Enzymatic activation of nitro-aryl fluorogens in live bacterial cells for enzymatic turnover-activated localization microscopy†
Marissa K.
Lee
a,
Jarrod
Williams
b,
Robert J.
Twieg
b,
Jianghong
Rao
c and
W. E.
Moerner
*a
aDepartment of Chemistry, Stanford University, Stanford, California 94305, USA. E-mail: wmoerner@stanford.edu
bDepartment of Chemistry and Biochemistry, Kent State University, Kent, Ohio 44240, USA
cDepartment of Radiology, Stanford University, Stanford, California 94305, USA
First published on 5th October 2012
Abstract
Many modern super-resolution imaging methods based on single-molecule fluorescence require the conversion of a dark fluorogen into a bright emitter to control emitter concentration. We have synthesized and characterized a nitro-aryl fluorogen which can be converted by a nitroreductase enzyme into a bright push–pull red-emitting fluorophore. Synthesis of model compounds and optical spectroscopy identify a hydroxyl-amino derivative as the product fluorophore, which is bright and detectable on the single-molecule level for fluorogens attached to a surface. Solution kinetic analysis shows Michaelis–Menten rate dependence upon both NADH and the fluorogen concentrations as expected. The generation of low concentrations of single-molecule emitters by enzymatic turnovers is used to extract subdiffraction information about localizations of both fluorophores and nitroreductase enzymes in cells. Enzymatic Turnover Activated Localization Microscopy (ETALM) is a complementary mechanism to photoactivation and blinking for controlling the emission of single molecules to image beyond the diffraction limit.
Introduction
Recently developed microscopies (e.g., PALM, FPALM, STORM) bypass the optical diffraction limit (DL) by successive imaging and localization of sparse subsets of single-molecule (SM) fluorophores.1–3 Using these “superresolution” (SR) imaging methods, structures can be imaged with spatial resolution 5–10 times smaller than the DL of ≈200 nm for visible wavelengths. Bright emitters are essential for this SR strategy, because the localization resolution improves as 1/√N, with N the number of photons detected before photobleaching or per switching cycle.4 Using emitters whose concentration can be controlled ensures that each frame during an acquisition has only a sparse set of emitting molecules, even if the object of interest is densely labelled.5 Though most previous SR experiments in cells have used photocontrollable fluorescent proteins (FPs)6–8 because of their genetic targeting capabilities, these emit (on average) 10-fold fewer photons before photobleaching than good small-molecule fluorophores.5 Targeting small organic fluorophores to specific cellular locations9,10 has been developed as an alternative to FPs, and although challenges remain for cell entry and washout, the chemical properties, spectral range, and photophysics are more readily modified by organic synthesis.11For SR imaging, the emission from organic fluorophores can be controlled optically by reversible photoswitching12 (often in the presence of additives like thiols, or oxidizing/reducing agents)3,13,14 or by irreversible photoactivation.15 Non-optical methods to control fluorescence emission include ligand binding,16 and fluorescence quenchers.17 Fluorogenic substrates have enabled detailed single-molecule enzymology studies18 but without extracting subdiffraction information. We present a small-molecule fluorophore synthetically modified to form an enzymatically activated and cell-permeable fluorogen, and demonstrate Enzymatic Turnover Activated Localization Microscopy (ETALM), a new SR imaging method, in bacterial cells. ETALM differs from other fluorescence modulation strategies using a chemical reduction19 because the enzymatic turnover continually generates new fluorophores. This activation is irreversible and the new fluorophores do not undergo multiple switching cycles. The emitting concentration can be set at the required low level for SR imaging simply by limiting the fluorogen concentration or slowing the enzymatic rate using inhibitors.
