
Recent progress towards the development of fluorescent probes for the detection of disease-related enzymes
Lopamudra
Mishra
 and
Monalisa
Mishra
*
and
Monalisa
Mishra
*
Neural Developmental Biology Lab, Department of Life Sciences, National Institute of Technology, Rourkela, Odisha 769008, India. E-mail: mishramo@nitrkl.ac.in; monalisam@nitrkl.ac.in
First published on 6th December 2024
Abstract
Normal physiological functions as well as regulatory mechanisms for various pathological conditions depend on the activity of enzymes. Thus, determining the in vivo activity of enzymes is crucial for monitoring the physiological metabolism and diagnosis of diseases. Traditional enzyme detection methods are inefficient for in vivo detection, which have different limitations, such as high cost, laborious, and inevitable invasive procedures, low spatio-temporal resolution, weak anti-interference ability, and restricted scope of application. Because of its non-destructive nature, ultra-environmental sensitivity, and high spatiotemporal resolution, fluorescence imaging technology has emerged as a potent tool for the real-time visualization of live cells, thereby imaging the motility of proteins and intracellular signalling networks in tissues and cells and evaluating the binding and attraction of molecules. In the last few years, significant advancements have been achieved in detecting and imaging enzymes in biological systems. In this regard, the high sensitivity and unparalleled spatiotemporal resolution of fluorescent probes in association with confocal microscopy have garnered significant interest. In this review, we focus on providing a concise summary of the latest developments in the design of fluorogenic probes used for monitoring disease-associated enzymes and their application in biological imaging. We anticipate that this study will attract considerable attention among researchers in the relevant field, encouraging them to pursue advances in the development and application of fluorescent probes for the real-time monitoring of enzyme activity in live cells and in vivo models while ensuring excellent biocompatibility.
 Lopamudra Mishra | Lopamudra Mishra is a PhD scholar (CSIR-JRF fellow) at the National Institute of Technology, Rourkela under the supervision of Prof. Monalisa Mishra. Her current research includes the detection of protein aggregation and neurotransmitters in the brain of the Drosophila disease model. |
 Monalisa Mishra | Dr Monalisa Mishra currently holds the position of Associate Professor at the National Institute of Technology, Rourkela, India. She obtained her PhD degree from Jacobs University Bremen, Germany. She was a Post-Doctoral Fellow at the University of California (San Diego, USA), Indiana University (Bloomington, USA) and MPI-CBG Dresden, Germany. Her research focuses on the in vivo toxicological analysis of fluorescent probes and biological applications of probes in disease detection in model organisms. |
1. Introduction
Enzymes are macromolecular biological catalysts that play a crucial role as biomarkers for the pathophysiology of several illnesses. They are engaged in numerous physiological and biological processes. Diseases including Alzheimer's disease, Parkinson's disease, cancer, diabetes, cardiovascular disease, and arthritis are associated with abnormal enzyme activity.1,2 The level of expression and activity of these enzymes must be determined to diagnose diseases in their early stages, increase the likelihood of survival, and aid in developing treatment strategies. Consequently, it is crucial to monitor the enzyme levels in living organisms in real-time and use enzyme detection to predict illness.Numerous techniques have been developed to measure the enzyme activity of various enzyme families. Traditional methods for the analysis of enzyme activity typically involve colorimetric assays,3–6 electrochemical assays,7–9 and luminescence assays.10–12 Despite of the great potential value of these technologies for determining the activities of enzymes, many researchers need help to measure specific enzyme activity in real time. Additionally, finding a non-destructive way to test enzyme activity in vivo still remains challenging.13 Usually, these techniques encounter several obstacles, such as preventing artificial factors from impacting the enzyme activity in living organisms and achieving fast, selective, and better resolution detection in vivo, especially in deeper tissues.2
Optical imaging technologies play a crucial role in fundamental research and clinical medicine.14 Fluorescence-based optical probes are excellent choices for monitoring intracellular enzymes. They offer high spatio-temporal resolution, non-invasive properties, real-time imaging capabilities, and operational simplicity. Previously, numerous studies have revealed significant advancements and promising opportunities in this area. Fluorescent probes have been effectively utilized in live-cell imaging, immunofluorescence staining, fluorescence-guided surgery and drug delivery (theranostics).15–20 Numerous experiments have been conducted to develop fluorescent sensors for the detection of enzyme activity. However, the effectiveness of the latest fluorescent detectors in precisely identifying the activity of enzymes in living organisms still has to be improved for in vitro experiments and histological studies. This is because certain limitations such as the presence of auto-fluorescence in the biological environment and the inadequate penetration of light in tissue make it laborious to measure enzyme activity accurately. Thus, to realize the optimal performance, a fluorescent probe should possess specific characteristics, including the ability to target cells or tissues specifically, selectivity for the desired biomarker even in the presence of other similar species resistance (anti-interference ability), stability upon expose to light, compatibility with biological systems, and the ability to be excited by long-wavelength light, which are essential for in vivo imaging.1 The construction of fluorescence sensors to track enzymes is a fascinating and engaging field of study.
Emerging techniques for imaging, the early identification of illness, and therapy are anticipated as a result of the advancement of enzyme-targeting fluorescent probes. For researchers studying organisms and environment, the development of NIR region fluorophores enabling two-photon fluorescence for efficient biomarker detection is incredibly exciting. These advancements have the potential to greatly enhance our ability to monitor enzyme activities in real time. Furthermore, researchers are dedicated to working on fluorescent chemosensors that possess specific characteristics such as water solubility, photostability, large Stokes shifts, high quantum yields, high signal-to-noise ratio, and improved biocompatibility. These properties are crucial for the safe and effective monitoring of enzymes in living organisms (Fig. 1). This review highlights the latest advancements in the development of enzyme-responsive fluorescent probes for disease diagnostics. It includes the molecular structure of probes and their functional mechanisms and applications in the imaging of live cells and models.
 | ||
| Fig. 1 Diagrammatic representation of fluorescent probes involved in enzymatic detection. | ||
2. Development of an ideal fluorescent probe
A fluorescent probe has the ability to bind to specific regions or functional groups in targeted biomolecules, resulting in an alteration in their fluorescence signal.21–24 It typically includes three components. Firstly, there is a signal/fluorophore component that undergoes a significant change in its spectroscopic property when it reacts with the desired analyte. Secondly, there is a recognition/labeling component that interacts specifically with the target. Finally, there is a suitable linker that connects these two components, although in some cases, they are directly integrated without a linker.25 The recognition groups have the ability to selectively bind to the analyte via various mechanisms such as alterations in their chemical environment and covalent and non-covalent bonds.26,27 An important part of the function of the recognition moiety is to interact with the target and adjust the fluorescence signal.28 The connecting group facilitates the connection and transmission of the signal between the reporter and recognition group.29A fluorophore or fluorescent dye may produce light at a different wavelength than it absorbs when exposed to light of a specific wavelength.30,31 The fluorescence characteristics of a probe are predominantly affected by its fluorophore, both before and after it interacts with the target.32 The photophysical properties of fluorophores include a maximum absorption (λabs), a maximum emission (λem), an extinction coefficient (ε), which is reported at λabs, and a fluorescence quantum yield (Φ). Another noteworthy characteristic is photo-stability or photo-bleaching, which illustrates the resilience of a dye to continuous irradiation. At last, the important chemical characteristics of fluorophores include sensitivity to the pH or polarity of the solvent, aggregation-induced characteristics, solubility, and cell permeability.33
Typically, fluorescent dyes possess a significant conjugated π-bond structure and a rigid planar arrangement, resulting in the manifestation of the lowest single-line electron excited state.34 The frequently used fluorophores in various applications include pyrene, coumarin, phthalimide, fluorescein, rhodamine, cyanine, and BODIPY. Among them, coumarin dyes often exhibit shorter emission wavelengths35 and can be less stable under alkaline conditions. However, they are relatively simple to synthesize and possess excellent cell permeability. In this case, the addition of a power-supplying group leads to a red shift in the absorption and emission wavelength of naphthalimides, resulting in a high quantum yield and excellent biological stability.36–38 Near-infrared (NIR) fluorescent probes absorb and emit light in the NIR spectrum.39,40 Light emitted by NIR-II probes is often in the wavelength range of 900 to 1700 nm, whereas that emitted by NIR-I probes is 700 to 900 nm. Thus, these two probes are utilized in different types of biological imaging. Imaging deeper into living organisms with less background auto-fluorescence is possible using NIR light because it enters biological tissues better than visible light.41 The common NIR fluorescent dyes (>600 nm) include cyanine dyes, and altering their structural makeup is a simple way to alter their biological stability. The fluoropyrrole dye has seen rapid development, which is a high-quantum yield near-infrared dye (>600 nm).1
3. Classification and design approaches for fluorescent probes
Fluorophores are employed for the detection of the fluorescence characteristics of probes. To produce various types of probes, fluorophores must be reasonably modified to allow acceptable optical alterations both before and after binding to the target.42–44 Fluorescence probes can be classified into many types based on various standards.3.1. Based on mechanism
The various types of fluorescence quenching can be categorised based on their mechanism, which includes aggregation-induced emission (AIE),45 photoinduced electron transfer (PET),46 fluorescence resonance energy transfer (FRET),47 intramolecular charge transfer (ICT),48 and excited-state intramolecular proton transfer (ESIPT),49 as illustrated (Fig. 2A). PET is frequently utilised in the development of fluorescence sensors for the detection of metal ions and protons. In PET, the transfer of an electron takes place between an electron-rich donor (D) and an electron-deficient acceptor (A), starting with light absorption. Two fluorophores (donor and acceptor) may be linked in a single molecule using a fluorescent probe that operates based on the FRET process. Radiation at the excitation wavelength is absorbed by the donor, D, which then conveys this energy to the acceptor, A, emitting at an extended wavelength. ICT provides an alternative method for creating chemo-sensors based on the detection of hazardous ions. A strong push–pull electron system is formed by the combination of a strong electron acceptor, strong electron donor, and fluorophore in ICT fluorescent molecular probes. Charge transfer from D to A happens during photoexcitation. Molecules have dual emission features in the ESIPT process. ESIPT emission is produced by the excited keto form (K*), while short-wavelength emission is produced by the excited enol form (E*). The D and A present within the molecule, H-bond forming ability, the basicity/acidity of the solvent, and the polymerization process are some of the elements that affect the spectral characteristics of ESIPT fluorophores. The restricted intramolecular motion, rotation or vibration (RIM, RIR, or RIV, respectively) inside aggregates is responsible for the AIE phenomena. Because of their unconstrained intramolecular movements, AIE-active molecules produce modest emissions in solution. However, when they aggregate in an appropriate environment, RIM, RIR, or RIV activation processes in the excited state cause them to become extremely emissive.50 | ||
| Fig. 2 Classification and design strategies: (A) based on mechanism; (B) based on response mode; and (C) based on interaction with the target molecules. | ||
3.2. Based on response mode
Fluorescent probes may exhibit three primary response modes including ratiometric, on–off (also known as fluorescence enhancement), and off–on (also known as fluorescence quenching) modes. These modes arise due to disruptions in different photo-physical processes.25,51–57 Fluorescent off–on probes are created by adding a recognition moiety to fluorophores, as seen in Fig. 2B(a), where the recognition moiety quenches the fluorescence of the fluorophore. This may be accomplished by substituting the free –OH or –NH2 groups in certain fluorophores with an enzyme recognition moiety or by protection–deprotection. Similarly, Fig. 2B(b) shows how to connect a fluorophore and recognition site to create a fluorescent off–on probe using the 4-hydroxybenzyl alcohol unit as a linker. This type of probe shows low background fluorescence, which is recovered to intense fluorescence when the recognition moiety or linker is cleaved or released by an enzyme during activation.25,51,55,58 As shown in Fig. 2B(c), a ratiometric fluorophore (typically a 1,8-naphthalimide skeleton) may be linked to a recognition moiety to create a ratiometric probe.3.3. Based on interaction with target molecules
There are three primary approaches for designing fluorescent probes based on how the probes interact with the target molecules, as follows: (a) binding of the probes, (b) cleaving ability of the probes, and (c) stoichiometric of the probes (Fig. 2C). Binding fluorescent probes are very popular, which often connect a fluorophore and a recognition group using a linker. By observing the change in the fluorescence intensity prior to and during incubation with the analyte, change in spectral location and fluorescence intensity, this type of probe can identify the analyte. Such fluorescent probe binds to the target analyte and releases a fluorophore, which generates a fluorescent signal. This approach for designing cleavable fluorescent probes allows the recognition group to attach both the analyte and the fluorescent indicator, therefore realising the detection of the analyte. The creation of biosensors that target enzymes is the primary application for this type of design methodology. A form of probe known as a stoichiometric fluorescent probe utilizes the variation in the fluorescence signal produced between the probe and the recognition moiety before and after a chemical reaction.1,59,604. Fluorescent probes for the detection of disease-related enzyme activities
Enzyme-responsive fluorescent probes have attracted keen interest among the numerous fluorescence sensing platforms for monitoring intracellular enzymes because of their high specificity, high spatiotemporal resolution, and non-invasive design. With the ongoing advancements in fluorescent probe technology, important disease biomarkers can soon be detected at lower concentrations, in deeper biological sites, and earlier in the progression of diseases. This will lead to more precise clinical diagnosis and insight into how bioactive chemicals function physiologically in certain illnesses. The enzyme targets that are involved in detection of Parkinson's disease (PD), Alzheimer's disease (AD), and cancer include oxidoreductases, glycosidases, phosphatases, cholinesterase, and proteases. Several other probes that are also involved in diagnosis and imaging studies in living systems are listed in Table 1.| Enzyme | Probe name | Fluorophore | Recognition unit | Limit of detection (LOD) | Overexpression cell type | Associated disease | Application | Ref. |
|---|---|---|---|---|---|---|---|---|
| Tyrosinase (TYR) | Probe 1 | Rhodamine | 3-Hydroxybenzyl oxygen | 0.45 U mL−1 | B16F10 (mouse melanoma cells), A375 cells (human malignant melanoma cells), MNT1 (human melanoma cell) | Pigmentary disorders, melanoma, melasma, vitiligo | Intracellular TYR imaging in B16F10 cells, indicating that the probe can be a promising tool for the detection of endogenous TYR activity. | 61 |
| MU | 4-Methylumelliferone | 3-Bromomethylphenol | 0.2 U mL−1 | Used for determination of TYR activity in human serum. | 62 | |||
| NITYO | 1,8-Naphthalimide | 3-Hydroxy benzyloxy | 0.7 U mL−1 | Imaging exogenous tyrosinase in vivo in zebrafish model. | 63 | |||
| CHMC-DOPA | Chloro-hydroxyl-merocyanine (CHMC) | Dopamine (DOPA) | 0.003 U mL−1 | Exhibits excellent cell membrane permeability and low cytotoxicity, which is successfully used to detect TYR activity in living cancer cells (HepG2) and zebrafish models. | 64 | |||
| EQR-TYR | 3-Hydroxy benzyl | EQR | 0.035 U mL−1 | EQR-TYR on imaging TYR in living cells (B16F10 melanoma) and xenograft tumor mouse model. | 65 | |||
| Monoamine oxidase-A (MAO-A) | ANET | EHBT | Propylamine | 19.8 ng mL−1 | SH-SY5Y cells (human neuro-blastoma cells), U87 cells (human derived glioma), PC-3 cells (human prostate cancer cells), H9c2(rat cardio-myocyte cell line) | Major depressive disorder, emotional disorders, prostate cancer, heart disorders | Ability to localize to mitochondria due to the “dual-targeting” strategy. Used to specifically image MAO-A in SH-SY5Y, PC-3, and GD H9c2 cardiac cells. | 66 |
| KXS-M2 | Chlorine-substituted dicyanoisophorone (DCF) | Propylamine | 9.8 ng mL−1 | Detection of MAO-A activity in glucose-deprived H9c2 cardiac cells, zebrafish and ISO-induced failing heart tissues. | 67 | |||
| Rma-1,2 | Hemicyanine, HXPI | 3-(2-Chlorophenoxy)propan-1-amine | 4.5 ng mL−1 | Imaging MAO-A in HeLa cells, zebrafish and mice in vivo. | 68 | |||
| β-Galactosidase (β-gal) | CPD-gal | CPD-OH | Galactosyl | 6.0 × 10−4 U mL−1 | SK-OV-3 (human ovarian cancer cell), OVCAR-3 (human ovarian cancer cell) | Primary ovarian cancer | Visualizing endogenous β-gal, which has been successfully demonstrated in SK-OV-3 cells, HeLa cells and tumor-bearing mice. | 69 |
| Gal-HCA | 2-Hydroxy-4′-dimethylamino-chalcone (HCA) | D-Galactose | 0.0122 U mL−1 | Monitoring senescent and ovarian cancer cells with overexpressed β-gal. | 70 | |||
| MLC | MLC-OH | Galactose | 4.0 × 10−3 U mL−1 | First pH-sensitive β-gal fluorescent probe in lysosomes, imaging in OVCAR-3 cells. | 71 | |||
| CB-FR | CB-OH | β-D-Galactosidase | 0.0035 U mL−1 | Imaging in OVCAR-3, track the endogenous β-Gal in zebrafish in real-time. | 72 | |||
| Alkaline phosphastase (ALP) | APN | 4-Amine-1,8-naphthalimide (NIN) | Phosphate-derived benzyl carbamate | 0.16 U L−1 | HepG2 cells (human hepatoma cells), HeLa cells (human cervical cancer cells) | Cholestatic liver injury, cervical cancer | Visualise endogenous ALP in cervical cancer cells (HeLa cells). | 73 |
| SWJT-3 | Dicyanoisophorone | Phosphate ester | 0.87 U L−1 | First used for bio imaging of endogenous ALP in both HeLa cells and mice. | 74 | |||
| YJ | T-OH | Phosphate | 2.36 U L−1 | Used to image endogenous ALP in vitro. Track and detect the elevated endogenous ALP levels caused by APAP-induced liver injury in zebrafish. | 75 | |||
| Acetylcholinesterase (AChE) | EW3 | TCF | N,N-Dimethyl carbamyl unit | 0.17 U mL−1 | PC-12 cells (pheochromocytoma cells), U-87MG (human glioblastoma cell) | Alzheimer's disease, Parkinson's disease | Study the fluctuation of AChE levels in zebrafish, imaging AChE in Neuro-2A cells. | 76 |
| NFL-SF | NFL-OH | 2-Thienylformyl chloride | 0.2 mU mL−1 | Monitor exogenous and endogenous AChE activity of chemotherapeutic drug-induced cell apoptosis in living cells. | 77 | |||
| Naph-3 | 1,8-Naphthalimide | N,N-Dimethylcarbamate group | 0.18 ± 0.01 U mL−1 | Tracing AChE in the Neuro-2a cells, detect human brain AChE in E. coli. | 78 | |||
| Butyrylcholinesterase (BChE) | DCD-P1 and DCD-P2 | DCD-based (2-dicyano-methyldiene-3-cyano-2,5-dihydrofuran)-(DCD-OH) | Chlorides (cyclopropanecarboxylic acid chloride and butanoyl chloride) | 0.12 μg mL−1 | HepG2 cells (human hepatoma cells), HT22 cells (mouse hippocampal neuron cells), U-87MG cells (human glioblastoma cells) | Liver cancer, liver injury, non-alcoholic fatty liver disease, Alzheimer's disease, Parkinson's disease | Real-time tracking of BChE activity in a mouse model. | 79 |
| Chy-1 | Chromene-benzoindolium | Propanecarboxylic acid chloride | 0.12 ng mL−1 | Imaging of tumor-bearing models, successful application for endogenous BChE detection in AD mouse models and brain slices revealed the enormous feasibility of Chy-1 in the diagnosis of Alzheimer's disease. | 80 | |||
| Aminopeptidase-N (APN) | TMN-PCPA | TMN-NH2 | PCPA | 7.5 ng mL−1 | TPC-1 cells (human papillary thyroid cancer cell), 786-O and ACHN cells (kidney cancer cells) | Thyroid cancer, renal cancer | Imaging-guided surgery, discrimination tumor bio-samples from normal bio-samples, endogenous APN activity in different normal and cancer cell lines. | 81 |
| TMN-Abu | Dicyanoisophorone derivatived NIR fluorophore (TMN) | Abu | 0.57 ng mL−1 | Used to discriminate tumor cells from normal cells and tracing tumor in nude mice, imaging in type 2 diabetic model mice. | 82 | |||
| Caspase | Ac-DEVD-PABC-Naph | Naphthalimide | Ac-DEVD peptide | 4.96 ng mL−1 | Staurosporine-induced HK-2 apoptotic cells (human kidney cells) | Acute kidney injury | Compare caspase-3 activity to other endogenous species, such as small biomolecules (cysteine, glucose, glutathione, glycine, and ascorbic acid) and proteins (lysozyme, trypsin, BSA, subtilisin, and pepsin). | 83 |
| Ac-Tat-DEVD-CV | Cresyl violet (CV) | Asp–Glu–Val–Asp (DEVD) | 5.3 pM | Quantitative visualization of caspase-3 activity and indicates the apoptosis stages of cells. | 84 | |||
4.1. Probes for oxidoreductases
Enzymes that catalyze electron transfer or oxidation–reduction processes are members of the incredibly diversified class known as oxidoreductases. Their catalytic activity facilitates oxidation–reduction processes, in which a reduced substance (O2) transfers oxygen atoms to an organic molecule that is already oxidized, or electrons are transferred from an oxidized donor (reducing agent) to an oxidized acceptor (oxidizing agent). On a fundamental physiological level, oxidoreductases are responsible for initiating processes that are essential to the functioning of all biological systems.85 These enzymes are classified into four primary categories including dehydrogenases, oxidases, peroxidases, and oxygenases. They are responsible for regulating the redox homeostasis within organisms. Accordingly, many diseases are linked to their abnormal levels.86Recent studies confirmed that TYR plays a role in wound healing. To monitor TYR in the wounded area of the zebrafish tail, Chen et al.101 synthesized a benzindole-based “turn-on” red fluorescent probe, Pro-OH. This probe is a hybrid of p-aminostyryl benzindole (Pro-NH) (fluorophore) and 3-hydroxyphenol (recognition site), which can avoid interference from ROS at the response site. Pro-OH was first oxidised to o-diphenols in the presence of TYR. Subsequently, these o-diphenols formed o-quinones, which in turn released Pro-NH (Fig. 3A). Furthermore, Pro-OH had very little fluorescence on its own; nevertheless, when it interacted with TYR, the fluorescence intensity increased by 14-fold at 580 nm (Fig. 3B). The results showed that the fluorescence intensity was directly proportional to TYR concentration (Fig. 3C). The fluorescence intensity and TYR concentration showed a strong linear relationship in the range of 5–175 U mL−1, with an estimated detection limit of 1.024 U mL−1. Upon interaction with Pro-OH, the zebrafish displayed red fluorescence throughout their bodies, with the exception of their yolk sac, indicating the effectiveness of this probe in detecting endogenous TYR (Fig. 3D). Bright-red fluorescence was seen in the region of zebrafish tail breakage, indicating the aggregation action of TYR (Fig. 3E). Additionally, the probe was used for detecting TYR in A549 human lung cancer cells and A375 human malignant melanoma cells. After being cultured with Pro-OH, the A549 cells showed faint red fluorescence, with a much lower fluorescence intensity than that in the A375 cells. Consequently, this probe is a trustworthy monitoring tool for endogenous TYR in live cells and at the location of zebrafish injuries.
