, 8871–8876 CrossRef CAS PubMed;
(c) Z. Zheng, J. Han, W. Pang and J. Hu, Sensors, 2013, 13, 1064–1075 CrossRef CAS PubMed;
(d) A. Bourdoncle, A. E. Torres, C. Gosse, L. Lacroix, P. Vekhoff, T. Le Saux, L. Jullien and J. L. Mergny, J. Am. Chem. Soc., 2006, 128, 11094–11105 CrossRef CAS PubMed.
-
(a) Z. Cheglakov, Y. Weizmann, B. Basnar and I. Willner, Org. Biomol. Chem., 2007, 5, 223–225 RSC;
(b) Y.-P. Zeng, J. Hu, Y. Long and C.-Y. Zhang, Anal. Chem., 2013, 85, 6143–6150 CrossRef CAS PubMed.
-
(a) J. Chen, J. Zhang, Y. Guo, J. Li, F. Fu, H.-H. Yang and G. Chen, Chem. Commun., 2011, 47, 8004–8006 RSC;
(b) C. Zhao, L. Wu, J. Ren and X. Qu, Chem. Commun., 2011, 47, 5461–5463 RSC.
- M. N. Stojanovic, P. de Prada and D. W. Landry, ChemBioChem, 2001, 2, 411–415 CrossRef CAS.
- J. S. Hartig, I. Grune, S. H. Najafi-Shoushtari and M. Famulok, J. Am. Chem. Soc., 2004, 126, 722–723 CrossRef CAS PubMed.
- S. Sando, T. Sasaki, K. Kanatani and Y. Aoyama, J. Am. Chem. Soc., 2003, 125, 15720–15721 CrossRef CAS PubMed.
- R. R. Breaker and G. F. Joyce, Chem. Biol., 1995, 2, 655–660 CrossRef CAS.
-
(a) E. Mokany, S. M. Bone, P. E. Young, T. B. Doan and A. V. Todd, J. Am. Chem. Soc., 2010, 132, 1051–1059 CrossRef CAS PubMed;
(b) F. Wang, J. Elbaz, C. Teller and I. Willner, Angew. Chem., Int. Ed., 2011, 50, 295–299 CrossRef CAS PubMed.
- D. M. Kolpashchikov, ChemBioChem, 2007, 8, 2039–2042 CrossRef CAS PubMed.
- Y. V. Gerasimova, E. Cornett and D. M. Kolpashchikov, ChemBioChem, 2010, 11, 811–817 CrossRef CAS PubMed.
- M. Levy and A. D. Ellington, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 6416–6421 CrossRef CAS PubMed.
- J. Elbaz, M. Moshe, B. Shlyahovsky and I. Willner, Chem.–Eur. J., 2009, 15, 3411–3418 CrossRef CAS PubMed.
- R. M. Franzini and E. T. Kool, J. Am. Chem. Soc., 2009, 131, 16021–16023 CrossRef CAS PubMed.
- P. Silverman and E. T. Kool, Chem. Rev., 2006, 106, 3775–3789 CrossRef PubMed.
-
(a) Y. Z. Xu, N. B. Karalkar and E. T. Kool, Nat. Biotechnol., 2001, 19, 148–152 CrossRef CAS PubMed;
(b) E. Jentzsch and A. Mokhir, Inorg. Chem., 2009, 48, 9593–9595 CrossRef CAS PubMed;
(c) A. H. El-Sagheer, V. V. Cheong and T. Brown, Org. Biomol. Chem., 2011, 9, 232–235 RSC;
(d) S. M. Gryaznov and R. L. Letsinger, J. Am. Chem. Soc., 1993, 115, 3808–3809 CrossRef CAS;
(e) A. Mattes and O. Seitz, Chem. Commun., 2001, 2050–2051 RSC;
(f) J. L. Czlapinski and T. L. Sheppard, ChemBioChem, 2004, 5, 127–129 CrossRef CAS PubMed;
(g) S. M. Gryaznov and R. L. Letsinger, Nucleic Acids Res., 1993, 21, 1403–1408 CrossRef CAS PubMed;
(h) Y. Z. Xu and E. T. Kool, Nucleic Acids Res., 1999, 27, 875–881 CrossRef CAS PubMed;
(i) K. Fujimoto, S. Matsuda, N. Takahashi and I. Saito, J. Am. Chem. Soc., 2000, 122, 5646–5647 CrossRef CAS.