Nitroreductase (NTR) enzymes are a family of NAD(P)H-dependent flavoproteins ubiquitous in bacteria and capable of reducing aryl-nitro compounds to their corresponding hydroxyl-amino or amino derivatives.20,21 Previous work has used a NTR enzyme to reduce a nitro-fluorogen into a cancer therapeutic22–24 but the resulting fluorophore was not sufficiently bright or emitted at too short a wavelength to allow SM imaging in cells. This study uses the commercially available E. coli nsfB NTR21 (>90 % pure, see ESI Fig. S1†) which has been well-studied because of its potential use in cancer therapeutic delivery.25
We show below that NTR converts the nitro-aryl fluorogen of the present study 1 into a bright organic fluorophore, suitable for SR in live bacterial cells (Scheme 1). The emissive product belongs to the dicyanomethylenedihydrofuran (DCDHF) class of push–pull fluorophores where an electron-poor DCDHF acceptor head is connected to an electron-rich donor, usually an amine, through a conjugated π network.26 The DCDHF fluorophores are detectable on the SM level because they emit millions of photons before photobleaching and have high fluorescence quantum yields in viscous environments, including membranes and gelatin.27 Importantly, DCDHF fluorophores can enter living cells fairly easily because they are neutral, and they emit at long wavelengths to avoid autofluorescence.27
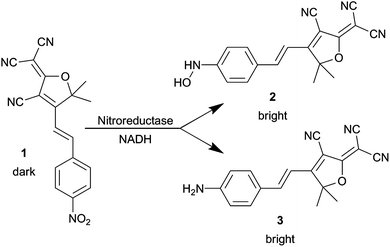 | ||
| Scheme 1 Reaction of 1 with NTR and NADH. | ||
Results and discussion
We previously reported a photoactivatable azido-DCDHF15,28 suitable for SR in bacterial and mammalian cells.29 To create another dark DCDHF fluorogen, the electron-rich amine of the emissive form can be replaced with an electron-poor nitro group which eliminates fluorescence and shifts the absorbance to the blue. Then, reduction of 1 in buffer by the NTR enzyme in the presence of NADH creates the red-shifted emitter (Fig. 1).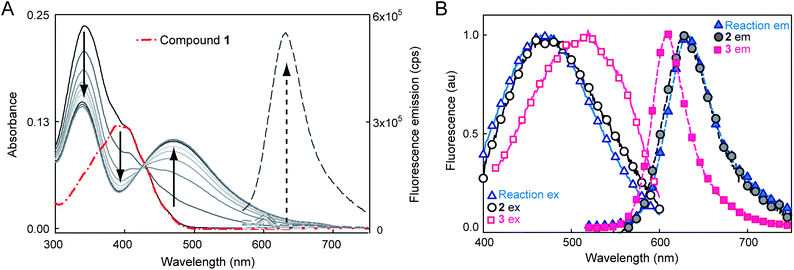 | ||
| Fig. 1 (A) Spectral change during the NTR reaction with 1 and NADH over ≈10 minutes showing red-shifted absorption and fluorescence increase. Solid curves show absorbance of the reaction mixture at different time points, broken red curve is absorbance of 1 alone. Dashed lines are fluorescence emission (500 nm excitation) before and after the NTR reaction. No fluorescence turn-on or absorbance change is observed without all three components, see ESI Fig. S2 and S3.† (B) Normalized excitation (solid curves and empty symbols, collected at emission max) and emission (dashed curves and solid symbols, 500 nm excitation) spectra of NTR reaction mixture (triangles) compared to separately synthesized 2 (circles) and 3 (squares). All spectra measured in 10 mM Tris–HCl (pH 7.5), for more details, see ESI.† | ||
The product fluorophore in these bulk spectroscopic measurements could be either the hydroxyl-amino-DCDHF 2 or the amino-DCDHF 3 (Scheme 1). The product of the NTR reaction was determined to be 2 instead of 3 by comparing the bulk emission and excitation spectra (in buffer) of separately synthesized 2 and 3 with the reaction mixture (Fig. 1B). There is close spectral overlap between the NTR reaction mixture product and 2.
