 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Phage-based detection of bacterial pathogens
R. G.
van der Merwe
*,
P. D.
van Helden
,
R. M.
Warren
,
S. L.
Sampson
and
N. C.
Gey van Pittius
DST/NRF Centre of Excellence for Biomedical Tuberculosis Research/MRC Centre for Molecular and Cellular Biology, Division of Molecular Biology and Human Genetics, Faculty of Medicine and Health Sciences, Stellenbosch University, Cape Town, South Africa. E-mail: rubengvdm@hotmail.com
First published on 13th March 2014
Abstract
Bacterial pathogens cause significant morbidity and mortality annually to both humans and animals. With the rampant spread of drug resistance and the diminishing effectiveness of current antibiotics, there is a pressing need for effective diagnostics for detection of bacterial pathogens and their drug resistances. Bacteriophages offer several unique opportunities for bacterial detection. This review highlights the means by which bacteriophages have been utilized to achieve and facilitate specific bacterial detection.
 Ruben van der Merwe | Ruben van der Merwe is currently a Postdoctoral Research Fellow in Professor Paul van Helden’s group. He obtained his Bsc and Honns Bsc at Stellenbosch University, South Africa. He obtained his PhD degree in medical biochemistry in 2012 which focused on the development of reporter mycobacteriophages for the rapid diagnosis of Mycobacterium tuberculosis. His current post-doctoral research is focused on investigating the evolution of drug resistance of clinical isolates of Mycobacterium tuberculosis by means of whole genome sequencing. |
 Paul van Helden | Paul van Helden obtained his doctorate in Biochemistry in 1978 and has been at the Faculty of Health Sciences at Stellenbosch University, Cape Town, since January 1979. He is professor and head of the division of Molecular Biology and Human Genetics and also Director of the MRC Centre for Molecular and Cellular Biology, as well as Director of the DST/NRF Centre of Excellence for Biomedical TB Research. His interests range from basic and clinical research in human and animal TB, and include research at both ends of the spectrum. |
 Rob Warren | Professor Rob Warren obtained a PhD degree in Biochemistry from the University of Cape Town in 1995 and subsequently joined the Department of Medical Biochemistry, Stellenbosch University. Under his guidance the study of the molecular epidemiology of Mycobacterium tuberculosis in a high incidence setting (Cape Town, South Africa) has been brought to the forefront of international tuberculosis research. Rob Warren's work has provided new understanding, which has allowed long standing dogmas to be challenged. He has published more than 180 papers in international peer reviewed journals in the fields of molecular epidemiology, drug resistance and bacterial evolution. |
 Samantha Sampson | Samantha Sampson is an Associate Professor in the Department of Biomedical Sciences, Faculty of Medicine and Health Sciences, Stellenbosch University. She received a BSc in Chemistry and Biochemistry, BSc Hons (cum laude), MSc (cum laude) and finally a PhD in Medical Biochemistry in 2002 from Stellenbosch University. She worked as a post-doctoral Research Fellow at the Harvard University School of Public Health, Boston, USA, and at Imperial College London, UK. She returned to South Africa in 2013, starting up her own research group exploiting high-throughput "omics" approaches to probe TB host-pathogen interactions. |
 Nico C. Gey van Pittius | Prof. Nico C. Gey van Pittius is the Deputy Dean: Research of the Faculty of Medicine and Health Sciences, and an Associate Professor in Molecular Biology and Human Genetics in the Department of Biomedical Sciences at Stellenbosch University. He received his BSc, Honns BSc and MSc degrees from the University of Potchefstroom and his PhD degree from Stellenbosch University, South Africa. As a molecular biologist, he has worked on tuberculosis for the past 16 years, focusing on the mechanisms of evolution of the mycobacteria and the development of mycobacterial pathogenicity and drug resistance. |
Introduction
Pathogenic bacteria pose a global health threat and cause extensive morbidity and mortality each year. The tuberculosis (TB) epidemic, caused by Mycobacterium tuberculosis was responsible for 1.4 million deaths world-wide in 2011 and 8.7 million new cases in the same year.1 Contaminated food and water is a major source for infection by bacterial pathogens, an estimated 1.2 million cases of Salmonella infection occurs annually in the USA alone, with far more cases likely in third world countries.2,3 Emergence and spread of drug resistance among bacterial pathogens is a cause for concern and has been observed among various pathogens such as the gram negatives Citrobacter freundii, Klebsiella pneumoniae, Acinetobacter, Proteus mirabilis, Pseudomonas aeruginosa and Escherichia coli as well as the gram positives Staphylococcus aureus and Enterococci.2,4–6 The rise in drug resistance and dwindling drug treatment options, best exemplified by the current Mycobacterium tuberculosis epidemic, emphasize the need for rapid and effective diagnostics to contain the spread of bacterial pathogens. Culture remains one of the most common methods for bacterial detection and drug resistance profiling. However, this leads to a diagnostic time delay for bacteria with a slow growth rate. Furthermore, certain pathogens require specialized biohazard facilities, prohibiting its wide-spread use. Culture is also limited to culturable bacteria, however viable but non-culturable (VBNC) cells could escape detection. Although molecular methods provide rapid alternatives to culture, their wide-spread use is limited by cost, infrastructure requirements and the need for skilled operators. Various bacteriophage-based assays have been investigated for specific host detection. These assays offer the advantages of rapid, sensitive and specific host detection, cheap production costs as well as detecting only viable cells. In this review, we consider how bacteriophages offer unique features that could provide simple solutions for bacterial detection.Bacteriophage detection assays
Phage typing as a diagnostic tool
As early as 1938, bacteriophages were utilized to determine bacterial taxonomy by phage typing.7,8 Phage typing exploits differential susceptibility of bacteria to various bacteriophages to enable the determination of bacterial genus and species.9 The method, shown in Fig. 1, is based on detection of plaques on bacterial lawns following bacteriophage replication and bacterial cell lysis and has been applied to several bacteria. The main restriction of using phage typing assays as a diagnostic is that they rely on the host bacterial replication rate for lawn formation, which can be time consuming for slow growing bacteria such as Mycobacteria.10,11 This limitation has since been addressed via surrogate fast growing bacteria in bacteriophage replication assays.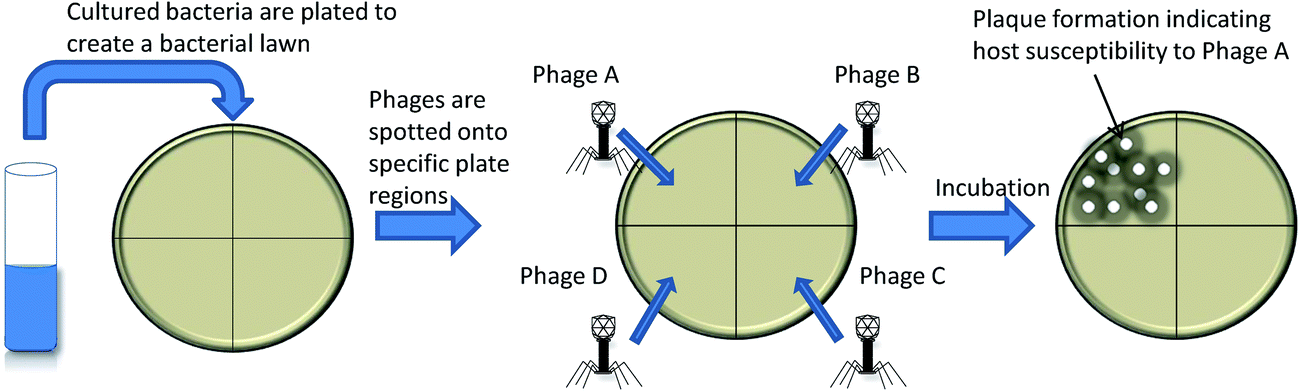 | ||
