 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A copper complex supported by an N2S-tridentate ligand inducing efficient heterolytic O–O bond cleavage of alkylhydroperoxide†
Tetsuro
Tano
a,
Kaoru
Mieda
b,
Hideki
Sugimoto
a,
Takashi
Ogura
b and
Shinobu
Itoh
*a
aDepartment of Material and Life Science, Division of Advanced Science and Biotechnology, Graduate School of Engineering, Osaka University, 2-1 Yamada-oka, Suita, Osaka 565-0871, Japan. E-mail: shinobu@mls.eng.osaka-u.ac.jp
bResearch Institute of Picobiology, Graduate School of Life Science, University of Hyogo, 3-2-1 Kouto, Kamigori-cho, Ako-gun, Hyogo 678-1297, Japan
First published on 13th January 2014
Abstract
We have recently reported a copper(II)-superoxide complex supported by an N3-tridentate ligand (LN3), which exhibits a similar structure and reactivity to those of a putative reactive intermediate involved in the catalytic reactions of copper monooxygenases such as peptidylglycine α-hydroxylating monooxygenase (PHM) and dopamine β-monooxygenase (DβM). In this study, we have synthesised and characterised copper complexes supported by a related sulphur-containing ligand (LN2S) to get insight into the notable electronic effect of the sulphur donor atom in the reaction with cumene hydroperoxide, inducing efficient heterolytic O–O bond cleavage.
Introduction
Copper monooxygenases such as peptidylglycine α-hydroxylating monooxygenase (PHM), dopamine β-monooxygenase (DβM) and tyramine β-monooxygenase (TβM) employ two uncoupled, mononuclear copper sites referred to as CuM and CuH.1–6 CuM serves as the reaction site for dioxygen activation and substrate oxygenation, whereas CuH functions as an electron acceptor from the physiological reductant (ascorbate). The CuM site is ligated by two histidine imidazoles and one methionine sulphur (N2S donor set), whereas CuH is coordinated by three histidine imidazoles (N3 donor set).3,4 These coordination environments are somewhat unusual, since most of the reaction centres of O2-activating copper proteins (type-2 and type-3 Cu proteins) consist of nitrogen rich donor sets, whereas the large number of electron transfer proteins (type-1 Cu proteins) involve a sulphur-containing ligand environment.7,8 Thus, such a unique feature of these copper monooxygenases has long been a target in synthetic bioinorganic modelling studies.9–20So far, a series of mononuclear copper–dioxygen adducts have been characterised as model compounds of possible reactive intermediates of copper monooxygenases.21–25 Among them, a mononuclear copper(II)-superoxide complex supported by a simple N3-ligand LN3 (1-[2-(2-pyridyl)ethyl]-1,5-diazacyclooctane derivatives, Fig. 1) is a unique example, which exhibits both a similar structural feature (distorted tetrahedral copper(II) with an end-on superoxide ligand) and a reactivity (aliphatic hydroxylation) to those of the putative reactive intermediate involved in the enzymatic reactions.26,27 In this study, we have synthesised and characterised the copper(I) and copper(II) complexes supported by LN2S (Fig. 1) in order to get insights into the effects of the sulphur atom on the structure and reactivity. Ligand LN2S has the same molecular framework as that of LN3, but one of the alkylamine nitrogen atoms is replaced by a sulphur atom, allowing us to simply access the electronic effects of the donor atoms.
