
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUpdate for oxidopyridinium cycloadditions and their synthetic applications: advances after Katritzky's pioneering studies
Yoshihiko
Yamamoto

Department of Basic Medicinal Sciences, Graduate School of Pharmaceutical Sciences, Nagoya University, Chikusa, Nagoya 464-8601, Japan. E-mail: yamamoto.yoshihiko.y9@f.mail.nagoya-u.ac.jp
First published on 18th March 2025
Abstract
Since the pioneering studies of the Katritzky group, the cycloaddition of 3-oxidopyridinium betaines has been continuously studied because (1) the multifaceted reactivity of 3-oxidopyridiniums has attracted attention from the viewpoint of physical chemistry, and (2) the production of nitrogen heterocycles with three-dimensional frameworks is crucial for natural product chemistry and pharmaceutical sciences. In this review, the development of oxidopyridinium cycloadditions is discussed. First, the results of the seminal investigations on oxidopyridinium cycloadditions are briefly presented. Subsequently, the follow-up research conducted since the pioneering studies of Katritzky and others is discussed.
 Yoshihiko Yamamoto | Yoshihiko Yamamoto received a Ph.D. degree from Nagoya University in 1996, where he was appointed as an Assistant Professor in 1996, promoted to Associate Professor in 2003 and Full Professor in 2012. He also belonged to the Tokyo Institute of Technology as an Associate Professor from 2006 to 2008. His research interests are focused on the development of efficient transition-metal catalysis and new methods for synthesizing medicinally interesting molecules. |
1. Introduction: early discoveries of oxidopyridinium cycloadditions
Nitrogen heterocycles (N-heterocycles) are found in natural products, drug molecules, and functional materials. Although aromatic N-heterocycles are ubiquitous, saturated non-aromatic N-heterocycles have attracted the attention of medicinal chemists because their three-dimensional structures tend to exhibit higher biological activity and target selectivity than aromatic N-heterocycles with planar two-dimensional structures.1 Therefore, synthetic chemists have focused on the development of efficient methods to construct complex saturated N-heterocycles. The dearomative transformations of aromatic pyridines, quinolines, isoquinolines and their derivatives offer an attractive approach for saturated N-heterocycles.2 These dearomative transformations can be categorized into two major groups: catalytic hydrogenation and N-activation followed by sequential functionalization (Fig. 1A). In addition to these methods, a different synthetic approach for saturated N-heterocycles from pyridin-3-ols has been developed by the Katritzky group and others. In this approach, pyridin-3-ols are converted into oxidopyridinium betaines, which subsequently undergo cycloaddition with an appropriate dipolarophile (Fig. 1B).3 Because oxidopyridinium cycloadditions enable the streamlined construction of various bridged N-heterocyclic scaffolds, they have been applied to the synthesis of natural products and biologically relevant molecules. In this review, the development of oxidopyridinium cycloadditions will be discussed. First, pioneering investigations on oxidopyridinium cycloadditions will be briefly outlined and then the research after Katritzky's seminal studies will be discussed in the following sections. To date, in addition to Katritzky's review articles, only a few reviews have discussed oxidopyridinium cycloadditions,3 and, to our knowledge, no review article has summarized recent advances in this field.4 | ||
| Fig. 1 (A) Dearomative transformations of pyridine and derivatives to saturated N-heterocycles. (B) Dearomative transformation of pyridin-3-ol to bridged N-heterocycles via 3-oxidopyridinium. | ||
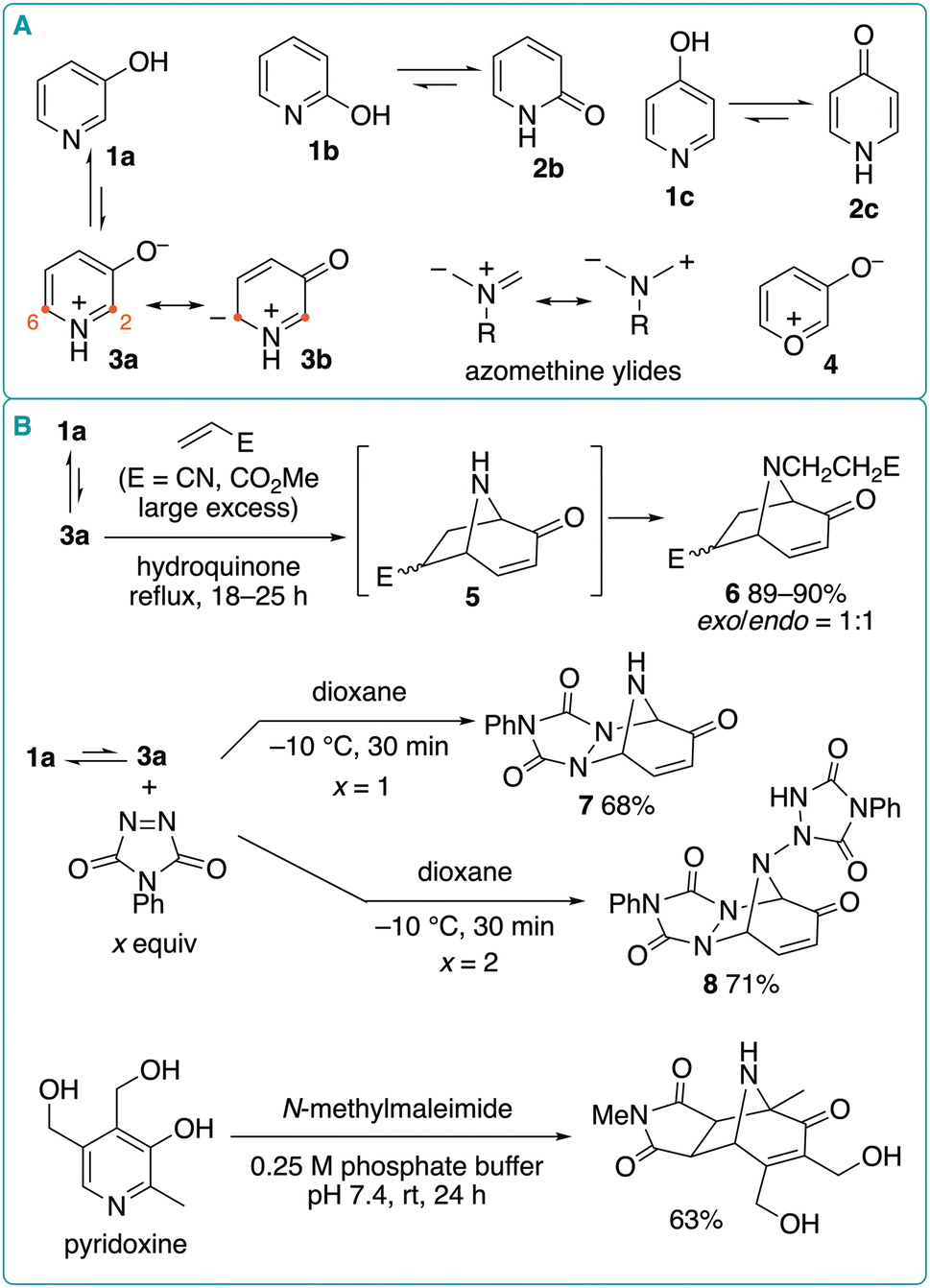
Pyridin-3-ol (1a) has a phenol-like structure unlike pyridin-2-ol (1b) and pyridin-4-ol (1c), which mainly exist as 2-pyridone (2b) and 4-pyridone (2c), respectively (Fig. 2A).5 In aqueous solution, 1a is in equilibrium with its tautomeric form (3a). Because 3b, one of the resonance structures of betaine 3a, is very similar to azomethine ylides, 3a exhibits 1,3-dipolar-cycloaddition reactivity similar to that of oxidopyrylium 4.6 Actually, Katritzky et al. reported that when 1a was heated under reflux in a large excess of acrylonitrile or methyl acrylate, tropane-like bicyclic compounds 6 were formed regioselectively in high yield via (5 + 2) cycloaddition at the 2- and 6-positions of 3a (Fig. 2B).7,8 It was proposed that the direct reaction of betaine 3a with electron-deficient alkenes initially formed N–H products 5, which then underwent aza-Michael reaction with the alkene to afford 6. However, an alternative pathway starting with the aza-Michael reaction of 1a cannot be excluded. Other electron-deficient alkenes, such as N-phenylmaleimide, diethyl maleate, and phenyl vinyl ketone, failed to afford the corresponding products. Later, El-Abbady et al. reported that the reaction of 1a with 1 equivalent of 4-phenyl-1,2,4-triazoline-3,5-dione proceeded in dioxane even at −10 °C to afford (5 + 2) cycloadduct 7 in 68% yield.9 On the other hand, increased amounts (2 equiv.) of the triazolinone led to the formation of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 adduct 8 in 71% yield. Moreover, the (5 + 2) cycloaddition of pyridoxine with N-methylmaleimide proceeded in pH 7.5 aqueous buffer at room temperature, affording the corresponding product in 63% yield.10
:2 adduct 8 in 71% yield. Moreover, the (5 + 2) cycloaddition of pyridoxine with N-methylmaleimide proceeded in pH 7.5 aqueous buffer at room temperature, affording the corresponding product in 63% yield.10
 | ||
| Fig. 2 (A) Structures of pyridinols. (B) (5 + 2) cycloaddition of pyridin-3-ol and pyridoxine. | ||