| Fig. 1 Phage typing. The bacterial sample to be typed is plated together with a series of bacteriophages to create a bacterial lawn where clearings/plaque formation is indicative of bacteriophage replication and host susceptibility to the specific bacteriophage. | ||
Bacteriophage replication assays
Use of mycobacteriophage D29, capable of infecting both the slow growing Mycobacterium tuberculosis and the fast growing Mycobacterium smegmatis, enabled development of a bacteriophage replication assay for M. tuberculosis detection.12,13 In their assay, the M. tuberculosis sample is incubated with mycobacteriophage D29, treated with an antiviral agent to kill excess mycobacteriophages and then plated onto an M. smegmatis lawn. Lytic phage replication, indicative of M. tuberculosis in the original sample, is detected by plaque formation. Fig. 2 shows an illustration of bacteriophage replication assays. A variation on this the assay substituted solid culture media with liquid culture media and made use of a multichannel series piezoelectric quartz crystal sensor to detect cellular lysis. The assay could detect as low as 100 c.f.u. ml−1 in 30 h.14 A similar bacteriophage replication assay was developed for Pseudomonas aeruginosa.15 The assay consisted of exposing the sample containing P. aeruginosa 10548, as well as control samples containing no cells, with bacteriophage NCIMB 101116 followed by addition of P. aeruginosa 10545 surrogate cells to allow bacteriophage replication and host lysis. The samples are then filtered, concentrated onto membranes and resuspended. The ratio of live to dead cells following fluorochromic staining and fluorescence measurement was then taken as a measure of bacteriophage amplification correlating to the initial bacterial concentration in each sample. The assay detected approximately 10 c.f.u. ml−1 in 4 hours.15 Bacteriophage replication assays have the advantage of detecting only viable cells, in contrast to PCR, makes them less prone to false positive case detection. | ||
| Fig. 2 Bacteriophage replication assays. | ||
Bacteriophage replication assays for detection of drug resistance
Mycobacteriophages gained popularity when they were shown to be able to discriminate between drug resistant and drug sensitive M. tuberculosis isolates for selected anti-mycobacterial drugs.12,13,16,17 This is possible since selected antibiotics block mycobacteriophage replication in susceptible strains and allow replication in drug resistant strains. Screening for ethambutol and isoniazid resistance, however, require several days' incubation of the sample with the drugs, since these drugs do not block mycobacteriophage replication directly and are active only in certain cell growth stages.12,18 A variation on this assay used a micro-well format of the replication assay described above, followed by an additional 16 h incubation step to allow detection of a colorimetric redox reaction indicative of M. smegmatis growth and thus drug resistant M. tuberculosis.19,20A meta-analysis of 31 studies investigated bacteriophage replication assays for M. tuberculosis drug susceptibility testing.21 The analysis determined that the commercial assays had an overall sensitivity and specificity of 96% and 95%, respectively, and in-house assays had a sensitivity and specificity of 99% and 98%. The contamination rates were initially 0–36% (higher in direct sputum samples), which leads to indeterminate results. The assays have since been updated to include an antibiotic which reduces indeterminate results by 68%, without reducing the sensitivity and specificity.21 The detection limit of mycobacteriophage replication assays is estimated at 100–300 bacilli ml−1 for a positive result,22 10 fold higher than for culture based techniques. With the current performance of mycobacteriophage replication assays it is clear that they cannot replace culture methods, due to inadequate sensitivity, specificity and minimal variety of drug susceptibility testing (DST). Bacteriophage replication assays do, however, offer a rapid and sensitive alternative to microscopy-based techniques for low resource settings.21,23–25
Detection of host lysis by using bacteriophages in conjunction with bioluminescence and electrochemical assays
Bacterial lysis, such as during the bacteriophage lytic cycle, releases cellular components that can be readily detected, including adenosine triphosphate (ATP), adenylate kinase (AK) and β-D-galactosidase.26,27 Bacteriophage strain-specificity permits specific detection of host lysis in mixed bacterial populations.26–28 Detection of host lysis and release of ATP have previously been performed following addition of luciferase and luciferin; this achieved a low detection limit of 103 to 104 cells.26 The method was improved by detection of AK released following bacteriophage-induced lysis of host bacterial cells. AK is an essential enzyme in most bacterial cells that catalyzes the equilibrium reaction:| ATP + AMP ↔ 2ADP |
Addition of ADP drives the reaction to produce ATP and this in turn can fuel light production via bioluminescence, creating a sensitive detection assay for AK. Blasco et al. investigated the use of bacteriophage for specific bacterial lysis in combination with the AK detection assay.26 The assay could detect fewer than 104 c.f.u. ml−1 in less than 1 h for E. coli and less than 2 h for Salmonella newport detection. Subsequent assay optimization improved sensitivity to 103 c.f.u. ml−1 for both species.29
Neufeld et al. developed an electrochemical assay to detect the release of the common cellular constituent, β-D-galactosidase.27 β-D-Galactosidase is released by E. coli K-12 MG1655 upon cell lysis by the bacteriophage lambda via gene product. The target sample is then filtered to separate the lysed cellular components from intact cells which potentially also contain β-D-galactosidase. The current resulting from the enzymatic activity of β-D-galactosidase is measured electrochemically in real-time. The assay was able to detect as little as 1 c.f.u. per 100 ml in 6–8 h. The assay has the advantage of detecting enzymatically active cells which are not necessarily culturable, in contrast to bacteriophage replication assays which can only detect viable cells. The assay requires a pre-incubation step for concentrations of bacteria lower than 2.5 × 103 c.f.u. ml−1, for example detecting 100 c.f.u. ml−1 requires 3–4 h pre-incubation. The electrochemical assay was later also adapted for detection of Bacillus cereus and M. smegmatis.28 The bacteriophage B1-7064 and substrate para-amino-phenyl-α-D-glucopyranoside was used for B. cereus whereas mycobacteriophage D29 and substrate para-amino-phenyl-β-D-glucopyranoside was used for M. smegmatis. The modified assay could detect 10 c.f.u. ml−1 in 8 h. The reporter enzyme alkaline phosphatase (AP), transferred to helper bacteriophage M13KO7, has also been exploited for the electrochemical detection of E. coli TG-1.30 AP is expressed by E. coli following infection by the helper bacteriophage and reacts with p-aminophenyl phosphate to produce p-aminophenol in an oxidation reaction which is measured using an electrochemical cell. The assay could detect 1 c.f.u. ml−1 in 2–3 h.30