The bulk and SM photophysical characteristics of molecules 1–3 were measured in solution and in a polymer film (Table 1). Solution measurements were performed in ethanol on purified compounds to allow comparison with our previously published work on DCDHF-type fluorophores.15 A poly(methylmethacrylate) (PMMA) polymer film in toluene was used in a bulk spectroscopic measurement to show the increase of fluorescence quantum yield (ΦF) of 2 and 3 in more rigid environments. PMMA polymers, though used previously to characterize the azido-DCDHF fluorogen,15 are less relevant to behaviour inside a cell, but measurements were performed in PMMA to compare 2 to prior work. As is the case for the other DCDHF fluorophores,26 the fluorescence quantum yield ΦF of 2 increases in the more constrained polymer environment. In order to assess the potential of 2 for cell imaging, more biologically relevant conditions were used for microscopy measurements of the photobleaching quantum yield (ΦB) and the average number of total photons emitted (Ntot). Here, we used a primarily aqueous polymer film (1% poly(vinyl alcohol) (PVA) in water) to immobilize the fluorophores. Other in vitro environments like poly-lysine or gelatin either did not provide sufficient immobilization, or had an unacceptably high fluorescence background at the wavelengths used.
| λ abs (nm), ε (dm3 mol−1 cm−1) | λ fl (nm) | Φ F | Φ B (×10−6) | SM Ntotc (×106) | |
|---|---|---|---|---|---|
| a See ESI† and ref. 28 for details on measurements and calculations. b Bulk quantum yield of permanent photobleaching, measured in aqueous PVA polymer film. c Average total number of photons emitted for SMs in PVA polymer film. d Fluorescence quantum yield in PMMA. | |||||
| 1 | 390, 17![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 020 020 |
n/a | n/a | n/a | n/a |
| 2 | 497, 15300 |
609 | 0.021, 0.27d | 5.6 | 1.7 |
| 3 | 570, 54100 |
613 | 0.025, 0.39d | 4.1 | 2.3 |
Importantly for single-molecule based superresolution imaging strategies, SMs of 2 were detectable when immobilized in the aqueous PVA polymer film (Fig. 2A), confirmed by the observation of single-step photobleaching to the background level. Movie S1† shows SMs of 2 in PVA, and representative time traces of the SMs appear in Fig. S7.† The average number of photons detected before photobleaching (Ntot) was measured for 2 and 3 (Fig. 2A and B, Table 1) using the survival probability. The survival probability PN (Fig. 2B) is the ratio of the number of SMs mn surviving after a given number of emission events N to the total number of molecules M in the measurement set (see ESI†).15,30 This approach gives comparable results to histogramming the detected photons31 but avoids binning artifacts. To convert photons detected to total emitted photons, the detected value is divided by the optical system collection efficiency32 of 0.04.
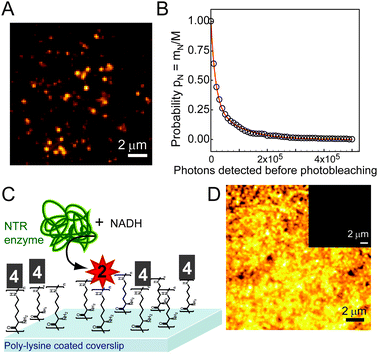 | ||
| Fig. 2 (A) SM fluorescent spots of 2 (verified by one-step photobleaching) in PVA film (532 nm excitation, 91 W cm−2). (B) Survival probability histogram of 2 doped into PVA films at nM concentrations with an average 71000 detected photons, corresponding to an average of 1.7 × 106 photons emitted by each SM. (C) Schematic of in vitro experiment where 4 (structure shown in Chart 1) was covalently attached to poly-lysine coverslips immersed in PBS buffer (pH 7.4) containing free NADH and NTR enzyme. Enzymatically generated fluorophores remained attached to surface lysines. (D) Bulk fluorescence at the surface after NADH and NTR addition. Inset: bulk fluorescence of 4 before NTR reaction (same contrast). Bulk fluorescence of all control incubation conditions is shown in Fig. S6.† sample and control details in ESI.† | ||
After photophysical characterization of separately synthesized 1–3, we used microscopy to measure the fluorescence generated by the NTR-reaction on a surface in vitro. Compound 4, a N-hydroxy-succinimide (NHS) derivative of 1 (Chart 1) was covalently attached to poly-lysine coated coverslips through reaction of NHS with the surface amines. After washing off the unreacted 4, NADH and NTR enzyme were added in buffer and allowed to react with the attached 4. Dramatic fluorescence turn-on was observed after this addition (Fig. 2D and inset).