Although the scope of dipolarophiles in the cycloaddition of 1a is severely limited, Katritzky et al. reported that N-substituted oxidopyridinium betaines underwent (5 + 2) cycloadditions with various alkenes. They prepared 1-methyl-3-oxidopyridinium 10 through the deprotonation of N-methylated pyridinium 9 with Amberlite IRA-401 (Fig. 3A).11 Betaine 10 underwent 1,3-dipolar cycloaddition at the 2- and 6-positions with activated alkenes to afford cycloadducts with the tropane scaffold, which is found in diverse bioactive molecules, such as cocaine. Typically, 10 and N-phenylmaleimide were heated in THF/dioxane (1:3) to stereoselectively afford (5 + 2) cycloadduct exo-11a in 72% yield. The reaction with a large excess of acrylonitrile stereo- and regioselectively afforded cycloadduct exo-11b, albeit in low yield (17%). In contrast, the reaction with methyl acrylate produced a mixture of the corresponding exo- and endo-cycloadducts 11c in 75% yield. Sasaki et al. also reported that betaine 10 underwent (5 + 2) cycloaddition with the strained cycloalkene, oxabenzonorbornadiene (12), to afford exo-13 in 80% yield.12 Notably, 5-methoxy-1-methyl-3-oxidopyridinium (14) exhibited high reactivity toward styrene in refluxing acetonitrile, affording endo-15 in 94% yield.13 The reaction with diethyl azodicarboxylate proceeded within 10 min in THF at room temperature to afford 16 in 71% yield.
 | ||
| Fig. 3 (A) Preparation of N-methylated 3-oxidopyridinium and (5 + 2) cycloaddition of N-methylated 3-oxidopyridinium betaines. (B) (5 + 2) Cycloaddition of 3-oxido-1-phenylpyridinium betaine. | ||
The N-substituent of 3-oxidopyridinium betaines affects both the reactivity and stereoselectivity of their (5 + 2) cycloaddition substantially. The reaction of 3-oxido-1-phenylpyridinium 17 with N-phenylmaleimide or acrylonitrile produced (5 + 2) cycloadducts 18a or 18b in 81% and 76% yields, respectively, with an exo/endo selectivity of 4:5 (Fig. 3B).14 The latter yield was substantially improved in relation to that of N-methyl analog 10 (Fig. 3A). The reaction of 17 with benzyne, derived from anthranilic acid, afforded the corresponding product 19 in 35% yield. The reaction of styrene with 17, which is generated in situ from the reaction of salt 17·HCl, afforded endo-18c in 50% yield, whereas a similar reaction with methyl acrylate produced exo-18d in 31% yield. Later, other groups reinvestigated the (5 + 2) cycloaddition of betaines 10 and 17 with 1,2-disubstituted electron-deficient alkenes.15
When the N-substituent was an electron-withdrawing 5-nitropyridin-2-yl group, the corresponding betaine 20 underwent facile dimerization in such a way that the 2- and 6-positions of one betaine (HOMO) and the 2′- and 4′-positions of the other betaine (LUMO) are involved, selectively producing 21 in 84% yield (Fig. 4A).16 Similarly, pyridinium 22·HCl, with a 4,6-dimethylpyrimidin-2-yl group at the 1-position, afforded the corresponding dimer 23 as a mixture of regioisomers (2:1) in 80% yield. Dimer 21 underwent retro-dimerization to generate two equivalents of betaine 20 at a higher temperature; (5 + 2) cycloaddition with dipolarophiles was carried out using 21 as the oxidopyridinium precursor in the absence of a base.
 | ||
| Fig. 4 (A) Dimerization of oxidopyridinium betaines bearing electron-withdrawing heteroaryl groups at the 1-position. (B) (5 + 2), (5 + 4), and (5 + 6) cycloadditions of 1-(5-nitropyridin-2-yl)-3-oxidopyridinium. (C) Oxidopyridinium precursors with diverse electron-withdrawing N-substituents. | ||
Dimer 21 and excess methyl acrylate were heated in THF to afford endo-24a and exo-24a in 25% and 75% yields, respectively (Fig. 4B).17 Similarly, the reaction with styrene selectively afforded endo-24b in 83% yield. The reaction with dimethyl maleate exclusively produced exo-24c in 90% yield, indicating that cis/trans isomerization of both maleate and cycloadduct was suppressed in the absence of a base. Moreover, the reaction with 2,3-dimethyl-1,3-butadiene afforded (5 + 4) cycloadduct 25 in 90% yield as a result of the cycloaddition at the 2- and 4-positions of betaine 20. This chemoselectivity can be rationalized as the cycloaddition proceeding via the interaction between the LUMO of betaine 20 and the HOMO of butadiene. In the presence of excess 6,6-dimethylfulvene, pyridinium salt 20·HCl was treated with triethylamine at 20 °C in diethyl ether for 2 h, affording (5 + 6) cycloadduct 26, albeit in moderate yield. In addition, 1-(4,6-dimethylpyrimidin-2-yl)-3-oxidopyridinium exhibited similar cycloaddition reactivities toward alkenes, 1,3-dienes, and fulvenes. Accordingly, Katritzky et al. demonstrated that oxidopyridinium betaines bearing an electron-withdrawing heteroaryl group at the 1-position are highly versatile 1,3-dipolar reagents. Moreover, the cycloaddition reactivities of 3-oxidopyridinium betaines with diverse electron-withdrawing N-substituents were investigated by the Katritzky group and others (Fig. 4C).18
Katritzky et al. introduced frontier molecular orbital theory to explain the experimentally observed stereo-, regio-, and periselectivities of oxidopyridinium cycloadditions.19 Later, several groups employed modern theoretical calculations based on density functional theory (DFT).20 Although the (5 + 2) cycloaddition of 1-methyl-3-oxidopyridinium with C70 fullerene has yet to be realized experimentally, it was theoretically investigated.21
Katritzky et al. reported the (3 + 2) cycloaddition of 3-oxidopyridinium betaines with haloketenes, in which the oxido moiety and C4 of the betaines were involved (Fig. 5).22 The reaction of 17·HCl with dichloroacetyl chloride in the presence of an excess of triethylamine generated oxidopyridinium 17 and dichloroketene 27a, whose (3 + 2) cycloaddition produced intermediate 28. Subsequent dehydrochlorination afforded the final product 29 in 57% yield. The yield of N-(4,6-dimethylpyrimidin-2-yl)-substituted analog 30 was higher (85%) than that of 29. Although the reaction with bromoketene 27b instead of 27a produced regioisomeric mixtures (e.g., 31 and 32), the (3 + 2) cycloaddition of 1-(p-chlorostyryl)-3-oxidopyridinium with 27b selectively afforded 33 in 61% yield.