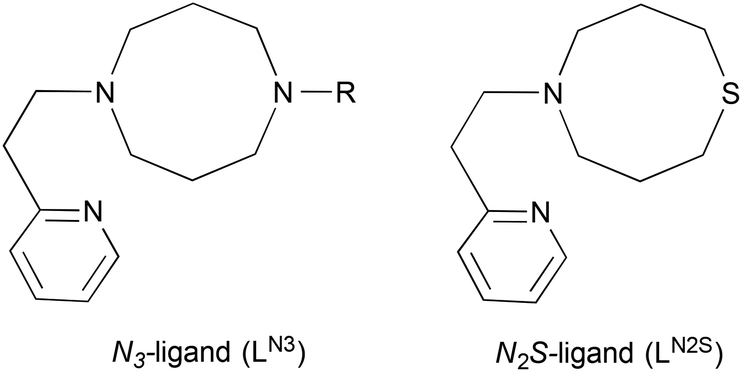 | ||
| Fig. 1 Ligand structures. | ||
Results and discussion
Synthesis and characterization
The ligand LN2S was synthesized in three steps as shown in Scheme 1. Treatment of 2-(2-aminoethyl)pyridine and 3-chloro-1-propanol in CH3CN gave a bis(3-hydroxypropyl)amine derivative, which was then converted to the dichloro derivative by the reaction with SOCl2. Ring closure using Na2S gave the ligand LN2S in a moderate yield.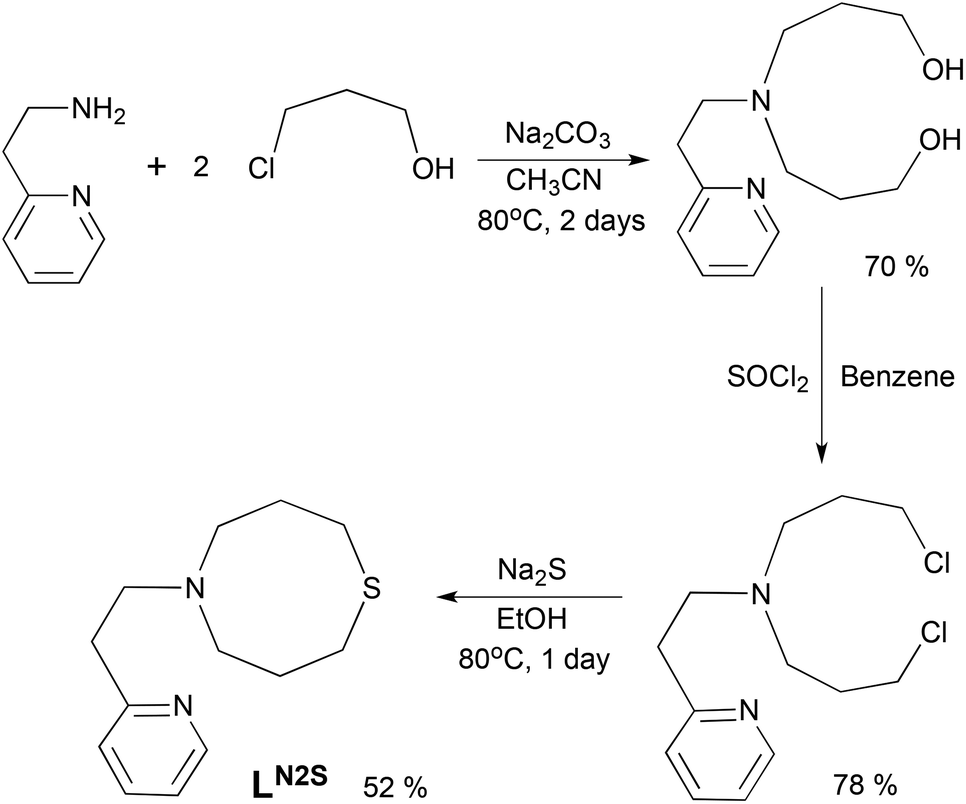 | ||
| Scheme 1 Ligand synthesis. | ||
The copper(I) complex [CuI(LN2S)](PF6) (1N2S) was prepared by treating LN2S with an equimolar amount of [CuI(CH3CN)4](PF6) in THF. The crystal structure of 1N2S is shown in Fig. 2. The crystallographic data and the selected bond lengths and angles of 1N2S are presented in Table S1† and the figure caption, respectively. 1N2S exhibits a slightly distorted T-shaped structure ligated by the two nitrogen atoms N(1) and N(2), and one sulphur atom S(1) of the supporting ligand. The sum of the three angles around the copper center [∠S(1)–Cu(1)–N(1), ∠S(1)–Cu(1)–N(2), ∠N(1)–Cu(I)–N(2)] is equal to 360°, indicating that the copper ion and the three donor atoms exist in the same plane. The copper(I) complex 1N3 supported by LN3 exhibited a similar T-shaped structure, demonstrating that the molecular framework of the ligands can highly stabilise such a three coordinate T-shaped structure of copper(I).26,27 Although the precise structure of the reduced enzyme is not known, the EXAFS data have suggested that the reduced CuM is 3-coordinate, or 4-coordinate with a weakly bound water molecule, where the CuM–SMet distance is 2.24 Å.4 In this context, 1N2S represents a good structural model of reduced CuM, even though the Cu–S distance in 1N2S (2.203 Å) is slightly shorter than that in the enzyme (2.24 Å).
 | ||
| Fig. 2 ORTEP drawing of 1N2S showing 50% probability thermal ellipsoids. Counter anion (PF6−) and hydrogen atoms are omitted for clarity. Cu(1)–S(1), 2.2028(4) Å; Cu(1)–N(1), 1.9295(13) Å; Cu(1)–N(2), 2.1662(13) Å; S(1)–Cu(1)–N(1), 163.02(4)°; S(1)–Cu(1)–N(2), 94.37(4)°; N(1)–Cu(1)–N(2), 102.60(6)°. | ||
1N2S showed a relatively intense absorption band at 375 nm (ε = 1058 M−1 cm−1) ascribable to a CuI to S charge transfer (MLCT) (Fig. 3), and exhibited a reversible CuI/CuII redox couple at 0.23 V vs. Ag/AgNO3 in acetone (Fig. 3, inset) that is higher than that of 1N3 (0.11 V for R = –CH2CH2Ph) under the same conditions.27
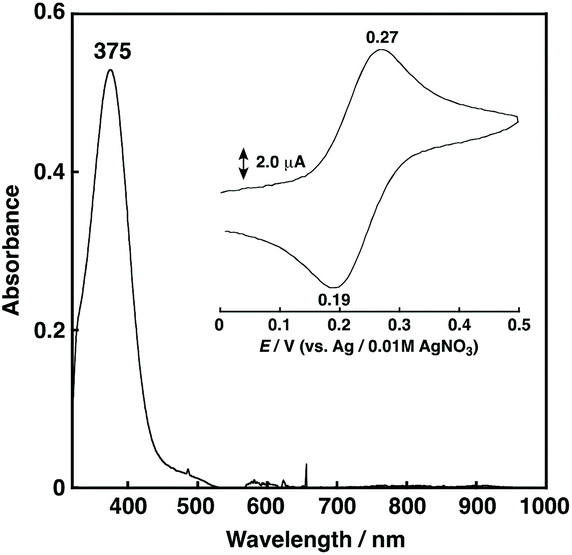 | ||
| Fig. 3 UV-vis spectrum of 1N2S (0.5 mM) in acetone at 25 °C. Inset: cyclic voltammogram of 1N2S (1.0 mM) in acetone containing 0.1 M TBAPF6; working electrode GC, counter electrode Pt, reference electrode Ag/0.01 M AgNO3, scan rate 50 mV s−1. | ||
Copper(II) complexes 2N2S·X (X = coordinating counter anion; Cl, N3, NO2, OTf) were also prepared as the model compounds of resting enzymes in the copper(II) oxidation state. The crystal structures of 2N2S·Cl, 2N2S·N3, 2N2S·NO2 and 2N2S·OTf have been determined as shown in Fig. S1, S2,† and Fig. 4. The crystallographic data and the selected bond lengths and angles are summarised in Tables S2 and S3,† respectively.