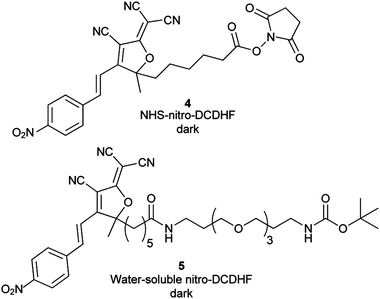 | ||
| Chart 1 Structures of nitro-DCDHF derivatives 4 and 5 used for surface immobilization and kinetic experiments, respectively. Bulk absorbance and fluorescence changes from the NTR reaction are shown in ESI Fig. S4 and Fig. S5, respectively.† Table S1 in ESI† summarizes photophysical characteristics for these molecules. | ||
Our microscopy-based experiments could detect SMs of 2 and see fluorescence from the NTR-reaction, but do not quantify the NTR reaction rate. We used bulk spectroscopic measurements in solution to find the NTR reaction kinetic rates (Fig. 3) and used the more water-soluble nitro derivative 5 (Chart 1) instead of 1 because 1 aggregates in higher concentration regimes. (Kinetic measurements using 1 at lower concentrations are shown in the ESI, Fig S8†). NTR follows a ping-pong bi–bi mechanism33 (eqn (1)) where the rate per enzyme (νi/E) depends on both NADH and nitro substrate 5 concentrations with two Michaelis constants:34,35
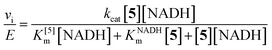 | (1) |
 | ||
| Fig. 3 3D Michaelis–Menten plot of initial rate of product formation (monitored by change in absorption at 500 nm) in PBS (pH 7.4) versus various starting concentrations of 5 and NADH. The structure of 5 is shown in Chart 1. The grid is the fit of eqn (1) to the data points. | ||
The initial rate of product formation, monitored by absorption at 500 nm, was measured for a range of initial concentrations of 5 and NADH (Fig. 3). These rates were fit to eqn (1), yielding fit parameters kcat of 224 ± 40 s−1 and KNADHM of 13 ± 3 μM and a K[5]M of 4.9 ± 1 μM, consistent with prior work.34
After confirming the NTR-reaction fluorescence turn-on on a surface and measuring the bulk NTR rate, we explored the NTR-reaction scheme for turnover of the fluorogen in bacterial cells. NTR enzymes are endogenous in many bacteria. When several bacterial species (E. coli, Caulobacter crescentus, Bacillus subtilis) are incubated with μM concentrations of 1, entire cells become fluorescent (Fig. 4A and ESI Fig S9†). This effect was most dramatic in wild type B. subtilis cells, which are known to have enhanced response to nitro compounds.36,37 To support the hypothesis that this fluorescence arises from a NTR reduction analogous to the reaction studied in vitro, 1 and an excess of menadione, a competitive NTR inhibitor, were added simultaneously to the bacteria. Under this incubation condition, the fluorescence of individual cells was reduced by 14× to just above background levels (Fig. 4B and E). No additives were needed to make 1 cell permeable.