 | ||
| Fig. 3 (A) Schematic of Pro-OH in detecting TYR. (B) Fluorescence spectra of Pro-NH and Pro-OH before and after reaction with TYR. (C) Fluorescence intensity changes incubated with different concentration of TYR. (D) Fluorescence images of zebrafish: (a) zebrafish only; (b) zebrafish incubated with Pro-OH; (c) zebrafish pre-treated with kojic acid, and then incubated with Pro-OH; and (d) zebrafish incubated with kojic acid. (E) Bright-field image and fluorescence image of 4 days old zebrafish tail: (a) tail-cutting zebrafish without Pro-OH (control); (b) intact zebrafish (without wound) incubated with Pro-Oh; (c) tail cutting zebrafish incubated with Pro-OH; and (d) tail-cutting zebrafish pretreated with kojic acid for 2 h, and then incubated with the Pro-OH. Chen et al.,101 Copyright 2024, Elsevier. | ||
For the purpose of sensitively and specifically monitoring the TYR dynamics with a large Stokes shift, Huang et al.102 devised and synthesised the first AIE fluorogenic probe, BTFTYR (Fig. 4A). In the presence of ROS, this enzyme exhibits good anti-interference capability in the cellular environment. It was found to recognise 3-hydroxybenzyloxy as its specific recognition substrate, and a benzothiazole derivative with ESIPT properties was selected as the fluorescence reporter.103 When TYR was added, a notable fluorescent intensity at 560 nm was seen, which progressively became stronger as the concentration of TYR increased (Fig. 4B). With a detection range of 3.94 U mL−1, BTFTYR could provide a strong linear response in the tyrosinase concentration range of 0–180 U mL−1. The stopping of the ESIPT process is likewise a function of the recognition group. Through effective 1,6-rearrangement-elimination, the tyrosinase-specific enzymatic reaction may be eliminated, allowing the ESIPT process to be restored to produce keto-emission.
 | ||
| Fig. 4 (A) Schematic of BTFTYR detecting TYR. (B) FL spectra of BTFTYR upon the addition of TYR. (C) In vivo imaging of TYR in zebrafish at 1 d, 3 d and 5 d post continuous treatment of PTU (1-phenyl-2-thiourea). (D) Imaging in different skin cell types: MNT1, B16F10, FB and HaCaT. MNT1 cells treated with different concentrations of (E) kojic acid and (F) α-MSH. (G) MNT1 cells treated with UVB radiation. Huang et al.,102 Copyright 2023, Elsevier. | ||
The in vivo tracking ability of the probe was confirmed by experiments in zebrafish model. There was a significant yellow fluorescence throughout the body, indicating the easy permeability of the probe into the skin due to the specific catalysis of TYR, whereas the time-dependent inhibition of TYR expression by PTU (1-phenyl-2-thiourea) showed a reduction in fluorescence signal (Fig. 4C). The imaging studies done in different skin cells showed that MNT1 (human melanoma cells) and B16F10 cells (mouse melanoma cells) emitted enhanced yellow fluorescence due to the catalytic activity of TYR to cleave BTFTYR in the presence of over-expressed TYR to release the fluorophore (Fig. 4D). Further, this probe could be employed to examine the efficacy of different medications related to pigmentary disorders. There was a drastic reduction in the fluorescence signal when MNT1 was treated with 100 μM kojic acid (Fig. 4E). This effect was reversed in the case of α-MSH (100 nM) with an enhanced fluorescence signal (Fig. 4F). Similarly, when it was exposed to 30 mJ cm−2 UVB, the upregulated expression of TYR was indicated by yellow emission (Fig. 4G). These results confirmed that upon UVB radiation and α-MSH treatment, the activity of tyrosinase was upregulated, whereas it was significantly lowered by kojic acid treatment.
The endoplasmic reticulum (ER) plays a special role in melanogenesis by trafficking the TYR produced by normal melanocytes to melanosomes.104 Sometimes, there is misdistribution of tyrosinase, where the mature molecule is missorted by the ER from melanosome to lysosome, causing it to undergo degradation. This causes imbalances in the level of TYR and leads to several disorders.105,106 Hence, it is necessary to visually monitor the mature TYR that has been sorted to ER. The fluorescent probe designed by Shu et al.107 is the first enzymatic probe that can target ER through the ICT mechanism, namely ER-Nap-TYR. The enzymatic site for TYR, i.e., 3-hydroxylbenzyl, and the ER-location moiety, i.e., p-toluene-sulfonamide groups, are attached towards the opposite ends of the naphthalimide fluorophore. This probe gets oxidized upon enzymatic reaction with TYR and produces a catechol intermediate and releases the red-shifted (absorption peak at 450 nm) fluorophore, i.e., ER-NAP-OH, via 1,6-rearrangement elimination (Fig. 5A). The ability of this probe to achieve ratiometric detection was suggested by the gradual decrease in its emission peak at 480 nm with an increase in the concentration of TYR (0–300 U mL−1) (Fig. 5B). The fluorescence response was further confirmed by yellow color. A good linear correlation was observed in the range of 30–150 U mL−1 with the detection limit of 5.64 U mL−1, indicating the enzymatic-activity-dependent ratiometric fluorescence of this probe.
 | ||
| Fig. 5 (A) Schematic of the ratiometric visualization of refluxed TYR by an endoplasmic reticulum-localized enzymatic probe, ER-Nap-TYR. (B) Fluorescence spectra of ER-Nap-TYR with different concentrations of TYR. (C) Fluorescence images of ER-Nap-TYR co-localized in the endoplasmic reticulum in B16 cells. (upper-left) 5 mM ER-Nap-TYR (yellow channel); (middle) 1 mM ER-tracker red (red channel); and (right) merged pattern of two channels. Bottom row: (left) Bright field pattern of cells; (middle) intensity scatter plot of two channels; and (right) intensity profile of ROI across the B16 cells costained with ER-Nap-TYR and ER-tracker red. (D) Fluorescence images of ER-Nap-TYR in living cells (left: HeLa cells, middle: B16 cells, and right: B16 cells + inhibitor). (E) Fluorescence microscopy images of B16 cells treated with different concentrations of H2O2 and 150 mM H2O2 with NAC using ER-Nap-TYR. Shu et al.,107 Copyright 2024, The Royal Society of Chemistry. | ||
The organelle level distribution of the enzyme in living cells was confirmed by co-localization experiments with ER-Tracker Red. In the red channel, the fluorescence of the probe overlapped with the ER-Tracker with a Pearson's coefficient of 0.84 (Fig. 5C). This result confirmed that ER-NAP-OH can be used for imaging the TYR activity inside the organelle. Further, the functionality of the probe was visualized in the ER of living cells. Murine melanoma (B16 cells) was detectable in the yellow channel, which proved the permeability and sensitivity of this probe to intracellular tyrosinase. However, upon treatment with kojic acid, this probe fluoresced in the blue channel (Fig. 5D). Further, the mechanism of the ER-associated degradation pathway in the H2O2-mediated inhibition of melanogenesis was studied. When the concentration of H2O2 increased, there was an increase in bright yellow fluorescence, indicating the uptake of ER-Nap-TYR by B16 cells (Fig. 5E).
4.1.2.1. Monoamine oxidase-A. Recently, Zhang et al.113 designed and synthesized the MAO-A-specific NIR-FP DDM-NH2 with a large Stokes shift (180 nm), which is extremely significant for applications in biological imaging. This probe has two main functional parts, i.e., dicyanoisophosphone (DDM, NIR dye), which serves as a fluorescent precursor group, and alanine as a recognition group, serving as the enzyme response and sharing structural similarities with the inhibitor. The DDM fluorescent group is released after detecting and binding to MAO-A; however, the NIR fluorescence emission of this probe was comparatively low (Fig. 6A). With an 11-fold enhancement in fluorescence (Fig. 6B) at 770 nm, the fluorescence intensity gradually increased from 50 to 551 nm, reaching its maximum after ∼1 h of incubation (Fig. 6C). Notably, the emission of DDM-NH2 showed a linear relationship with the dosage of MAO-A in the range of 4–10 μg mL−1, displaying a high correlation value of 0.9970.
 | ||
| Fig. 6 (A) Schematic of DDM-NH2 probe activated by MAO-A and its application in tumor visualization in a mouse diffused-tumor model. (B) Fluorescence emission spectra of DDM-NH2 before and after reaction with MAO-A. (C) Time-dependent changes in fluorescence emission of DDM-NH2 in the presence of MAO-A. (D) Confocal fluorescence images of the co-incubation of different cells with DDM-NH2: (a) HeLa cells incubated with DDM-NH2; (b) HeLa cells pretreated with clorgyline, and then incubated with DDM-NH2; (c) SH-SY5Y cells incubated with DDM-NH2; (d) SH-SY5Y cells pretreated with clorgyline and then incubated with DDM-NH2; (e) HepG-2 cells incubated with DDM-NH2; and (f) MCF-7 cells incubated with DDM-NH2. (E) (a) In situ spray of 500 μM DDM-NH2 applied to the tumor; (b) in vitro imaging performed by spraying DDM-NH2 in normal organs (1, 2, 3, 4, and 5) and tumor tissue (6); (c) statistical analysis of the differences in the fluorescence intensities of the organs detected in the images; (d) in situ administration of DDM-NH2 for the excision of multiple disseminated tumors in the subcutaneous tissue; and (e) fluorescence imaging of numerous subcutaneous diffused nodules via in situ spraying of DDM-NH2 for tumor identification. Zhang et al.,113 Copyright 2024, ACS. | ||
Strong fluorescence was seen when MAO-A was detected in HeLa and SH-SY5Y cells using DDM-NH2, suggesting that DDM-NH2 responded well to MAO-A. Furthermore, at the same dosage of DDM-NH2 in HeLa cells, the fluorescence intensity was substantially greater than that in SH-SY5Y cells. This is because the activity of MAO-A was inhibited when the cells were pre-processed with clorgyline, resulting in a noticeable decrease in cell fluorescence (Fig. 6D). The in situ spraying of DDM-NH2 allowed the detection of the tumor and its boundaries as well as tumors with a smaller size (∼2 mm). This suggests that DDM-NH2 can be used as an indispensable tool for surgical navigation (Fig. 6E). Also, owing to its exceptional biocompatibility, it is possible to monitor the endogenous MAO-A activity in tumor cells, zebrafish, and mice tumor tissues safely.
It was discovered that aberrant MAO-A expression is directly linked to the formation of gliomas, making it a unique biomarker for glioma diagnosis and therapy.114 An affinity-binding-based two-photon fluorogenic probe (TPFP), CD1, was created and synthesized by Zhang et al.115 to quickly identify the content of MAO-A in human glioma cells and tissues (Fig. 7A). Because of their higher spatial-temporal resolution and greater tissue penetration, TPFPs are a better choice for imaging tissues.116,117 Pre-mito or N,N-dimethyl-naphthalenamine derivative was functionalized with several substitutions to enable mitochondrial targeting and exploration of the MAO-A pocket with the use of a two-photon fluorophore and molecular docking data. Using the TICT effect, the fluorescence of CD1 is first quenched. Nevertheless, the confinement fluorescence effect (CFE) causes a conformational shift in CD1 after entering the MAO-A cavity, blocks the TICT effect and restores the fluorescence of CD1. With an limit of detection (LOD) of 1.4 nM, CD1 exhibited a linear relationship with the MAO-A concentration in the range of 0 to 500 nM (Fig. 7B). Consequently, this probe completed detection within 20 s.
 | ||
| Fig. 7 (A) Structural formula of probe CD1. (B) Schematic of MAO-A response mechanism of CD1. (C) Fluorescence spectra of CD1 incubated with different concentrations of MAO-A. (D) Fluorescence images SH-SY5Y cells upon incubation with CD1, MAO-A A gRNA1 was used to knock out endogenous MAO-A and Ctrl gRNA was used as a non-target control and fluorescence images HepG2 cells upon incubation with CD1, MAO-A POE (protein over-expression) vector was used to regulate endogenous MAO-A and POE Puro was used as an empty carrier. (E) Two-photon fluorescence images and the corresponding plots of endogenous MAO-A content with CD1-treated human glioma (top) and paracancerous tissues (bottom). Zhang et al.,115 Copyright 2023, Wiley. | ||
Experiments involving the imaging of living cells and tissues have shown that CD1 is capable of selectively identifying the endogenous MAO-A content in mitochondria under the intricate physiological conditions. The human neuroblastoma cell line (SH-SY5Y, high level of MAO-A) and hepatoma cell line (HepG2, high level of MAO-B) are two frequently utilized cell lines for investigating MAO. Confocal microscopy showed a distinct fluorescence signal in wild-type SH-SY5Y cells treated with CD1, but not in wild-type HepG2 cells. The precise cellular localization of CD1 was verified by co-incubating CD1 with Mito-Tracker Green (MTG, 200 nM) in live cells, which also confirmed the binding of CD1 to MAO (in situ binding). Overlapping of the green fluorescence (MTG) and the strong red fluorescence (CD1) was detected (Fig. 7C). Finally, CD1 could discriminate between the expression differences of MAO-A in numerous live cells and human glioma/para-cancerous tissues (Fig. 7D).
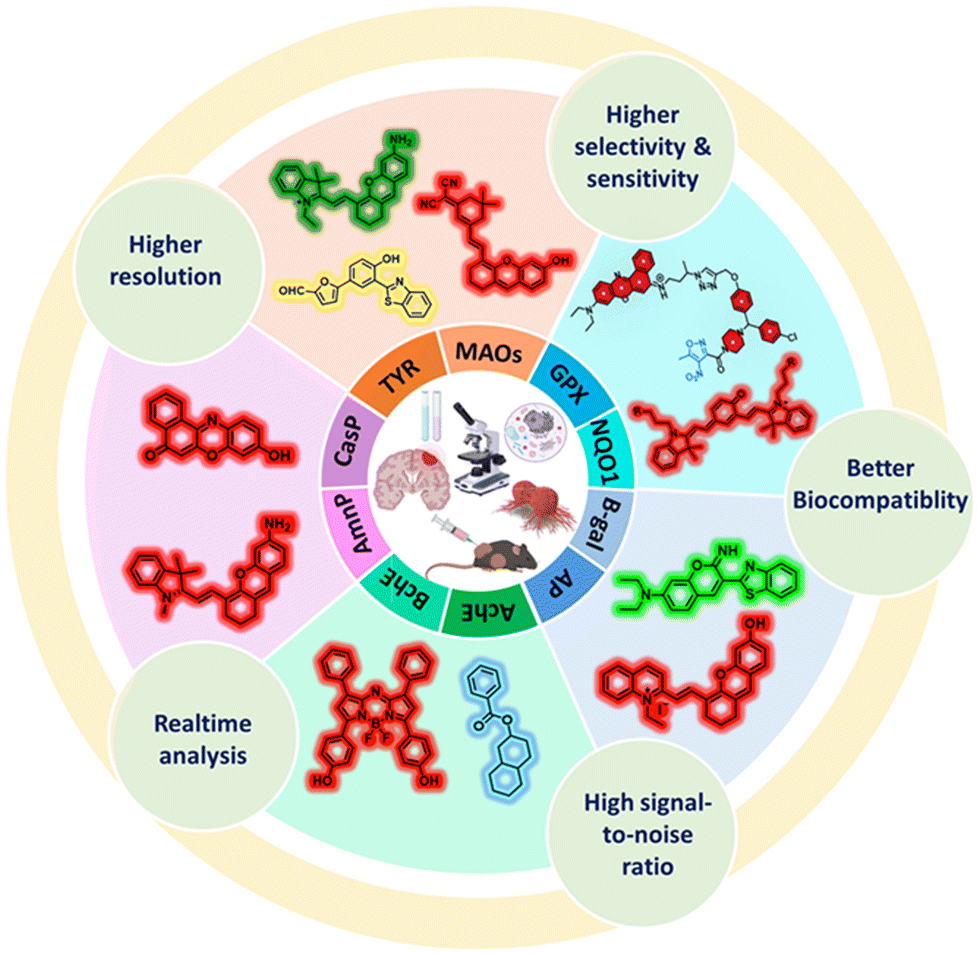
AIE-M, a red-emitting fluorescent probe that relies on the selectivity of MAO for the structural catalysis of propylamine group substrates, was successfully synthesized by Yang et al.118 Quinolone-malononitrile (QM) with AIE characteristics was used in this “turn-on” probe. The o-chloro-substituted propylamine unit functioned as the MAO-A-recognition group, while AIE-O provided the fluorescence signal for the AIE-M probe. AIE-M was involved in the process of propylamine elimination after its hydrolysis catalyzed by MAO-A to monitor the activity of MAO-A in situ. In an aqueous solution, AIE-M emitted very little fluorescence; however, inside the cell, it was broken down by MAO-A to produce AIE-O, which has AIE properties and fluoresces strongly as a result of in situ aggregation (Fig. 8A). Thus, it provides a more precise method for monitoring the real-time activity of MAO-A. Following a 120-min reaction with 50 g mL−1 of MAO-A, Stokes shift of this probe approached 170 nm (Fig. 8B). The minimum detection limit was found to be 46 ng mL−1, with a detection range of 0–50 μg mL−1.
 | ||
| Fig. 8 (A) Schematic of mechanism of AIE-M detecting MAO-A. (B) Fluorescence spectra of the probe AIE-M with MAO-A. (C) Confocal fluorescence images of cells: (a), (g) and (m), (d), (j) and (p) only SH-SY5Y and NIH-3T3 cells, respectively. (b), (h) and (n), (e) and (k) and (q) are probes AIE-M incubated with SH-SY5Y and NIH-3T3 cells, respectively. (c), (i), (o), (f), (l) and (r) Are SH-SY5Y and NIH-3T3 cells first incubated with clorgyline and then treated as in the (b), (h) and (n) group. (D) Fluorescence images of MAO-A in zebrafish. (a), (d) and (g) Zebrafish only; (b), (e) and (h) zebrafish treated with probe AIE-M; (c), (f) and (i) zebrafish pretreated with clorgyline, and then incubated with AIE-M. Yang et al.,118 Copyright 2024, Elsevier. | ||
Then, it was studied in two cell models, mouse fibroblasts (NIH-3T3) devoid of MAO-A expression and human neuroblastoma (SH-SY5Y) with significant endogenous MAO-A expression. Low cytotoxicity was observed for both cells. Although the SH-SY5Y-treated cells had a strong intracellular fluorescence, the NIH-3T3 cells did not exhibit any fluorescence (Fig. 8C). After two hours of incubation with AIE-M, red fluorescence was discovered in the head and gut of zebrafish, confirming the identification of MAO-A in live fish using AIE-M (Fig. 8D). Because AIE-M has high selectivity and little cytotoxicity in live cells and zebrafish, it may be utilized to identify MAO-A activity in real-time.
4.1.2.2. Monoamine oxidase-B. The overexpression of MAO-B in cells may damage mitochondria, increase the H2O2 and ROS levels, and create other problems linked to it.117,119–121 The identification, management, and prognosis of illnesses with elevated MAO-B expression may be greatly aided by the precise in situ tracking of MAO-B. Due to their limited specificity and tendency to react to MAO-A, the majority of MAO-B fluorescent probes on the market today are visible wavelength emitters that may readily cause photobleaching and phototoxicity issues.122–125 Consequently, their potential use in biological systems is restricted by these flaws. Therefore, to establish the role of MAO-B, it is essential to construct fluorescent probes.
Yang et al.126 created and synthesised FNJP, a red-emission fluorescent probe that selectively identifies MAO-B by using the di-cyano-methylene-di-hydro-furan (TCF) fluorescent moiety. Due to the presence of hydroxyl-substituted propylamine group in the benzene ring, the capacity of OH to push electrons decreased, which impeded intramolecular charge transfer (ICT), causing the fluorescence of the FNJP probe to be feeble. The propylamine was partly hydrolysed and eliminated when MAO-B interacted with FNJP, producing the fluorescent molecule FNJO. This increased the fluorescence intensity and triggered the ICT effect (Fig. 9A). The FNJP probe has special qualities, such as strong cell permeability, favourable reactivity (Km = 10.8 μM), and NIR characteristics (λem = 610 nm). When FJNP reacted with MAO-B, the I/I0 ratio was 28-fold (Fig. 9B).
 | ||
| Fig. 9 (A) Schematic of sensing mechanism of FNJP probe. (B) Fluorescence intensity of FJNP reacted with MAO-B. (C) (A1)–(F1) Fluorescence imaging of 5 μM FNJP probe incubated in LPS-stimulated NIH-3 T3, SH-SY5Y, and HepG2 cells at different times. (D) Deep scan imaging of mixed cultivation cells of HepG-2 (outside the green region) and NIH-3 T3 (green region). (E) Fluorescence microscopy images of MAO-B in zebrafish larvae. Yang et al.,126 Copyright 2024, Elsevier. | ||
This probe was used to investigate the expression of MAO-B in LPS-induced oxidative stress in NIH-3 T3, HepG2, and SH-SY5Y cells. When SH-SY5Y cells were triggered with LPS, it induced cellular oxidative stress, and the MAO-B level was elevated with a red fluorescence signal (Fig. 9C). This suggests that oxidative stress is related to MAO-B activity and it has a linear relationship with MAO-B and is associated with mitochondria dysfunction, neuronal apoptosis, and then brain injury. This probe showed the benefits of minimal cytotoxicity and good sensitivity and specificity. It could differentiate HepG2 cells from normal NIH-3 T3 cells in the population of cells (Fig. 9D). Additionally, this probe showed exceptional sensitivity and remarkable biocompatibility in zebrafish. Following incubation with the FNJP probe, the eyes and digestive system of zebrafish displayed a red fluorescence signal (Fig. 9E).