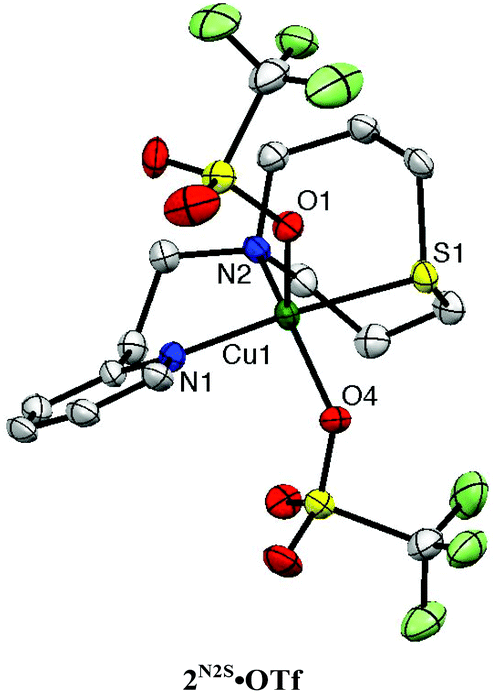 | ||
| Fig. 4 ORTEP drawings of 2N2S·OTf showing 50% probability thermal ellipsoids. Hydrogen atoms are omitted for clarity. Cu(1)–S(1), 2.2860(15) Å; Cu(1)–O(1), 2.247(4) Å; Cu(1)–O(4), 2.118(4); Cu(1)–N(1), 2.006(5) Å; Cu(1)–N(2), 2.040(6) Å; S(1)–Cu(1)–O(1), 84.84(11)°; S(1)–Cu(1)–O(4), 84.78(12)°; S(1)–Cu(1)–N(1), 171.15(16)°; S(1)–Cu(1)–N(2), 90.42(14)°; O(1)–Cu(1)–O(4), 95.93(15)°; O(1)–Cu(1)–N(1), 92.12(16)°; O(1)–Cu(1)–N(2), 109.86(17)°; O(4)–Cu(1)–N(1), 87.27(18)°; O(4)–Cu(1)–N(2), 153.25(16)°; N(1)–Cu(1)–N(2), 98.4(2)°. | ||
All the copper(II) complexes exhibit five-coordinate structures with τ = 0.01–0.55,28 among which 2N2S·Cl and 2N2S·N3 form coordination polymer chain structures, where the coordinating counter anion (Cl− and N3−) acts as the bridging ligand (Fig. S1†). On the other hand, 2N2S·OTf and 2N2S·NO2 exhibit a monomeric structure as shown in Fig. 4 and Fig. S2.† Thus, the structure and chemical properties were examined using 2N2S·OTf in more detail.
The copper(II) complex 2N2S·OTf exhibits a distorted square pyramidal structure coordinated by N(1), N(2), S(1) and O(4) in the equatorial plane and O(1) occupying the axial position (τ = 0.30), which is in accord with the observed ligand field absorption band at 660 nm (ε = 222 M−1 cm−1, Fig. 5) and a typical EPR signals of CuII with a tetragonal geometry (Fig. 6, the EPR parameters are provided in the figure caption). The structural feature is quite different from that of the copper(II) complexes supported by LN3, which enforces the copper(II) center having a four-coordinate distorted tetrahedral geometry.26,27 Thus, the replacement of the donor atom from nitrogen to sulphur resulted in a significant change in the coordination geometry of the metal center from the four-coordinate distorted tetrahedral to the five-coordinate square pyramid or trigonal bipyramidal. In this case, Cu–S distance is 2.286 Å, which is longer than that in the copper(I) complex (2.203 Å). Such an elongation of the CuII–S bond demonstrates the strong preference of the CuI–S bond as compared to the CuII–S bond. However, the CuII–S distance in the model complex (2.286 Å) is much shorter than that in the resting state (copper(II) oxidation state) of the enzyme, that is, 2.68 Å, although the coordination geometries are similar to each other.3
 | ||
| Fig. 5 The UV-vis spectrum of 2N2S·OTf (0.25 mM) in acetone, at −85 °C. λmax = 370 nm (ε = 5520 M−1 cm−1). Inset: an expanded UV-vis spectrum of 2N2S·OTf (0.60 mM). λmax = 660 nm (ε = 222 M−1 cm−1). | ||
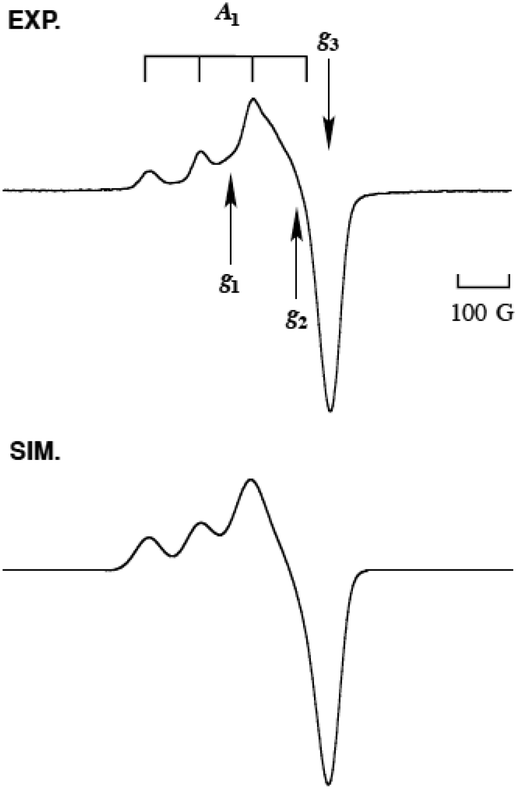 | ||
| Fig. 6 The EPR spectrum (EXP, top) of 2N2S·OTf (0.5 mM) in acetone at −196 °C, and its computer simulation spectrum (SIM, bottom) with the parameters of g1 = 2.275, g2 = 2.100, g3 = 2.015, A1 = 150 G, A2 = 59 G, and A3 = 16 G. | ||
Reactivity