Assays based on host cell lysis detection have the inherent risk of background signal from non-target bacteria. To overcome this, assays that monitor the release of bacteriophage components were investigated. One such method, for Yersinia pestis detection, involved real-time PCR-based detection of bacteriophage DNA.31 The assay consisted of a parallel infection assay using two bacteriophages to increase specificity.31 The samples, containing as little as a single cell of Yersinia pestis, were infected with bacteriophage φA1122 and L-431C respectively.31 Following incubation, bacteriophage specific DNA amplification was monitored using real-time PCR which could detect bacteriophage amplification in 4 h.31
Antibody-based host concentration in conjunction with bacteriophage assays
Immunomagnetic separation (IMS) utilizes antibody-coated magnetic beads as a means to capture, concentrate and purify bacteria from samples.32 Detection assays combining IMS with bacteriophage-dependent methodologies have been investigated for detection of several bacteria.32–34 Applied to the detection of Salmonella serovar Enteritidis in broth, using bacteriophage SJ2, the assay could detect less than 104 c.f.u. ml−1 in 4–5 h using either optical density or fluorescence measurement to detect bacteriophage amplification.34A further study applied the method to detection of E. coli O157:H7 and Salmonella enteritidis in food samples.33 The assay involved IMS of target bacterial cells, addition of bacteriophages (bacteriophage SJ2 for Salmonella enteritidis and bacteriophage LG1 for E. coli O157:H7), a wash step to remove unbound bacteriophages, followed by addition of surrogate cells and detection by measuring differences in optical density of the sample medium.33 The assay had a detection limit of less than 104 c.f.u. ml−1 in broth, could detect 3 c.f.u. of Salmonella enteritidis in 25 g or 25 ml food samples and could detect 2 c.f.u. g−1 of E. coli in food samples in 20 h.33 Applied to detection of E. coli, an assay consisting of IMS of host cells, infection by bacteriophage MS2 and detection of MS2 capsid protein from the assay medium by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOFMS) was shown to detect phage amplification from samples containing ∼5.0 × 104 cells per ml in less than 2 h.32 Antibody-based assays have the disadvantages of cross-reactivity of polyclonal antibodies and high production costs for monoclonal antibodies.35 Bacteriophages bind to their bacterial hosts with similar high specificity, however they have the advantages of reliable specific binding, significantly cheaper large scale production and their relative insensitivity to temperature and pH compared to antibodies.36
Bacteriophage biosorbents
Immuno-PCR relies on covering a surface with host/antigen specific antibodies, removal of unbound antibodies, addition of the sample to be probed, wash steps, followed by PCR based detection of target DNA.37 Analogous assays which utilize bacteriophages as biosorbent have been investigated38 as shown in Fig. 3. An assay that exploited sapphire phage (Amersham International) for specific capture of Salmonella followed by fluorescence-based microscopy detection was investigated.38 The authors reported poor performance due to inefficient phage immobilization, stating that their method allowed both head and tail to immobilize onto the solid surface due to passive adsorption.38 In lieu of this, oriented immobilization of a recombinant bacteriophage T4 was investigated for the specific capture and detection of E. coli.39 The study investigated biotin carboxyl carrier protein gene fusions to the T4 small outer capsid (SOC) protein gene, resulting in the ligands' localization on the bacteriophage head.39 The recombinant bacteriophages could be immobilized on streptavidin-coated magnetic beads and captured 72–99% of target bacteria from a 10–105 c.f.u. ml−1 sample in contrast to 10–30% for the non-labeled control bacteriophages.39 By means of real time PCR monitoring of bacteriophage replication, as low as 800 cells could be detected within 2 h.39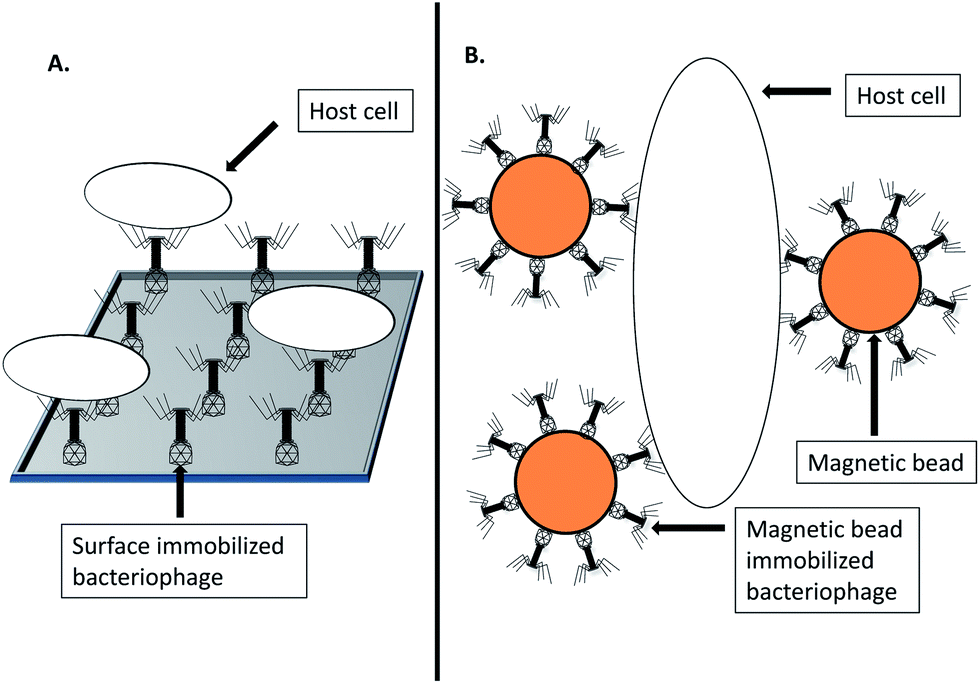 | ||
| Fig. 3 Bacteriophage biosorbents. (A) Surface immobilized bacteriophage biosorbent for specific host capture. (B) Magnetic bead immobilized bacteriophage biosorbent for specific host capture. | ||
Investigation of bacteriophage-encoded bacterial binding proteins has resulted in the discovery of several proteins responsible for specific host recognition. The cell wall-binding domains from the endolysins encoded by bacteriophages A118 and A500 were investigated for specific immobilization and separation of Listeria monocytogenes as an alternative to IMS.40 Recombinant cell wall-binding domains from the bacteriophages (named CBD118 and CBD500) were expressed in E. coli as a fusion protein to a histidine-tagged green fluorescence protein (GFP) reporter protein and used to coat magnetic-beads, followed by binding and separation of host Listeria monocytogenes cells from samples.40 The proteins showed specific host recognition without cross-reactivity as is frequently the case with antibodies.40 By means of fluorescence microscopy, the assay could detect as low as 100 c.f.u. g−1 after 6 h selective enrichment, and could detect 0.1 c.f.u. ml−1 after 24 h selective enrichment.40
Investigation of another bacteriophage-encoded bacterial binding protein followed the discovery that the C-terminal region of a bacterial binding protein from γ-phage specifically binds to the cell wall of Bacillus anthracis.41 This particular protein, Phage-Lysin-Gamma (PlyG), is a lysin protein. The region of PlyG conferring the binding activity was modified to incorporate a glutathione S-transferase (GST) tag. The bacterial detection assay consisted of blotting bacterial suspensions onto a nitrocellulose membrane, blocking the membrane and exposing it to the PlyG-GST fusion proteins, followed by horseradish peroxidase (HRP)-conjugated mouse anti-GST detection. This assay could detect 103 c.f.u. ml−1 in ∼3 h with superior sensitivity and similar specificity to plaque-based detection methods using γ-phage.41 The authors speculated that the assay could be improved by labeling the recombinant PlyG protein with stable quantum dot nanocrystals.41 To this end, a recombinant biotin-tagged PlyG together with streptavidin-conjugated quantum Dot nanocrystals was investigated for use as an improved diagnostic for Bacillus anthracis.42 The assay consisted of incubating biotin-tagged PlyG with bacterial samples, followed by addition of streptavidin-conjugated quantum dot nanocrystals. Fluorescence was measured by microscopy or fluorometry using a micro-plate reader. The assay was rapid and showed high sensitivity capable of detecting single cells.42
