 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Amino acid-accepting ketosynthase domain from a trans-AT polyketide synthase exhibits high selectivity for predicted intermediate†
Christoph
Kohlhaas‡
a,
Matthew
Jenner‡
b,
Annette
Kampa
a,
Geoff S.
Briggs
b,
José P.
Afonso
b,
Jörn
Piel
*a and
Neil J.
Oldham
*b
aInstitute of Microbiology, Eidgenössische Technische Hochschule (ETH) Zurich, Wolfgang-Pauli-Strasse 10, 8083, Zurich, Switzerland. E-mail: jpiel.ethz.ch
bSchool of Chemistry, University of Nottingham, University Park, Nottingham, NG7 2RD (UK). E-mail: neil.oldham@nottingham.ac.uk
First published on 31st May 2013
Abstract
The trans-acyltransferase (AT) polyketide synthases are a recently recognised group of bacterial enzymes that generate complex polyketides. A prerequisite for re-engineering these poorly studied systems is knowledge about the substrate specificity of their components. In this work, KS domain 1 from the bacillaene polyketide synthase has been shown to possess high specificity towards 2-amidoacetyl intermediates, which are derived from incorporation of alpha amino acids into the polyketide chain. N-Acetylcysteamine (SNAC) analogues of full-length substrates were synthesised and incubated with the KS1 domain. The natural glycine-derived acyl–SNAC was found to acylate KS1 with highest efficiency, as evidenced by mass spectrometry (MS). An alanine variant was also incorporated, but its valine equivalent was not, which indicated limited tolerance of substitution at the α-position. Substrate analogues without an amine or amide nitrogen substituted on the 2-position were not accepted by KS1 at the standard assay concentration of 0.5 mM. Moreover, removal of Asn-206 from the active site of KS1 by site-directed mutagenesis reduced kcat/Km by a factor of approx. 2. This residue is conserved in most known 2-amidoacetyl-accepting KS domains from trans-AT PKSs and we postulate an important interaction between Asn-206 and the amide nitrogen of the substrate.
Introduction
Polyketide synthases (PKSs) are responsible for the biosynthesis of an array of complex, biologically active compounds, many of which are employed in medicine.1,2 Type I PKS assembly lines are comprised of an assortment of domains organised into modules. Each module carries out a single cycle of chain elongation, and optional modification. Minimally, a module consists of an acyltransferase (AT) domain, a ketosynthase (KS) domain, and an acyl carrier protein (ACP). The AT domain catalyses transfer of a malonyl unit from a CoA thioester onto the phosphopantetheinyl moiety of the ACP. Chain elongation is achieved by Claisen condensation of the malonyl unit and an acyl chain attached to the KS domain. Further optional processing at the β-keto position is directed by the presence of ketoreductase (KR), dehydratase (DH) and enoylreductase (ER) domains, producing β-hydroxyl, olefinic and fully saturated moieties, respectively.3 Additional functionalisation may be incorporated into the product by, for example, AT domains that accept alternative, α-functionalised malonyl units or methyltransferase (MT) domains. Following the final elongation cycle, a thioesterase (TE) domain catalyses hydrolysis or lactonisation releasing the polyketide chain from the PKS.4Utilising the tendency for type I PKS genes to arrange into distinct clusters, many biosynthetic pathways have been successfully elucidated from genetic sequences alone. The principle of co-linearity between PKS and product has greatly assisted the analysis of numerous biosynthetic clusters. Our understanding of the co-linearity rules has come in large part from study of the deoxyerthryonolide-6-synthase (DEBS) cluster.5,6 An architectural variant of type I PKSs, known as trans-AT PKSs, has only relatively recently been discovered.7–9 These synthases employ discrete, free-standing AT enzymes, which are not integral to the PKS.2,8
The trans-AT PKSs are notorious for their high diversity of non-canonical modules that often contain novel enzymatic domains, which usually results in poor biosynthetic assignment of clusters using standard co-linearity rules. Similar to textbook (cis-AT) PKSs,10,11 many trans-AT systems include non-ribosomal peptide synthase (NRPS) modules that incorporate amino acid building blocks, resulting in NRPS–PKS hybrids.10,12 This property requires downstream KS domains to accept atypical, NRPS-derived intermediates. While for cis-AT systems insights exist into the factors governing physical association between PKS and NRPS modules,13 little information exists, for either PKS type, about the substrate requirements for a successful assembly line switch.14 This information, however, can provide crucial guidance to create functional hybrid assembly lines by metabolic engineering.