First, the reaction of 1N2S and O2 was examined in acetone at low temperature (−85 °C). However, no spectral change was observed. Even at room temperature or in other solvents such as THF and CH2Cl2, 1N2S showed no reactivity toward O2. This result is in sharp contrast to the high O2-reactivity of 1N3, which gives a mononuclear copper(II)-superoxide complex.26,27 Such a big difference in the O2-reactivity between the two ligand systems could be attributed to the higher oxidation potential of 1N2S (0.23 V) as compared to that of 1N3 (0.11 V). As mentioned above, the Cu–S distance in the enzyme is much longer, especially in the copper(II) oxidation state, which may cause the less copper(I)-stabilizing effect of the sulphur donor atom in the enzymatic system. Therefore, CuM may be able to activate O2 for the substrate oxygenation reaction even in the presence of the Met ligand.On the other hand, 1N2S smoothly reacted with cumene hydroperoxide (CmOOH) in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio to give CmOH (cumyl alcohol) as the major product (98%) together with a trace amount of acetophenone (PhC(O)Me), where CmOOH was completely consumed (Fig. S3†). In the case of 1N3, on the other hand, only 44% of CmOH was obtained together with a small amount of PhC(O)Me (6%), and 50% of CmOOH was recovered (Fig. S4†). Thus, the stoichiometry in the 1N3 case was 2:1.
:1 ratio to give CmOH (cumyl alcohol) as the major product (98%) together with a trace amount of acetophenone (PhC(O)Me), where CmOOH was completely consumed (Fig. S3†). In the case of 1N3, on the other hand, only 44% of CmOH was obtained together with a small amount of PhC(O)Me (6%), and 50% of CmOOH was recovered (Fig. S4†). Thus, the stoichiometry in the 1N3 case was 2:1.
We have recently examined the reactivity of a series of copper complexes using CmOOH as a proof for searching the O–O breaking pattern in the copper-peroxide adducts.‡29,30 It has been demonstrated that copper(I) complexes supported by nitrogen ligands react with CmOOH in a 2:1 ratio to induce the heterolytic O–O bond cleavage of the peroxide to give CmOH as the major product. In this case, each copper(I) complex formally acts as a one-electron donor to induce total two-electron reduction of CmOOH, thus providing the heterolytic cleavage product, CmOH, in a ∼50% yield based on the copper(I) starting material (stoichiometry of Cu:CmOOH is 2:1).30 Thus, 1N3 (N3-donor ligand) giving CmOH in a ∼50% yield is involved in this category.
In this respect, the reactivity of 1N2S (N2S-donor ligand) is different, showing the 1:1 stoichiometry to give CmOH in a ∼100% yield based on the copper(I) starting material. Such a unique reactivity of 1N2S can be attributed to the presence of the sulphur atom. In order to get insight into the effect of the sulphur atom, the reaction was examined by UV-vis, resonance Raman and EPR spectra as follows.
Fig. 7 shows the UV-vis spectral change for the reaction of 1N2S and an equimolar amount of CmOOH in acetone. After about 1000 s, an intermediate A exhibiting characteristic absorption bands at 336, 465 and 625 nm was developed (Fig. 7, solid line). This intermediate was gradually converted to another intermediate B having absorption bands at 336, 415, and 635 nm (Fig. 7, dashed line). When the solution was warmed to room temperature, the absorption bands in the visible region disappeared, suggesting formation of a copper(I) product C (Fig. 7, dotted line). The EPR spectrum of the final reaction solution was essentially EPR silent, being consistent with the formation of a copper(I) complex (Fig. S8†).