Another type of bacteriophage encoded bacterial binding protein called tailspike protein has been investigated for use in detection of Salmonella enterica serovar Typhimurium.43 Bacteriophage P22 tailspike proteins were engineered to express a truncated, cysteine-tagged recombinant protein in E. coli; this exhibited superior binding activity to target host cells compared to the wild-type tailspike protein as well as the intact bacteriophage P22.43 The assay consisted of immobilization of the recombinant tailspike proteins onto gold surfaces by thiol-chemistry (facilitated by the cysteine tags), addition of the sample containing host cells followed by detection using surface-plasmon resonance.43 The assay could detect concentrations of host bacteria as low as 103 c.f.u. ml−1. A similar assay made use of engineered receptor binding protein Gp48 from bacteriophage NCTC 12673 for specific detection of Campylobacter jejuni.44 The assay consisted of expressing and immobilizing the recombinant protein followed by detection of host cells using a surface plasmon resonance device, which could detect host cell concentrations as low as 102 c.f.u. ml−1.44 Following whole genome sequencing of phage NCTC12673, Gp48 was re-annotated as Gp047 and used to create two detection assays capable of detecting Campylobacter jejuni and Campylobacter coli.45 The first detection modality made use of mixing recombinant Gp047 with the sample followed by observing bacterial agglutination, whereas the second modality made use of EGFP-Gp047 fusion protein binding to bacterial host cells and detection of the labeled host cells by means of fluorescence microscopy.45
Labeled phage
Detection of host-bacteriophage binding has been facilitated through the use of various labeled bacteriophages. Detection of Shiga toxin-producing E. coli was investigated using HRP-labeled bacteriophages CBA120, AR1 and bacteriophage 56.46 The detection assay of inoculating a swab, culturing for 8 h in selective media, IMS of target cells, addition of HRP-labeled bacteriophages with subsequent colorimetric or luminescence detection following addition of a substrate. An 8 h enrichment step improved the assay's detection threshold from approximately 105 c.f.u. ml−1 to 1 c.f.u. ml−1.46 A similar method combined immuno-separation of E. coli O157 with flow-cytometry-based detection of bacteriophage-host binding.47 The study made use of bacteriophage LG1 stained with the nucleic acid dye YOYO-1 (Molecular probes, Inc., Eugene, Oreg.). The assay was able to detect 104 c.f.u. ml−1 in 8 h.47 The assay was also adapted to detect E. coli O157 in food samples such as ground beef (2.2 c.f.u. g−1 in 7 h) and raw milk (10 to 100 c.f.u. ml−1 in 12 h).48 Another labeled phage detection assay made use of bacteriophages labeled with radioactive isotopes.49 The assay consisted of propagating bacteriophage 53 in its host S. aureus which is cultured in media containing N15, producing N15 labeled progeny bacteriophages.49 The detection assay consisted of infecting S. aureus-containing samples with the N15 labeled bacteriophages, followed by detection of N14 labeled capsid proteins by means of mass spectrometry which indicate the presence of host cells in the sample.49 The assay could detect 6.7 × 106 c.f.u. ml−1 in 2 h and 6.7 × 105 c.f.u. ml−1 in 5 h.49As a means to improve bacteriophage labeling methods, use of recombinant bacteriophages with affinity fusions to structural components have been investigated.50,51 Affinity tags such as biotin and tetracysteine allow specific binding to detectable markers. A tetracysteine-tagged bacteriophage M13KE was constructed for specific detection of E. coli ER2738.51 The detection assay consisted of infection of host cells with the recombinant bacteriophages, incubation to allow bacteriophage progeny production, addition of a biarsenical dye and fluorescence detection. The dye, which bound to the tetracysteine-tags, allowed specific detection of host cells through detection of increased fluorescence levels using flow-cytometry and fluorescence microscopy.51
A recombinant E. coli phage T7 was constructed for detection of E. coli by fusing a biotinylation peptide tag to the T7 major capsid protein (named gp10a). The assay involved incubating the recombinant bacteriophage with the bacterial sample together with streptavidin-coated quantum dots. Following infection, the recombinant bacteriophage T7 becomes biotinylated by the native E. coli biotinylation enzymes. The biotinylated bacteriophages subsequently bind to the streptavidin-coated quantum dots, which can then be visualized by fluorescence microscopy. The diagnostics assay is illustrated in Fig. 4. Visual detection of target bacteria was possible within 1 h and could detect as little as 10 c.f.u. ml−1.52 The lytic nature of bacteriophage T7 has, however, been suggested to hamper the sensitive detection of single cells.50 To circumvent this problem, the use of non-lytic or conditionally replicating bacteriophages has been explored; for example, bacteriophage lambda gt11 was engineered to contain a biotin-binding peptide fused to the bacteriophage major coat protein gpD.50 The biotin-tagged bacteriophages were harvested following temperature-induced replication. The progeny bacteriophages were then biotinylated by the E. coli host biotinylation enzymes, after which the bacteriophages could be purified. The purified and biotinylated bacteriophage lambda gt11 was added, together with streptavidin coated quantum dots, to the bacterial sample and incubated at room temperature. Since lambda gt11 harbours mutations which render it temperature sensitive, the bacteria do not undergo lysis at room temperature and intact cells could be fluorescently visualized.50 Although the fluorescent properties of quantum dot nanocrystals are potentially superior to common fluorophores,53 they have some attributes that hinder their usefulness. Quantum dot nanocrystals display non-constitutional fluorescence or “blinking”, have solubility issues in polar solvents due to their inorganic nature which is problematic in biological samples and comprise structures an order of magnitude larger than other common fluorophores.53 These issues have been addressed with a varying degree of success.53 An alternative to assays which require the addition of substrate or dyes for detection is through the use of reporter phages which natively express a reporter signal.