A phylogenetic method for predicting the substrate of KS domains from trans-AT clusters has been developed recently. This approach, based on in silico analyses, provides improved assignment of biosynthetic clusters to their putative products when compared with the standard co-linearity rules.12 Recently, we have developed a mass-spectrometry-based method to functionally test substrate specificities of KS domains.15 This utilises measurement of the intact KS domain to detect acylation by N-acetylcysteamine (SNAC) thioester substrate analogues. Testing ketide-accepting KSs from the bacillaene (bae) and psymberin (psy) PKSs with short SNAC diketide mimics of full-length intermediates, we indeed observed for some KSs and substrates pronounced selectivity. Other KS–SNAC combinations exhibited significant promiscuity. However, since simplified analogues instead of full-length SNAC thioesters were tested, the possibility could not be excluded that remote regions of the substrate contribute to selectivity.
Herein we report the first study of the substrate specificity of a PKS KS domain immediately downstream of a NRPS module. Utilising full-length acyl precursors synthesized in this work and the previously reported MS methodology,15 we have examined KS1 from the BaeJ protein of the bacillaene trans-AT PKS (Scheme 1).16 In the biosynthesis of bacillaene (1), KS1 accepts a 2-amidoacetyl chain, which is derived from incorporation of a glycine unit into the polyketide intermediate. We demonstrate that the isolated KS1 domain is highly selective towards 2-amido substrates, and that its affinity can be effectively diminished by a single Asn → Ala point mutation in the active-site binding pocket.
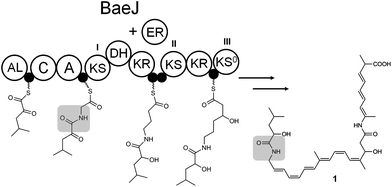 | ||
| Scheme 1 Partial proposed biosynthetic scheme for bacillaene (1). KS, ketosynthase; KS0, non-elongating ketosynthase; DH, dehydratase; KR, ketoreductase; ER, enoylreductase (trans-acting); C, NRPS condensation domain; A, NRPS adenylation domain; AL, acyl-CoA ligase, ●, acyl carrier protein domain. The amide region of the relevant intermediate is highlighted in grey. | ||
Results
Synthesis of N-acetylcysteamine thioesters
SNAC thioesters used in this study were synthesised to probe the specificity of BaeJ KS1 (Scheme 1). Although in silico analyses suggested that KS substrate specificity only extends as far as the β-position of the intermediate,12,17 longer-range interactions are also feasible. For this reason full length intermediates were produced. Synthesis of the SNAC thioesters 2–4 was performed as shown in Scheme 2. Following EDC/HOBt mediated peptide coupling of the O-benzyl protected amino acids 13a–c to acid 12, the benzyl group was cleaved by hydrogenation. Introduction of the SNAC moiety by coupling with carbonyldiimidazol gave the desired SNAC thioesters 2–4. Synthesis of the remaining SNAC thioesters 5–8, 10 and 11 was achieved by previously reported methods18–20 (see ESI†).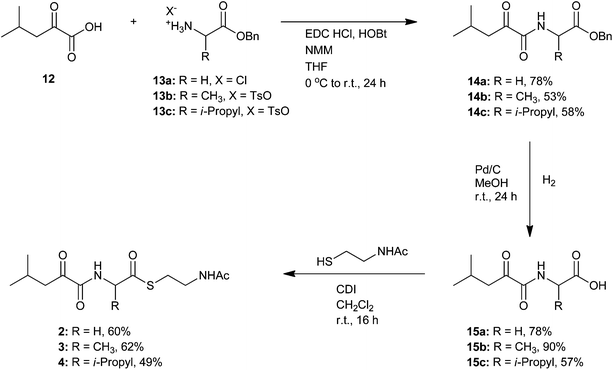 | ||
| Scheme 2 Synthesis of N-acetylcysteamine (SNAC) thioesters (2–4). | ||
Substrate tolerance of BaeJ KS1
BaeJ KS1 is located in a phylogenetic clade with other amino acid accepting KS domains, of which the majority are predicted to utilise glycine derived intermediates.12 Initially we aimed to test the full-length glycine-containing substrate and probe the tolerance of BaeJ KS1 towards substrates containing other amino acids. The glycine, alanine and valine derived intermediates 2–4, synthesised as described above, were incubated with BaeJ KS1 as described in the Experimental section. Electrospray mass spectra were recorded to monitor acylation of the KS domain. The unacylated mass of wild-type (WT) BaeJ KS1 was determined to be 73![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 507 Da, in good agreement with the theoretical value of 73509 Da (ESI S2†). Successful acylation of KS1 was observed by the appearance of additional peaks corresponding to acyl–KS1 (Fig. 1a).