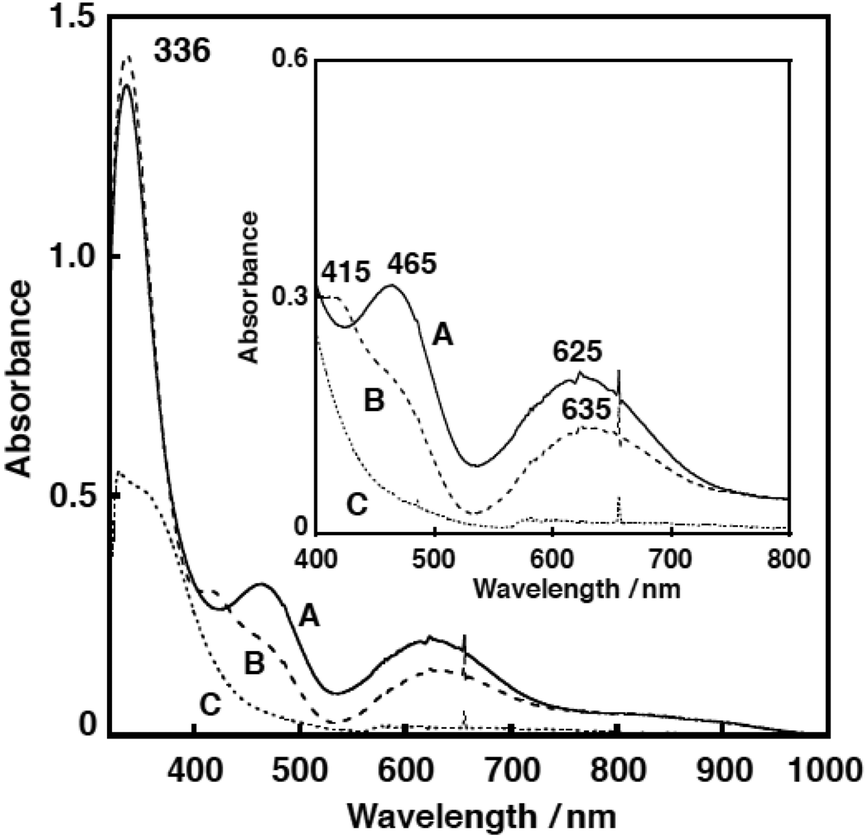 | ||
| Fig. 7 The UV-vis spectra of intermediate A (solid line; 1000 s after the reaction of 1N2S (0.5 mM) with an equimolar amount of CmOOH in acetone at −85 °C), B (dashed line; 1 h after the reaction), and C (dotted line; after warming to 30 °C). Inset: expanded spectra at the 400–800 nm region. | ||
Then, the reaction was examined by resonance Raman spectrum at the low temperature. As shown in Fig. S5,† isotope-sensitive Raman bands were observed at 887, 827 and 610 cm−1, which shifted to 794 and 603 cm−1, when 18O-labeled CmOOH was employed. The Raman bands at 887 and 827 cm−1 could be attributed to the O–O bond stretching vibration of a cumylperoxide adduct intermediate A and the band at 610 cm−1 may be due to the Cu–O stretching vibration.§31
On the basis of these results, a possible reaction mechanism is proposed in Scheme 2. Association of the copper(I) complex 1N2S and CmOOH may induce proton-coupled electron-transfer (PCET) to generate intermediate A, where a proton of CmOOH is transferred to the sulphur atom and an electron is transferred from copper(I) to the sulphur atom. In this case, an empty d orbital of the sulphur atom can accommodate the electron provided from copper(I). Then, nucleophilic attack of the sulphur atom to the proximal oxygen leads to S–O bond formation with concomitant proton migration to the distal oxygen atom. Eventually, O–O bond heterolytic cleavage takes place to give intermediate B and CmOH. Then, electronic rearrangement of B occurs upon warming to give a copper(I) complex C with an oxygenated ligand.
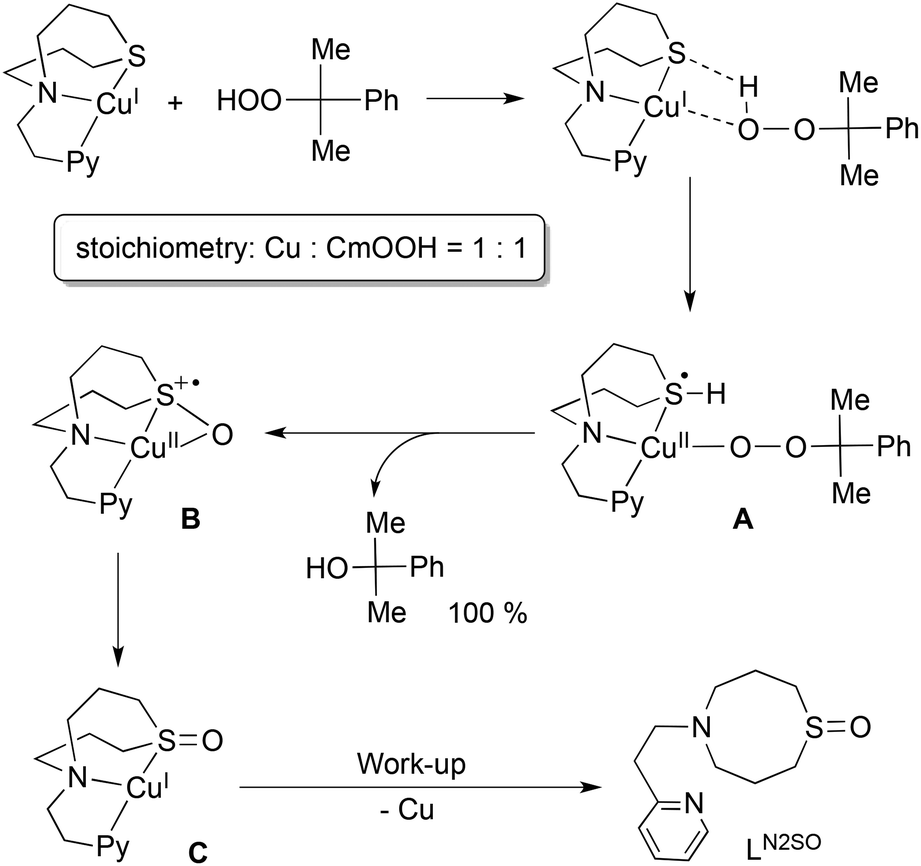 | ||
| Scheme 2 | ||
The sulphoxide ligand LN2SO was obtained nearly quantitatively after the work-up treatment of the reaction mixture with NH4OH aq (demetalation). The formation of LN2SO was confirmed by 1H-NMR, IR and ESI-mass spectra (Fig. S5 and S6,† also see the Experimental section), and the isotope labeling experiment using Cm18O2H confirmed that the oxygen atom incorporated into the ligand originated from the CmOOH used (Fig. S6†). Oxygenated product LN2SO was not obtained at all in the reaction of ligand itself and CmOOH under the same experimental conditions. Although mechanistic details of the enzymatic reaction remain obscure, such a role of sulphur ligand could be involved in enhancing the O–O bond cleavage of the peroxide intermediate in the copper monooxygenases.