- J. F. Cai, X. X. Li, X. Yue and J. S. Taylor, J. Am. Chem. Soc., 2004, 126, 16324–16325 CrossRef CAS PubMed.
- Z. C. Ma and J. S. Taylor, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 11159–11163 CrossRef CAS PubMed.
- Z. C. Ma and J. S. Taylor, Bioorg. Med. Chem., 2001, 9, 2501–2510 CrossRef CAS.
- S. Sando and E. T. Kool, J. Am. Chem. Soc., 2002, 124, 9686–9687 CrossRef CAS PubMed.
- H. Abe and E. T. Kool, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 263–268 CrossRef CAS PubMed.
- Y. Huang and J. M. Coull, J. Am. Chem. Soc., 2008, 130, 3238–3239 CrossRef CAS PubMed.
-
(a) H. Abe, J. Wang, K. Furukawa, K. Oki, M. Uda, S. Tsuneda and Y. Ito, Bioconjugate Chem., 2008, 19, 1219–1226 CrossRef CAS PubMed;
(b) L. Pianowski and N. Winssinger, Chem. Commun., 2007, 3820–3822 RSC;
(c) Z. Pianowski, K. Gorska, L. Oswald, C. A. Merten and N. Winssinger, J. Am. Chem. Soc., 2009, 131, 6492–6497 CrossRef CAS PubMed;
(d) R. M. Franzini and E. T. Kool, ChemBioChem, 2008, 9, 2981–2988 CrossRef CAS PubMed.
-
(a) J. F. Cai, X. X. Li and J. S. Taylor, Org. Lett., 2005, 7, 751–754 CrossRef CAS PubMed;
(b) R. M. Franzini and E. T. Kool, Org. Lett., 2008, 10, 2935–2938 CrossRef CAS PubMed.
- T. N. Grossmann and O. Seitz, J. Am. Chem. Soc., 2006, 128, 15596–15597 CrossRef CAS PubMed.
- T. N. Grossmann and O. Seitz, Chem.–Eur. J., 2009, 15, 6723–6730 CrossRef CAS PubMed.
- T. N. Grossmann, L. Roeglin and O. Seitz, Angew. Chem., Int. Ed., 2008, 47, 7119–7122 CrossRef CAS PubMed.
-
(a) A. P. Silverman and E. T. Kool, Nucleic Acids Res., 2005, 33, 4978–4986 CrossRef CAS PubMed;
(b) S. Sando, H. Abe and E. T. Kool, J. Am. Chem. Soc., 2004, 126, 1081–1087 CrossRef CAS PubMed;
(c) A. P. Silverman, E. J. Baron and E. T. Kool, ChemBioChem, 2006, 7, 1890–1894 CrossRef CAS PubMed.
- H. Abe and E. T. Kool, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 263–268 CrossRef CAS PubMed.
- G. P. Miller, A. P. Silverman and E. T. Kool, Bioorg. Med. Chem., 2008, 16, 56–64 CrossRef CAS PubMed.
- A. P. Silverman, H. Abe and E. T. Kool, Methods Mol. Biol., 2008, 429, 161–170 CAS.