507 Da, in good agreement with the theoretical value of 73509 Da (ESI S2†). Successful acylation of KS1 was observed by the appearance of additional peaks corresponding to acyl–KS1 (Fig. 1a).
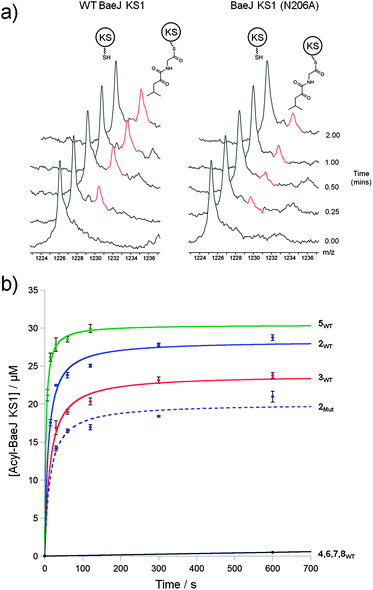 | ||
| Fig. 1 Acylation of Bae KS1 using SNAC thioesters (2–8). (a) Stacked mass spectrum of the 60+ charge state of WT-KS1 and KS1(N206A) showing the difference in acylation by SNAC 2. (b) Kinetic plot of WT-KS1 with SNACs (2–8) (solid trace), and KS1(N206A) with SNAC 2 (dashed trace). | ||
Kinetic analysis of KS1 with the SNAC substrate analogues (2–4) revealed that the cognate glycine-derived SNAC (2) was a better substrate than the alanine variant (3) (Table 1 and Fig. 1b), suggesting that the presence of an α-Me branch may reduce acylation efficiency on steric grounds. Incubation of the valine derivative (4) with KS1 resulted in no observable acylation after 10 min, indicating that the KS1 binding pocket exerts significant selectivity against bulkier substituents at the α-position of the substrate.
| Rate/× 10−6 mol dm−3 min−1 | |||
|---|---|---|---|
| Substrate | WT BaeJ KS1 | BaeJ KS1 (N206A) | |
a R = 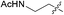 . Initial rate was calculated from a ln[KS]/[KS0] vs. t plot, given to 2 significant figures. ND – no acylation was detected over the tme scale of the incubation (10 min). Estimated error in measurements: ±0.005 × 10−6 mol dm−3 min−1. . Initial rate was calculated from a ln[KS]/[KS0] vs. t plot, given to 2 significant figures. ND – no acylation was detected over the tme scale of the incubation (10 min). Estimated error in measurements: ±0.005 × 10−6 mol dm−3 min−1.