Conclusions
In summary, copper(I) and copper(II) complexes of the N2S ligand (LN2S) have been synthesised and characterised in order to get insights into the effects of the sulphur atom of the Met ligand in the enzyme active site. Structural examinations have suggested that the sulphur coordination in the present model system is stronger than that in the enzymatic system, causing higher stability of the copper(I) oxidation state toward O2. Nonetheless, a reactivity study using cumen hydroperoxide has clearly indicated that the sulphur atom helps in the O–O bond heterolysis of the peroxide intermediate. Such an effect of sulphur might be important to facilitate the enzymatic reactions. The present result also suggests that the sulphur atom of the Met ligand in the enzymatic system can be oxygenated to sulphoxide during the catalytic cycles. In other words, posttranslational modification of the Met sulphur could occur in the enzyme active site.Experimental section
Materials and physical measurements
The reagents and the solvents used in this study, except the ligands, cumene hydroperoxide and the copper complexes, were commercial products of the highest available purity and were further purified by standard methods, if necessary.32 Cumene hydroperoxide (CmOOH) used in this study was prepared by a reported procedure,33 and purified by silica gel column chromatography (eluent: AcOEt–Hexane). 18O-labeled (Cm18O18OH) was synthesized using 18O2 instead of 16O2 by the same procedure.UV-visible spectra were taken in acetone or in acetonitrile on a Hewlett Packard 8453 photodiode array spectrophotometer equipped with a Unisoku thermostated cell holder designed for low temperature measurements (USP-203). 1H-NMR spectra were recorded on a JEOL ECS 400 or a Varian UNITY INOVA 600 MHz. ESR spectra were recorded on a BRUKER E-500 spectrometer at −196 °C equipped with a variable temperature cell holder or a JEOL JES-FA100. A MnII-maker was used as the reference, and experimental error in the EPR parameter is ±0.001. Mass spectra were recorded with a JEOL JMS-700T Tandem MS station or a JEOL JMS-700. ESI-MS (electrospray ionization mass spectra) measurements were performed on a Mariner ESI-TOF instrument. Cyclic voltammetric measurements were performed with a Hokuto Denko HZ-3000 or HZ-7000 in deaerated solvent containing 0.10 M n-Bu4PF6 as a supporting electrolyte. A GC working (BAS) electrode was polished with BAS polishing alumina suspension and rinsed with acetone before use. The counter electrode was a platinum wire. The measured potentials were recorded with respect to an Ag/AgNO3 (0.01 M) reference electrode.
Resonance Raman scattering was excited 441.6 nm from a He–Cd laser (KIMMON KOHA, K5651R). Resonance Raman scattering was dispersed by a single polychromator (Ritsu Oyo Kogaku, MC-100) and was detected by a liquid nitrogen cooled CCD detector (HORIBA JOBIN YVON, Symphony 1024 × 128 Cryogenic Front Illuminated CCD Detector). The resonance Raman measurements were carried out using a rotated cylindrical cell thermostated at −80 °C or a rotating NMR tube (outer diameter = 5 mm) thermostated at −40 °C by flashing cold nitrogen gas. A 135° back-scattering geometry was used.
Synthesis
:5) in 70% (6.5 g). 1H-NMR (CDCl3) δ 1.67 (4 H, pentet, J = 6.0 Hz, CH2–CH2–CH2), 2.65 (4 H, t, J = 6.0 Hz, N–CH2–CH2–CH2–OH), 2.82 (2 H, t, J = 7.2 Hz, Py–CH2–CH2–), 3.00 (2 H, t, J = 7.2 Hz, Py–CH2–CH2–), 3.61 (4 H, t, J = 5.6 Hz, –CH2–OH), 7.15–7.18 (1 H, m, PyH5), 7.20 (1 H, d, J = 7.6 Hz, PyH3), 7.66 (1 H, td, J = 1.6 Hz and J = 7.2 Hz, PyH4), 8.50 (1 H, d, J = 2.4 Hz, PyH6); HRMS (EI+) m/z = 238.1680, calcd for C13H22N2O2 = 238.1681.