 | ||
| Fig. 5 (3 + 2) cycloaddition of 3-oxidopyridinium betaines with haloketenes. | ||
Although less extensively investigated, oxidoisoquinolinium betaines have also been used for (5 + 2) cycloaddition. Katritzky et al. reported that the reaction of 2-methyl-4-oxidoisoquinolinium (34) with acrylonitrile afforded (5 + 2) cycloadduct endo-35a in 18% yield (Fig. 6A).23 Interestingly, the observed stereoselectivity was opposite to that observed for the formation of exo-11b (Fig. 3A). The reaction with methyl acrylate afforded exo- and endo-35b in 13% and 10% yields, respectively. This loss of stereoselectivity was similar to that observed in the reaction of 10 with methyl acrylate (Fig. 3A). Moreover, the reaction of N-(2,4-dinitrophenyl)-substituted oxidoisoquinolinium 36 with N-phenylmaleimide exclusively produced endo-37a in 72% yield.24 In striking contrast, the reaction with acrylonitrile afforded endo-37b in a low yield. Katritzky et al. transformed oxidopyridinium dimer 23, with N-(4,6-dimethylpyrimidin-2-yl) group, into mixed dimer 38 (Fig. 6B).25 Upon heating, 38 underwent retrodimerization to generate 4-oxidoisoquinolinium 39, which could be trapped by N-phenylmaleimide; however, the reaction conditions and yield of exo-40 were not reported. The (5 + 4) cycloaddition of 3-hydroxyquinolinium-derived betaine with 1,3-butadienes was also described; however, neither details of the reaction conditions nor product characterization data were provided (Fig. 6C).26
 | ||
| Fig. 6 (A) (5 + 2) cycloaddition of 2-methyl- and 2-(2,4-dinitrophenyl)-4-oxidoisoquinoliniums. (B) Generation of 2-(4,6-dimethylpyrimidin-2-yl)-4-oxidoisoquinolinium and its (5 + 2) cycloaddition with N-phenylmaleimide. (C) (5 + 4) cycloaddition of 1-methyl-3-oxidoquinolinium with 1,3-butadienes. | ||
In this section, the early discoveries and development of oxidopyridinium cycloaddition have been briefly reviewed. In seminal studies by the Katritzky group, the divergent 1,3-dipolar-cycloaddition reactivity of oxidopyridinium betaines was disclosed. However, because these studies focused on establishing the reaction profiles of oxidopyridinium betaines, their applications in the synthesis of bioactive compounds and natural products lagged. In the following section, advances in the oxidopyridinium cycloadditions after the pioneering discoveries by Katritzky and others are discussed.
2. Advances in (5 + 2) cycloaddition
2.1 Methodological developments
 | ||
| Fig. 7 (A) Intramolecular (5 + 2) cycloaddition of oxidopyridinium betaines with a tethered alkene. (B) Intramolecular (5 + 2) cycloaddition of oxidopyridinium betaine derived from kojic acid. | ||
Mascareñas et al. investigated the intramolecular (5 + 2) cycloaddition of precursors 52, which were prepared from commercially available kojic acid (Fig. 7B).30 Although 4-pyrone 52a, with a sulfur-tethered alkene, was heated in toluene at 140 °C to afford the desired product 54avia 4-siloxy-3-oxidopyrylium 53a, similar 4-pyridone 52b failed to produce the corresponding product 54b. After searching for different reaction conditions, it was found that the treatment of 55 with methyl triflate in chloroform under reflux followed by heating in acetonitrile at 110 °C generated 4-silyloxy-3-oxidopyridinium 56, which underwent the smooth (5 + 2) cycloaddition to afford 57 in 92% yield.
:1 regioselectivity. Among the four possible transition states, TS2 and TS4 are disfavored because of the steric repulsion between one sulfoxide moiety and the C5–H or oxido moiety, whereas TS1 and TS2 are comparably favored, leading to the formation of the major and minor regioisomers of 62, respectively. Similarly, the use of 2-methylated precursor 63a·HCl resulted in the formation of regioisomers of 64a with an improved selectivity of 8:1, albeit in low yield (25%). In contrast, three isomers of 64b were obtained in a 5.5:4.4:1 ratio from 6-methylated precursor 63b·HCl. In addition, the diastereoselective (5 + 2) cycloaddition was studied using an optically active acrylate derived from (S)-methyl lactate (see Section 2.2.1).
 | ||
| Fig. 8 (A) (5 + 2) Cycloaddition of 1-methyl-3-oxidopyridinium with optically active vinyl sulfone. (B) (5 + 2) cycloaddition of 1-benzyl-3-oxidopyridiniums with racemic 2-methylene-1,3-dithiolane 1,3-dioxide. | ||
Oxidopyridinium betaine 66, bearing a chiral auxiliary, was prepared by the Curtis group (Fig. 9).34 Starting from 2-furyl phenyl ketone, titanium-mediated condensation with (S)-phenethylamine was followed by the reduction of the resultant imine to afford amine 65 in high yield. The subsequent treatment of 65 with Br2 in aqueous THF produced the desired oxidopyridinium 66 in 52% yield. Its (5 + 2) cycloaddition with tert-butyl acrylate was conducted in toluene at 95 °C for 4 days, affording four diastereomers of the expected cycloadduct 67. Among these, (1S,5R,6S,9S)-67 was obtained as the major diastereomer in 43% yield.
 | ||
| Fig. 9 Preparation of an oxidopyridinium betaine, bearing a chiral auxiliary, and its (5 + 2) cycloaddition with tert-butyl acrylate. | ||
Jørgensen et al. developed the catalytic enantioselective (5 + 2) cycloaddition using a proline-derived organocatalyst (Fig. 10).35 A challenge with this method is that oxidopyridinium and chiral dienamine should be simultaneously generated in situ. To this end, the authors used dimer 23 as the oxidopyridinium precursor in the presence of catalytic amounts (20 mol%) of organocatalyst 69, pyridinium salt 22·HCl, and diphenyl phosphate (DPP). The reaction with crotonaldehyde 68 quantitatively afforded endo-70 with 96% enatiomeric excess (ee) and high endo/exo- and regioisomeric ratios (Fig. 10A). This method has a broad scope for α,β-unsaturated aldehydes and oxidopyridinium betaines; however, the yield and selectivity altered depending on the substrates (Fig. 10B). Based on the observed stereoselectivity and the results of DFT calculations, a stepwise mechanism was proposed, as outlined in Fig. 10C. The condensation of enal 68 with catalyst 69 generates dienamine 71. The s-cis form of nucleophilic dienamine 71 attacks 22 such that the C(2)–C(γ) bond is formed via TS1. The resultant intermediate 72 undergoes ring closure via TS2, where a second C–C bond is formed between C(6) and C(β) to produce 73. The subsequent hydrolysis of 73 affords endo-70 with the restoration of 69.