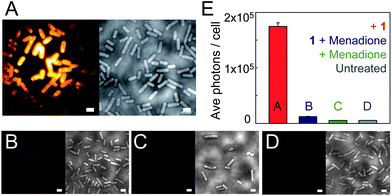 | ||
| Fig. 4 (A) B. subtilis incubated with 5 μM of 1. Left image: average fluorescence; right image: corresponding white light image of cells. All cells become fluorescent; cells in the center are brighter because the laser excitation spot is more intense in the center. Images in the controls (B)–(D) are structured similarly: left image is average fluorescence at same contrast scale as (A), and right image is corresponding white light image. (B) B. subtilis incubated with both 1 (5 μM) and menadione (0.2 mM), showing a dramatic fluorescence reduction. The images in (C) show cells incubated with only menadione, and part (D) shows untreated B. subtilis. (E) Quantification of number of photons detected per cell (∼300 cells per condition) for each case. All scale bars are 2 μm, imaging intensity is 2.5 kW cm−2. All conditions were incubated for 30 minutes. For more details on incubation and imaging conditions, see ESI.† | ||
When B. subtilis cells are incubated with 5 μM concentrations of 1, the emission spots from the large number of enzymatically generated diffusing fluorophores overlap, and the cells appear bright so that distinct single molecules cannot be observed. By lowering the incubation concentration of 1 to <nM levels, separated fluorescent spots can be observed turning on and subsequently photobleaching (Fig. 5A and insets). Even though the true size of the emitting molecule is ≈1 nm, the diameter of the SM fluorescent spot is ≈200 nm, on the order of the bacterial cell width, due to the DL. These fluorescent spots correspond to SMs, verified by one step photobleaching to background levels. At the laser intensity used here (15 kW cm−2), photobleaching is fast relative to the 100 ms exposure time, as shown by representative time-traces of the SM emission (Fig. 5A).
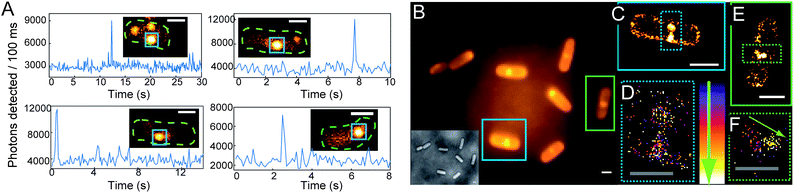 | ||
| Fig. 5 (A) Representative SM emission traces of enzymatically generated fluorophores in B. subtilis showing photons detected per 100 ms bin vs. time. For each graph, the inset is a single imaging frame from the specific B. subtilis cell (cell outline in dashed green, SM plotted is in blue box). (B) DL average image of 3000 frames (24 s) of B. subtilis incubated with 1 and corresponding white light image (inset). (C) SR reconstruction (average 17.9 ± 0.3 nm statistical precision) from the cell boxed in blue with both membrane localizations and punctate spots (blue dashed) consisting of many overlapping localizations. (D) Temporal progression of the localizations in the punctate spots, with early localizations colored purple, and later localizations colored white. The lack of temporal progression in the localizations suggests that these punctate spots correspond to the fluorescent cloud generated by two relatively stationary enzymes. (E) SR reconstruction from the green-boxed cell. (F) Temporal progression of localizations in the green dashed box of (E) showing a defined temporal progression with later localizations toward the center of the cell (shown by arrow), suggesting that this punctate spot is a single enzyme diffusing on the time scale of our experiment. Scale bars: 1 μm except 0.5 μm in (D and F). For more details on incubation, imaging, fitting, and display conditions, see the ESI.† | ||
We now seek to demonstrate that SR information can be extracted from the enzyme turnovers using ETALM.‡ As is well-known, with separated SM fluorophores, the fitted center of the DL spot is an estimate of the true position of the emitting molecule. By lowering the incubation concentration, the total number of enzymatically generated fluorophores is easily decreased. The incubation concentration of 1 used to create Fig. 5A was slightly too low for SR imaging, as only one SM spot would appear in a given DL region every 8–30 seconds. The incubation concentration of 1 was increased very slightly to 1.5 nM, so new SMs were generated at a reasonable rate during continuous imaging. A DL image composed of the average of many imaging frames is shown in Fig. 5B. It should be noted that our interpretation of SR reconstructions could still be complicated by blurred spots arising from diffusion of the free fluorophores after catalysis. To minimize this effect, the number of long-lived fluorophores is reduced by using high intensity reading light, and the camera exposure time is reduced to 8 ms per frame. In the limit of very high laser