[CuI(CH3CN)](PF6) (24 mg, 64 μmol) was added to a THF solution of LN2S (15 mg, 64 μmol). After stirring the solution for 30 min at room temperature, the insoluble material was removed by filtration. Addition of ether (50 mL) to the filtrate gave yellow-green solids that were isolated by decantation, washed with ether three times, and dried (45% yield, 13 mg). Single crystals were obtained for the X-ray analysis by vapor diffusion of ether into a THF solution of the complex. FT-IR (KBr) 841 cm−1 (PF6−); ESI-MS (pos.) m/z = 299.3, calcd for [CuI(LN2S)]+ 299.1; Anal. Calcd for [CuI(LN2S)]PF6 (C13H20CuF6N2PS): C, 35.10; H, 4.53; N, 6.30. Found: C, 35.43; H, 4.64; N, 6.30.
Other copper(II) complexes 2N2S·Cl, 2N2S·N3, and 2N2S·NO2 were prepared in a similar manner, as described in the ESI.†
Product analysis
:60) mixed solvent as the mobile phase at a constant flow rate of 0.5 mL min−1. The yields of products were determined by comparing the integrated peak areas of the products with that of the internal standard (anisole) using calibration lines.†
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O).†
O).†
X-ray crystal structure determination†
Each single crystal obtained was mounted on a CryoLoop (Hampton Research Co.) with a mineral oil, and all data of X-ray diffraction were collected at −170 °C on a Rigaku R-AXIS-RAPID diffractometer using filtered Mo-Kα radiation. The structures were solved by direct methods SIR 92 or SIR 2008 and expanded using Fourier techniques.34,35 The non-hydrogen atoms were refined anisotropically by full-matrix least-squares on F2. The hydrogen atoms were attached at idealized positions on carbon atoms and were not refined. All structures in the final stages of refinement showed no movement in the atom positions. The calculations were performed using Single-Crystal Structure Analysis Software, version 4.0.Acknowledgements
This work was supported by a Grant-in-Aid for Scientific Research on Innovative Areas “Molecular Activation Directed toward Straightforward Synthesis” (no. 22105007) and “Stimuli-responsive Chemical Species” (no. 24109015) and a Grant-in-Aid for Exploratory Research (no. 25620044) from the Ministry of Education, Culture, Sports, Science and Technology, Japan.Notes and references
- J. P. Klinman, Chem. Rev., 1996, 96, 2541–2562 CrossRef CAS PubMed.
- S. T. Prigge, R. E. Mains, B. A. Eipper and L. M. Amzel, Cell. Mol. Life Sci., 2000, 57, 1236–1259 CrossRef CAS.
- S. T. Prigge, A. S. Kolhekar, B. A. Eipper, R. E. Mains and L. M. Amzel, Science, 1997, 278, 1300–1305 CrossRef CAS.
- N. J. Blackburn, F. C. Rhames, M. Ralle and S. Jaron, J. Biol. Inorg. Chem., 2000, 5, 341–353 CrossRef CAS.
- N. J. Blackburn, S. S. Hasnain, T. M. Pettingill and R. W. Strange, J. Biol. Chem., 1991, 266, 23120–23127 CAS.
- C. Hess, J. Klinman and N. Blackburn, J. Biol. Inorg. Chem., 2010, 15, 1195–1207 CrossRef CAS PubMed.
- E. I. Solomon, M. J. Baldwin and M. D. Lowery, Chem. Rev., 1992, 92, 521–542 CrossRef CAS.
- E. I. Solomon, F. Tuczek, D. E. Root and C. A. Brown, Chem. Rev., 1994, 94, 827–856 CrossRef CAS.
- L. Casella, M. Gullotti, M. Bartosek, G. Pallanza and E. Laurenti, J. Chem. Soc., Chem. Commun., 1991, 1235–1237 RSC.
- G. Alzuet, L. Casella, M. Laura Villa, O. Carugo and M. Gullotti, J. Chem. Soc., Dalton Trans., 1997, 4789–4794 RSC.
- F. Champloy, N. Benali-Cherif, P. Bruno, I. Blain, M. Pierrot, M. Reglier and A. Michalowicz, Inorg. Chem., 1998, 37, 3910–3918 CrossRef CAS PubMed.
- T. Ohta, T. Tachiyama, K. Yoshizawa, T. Yamabe, T. Uchida and T. Kitagawa, Inorg. Chem., 2000, 39, 4358–4369 CrossRef CAS.
- M. Kodera, T. Kita, I. Miura, N. Nakayama, T. Kawata, K. Kano and S. Hirota, J. Am. Chem. Soc., 2001, 123, 7715–7716 CrossRef CAS.
- N. W. Aboelella, B. F. Gherman, L. M. R. Hill, J. T. York, N. Holm, V. G. Young, C. J. Cramer and W. B. Tolman, J. Am. Chem. Soc., 2006, 128, 3445–3458 CrossRef CAS PubMed.
- L. Zhou, D. Powell and K. M. Nicholas, Inorg. Chem., 2006, 45, 3840–3842 CrossRef CAS PubMed.
- L. Zhou, D. Powell and K. M. Nicholas, Inorg. Chem., 2007, 46, 7789–7799 CrossRef CAS PubMed.
- Y. Lee, D.-H. Lee, A. A. Narducci Sarjeant, L. N. Zakharov, A. L. Rheingold and K. D. Karlin, Inorg. Chem., 2006, 45, 10098–10107 CrossRef CAS PubMed.