 | ||
| Fig. 10 (A) Catalytic enantioselective (5 + 2) cycloaddition with enals using proline-based organocatalyst. (B) Reaction scope. (C) Proposed mechanism. | ||
 | ||
| Fig. 11 (A) (5 + 2) cycloaddition of 3-hydroxy-5-methoxypyridinium N-imine with N-phenylmaleimide. (B) Sequential (5 + 2) cycloaddition and intramolecular oxy-Michael addition. | ||
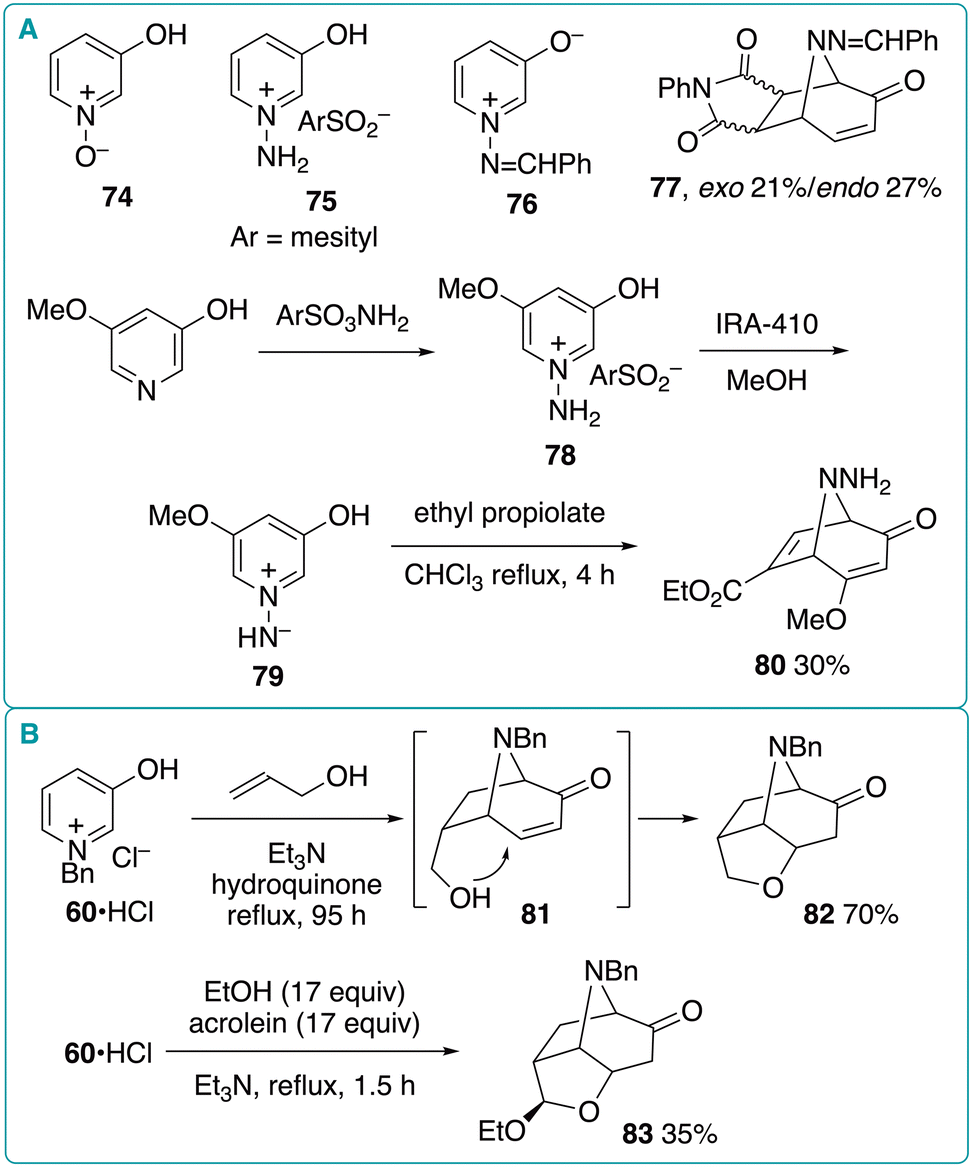
Chavignon et al. reported that the reaction of 1-benzyl-3-oxidopyridinium (60) with allyl alcohol produced tricyclic product 82 in 70% yield, via the intramolecular oxy-Michael addition of (5 + 2) cycloadduct 81 (Fig. 11B).37 The authors also established a three-component reaction of 60, ethanol, and acrolein, which diastereoselectively afforded similar tricyclic product 83, albeit in moderate yield (35%).
Katritzky et al. achieved the (5 + 2) cycloaddition of 1-phenyl-3-oxidopyridinium with benzyne, albeit in moderate yield (Fig. 3B).14a Moreover, the reaction of 1-methyl-3-oxidopyridinium with benzyne produced a 1:2 product. In this study, anthranilic acid was used as the benzyne precursor. Shi et al. reported that the use of Kobayashi reagent 84 as the benzyne precursor improved the yield of the (5 + 2) cycloadducts (Fig. 12A).38 In the presence of CsF (2.6 equiv.), 1-benzyl-3-oxidopyridinium (60) and 84 were stirred in acetonitrile at 17 °C for 18 h, affording the desired (5 + 2) cycloadduct 85 in 78% yield. This method allowed the use of various 1-benzylated oxidopyridinium betaines substituted with methyl, chloro, and hydroxymethyl groups. Moreover, aryl and pyridyl groups as well as various alkyl groups were tolerated as N-substituents. Symmetrically substituted benzyne could be used; however, unsymmetrical benzynes produced regioisomers.
 | ||
| Fig. 12 (A) (5 + 2) cycloaddition of 1-benzyl-3-oxidopyridinium with benzyne generated using the Kobayashi reagent. (B) (5 + 2) cycloaddition of 1-methyl-4-methoxycarbonyl-3-oxidopyridinium with maleimides. | ||
Because the use of 5-methoxycarbonyl-1-methyl-3-oxidopyridinium significantly improved the (5 + 4) cycloaddition efficiency (see Section 3.1), Wang et al. optimized the reaction of pyridinium salt 86·HOTf, derived from methyl 5-hydroxynicotinate, with N-phenylmaleimide to improve the reaction conditions (Fig. 12B).39 They established the optimal conditions involved the use of NaHCO3 as the base in acetonitrile at 80 °C. A 1:1 stoichiometric reaction exclusively afforded the desired product exo-87 in excellent yield (95%). Various N-arylmaleimides and N-alkylmaleimides were used as dipolarophiles; however, the reaction with ortho-substituted phenyl and 1-naphthyl derivatives produced diastereoisomers owing to their axial chirality. Several N-alkyl and ester alkyl groups of the oxidopyridinium betaines were also well tolerated.