intensity, each newly generated SM fluorophore should be photobleached before it has time to diffuse away from the NTR enzyme. In this limit, the extracted SM positions would be samplings of the NTR enzyme position over time as it moves through the cell, generating fluorescent molecules. In this initial demonstration, we only approached this limit in intensity, thus in our SR reconstructions (Fig. 5C–F and Movie S2†), two populations of localizations are observed: (i) punctate spots (dashed boxes in Fig. 5C and E) and (ii) membrane localizations (e.g., seen easily in Fig. 5C). For case (i), aggregation could cause the punctate spots, but 1 is added at the nM level and was only found to aggregate at the μM regime in separate experiments. We hypothesize that the punctate spots arise from a “cloud” of enzymatically generated fluorophores photobleached before they could travel very far from the enzyme. Notably, time-coloring of the successive SM localizations from the fluorescence movie sometimes shows apparent motion of the enzyme (Fig. 5F). Considering the second population (ii), some emitters can apparently survive for a longer time and are able to diffuse to reach the membrane which represents a sink for the relatively nonpolar 2. It is reasonable that a small fraction of fluorophores could reach the protective environment of the membrane without photobleaching given that the expected diffusion coefficient for a small molecule in a bacterial cell is on the order of 5.0 × 10−7 cm2 s−1,38 which could easily result in an rms displacement of 1.5 microns during the 8 ms imaging time. Membrane-bound fluorophores are more easily localized because diffusion is slower39 and the DCDHF-type fluorophores are harder to bleach (since ΦF is larger in the membrane).27
Conclusions
We have synthesized and characterized a nitro-fluorogen which can be enzymatically activated to produce a bright SM emitter. The fluorescent product was identified and characterized on a bulk and SM-level in vitro and in cells. Unlike previous work using enzymatic turnovers to generate fluorescent SM products,40–42 we used the nascent fluorescent molecules from the NTR enzyme on the single-molecule level to explore the subdiffraction-scale organization of a cytosolic bacterial enzyme in B. subtilis as well as the membrane locations where some of the product molecules are found.Considering extensions beyond bacteria, mammalian cells often have much lower levels of NTR-type enzymes20 or activity and would be interesting targets for future work. In cells with suitably low levels of endogenous NTR activity, the ETALM scheme could be used as an orthogonal genetically encoded strategy for SR imaging of protein structures of interest fused to NTR. One particular advantage of this approach arises from the fact that the precursor nitro-fluorogen 1 is not fluorescent, so no washing steps are required. The activation of 1 is not light-dependent, and samples do not require special handling under red lights or in the dark. It may be useful in some contexts to use the NTR scheme as a generator of fluorescent molecules from a source inside the bacterial cell to obtain the position of the inner membrane.43 At the same time, one of the challenges of this system is controlling the total amount of activated fluorophores. We used high intensity laser light to rapidly bleach nascent fluorophores, but the use of an inhibitor could also allow temporary suppression and tuning of the fluorescence generation rate.
Acknowledgements
This work was supported in part by grant no. R01-GM086196 from the National Institute of General Medical Sciences. Samples of wild-type Bacillus subtilis, E. coli, and Caulobacter crescentus were obtained from Jerod Ptacin in the Shapiro lab at Stanford University. The protein gel was run with the help of Steffi Duttler in the Frydman lab at Stanford University.Notes and references
- E. Betzig, G. H. Patterson, R. Sougrat, O. W. Lindwasser, S. Olenych, J. S. Bonifacino, M. W. Davidson, J. Lippincott-Schwartz and H. F. Hess, Science, 2006, 313, 1642–1645 CrossRef CAS.
- S. T. Hess, T. P. K. Girirajan and M. D. Mason, Biophys. J., 2006, 91, 4258–4272 CrossRef CAS.
- M. J. Rust, M. Bates and X. Zhuang, Nat. Methods, 2006, 3, 793–796 CrossRef CAS.
- R. E. Thompson, D. R. Larson and W. W. Webb, Biophys. J., 2002, 82, 2775–2783 CrossRef CAS.
- S. J. Lord, H. D. Lee and W. E. Moerner, Anal. Chem., 2010, 82, 2192–2203 CrossRef CAS.