- L. Q. Hatcher, D. H. Lee, M. A. Vance, A. E. Milligan, R. Sarangi, K. O. Hodgson, B. Hedman, E. I. Solomon and K. D. Karlin, Inorg. Chem., 2006, 45, 10055–10057 CrossRef CAS PubMed.
- D.-H. Lee, L. Q. Hatcher, M. A. Vance, R. Sarangi, A. E. Milligan, A. A. Narducci Sarjeant, C. D. Incarvito, A. L. Rheingold, K. O. Hodgson, B. Hedman, E. I. Solomon and K. D. Karlin, Inorg. Chem., 2007, 46, 6056–6068 CrossRef CAS PubMed.
- G. Y. Park, Y. Lee, D.-H. Lee, J. S. Woertink, A. A. N. Sarjeant, E. I. Solomon and K. D. Karlin, Chem. Commun., 2010, 46, 91–93 RSC.
- S. Itoh, Curr. Opin. Chem. Biol., 2006, 10, 115–122 CrossRef CAS PubMed.
- L. M. Mirica, X. Ottenwaelder and T. D. P. Stack, Chem. Rev., 2004, 104, 1013–1045 CrossRef CAS PubMed.
- E. A. Lewis and W. B. Tolman, Chem. Rev., 2004, 104, 1047–1076 CrossRef CAS PubMed.
- S. Itoh, in Copper-Oxygen Chemistry, ed. K. D. Karlin and S. Itoh, John Wiley & Sons, Hoboken, 2011, vol. 4, pp. 225–282 Search PubMed.
- R. L. Peterson, S. Kim and K. D. Karlin, in Comprehensive Inorganic Chemistry II, ed. J. Reedijk and P. K. Elsevier, Oxford, 2013, vol. 3, pp. 149–177 Search PubMed.
- A. Kunishita, M. Kubo, H. Sugimoto, T. Ogura, K. Sato, T. Takui and S. Itoh, J. Am. Chem. Soc., 2009, 131, 2788–2789 CrossRef CAS PubMed.
- A. Kunishita, M. Z. Ertem, Y. Okubo, T. Tano, H. Sugimoto, K. Ohkubo, N. Fujieda, S. Fukuzumi, C. J. Cramer and S. Itoh, Inorg. Chem., 2012, 51, 9465–9480 CrossRef CAS PubMed.
- A. W. Addison, T. N. Rao, J. Reedijk, J. van Rijn and G. C. Verschoor, J. Chem. Soc., Dalton Trans., 1984, 1349–1356 RSC.
- T. Tano, M. Z. Ertem, S. Yamaguchi, A. Kunishita, H. Sugimoto, N. Fujieda, T. Ogura, C. J. Cramer and S. Itoh, Dalton Trans., 2011, 40, 10326–10336 RSC.
- T. Tano, H. Sugimoto, N. Fujieda and S. Itoh, Eur. J. Inorg. Chem., 2012, 2012, 4099–4103 CrossRef CAS.
- A. Kunishita, H. Ishimaru, S. Nakashima, T. Ogura and S. Itoh, J. Am. Chem. Soc., 2008, 130, 4244–4245 CrossRef CAS PubMed.
- D. D. Perrin, W. L. F. Armarego and D. R. Perrin, Purification of Laboratory Chemicals 4th Edition, Pergamon Press, Elmsford, NY, 4th edn, 1996 Search PubMed.
- M. G. Finn and K. B. Sharpless, J. Am. Chem. Soc., 1991, 113, 113–126 CrossRef CAS.
- A. Altomare, G. Cascarano, C. Giacovazzo, A. Guagliardi, M. Burla, G. Polidori and M. Camalli, J. Appl. Crystallogr., 1994, 27, 435–436 Search PubMed.
- M. C. Burla, R. Caliandro, M. Camalli, B. Carrozzini, G. L. Cascarano, L. D. Caro, C. Giacovazzo, G. Polidori, D. Siliqi and R. Spagna, J. Appl. Crystallogr., 2007, 40, 609–613 CrossRef CAS.
- S. Adachi, S. Nagano, K. Ishimori, Y. Watanabe, I. Morishima, T. Egawa, T. Kitagawa and R. Makino, Biochemistry, 1993, 32, 241–252 CrossRef CAS.
- E. Baciocchi, M. Bietti, M. Salamone and S. Steenken, J. Org. Chem., 2002, 67, 2266–2270 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures and characterization details. CCDC 959052–959056. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3dt52952e |
| ‡ It has been reported that the products obtained from a cumylperoxo complex largely depend on the O–O bond cleavage pattern. If homolytic O–O bond cleavage occurs, acetophenone (PhCOMe) is produced from cumyloxyl radical via β-scission releasing methyl radical (CH3˙). In contrast, heterolytic O–O bond cleavage gives cumyl alcohol (CmOH) after oxyanion protonation.36,37 |
| § The resonance Raman bands due to the C–C–C and C–C–O deformation modes of the alkylperoxide moiety were also observed at 540, 490 and 436 cm−1 in the 16O-derivative (see Fig. S5†). However, their counterparts in the 18O-derivative became obscure due to low quality of the spectrum. The two peaks at 887 and 827 cm−1 for the O–O vibration of the 16O-derivative may be Fermi doublet, which collapsed into one peak at 794 cm−1 in the 18O-derivative. More detailed examination is required for the complete assignment of the resonance Raman spectrum. |
| This journal is © The Royal Society of Chemistry 2014 |