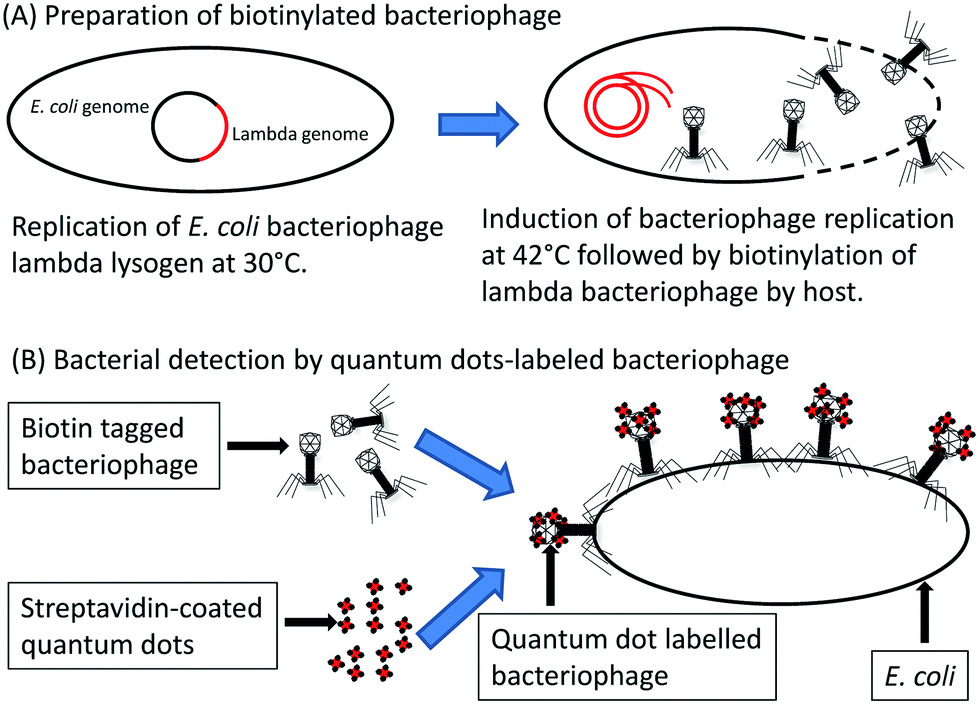 | ||
| Fig. 4 Quantum dot labelled-bacteriophage detection of E. coli host cells. E. coli lysogens, containing recombinant bacteriophage lambda, replicates at 30 °C and releases biotinylated progeny bacteriophage following induction at 42 °C. Biotinylated bacteriophage lambda and streptavidin coated quantum dots are added to the bacterial sample which allow detection of host cells by means of fluorescence detection. | ||
Ice nuclease reporter bacteriophages
Super-cooled water can remain liquid below 0 °C, but can rapidly undergo a chain reaction of freezing when ice nuclei, or a nucleating agent is introduced to it.54 Various organisms have the ability to cause nucleation of ice in supercooled water, such as Pseudomonas, Erwinia and Xanthomona.55 The first recombinant bacteriophage to be constructed with ice nucleation as reporter signal, utilized the inaW gene from Pseudomonas fluorescens.56 The inaW gene was transferred to bacteriophage P22 for specific detection of Salmonella. Expression of the inaW gene causes ice nucleation at temperatures below −9.3 °C, which allows the cells to freeze. Detection of ice formation is aided by making use of a fluorescent freezing-indicator dye.56 When tested on Salmonella, the assay could detect as low as 2 c.f.u. ml−1 in mixed bacterial populations, indicating that an enrichment step was not required.57 A commercial assay based on the ice nuclease reporter bacteriophage P22 was also developed (called the Bacterial Ice Nucleation Diagnostic, (BIND) assay), which could detect less than 3 c.f.u. ml−1 in 3 h.58 Assays based on ice nucleation have the advantage that background bacteria are unlikely to contain similar genes to inaW, which makes the assay specific.Bioluminescence
The firefly luciferase gene (Fflux) and the Vibrio fischeri luxCDABE operon have been extensively investigated and utilized to create luciferase reporter phage (LRP) assays (Table 1). The luxAB component of the luxCDABE operon synthesizes the luciferase enzyme and is under control of the regulatory genes luxI and luxR. The luxC luxD and luxE components produce an aldehyde substrate required for luciferase to produce light. luxI codes for a regulatory protein called an auto-inducer, which interacts with luxR which in turn stimulates transcription of luxCDABE and luxI. As the concentration of autoinducer rises, so too does transcription of luciferase, luxI and luxR binding which creates an auto-amplified loop generating increasing levels of bioluminescence measurable by means of a luminometer.| LRP construct | Bacterial host | Luciferese origen | Substrate | Detection limit | Detection time | Reference |
|---|---|---|---|---|---|---|
| lambda::lux | Escherichia coli | Vibrio fischeri | n/a | 10–100 c.f.u. ml−1 | 1 h | 59 |
| lambda::luxI | Escherichia coli XL1-Blue | Vibrio fischeri | n/a | 1 c.f.u. ml−1 | 10 h | 60 |
| lambda::luxI | Escherichia coli O157:H7 | Vibrio fischeri | n/a | 1 c.f.u. ml−1 | 6–6.5 h | 61 |
| A511::luxAB | Listeria monocytogenes | Vibrio harveyi | Aldehyde | 1 c.f.u. ml−1 | 24 h | 64 and 65 |
| ΦV10::luxAB | Escherichia coli O157:H7 | Vibrio harveyi | n-Decanal | 106 c.f.u. ml−1 | ∼1 h | 63 |
| Felix 01::luxAB | Salmonella | Vibrio harveyi | Aldehyde | — | — | 66 |
| A2211:luxAB | Yersinia pestis | Vibrio harveyi | n-Decanal | 100 c.f.u. ml−1 | 1 h | 67 |
| Wβ::luxAB: | Bacillus anthracis | Vibrio harveyi | n-Decanal | 103 c.f.u. ml−1 | 1 h | 67 |
| PBSPCA1::luxAB | Pseudomonas cannabina pv. Alisalensis | Vibrio harveyi | n-Decanal | 1.3 × 103 c.f.u. ml−1 | 2 h | 62 |
| L5::hsp60::Fflux | Mycobacterium tuberculosis | Firefly luciferase | Luciferin | n/a | n/a | 85 |
| D29::hsp60::Fflux | Mycobacterium bovis BCG, Mycobacterium smegmatis | Firefly luciferase | Luciferin | 13–125 c.f.u. ml−1 (tested on M. smegmatis0 | 21 h (tested on M. smegmatis) | 86 |
| phAE88::hsp60::Fflux | Mycobacterium bovis BCG | Firefly luciferase | Luciferin | 120 c.f.u. ml−1 | 12 h | 69 |
| phAE85::hsp60::Fflux | Mycobacterium avium spp paratuberculosis | Firefly luciferase | Luciferin | >1000 c.f.u. ml−1 | 24–48 h | 87 |
The very first LRP was constructed by introducing the entire lux operon from Vibrio fischeri into bacteriophage lambda Charon 30.59 In contrast to using the entire lux operon, LRPs have also been investigated which make use of the luxI auto-inducer.60,61 These assays make use of LRPs which express luxI in their hosts, which in turn induce luciferase and substrate production by phage-immune cells which contain luxCDABE and luxR (but not luxI). LRP assays have also been investigated which make use of expressing luxAB in their hosts to produce luciferase,62–67 however, this requires the manual addition of substrate for light production.