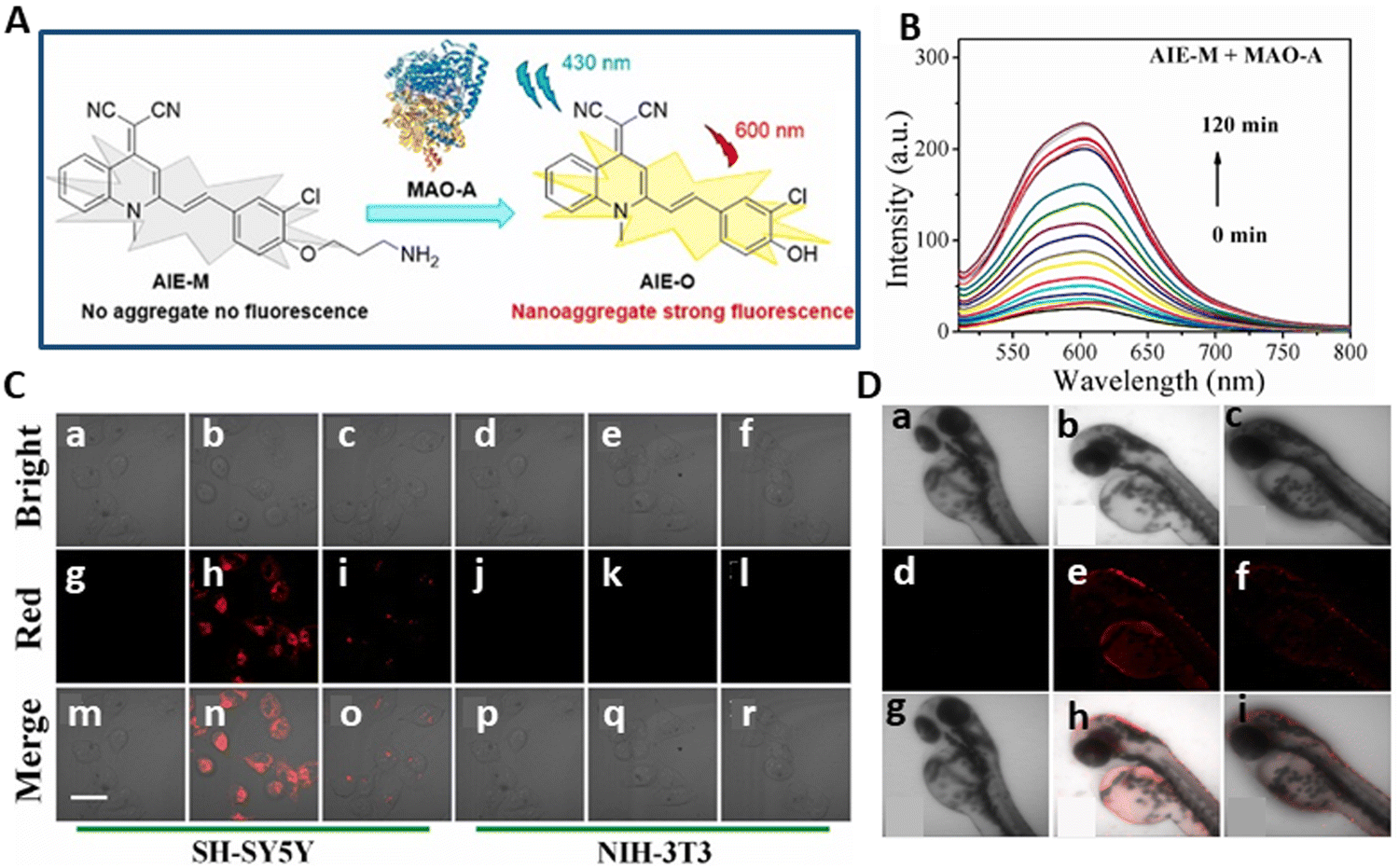
Many studies have shown that early-stage liver fibrosis patients have high blood levels of MAO-B, which is regarded as the perfect marker for the diagnosis of liver fibrosis.127,128 Consequently, the diagnosis and prevention of liver fibrosis depend on the efficient implementation of detecting changes in MAO-B. Thus, effective fluorescent probes must be created to image the alterations in MAO-B that occur during liver fibrosis. Sun et al.129 created and synthesised the YXHcy-NH2 fluorescent probe using carbamate to connect the hemi-carbocyanine fluorophore to propylamine, which serves as the recognition group. When MAO-B oxidises amine to aldehyde, the propylamine part is released by β-elimination. This step results in the elimination of CO2 and electrons and enhancement in fluorescence (Fig. 10A). It was discovered that the chemical makeup of this probe is unique to the enzyme activity centre. Presently, available research indicates that hemi-carbocyanine is in a “off” state because alanine is connected to the fluorophore by carbamate.130 When the spectral properties were further examined, it was shown that the emission wavelength of YXHcy-NH2 shifted from 600 nm to 680 nm due to the presence of recombinant human MAO-B. The corresponding fluorescence intensity increased with an increase in YXHcy-NH2, having a peak at 715 nm (Fig. 10B). This probe was highly responsive for MAO-B, with a concentration-dependent linear response of 0–10 μg mL−1 and LOD of YXHcy-NH2 for MAO-B was 0.10 μg mL−1.
 | ||
| Fig. 10 (A) Schematic of YXHCy-NH2 used to detect MAO-B changes in hepatic fibrotic cells and hepatic fibrotic mice. (B) Fluorescence emission spectra of YXHcy-NH2. (C) Fluorescence images of YXHcy-NH2 in LX-2 cells. (a) Normal LX-2 cells were cultured with YXHcy-NH2 for 100 min; normal LX-2 cells were incubated with PA for 120 min, followed by YXHcy-NH2 for 100 min; (b) TGF-β1 induced LX-2 cells were incubated with YXHcy-NH2 for 100 min; TGF-β1 induced LX-2 cells were incubated with PA for 120 min, followed by YXHcy-NH2 for 100 min. (D) In vivo imaging of MAO-B in the mice with different degrees of fibrosis. All mice were administrated with YXHcy-NH2 in DMSO/saline solution and subjected to liver injection in situ. (a) Imaging of control group mice, CCl4 was orally administered at a dose a week for 21 days and 42 days. (b) Fluorescence imaging of isolated organs in three groups of mice corresponding to (a) Sun et al.,129 Copyright 2024, Elsevier. | ||
The benefits of this probe include its excellent specificity, high detection sensitivity, and minimal toxicity. In both liver fibrosis mouse and cell (LX-2 cells) models, the activity of MAO-B could be measured using YXHcy-NH2. The findings of the experiment indicated that the intensity in LX-2 cells caused by TGF-β1 was more than that in normal LX-2 cells. This showed that the amount of MAO-B in the fibrotic liver cells induced by TGF-β was much high in comparison to normal liver cells (Fig. 10C). In the mouse model, the liver tissue fluoresced strongly during the time that CCl4 was introduced. As the experimental group, an inhibitor of MAO-B, i.e., pargyline (PA), was utilised to block the activity of MAO-B. Consequently, the level of fluorescence in the mice administered with PA injection was comparable with that of the control group, which was much lower than that of the fibrotic animals. With the exception of the liver, almost none of the separated organs, including heart, spleen, lungs, and kidneys, fluoresced (Fig. 10D). As a result, this probe can be used as a support instrument for drug screening and is a useful tool for the early identification of liver fibrosis.
Lung cancer is the most malignant cancer in the world.131 Around 80% of lung cancer patients are non-small cell lung cancer (NSCLC), with approximately 75% of patients having this disease in its medium or late stages. The antioxidant enzyme GPX4 is selenoprotein-based and has significance in the regulation of ferroptosis.132,133 It has the ability to preferentially convert lipid peroxides into lipid alcohols.134 GPX4 is overexpressed in NSCLC135 and acts as a target probe for NSCLC imaging. ENBO-ML210, an NIR fluorescent probe, was designed by Hu et al.136 for imaging NSCLC, in combination with an NIR fluorophore, Nile blue (NB), and a GPX4 inhibitor, ML210yne. Targeted imaging of GPX4 was possible due to the ability of ENBO-ML210 to attach to the protein with selectivity. This process transforms the nitrosoisoxazole-containing moiety into a nitrile electrophile, which then covalently bonds to the selenocysteine of GPX4 (Fig. 11A). The fluorescence intensity of ENBO-ML210 increased linearly in the range of 0–12 μM (Fig. 11B).
 | ||
| Fig. 11 (A) Schematic of fluorescent probe ENBO-ML210 for the detection of non-small cell lung cancer by targeting GPX4. (B) Fluorescence spectra of ENBO-ML210 as a function of its concentration in MeOH/PBS solution. (C) Cell uptake assay and time-dependent fluorescence imaging of ENBO-ML210 in H1299, LO2, 4T1, and MCF-7 cells. (D) Fluorescence imaging of tumor-bearing mice by tail vein injection of ENBO-ML210. Hu et al.,136 Copyright 2023, The Royal Society of Chemistry. | ||
Rapid membrane penetrating ability is an essential factor for bio-imaging. Real-time uptake imaging was performed in different cells. Obvious red fluorescence was seen during the imaging experiments conducted on H1299 cells, which are human NSCLC cells. In contrast, the control groups showed a minimal fluorescence signal. The H1299 cells displayed red fluorescence for a longer period and reached saturation at 1.5 h after a longer incubation period, while the 4T1 and MCF-7 cells needed 3–4 h (Fig. 11C). This demonstrated that the H1299 cells with high GPX4 expression could absorb ENBO-ML210 more quickly and maintain it for a longer period. An in vivo model of H1299 NSCLC xenograft Balb/c nude mice was employed to assess the efficacy of ENBO-ML210. After injecting ENBO-ML210 intravenously, the tumor showed distinct fluorescence when its volume approached approximately 100 mm3. The fluorescence intensity gradually increased and reached its maximum at 3 h, lasting for 12 h (Fig. 11D). According to the findings, it can be deduced that ENBO-ML210 is a selective probe to target NSCLC.
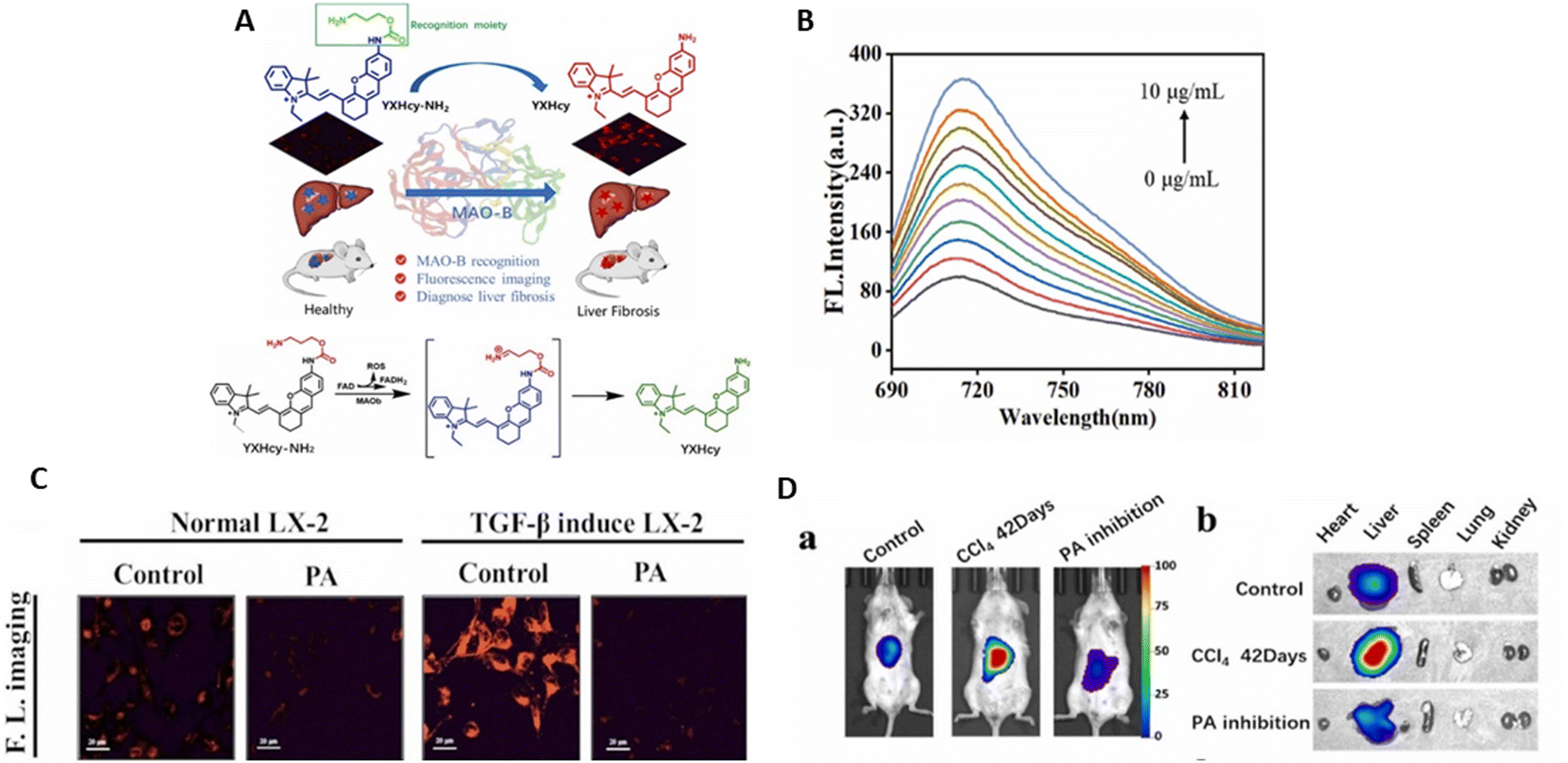
Two NAD(P)H: quinone oxidoreductase-1 (NQO1)-activated NIR fluorogenic probes, S-QCy7-NQO1 and QCy7-NQO1, were reported by Zhang et al.144 for the detection and assessment of NQO1 in cell and tumor xenograft mouse models. These probes were composed of a hydrophilic substituent (pre-QCy7) fluorophore and a tri-methyl locked quinone propionate (Q3PA) cage. Through the intramolecular PET process, Q3PA, the particular trigger group of NQO1, could efficiently suppress the fluorescence of the probe due to its electron-poor nature.145,146 Concurrently, the fluorescence of S-QCy7-NQO1 and QCy7-NQO1 diminished in the NIR spectrum owing to the pre-QCy7 skeleton breakdown of the π-electron conjugation. Under NAD(P)H, the quinone in Q3PA was reduced by NQO1 to hydroquinone; subsequently, the linker was removed and the reporter was released.147 This releasing technique produced the rebuilding of the QCy7-like structure with an enlarged π-electron system, thereby restoring the NIR signal (Fig. 12A).148,149
 | ||
| Fig. 12 (A) Proposed sensing mechanism of S-QCy7-NQO1 and QCy7-NQO1 for NQO1. (B) Comparison of the response of S-QCy7-NQO1 and QCy7-NQO1 to NQO1 and schematic of the chemical structures of S-QCy7-NQO1 and QCy7-NQO1 and their fluorescent response to NQO1. (C) Changes in the fluorescence spectra and fluorescence emission at 695 nm of S-QCy7-NQO1 with different concentrations of NQO1. (D) Fluorescence images of HT-29 cells, pretreated with dicoumarol and H596 cells after incubation with S-QCy7-NQO1. (E) In vivo time-dependent fluorescence imaging of endogenous NQO1 activity in HT-29 tumor-bearing mice after intratumoral administration of S-QCy7-NQO1 and QCy7-NQO1. Zhang et al.,144 Copyright 2022, Elsevier. | ||
The addition of two extra hydrophilic sulfonate moieties to S-QCy7-NQO1 makes the two probes different (Fig. 12B). Notably, S-QCy7-NQO1 demonstrated a favorable reaction to NQO1, displaying an impressive 105 nm Stokes shift and an increase in fluorescence by up to 245-fold at 695 nm. The fluorescence intensity practically reached the maximum when the concentration of NQO1 was extended to 0.4 μg mL−1 (Fig. 12C). The intensity at 695 nm and various NQO1 concentrations (0.0–0.16 μg mL−1) showed a linear relationship with the minimum detectable level of 0.05 μg mL−1. S-QCy7-NQO1 was very sensitive to NQO1 in vitro, and its short response time and low detection limit suggest that it can be used to track traces of NQO1 in cells or in vivo.
The addition of S-QCy7-NQO1 to NQO1-positive HT-29 cells resulted in an increase in fluorescence in the NIR channel. In contrast, the probe-treated NQO1-negative H596 and MDA-MB-231 cells displayed a negligible NIR signal. However, the fluorescence was significantly reduced when the cell groups were pretreated with a dicoumarol,150 and subsequently incubated with S-Q Cy7-NQO1 (Fig. 12D). Furthermore, a similar fluorescence level was detected with QCy7-NQO1. The potential of S-Q Cy7-NQO1 was assessed for the in situ monitoring of the NQO1 contents in vivo. To confirm the specific activity of NQO1, different sets of NOQ1-positive tumor xenografts (HT-29) were established subcutaneously. The HT-29 tumor showed strong fluorescence with S-QCy7-NQO1 injection. The intensity of the NIR signal of S-QCy7-NQO1 progressively increased with time in response to NQO1 activation. It peaked within ten minutes and was only seen at the tumor site. Alternatively, the signal in the mice given QCy7-NQO1 was significantly reduced compared to S-QCy7-NQO1 (Fig. 12E). The outcomes revealed that S-QCy7-NQO1 has great potential to activate NQO1 in vivo in real time.
4.2. Probes for glycosidase
Enzymes belonging to the class known as glycosidases are responsible for the degradation of oligosaccharides via the hydrolytic catalysis of glycosidic linkages. Because of the prominent role of these enzymes, understanding their function is essential for both the prevention and treatment of diseases. Among the various glycosidases found in mammals, galactosidase and glucosidase stand out as potentially their most crucial forms. Galactosidase is a typical indicator for illnesses or ageing, in contrast to glucosidases, which decompose complex carbohydrates such as starch and glycogen into their monosaccharide components. Galactosidases are enzymes that act as reporters to monitor the expression of genes. Although galactosidases do not have a direct correlation with cellular senescence, their reliability concerns and ease of detection make them useful biomarkers for cellular senescence and ageing.58To identify β-galactosidase activity in living systems, Lo et al.161 designed PTA-gal, an NIR fluorescent probe. The β-galactose reaction motif and PTA chromophore are used by this probe to detect β-galactosidase. The electron acceptors in PTA are pyridine cations and the donor electrons are N,N-dimethylamine groups. In the presence of β-galactosidase, PTA is formed through 1,6-elimination of the intermediate by the p-hydroxybenzyl group (Fig. 13A), resulting in the fluorescence of probe PTA-gal. The intramolecular charge transfer (ICT) effect is caused by the electron push–pull mechanism, which has a large Stokes shift. Besides β-galactosidase, the galactosidic bond of PTA-gal can be hydrolyzed, releasing the fluorescent chromophore PTA, which emits light at 606 nm. Thus, to measure the increase in fluorescence intensity, various β-galactosidase concentrations (0–5 U mL−1) were incubated with the PTA-gal probe. A direct correlation between β-galactosidase concentration and the fluorescence intensity was established (Fig. 13B). The fluorescence quantum yield was 0.30 and the LOD was 2.15 × 10−5 U mL−1.
 | ||
| Fig. 13 (A) Schematic of sensing mechanism of PTA-gal. (B) Fluorescence spectra of PTA-gal (10 μM) in the presence of different concentration of β-galactosidase. (C) Imaging studies of A2780 cells treated (a)–(c) without and (d)–(f) with PTA-gal. (g)–(i) A2780 cells pretreated with D-galactose (100 μM) for 1 h, and then treated with PTA-gal. (D) Fluorescence figures for monitoring endogenous β-galactosidase of zebrafish via PTA-gal: (a)–(c) zebrafish without any treatment. (d)–(i) zebrafish injected with A2780 cancer cells and treated with (d)–(f) PTA-gal and (g)–(i) pretreated with D-galactose. (E) Fluorescence imaging of tumor-bearing BALB/c nude mice. PTA-gal injected intratumorally into tumor-bearing mice: (a) control (before injection), (b) 30 min after injection, (c) 1 h after injection and (d) 1 h after injection; the A549 control tumor is on the left and the A2780 tumor is on the right. Lo et al.,161 Copyright 2024, Elsevier. | ||
A2780 cells (ovarian cancer cells) showed red fluorescence upon treatment with PTA-gal. When the cells were pre-incubated with D-galactose before incubation with PTA-gal, the fluorescence intensity of the probe decreased (Fig. 13C). PTA-gal was studied in biological systems by treating zebrafish to detect β-galactosidase through in vivo experiments. Zebrafish injected with A2780 cells showed red fluorescence after the addition of PTA-gal, indicating the presence of endogenous β-galactosidase, whereas a reduction in fluorescence was detected when they were pre-treated with D-galactose (Fig. 13D). BALB/c nude mice with tumors were used for the further identification of ovarian tumors. Following the intra-tumoral injection of PTA-gal into a mouse with tumor, fluorescence was detected at the right flank tumor site of A2780 cells for 30 min, while the A549 tumor (left flank) showed faint fluorescence (Fig. 13E). The results of these investigations showed that PTA-gal has strong β-galactosidase selectivity and sensitivity. Its good biocompatibility allows the detection of endogenous β-galactosidase in both living animals and A2780 cells.
Liu et al.162 created a ratiometric fluorescent probe (P1) to sense the activity of β-gal in lysosomes. The fluorophore used in its construction was cyanovinylidene dye (BMZ), which consisted of an electron donor (N,N-diethylaniline), π-conjugated bridge (cyanovinyl), electron acceptor (benzothiazole), and β-galactoside bond. The designed probe emitted excimers at 620 nm and showed the maximum absorption at 470 nm (Fig. 14A).163 Upon incubation with β-gal, there was a blue-shift in its fluorescence, with the maximum at 533 nm. This probe could sense β-gal with high accuracy, as seen by the easy capture of the evident fluorescence response of P1 when added to 0.1 U mL−1 β-gal (Fig. 14B). After being incubated with β-gal, the fluorescence spectrum of probe P1 changed, mostly due to the enzyme cleaving the glycoside bond, and then eliminating the cyclocycles intramolecularly to create a new fluorophore called PF. I533nm/I620nm and the β-gal concentration in the range of 0–1.0 U mL−1 showed a reasonable linear relationship, yielding a high correlation coefficient and a minimal detection range of 2.7 mU mL−1.
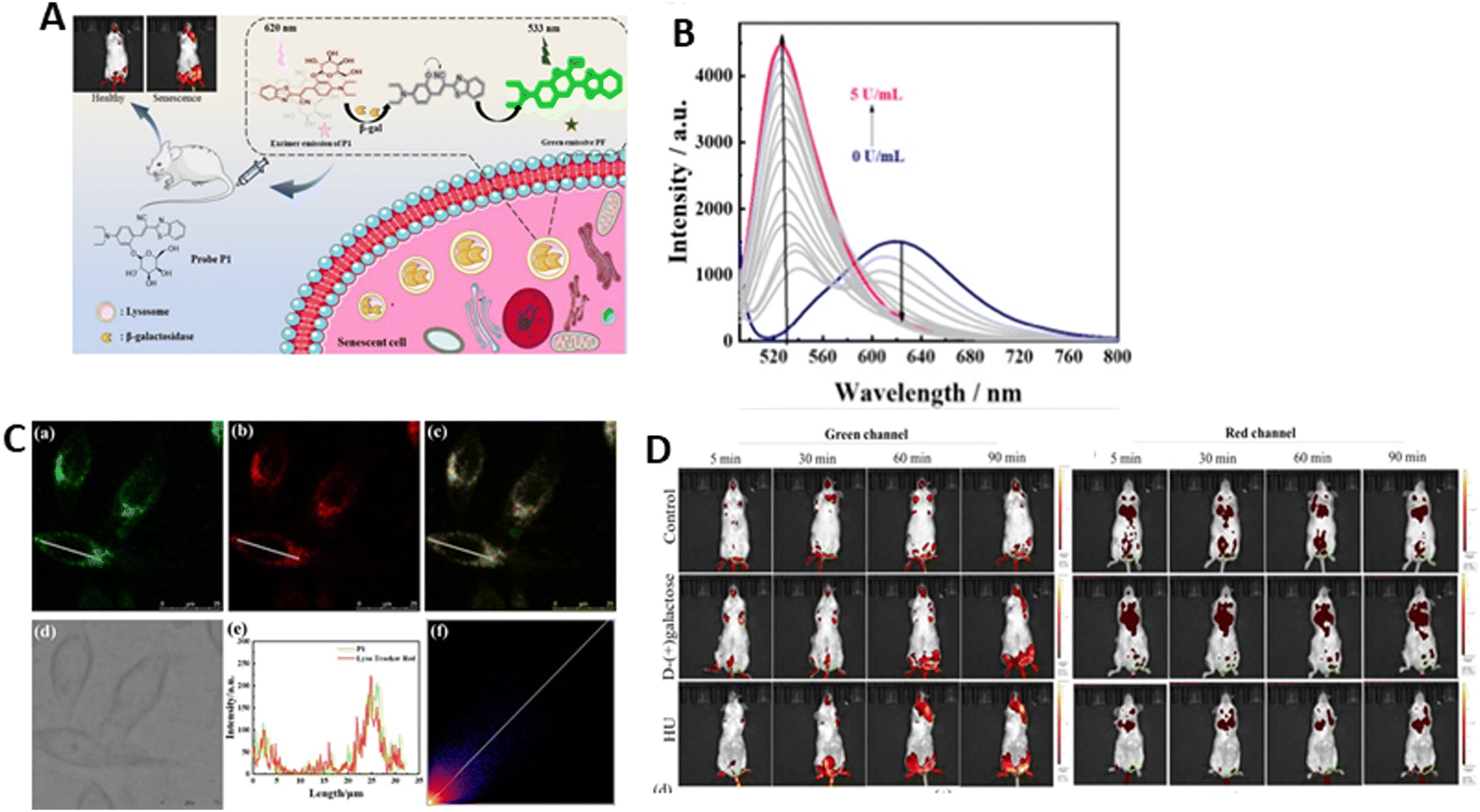 | ||
| Fig. 14 (A) Rational design of probe P1 for the detection of β-gal activity in living cells and in vivo. (B) Fluorescence spectra of β-gal concentration after incubation with P1. (C) Fluorescence localization of lysosomes in drug-induced senescent Hep G2 cells. (a) Fluorescence emission of P1 captured from the green channel; (b) fluorescence emission of LysoTracker Red captured from the red channel; (c) superposition emission of red and green channels; (d) image of bright field; (e) emission intensity of ROI on the selected cells; and (f) confocal area of the selected red and green channels. (D) Fluorescence images of the mice after the injection of P1 from green and red channel. Liu et al.,162 Copyright 2024, ACS. | ||
The subcellular localization of β-gal was detected by P1 in senescent Hep G2 cells. The overlapping green fluorescence (490–540 nm) with red emission (590–640 nm) of LysoTracker Red suggested the localization of β-gal in lysosomes. The co-localization coefficient was 0.87. Furthermore, there was a good fit observed between the P1 and commercially available red lysosome fluorescence intensity curves in the ROI (Fig. 14C). Probe P1 was used for imaging drug-induced senescence. The senescence models were found in mice produced with D-galactose. The mice were grouped into 3 categories, i.e.D-galactose (group 1), HU (group 2), and control group (PBS). The senescence groups (1 and 2) showed a clear fluorescence signal in their brain and muscle, indicating the presence of highly active β-gal in the senescent groups. The control group did not exhibit any noticeable elevation in the green channel. The heart, liver and kidney showed prominent fluorescence in the red channel (Fig. 14D). These findings demonstrate that β-gal activity can be monitored in vivo using probe P1 as a senescence indicator.