|
|||
| 2 |
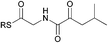
|
0.73 | 0.26 |
| 3 |
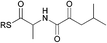
|
0.31 | 0.14 |
| 4 |
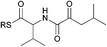
|
ND | ND |
| 5 |
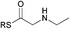
|
0.90 | 0.39 |
| 6 |
|
ND | ND |
| 7 |
|
ND | ND |
| 8 |
|
ND | ND |
| 9 |
|
0.60 | 0.14 |
| 10 |
|
0.06 | 0.06 |
| 11 |
|
0.06 | 0.04 |
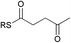
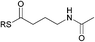
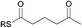
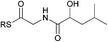
To interrogate further the structural features that are important for substrate recognition by KS1, a range of SNAC-thioesters (5–8) was synthesised. Each SNAC was designed to possess partial functionality of the natural acyl substrate (2). SNACs 5 and 6 were used to dissect the individual importance of the nitrogen and carbonyl within the amide group. SNAC 7 was employed to examine the effect of moving the amide to the 4-position (corresponding to a γ-amino acid-derived chain). Finally, SNAC 8 was used to test the effect of any long-range interactions involving the carbonyl group in the 2-keto-4-methylpentanoyl moiety of 2. No acylation was observed using SNAC 7, demonstrating that the amide substrate should be derived from an α-amino acid. Equally, no acylation was seen with SNAC 8 indicating that the distal carbonyl was not key to substrate viability. Previous studies by Calderone et al. showed that reduction of the 2-keto-4-methylpentanoyl moiety of bacillaene to its 2-hydroxy-analogue is performed by the KR domain of module 3 (Scheme 1).21 Since the reduction step is predicted to occur after KS1-catalysed chain elongation, this result implies that the oxidation level of SNAC 2 is correct for the natural substrate of KS1. To test this hypothesis further, we examined the tolerance of KS1 towards a 2-hydroxy-4-methylpentanoyl moiety using SNAC 9. Upon incubation with KS1, SNAC 9 exhibited a slightly decreased acylation rate (ca. 20%) compared to SNAC 2. This effect could be attributed to an unfavourable loss in planarity of the substrate, or a reduction in the intrinsic reactivity of the SNAC thioester. Given the similarity in acylation rates of SNACs 2 and 9, this result did not provide any new insights into our understanding of the oxidation level of this bacillaene intermediate. These observations suggests that the selectivity of BaeJ KS1 may not extend beyond the β-position, which is at least for this example in agreement with the general prediction that KS domain substrate specificity is dictated by the first 3–4 atoms of the acyl chain.12 Interestingly, SNAC 2 is also capable of acylating BaeL KS5, despite the latter's presence in a different phylogenetic clade from KS1.12 KS5 clusters with other domains predicted to accept unbranched, unsaturated acyl chains. Our previous functional study has shown that KS5 does not tolerate β-methyl branched intermediates, but does accept saturated chains.15 On this basis, SNAC 2 might be expected to be a reasonable substrate of KS5.
Incubation of KS1 with the 2-amino SNAC (5) and the 4-keto SNAC (6) yielded extremely valuable information. Although SNAC 5 was able to acylate KS1 efficiently, when SNAC 6 was incubated with KS1 no acylation was observed suggesting that the carbonyl is not important for substrate recognition (Fig. 1 and Table 1). These observations indicate that the nitrogen atom of the amide functionality in 2 is a crucial feature of an effective substrate. Although the amino nitrogen of 5 and the amido nitrogen of 2 will possess markedly different pKa values, both would be expected to act as effective hydrogen bond donors. Although we could not detect the effect of any other favourable interactions apart from the nitrogen at the 2-position, this result does not rule out the potential for other stabilising interactions between the KS and the substrate.
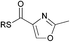
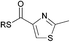
NRPS modules that incorporate amino acid building blocks can also harbour heterocyclisation (HC) and oxidation (OX) domains, which in concert catalyse the production of thiazole and oxazole moieties from N-acetylcysteine and N-acetylserine respectively.22,23 We therefore synthesised 2-methyloxazole and thiazole SNAC thioesters (10 and 11) to examine whether these cyclic, non-amide substrates could acylate KS1. Incubation of SNACs 10 and 11 with KS1 yielded low levels of acylation, at a far reduced rate than that seen for the straight chain amido-SNACs (see Table 1). This is likely due to the increased steric effects of the ring.
In an effort to promote acylation of KS1 with SNAC 4, two mutants were constructed. Guided by homology modelling, Met268 and Leu450 residues were identified as potentially blocking the KS active site from acylation by α-branched SNACs (ESI S6†). Both M268A and L450A mutants were produced, however no acylation was observed with SNAC 4 in both cases. In addition, no difference in acylation rate was observed between SNACs 2 and 3 for each of the mutants. These observations suggest that M268 and L450 are not the limiting factor for KS1 acylation by these substrates, and that intrinsic differences in reactivity of the various α-branched SNACs may be a significant factor.