Mycobacteriophage-based LRPs which utilize Fflux, and the addition of its substrate luciferin, have been utilized for mycobacterial detection as shown in Table 1 as well as for DST to varying degrees of success. Initially a mycobacteriophage Tm4-based LRP was constructed to express Fflux under control of the Mycobacterium bovis BCG hsp60 promoter. The assay consisted of 7–8 days culture, 48 h of pre-incubation of the sample with antibiotics, 1–5 h infection with the LRP followed by addition of the substrate luciferin and detection using a luminometer resulting in a detection limit of 104 c.f.u. ml−1.68 It was later discovered that the temperature sensitive and conditionally replicating mycobacteriophage Tm4-based LRP (phAE88), which granted control over the timing of cellular lysis, produced a superior buildup of luciferase and thus a superior detection limit.69 Use of a selective growth inhibitor (p-nitro-α-acetylamino-β-hydroxy propiophenone) was later incorporated to reduce false positives due to the large host range of the LRPs and enabled the assay to discriminate between M. tuberculosis and non-tuberculous mycobacteria.70 A further improvement on these assays was the development of LRP phAE142 which expresses Fflux under control of the Pleft promoter from mycobacteriophage L5.71 The phAE142 based LRP assay was tested in several studies, which showed a high sensitivity and specificity.71–73 The assay was performed on cultured samples, required a median of 3 days for DST and had a detection limit of 0.5 × 105 to 1 × 105 c.f.u. ml−1.71,72
The ability to detect both dormant and active M. tuberculosis bacilli was investigated by testing various mycobacterial promoters that are potentially active during dormancy in LRP assays.74 The promoters hsp60, isocitrate lyase (icl) and alpha crystalline (acr) genes from M. tuberculosis were cloned to drive Fflux expression in mycobacteriophages Tm4 and Che12. The authors reported success in detecting dormant and active M. tuberculosis in clinical samples using mycobacteriophage Tm4-based constructs with a detection limit of 105 c.f.u. ml−1 in clinical samples.74 The ability of Tm4 to infect dormant cells has been attributed to motifs in the Tm4 structure (Mt3 motif in the tape measure protein) allowing infection of stationary phase cells.75
Fluorescent protein expression
In contrast to stained bacteriophages, recombinant bacteriophages expressing fluorescent proteins reduce production steps and cost. An example of this was the use of PP01, a recombinant T-even type bacteriophage, for detection of E. coli strain O157:H7.76 A GFP fusion-tag was added to the PP01 SOC protein.76 Culturable and VBNC E. coli O157:H7 cells could be detected in 1–3 h using a high multiplicity of infection and observing adsorbed phages to the host cell membrane by fluorescence microscopy.76 In lieu of the lytic nature of PP01, which could potentially decrease sensitivity in microscopy-based bacterial detection, a recombinant lysis-deficient T4 bacteriophage was investigated for E. coli detection.77 A recombinant T4 was engineered with a mutated lysozyme gene and a GFP fusion to its SOC protein. The recombinant bacteriophage maintained infectivity and host fertility, but lacked the ability to lyse the host. The assay required a detection time of 10–30 min for both viable and VBNC E. coli and could discriminate the two cellular states in under 1 hour.77 However, the assay could not infect all strains of E. coli when tested on sewage influent, indicating the need for a bacteriophage with a wider host range.78 This lead to the investigation of a variety of bacteriophages that infect environmental E. coli, two of which were found to have a wider host range.78 The two bacteriophages, IP008 and IP052, were modified to replace their lytic genes with a GFP gene in addition to a GFP fusion to their SOC genes.78 The combined host range of the recombinant bacteriophages allowed the assay to detect 35 out of 70 tested strains of E. coli in a few hours using fluorescence microscopy.78Recombinant bacteriophages which express non-structurally-bound fluorescent proteins in their natural hosts' cellular milieu, have also been investigated. A recombinant GFP-expressing bacteriophage lambda was developed for detection of E. coli XL1-blue in 4–6 h using fluorescence microscopy.79 Similarly, recombinant mycobacteriophage Tm4 (phAE87) was created for detection of M. tuberculosis80 by introducing enhanced green fluorescence protein (EGFP) and ZsYellow fluorescent proteins under control of the M. bovis BCG Hsp60 promoter. The diagnostic assay consisted 16–30 h infection of the cultured samples with the mycobacteriophgaes, a paraformaldehyde fixation step, then a wash step followed by fluorescence microscopy. DST was also possible by means of incubating the samples in the presence of antibiotics prior to infection, after which fluorescence is indicative of drug resistance. The assay could detect drug resistance to several antibiotics; The specificity was 90%, 93% and 95%, sensitivity was 94% for isoniazid, rifampicin and streptomycin, respectively, while the sensitivity was 94% for all 3 drugs using the proportion method.81 The time to detection was 2 days post-culture for rifampicin, streptomycin and ofloxacin and 3 days post-culture for isoniazid resistance detection.81 The hsp60 promoter and EGFP cassette also showed detectable fluorescence levels when transferred to mycobacteriophage D29 and used to infect M. smegmatis.82
Another study showed that addition of a Strep-tag II fusion to the phAE87:hsp60-EGFP gp9 C-terminus enabled affinity purification of host bacterial complexes.83 One study involved the development of a fluorescent mycobacteriophage assay with a 100 fold increase in fluorescence per cell over phAE87:hsp60-EGFP.84 The recombinant mycbacteriophage made use of the mycobacteriophage L5 pleft promoter and the monomeric fluorescent protein mVenus cloned into mycobacteriophage phAE159 to create ϕ2GFP10.84 phAE159 contains a TM4-gp49 gene deletion, which was suggested to allow bacteriophage superinfection and could possibly be responsible for the increased fluorescence of ϕ2GFP10.84 The assay enabled detection of M. tuberculosis cells directly from sputum samples within 1 h with 90% of the cells fluorescing after 4.5 h.84 The assay allowed DST on cultured samples for rifampin and kanamycin after 12 h incubation and 36 h incubation for isoniazid and ofloxacin.84 In contrast to bioluminescence, which has a time-dependent reporter signal, fluorescent proteins have the benefit that the fluorescent signal remains detectable for weeks after the diagnostic assay is performed, also they do not require substrate addition as with many LRP assays.80
Concluding remarks
Effective diagnostics are required for detection and DST of bacterial pathogens, especially considering the diminishing effective treatment options owing to the rampant spread of drug resistance among pathogenic bacteria. Current routine diagnostics tend to be time consuming and require infrastructure which is not ideal for field use. Rapid molecular diagnostics have cost, skills and infrastructure requirements which limits their use. Bacteriophages offer unique features which could be utilized to create novel, cheap and effective diagnostics for bacterial pathogens. The assays discussed in this review have the advantage of high host-specificity, similar to antibody-based detection assays, although with significantly cheaper large-scale production costs. Current bacteriophage-based assays are designed to detect a specific host, however, diagnostic assays capable of detecting and discerning between a wide range of pathogens simultaneously is of great clinical and industrial utility. Future research on bacteriophage diagnostics could yield assays capable of detecting and discerning between multiple bacterial pathogens. This could be achieved through use of bacteriophages with altered host ranges or simulations use of multiple bacteriophages to encompass a larger host range in combination with various detection modalities. Bacteriophages represent an untapped biomass which offer novel tools for bacterial detection, as the global amount of bacteriophage particles has been estimated at 1031. The enormous potential offered by phage-based diagnostics warrants further optimization and development. With the emerging field of nanotechnology, it is possible that future research on bacteriophages and their components will yield a greater set of bacteriophage-based tools for the detection of pathogenic bacteria.References
- World Health Organization, 2012.
- Centers for Disease Control and Prevention (CDC), MMWR Morb. Mortal. Wkly. Rep., 2010, 59, 750.
- A. C. Voetsch, T. J. Van Gilder, F. J. Angulo, M. M. Farley, S. Shallow, R. Marcus, P. R. Cieslak, V. C. Deneen and R. V. Tauxe, Emerging Infections Program FoodNet Working Group, Clin. Infect. Dis., 2004, 38(suppl 3), S127–S134, DOI:10.1086/381578
.