To distinguish senescence-related β-gal (SA-β-gal) from other β-gal (such as ovarian cancer), a unique fluorescent receptor, SA-HCy-1, was designed by Liu et al.164 for the accurate detection of senescence. The skeleton of the probe is made of NIR-emissive cyanine and is embellished with galactose via a self-immolative linker. It is anticipated that SA-HCy-1 will respond more quickly and with less spatial site resistance in living cells due to the addition of 4-(hydroxyl methyl) phenol, a linker between galactose and the NIR fluorescent dye HCy-OH. When the β-galactopyranoside derivative blocks the phenolic hydroxyl moiety of HCy-OH, SA-HCy-1 displays mild fluorescence in water medium. In SA-HCy-1, β-gal may induce the cleavage of the galactopyranoside group. This probe is expected to develop a phenolate intermediate once the galactose residue is removed, and then spontaneously undergo 1,6-elimination of 4-hydroxybenzyl to produce the fluorophore HCy-OH, which has a high NIR emission (Fig. 15A). It is noteworthy that the 1,6-elimination of 4-hydroxybenzyl is dependent on pH.54 The xanthene derivative HCy-XTD with cyan emission (468 nm) is produced by the oxidative cleavage of the alkene linkage of HCy-OH in the subsequent reaction with ROS (ClO− or 1O2). This significant spectrum shift made it possible to develop a great ratiometric pattern for ROS detection. Throughout the experiment, a simultaneous increase toward the β-gal level was seen, and it reached the saturated condition in 100 mU mL−1 (Fig. 15B). The limit of detection (LOD) was 0.25 mU mL−1, with a well-defined linearity within the range of 0–30 mU mL−1.
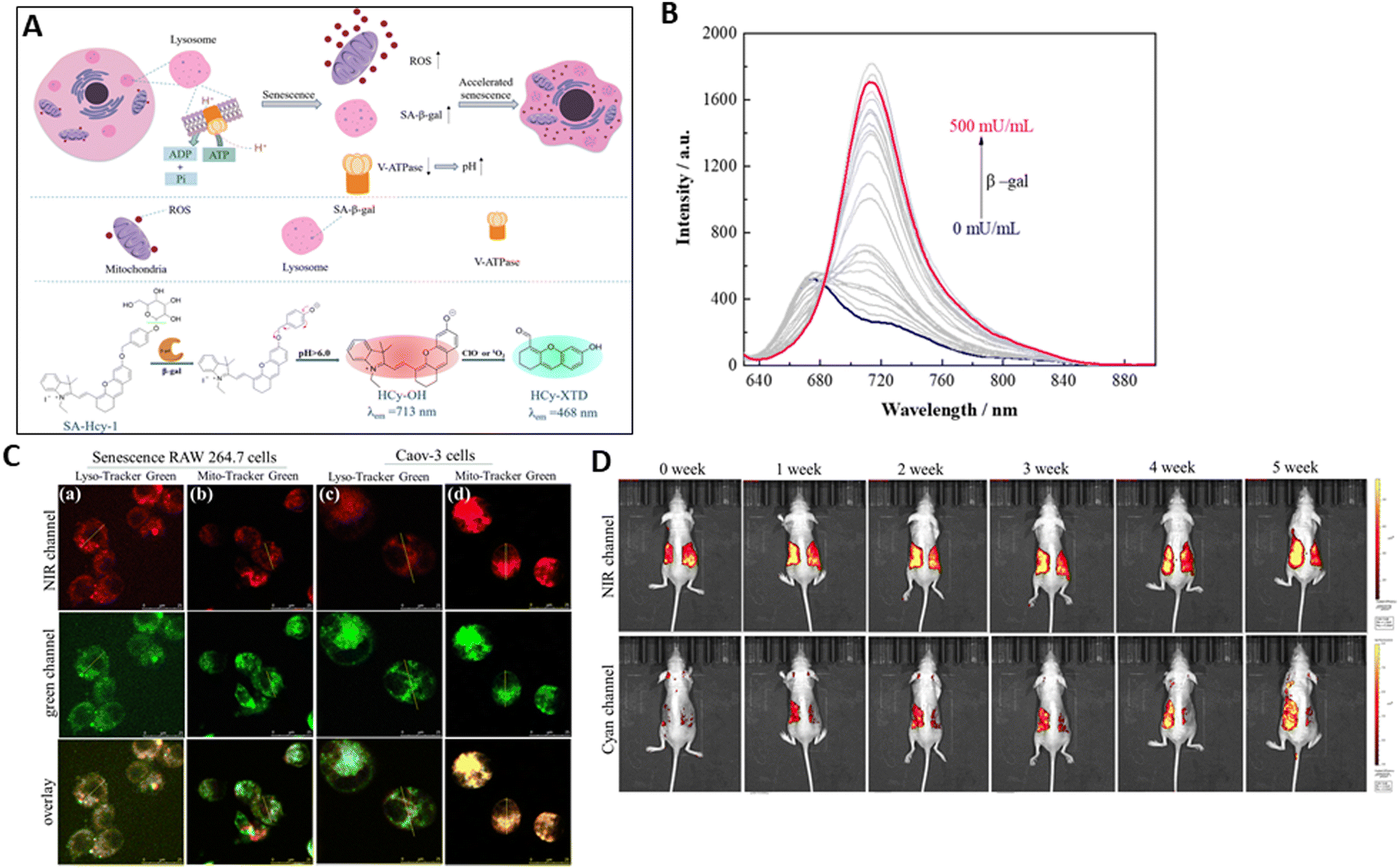 | ||
| Fig. 15 (A) Schematic of signaling pathways for cellular senescence and proposed sensing mechanism of SA-HCy-1 probe for β-gal and ROS in a high lysosomal pH environment. (B) Fluorescence spectra of SA-HCy-1 upon incubation with increasing concentration of β-gal. (C) Fluorescence co-localization images of senescence RAW 264.7 cells (a) and (b) and Caov-3 cells (c) and (d). (D) Fluorescence imaging of the mouse model of ultraviolet light-induced skin photo-damage from 0 to 5 weeks. Liu et al.,164 Copyright 2024, The Royal Society of Chemistry. | ||
The discriminative power of probe SA-HCy-1 probe against tumour cells and senescence cells, in particular ovarian cancer cells (Caov-3 cells) was investigated. SA-β-gal activity was primarily found in lysosomes. This was mostly because Caov-3 cells had distinct sites for the β-gal protein. These co-localization findings suggested that lysosomal β-gal in senescence cells and mitochondrial β-gal in Caov-3 cells may particularly light up the NIR emission of SA-HCy-1 (Fig. 15C). To assess skin senescence in a mouse model of skin photo-aging, the mice were coated with 3% isoflavone (to prevent skin aging) and sesame oil, a pure substrate as a reference, on each side of the bare back, and then exposed to UV irradiation for varying intervals (Fig. 15D). The imaging was monitored for 5 weeks following in situ treatment, which indicated that during the irradiation, the content of SA-β-gal and ROS increased, leading to skin photo-aging with overexpressed SA-β-gal and ROS. Thus, it was shown that the SA-HCy-1 receptor could detect ROS and SA-β-gal via the cyan and NIR channels, respectively. SA-HCy-1 simultaneously analysed ROS, SA-β-gal activity, and the surrounding microenvironment, enabling it to track senescence and assess the impact of anti-aging medications. Furthermore, in vivo investigations have demonstrated its tremendous potential in assessing UV-induced skin photo-aging and anti-aging reagents.
4.3. Probes for phosphatase
During the process of drug discovery and development, phosphatases are frequently regarded as the “black sheep” or “strangers” of phosphorylation-mediated signal transduction. Together with kinases, phosphatases control the equilibrium of the physiological phosphorylation state by removing the phosphate groups from biomolecules. The regulation of phosphorylation and dephosphorylation is extremely critical for therapeutic targeting, given that an imbalance in this process can result in the development and progression of a diverse array of disorders. Kinases and phosphatases play critical roles.165Clinical investigations have shown that serum ALP is a useful biomarker of liver function. ALP is reportedly highly released into the bile and primarily overexpressed at the apical region of hepatocytes and bile duct cells. To assess liver health more accurately, a novel method that concentrates on monitoring the level of ALP and its localization in liver tissue will be preferable, particularly for the early detection of cholestatic liver injury (CLI). Therefore, Chen et al.171 produced TX-PS, an ICT-based activatable NIR fluorescent probe with a large Stokes shift (198 nm), which demonstrated excellent sensitivity and specificity for the detection of ALP. This probe was utilized for the early diagnosis of CLI and screening of ALP inhibitors or CLI drug candidates. Dicyanoisophorone was conjugated with a hydroxyl-substituted coumarin derivative to yield TX-OH, a typical ICT-based biocompatible fluorophore with comparatively long emission wavelength (720 nm) and high quantum yields (9.5%). To build the target probe TX-PS, the phosphate group was chosen as the ALP recognition site, and TX-OH was used as the fluorescence reporter. The phosphate ester link was broken upon binding to ALP, releasing the fluorophore TX-OH (Fig. 16A). The addition of ALP resulted in a stunning “turn-on” fluorescence, with a 92-fold increase in fluorescence intensity at 720 nm and a clear red-shift with absorption in the range of 425 nm to 520 nm. Meanwhile, the change in the colour of the TX-PS solution was dependent on ALP. Under white light, it turned pink, while under 365 nm irradiation, it turned orange-yellow. An increase in the ALP concentration (0–100 U L−1) showed a strong linear correlation with variations in fluorescence intensity (Fig. 16B), which implies that TX-PS is capable of quantitative detection. It was determined that its LOD may be as low as 0.079 U L−1, indicating that TX-PS is sensitive to ALP.
 | ||
| Fig. 16 (A) Graphical representation of TX-PS involved in the diagnosis of cholestatic liver injury and sensing mechanism of TX-PS. (B) Fluorescence spectra of TX-PS (10 μM) upon addition of ALP (0–100 U L-1). (C) Confocal images of ALP activities in 293 T, HeLa, A549, and HepG2 cells stained with TX-PS. (D) Fluorescence images of HepG2 cells co-stained with TX-PS (10 μM) and Hoechst 33342 (5 μg mL−1) and Lysotracker Green (1 μM) or MitoTracker Green (1 μM). (E) Fluorescence imaging of ANIT-treated HepG2 cells and remediation: (a1) cells incubated with TX-PS; (a2)–(a4) cells pretreated with ANIT (500 μM) for 2 h (a2); and then incubated with UDCA (500 μM) (a3) or PDN (500 μM) (a4) for 2 h before incubation of TX-PS (10 μM, 30 min). (F) Confocal imaging of zebrafish incubated with TX-PS. Chen et al.,171 Copyright 2024, Elsevier. | ||
The capacity of TX-PS to image and identify ALP in cancerous (HeLa, A549, and HepG2 cells) and normal cells (293 T) was studied. ALP is overexpressed in cancer cells and HepG2 cells (human hepatoma cell) exhibited the strongest fluorescence signals compared to normal cells (Fig. 16C). HepG2 cells were co-stained with TX-PS, Hoechst 33342, LysoTracker Green, and MitoTracker Green. As demonstrated in Fig. 16D, the green channels of Lysotracker Green and MitoTracker Green and red fluorescence of TX-red PS overlapped extremely well, with co-localization coefficients of P = 0.93 and 0.99, respectively. These findings suggested that in living cells, the activated TX-PS was found in the lysosome and mitochondria. Additionally, the pretreated cells showed enhanced fluorescence with α-naphthylisothiocyanate (ANIT) drug for cholestasis. The cells pretreated with ursodeoxycholic acid (UDCA), a drug used for treating cholestasis, showed a decreased fluorescent signal in comparison with that pretreated with prednisone (PDN), a drug for treating cholestasis, suggesting the efficacy of UDCA towards treating cholestasis (Fig. 16E). After incubation with TX-PS for 60 min, the fluorescent signals in zebrafish were primarily seen in the intestine region, indicating that the intestine has an abundance of ALP and TX-PS can be metabolized through the enteric canal (Fig. 16F). Furthermore, the use of TX-PS allowed considerable differences in the dynamic fluctuations of the ALP levels in liver tissue, where the ALP levels in the CLI tissues increased significantly and clearly decreased following treatment. This probe offers a precise method for the early diagnosis of CLI and is regarded as a useful tool for screening ALP inhibitors or CLI medications.
Wang et al.172 developed and manufactured QX-P, a water-soluble NIR fluorescent that connects a phosphate to the hydroxyl group of QX-OH. Quinolinium serves as a substitute for indole in hemicyanine to create the fluorophore QX, which exhibits longer absorption and emission wavelengths than the traditional hemicyanine dye. Quinolinium has a larger conjugated structure and strong water solubility. Given that the phosphate group prevents the intramolecular charge transfer (ICT) phenomenon, QX-P itself is non-fluorescent. The phosphate component of QX-P hydrolyzes and releases the hydroxyl group upon combining with ALP (Fig. 17A). This initiates the ICT process again and produces the brightest fluorescence signal at 770 nm. An increase in fluorescence intensity by approximately seven times was detected when the ALP activity increased from 0.0 to 1.0 U mL−1 (Fig. 17B). The LOD was 0.017 U mL−1, and the fluorescence intensity and ALP activity showed a linear relationship in the range of 0.05–1.0 U mL−1. This suggests that QX-P has high sensitivity for quantitatively detecting ALP activity.
 | ||
| Fig. 17 (A) Design of probe QX-P for ALP. (B) Fluorescence spectra of QX-P (10 mM) in the presence of various activities of ALP. (C) Fluorescence images of mice from the normal group, diabetes group, and treatment group after injection of QX-P (200 mM) with time. (D) Fluorescence images of major organs, including the liver, heart, spleen, lungs, and kidneys and fluorescence images of blood from the mice. Wang et al.,172 Copyright 2021, The Royal Society of Chemistry. | ||
For the in vivo fluorescence imaging of ALP activity, the QX-P probe was utilized. The efficacy of this probe to visualize ALP activity in normal mice was demonstrated by the observation that the fluorescence intensity in the mice in the normal group increased progressively with time. The fluorescence signal was comparatively greater in the diabetic mice administered with streptozotocin compared to the normal mice. This suggests that in diabetic mice, the activity of ALP is elevated. The downregulation of ALP caused a considerable reduction in fluorescence signal in the animals following metformin treatment (Fig. 17C). In the case of diabetic patients, significant ALP levels accumulated in their liver and kidney. The kidneys and liver of the mice with diabetes showed comparatively more fluorescence than the other organs, whereas the mice without diabetes showed comparatively less fluorescence. In addition, compared to the diabetic mice, the liver and kidneys of the therapy group mice showed a reduced fluorescent signal. When metformin was administered to the diabetic mice, the fluorescence signal was reduced, but it was stronger in the diabetic mouse blood (Fig. 17D). This finding suggests that QX-P may be used to track ALP activity in the course of treating and diagnosing diabetic mice.
4.4. Probes for cholinesterase
The enzyme cholinesterase (ChE) is generally classified into two categories, true cholinesterase and pseudocholinesterase. Acetylcholinesterase (AChE), which is also known as true choline esterase, is primarily found in the synaptic region of cholinergic neurons, choline nerve endings, and red blood cells. Although it is found in trace levels within muscle and brain tissue, pseudocholinesterase, sometimes referred to as butyrylcholinesterase, is mostly found in the serum and liver. By breaking down acetylcholine between cholinergic synapses, AChE, a crucial enzyme in biological nerve transmission, may disrupt the neurotransmitter process. It can enhance neuronal growth and nerve regeneration and exhibits carboxypeptidase and aminopeptidase action. Butyrylcholinesterase (BChE), belonging to the serine hydrolase family, is distributed throughout the human body. Lipid metabolism and several human disorders, including liver damage and metastases, diabetes and AD have been linked to BChE. The three-dimensional structures of human BChE and AChE are quite similar, sharing 55% of their sequence. One key distinction is the precise substrates on which they interact.1Oxidative stress in cells is believed to be significantly influenced by the activity of AChE and it has been shown to be a contributing factor for cancer,189 cardiovascular disease,190,191 and inflammatory diseases.192,193 Two NIR fluorescent probes, SNCN-AE and SNC-AE, were created by Wei et al.194 to track AChE activity; these probes combine ESIPT and ICT methods (Fig. 18A). The electron acceptor (the dicyanoisophorone moiety, CN) was conjugated with the conventional ESIPT fluorophore 2-(2′-hydroxyphenyl) benzothiazole (SN-1) to construct the NIR fluorophores (SNCN and SNC) and obtain a significant Stokes shift and extended emission wavelength. Dimethyl carbamate was used as the recognition unit for the construction of the SNCN-AE and SNC-AE probes. Dimethyl carbamate exhibited strong fluorescence quenching and blocked ESIPT and ICT in the absence of AChE by hindering the phenolic hydroxyl group. In the presence of AChE, dimethyl carbamate is recognized by the hydrolytic centre of AChE, which leads to cleavage of the ester bond to generate a free fluorophore. Afterwards, the phenolic hydroxyl group and malononitrile moiety have a strong electron push–pull action, which improves the ICT process and may further boost the ESIPT process, resulting in a spectacular fluorescence signal. This probe showed a linear increase with the AChE concentration at 710 nm, which was 243-fold stronger than its original state (Fig. 18B). The emission intensity was strongly correlated with 0–50 U mL−1 AChE. The lowest concentration that could be detected was 0.13 U mL−1.
 | ||
| Fig. 18 (A) Molecular structures of SNCN-AE and SNC-AE probes and their sensing mechanism for detecting AChE. (B) Fluorescence spectra of SNCN-AE with different concentrations of AChE. (C) Fluorescence imaging of cells incubated with SNCN-AE probe (10 μM) and pretreated with AChE inhibitor neostigmine with different concentrations. (D) Confocal fluorescence images of endogenous AChE. Control group (a) treated with SNCN-AE probe. Oxidative stress group (b) treated with glutamate, and then SNCN-AE. Inhibition group (c) pretreated with glutamate, and neostigmine, then SNCN-AE. (E) Fluorescence imaging of AChE in zebrafish: (a) zebrafish incubated with probe SNCN-AE; (b) zebrafish treated with neostigmine followed by the addition of SNCN-AE; (c) zebrafish treated with glutamate followed by the addition of SNCN-AE for 2 h; and (d) zebrafish incubated without probe. Wei et al.,194 Copyright 2023, Elsevier. | ||
Rat adrenal chromaffin tumour cells (PC12) have been shown to overexpress AChE.195 A red fluorescence signal was seen when PC12 cells were treated with SNCN-AE, and this signal was positively correlated with the length of incubation. Nevertheless, pre-treating PC12 cells with neostigmine (an AChE inhibitor) decreased the intensity of the fluorescence (Fig. 18C). Moreover, AChE expression in live cells under oxidative stress was monitored using SNCN-AE. Following glutamate pre-treatment, SNCN-AE-loaded PC12 cells showed noticeably increased fluorescence signal, indicating that greater amounts of endogenous AChE were found in glutamate-stimulated cells. However, a significant drop in intensity was seen when the cells were pre-treated with glutamate followed by neostigmine, indicating that the shift was, in fact, caused by the activity of AChE (Fig. 18D). Consequently, under oxidative stress, AChE activity was monitored using SNCN-AE. Zebrafish were treated with SNCN-AE for the purpose of detecting AChE activity in vivo. This resulted in weak red fluorescence, but neostigmine did not produce any fluorescence signal. Alternatively, a significant increase in NIR fluorescence was seen when glutamate was pretreated (Fig. 18E). Consequently, the results verified that SNCN-AE demonstrated superior in vivo detection capabilities and was effectively used to observe AChE fluctuations in oxidative stress under external stimulation and track its baseline levels in cells and zebrafish.