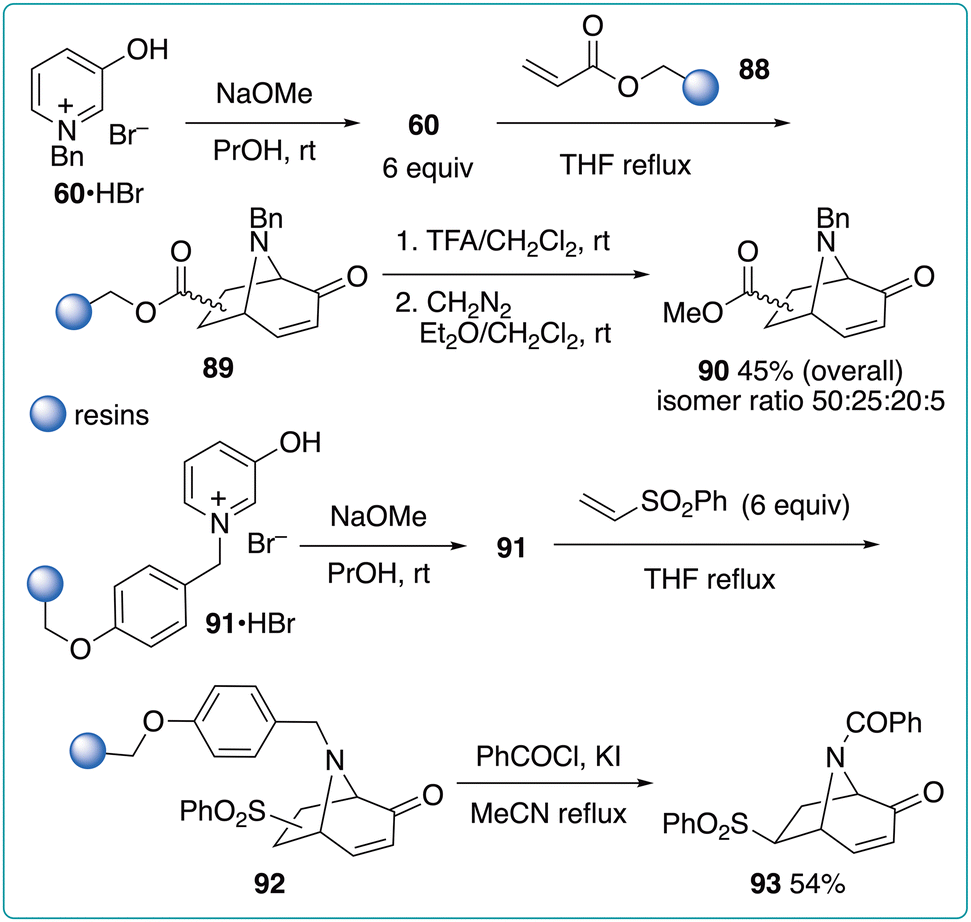
Hanna et al. developed a solid-phase (5 + 2) cycloaddition using resin-supported acrylate or oxidopyridinium betaine (Fig. 13).40 The reaction of resin-supported acrylate 88 with 1-benzyl-3-oxidopyridinium (60, 6 equiv.) produced 89. The acidic cleavage from the resin support and subsequent methylation using diazomethane afforded the final product 90 as a mixture of regio- and stereoisomers in 45% overall yield. In contrast, the reaction of resin-supported oxidopyridinium 91 with phenyl vinyl sulfone and the cleavage from the resin support of the resultant cycloadduct 92 exclusively afforded the final product 93 after the benzoylation of the bridged nitrogen atom. However, although 92 is the common intermediate, methylation and vinylation, rather than benzoylation, produced regioisomeric mixtures.
 | ||
| Fig. 13 Solid-phase (5 + 2) cycloaddition using resin-supported substrates. | ||
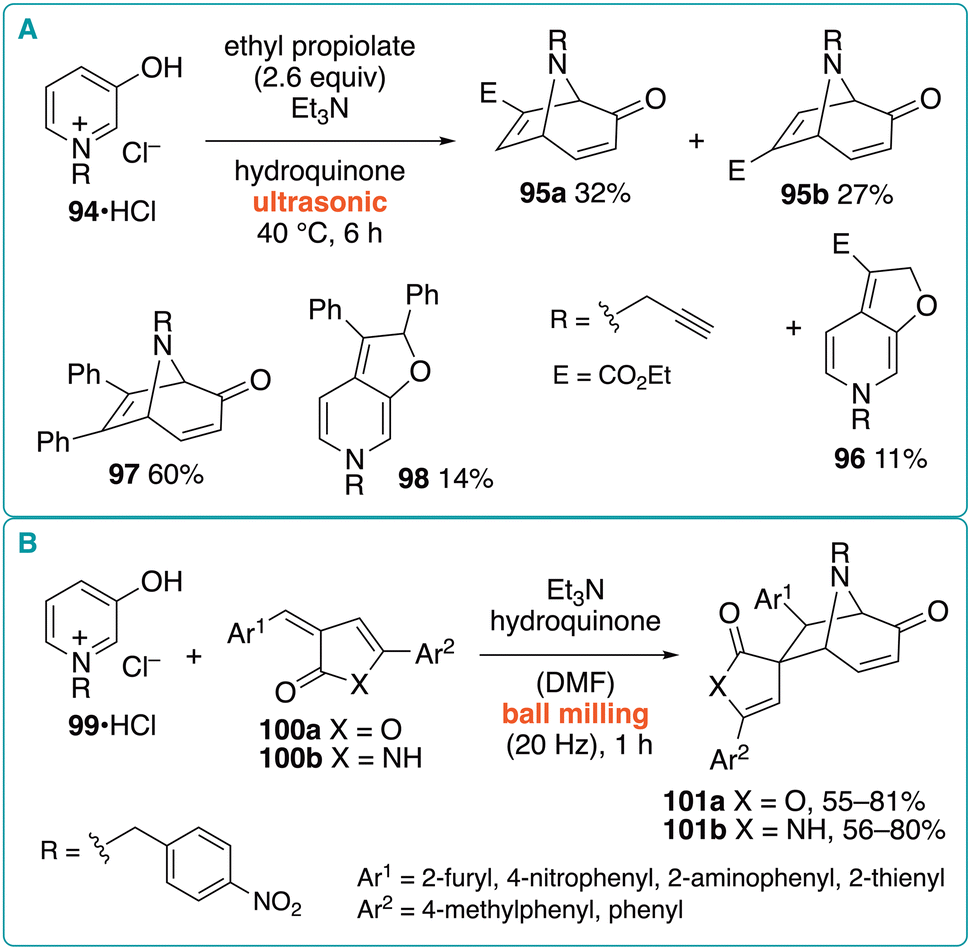
The (5 + 2) cycloaddition using ultrasound irradiation conditions was reported by Hagar et al. (Fig. 14A).41 They investigated the reaction of N-propargylpyridinium chloride 94·HCl and ethyl propiolate in the presence of hydroquinone and triethylamine at 40 °C with ultrasonic irradiation for 6 h, which afforded regioisomeric (5 + 2) cycloadducts 95a and 95b in 32% and 27% yields, respectively. Additionally, (3 + 2) cycloaddition product 96 was obtained in 11% yield. By substituting diphenylacetylene for ethyl propiolate, (5 + 2) cycloadduct 97 was obtained in 60% yield along with (3 + 2) cycloadduct 98 (14% yield). However, because no results from conventional heating experiments were reported, the efficacy of the ultrasound irradiation conditions is unclear. No comment was made regarding the role played by the N-propargyl group.
 | ||
| Fig. 14 (A) (5 + 2) cycloaddition upon ultrasonic irradiation. (B) (5 + 2) cycloaddition using the ball-milling method. | ||
Aboelnaga and Abbady reported the (5 + 2) cycloaddition of oxidopyridinium betaine 99 with benzylidenefuranones 100a or benzylidenepyrrolinones 100b using the ball-milling method (Fig. 14B).42 In the presence of triethylamine and hydroquinone, N-(p-nitrobenzyl)pyridinium salt 99·HCl and furanones 100a (1:1) were allowed to react under ball-milling conditions (20 Hz, stainless-steel vial) for 1 h to afford (5 + 2) cycloadducts 101a as single isomers in 55–81% yield. Notably, chromatographic purification was not required. Although N,N-dimethylformamide (DMF) was used as the solvent, a similar ball-milling reaction using pyrrolinones 100b afforded the corresponding products 101b in 56–80% yield. Because these results were not compared with those of conventional methods, the efficacy of the ball-milling conditions has not been clarified.
 | ||
| Fig. 15 Rh-catalyzed reaction of α-diazo esters bearing a pendant imine with N-phenylmaleimide and DMAD. | ||
Shin et al. reported that when o-alkynylphenyl nitrone 109, with a pendant alkene, was heated in the presence of 2 mol% AuCl3 at 70 °C in nitromethane for 1 h, tetracyclic product 113 was obtained in 82% yield (Fig. 16).44 It was proposed that the 6-exo-cyclization of the nitrone oxygen to the Au-activated alkyne moiety generates vinyl gold intermediate 110, which undergoes internal redox isomerization to generate α-oxo gold carbene complex 111. The subsequent attack of the imine nitrogen on the gold carbene moiety in 111 generates an Au complex of a 4-oxidoisoquinolinium (112). The final intramolecular (5 + 2) cycloaddition of 112 affords 113. This reaction tolerated both the internal and terminal substituents on the pendant alkene moiety. Alkynes could also be used as dipolarophiles, with 114 being obtained in high yield. A tosylamide was compatible as a tether between the alkyne and alkene moieties; however, the ether-tethered substrate underwent decomposition. The phenylene moiety between the nitrone and alkyne moieties was not essential as shown by the reactions of alkenyl nitrones, which afforded similar products 115 and 116 in moderate yields. Moreover, the intermolecular reaction of nitrone 117 with diethyl acetylenedicarboxylate produced the corresponding cycloadduct 118 in 77% yield. This method was extended to an asymmetric cycloaddition using similar substrates bearing a chiral auxiliary on the nitrone moiety.45 Representative examples are shown in Fig. 16B. Intramolecular reactions afforded single diastereomers with a high enantiomeric excess >95%. In contrast, the intermolecular reactions with diethyl acetylenedicarboxylate produced diastereomeric mixtures.