- B. V. Krishna, Indian. J. Med. Microbiol., 2010, 28, 265–266, DOI:10.4103/0255-0857.66477
- C. Walsh and S. Fanning, Curr. Drug Targets, 2008, 9, 808–815 CrossRef CAS PubMed
- A. de Costa and D. Mavalankar, Lancet Infect. Dis., 2010, 10, 752, DOI:10.1016/S1473-3099(10)70244-9
- J. Craigie and C. H. Yen, Can. J. Public Health, 1938, 29, 448–484 Search PubMed
- J. Craigie and C. H. Yen, Can. J. Public Health, 1938, 29, 484–496 Search PubMed
- W. B. Redmond and D. M. Ward, Bull. W. H. O., 1966, 35, 563–568 CAS
- R. McNerney and H. Traore, J. Appl. Microbiol., 2005, 99, 223–233 CrossRef CAS PubMed
- T. G. Abshire, J. E. Brown and J. W. Ezzell, J. Clin. Microbiol., 2005, 43, 4780–4788 CrossRef CAS PubMed
- H. L. David, S. Clavel, F. Clement and J. Moniz-Pereira, Antimicrob. Agents Chemother., 1980, 18, 357–359 CrossRef CAS PubMed
- W. D. Jones Jr and H. L. David, Am. Rev. Respir. Dis., 1971, 103, 618–624 CAS
- X. Mi, F. He, M. Xiang, Y. Lian and S. Yi, Anal. Chem., 2012, 84, 939–946, DOI:10.1021/ac2020728; 10.1021/ac2020728
- S. A. Jassim and M. W. Griffiths, Lett. Appl. Microbiol., 2007, 44, 673–678, DOI:10.1111/j.1472-765X.2007.02115.x
- T. Tokunaga and M. I. Sellers, J. Bacteriol., 1965, 89, 537–538 CAS
- R. M. Nakamura, T. Tokunaga and T. Murohashi, Am. Rev. Respir. Dis., 1967, 96, 542–544 CAS
- S. M. Wilson, Z. al-Suwaidi, R. McNerney, J. Porter and F. Drobniewski, Nat. Med., 1997, 3, 465–468 CrossRef CAS PubMed
- N. Gali, J. Dominguez, S. Blanco, C. Prat, F. Alcaide, P. Coll and V. Ausina, J. Clin. Microbiol., 2006, 44, 201–205, DOI:10.1128/JCM.44.1.201-205.2006
- R. McNerney, P. Kiepiela, K. S. Bishop, P. M. Nye and N. G. Stoker, Int. J. Tuberc. Lung Dis., 2000, 4, 69–75 CAS
- J. Minion and M. Pai, Int. J. Tuberc. Lung Dis., 2010, 14, 941–951 CAS
- A. Albay, O. Kisa, O. Baylan and L. Doganci, Diagn. Microbiol. Infect. Dis., 2003, 46, 211–215 CrossRef CAS PubMed
- H. Albert, A. Heydenrych, R. Brookes, R. J. Mole, B. Harley, E. Subotsky, R. Henry and V. Azevedo, Int. J. Tuberc. Lung Dis., 2002, 6, 529–537 CAS
- S. Kalantri, M. Pai, L. Pascopella, L. Riley and A. Reingold, BMC Infect. Dis., 2005, 5, 59, DOI:10.1186/1471-2334-5-59
- H. Traore, S. Ogwang, K. Mallard, M. L. Joloba, F. Mumbowa, K. Narayan, S. Kayes, E. C. Jones-Lopez, P. G. Smith, J. J. Ellner, R. D. Mugerwa, K. D. Eisenach and R. McNerney, Ann. Clin. Microbiol. Antimicrob., 2007, 6, 1, DOI:10.1186/1476-0711-6-1
- R. Blasco, M. J. Murphy, M. F. Sanders and D. J. Squirrell, J. Appl. Microbiol., 1998, 84, 661–666 CAS
- T. Neufeld, A. Schwartz-Mittelmann, D. Biran, E. Z. Ron and J. Rishpon, Anal. Chem., 2003, 75, 580–585 CrossRef CAS PubMed
- M. Yemini, Y. Levi, E. Yagil and J. Rishpon, Bioelectrochemistry, 2007, 70, 180–184, DOI:10.1016/j.bioelechem.2006.03.014
- Y. Wu, L. Brovko and M. W. Griffiths, Lett. Appl. Microbiol., 2001, 33, 311–315 CrossRef CAS PubMed
- T. Neufeld, A. S. Mittelman, V. Buchner and J. Rishpon, Anal. Chem., 2005, 77, 652–657, DOI:10.1021/ac0488053
- K. V. Sergueev, Y. He, R. H. Borschel, M. P. Nikolich and A. A. Filippov, PLoS One, 2010, 5, e11337, DOI:10.1371/journal.pone.0011337; 10.1371/journal.pone.0011337
- A. J. Madonna, S. Van Cuyk and K. J. Voorhees, Rapid Commun. Mass Spectrom., 2003, 17, 257–263, DOI:10.1002/rcm.900
- S. J. Favrin, S. A. Jassim and M. W. Griffiths, Int. J. Food Microbiol., 2003, 85, 63–71 CrossRef PubMed
- S. J. Favrin, S. A. Jassim and M. W. Griffiths, Appl. Environ. Microbiol., 2001, 67, 217–224, DOI:10.1128/AEM.67.1.217-224.2001
- A. Singh, S. Poshtiban and S. Evoy, Sensors, 2013, 13, 1763–1786, DOI:10.3390/s130201763; 10.3390/s130201763
- R. Naidoo, A. Singh, S. K. Arya, B. Beadle, N. Glass, J. Tanha, C. M. Szymanski and S. Evoy, Bacteriophage, 2012, 2, 15–24, DOI:10.4161/bact.19079
- N. Malou and D. Raoult, Trends Microbiol., 2011, 19, 295–302, DOI:10.1016/j.tim.2011.03.004; 10.1016/j.tim.2011.03.004
- A. R. Bennett, F. G. Davids, S. Vlahodimou, J. G. Banks and R. P. Betts, J. Appl. Microbiol., 1997, 83, 259–265 CAS
- M. Tolba, O. Minikh, L. Y. Brovko, S. Evoy and M. W. Griffiths, Appl. Environ. Microbiol., 2010, 76, 528–535, DOI:10.1128/AEM.02294-09; 10.1128/AEM.02294-09
- J. W. Kretzer, R. Lehmann, M. Schmelcher, M. Banz, K. P. Kim, C. Korn and M. J. Loessner, Appl. Environ. Microbiol., 2007, 73, 1992–2000, DOI:10.1128/AEM.02402-06
- Y. Fujinami, Y. Hirai, I. Sakai, M. Yoshino and J. Yasuda, Microbiol. Immunol., 2007, 51, 163–169 CrossRef CAS PubMed
- S. Sainathrao, K. V. Mohan and C. Atreya, BMC Biotechnol., 2009, 9, 67 CrossRef PubMed
- A. Singh, S. K. Arya, N. Glass, P. Hanifi-Moghaddam, R. Naidoo, C. M. Szymanski, J. Tanha and S. Evoy, Biosens. Bioelectron., 2010, 26, 131–138, DOI:10.1016/j.bios.2010.05.024; 10.1016/j.bios.2010.05.024
- A. Singh, D. Arutyunov, M. T. McDermott, C. M. Szymanski and S. Evoy, Analyst, 2011, 136, 4780–4786, 10.1039/c1an15547d; 10.1039/c1an15547d
- M. A. Javed, S. Poshtiban, D. Arutyunov, S. Evoy and C. M. Szymanski, PLoS One, 2013, 8, e69770, DOI:10.1371/journal.pone.0069770; 10.1371/journal.pone.0069770
- J. D. Willford, B. Bisha, K. E. Bolenbaugh and L. D. Goodridge, Bacteriophage, 2011, 1, 101–110, DOI:10.4161/bact.1.2.15666
- L. Goodridge, J. Chen and M. Griffiths, Appl. Environ. Microbiol., 1999, 65, 1397–1404 CAS
- L. Goodridge, J. Chen and M. Griffiths, Int. J. Food Microbiol., 1999, 47, 43–50 CrossRef CAS PubMed
- C. L. Pierce, J. C. Rees, F. M. Fernandez and J. R. Barr, Anal. Chem., 2011, 83, 2286–2293, DOI:10.1021/ac103024m; 10.1021/ac103024m