- R. M. Dickson, D. J. Norris, Y. L. Tzeng and W. E. Moerner, Science, 1996, 274, 966–969 CrossRef CAS.
- G. H. Patterson and J. Lippincott-Schwartz, Science, 2002, 297, 1873–1877 CrossRef CAS.
- R. Ando, H. Mizuno and A. Miyawaki, Science, 2004, 306, 1370–1373 CrossRef CAS.
- M. Fernandez-Suarez and A. Y. Ting, Nat. Rev. Mol. Cell Biol., 2008, 9, 929–943 CrossRef CAS.
- N. George, H. Pick, H. Vogel, N. Johnsson and K. Johnsson, J. Am. Chem. Soc., 2004, 126, 8896–8897 CrossRef CAS.
- D. Maurel, S. Banala, T. Laroche and K. Johnsson, ACS Chem. Biol., 2010, 5, 507–516 CrossRef CAS.
- J. Fölling, V. Belov, R. Kunetsky, R. Medda, A. Schönle, A. Egner, M. Bossi and S. W. Hell, Angew. Chem., Int. Ed., 2007, 46, 6266–6270 CrossRef.
- G. T. Dempsey, J. C. Vaughan, K. H. Chen, M. Bates and X. Zhuang, Nat. Methods, 2011, 8, 1027–1036 CrossRef CAS.
- M. Heilemann, S. van de Linde, M. Schüttpelz, R. Kasper, B. Seefeldt, A. Mukherjee, P. Tinnefeld and M. Sauer, Angew. Chem., Int. Ed., 2008, 47, 6172–6176, DOI:10.1002/anie.200802376.
- S. J. Lord, N. R. Conley, H. D. Lee, R. Samuel, N. Liu, R. J. Twieg and W. E. Moerner, J. Am. Chem. Soc., 2008, 130, 9204–9205, DOI:10.1021/ja802883k.
- A. Sharonov and R. M. Hochstrasser, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 18911–18916 CrossRef CAS.
- M. Schwering, A. Kiel, A. Kurz, K. Lymperopoulos, A. Sprödefeld, R. Kramer and D. Herten, Angew. Chem., Int. Ed., 2011, 50, 2940–2945 CrossRef CAS.
- B. P. English, W. Min, A. M. van Oijen, K. T. Lee, G. Luo, H. Sun, B. J. Cherayil, S. C. Kou and X. S. Xie, Nat. Chem. Biol., 2006, 2, 87–94 CrossRef CAS.
- J. Vogelsang, T. Cordes, C. Forthmann, C. Steinhauer and P. Tinnefeld, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 8107–8112, DOI:10.1073/pnas.0811875106.
- M. D. Roldán, E. Pérez-Reinado, F. Castillo and C. M. Vivián, FEMS Microbiol. Rev., 2008, 32, 474–500 CrossRef.
- G. Anlezark, R. Melton, R. Sherwood, B. Coles, F. Friedlos and R. Knox, Biochem. Pharmacol., 1992, 44, 2289–2295 CrossRef CAS.
- R. J. Hodgkiss, A. C. Begg, R. W. Middleton, J. Parrick, M. L. Stratford, P. Wardman and G. D. Wilson, Biochem. Pharmacol., 1991, 41, 533–541 CrossRef CAS.
- Y. Liu, Y. Xu, X. Qian, Y. Xiao, J. Liu, L. Shen, J. Li and Y. Zhang, Bioorg. Med. Chem. Lett., 2006, 16, 1562–1566 CrossRef CAS.
- S. H. Thorne, Y. Barak, W. Liang, M. H. Bachmann, J. Rao, C. H. Contag and A. Matin, Mol. Cancer Ther., 2009, 8, 333–341 CrossRef CAS.
- N. K. Green, D. J. Kerr, V. Mautner, P. A. Harris and P. F. Searle, Methods Mol. Med., 2004, 90, 459–477 CAS.
- K. A. Willets, S. Y. Nishimura, P. J. Schuck, R. J. Twieg and W. E. Moerner, Acc. Chem. Res., 2005, 38, 549–556 CrossRef CAS.