Exploring the nature of the X-Cys residue
Within trans-AT KS domains, our previous work has demonstrated the importance of the amino acid residue X immediately preceding the active site Cys. A bulky amino acid, such as Met, prevents incorporation of a β-carbon branched substrate, whilst carbon branching is tolerated when X = Ala.15 Sequence alignment of all 17 known amino acid-accepting KS domains from trans-AT PKSs revealed that an Asn residue occupied the X position in 11 cases (see ESI S7†), suggesting that the residue is of biochemical significance. KS domains predicted to accept oxazole and thiazole intermediates were included in the sequence alignment due to the amino acid origin of the substrate and the resulting presence of a nitrogen atom. For these KSs none were found to possess an Asn residue at the X-Cys position. In most cases the smaller Ser or Gly residues were present, possibly to provide the necessary space for the heterocycle within the binding pocket. A phylogenetic tree was constructed using the sequence alignment, which grouped BaeJ KS1 together with exclusively KS domains containing X = Asn. In addition, a separate clade of amino acid-accepting KSs exists where X = Ala (see ESI S8†). It is notable that no other known KS domain possesses an Asn at the X position in cis or trans-AT PKSs. We therefore postulated that in the BaeJ clade, Asn may form an important interaction with the 2-amidoacetyl substrates of KS1, which could assist the acylation reaction. To test this hypothesis a BaeJ KS1 (N206A) mutant was constructed, which was predicted to reduce the efficiency of KS1. Incubation of KS1(N206A) with SNAC 2 resulted in markedly reduced levels of acylation, with the initial rate reduced by approx. 65%. This observation, combined with the previous substrate-based analysis leads us to the conclusion that the presence of an Asn in the X position increases the rate of incorporation of a 2-amidoacetyl substrate in this KS domain. It is tempting to speculate that such an effect may hold for all members of the BaeJ clade, but the existence of other clades where X = Ala indicate that the presence of Asn in this position is not essential.In addition to the sequence analysis described above, a homology model of BaeJ KS1 was constructed to visualise the potential interactions between SNAC 2 and Asn206. Two models of BaeJ KS1 were produced, the first of which shows a potential hydrogen bond between the Asn residue and the δ-carbonyl of SNAC 2 (ESI S9A†). The substrate specificity measurements described above revealed no experimental evidence for a significant role of the δ-carbonyl function. Rotation of the carboxamide region of Asn206 by approx. 90° anticlockwise, however, allows the formation of a hydrogen bond between the amide group of SNAC 2 and the carboxyl of Asn206 (ESI S9B†), and the significance of this interaction is supported by the mutagenesis data above. A second finding of the homology modelling was the putative existence of a pocket in the KS binding cleft which was able to accommodate the terminus of the acyl chain (ESI S9C†). The role of these potential long-range interactions in KS–acyl binding remains unclear, but this model may indicate the benefit of longer chain intermediates in KS enzyme assays.