- P. B. Yim, M. L. Clarke, M. McKinstry, S. H. De Paoli Lacerda, L. F. Pease 3rd, M. A. Dobrovolskaia, H. Kang, T. D. Read, S. Sozhamannan and J. Hwang, Biotechnol. Bioeng., 2009, 104, 1059–1067, DOI:10.1002/bit.22488
- L. Wu, T. Huang, L. Yang, J. Pan, S. Zhu and X. Yan, Angew. Chem., Int. Ed., 2011, 50, 5873–5877, DOI:10.1002/anie.201100334; 10.1002/anie.201100334
- R. Edgar, M. McKinstry, J. Hwang, A. B. Oppenheim, R. A. Fekete, G. Giulian, C. Merril, K. Nagashima and S. Adhya, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 4841–4845 CrossRef CAS PubMed
- M. A. Walling, J. A. Novak and J. R. Shepard, Int. J. Mol. Sci., 2009, 10, 441–491, DOI:10.3390/ijms10020441; 10.3390/ijms10020441
- F. Franks, Philos. Trans. R. Soc., A, 2003, 361, 557–574, DOI:10.1098/rsta.2002.1141
- L. V. Corotto, P. K. Wolber and G. J. Warren, EMBO J., 1986, 5, 231–236 CAS
- P. K. Wolber and R. L. Green, Trends Biotechnol., 1990, 8, 276–279 CrossRef CAS PubMed
- P. K. Wolber, Adv. Microb. Physiol., 1993, 34, 203–237 CrossRef CAS PubMed
- P. Irwin, A. Gehring, S. I. Tu, J. Brewster, J. Fanelli and E. Ehrenfeld, J. AOAC Int., 2000, 83, 1087–1095 CAS
-
S. Ulitzur, J. Kuhn, J. Scholmerich, P. Andreesen, A. Kapp, M. Ernst and W. G. Woods, Proceedings of the 4th International Bioluminescence and Chemiluminescence Symposium held in Freiburg, Chichester, U.K., 1987, pp. 463–472 Search PubMed
- S. Ripp, P. Jegier, M. Birmele, C. M. Johnson, K. A. Daumer, J. L. Garland and G. S. Sayler, J. Appl. Microbiol., 2006, 100, 488–499, DOI:10.1111/j.1365-2672.2005.02828.x
- S. Ripp, P. Jegier, C. M. Johnson, J. R. Brigati and G. S. Sayler, Anal. Bioanal. Chem., 2008, 391, 507–514, DOI:10.1007/s00216-007-1812-z
- D. A. Schofield, C. T. Bull, I. Rubio, W. P. Wechter, C. Westwater and I. J. Molineux, Appl. Environ. Microbiol., 2012, 78, 3592–3598, DOI:10.1128/AEM.00252-12; 10.1128/AEM.00252-12
- T. E. Waddell and C. Poppe, FEMS Microbiol. Lett., 2000, 182, 285–289 CrossRef CAS PubMed
- M. J. Loessner, C. E. Rees, G. S. Stewart and S. Scherer, Appl. Environ. Microbiol., 1996, 62, 1133–1140 CAS
- M. J. Loessner, M. Rudolf and S. Scherer, Appl. Environ. Microbiol., 1997, 63, 2961–2965 CAS
- J. Kuhn, M. Suissa, J. Wyse, I. Cohen, I. Weiser, S. Reznick, S. Lubinsky-Mink, G. Stewart and S. Ulitzur, Int. J. Food Microbiol., 2002, 74, 229–238 CrossRef CAS PubMed
- D. A. Schofield, I. J. Molineux and C. Westwater, J. Clin. Microbiol., 2009, 47, 3887–3894 CrossRef CAS PubMed
- W. R. Jacobs Jr, R. G. Barletta, R. Udani, J. Chan, G. Kalkut, G. Sosne, T. Kieser, G. J. Sarkis, G. F. Hatfull and B. R. Bloom, Science, 1993, 260, 819–822 CAS
- C. Carriere, P. F. Riska, O. Zimhony, J. Kriakov, S. Bardarov, J. Burns, J. Chan and W. R. Jacobs Jr, J. Clin. Microbiol., 1997, 35, 3232–3239 CAS
- P. F. Riska, W. R. Jacobs Jr, B. R. Bloom, J. McKitrick and J. Chan, J. Clin. Microbiol., 1997, 35, 3225–3231 CAS
- S. Bardarov Jr, H. Dou, K. Eisenach, N. Banaiee, S. Ya, J. Chan, W. R. Jacobs Jr and P. F. Riska, Diagn. Microbiol. Infect. Dis., 2003, 45, 53–61 CrossRef
- N. Banaiee, M. Bobadilla-del-Valle, P. F. Riska, S. Bardarov Jr, P. M. Small, A. Ponce-de-Leon, W. R. Jacobs Jr, G. F. Hatfull and J. Sifuentes-Osornio, J. Med. Microbiol., 2003, 52, 557–561 CrossRef CAS PubMed
- M. H. Hazbon, N. Guarin, B. E. Ferro, A. L. Rodriguez, L. A. Labrada, R. Tovar, P. F. Riska and W. R. Jacobs Jr, J. Clin. Microbiol., 2003, 41, 4865–4869 CrossRef CAS PubMed
- A. Dusthackeer, V. Kumar, S. Subbian, G. Sivaramakrishnan, G. Zhu, B. Subramanyam, S. Hassan, S. Nagamaiah, J. Chan and N. Paranji Rama, J. Microbiol. Methods, 2008, 73, 18–25, DOI:10.1016/j.mimet.2008.01.005
- M. Piuri and G. F. Hatfull, Mol. Microbiol., 2006, 62, 1569–1585, DOI:10.1111/j.1365-2958.2006.05473.x
- M. Oda, M. Morita, H. Unno and Y. Tanji, Appl. Environ. Microbiol., 2004, 70, 527–534 CrossRef CAS PubMed
- Y. Tanji, C. Furukawa, S. H. Na, T. Hijikata, K. Miyanaga and H. Unno, J. Biotechnol., 2004, 114, 11–20 CrossRef CAS PubMed
- M. Namura, T. Hijikata, K. Miyanaga and Y. Tanji, Biotechnol. Prog., 2008, 24, 481–486 CrossRef CAS PubMed
- T. Funatsu, T. Taniyama, T. Tajima, H. Tadakuma and H. Namiki, Microbiol. Immunol., 2002, 46, 365–369 CrossRef CAS PubMed
- M. Piuri, W. R. Jacobs Jr and G. F. Hatfull, PLoS One, 2009, 4, e4870, DOI:10.1371/journal.pone.0004870
- L. Rondon, M. Piuri, W. R. Jacobs Jr, J. de Waard, G. F. Hatfull and H. E. Takiff, J. Clin. Microbiol., 2011, 49, 1838–1842, DOI:10.1128/JCM.02476-10
- J. L. da Silva, M. Piuri, G. Broussard, L. J. Marinelli, G. M. Bastos, R. D. Hirata, G. F. Hatfull and M. H. Hirata, FEMS Microbiol. Lett., 2013, 344, 166–172, DOI:10.1111/1574-6968.12171; 10.1111/1574-6968.12171
- M. Piuri, L. Rondon, E. Urdaniz and G. F. Hatfull, Appl. Environ. Microbiol., 2013, 79, 5608–5615, DOI:10.1128/AEM.01016-13; 10.1128/AEM.01016-13
- P. Jain, T. E. Hartman, N. Eisenberg, M. R. O'Donnell, J. Kriakov, K. Govender, M. Makume, D. S. Thaler, G. F. Hatfull, A. W. Sturm, M. H. Larsen, P. Moodley and W. R. Jacobs Jr, J. Clin. Microbiol., 2012, 50, 1362–1369, DOI:10.1128/JCM.06192-11; 10.1128/JCM.06192-11
- G. J. Sarkis, W. R. Jacobs Jr and G. F. Hatfull, Mol. Microbiol., 1995, 15, 1055–1067 CrossRef CAS PubMed
- R. E. Pearson, S. Jurgensen, G. J. Sarkis, G. F. Hatfull and W. R. Jacobs Jr, Gene, 1996, 183, 129–136 CrossRef CAS PubMed
- K. C. Sasahara, M. J. Gray, S. J. Shin and K. J. Boor, Foodborne Pathog. Dis., 2004, 1, 258–266, DOI:10.1089/fpd.2004.1.258
| This journal is © The Royal Society of Chemistry 2014 |