AChE plays a major role in the choline alterations that occur with ageing. Dietary restriction (DR) is a modest intervention that possesses antiaging properties in addition to a positive role on the neuroendocrine system. He et al.180 described the use of an NIR fluorescent probe, BD-AChE, as a chemical bio-imaging tool to scan and detect AChE in live cells and in vivo, with the aim to investigate the relationship between DR and AChE. The relationship between the AChE level and DR in ageing mice was well shown by BD-AChE, the first probe reported for imaging AChE in aged DR animals. The fluorophore unit was BODIPY dye, which produces a fluorescence signal with an emission wavelength in the near-infrared spectrum. The dimethyl carbamate group, or recognition moiety, was combined with the fluorophore unit. The dimethyl carbamate group has binding affinity to the AChE active site at the serine residues. This carbamylation of the serine in the active site of AChE causes AChE to become inactive. Because the carbonyl group of BD-AChE is attached to an oxygen atom and the dimethyl carbamate group suppresses the emission of BODIPY via the PET, BD-AChE exhibited modest fluorescence. Following the recognition of the dimethyl carbamate by AChE in BD-AChE, the ester link is broken, converting BD-AChE into free BODIPY and enhancing its fluorescence (Fig. 19A). With peaks centred at 740 nm, the associated fluorescence emission profiles increased (Fig. 19B). Also, the quantum yield of BD-AChE increased from 0.03 to 0.27. The fluorescence intensity of AChE and BD-AChE showed a linear relationship in a concentration-dependent manner in the range of 0–20.0 U mL−1. The calculated detection limit towards AChE, which was 0.21 U mL−1, showed the high sensitivity of the BD-AChE probe to AChE under simulated conditions.
 | ||
| Fig. 19 (A) Detection mechanism of BD-AChE towards AChE. (B) Variations in the fluorescence emission spectrum of BD-AChE with a wide range of AChE concentrations. (C) Confocal microscopy images of PC12 cells for measuring AChE levels in living cells: (a) AChE imaging with BD-AChE in PC12 cells at different time points as the control. (b) PC12 cells were subjected to pre-treatment with neostigmine. (c) PC12 cells were treated as described for the control after AChE transfection. (D) Imaging of AChE levels in the brains and brain slices of mouse aging models according to distinct ages: 1, 6, 12 and 18 months old. The mice in the four groups were pre-treated with intracranial injection of BD-AChE. (E) Effects of DR on the AChE levels in the brains of aging mice: imaging of AChE levels in the brains and brain slices of mice after short-term 24 h fasting. All the aging mice were treated with BD-AChE by intracranial injection. (F) Effects of DR on the AChE levels in the brains of aging mice: imaging of AChE levels in the mice brains and brain slices in control group after continuous supply of food and after long-term DR (supplied with food on alternate days for 1, 2 and 3 months, respectively). He et al.180 Copyright 2021, The Royal Society of Chemistry. | ||
BD-AChE was used to identify AChE in live brain cells because of its exceptional selectivity towards AChE. BD-AChE was incubated with PC12 cells prior to fluorescence imaging. Because neostigmine inhibits AChE, the fluorescence in the control group steadily increased, while the fluorescence intensity of the neostigmine-treated cells decreased. In the PC12 cells transfected with AChE-RNA, the fluorescence intensity was higher (Fig. 19C). The variations in AChE levels in cell ageing models were examined. When the mice were pre-treated with BD-AChE, the fluorescence intensity within the brain decreased with age, indicating a reduction in AChE levels (Fig. 19D). Subsequently, the fluorescence imaging of brain slices was demonstrated. These results were sufficient to explain that BD-AChE efficiently tracked the AChE level in vivo, but they also showed that the AChE level fell with advancing age. Additionally, the impact of DR on the AChE levels in the brains of aged mice was investigated. The AChE alterations were identified by pretreating the mice with BD-AChE in the 24-h deprived group (18 months) and in the control group with continuous food supply. In these two groups, no discernible changes in fluorescence were seen in the brain (Fig. 19E). To verify the amount of AChE, the brain slices were imaged using fluorescence. Subsequently, it was studied in mice that had received long-term DR. The fluorescence intensity of the DR group decreased in comparison with the control in the third month of food restriction in mice, and fluorescence imaging of the brain slices further supported this finding (Fig. 19F). The findings demonstrated the close relationship between DR and AChE and the decline in AChE after long-term DR.
The “turn-on” probe TCFPB-AChE with characteristics of AIE demonstrating great selectivity, sensitivity, cell permeability, and photo-stability was described by Xiang et al.185 TCFPB-AChE was built based on the tricyanofuranyl imino-benzaldehyde core (TCFIS), which is recognised for its ability to function as an AIEgen. Additionally, dimethyl carbamate, when coupled to a benzylic linker containing an alkylated ammonium group, acts as an AChE-responsive cage unit, camouflaging the hydroxyl group in the parent core. TCFPB-AChE, when caged, was non-emissive in PBS and had a mild ICT impact. A TCFIS core that was highly emissive was produced in PBS with the ICT effect by AChE-induced selective cleavage of the dimethyl carbamate group, followed by spontaneous 1,6-elimination (Fig. 20A). The emission signal increased linearly with the addition of AChE (0–0.2 U mL−1). A turn-on response of 8.7-fold was detected with the concentration of 0.2 U mL−1 AChE (Fig. 20B). The detection limit of AChE fell with long-term DR and was determined to be 0.009355 U mL−1. Among the fluorescent probes that have been shown to respond to AChE, this one has the lowest value.
 | ||
| Fig. 20 (A) Structure of TCFPB-AChE and its activation mechanism. (B) PL spectra of TCFPB-AChE (5 mM) after incubation with AChE. (C) Confocal images of U87MG cells. First row: cells untreated with TCFPB-AChE; second row: cells treated with TCFPB-AChE; and third row: cells pretreated with 1 mM donepezil, and then incubated with TCFPB-AChE. (D) Fluorescence images of Alzheimer's disease (AD) mouse brain tissues obtained by treatment with TCFPB-AChE + donepezil and with TCFPB-AChE only. (E) Time-dependent fluorescence image of AChE in live mice using TCFPB-AChE probe, upper row: mice were injected with TCFPB-AChE intracranially and then injected with PBS and lower row: mice were intracranially injected with TCFPB-AChE, and then injected with AChE. Xiang et al.,185 Copyright 2022, The Royal Society of Chemistry. | ||
The in vitro model used for imaging was the U87 glioblastoma cell line, given that AChE is overexpressed in these cell lines. Compared to the cells that were not treated, the TCFPB-AChE-incubated cells showed a notable fluorescence in the red channel. When the cells were treated with donepezil, the fluorescence intensity significantly decreased. These findings showed that endogenous AChE in malignant U87 cells triggered TCFPB-AChE, which may be utilised to observe variations in intracellular AChE levels (Fig. 20C). The red emission was strong when TCFPB-AChE was incubated with brain tissues from AD mice, suggesting that TCFPB-AChE was stimulated in the AD brain. In brain tissues treated with donepezil, the fluorescence signal was observed to be greatly diminished (Fig. 20D). For in vivo imaging, the brains of living mice were used. The mice were pre-injected with TCFPB-AChE and divided into two groups, i.e., one with PBS and other with AChE. A notable low signal was detected in the PBS-injected group, whereas a significantly stronger signal was detected in the AChE group with respect to time (Fig. 20E).
Yu et al.208 developed a bioinspired probe called PM1 that is highly selective for BChE. PM1 was synthesized with benzoyl as the recognition moiety, given that it is hydrolyzed by BChE more efficiently than AChE (9.9-fold). PM1 was created by enclosing a fluorophore with the benzoyl group, taking inspiration from the BChE-catalyzed hydrolysis of certain substrates containing the same group under physiological conditions. For the purpose of developing a BChE probe that is resistant to AChE interference, the fluorophore hydroxyl styrylpyridinium was chosen in conjunction with the large benzoyl group. Benzoylcholine, cocaine, and tetracaine, which all include a benzoyl group in their structure, are widely recognized as particular substrates of BChE under normal physiological circumstances. The hydrolysis particularly catalyzed by BChE was demonstrated using benzoyl choline as the substrate.209 Experimental probes PM2–5 were also developed, which exhibited lower specificity towards BChE due to their smaller recognition groups. Benzoylcholine and butyryl-cholinesterase produce a complex when the –OH of serine in the esterification site of butyryl-cholinesterase forms a covalent bond using the carbonyl carbon of benzoylcholine. Subsequently, butyrylcholinesterase, choline, and benzoic acid will dissolve from the complex (Fig. 21A). Nevertheless, AChE cannot hydrolyze this substance. Under physiological conditions, BChE may be detected using probe PM1. There was a progressive decrease in the emission band, which was previously at 466 nm by adding varying concentrations (0 to 1 U mL−1) of BChE to PM1. Concurrently, the conversion of PM1 to PMOH resulted in the appearance of a new peak at 522 nm (Fig. 21B). The LOD of 0.05 U mL−1 demonstrates its sensitivity for BChE detection.
 | ||
| Fig. 21 (A) Structure of BChE specific probe PM1. (B) Bioinspired design of BChE-specific probe. (a) Specific substrates of BChE in the physiological environment. (b) Schematic of BChE and AChE hydrolyzing substrate benzoylcholine. (c) Bioinspired design of PM1 probe and its activation for detecting BChE. (C) Concentration-dependent reaction of PM1 with BChE (0–1 U mL−1). (D) Fluorescence imaging of BChE in live cells. First row (Probe): MCF-7 cells + PM1–PM5. Second row (+donepezil): MCF-7 cells + donepezil + PM1–PM5. (E) MCF-7 cells were treated with different concentrations of neostigmine + PM1 and iso-OMPA + PM1. (F) Interaction between Aβ and BChE in living cells. (a) MCF-7 cells were treated with different treatments for three days, and then incubated with PM1. (b)–(d) MCF-7 cells were treated with different concentrations of hydrogen peroxide, oxygen, and PMA, and then incubated with PM1, respectively. Yu et al.,208 Copyright 2024, Elsevier. | ||
Probes PM2–5 were employed as the control in live cells to determine the specificity of PM1 to BChE. The MCF-7 cells pre-treated with donepezil (AChE-specific inhibitor) showed no noticeable fluorescence changes with PM1, whereas the control groups showed considerable changes, particularly PM2–5. PM1 was unresponsive to the reduction in AChE level caused by an AChE-specific inhibitor (Fig. 21C). In these cells, the fluorescence signal of PM1 was shown to be diminished when neostigmine and tetraisopropyl pyrophosphoramide (iso-OMPA) were utilised as BChE inhibitors (Fig. 21D). An essential characteristic of AD is the emergence of Aβ senile plaques.210,211 The cholinergic neurons can be damaged by Aβ, which can induce the proliferation of glial cells (BChE is a secretory product of glial cells), and thereby increase the activity of BChE.212,213 The fluorescence intensity increased and the BChE level was elevated in the MCF-7 cells during pretreatment with varying amounts of Aβ. The fluorescence signals were significantly diminished due to the BChE inhibitor, suggesting that the up-regulation of BChE in living cells may be triggered by the formation of Aβ in AD. Additionally, the level of BChE was examined in MCF-7 cells that were stained with PM1 under conditions of minimal oxygen, H2O2, and PMA concentration. The results indicated an increase in the fluorescence signals (Fig. 21E). Therefore, PM1 can be considered a reliable tool to detect BChE in AD pathogenesis.
Recent research has indicated that the BChE levels are consistently raised in non-alcoholic fatty liver disease (NAFLD) patients.214–217 Thus, to quantify and image BChE in vitro and in vivo, Guo et al.218 synthesized a series of NIR fluorescent probes, FRBN-B, NF-SB, and NF-B, all produced using benzorhodol. After being incubated with BChE, FRBN-B and NF-SB emitted high FR/NIR fluorescence. These two probes exhibited a relatively low detection limit and exceptional selectivity for BChE. BChE cleaved the cyclopropanecarbonyl groups of FRBN-B and NF-SB, resulting in activated fluorescence in the spiro ring-opening process (Fig. 22A). FRBN-B and NF-SB showed a considerable improvement in fluorescence signal when the concentration of BChE was in the range of 0.015 to 0.15 U mL−1. After half an hour, the BChE-catalyzed reaction caused FRBN-B and NF-SB to display substantial fluorescence, reaching a peak at 642 nm for far-red emissions and 671 nm for near-infrared emissions (Fig. 22B and C), respectively. In contrast, NF-B exhibited little fluorescence activation, which is most likely because of the dual-lock of cyclopropanecarboxylate. Both probes demonstrated a stable linear relationship in the range of 0.015–0.15 U mL−1 for BChE. They were shown to be extremely sensitive to BChE, with FRBN-B exhibiting an LOD of 7.2 × 10−3 U mL−1 and NF-SB exhibiting an LOD of 1.9 × 10−3 U mL−1. Then, utilizing tacrine as the model inhibitor, the possibilities of the probes in evaluating the efficacy of BChE inhibitors were examined. The addition of BChE inhibitor (tacrine) led to a diminished emission from the probes.
 | ||
| Fig. 22 (A) Fluorescent probe design and mechanism of FRBN-B and NF-SB for BChE detection. (B) Fluorescence spectra of a mixture containing FRBN-B and various concentrations of BChE. (C) Fluorescence spectra of a mixture containing NF-SB and various concentrations of BChE. (D) HeLa cell imaging of FRBN-B with (a) endogenous and (b) exogenous BChE and (c) tacrine. (E) Confocal imaging of part of the liver of zebrafish using FRBN-B with (a) normal zebrafish and (b) NAFLD zebrafish. (c) NAFLD zebrafish were injected with tacrine, and then injected with FRBN-B. Guo et al.,218 Copyright 2024, The Royal Society of Chemistry. | ||
By employing the two probes, the activity of BChE was observed in HeLa cells. The fluorescence of FRBN-B was due to the endogenous BChE content in HeLa cells. The addition of 1 U mL−1 BChE increased the fluorescence intensity, which verified that the activity of BChE initiated the signal. However, the fluorescent signal of FRBN-B in live cells was significantly reduced after incubation with tacrine, which suppressed the intracellular activity of BChE (Fig. 22D). In addition, zebrafish with NAFLD were employed to monitor the BChE activity in vivo using this probe. The liver tissue of the normal zebrafish showed slight red fluorescence with FRBN-B treatment due to the low level of BChE activity. Alternatively, the fluorescence signal of FRBN-B was much stronger in NAFLD zebrafish, suggesting that disease-associated BChE activity levels were higher. Crucially, a comparable decrease in fluorescence intensity showed that treating NAFLD zebrafish with tacrine may reduce this effect (Fig. 22E). Thus, the use of the FRBN-B probe is an active method for imaging BChE intracellularly in live HeLa cells and measuring the BChE activity levels in normal and NAFLD zebrafish liver tissues.
Certain investigations219–221 have indicated that BChE is a feasible biomarker for the diagnosis of diabetes. To track the activity of BChE, Yang et al.222 created the CHQ-B “turn-on” fluorescent probe, which was generated from dihydroxanthene. This probe showed NIR excitation at 715 nm and emission maximum at 775 nm. This probe is composed of CHQ as the NIR fluorophore and a cyclopropanecarbonyl (a recognition group) that is specifically cleaved by BChE.196,202,206 When BChE hydrolyzed CHQ-B, the UV-vis absorption maximum of the testing solutions changed from 560 nm to 715 nm, which was red-shifted, and their color clearly changed from purple to blue. Similarly, excitation at 715 nm greatly increased the fluorescence emission at 775 nm (Fig. 23A). After CHQ-B was hydrolyzed, which was catalyzed by BChE, it exhibited the same bands as CHQ. Within the D–π–A structure of CHQ, the –OH group acts as a potent electron donor, promoting the intramolecular charge transfer (ICT) action of CHQ and producing an increased fluorescence emission as well as a red shift in the absorption maximum. In the presence of BChE, the fluorescence spectrum of CHQ-B was very similar to that of CHQ (Fig. 23B). As the activities of BChE increased, the fluorescence intensities of the experimental solutions progressively increased (Fig. 23C). Furthermore, a strong linear association was seen between fluorescence intensity of the peak at 775 nm and BChE activity (0.5–18 U L−1), with an LOD of 0.032 U L−1.
 | ||
| Fig. 23 (A) Sensing mechanism of CHQ-B towards BChE. (B) Fluorescence spectra of CHQ-B (10 μM), CHQ-B (10 μM) in the presence of BChE, and CHQ (10 μM). (C) Fluorescence responses of CHQ-B (10 μM) towards BChE. (D) Confocal imaging of endogenous BChE in living HepG2 cells pretreated with different reagents before incubation with CHQ-B. (E) Time-dependent fluorescence imaging of normal mouse and diabetic mouse model with injection of CHQ-B (200 μL, 300 μM). Excitation and emission wavelengths were set at 710 nm and 760 nm, respectively. Yang et al.,222 Copyright 2024, Elsevier. | ||
The effectiveness of CHQ-B for detecting the BChE level in live cells treated with various chemicals was examined. Tacrine is a broad-spectrum inhibitor for cholinesterases, while donepezil solely inhibits the level of acetylcholinesterase. Conversely, tetraisopropyl pyrophosphoramide (iso-OMPA) selectively suppresses the BChE level. In the control group, the cells exhibited clear red fluorescence when incubated with CHQ-B, whereas pre-treatment with tacrine prior to incubation with CHQ-B showed faint red fluorescence. In addition, the cells treated with both donepezil and CHQ-B showed a non-significant fluorescence change compared with the control group (Fig. 23D). The fluorescence intensity decreased significantly in the cells treated with iso-OMPA and CHQ-B. Streptozotocin-induced diabetic mice were also employed to investigate the efficacy of the probe. The diabetic mouse model showed more intense fluorescence responses in the abdomen region compared to the normal animal (Fig. 23E). These findings clearly show that BChE is up-regulated in diabetes and highlight the wide range of applications for CHQ-B in diabetes diagnostic support.
4.5. Probes for proteases
Proteases are a class of large, intricate enzyme molecules that are responsible for highly focused proteolytic functions. Amino acid-protein peptide linkages are hydrolyzed by proteases. Approximately 60% of the enzymes in the global market are proteases. Apoptosis, digestion, and other activities are all impacted by the catalytic activity of proteases. To synthesize essential biomolecules and regulate the size, shape, turnover, and composition of basic proteins, proteases are extremely important in all living things. One of the most important metabolic events in all living things is the very precise and restricted cleavage of the substrate by the protease. Several biological processes, including DNA replication and the cell cycle, begin with hydrolysis by protease. Depending on the type of reaction, proteases can be further classified as either exopeptidases or endopeptidases, two classes of enzymes that hydrolyze peptide bonds.223An essential feature of the pathogenesis of solid cancers, APN is an overexpressed tumor biomarker on a variety of malignant tumor cells. In addition, APN is critical for the development, invasion, and spread of cancer, in addition to the development of new blood vessels. Therefore, Yang et al.242 developed an aminopeptidase-N (APN/CD13)-activable probe, APN-SUB, with a quinoxalinone scaffold that contains leucine, which has the ability to hydrolyze leucine at the N-terminus of peptides and small proteins (Fig. 24A). APN-SUB can undergo catalysis by APN, resulting in the hydrolysis of leucine and subsequent disclosure of the amino group to produce APN-DEG. Using an amino group as a donor group allows color-convertible detection by intramolecular charge transfer (ICT), which causes a red shift in the fluorescence emission. Simultaneously, a pH-sensitive nano-carrier system with acylhydrazone-connected block amphiphilic copolymers (PEG–Hz1–PLA) was synthesized for the administration of the fluorescent molecule to enhance the tumor passive targeting and quick response of APN located within the cell membrane. Upon the incubation of APN-SUB with various concentrations of APN, the fluorescence intensity at 495 nm decreased, while a new peak emerged at 600 nm and simultaneously increased (Fig. 24B). The relationship was linear between the proportion of fluorescence intensity (F600/F495) and APN concentration in the range of 0 to 0.96 mU mL−1. This nanoprobe demonstrated remarkable sensitivity with an LOD of 0.14 nU mL−1.
 | ||
| Fig. 24 (A) Illustration demonstrating the formation of the APN-SUB nanoprobe and the response mechanism of APN-SUB toward APN. (a) Self-assembly and disassembly of the APN-SUB nanoprobe. (b) In vivo recognition mechanism of APN-SUB as a color-convertible probe toward APN. (B) Fluorescent emission spectra of the APN-SUB nanoprobe treated with different concentrations of APN. (C) Confocal images of HeLa cells treated with the APN-SUB nanoprobe. (a) Imaging in HeLa cells incubated with the APN-SUB nanoprobe. (b) Imaging in HeLa cells pre-treated with bestatin and incubated with APN-SUB. (D) In vivo imaging of the APN-SUB nanoprobe in nude BABL/c mice bearing HeLa xenograft tumor. (E) Imaging of tumor slices after treatment with APN-SUB and APN-SUB nanoprobe. Yang et al.,242 Copyright 2023, The Royal Society of Chemistry. | ||
Two cancer cell lines, HeLa and BT-549, which exhibit the overexpression of APN, and one normal cell line, BRL 3A, derived from the liver, were used for the imaging studies. After a prolonged incubation duration of the APN-SUB nanoprobe with HeLa cells, notable red fluorescence was detected, accompanied by a corresponding reduction in the green channel. The signal in the red channel was diminished once the cells were pre-treated with bestatin, an APN inhibitor (Fig. 24C). The APN-SUB nanoprobe was injected intravenously for variable durations and in vivo fluorescence imaging was performed on xenograft tumor mice to assess its passive tumor targeting efficacy. The accumulation of APN-SUB at the tumor site was demonstrated by the strongest fluorescent signal (Fig. 24D). Afterwards, the tumor tissue was excised, cut and stained with Hoechst 33342. The APN-SUB nanoprobe-treated group of mice demonstrated an increase in red fluorescence intensity and decrease in green fluorescence in the tumor compared to the animals treated with APN-SUB. This indicates the presence of a considerable amount of the nanoprobe at the tumor site and demonstrates the usual APN-responsive action of the APN-SUB nanoprobe (Fig. 24E). Therefore, APN-SUB can be considered as a probable probe for tracking cancer through amino-peptidase detection.
Xu et al.243 developed a very delicate NIR fluorescent probe called LAN-apn. Its purpose was to examine the amount of APN expressed in thyroid cancer and determine its potential as a diagnostic for this type of cancer. It was employed for visualizing APN in a living organism, confirming the elevation of APN levels in thyroid carcinoma. APN can potentially be detected by LAN-apn via the selective digestion of alanine, therefore enabling the detachment of the p-amino benzyl alcohol linker to release LAN-OH (Fig. 25A). The fluorescence spectrum revealed that the presence of APN substantially enhanced the strength of the emission peak of LAN-apn at 642 nm (excitation wavelength of 580 nm) and resulted in a color change from yellow to bright red at 365 nm. As the concentration of APN (λex = 580 nm) increased, the fluorescence spectrum showed a progressive upsurge in the fluorescence intensity of the reaction system at 642 nm. Furthermore, a positive linear correlation was identified between the fluorescence intensity and APN concentration in the range of 1–400 ng mL−1, with an LOD of 0.069 ng mL−1 (Fig. 25B).
 | ||
| Fig. 25 (A) Proposed detection mechanism of LAN-apn towards APN. (B) Fluorescence spectra of 10 μM LAN-apn in the presence of APN at various concentrations. (C) Fluorescence images of endogenous APN in living cells. (D) Fluorescence images of control mouse and TPC-1 tumor-bearing mouse injected with LAN-apn. (E) In vitro imaging of living tissues incubated with LAN-apn. Xu et al.,243 Copyright 2024, Elsevier. | ||
TPC-1 cells (thyroid cancer cells with endogenous APN) and Nthy-ori3-1 cells (normal thyroid cells) were evaluated by fluorescence imaging using LAN-apn. Although the TPC-1 cells (b) displayed significant fluorescence, the Nthy-ori3-1 cells (e) showed low fluorescence signals. Moreover, the intracellular fluorescence signals were markedly reduced when bestatin, an APN inhibitor, was added. This finding suggests that the APN levels are elevated in thyroid cancer cells, and that LAN-apn can analyze endogenous APN in cells (Fig. 25C). In contrast to the mouse that was carrying the TPC-1 tumor, which had a substantial fluorescence signal at the tumor location, no fluorescence signal was detected in the control group. A similar result was obtained in the cell imaging (Fig. 25D). The findings indicated that the level of APN increased in thyroid tumors, and the probe can be employed to detect early-stage thyroid tumors. No visible fluorescent signals were detected in the tissues of the heart, spleen, lungs, kidneys, or muscles. Alternatively, the liver and TPC-1 tumor exhibited prominent fluorescent signals, suggesting an increase in APN levels in the TPC-1 tumor (Fig. 25E). These outcomes highlight the effectiveness of LAN-apn for the in vitro detection of APN levels. Consequently, the overexpression of APN in thyroid cancer has been validated by using LAN-apn to selectively “light up” APN in cells and tumors both in vivo and in vitro. Consequently, it is obvious that LAN-apn is beneficial for new imaging technology involved in the early identification of thyroid cancer.