Michaelis–Menten treatment of WT KS1 and KS1(N206A)
To provide more insight into the interaction between the active site Asn residue of KS1 and SNAC 2, a Michaelis–Menten treatment was performed on WT KS1 and KS1(N206A). The KS domains were incubated with concentrations of SNAC 2 ranging from 0.250 mM to 2 mM, and their initial velocities were used to construct Lineweaver–Burk plots (ESI S10†). Due to the observation of a small amount of secondary, non-specific, acylation of KS1 at higher concentrations of SNAC 2 we constructed a mutant lacking the active site Cys (C207A). Using this as a control to monitor the degree of non-specific acylation we observed that 5% non-specific acylation occurred at 1 mM 2, and 8.5% at 2 mM 2. These values were subtracted from the percentage acylation detected for the WT and N206A mutant of KS1. Presumably this non-specific acylation took place at one of the three non-active site Cys residues of KS1. From our analysis a Km value of 1.8 mM ± 0.4 was obtained for WT KS1. In comparison, the Km for KS1(N206A) was 3.7 mM ± 1.3, indicating a decreased affinity towards SNAC 2 upon removal of the Asn residue. In addition, kcat/Km was reduced by a factor of approx. 2 from WT to mutant, revealing that the catalytic efficiency of KS1(N206A) has also been reduced by the removal of the Asn residue. However, this reduction in kcat/Km is largely due to the difference between Km values, as the values for kcat were similar for both WT and mutant KS1 (Table 2).| K m (mM) | k cat (min−1) | V max (mM min−1) | |
|---|---|---|---|
| WT KS1 | 1.8 ± 0.4 | 3.0 ± 0.6 | 0.24 ± 0.05 |
| KS1(N206A) | 3.7 ± 1.3 | 2.8 ± 0.9 | 0.22 ± 0.07 |
We propose that the presence of an Asn residue at the X-Cys position enhances the preference of KS1 towards 2-amidoacetyl substrates. These intermediates occur following the presence of NRPS domains within the PKS. To date there are ten confirmed NRPS-containing trans-AT PKSs that incorporate amino acids into the intermediate, generating an amide bond,2 and a total of 17 KS domains within these PKSs that immediately follow NRPS modules.
Of the 17 KSs, 11 of them exhibit an X = Asn residue, whilst the remaining 6 contain an Ala. This indicates that an active site Asn residue is not essential for the function of such ketosynthases (as mentioned above and in agreement with our results for the BaeJ KS1 mutant). It is noteworthy, however, that for all the known KSs Asn is found in the X position only when a 2-amidoacyl intermediate is the predicted substrate. Moreover, those five amino acid-accepting KS domains with X = Ala exhibit other sequence differences from the X = Asn containing group, such that a phylogenetic analysis based on total sequence similarity places them in separate clades (see ESI S8†).
Conclusions
In this study we have described and rationalised the substrate preference of a KS domain for 2-amidoacetyl intermediates. Using full-length SNAC mimics of the acyl chain, we have shown that Bae KS1 will readily accept the natural glycine substrate and an alanine-derived analogue. It is notable that PKSs do exist which naturally incorporate Ala-containing acyl chains into their products, including KS10 within the bacillaene cluster, and KS5 of the thailandamide PKS. In our experiments a valine-containing SNAC thioester variant was not accepted, suggesting that steric effects restrict the incorporation of substrates with bulky substituents at the α-position. This constraint might be a reason why, to our knowledge, only Gly, Ala, and Ser were found to be incorporated by trans-AT PKSs unless the amino acid is processed by components other than trans-AT PKS modules.24,25 Under our assay conditions, conducted at 0.5 mM substrate, we observed that SNAC thioesters lacking an amide or amine nitrogen on the 2-position failed to acylate KS1. We have demonstrated that mutation of Asn206 to Ala reduced the affinity of KS1 towards 2-amidoacetyl substrates (kcat/Km lowered by a factor of ca. 2).An understanding of factors dictating specificity and selectivity of KS domains can aid both assignment of uncharacterised clusters and potential PKS engineering approaches to PKSs. Using the data from this study it may be possible to enhance the selectivity of other KS domains towards 2-amidoacetyl intermediates, and further explore the potential to incorporate unnatural amino acids into biosynthetic pathways. In addition, our data suggest that modifying adenylation domains in trans-AT PKS systems to engineer bulkier amino acids into metabolites will be prohibited by the substrate specificity of the downstream KS domain. In this case, inserting a downstream PKS module from cis-AT PKS–NRPS hybrid systems, which generate compounds with a wider range of amino acids, might be a way to bypass this restriction.
Acknowledgements
We are grateful for financial support from the Biotechnology and Biological Science Research Council (BBSRC) for studentship to M.J., the Leverhulme Trust for funding (code RPG-2012-578) to J.A. and N.J.O., and the DFG (SFB 642) to C.K., A.K. and J.P.Notes and references
- C. Hertweck, Angew. Chem., Int. Ed., 2009, 48, 4688–4716 CrossRef CAS.
- J. Piel, Nat. Prod. Rep., 2010, 27, 996–1047 RSC.