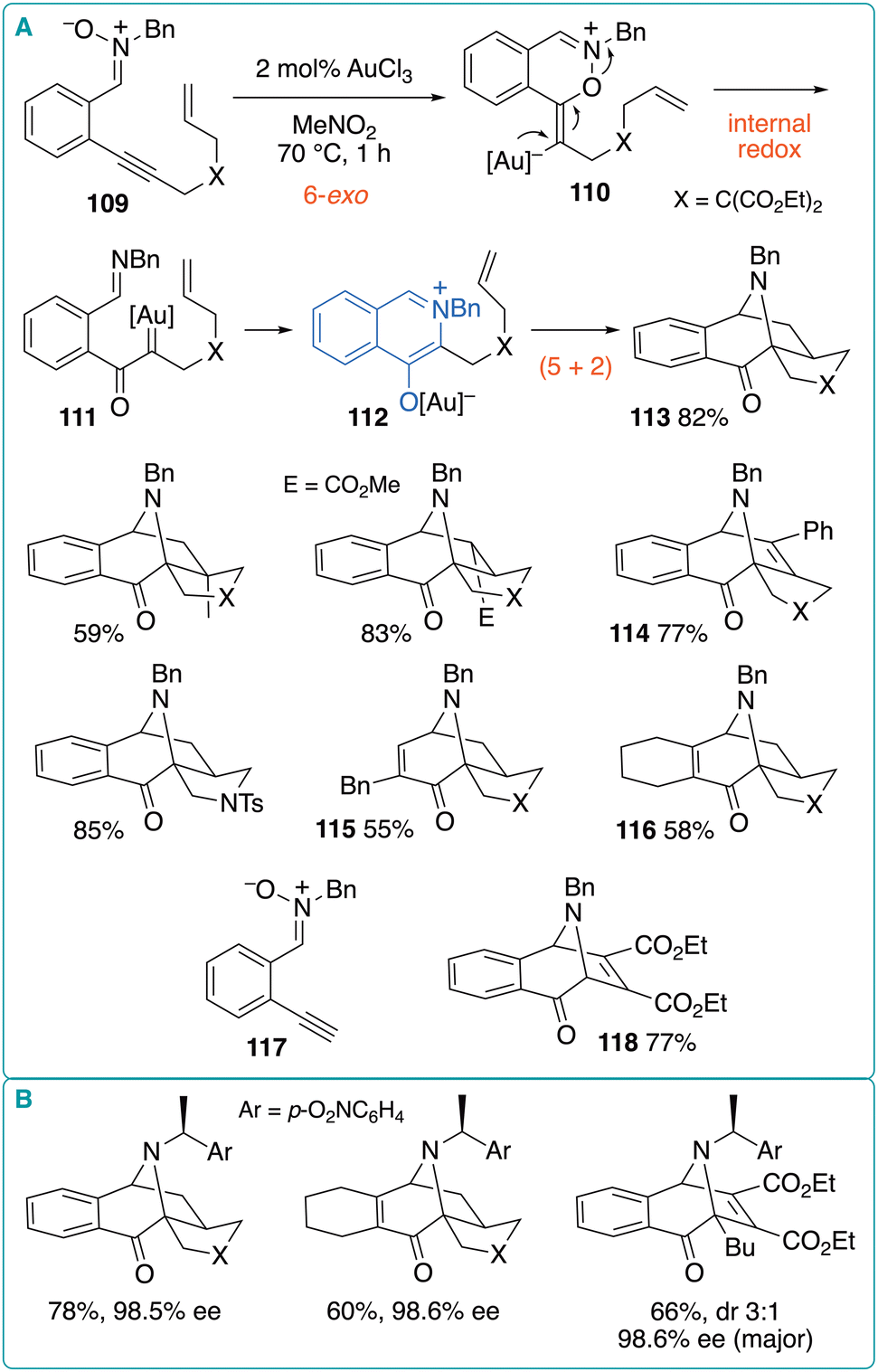 | ||
| Fig. 16 (A) Au-catalyzed cycloisomerization of o-alkynylphenyl nitrones bearing a pendant alkene. (B) Diastereoselective cycloisomerization using a chiral auxiliary. | ||
The formation of 4-oxidoisoquinolinium intermediates and its transition-metal complexes was confirmed by Jia, Li, and their coworkers (Fig. 17).46 The stoichiometric reaction of o-ethynylphenyl nitrone 119 with [Cp*IrCl2]2 in acetonitrile at 0–55 °C afforded the O-bound iridium complex of 4-oxidoisoqunolinium (120) in 84% yield. Notably, its structure was unambiguously confirmed by X-ray crystallography. A similar ruthenium complex was also obtained from 119 and [(p-cymene)RuCl2]2. Moreover, the catalytic isomerization of 119 also occurred in the presence of 1 mol% [Cp*IrCl2]2 in CH2Cl2 at room temperature, affording 4-oxidoisoquinolinium 121 in 87% yield. The catalytically generated 121 was further subjected to the (5 + 2) cycloaddition with N-methylmaleimide at room temperature for 4 h to afford endo-cycloadduct 122 in 85% yield. Similarly, the reaction of 121 with other electron-deficient alkenes such as ethyl acrylate and diethyl fumalate/maleate afforded the corresponding (5 + 2) cycloadducts with varied stereoselectivity. The intermolecular cycloaddition of o-alkynylphenyl nitrones with electron-deficient alkenes was also catalyzed by Pd(OAc)2 (10 mol%) to afford (5 + 2) cycloadducts in good yields.47
 | ||
| Fig. 17 Ir-mediated cycloisomerization of o-ethynylphenyl nitrone leading to 4-oxidoisoquinolinium and its O-bound iridium complex. | ||
The catalytic enantioselective reaction of o-ethynylphenyl nitrones with alkylideneindolinones was reported by Feng et al. (Fig. 18).48 They developed a cooperative catalytic system involving a Pd catalyst for the cycloisomerization of o-ethynylphenyl nitrones and a chiral Co catalyst as a Lewis-acid activator of alkylideneindolinones. Typically, in the presence of catalysts (10 mol% each), the reaction of nitrone 123 and indolinone 124 was performed in CH2Cl2 at −10 °C for 8 h, affording 125 in 78% yield, with diastereomeric ratio of >19:1 and 95% enantiomeric excess. In the absence of a Co catalyst, 123 was converted into 4-oxidoisoquinolinium 126, which was then converted into isolable methyl ether 127. According to the X-ray analysis of the CoL(THF)2 complex and the mechanism underlying the formation of 125, the observed high selectivity was ascribed to the favored TS. This method can be applied to a variety of substrates without lowering the enantioselectivity. However, diminished diastereomeric ratios were observed when nitrones and indolinones bearing substituents on their aromatic rings were used (e.g., 128 and 129). In contrast, aryl substituents on the nitrone moiety had no impact on the stereoselectivity.