Ren et al.255 designed two caspase-1-responsive biosensors, i.e., WEHD-HCy and YVAD-HCy to selectively detect inflammasome activity in an inflammatory disease model. WEHD-HCy and YVAD-HCy consisted of two parts (Fig. 26A), as follows: (1) peptide sequence Trp–Glu–His–Asp (WEHD) and Tyr–Val–Ala–Asp (YVAD) and (2) hemicyanine (HCy) for fluorescence. The fluorescence signal of the probe is “off” because amino group of HCy is in the caged condition. Inflammasome activation triggers caspase-1 activation, which selectively cleaves the probe, releasing free HCy and activating a fluorescence signal. Furthermore, HCy could be detected in mitochondria, blocking the fluorescence signal from entering other cells that do not have activated caspase-1 and generating a non-specific fluorescence signal.
 | ||
| Fig. 26 (A) Chemical structures of WEHD-HCy and YVAD-HCy and schematic of mitochondria-targeted WEHD-HCy and YVAD-HCy response to inflammasome activation. (B) Fluorescence spectra of 25 μM WEHD-HCy or YVAD-HCy in the absence or presence of caspase-1 (25 U mL−1). (C) In vivo imaging of caspase-1 activation in DSS-induced mouse colitis model. Time-course fluorescence imaging in the abdomen of mice in DSS groups, control groups and caspase-1−/− + DSS groups. Ex = 640 nm, Em = 710 nm. (D) Ex vivo fluorescence imaging of intestinal tract from mice in DSS groups and control groups. (E) Monitoring caspase-1 biosensors activation during S. typhimurium infection. Time-course fluorescence imaging in the abdomen of mice in S. typhimurium infection groups, control groups and caspase-1−/− groups. (F) Ex vivo fluorescence imaging of intestinal tract and spleen from mice in S. typhimurium infection groups and control groups. Ren et al.,255 Copyright 2023, Elsevier. | ||
The intensity of 25 μM WEHD-HCy increased by 11.8-fold in the presence of 25 U mL−1 caspase-1 in working buffer, whereas that of 25 μM YVAD-HCy increased by 5.1-fold (Fig. 26B). The fluorescence intensity of WEHD-HCy or YVAD-HCy increased in a linear manner when the amount of caspase-1 increased in the range of 0 to 5 U mL−1. Furthermore, the LOD of WEHD-HCy was 0.033 U mL−1 compared to that of YVAD-HCy of 0.217 U mL−1, suggesting that WEHD-HCy had a better detection sensitivity than YVAD-HCy.
The efficacy of the WEHD-HCy and YVAD-HCy biosensors for monitoring inflammasome activation in vivo has been confirmed in an inflammatory disease model. Ulcerative colitis (UC) and Crohn's disease (CD) and autoimmune conditions in inflammatory bowel disease (IBD) that affect the colon and the whole gastrointestinal system. A major element in IBD pathogenesis is NLRP3 inflammasome action.256 The DSS-induced mouse colitis model was selected due to its simplicity and several similarities to human UC. The mice in the DSS groups exhibited a brighter fluorescence signal in their abdomen after being injected with WEHD-HCy or YVAD-HCy. This signal steadily intensified within 4 h, and then reduced after 6 h. The caspase-1−/− + DSS groups of mice did not exhibit a fluorescence signal (Fig. 26C). In the DSS group, the intestinal tract showed a higher fluorescence signal than that in control groups (Fig. 26D). These results proved that the caspase-1 biosensors WEHD-HCy and YVAD-HCy are suitable for the specific tracking of UC and CD. WEHD-HCy is extra-sensitive compared to YVAD-HCy and has an additional 1.5-fold enhanced fluorescence signal as a result of its higher caspase-1 catalytic efficiency than YVAD-HCy.
The activation of the NLRC4 and NLRP3 inflammasomes by S. typhimurium infection may lead to the activation of the effector protein caspase 1, which will cleave pro-IL-1β, pro-IL-18, and pro-GSDMD, resulting in the formation of bioactive proteins.257 The intragastric delivery of S. typhimurium (or PBS control) to the mice caused infection. The mice infected with S. typhimurium attained a plateau in fluorescence signal 4 h after the intraperitoneal injection of WEHD-HCy or YVAD-HCy, in contrast to the little detectable fluorescence signal in the PBS control group. There was essentially little fluorescence signal recorded in the caspase-1−/− group of mice, suggesting that WEHD-HCy or YVAD-HCy was sensitive to caspase-1 activation, even if the S. typhimurium infection titer was higher in these groups (Fig. 26E). Moreover, the fluorescence signal detected in the intestine and spleen of the S. typhimurium infectious groups was significantly higher than in the control groups (Fig. 26F). Thus, the WEHD-HCy and YVAD-HCy biosensors accurately detected caspase-1 activity during inflammatory responses in bacterial infections. However, the efficacy of WEHD-HCy was better than YVAD-HCy, showing a 1.6-fold increase in fluorescence due to the catalytic activity of caspase-1 towards WEHD-HCy compared to YVAD-HCy.
Fluorescent/photoacoustic (FL/PA) imaging has gained significant interest recently due to its exceptional sensitivity, impressive spatial resolution and penetration depth. To detect caspase-3 in tumor apoptosis, Liu et al.258 designed a tumor-targeted FL/PA probe (Bio-DEVD-HCy). The tripartite structure of the tumor-targeted FL/PA probe (Bio-DEVD-HCy) was as follows: (a) a tumor targeting component;259,260 (b) the peptide component Asp–Glu–Val–Asp (DEVD) for specific caspase-3 cleavage;261,262 and (c) a hemi-cyanine dye (HCy) segment with exceptional NIR properties for FL/PA imaging. Because of the caged amino group of HCy, which inhibits ICT in Bio-DEVD-HCy, the FL/PA signal is “off.” Upon encountering tumor cells, Bio-DEVD-HCy attaches to the highly expressed biotin receptor on their surface, allowing it to penetrate and concentrate within the tumor cells. Subsequently, caspase-3, which is overexpressed in apoptotic tumor cells, cleaves Bio-DEVD-HCy in a particular manner, which results in the generation of HCy with a free amino group. Strong NIR FL/PA signals are generated by HCy as a result of the ICT process. Consequently, Bio-DEVD-HCy could visualize tumor apoptosis with absolute precision (Fig. 27A).
 | ||
| Fig. 27 (A) Schematic of Bio-DEVD-HCy and Ac-DEVD-HCy for FL/PA imaging of caspase-3 in apoptotic tumor cells. (B) FL intensity of 25 μM Bio-DEVD-HCy or Ac-DEVD-HCy after incubation without or with 200 μg mL−1 caspase-3 for 8 h. (C) PA spectra of 25 μM Bio-DEVD-HCy or Ac-DEVD-HCy without or with 200 μg mL−1 caspase-3 for 8 h. Inset: PA images at 675 nm. (D) Confocal FL images (a) and PA images at 675 nm (b) of 4T1 cells in five groups. (E) Time-course FL (a) and PA (b) images of 4T1 tumor-bearing mice in five groups. Liu et al.,258 Copyright 2023, ACS. | ||
After the incubation of Bio-DEVD-HCy with caspase-3, there was an increase in fluorescence intensity by 5.5 times, whereas that for Ac-DEVD-HCy increased by 4.6 times at 720 nm. Ac-DEVD-HCy, which lacks biotin (tumor-targeted component), was employed as a control probe (Fig. 27B). At 720 nm, there was a linear enhancement in the FL intensity for Bio-DEVD-HCy or Ac-DEVD-HCy as the quantity of caspase-3 increased from 0 to 8 μg mL−1. In the case of Bio-DEVD-HCy and Ac-DEVD-HCy, their corresponding LOD was determined to be 0.035 and 0.084 μg mL−1, respectively. Furthermore, at 675 nm, the PA intensity increased by 3.1-fold for Bio-DEVD-HCy, whereas in the case of Ac-DEVD-HCy, it increased by 2.5-fold (Fig. 27C). The PA intensity is linearly related to the concentration (0 to 10 μg mL−1) of caspase-3 at 675 nm. The LOD was 0.14 for Bio-DEVD-HCy and 0.28 μg mL−1 for Ac-DEVD-HCy, respectively.
These two probes were used for the visualization of cell apoptosis and triggered by caspase-3. Two cell lines were employed, namely mouse breast cancer (4T1) cells with a positive biotin receptor, and murine fibroblast (NIH-3T3) cells without a biotin receptor. The FL intensity of the 4T1 cells in the DOX + Bio-DEVD-HCy group was 4.3 fold and DOX + Ac-DEVD-HCy group was 3.4-fold. The DOX + Biotin + Bio-DEVD-HCy group had a 4.1-fold reduction in FL intensity in the 4T1 cells as compared to the DOX + Bio-DEVD-HCy group. In addition, at 675 nm, the PA intensity in the 4T1 cells increased by 3.5-fold for DOX + Bio-DEVD-HCy and 1.5-fold for DOX + Ac-DEVD-HCy (Fig. 27D). Bio-DEVD-HCy together with tumor-targeted biotin enter and aggregate in tumour cells with an increased FL/PA signal, as evidenced by the 3.5-fold decrease in PA intensity at 675 nm for the 4T1 cells in the DOX + Biotin + Bio-DEVD-HCy group compared to the DOX + Bio-DEVD-HCy group. These data verified the potential of these two probes to detect caspase-3 in apoptotic cells, and Bio-DEVD-HCy outperformed Ac-DEVD-HCy.
The saline-treated mouse group was given either Bio-DEVD-HCy or Ac-DEVD-HCy intravenously. The fluorescence (FL) signal in the DOX group exhibited a gradual increase over a period of 4 h, followed by a subsequent drop in the tumor site. Furthermore, the photoacoustic (PA) signal in the group treated with DOX was significantly increased after 4 h, and then decreased (Fig. 27E). The results confirmed that these two probes, which respond to caspase-3, are appropriate for observing tumour cell death in living organisms. Additionally, Bio-DEVD-HCy was shown to be better than Ac-DEVD-HCy.
5. Conclusion and future perspectives
Variations in the degree of enzyme expression indicate the onset and development of human illnesses. Enzyme activities are necessary for the maintenance of normal biological processes and play a role in the management of many abnormal circumstances. Therefore, monitoring the activity of some essential enzymes is very significant in the context of theranostics and diagnosis. For instance, tyrosinases are involved in PD and melanoma malignancy. The neurodegenerative disorders PD and AD are associated with MAOs A and B, whereas NQO1 is a key biomarker for a variety of human cancer cells, including those found in the breast, lung, ovary, stomach, pancreas, colon and kidney. Cholestatic disease is caused by the abnormal production of alkaline phosphatase and is linked to β-galactosidase and ovarian cancer. APN acts as a biomarker of tumour identification, and caspase participates in the process of apoptosis. One unique method to study the activity of biomarkers is fluorescence imaging. When imaging endogenous biomarkers in live cells, it is typically necessary to use fluorescent probes that have strong water solubility and high selectivity for accurate imaging. The existence of enzymes and their behaviour during physiological processes may both be inferred from the fluorescence signal. New fluorescent enzyme probes will be developed with the help of developments in biochemistry and biomedicine, which will expand our understanding of particular reaction sites in enzymes and biomarkers. The fact that the processes selectively catalyzed by fluorescent enzyme probes result in fluorescence responses emphasizes the significance of recognition moieties as a key component of their design.Therefore, we aim to offer guidance concerning suitable research domains that necessitate the advancement of fluorogenic probes for biomedical applications with improved functionalities. Ideally, these enhanced probe systems would:
(1) Have the capability to penetrate deeper into tissues than visible fluorescence to image in vivo biomarkers. Creating activable probes with extremely long fluorescence emission wavelengths and auto-fluorescence is one promising strategy. Recent advancements in emission wavelengths have allowed organic dyes to achieve much improved imaging features, such as in-depth tissue penetration, better imaging contrast, higher spatial and temporal resolution and biocompatibility. One example is the expansion of emission wavelengths in the range of 1000–1700 nm (NIR-II range). Furthermore, the imaging accuracy of fluorescence may be enhanced by combining it with imaging modalities such as computed tomography and magnetic resonance imaging, which have tissue penetration benefits for multimodal imaging. This will allow the precise tracking and labelling of disease-related bioactive molecules in living systems.
(2) Currently, various probes have been utilised for the development of theranostic probes aimed at integrating the approach of disease diagnosis and treatment. An assessment of the advancements in this domain reveals the significant advances in tumour ablation by photodynamic treatment, sonodynamic therapy, chemodynamic therapy, photothermal therapy, and the investigation of major organ ailments. In the field of cancer theranostics, fluorescent probes have been shown to be effective because of their advantages in terms of flexibility in structure, simple application, minimum invasiveness and biocompatibility. Nevertheless, their clinical implementation is still constrained by the reduced penetration capacity of their excitation light. Thus, there is an immediate need to create theranostic agents with improved penetrating capability.
(3) In the near future, new fluorescent probes with the capability to differentiate between normal and malignant tissue will likely be given the green light for clinical use. The enhanced accuracy is expected to markedly enhance the prognosis and post-operative recovery of individuals diagnosed with cancer.
(4) In the case of molecular probes and nano-probes containing heavy metal ions, concerns regarding their metabolism and bio-toxicity in vivo must be addressed. When designing these systems, toxicology and pharmacokinetic investigations should be routinely carried out to assure their biocompatibility and maximise the possibility for their eventual clinical acceptance. Their biocompatibility with the tissues being tested in a clinical setting should be thoroughly investigated to ensure they are safe and friendly. It is imperative to maintain consistent efforts to minimise the quantity and size of fluorescent-based nanomaterials, while simultaneously ensuring that they are biodegradable, swiftly metabolised, and clearable.
(5) To strategize and focus on the design of probes for other enzymes that act as diagnostic markers. Elevated levels of these enzymes in the blood indicate various disease conditions linked to them. Thus far, limited fluorescent probes have been developed for enzymes such as lactate dehydrogenase (marker of cardiac injury), creatine kinase (marker for muscle or heart damage), and gamma-glutamyl transpeptidase (chronic hepatitis infections).
(6) With the goal of developing a more intrinsic targeting approach, it is crucial to do more research into enhancing the targeting of probe towards certain tissue types. This will help to both improve the targeting groups and expand beyond specific targeting motifs.
(7) The creation of innovative specialised recognition responses for both existing and newly discovered disease biomarkers is essential to further explore the application of activatable fluorescence probes. For more accurate diagnosis, it is essential to develop probes by incorporating new and improved fluorophores with higher quantum yields. In summary, the process of developing environmentally friendly manufacturing methods that are capable of satisfying clinical requirements and fostering the commercialisation and industrialisation of fluorescence probes is necessary for their widespread application in clinical settings. These strategies should be economical, highly effective, and environmentally sustainable.
This review summarized the latest progress in fluorescence imaging, primarily focusing on design, imaging techniques and in vivo enzyme activity detection. We expect that enzymatic fluorescent probes are highly required in theranostics and diagnostics, and we hope that our review will help the next generation of researchers working in this area.
Data availability
This is a review article and no unpublished data or software were used to prepare this article.Conflicts of interest
There are no conflicts to declare.Acknowledgements
LM is thankful to UGC (NTA reference number 211610083530) for the financial support that she has received for her doctoral work.References
- A. A. Rajapaksha,
et al., Review on the recent progress in the development of fluorescent probes targeting enzymes, Methods Appl. Fluoresc., 2021, 9(3), 032001 CrossRef CAS PubMed
.
- C. Yang, Q. Wang and W. Ding, Recent progress in the imaging detection of enzyme activities in vivo, RSC Adv., 2019, 9(44), 25285–25302 RSC
- J. Wang,
et al., Visualizing human telomerase activity with primer-modified Au nanoparticles, Small, 2012, 8(2), 259–264 CrossRef CAS PubMed
- X. Xie, W. Xu and X. Liu, Improving colorimetric assays through protein enzyme-assisted gold nanoparticle amplification, Acc. Chem. Res., 2012, 45(9), 1511–1520 CrossRef CAS
- C. Jiang,
et al., Colorimetric assay for T4 polynucleotide kinase activity based on the horseradish peroxidase-mimicking DNAzyme combined with λ exonuclease cleavage, Anal. Chim. Acta, 2013, 766, 88–93 CrossRef CAS
- A. Hayat, G. Bulbul and S. Andreescu, Probing phosphatase activity using redox active nanoparticles: a novel colorimetric approach for the detection of enzyme activity., Biosens. Bioelectron., 2014, 56, 334–339 CrossRef CAS PubMed
- B. V. Chikkaveeraiah,
et al., Electrochemical immunosensors for detection of cancer protein biomarkers, ACS Nano, 2012, 6(8), 6546–6561 CrossRef CAS
- L. Wu,
et al., Label-free ultrasensitive detection of human telomerase activity using porphyrin-functionalized graphene and electrochemiluminescence technique, Adv. Mater., 2012, 24(18), 2447–2452 CrossRef CAS
- G. Wang,
et al., Detection of T4 polynucleotide kinase activity with immobilization of TiO2 nanotubes and amplification of Au nanoparticles, Biosens. Bioelectron., 2013, 43, 125–130 CrossRef CAS
-
L. J. Kricka, J. C. Voyta and I. Bronstein, Chemiluminescent methods for detecting and quantitating enzyme activity, 2000 Search PubMed
- C. Wang,
et al., A turn-on fluorescent probe for detection of tyrosinase activity, Analyst, 2013, 138(10), 2825–2828 RSC
- Y. Su,
et al., In vitro and in vivo enzyme activity screening via RNA-based fluorescent biosensors for S-adenosyl-L-homocysteine (SAH), J. Am. Chem. Soc., 2016, 138(22), 7040–7047 CrossRef CAS PubMed
- C.-H. Tung,
et al., In vivo imaging of proteolytic enzyme activity using a novel molecular reporter, Cancer Res., 2000, 60(17), 4953–4958 CAS
- A. J. Wilson, D. Devasia and P. K. Jain, Nanoscale optical imaging in chemistry, Chem. Soc. Rev., 2020, 49(16), 6087–6112 RSC
- Z. Hu,
et al., First-in-human liver-tumour surgery guided by multispectral fluorescence imaging in the visible and near-infrared-I/II windows, Nat. Biomed. Eng., 2020, 4(3), 259–271 CrossRef PubMed
- X. Huang,
et al., Ratiometric optical nanoprobes enable accurate molecular detection and imaging, Chem. Soc. Rev., 2018, 47(8), 2873–2920 RSC
- X. Zhao,
et al., Recent progress in photosensitizers for overcoming the challenges of photodynamic therapy: from molecular design to application, Chem. Soc. Rev., 2021, 50(6), 4185–4219 RSC
- J. Yin,
et al., Small molecule based fluorescent chemosensors for imaging the microenvironment within specific cellular regions, Chem. Soc. Rev., 2021, 50(21), 12098–12150 RSC
- L. Wu,
et al., Dual-locked spectroscopic probes for sensing and therapy, Nat. Rev. Chem., 2021, 5(6), 406–421 CrossRef CAS PubMed
- W. Xu,
et al., Discerning the chemistry in individual organelles with small-molecule fluorescent probes, Angew. Chem., Int. Ed., 2016, 55(44), 13658–13699 CrossRef CAS PubMed
- L. Feng,
et al., Fluorescent probes for bioactive detection and imaging of phase II metabolic enzymes, Coord. Chem. Rev., 2019, 399, 213026 CrossRef CAS
- T. D. Ashton, K. A. Jolliffe and F. M. Pfeffer, Luminescent probes for the bioimaging of small anionic species in vitro and in vivo, Chem. Soc. Rev., 2015, 44(14), 4547–4595 RSC
- P. Kaur and K. Singh, Recent advances in the application of BODIPY in bioimaging and chemosensing, J. Mater. Chem. C, 2019, 7(37), 11361–11405 RSC
- D. Wu,
et al., Recent progress in the development of organic dye based near-infrared fluorescence probes for metal ions, Coord. Chem. Rev., 2018, 354, 74–97 CrossRef CAS
- X. Li,
et al., Design strategies for water-soluble small molecular chromogenic and fluorogenic probes, Chem. Rev., 2014, 114(1), 590–659 CrossRef CAS
- X. Yang,
et al., A multi-signal mitochondria-targeted fluorescent probe for real-time visualization of cysteine metabolism in living cells and animals, Chem. Commun., 2018, 54(81), 11387–11390 RSC
- W. Li,
et al., Recent advances in molecular fluorescent probes for organic phosphate biomolecules recognition, Chin. Chem. Lett., 2019, 30(10), 1775–1790 CrossRef CAS
- X. Wu,
et al., Recognition moieties of small molecular fluorescent probes for bioimaging of enzymes, Acc. Chem. Res., 2019, 52(7), 1892–1904 CrossRef CAS
- J. Bell,
et al., Synthesis and complexing properties of molecular probes linked with fluorescent phosphane oxide derivatives, J. Photochem. Photobiol., A, 2016, 318, 25–32 CrossRef CAS