- S.-C. Tsai and B. D. Ames, Complex Enzymes in Microbial Natural Product Biosynthesis, Part B: Polyketides, Aminocoumarins and Carbohydrates, 2009, vol. 459 Search PubMed.
- L. Du and L. Lou, Nat. Prod. Rep., 2010, 27, 255–278 RSC.
- C. Khosla, Y. Tang, A. Y. Chen, N. A. Schnarr and D. E. Cane, Annu. Rev. Biochem., 2007, 76, 195–221 CrossRef CAS.
- S. Kapur, B. Lowry, S. Yuzawa, S. Kenthirapalan, A. Y. Chen, D. E. Cane and C. Khosla, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 4110–4115 CAS.
- J. Piel, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 14002–14007 CrossRef CAS.
- Y. Q. Cheng, G. L. Tang and B. Shen, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 3149–3154 CrossRef CAS.
- A. K. El-Sayed, J. Hothersall, S. M. Cooper, E. Stephens, T. J. Simpson and C. M. Thomas, Chem. Biol., 2003, 10, 419–430 CrossRef CAS.
- L. H. Du, C. Sanchez and B. Shen, Metab. Eng., 2001, 3, 78–95 CrossRef CAS.
- S. C. Wenzel and R. Müller, Curr. Opin. Chem. Biol., 2005, 9, 447–458 CrossRef CAS.
- T. Nguyen, K. Ishida, H. Jenke-Kodama, E. Dittmann, C. Gurgui, T. Hochmuth, S. Taudien, M. Platzer, C. Hertweck and J. Piel, Nat. Biotechnol., 2008, 26, 225–233 CrossRef CAS.
- K. J. Weissman and R. Müller, ChemBioChem, 2008, 9, 826–848 CrossRef CAS.
- S. J. Admiraal, C. Khosla and C. T. Walsh, J. Am. Chem. Soc., 2003, 125, 13664–13665 CrossRef CAS.
- M. Jenner, S. Frank, A. Kampa, C. Kohlhaas, P. Pöplau, G. S. Briggs, J. Piel and N. J. Oldham, Angew. Chem., Int. Ed., 2013, 52, 1143–1147 CrossRef CAS.
- J. Moldenhauer, X. H. Chen, R. Borriss and J. Piel, Angew. Chem., Int. Ed., 2007, 46, 8195–8197 CrossRef CAS.
- R. Teta, M. Gurgui, E. J. N. Helfrich, S. Kuenne, A. Schneider, G. Van Echten-Deckert, A. Mangoni and J. Piel, ChemBioChem, 2010, 11, 2506–2512 CrossRef CAS.
- J. Deska, S. Haehn and U. Kazmaier, Org. Lett., 2011, 13, 3210–3213 CrossRef CAS.
- H. Elokdah, T. S. Sulkowski, M. Abou-Gharbia, J. A. Butera, S. Y. Chai, G. R. McFarlane, M. L. McKean, J. L. Babiak, S. J. Adelman and E. M. Quinet, J. Med. Chem., 2004, 47, 681–695 CrossRef CAS.
- B. Kusebauch, N. Brendel, H. Kirchner, H.-M. Dahse and C. Hertweck, ChemBioChem, 2011, 12, 2284–2288 CrossRef CAS.
- C. T. Calderone, S. B. Bumpus, N. L. Kelleher, C. T. Walsh and N. A. Magarvey, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 12809–12814 CrossRef CAS.
- H. W. Chen, S. O'Connor, D. E. Cane and C. T. Walsh, Chem. Biol., 2001, 8, 899–912 CrossRef CAS.
- T. L. Schneider, B. Shen and C. T. Walsh, Biochemistry, 2003, 42, 9722–9730 CrossRef CAS.
- J. Piel, D. Q. Hui, G. P. Wen, D. Butzke, M. Platzer, N. Fusetani and S. Matsunaga, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 16222–16227 CrossRef CAS.
- N. Pulsawat, S. Kitani and T. Nihira, Gene, 2007, 393, 31–42 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3sc50540e |
| ‡ These authors contributed equally to the work. |
| This journal is © The Royal Society of Chemistry 2013 |