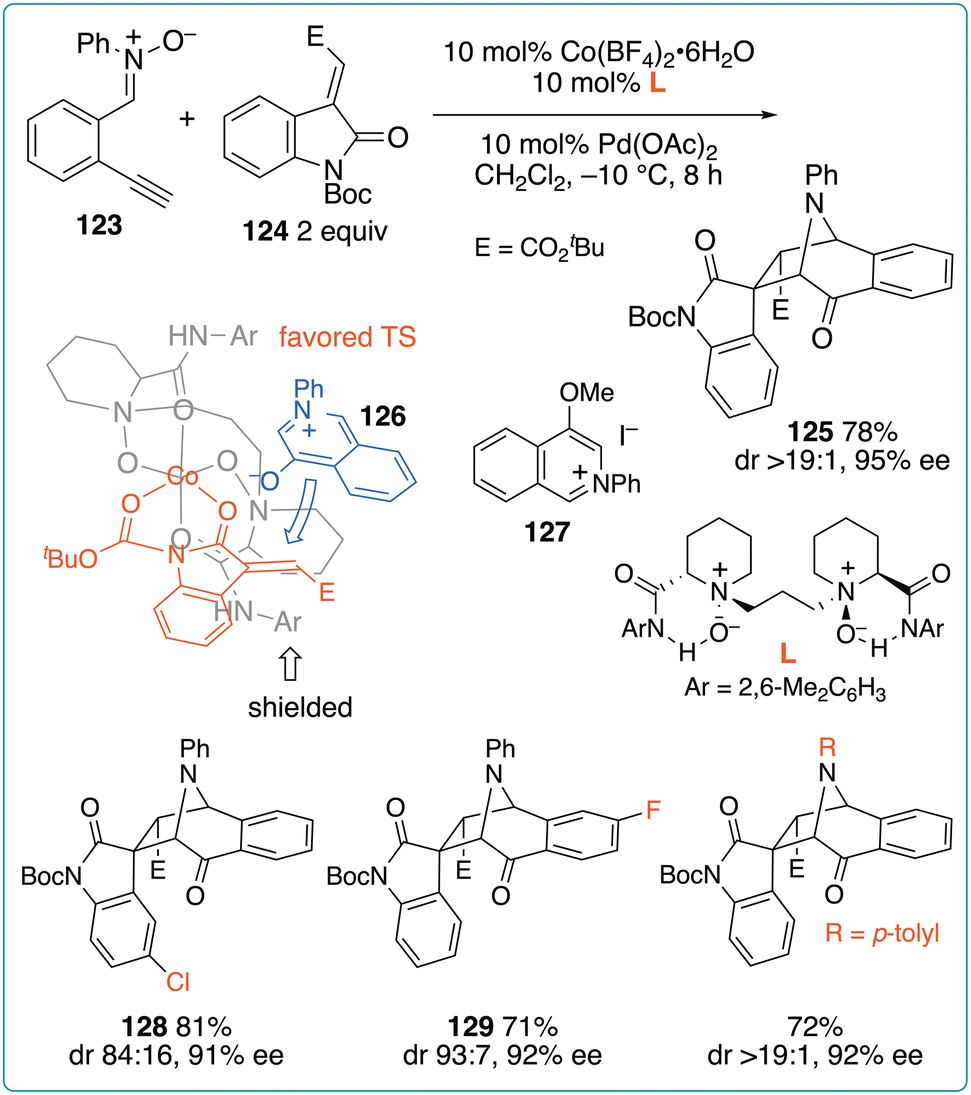 | ||
| Fig. 18 Enantioselective reaction of o-ethynylphenyl nitrones with alkylideneindolinones using a Pd/Co cooperative catalyst system. | ||
2.2 Applications of (5 + 2) cycloaddition
 | ||
| Fig. 19 Synthesis of stipitatic acid and hinokitiol via oxidopyridinium (5 + 2) cycloaddition with propiolates. | ||
The total synthesis of a natural antiglaucoma compound, bao gong teng A, was achieved by Jung et al. (Fig. 20A).50 The authors performed the (5 + 2) cycloaddition of 1-benzyl-3-oxidopyridium (60) with acrylonitrile to obtain endo- and exo-135. The desired isomer, exo-135, which was isolated in 54% yield, was hydrogenated using palladium black to afford 136 in 79% yield. Subsequent ketone reduction was performed using NaBH4 to obtain the desired alcohol 137 in 56% yield along with its epimer (3% yield). Next, the cyano moiety of 137 was transformed into an acetoxy group via the silylation of the hydroxy group (100%), addition of MeMgI to the cyano group (71%), and Baeyer–Villiger oxidation (54%). The final debenzylation of 138 afforded bao gong teng A in 74% yield. The synthesis of C6-epimer of bao gong teng A through the oxidopyridinium (5 + 2) cycloaddition with 2-chloroacrylonitrile was also reported by Pei and Shen.51 Subsequently, the asymmetric synthesis of (–)-bao gong teng A using a chiral auxiliary was reported by Pham and Charlton (Fig. 20B).52 In this study, diastereoselective (5 + 2) cycloaddition of oxidopyridinium 60 with the acrylate of (S)-lactate (139) was developed. Although a very long reaction time (10 days) was required, the desired isomer, exo-140, was obtained with 65% selectivity. Subsequent transformations are similar to those employed in the racemic synthesis by Jung et al. (Fig. 20A). The hydrogenation of exo-140 afforded 141, which was subjected to reduction using a bulky hydride reagent, LiAl(OtBu)3H. The desired product 142 was obtained in 62% yield, along with its epimer (21% yield). After the silylation of the hydroxy group and the benzyl-to-Boc exchange, the chiral auxiliary was removed to afford carboxylic acid 143. The final functional group manipulations gave (–)-bao gong teng A.
 | ||
| Fig. 20 (A) Racemic total synthesis of bao gong teng A. (B) Asymmetric total synthesis of bao gong teng A. | ||
Peese and Gin achieved the total synthesis of the hetisine C20-diterpenoid alkaloid nominine via an intramolecular (5 + 2) cycloaddition of an oxidoisoquinolinium (Fig. 21).53 They prepared oxidoisoquinolinium 147, bearing a pendant cyclohexene moiety on its nitrogen atom, from cyanocyclohexene 144 and azide 145. Tandem aza-Wittig reaction/reduction of 144 and 145 afforded 146 in 79% yield with a diastereomeric ratio of 3:3:2:2. The treatment of 146 with 10% trifluoroacetic acid in methanol at 0 °C generated 147 in 93% yield. Heating 147 in THF at 180 °C promoted intramolecular (5 + 2) cycloaddition via TS1 and TS2, producing 148 and 149 with a 1:3.6 ratio. Because 148 and 149 were in equilibrium under the cycloaddition conditions, the undesired product 149 could be recycled by partial conversion into 148. The desired isomer 148 was transformed into cyclohexenone 150 in several steps. Pyrrolidine-promoted intramolecular Diels–Alder cycloaddition occurred via dienamine 151, affording 152 in 78% yield. After additional steps, 152 was converted into nominine. The same group also accomplished the enantioselective synthesis of nominine by employing enantioenriched 144.54
 | ||
| Fig. 21 Gin's total synthesis of nominine. | ||
The asymmetric total syntheses of three Sarpagine alkaloids were accomplished through the diastereoselective (5 + 2) cycloaddition of an oxidopyridinium with optically active 2-methylene-1,3-dithiolane 1,3-dioxide 61 by Krüger and Gaich (Fig. 22).55 In the presence of Hünig's base, pyridinium salt 153 and 61 (93% ee) were allowed to react in dichloromethane at room temperature for 36 h, affording 154 in 77% yield with a 2:1 regioisomeric ratio. After the reduction of the sulfoxide and enone moieties, the resultant 155 was subjected to Pd-catalyzed cyclization to afford 156 in 88% yield. Subsequent manipulations converted 156 into the key intermediate 157, which was transformed into (+)-vellosomine, (+)-N-methylvellosomine, and (+)-10-methoxyvellosomine, through Fisher indole synthesis using N-phenylhydrazones 158. In the final step, the methyl vinyl ether moiety underwent hydrolysis to give the aldehyde with the desired configuration at the C16-position. Using this strategy, the same group completed the formal total synthesis of 16-epinormacusine B.56 Taking advantage of the flexibility of this strategy, the Gaich group also synthesized non-natural analogs of Sarpagine alkaloids.57
 | ||
| Fig. 22 Gaich's total synthesis of Sarpagine alkaloids. | ||
Gaich et al. also applied their strategy to the total synthesis of a Stemona alkaloid, parvineostemonine (Fig. 23).58 In this case, pyridinium salt 159 was used as an oxidopyridinium precursor to obtain (5 + 2) cycloadduct 160. After the deoxygenation of the sulfoxide moieties, the desired product 161 was obtained in 86% yield with a 5.4:1 regioisomeric ratio. The conjugate allylation of the major isomer of 161 afforded 162 in 83% yield, which was then subjected to ring-closing metathesis using the Grubbs II catalyst to afford 163 in 77% yield. The subsequent removal of the dithioketal moiety and alkene hydrogenation afforded the key intermediate (–)-164, which was subjected to two-step lactonization to afford (+)-parvineostemonine in 62% yield. The minor isomer of 161 was similarly converted into (–)-parvineostemonine.