- S. Park,
et al., A white-light-emitting molecule: frustrated energy transfer between constituent emitting centers, J. Am. Chem. Soc., 2009, 131(39), 14043–14049 CrossRef CAS PubMed
- M. Guerra,
et al., Protease FRET reporters targeting neutrophil extracellular traps, J. Am. Chem. Soc., 2020, 142(48), 20299–20305 CrossRef CAS
- N. Hirata,
et al., Preparation and fluorescence properties of fluorophore-labeled avidin–biotin system immobilized on Fe3O4 nanoparticles through functional indolequinone linker, Bioorg. Med. Chem., 2009, 17(11), 3775–3781 CrossRef CAS PubMed
- J. V. Jun, D. M. Chenoweth and E. J. Petersson, Rational design of small molecule fluorescent probes for biological applications, Org. Biomol. Chem., 2020, 18(30), 5747–5763 RSC
- M. Swierczewska, S. Lee and X. Chen, The design and application of fluorophore–gold nanoparticle activatable probes, Phys. Chem. Chem. Phys., 2011, 13(21), 9929–9941 RSC
- G.-J. Shi,
et al., Hybrid coumarin-difluoroboron dyes for mitochondrial staining, Dyes Pigm., 2020, 179, 108430 CrossRef CAS
- G. Li,
et al., Charge Transfer Switching in Donor–Acceptor Systems Based on BN-Fused Naphthalimides, J. Org. Chem., 2018, 83(10), 5577–5587 CrossRef CAS
- H. Park and S.-K. Chang, Signaling of water content in organic solvents by solvatochromism of a hydroxynaphthalimide-based merocyanine dye, Dyes Pigm., 2015, 122, 324–330 CrossRef CAS
- J. Lee,
et al., A red-emitting styrylnaphthalimide-based fluorescent probe providing a ratiometric signal change for the precise and quantitative detection of H2O2, Anal. Chim. Acta, 2019, 1080, 153–161 CrossRef CAS PubMed
- Y. Chen, S. Wang and F. Zhang, Near-infrared luminescence high-contrast in vivo biomedical imaging, Nat. Rev. Bioeng., 2023, 1(1), 60–78 CrossRef CAS
- R. Zhang,
et al., Blood Circulation Assessment by Steadily Fluorescent Near-Infrared-II Aggregation-Induced Emission Nano Contrast Agents, ACS Nano, 2023, 17(19), 19265–19274 CrossRef CAS
- L. Lu,
et al., Illuminating the invisible: advancing bio-imaging and diagnosis with modified near-infrared fluorescents, Appl. Mater. Today, 2024, 38, 102210 CrossRef
- A. P. De Silva,
et al., Signaling recognition events with fluorescent sensors and switches, Chem. Rev., 1997, 97(5), 1515–1566 CrossRef CAS PubMed
- M. Schäferling, The art of fluorescence imaging with chemical sensors, Angew. Chem., Int. Ed., 2012, 51(15), 3532–3554 CrossRef
- J. Wu,
et al., New sensing mechanisms for design of fluorescent chemosensors emerging in recent years, Chem. Soc. Rev., 2011, 40(7), 3483–3495 RSC
- Y. Hong, J. W. Lam and B. Z. Tang, Aggregation-induced emission, Chem. Soc. Rev., 2011, 40(11), 5361–5388 RSC
- S. Doose, H. Neuweiler and M. Sauer, Fluorescence quenching by photoinduced electron transfer: a reporter for conformational dynamics of macromolecules, ChemPhysChem, 2009, 10(9–10), 1389–1398 CrossRef CAS PubMed
- K. E. Sapsford, L. Berti and I. L. Medintz, Materials for fluorescence resonance energy transfer analysis: beyond traditional donor–acceptor combinations, Angew. Chem., Int. Ed., 2006, 45(28), 4562–4589 CrossRef CAS PubMed
- M. Hao,
et al., Molecular origins of photoinduced backward intramolecular charge transfer, J. Phys. Chem. C, 2020, 124(31), 16820–16826 CrossRef CAS
- J. E. Kwon and S. Y. Park, Advanced organic optoelectronic materials: harnessing excited-state intramolecular proton transfer (ESIPT) process, Adv. Mater., 2011, 23(32), 3615–3642 CrossRef PubMed
- D. Udhayakumari, Mechanistic Innovations in Fluorescent Chemosensors for Detecting Toxic Ions: PET, ICT, ESIPT, FRET and AIE Approaches, J. Fluoresc., 2024, 1–30 Search PubMed
- H.-W. Liu,
et al., Recent progresses in small-molecule enzymatic fluorescent probes for cancer imaging, Chem. Soc. Rev., 2018, 47(18), 7140–7180 RSC
- W. Shi and H. Ma, Spectroscopic probes with changeable π-conjugated systems, Chem. Commun., 2012, 48(70), 8732–8744 RSC
- H. Chen,
et al., A unique “integration” strategy for the rational design of optically tunable near-infrared fluorophores, Acc. Chem. Res., 2017, 50(6), 1410–1422 CrossRef CAS
- S. Gnaim and D. Shabat, Quinone-methide species, a gateway to functional molecular systems: from self-immolative dendrimers to long-wavelength fluorescent dyes, Acc. Chem. Res., 2014, 47(10), 2970–2984 CrossRef CAS
- J. Yan,
et al., Self-immolative colorimetric, fluorescent and chemiluminescent chemosensors, Chem. Soc. Rev., 2018, 47(18), 6900–6916 RSC
- M. Rolff,
et al., Copper–O2 reactivity of tyrosinase models towards external monophenolic substrates: molecular mechanism and comparison with the enzyme, Chem. Soc. Rev., 2011, 40(7), 4077–4098 RSC
- J. Zhou and H. Ma, Design principles of spectroscopic probes for biological applications, Chem. Sci., 2016, 7(10), 6309–6315 RSC
- J. Zhang,
et al., Fluorogenic probes for disease-relevant enzymes, Chem. Soc. Rev., 2019, 48(2), 683–722 RSC
- R. Friedman Ohana,
et al., Utilizing a simple method for stoichiometric protein labeling to quantify antibody blockade, Sci. Rep., 2019, 9(1), 7046 CrossRef PubMed
- P. Agrawal,
et al., Stoichiometric analyses of soluble CD4 to native-like HIV-1 envelope by single-molecule fluorescence spectroscopy, Cell Rep., 2019, 29(1), 176–186.e4 CrossRef CAS
- J. Song,
et al., A high quantum yield xanthene-based fluorescent probe for the specific detection of tyrosinase and cell imaging, J. Photochem. Photobiol., A, 2023, 441, 114693 CrossRef CAS
- Y. Ding,
et al., A novel fluorescent off–on probe based on 4-methylumbelliferone for highly sensitive determination of tyrosinase, New J. Chem., 2022, 46(20), 9923–9930 RSC
- J. Cao, J. Gong and N. Fu, A 1,8-naphthalimide based fluorescent probe for sensing tyrosinase in zebrafish, Microchem. J., 2022, 173, 107007 CrossRef CAS
- Z. Li,
et al., Fast-response fluorescent probe with favorable water solubility for highly sensitive imaging of endogenous tyrosinase in living cells and zebrafish model, Chin. Chem. Lett., 2021, 32(5), 1785–1789 CrossRef CAS
- X. Wang and C. Ma, A near-infrared fluorescent chemosensor with a remarkably large Stokes shift for the ultrasensitive detection of tyrosinase activity and bioimaging in living cells and mouse xenograft model, Sens. Actuators, B, 2022, 354, 131211 CrossRef CAS
- Y. Shu,
et al., pH-Triggered mitochondria targeting fluorescent probe for detecting Monoamine Oxidases A in living cells, Sens. Actuators, B, 2023, 390, 133912 CrossRef CAS
- X. Li,
et al., Specific tracking of monoamine oxidase A in heart failure models by a far-red fluorescent probe with an ultra large Stokes shift, Chin. Chem. Lett., 2022, 33(3), 1572–1576 CrossRef CAS
- J. Shang,
et al., Water-soluble near-infrared fluorescent probes for specific detection of monoamine oxidase A in living biosystems, Anal. Chem., 2021, 93(9), 4285–4290 CrossRef CAS
- J. Gao,
et al., β-Galactosidase activity monitoring and bioimaging by a novel ICT mechanism-based NIR fluorescent probe with large Stokes shift, Sens. Actuators, B, 2024, 398, 134696 CrossRef CAS
- Y. Hu,
et al., A chalcone-based ESIPT and AIE fluorophore for β-gal imaging in living cells, Org. Biomol. Chem., 2024, 22(9), 1850–1858 RSC
- S. Chen,
et al., Design and application of lysosomal targeting pH-sensitive β-galactosidase fluorescent probe, Sens. Actuators, B, 2023, 379, 133272 CrossRef CAS
- S. Chen,
et al., Construction of an intelligent fluorescent probe that can accurately track β-galactosidase activity in fruits and living organisms, Sens. Actuators, B, 2023, 387, 133787 CrossRef CAS
- X. Huang,
et al., Novel ratiometric fluorescent probe for real-time detection of alkaline phosphatase and its application in living cells, Spectrochim. Acta, Part A, 2021, 260, 119953 CrossRef CAS PubMed
- Y.-A. Feng,
et al., Ratiometric detection and bioimaging of endogenous alkaline phosphatase by a NIR fluorescence probe, Sens. Actuators, B, 2022, 358, 131505 CrossRef CAS
- L. Wang,
et al., Near-infrared fluorescent probe with a self-immolative spacer for ratiometric imaging alkaline phosphatase in cell and zebrafish, Dyes Pigm., 2023, 209, 110910 CrossRef CAS
- M. M. Fortibui,
et al., Near-infrared fluorescence probe for specific detection of acetylcholinesterase and imaging in live cells and zebrafish, ACS Appl. Bio Mater., 2022, 5(5), 2232–2239 CrossRef CAS PubMed
- C. Zhao,
et al., Near-infrared fluorescent probe for in vivo monitoring acetylcholinesterase activity, Sens. Actuators, B, 2022, 360, 131647 CrossRef CAS
- J. S. Sidhu,
et al., Acetylcholine structure-based small activatable fluorogenic probe for specific detection of acetylcholinesterase, Anal. Chem., 2023, 95(19), 7594–7602 CrossRef CAS
- Y. Yang,
et al., Real-time fluorescent determination and biological imaging in living models via a butyrylcholinesterase-activated fluorescent probe, Dyes Pigm., 2022, 206, 110596 CrossRef CAS
- Y. Yang,
et al., Diagnosis of Alzheimer's disease and in situ biological imaging via an activatable near-infrared fluorescence probe, Anal. Chem., 2022, 94(39), 13498–13506 CrossRef CAS PubMed
- X. Gao,
et al., High-contrast tumor imaging via a de novo designed aminopeptidase N targeted fluorogenic probe, Sens. Actuators, B, 2024, 401, 135097 CrossRef CAS
- S.-Y. Liu,
et al., De novo design of an ultrasensitive fluorogenic probe for aminopeptidase N sensing in living system, Sens. Actuators, B, 2022, 363, 131828 CrossRef CAS
- C. Wynne and R. B. Elmes, Utilising a 1, 8-naphthalimide probe for the ratiometric fluorescent visualisation of caspase-3, Front. Chem., 2024, 12, 1418378 CrossRef CAS
- Y. Jin,
et al., Activity-based probe for ratiometric fluorescence imaging of caspase-3 in living cells, Anal. Chem., 2020, 93(4), 2045–2052 CrossRef PubMed
- Y. Cárdenas-Moreno,
et al., Oxidoreductase enzymes: characteristics, applications, and challenges as a biocatalyst, Biotechnol. Appl. Biochem., 2023, 70(6), 2108–2135 CrossRef
- J. Zhou, Y. Geng and Z. Wang, Fluorescent molecular probes for imaging and detection of oxidases and peroxidases in biological samples, Methods, 2023, 210, 20–35 CrossRef CAS PubMed
- A. Slominski,
et al., Melanin pigmentation in mammalian skin and its hormonal regulation, Physiol. Rev., 2004, 84(4), 1155–1228 CrossRef CAS
- M. Friedman, Food browning and its prevention: an overview, J. Agric. Food Chem., 1996, 44(3), 631–653 CrossRef CAS
- G. Battaini,
et al., Tyrosinase-catalyzed oxidation of fluorophenols, J. Biol. Chem., 2002, 277(47), 44606–44612 CrossRef CAS
- X. Lai,
et al., Structure and function of human tyrosinase and tyrosinase-related proteins, Chem. – Eur. J., 2018, 24(1), 47–55 CrossRef CAS PubMed
- Y. Chen, Advances in fluorescent probes for detection and imaging of endogenous tyrosinase activity, Anal. Biochem., 2020, 594, 113614 CrossRef CAS PubMed
- X. Wang and C. Ma, A near-infrared fluorescent chemosensor with a remarkably large Stokes shift for the ultrasensitive detection of tyrosinase activity and bioimaging in living cells and mouse xenograft model, Sens. Actuators, B, 2022, 354, 131211 CrossRef CAS
- O. Merimsky,
et al., Reactivity to tyrosinase: expression in cancer (melanoma) and autoimmunity (vitiligo), Hum. Antibodies, 1996, 7(4), 151–156 CAS
- A. Gargiulo,
et al., AAV-mediated tyrosinase gene transfer restores melanogenesis and retinal function in a model of oculo-cutaneous albinism type I (OCA1), Mol. Ther., 2009, 17(8), 1347–1354 CrossRef CAS PubMed
- W. S. Oetting and R. A. King, Molecular basis of oculocutaneous albinism, J. Invest. Dermatol., 1994, 103(5), 131s–136s CrossRef CAS PubMed
- C. Angeletti,
et al., Novel tyramide-based tyrosinase assay for the detection of melanoma cells in cytological preparations, Diagn. Cytopathol., 2004, 31(1), 33–37 CrossRef PubMed
- J. Y. Lin and D. E. Fisher, Melanocyte biology and skin pigmentation, Nature, 2007, 445(7130), 843–850 CrossRef CAS
- C. Zhan,
et al., A fluorescent probe for early detection of melanoma and its metastasis by specifically imaging tyrosinase activity in a mouse model, Anal. Chem., 2018, 90(15), 8807–8815 CrossRef CAS
- I. Carballo-Carbajal,
et al., Brain tyrosinase overexpression implicates age-dependent neuromelanin production in Parkinson's disease pathogenesis, Nat. Commun., 2019, 10(1), 973 CrossRef PubMed
- T. Hasegawa,
et al., Parkin protects against tyrosinase-mediated dopamine neurotoxicity by suppressing stress-activated protein kinase pathways, J. Neurochem., 2008, 105(5), 1700–1715 CrossRef CAS
- D. Chen,
et al., A turn-on fluorescence probe for imaging tyrosinase at the wound site in broken tail of zebrafish, Bioorg. Chem., 2024, 146, 107298 CrossRef CAS
- X. Huang,
et al., Debut of a novel AIE-based fluorescent probe as tyrosinase tracer to image skin pigmentary disorders, Sens. Actuators, B, 2023, 389, 133889 CrossRef CAS
- X. Wu,
et al., Near-infrared fluorescent probe with new recognition moiety for specific detection of tyrosinase activity: design, synthesis, and application in living cells and zebrafish, Angew. Chem., Int. Ed., 2016, 55(47), 14728–14732 CrossRef CAS
- N. Wang and D. N. Hebert, Tyrosinase maturation through the mammalian secretory pathway: bringing color to life, Pigm. Cell Res., 2006, 19(1), 3–18 CrossRef CAS PubMed
- H. Ando,
et al., Approaches to identify inhibitors of melanin biosynthesis via the quality control of tyrosinase, J. Invest. Dermatol., 2007, 127(4), 751–761 CrossRef CAS PubMed
- R. Halaban,
et al., Aberrant retention of tyrosinase in the endoplasmic reticulum mediates accelerated degradation of the enzyme and contributes to the dedifferentiated phenotype of amelanotic melanoma cells, Proc. Natl. Acad. Sci. U. S. A., 1997, 94(12), 6210–6215 CrossRef CAS
- D. Shu,
et al., Fluorescence monitoring of refluxed tyrosinase using endoplasmic reticulum-localized enzymatic activity-based sensing, Chem. Commun., 2024, 60(43), 5618–5621 RSC
- M. Berry, A. Juorio and I. Paterson, The functional role of monoamine oxidases A and B in the mammalian central nervous system, Prog. Neurobiol., 1994, 42(3), 375–391 CrossRef CAS PubMed
- D. Edmondson,
et al., Structure and mechanism of monoamine oxidase, Curr. Med. Chem., 2004, 11(15), 1983–1993 CrossRef CAS
- S. Singha,
et al., An endeavor in the reaction-based approach to fluorescent probes for biorelevant analytes: challenges and achievements, Acc. Chem. Res., 2019, 52(9), 2571–2581 CrossRef CAS PubMed
-
H. Gaweska and P. F. Fitzpatrick, Structures and mechanism of the monoamine oxidase family, 2011 Search PubMed
- M. S. Benedetti and P. Dostert, Monoamine oxidase, brain ageing and degenerative diseases, Biochem. Pharm., 1989, 38(4), 555–561 CrossRef PubMed
- L. Zhang,
et al., Near-Infrared Fluorescence Probe for Monoamine Oxidase A with a Large Stokes Shift for Intraoperative Navigation, ACS Appl. Bio Mater., 2024, 7(2), 1115–1124 CrossRef CAS
- H. Fang,
et al., A fluorogenic-inhibitor-based probe for profiling and imaging of monoamine oxidase A in live human glioma cells and clinical tissues, Sci. China: Chem., 2023, 66(7), 2053–2061 CrossRef CAS
- C. Zhang,
et al., Ultrafast Detection of Monoamine Oxidase A in Live Cells and Clinical Glioma Tissues Using an Affinity Binding-Based Two-Photon Fluorogenic Probe, Angew. Chem., Int. Ed., 2023, 62(42), e202310134 CrossRef CAS
- H. Fang,
et al., Intramolecular charge transfer enhancing strategy based MAO-A specific two-photon fluorescent probes for glioma cell/tissue imaging, Chem. Commun., 2021, 57(85), 11260–11263 RSC
- D. Kim,
et al., Close correlation of monoamine oxidase activity with progress of Alzheimer's disease in mice, observed by in vivo two-photon imaging, ACS Cent. Sci., 2016, 2(12), 967–975 CrossRef CAS
- Z. Yang,
et al., Monoamine oxidase A probe synthesis and fluorescence imaging in cells and zebrafish based on AIE mechanism, Mater. Today Chem., 2024, 35, 101890 CrossRef CAS
- S. Moriguchi,
et al., Monoamine oxidase B total distribution volume in the prefrontal cortex of major depressive disorder: an [11C] SL25. 1188 positron emission tomography study, JAMA Psychiatry, 2019, 76(6), 634–641 CrossRef
- S. M. Graves,
et al., Dopamine metabolism by a monoamine oxidase mitochondrial shuttle activates the electron transport chain, Nat. Neurosci., 2020, 23(1), 15–20 CrossRef CAS PubMed
- K. A. Jellinger, Brain monoamine oxidases in human parkinsonian disorders, Brain, 2017, 140(9), 2262–2264 CrossRef PubMed
- Y. Zhang,
et al., Rapid and sensitive fluorescent probes for monoamine oxidases B to A at low concentrations, Tetrahedron Lett., 2012, 53(51), 6881–6884 CrossRef CAS
- X. Li,
et al., Fluorescent probes for detecting monoamine oxidase activity and cell imaging, Org. Biomol. Chem., 2014, 12(13), 2033–2036 RSC
- S. Long,
et al., An activity-based fluorogenic probe for sensitive and selective monoamine oxidase-B detection, Chem. Commun., 2012, 48(57), 7164–7166 RSC
- L. Li,
et al., A small-molecule probe for selective profiling and imaging of monoamine oxidase B activities in models of Parkinson's disease, Angew. Chem., Int. Ed., 2015, 54(37), 10821–10825 CrossRef CAS PubMed
- Z. Yang,
et al., Monoamine oxidase B activatable red fluorescence probe for bioimaging in cells and zebrafish, Bioorg. Chem., 2024, 145, 107156 CrossRef CAS
- S. Dragoni,
et al., CYP-dependent metabolism of PF9601N, a new monoamine oxidase-B inhibitor, by C57BL/6 mouse and human liver microsomes, J. Pharm. Pharm. Sci., 2007, 10(4), 473–485 CAS
- P. Jarčuška,
et al., Circulating markers of liver fibrosis progression, Clin. Chim. Acta, 2010, 411(15–16), 1009–1017 CrossRef
- M. Sun,
et al., Evaluation of Monoamine Oxidase B Fluctuation in Liver Fibrosis Cell and Mice Models via a Specificity Fluorescent Probe, Sens. Actuators, B, 2024, 136111 CrossRef CAS
- L. Li,
et al., A sensitive two-photon probe to selectively detect monoamine oxidase B activity in Parkinson's disease models, Nat. Commun., 2014, 5(1), 3276 CrossRef PubMed
- J. F. Gainor, Adjuvant PD-L1 blockade in non-small-cell lung cancer, The Lancet, 2021, 398(10308), 1281–1283 CrossRef CAS
- M. Maiorino, M. Conrad and F. Ursini, GPx4, lipid peroxidation, and cell death: discoveries, rediscoveries, and open issues, Antioxid. Redox Signaling, 2018, 29(1), 61–74 CrossRef CAS
- R. Brigelius-Flohé and M. Maiorino, Glutathione peroxidases, Biochim. Biophys. Acta, Gen. Subj., 2013, 1830(5), 3289–3303 CrossRef PubMed
- X. Jiang, B. R. Stockwell and M. Conrad, Ferroptosis: mechanisms, biology and role in disease, Nat. Rev. Mol. Cell Biol., 2021, 22(4), 266–282 CrossRef
- Y. Shi,
et al., Integration of metabolomics and transcriptomics to reveal metabolic characteristics and key targets associated with cisplatin resistance in nonsmall cell lung cancer, J. Proteome Res., 2019, 18(9), 3259–3267 CrossRef CAS PubMed
- Q. Hu,
et al., A near-infrared GPX4 fluorescent probe for non-small cell lung cancer imaging, Chem. Commun., 2023, 59(3), 294–297 RSC
- M. H. Lee,
et al., Fluorogenic reaction-based prodrug conjugates as targeted cancer theranostics, Chem. Soc. Rev., 2018, 47(1), 28–52 RSC
- P. Talalay and A. T. Dinkova-Kostova, Role of nicotinamide quinone oxidoreductase 1 (NQO1) in protection against toxicity of electrophiles and reactive oxygen intermediates, Methods Enzymol., 2004, 382, 355–364 CAS
- D. Ross,
et al., NAD (P) H: quinone oxidoreductase 1 (NQO1): chemoprotection, bioactivation, gene regulation and genetic polymorphisms, Chem.-Biol. Interact., 2000, 129(1–2), 77–97 CrossRef CAS PubMed
- D. Thapa,
et al., NQO1 suppresses NF-κB-p300 interaction to regulate inflammatory mediators associated with prostate tumorigenesis, Cancer Res., 2014, 74(19), 5644–5655 CrossRef CAS
- A. T. Dinkova-Kostova and P. Talalay, NAD (P) H: quinone acceptor oxidoreductase 1 (NQO1), a multifunctional antioxidant enzyme and exceptionally versatile cytoprotector, Arch. Biochem. Biophys., 2010, 501(1), 116–123 CrossRef CAS PubMed
- B. Lyn-Cook,
et al., Increased levels of NAD (P) H: quinone oxidoreductase 1 (NQO1) in pancreatic tissues from smokers and pancreatic adenocarcinomas: a potential biomarker of early damage in the pancreas, Cell Biol. Toxicol., 2006, 22, 73–80 CrossRef CAS PubMed
- S. Y. Sharp,
et al., Establishment of an Isogenic Human Colon Tumor Model forNQO1 Gene Expression: Application to Investigate the Role of DT-Diaphorase in Bioreductive Drug Activation In Vitro and In Vivo, Mol. Pharmacol., 2000, 58(5), 1146–1155 CrossRef CAS PubMed
- Y. Zhang,
et al., A high-performance enzyme-activated near-infrared probe for the sensing and tracking of tumor-related NQO1 in cells and in vivo, Sens. Actuators, B, 2022, 354, 131129 CrossRef CAS
- M. F. Mendoza,
et al., Human NAD (P) H: quinone oxidoreductase type I (hNQO1) activation of quinone propionic acid trigger groups, Biochemistry, 2012, 51(40), 8014–8026 CrossRef CAS
- G. G. Dias,
et al., Quinone-based fluorophores for imaging biological processes, Chem. Soc. Rev., 2018, 47(1), 12–27 RSC