- S. J. Lord, N. R. Conley, H. D. Lee, S. Y. Nishimura, A. K. Pomerantz, K. A. Willets, Z. Lu, H. Wang, N. Liu, R. Samuel, R. Weber, A. N. Semyonov, M. He, R. J. Twieg and W. E. Moerner, ChemPhysChem, 2009, 10, 55–65, DOI:10.1002/cphc.200800581.
- S. J. Lord, H. D. Lee, R. Samuel, R. Weber, N. Liu, N. R. Conley, M. A. Thompson, R. J. Twieg and W. E. Moerner, J. Phys. Chem. B, 2010, 114, 14157–14167 CrossRef CAS.
- H. D. Lee, S. J. Lord, S. Iwanaga, K. Zhan, H. Xie, J. C. Williams, H. Wang, G. R. Bowman, E. D. Goley, L. Shapiro, R. J. Twieg, J. Rao and W. E. Moerner, J. Am. Chem. Soc., 2010, 132, 15099–15101, DOI:10.1021/ja1044192.
- A. Molski, J. Chem. Phys., 2001, 114, 1142–1147, DOI:10.1063/1.1333760.
- B. L. Lounis and M. Orrit, Rep. Prog. Phys., 2005, 68, 1129–1179 CrossRef CAS.
- W. E. Moerner and D. P. Fromm, Rev. Sci. Instrum., 2003, 74, 3597–3619 CrossRef CAS.
- W. Cleland, Biochim. Biophys. Acta, 1963, 67, 104–137 CrossRef CAS.
- D. Jarrom, M. Jaberipour, C. Guise, S. Daff, S. White, P. Searle and E. Hyde, Biochemistry, 2009, 48, 7665–7672 CrossRef CAS.
- P. Race, A. Lovering, R. Green, A. Ossor, S. White, P. Searle, C. Wrighton and E. Hyde, J. Biol. Chem., 2005, 280, 13256–13264 CrossRef CAS.
- J. Mostertz, C. Scharf, M. Hecker and G. Homuth, Microbiology, 2003, 150, 497–512 CrossRef.
- N. Van Duy, C. Wolf, U. Mäder, M. Lalk, P. Langer, U. Lindequist, M. Hecker and H. Antelmann, Proteomics, 2007, 7, 1391–1408 CrossRef CAS.
- J. T. Mika, G. van den Bogaart, L. Veenhoff, V. Krasnikov and B. Poolman, Mol. Microbiol., 2010, 77, 200–207 CrossRef CAS.
- S. Y. Nishimura, S. J. Lord, L. O. Klein, K. A. Willets, M. He, Z. Lu, R. J. Twieg and W. E. Moerner, J. Phys. Chem. B, 2006, 110, 8151–8157, DOI:10.1021/jp0574145.
- R. D. Smiley and G. G. Hammes, Chem. Rev., 2006, 106, 3080–3094, DOI:10.1021/cr0502955 er.
- M. Roeffaers, G. D. Cremer, J. Libeert, R. Ameloot, P. Dedecker, A. Bons, M. Bückins, J. Martens, B. Sels, D. De Vos and J. Hofkens, Angew. Chem., Int. Ed., 2009, 121, 9449–9453 CrossRef.
- H. Yoon, S. Shim, L. Baek and J. Hong, Bioorg. Med. Chem. Lett., 2011, 21, 2403–2405 CrossRef CAS.
- M. D. Lew, S. F. Lee, J. L. Ptacin, M. K. Lee, R. J. Twieg, L. Shapiro and W. E. Moerner, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, E1102–E1110, DOI:10.1073/pnas.1114444108.
Footnotes |
| † Electronic supplementary information (ESI) available: Full experimental details, product characterization, selected NMR spectra, and additional figures are provided. See DOI: 10.1039/c2sc21074f |
| ‡ Since many nitro compounds are known to be toxic to bacterial cells,20 the total time of SR experiments from addition of 1 to imaging was kept as short as possible (<10 minutes). |
| This journal is © The Royal Society of Chemistry 2013 |