-
(a) K. Furukawa, H. Abe, K. Hibino, Y. Sako, S. Tsuneda and Y. Ito, Bioconjugate Chem., 2009, 20, 1026–1036 CrossRef CAS PubMed;
(b) H. Li, R. M. Franzini, C. Bruner and E. T. Kool, ChemBioChem, 2010, 11, 2132–2137 CrossRef CAS PubMed;
(c) A. Okamoto, K. Tanabe, T. Inasaki and I. Saito, Angew. Chem., Int. Ed., 2003, 42, 2502–2504 CrossRef CAS PubMed;
(d) K. Tanabe, H. Nakata, S. Mukai and S. Nishimoto, Org. Biomol. Chem., 2005, 3, 3893–3897 RSC;
(e) M. Roethlingshoefer, K. Gorska and N. Winssinger, J. Am. Chem. Soc., 2011, 133, 18110–18113 CrossRef PubMed;
(f) D. Arian, E. Clo, K. V. Gothelf and A. Mokhir, Chem.–Eur. J., 2010, 16, 288–295 CrossRef CAS PubMed;
(g) D. Arian, L. Kovbasyuk and A. Mokhir, Inorg. Chem., 2011, 50, 12010–12017 CrossRef CAS PubMed;
(h) M. Roethlingshoefer, K. Gorska and N. Winssinger, Org. Lett., 2012, 14, 482–485 CrossRef PubMed;
(i) D. K. Prusty and A. Herrmann, J. Am. Chem. Soc., 2010, 132, 12197–12199 CrossRef CAS PubMed;
(j) D. K. Prusty, M. Kwak, J. Wildeman and A. Herrmann, Angew. Chem., Int. Ed., 2012, 51, 11894–11898 CrossRef CAS PubMed;
(k) T. N. Grossmann, A. Strohbach and O. Seitz, ChemBioChem, 2008, 9, 2185–2192 CrossRef CAS PubMed.
- P. Yin, H. M. T. Choi, C. R. Calvert and N. A. Pierce, Nature, 2008, 451, 318–322 CrossRef CAS PubMed.
-
(a) D. Y. Zhang, A. J. Turberfield, B. Yurke and E. Winfree, Science, 2007, 318, 1121–1125 CrossRef CAS PubMed;
(b) B. Li, A. D. Ellington and X. Chen, Nucleic Acids Res., 2011, 39, e110 CrossRef CAS PubMed;
(c) A.-X. Zheng, J. Li, J.-R. Wang, X.-R. Song, G.-N. Chen and H.-H. Yang, Chem. Commun., 2012, 48, 3112–3114 RSC;
(d) D. Y. Zhang and E. Winfree, Nucleic Acids Res., 2010, 38, 4182–4197 CrossRef CAS PubMed.
- F. A. Wang, J. Elbaz and I. Willner, J. Am. Chem. Soc., 2012, 134, 5504–5507 CrossRef CAS PubMed.
- R. M. Dirks and N. A. Pierce, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 15275–15278 CrossRef CAS PubMed.
- H. M. T. Choi, J. Y. Chang, L. A. Trinh, J. E. Padilla, S. E. Fraser and N. A. Pierce, Nat. Biotechnol., 2010, 28, 1208–1212 CrossRef CAS PubMed.
- J. Huang, Y. Wu, Y. Chen, Z. Zhu, X. Yang, C. J. Yang, K. Wang and W. Tan, Angew. Chem., Int. Ed., 2011, 50, 401–404 CrossRef CAS PubMed.
-
(a) F. Wang, J. Elbaz, R. Orbach, N. Magen and I. Willner, J. Am. Chem. Soc., 2011, 133, 17149–17151 CrossRef CAS PubMed;
(b) S. Shimron, F. Wang, R. Orbach and I. Willner, Anal. Chem., 2012, 84, 1042–1048 CrossRef CAS PubMed.
- B. Li, Y. Jiang, X. Chen and A. D. Ellington, J. Am. Chem. Soc., 2012, 134, 13918–13921 CrossRef CAS PubMed.
- C. Lin, Y. Liu and H. Yan, Nano Lett., 2007, 7, 507–512 CrossRef CAS PubMed.
-
(a) Y. G. Li, Y. D. Tseng, S. Y. Kwon, L. D'Espaux, J. S. Bunch, P. L. McEuen and D. Luo, Nat. Mater., 2004, 3, 38–42 CrossRef CAS PubMed;
(b) A. L. Benvin, Y. Creeger, G. W. Fisher, B. Ballou, A. S. Waggoner and B. A. Armitage, J. Am. Chem. Soc., 2007, 129, 2025–2034 CrossRef CAS PubMed;
(c) U. Feldkamp, B. Sacca and C. M. Niemeyer, Angew. Chem., Int. Ed., 2009, 48, 5996–6000 CrossRef CAS PubMed.
- Y. G. Li, Y. T. H. Cu and D. Luo, Nat. Biotechnol., 2005, 23, 885–889 CrossRef CAS PubMed.
- Y. Yan, W. Sha, J. Sun, X. Shu, S. Sun and J. Zhu, Chem. Commun., 2011, 47, 7470–7472 RSC.
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here.  Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a