- S. U. Hettiarachchi, B. Prasai and R. L. McCarley, Detection and cellular imaging of human cancer enzyme using a turn-on, wavelength-shiftable,
self-immolative profluorophore, J. Am. Chem. Soc., 2014, 136(21), 7575–7578 CrossRef CAS
- N. Karton-Lifshin,
et al., “Donor–two-acceptor” dye design: a distinct gateway to NIR fluorescence, J. Am. Chem. Soc., 2012, 134(50), 20412–20420 CrossRef CAS PubMed
- N. Karton-Lifshin,
et al., A unique paradigm for a Turn-ON near-infrared cyanine-based probe: noninvasive intravital optical imaging of hydrogen peroxide, J. Am. Chem. Soc., 2011, 133(28), 10960–10965 CrossRef CAS PubMed
- G. Asher,
et al., The crystal structure of NAD (P) H quinone oxidoreductase 1 in complex with its potent inhibitor dicoumarol, Biochemistry, 2006, 45(20), 6372–6378 CrossRef CAS PubMed
- Q. Husain, β Galactosidases and their potential applications: a review, Crit. Rev. Biotechnol., 2010, 30(1), 41–62 CrossRef CAS PubMed
- G. P. Dimri,
et al., A biomarker that identifies senescent human cells in culture and in aging skin in vivo, Proc. Natl. Acad. Sci. U. S. A., 1995, 92(20), 9363–9367 CrossRef CAS
- J. Zhang, P. Cheng and K. Pu, Recent advances of molecular optical probes in imaging of β-galactosidase, Bioconjugate Chem., 2019, 30(8), 2089–2101 CrossRef CAS PubMed
- K. Ishikawa,
et al., Crystal structure of β-galactosidase from Bacillus circulans ATCC 31382 (BgaD) and the construction of the thermophilic mutants. The, FEBS J., 2015, 282(13), 2540–2552 CrossRef CAS
- L. Shi,
et al., In vivo ratiometric tracking of endogenous β-galactosidase activity using an activatable near-infrared fluorescent probe, Chem. Commun., 2019, 55(82), 12308–12311 RSC
- X. Li,
et al., Specific near-infrared probe for ultrafast imaging of lysosomal β-galactosidase in ovarian cancer cells, Anal. Chem., 2020, 92(8), 5772–5779 CrossRef CAS PubMed
- B. Lozano-Torres,
et al., An OFF–ON two-photon fluorescent probe for tracking cell senescence in vivo, J. Am. Chem. Soc., 2017, 139(26), 8808–8811 CrossRef CAS
- Z. Li,
et al., Aging diagnostic probe for research on aging and evaluation of anti-aging drug efficacy, Anal. Chem., 2021, 93(41), 13800–13806 CrossRef CAS PubMed
- B. Lozano-Torres,
et al., A two-photon probe based on naphthalimide-styrene fluorophore for the in vivo tracking of cellular senescence, Anal. Chem., 2021, 93(5), 3052–3060 CrossRef CAS
- D. Liu,
et al., A turn on fluorescent assay for real time determination of β-galactosidase and its application in living cell imaging, Spectrochim. Acta, Part A, 2022, 265, 120345 CrossRef CAS PubMed
- Y.-P. Lo,
et al., A near-infrared fluorescent probe with a substantial Stokes shift designed for the detection and imaging of β-galactosidase within living cells and animals, Methods, 2024, 222, 10–18 CrossRef CAS
- C. Liu,
et al., Ratiometric Fluorescent Probe for Real-Time Detection of β-Galactosidase Activity in Lysosomes and Its Application in Drug-Induced Senescence Imaging, Anal. Chem., 2024, 96(7), 3223–3232 CAS
- G. Han,
et al., Excimers beyond pyrene: a far-red optical proximity reporter and its application to the label-free detection of DNA, Angew. Chem., 2015, 127(13), 3984–3988 CrossRef
- H. Liu,
et al., A near-IR ratiometric fluorescent probe for the precise tracking of senescence: a multidimensional sensing assay of biomarkers in cell senescence pathways, Chem. Sci., 2024, 15(15), 5681–5693 RSC
- M. Köhn, Turn and face the strange: § a new view on phosphatases, ACS Cent. Sci., 2020, 6(4), 467–477 CrossRef
- J. E. Coleman, Structure and mechanism of alkaline phosphatase, Annu. Rev. Biophys. Biomol. Struct., 1992, 21(1), 441–483 CrossRef CAS PubMed
- N. K. Tonks, Protein tyrosine phosphatases: from genes, to function, to disease, Nat. Rev. Mol. Cell Biol., 2006, 7(11), 833–846 CrossRef CAS PubMed
- D. M. Zaher,
et al., Recent advances with alkaline phosphatase isoenzymes and their inhibitors, Arch. Pharm., 2020, 353(5), e2000011 CrossRef CAS
- K. Kalantar-Zadeh,
et al., Kidney bone disease and mortality in CKD: revisiting the role of vitamin D, calcimimetics, alkaline phosphatase, and minerals, Kidney Int., 2010, 78, S10–S21 CrossRef PubMed
- H. Zhang,
et al., Recent progress of fluorescent probes for the detection of alkaline phosphatase (ALP): a review, Dyes Pigm., 2021, 194, 109569 CrossRef CAS
- J. Chen,
et al., Alkaline phosphatase activatable near-infrared fluorescent probe for in situ diagnosis of cholestatic liver injury, Sens. Actuators, B, 2024, 413, 135896 CrossRef CAS
- W.-X. Wang,
et al., Real-time imaging of alkaline phosphatase
activity of diabetes in mice via a near-infrared fluorescent probe, Chem. Commun., 2021, 57(4), 480–483 RSC
- C. Lei,
et al., Resurfaced fluorescent protein as a sensing platform for label-free detection of copper(II) ion and acetylcholinesterase activity, Anal. Chem., 2015, 87(3), 1974–1980 CrossRef CAS
- A. Contestabile, The history of the cholinergic hypothesis, Behav. Brain Res., 2011, 221(2), 334–340 CrossRef CAS
- D. M. Quinn, Acetylcholinesterase: enzyme structure, reaction dynamics, and virtual transition states, Chem. Rev., 1987, 87(5), 955–979 CrossRef CAS
- M. M. Fortibui,
et al., Near-infrared fluorescence probe for specific detection of acetylcholinesterase and imaging in live cells and zebrafish, ACS Appl. Bio Mater., 2022, 5(5), 2232–2239 CrossRef CAS
- A. M. Baig,
et al., Traced on the timeline: discovery of acetylcholine and the components of the human cholinergic system in a primitive unicellular eukaryote Acanthamoeba spp, ACS Chem. Neurosci., 2017, 9(3), 494–504 CrossRef
- C.-I. Wang, A. P. Periasamy and H.-T. Chang, Photoluminescent C-dots@ RGO probe for sensitive and selective detection of acetylcholine, Anal. Chem., 2013, 85(6), 3263–3270 CrossRef CAS
- T. Miyakawa,
et al., Hyperactivity and intact hippocampus-dependent learning in mice lacking the M1 muscarinic acetylcholine receptor, J. Neurosci., 2001, 21(14), 5239–5250 CrossRef CAS PubMed
- N. He,
et al., Near-infrared fluorescent probe for evaluating the acetylcholinesterase effect in the aging process and dietary restriction via fluorescence imaging, J. Mater. Chem. B, 2021, 9(11), 2623–2630 RSC
- V. N. Talesa, Acetylcholinesterase in Alzheimer's disease, Mech. Ageing Dev., 2001, 122(16), 1961–1969 CrossRef CAS PubMed
- S. Ahmed,
et al., Potential therapeutic natural products against Alzheimer's disease with Reference of Acetylcholinesterase, Biomed. Pharmacother., 2021, 139, 111609 CrossRef CAS
- X. Wang,
et al., Observation of acetylcholinesterase in stress-induced depression phenotypes by two-photon fluorescence imaging in the mouse brain, J. Am. Chem. Soc., 2019, 141(5), 2061–2068 CrossRef CAS
- J. R. Suarez-Lopez,
et al., Associations of acetylcholinesterase activity with depression and anxiety symptoms among adolescents growing up near pesticide spray sites, Int. J. Hyg. Environ. Health, 2019, 222(7), 981–990 CrossRef CAS PubMed
- C. Xiang,
et al., A responsive AIE-active fluorescent probe for visualization of acetylcholinesterase activity in vitro and in vivo, Mater. Chem. Front., 2022, 6(11), 1515–1521 RSC
- C. Perry, E. H. Sklan and H. Soreq, CREB regulates AChE-R-induced proliferation of human glioblastoma cells, Neoplasia, 2004, 6(3), 279–286 CrossRef CAS
- H. Soreq, Y. Lapidot-Lifson and H. Zakut, A role for cholinesterases in tumorigenesis?, Cancer Cells, 1991, 3(12), 511–516 CAS
- N. Razon,
et al., Characterization of activities and forms of cholinesterases in human primary brain tumors, Exp. Neurol., 1984, 84(3), 681–695 CrossRef CAS PubMed
- H. E. Greenwood and T. H. Witney, Latest advances in imaging oxidative stress in cancer, J. Nucl. Med., 2021, 62(11), 1506–1510 CrossRef CAS
- A. J. Kattoor,
et al., Oxidative stress in atherosclerosis, Curr. Atheroscler. Rep., 2017, 19, 1–11 CrossRef CAS
- E. Dubois-Deruy,
et al., Oxidative stress in cardiovascular diseases, Antioxidants, 2020, 9(9), 864 CrossRef CAS PubMed
- S. M. Wu and S. V. Pizzo, α2-Macroglobulin from rheumatoid arthritis synovial fluid: functional analysis defines a role for oxidation in inflammation, Arch. Biochem. Biophys., 2001, 391(1), 119–126 CrossRef CAS PubMed
- P. Li and M. Chang, Roles of PRR-mediated signaling pathways in the regulation of oxidative stress and inflammatory diseases, Int. J. Mol. Sci., 2021, 22(14), 7688 CrossRef CAS
- X. Wei,
et al., Monitoring acetylcholinesterase level changes under oxidative stress through ESIPT-ICT-based near-infrared fluorescent probe, Sens. Actuators, B, 2023, 380, 133392 CrossRef CAS
- K. P. Das and S. Barone Jr, Neuronal differentiation in PC12 cells is inhibited by chlorpyrifos and its metabolites: is acetylcholinesterase inhibition the site of action?, Toxicol. Appl. Pharmacol., 1999, 160(3), 217–230 CrossRef CAS
- S.-Y. Liu,
et al., Discovery of butyrylcholinesterase-activated near-infrared fluorogenic
probe for live-cell and in vivo imaging, ACS Sens., 2018, 3(10), 2118–2128 CrossRef CAS
- L. Fang,
et al., Active site gating and substrate specificity of butyrylcholinesterase and acetylcholinesterase: insights from molecular dynamics simulations, J. Phys. Chem. B, 2011, 115(27), 8797–8805 CrossRef CAS
- Y. Nicolet,
et al., Crystal structure of human butyrylcholinesterase and of its complexes with substrate and products, J. Biol. Chem., 2003, 278(42), 41141–41147 CrossRef CAS
- S. Darvesh, D. A. Hopkins and C. Geula, Neurobiology of butyrylcholinesterase, Nat. Rev. Neurosci., 2003, 4(2), 131–138 CrossRef CAS
- D. Kolarich,
et al., Glycoproteomic characterization of butyrylcholinesterase from human plasma, Proteomics, 2008, 8(2), 254–263 CrossRef CAS
- J. Z. Long and B. F. Cravatt, The metabolic serine hydrolases and their functions in mammalian physiology and disease, Chem. Rev., 2011, 111(10), 6022–6063 CrossRef CAS PubMed
- Q. Zhang,
et al., Fluorescent determination of butyrylcholinesterase activity and its application in biological imaging and pesticide residue detection, ACS Sens., 2021, 6(3), 1138–1146 CrossRef CAS PubMed
- G. Chen,
et al., Redox-controlled fluorescent nanoswitch based on reversible disulfide and its application in butyrylcholinesterase activity assay, Anal. Chem., 2018, 90(3), 1643–1651 CrossRef CAS
- J. Ma,
et al., Rational design of a near-infrared fluorescence probe for highly selective sensing butyrylcholinesterase (BChE) and its bioimaging applications in living cell, Talanta, 2020, 219, 121278 CrossRef CAS
- M. Stojanov,
et al., Butyrylcholinesterase activity in young men and women: association with cardiovascular risk factors, Clin. Biochem., 2011, 44(8–9), 623–626 CrossRef CAS PubMed
- Y. Yang,
et al., Diagnosis of Alzheimer's disease and in situ biological imaging via an activatable near-infrared fluorescence probe, Anal. Chem., 2022, 94(39), 13498–13506 CrossRef CAS PubMed
- V. P. Chen,
et al., Plasma butyrylcholinesterase regulates ghrelin to control aggression, Proc. Natl. Acad. Sci. U. S. A., 2015, 112(7), 2251–2256 CrossRef CAS
- Z.-Q. Yu,
et al., Bioinspired design of highly specific fluorescent probe for butyrylcholinesterase imaging in living cells and Alzheimer's disease model, Sens. Actuators, B, 2024, 410, 135662 CrossRef CAS
- A. R. Mukhametgalieva,
et al., Activation/Inhibition of Cholinesterases by Excess Substrate: Interpretation of the Phenomenological b Factor in Steady-State Rate Equation, Int. J. Mol. Sci., 2023, 24(13), 10472 CrossRef CAS
- C. Leistner,
et al., The in-tissue molecular architecture of β-amyloid pathology in the mammalian brain, Nat. Commun., 2023, 14(1), 2833 CrossRef CAS
- T. Zhang,
et al., Near-Infrared Aggregation-Induced Emission Luminogens for In Vivo Theranostics of Alzheimer's Disease, Angew. Chem., Int. Ed., 2023, 62(2), e202211550 CrossRef CAS
- P. Zhang,
et al., AND-logic strategy for accurate analysis of Alzheimer's disease via fluorescent probe lighted up by two specific biomarkers, Anal. Chem., 2021, 93(32), 11337–11345 CrossRef CAS PubMed
- I. R. Macdonald,
et al., Quantification of butyrylcholinesterase activity as a sensitive and specific biomarker of Alzheimer's disease, J. Alzheimer's Dis., 2017, 58(2), 491–505 CAS
- C. Xiang,
et al., Ratiometric imaging of butyrylcholinesterase activity in mice with nonalcoholic fatty liver using an AIE-based fluorescent probe, J. Mater. Chem. B, 2022, 10(22), 4254–4260 RSC
- D. Wang,
et al., Hepatocellular BChE as a therapeutic target to ameliorate hypercholesterolemia through PRMT5 selective degradation to restore LDL receptor transcription, Life Sci., 2022, 293, 120336 CrossRef CAS
- O. Ogunkeye, E. Chuhwak and A. Otokwula, Serum cholinesterase activity in the diagnosis of nonalcoholic fatty liver disease in type 2 diabetic patients, Pathophysiology, 2010, 17(1), 29–32 CrossRef CAS PubMed
- S. Katoh,
et al., Fatty liver and serum cholinesterase are independently correlated with HbA1c levels: cross-sectional analysis of 5384 people, J. Int. Med. Res., 2014, 42(2), 542–553 CrossRef CAS PubMed
- Z. Guo,
et al., Benzorhodol derived far-red/near-infrared fluorescent probes for selective and sensitive detection of butyrylcholinesterase activity in living cells and the non-alcoholic fatty liver of zebrafish, Anal. Methods, 2024, 16(25), 4054–4059 RSC
- C. Abbott,
et al., Relationship between serum butyrylcholinesterase activity, hypertriglyceridaemia and insulin sensitivity in diabetes mellitus, Clin. Sci., 1993, 85(1), 77–81 CrossRef CAS
- G. I. Lunkes,
et al., Serum cholinesterase activity in diabetes and associated pathologies, Diabetes Res. Clin. Pract., 2006, 72(1), 28–32 CrossRef CAS
- B. N. Patel,
et al., Serum esterase activities and hyperlipidaemia in the streptozotocin-diabetic rat, Biochim. Biophys. Acta, Gen. Subj., 1990, 1035(1), 113–116 CrossRef CAS PubMed
- B. Yang,
et al., Exploring butyrylcholinesterase expression in living cells and diabetic mouse models using a fluorescent probe with near-infrared excitation and emission maxima, Dyes Pigm., 2024, 112295 CrossRef CAS
- P. Gurumallesh,
et al., A systematic reconsideration on proteases, Int. J. Biol. Macromol., 2019, 128, 254–267 CrossRef CAS
- A. Addlagatta, L. Gay and B. W. Matthews, Structure of aminopeptidase N from Escherichia coli suggests a compartmentalized, gated active site, Proc. Natl. Acad. Sci. U. S. A., 2006, 103(36), 13339–13344 CrossRef CAS
- A. H. Wong, D. Zhou and J. M. Rini, The X-ray crystal structure of human aminopeptidase N reveals a novel dimer and the basis for peptide processing, J. Biol. Chem., 2012, 287(44), 36804–36813 CrossRef CAS
- C. Schmitt,
et al., Selective aminopeptidase-N (CD13) inhibitors with relevance to cancer chemotherapy, Bioorg. Med. Chem., 2013, 21(7), 2135–2144 CrossRef CAS
- S.-Y. Liu,
et al., De novo design of an ultrasensitive fluorogenic probe for aminopeptidase N sensing in living system, Sens. Actuators, B, 2022, 363, 131828 CrossRef CAS
- B. Bauvois and D. Dauzonne, Publications de l’U 507 (Inserm), Med. Res. Rev., 2006, 26(1), 88–130 CrossRef CAS PubMed
- P. Mina-Osorio, The moonlighting enzyme CD13: old and new functions to target, Trends Mol. Med., 2008, 14(8), 361–371 CrossRef CAS PubMed
- C. L. Yeager,
et al., Human aminopeptidase N is a receptor for human coronavirus 229E, Nature, 1992, 357(6377), 420–422 CrossRef CAS PubMed
- D. E. Wentworth,
et al., Cells of human aminopeptidase N (CD13) transgenic mice are infected by human coronavirus-229E in vitro, but not in vivo, Virology, 2005, 335(2), 185–197 CrossRef CAS PubMed
- D. Reinhold,
et al., DP IV/CD26, APN/CD13 and related enzymes as regulators of T cell immunity: implications for experimental encephalomyelitis and multiple sclerosis, Front. Biosci., 2008, 13, 2356–2363 CrossRef CAS PubMed
- L. Guzman-Rojas,
et al., Cooperative effects of aminopeptidase N (CD13) expressed by nonmalignant and cancer cells within the tumor microenvironment, Proc. Natl. Acad. Sci. U. S. A., 2012, 109(5), 1637–1642 CrossRef CAS
- S.-X. Cui,
et al., Targeting aminopeptidase N (APN/CD13) with cyclic-imide peptidomimetics derivative CIP-13F inhibits the growth of human ovarian carcinoma cells, Cancer Lett., 2010, 292(2), 153–162 CrossRef CAS
- W. Liang,
et al., Possible contribution of aminopeptidase N (APN/CD13) to migration and invasion of human osteosarcoma cell lines, Int. J. Oncol., 2014, 45(6), 2475–2485 CrossRef CAS
- S. Nohara,
et al., Aminopeptidase N (APN/CD13) as a target molecule for scirrhous gastric cancer, Clin. Res. Hepatol. Gastroenterol., 2016, 40(4), 494–503 CrossRef CAS
- J. I. Scott, Q. Deng and M. Vendrell, Near-infrared fluorescent probes for the detection of cancer-associated proteases, ACS Chem. Biol., 2021, 16(8), 1304–1317 CrossRef CAS PubMed
- Y. Luan and W. Xu, The structure and main functions of aminopeptidase N, Curr. Med. Chem., 2007, 14(6), 639–647 CrossRef CAS PubMed
- Y. Liu,
et al., Precipitated fluorophore-based molecular probe for in situ imaging of aminopeptidase N in living cells and tumors, Anal. Chem., 2021, 93(16), 6463–6471 CrossRef CAS PubMed
- U. Lee,
et al., Native Chemical Ligation-Based Fluorescent Probes for Cysteine and Aminopeptidase N Using meso-thioester-BODIPY, Chem. – Eur. J., 2021, 27(49), 12545–12551 CrossRef CAS PubMed
- M.-Y. Tang,
et al., A novel aminopeptidase N triggered near-infrared fluorescence probe for imaging enzyme activity in cells and mice, Sens. Actuators, B, 2024, 404, 135290 CrossRef CAS
- J. Yang,
et al., An aminopeptidase N-based color-convertible fluorescent nano-probe for cancer diagnosis, Biomater. Sci., 2023, 11(8), 2809–2817 RSC
- L. Xu,
et al., Exploration of aminopeptidase N as new biomarker for early diagnosis of thyroid cancer, Biosens. Bioelectron., 2024, 244, 115808 CrossRef CAS
- A. G. Porter and R. U. Jänicke, Emerging roles of caspase-3 in apoptosis, Cell Death Differ., 1999, 6(2), 99–104 CrossRef CAS
- M. D’Amelio, M. Sheng and F. Cecconi, Caspase-3 in the central nervous system: beyond apoptosis, Trends Neurosci., 2012, 35(11), 700–709 CrossRef
- R. K. Graham,
et al., Cleavage at the caspase-6 site is required for neuronal dysfunction and degeneration due to mutant huntingtin, Cell, 2006, 125(6), 1179–1191 CrossRef CAS
- M. J. Leyva,
et al., Identification and evaluation of small molecule pan-caspase inhibitors in Huntington's disease models, Chem. Biol., 2010, 17(11), 1189–1200 CrossRef CAS PubMed
- K. S. Putt,
et al., Small-molecule activation of procaspase-3 to caspase-3 as a personalized anticancer strategy, Nat. Chem. Biol., 2006, 2(10), 543–550 CrossRef CAS PubMed
- D. W. Wolan,
et al., Small-molecule activators of a proenzyme, Science, 2009, 326(5954), 853–858 CrossRef CAS
- C. J. Vickers,
et al., Small-Molecule Procaspase Activators Identified Using Fluorescence Polarization, ChemBioChem, 2013, 12, 1419–1422 CrossRef PubMed
- C. Communal,
et al., Functional consequences of caspase activation in cardiac myocytes, Proc. Natl. Acad. Sci. U. S. A., 2002, 99(9), 6252–6256 CrossRef CAS
- R. S. Hotchkiss and D. W. Nicholson, Apoptosis and caspases regulate death and inflammation in sepsis, Nat. Rev. Immunol., 2006, 6(11), 813–822 CrossRef CAS PubMed
- K. M. Boatright and G. S. Salvesen, Mechanisms of caspase activation, Curr. Opin. Cell Biol., 2003, 15(6), 725–731 CrossRef CAS
- S. M. Man and T.-D. Kanneganti, Converging roles of caspases in inflammasome activation, cell death and innate immunity, Nat. Rev. Immunol., 2016, 16(1), 7–21 CrossRef CAS PubMed
- X. Ren,
et al., Caspase-1-responsive fluorescence biosensors for monitoring endogenous inflammasome activation, Biosens. Bioelectron., 2023, 219, 114812 CrossRef CAS PubMed
- V. Khatri and R. Kalyanasundaram, Therapeutic implications of inflammasome in inflammatory bowel disease, FASEB J., 2021, 35(5), e21439 CrossRef CAS PubMed
- B. Clare, Inflammasome activation by Salmonella, Curr. Opin. Microbiol., 2021, 64, 27–32 CrossRef CAS
- J. Liu,
et al., Caspase-3-responsive fluorescent/photoacoustic imaging of tumor apoptosis, Anal. Chem., 2023, 95(25), 9404–9408 CrossRef CAS
- N. Gholipour,
et al., Development of Ga-68 labeled, biotinylated thiosemicarbazone dextran-coated iron oxide nanoparticles as multimodal PET/MRI probe, Int. J. Biol. Macromol., 2020, 148, 932–941 CrossRef CAS
- W. Li,
et al., Molecular engineering of pH-responsive NIR oxazine assemblies for evoking tumor ferroptosis via triggering lysosomal dysfunction, J. Am. Chem. Soc., 2023, 145(6), 3736–3747 CrossRef CAS PubMed
- L. He,
et al., Real-time visualization of embryonic apoptosis using a near-infrared fluorogenic probe for embryo development evaluation, Anal. Chem., 2021, 93(35), 12122–12130 CrossRef CAS PubMed
- T. Ma,
et al., Morphological Transformation and In Situ Polymerization of Caspase-3 Responsive Diacetylene-Containing Lipidated Peptide Amphiphile for Self-Amplified Cooperative Antitumor Therapy, Small, 2022, 18(48), 2204759 CrossRef CAS PubMed
| This journal is © The Royal Society of Chemistry 2025 |