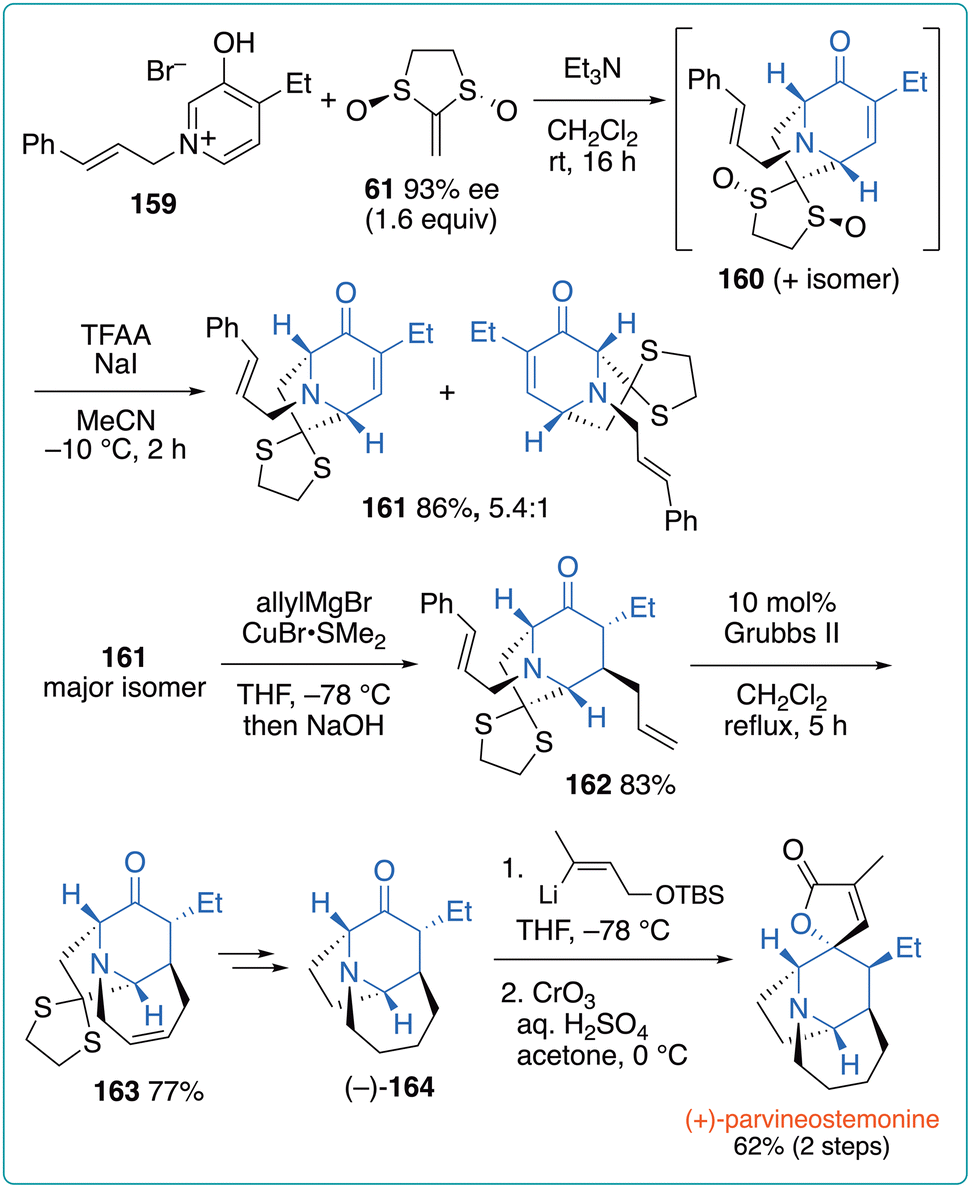 | ||
| Fig. 23 Gaich's total synthesis of both enantiomers of parvineostemonine. | ||
The studies by the Gin and Gaich groups demonstrated that the (5 + 2) cycloaddition of oxidopyridinium and oxidoisoquinolinium betaines serves as an effective strategy for the efficient construction of complex natural products. In the next section, the approach that has been used to create biologically active compounds is discussed.
 | ||
| Fig. 24 (A) (5 + 2) cycloaddition of oxidoisoquinolinium with 1,4-quinones. (B) Synthesis of MK-801 via the (5 + 2) cycloaddition of oxidoisoquinolinium with benzyne. | ||
Cocaine is a potent stimulant of the central nervous system. It has been suggested that cocaine binds to dopamine transporters to inhibit dopamine reuptake, resulting in the reinforcing properties of the drug. Replacing the C3 benzoate in cocaine with an aryl group led to more potent analogs 169 (WIN series, Fig. 25A).61 Kozikowski et al. developed similar compounds 170, bearing an alkyl group instead of a C2 methoxycarbonyl group.62 To synthesize 170, they investigated the (5 + 2) cycloaddition of 1-methyl-3-oxido-4-phenylpyridinium (171) with chiral vinyl sulfoxide (R)-58 in dioxane under reflux (Fig. 25A). Consequently, three diastereomers 172, 173, and 174 were obtained in 44%, 11%, and 22% yields, respectively. The Luche reduction of the major product 172 was followed by acetylation and deoxygenation to afford allylic acetate 175 in 88% yield. Subsequent SN2′ alkylation followed by the treatment with RANEY®-Ni converted 175 into 170 (12–13% yield) and 176 (40–43% yield). Alternatively, a diastereomeric mixture of 172/173 was subjected to conjugate alkylation to afford 177 and 178 (Fig. 25B). These intermediates were separately converted into 170a and its enantiomer via a five-step transformation sequence. Using this strategy, Kozikowski et al. synthesized a series of C2/C3-modified cocaine analogs and evaluated their biological activity.63 There are several reports on the synthesis of biologically interesting tropanes via oxidopyridinium (5 + 2) cycloaddition with activated alkenes.64
 | ||
| Fig. 25 (A) Enantioselective synthesis of 2-alkyl-3-phenyltropanes via the (5 + 2) cycloaddition of oxidopyridinium with chiral vinyl sulfoxide. (B) Alternative late-stage transformations of (5 + 2) cycloadducts. | ||
To gain insights into the effect of the orientation of the nitrogen lone pair on binding affinity, Kozikowski et al. developed tricyclic cocaine analogs, in which the conformation of the N-lone pairs is restricted by introducing an additional ring moiety (Fig. 26).65 Pyridinium salts 171·HBr, bearing a 2-bromopropenyl substituent on the nitrogen atom, were subjected to (5 + 2) cycloaddition with phenyl vinyl sulfone to obtain 172 in 21–60% yield as a mixture of regioisomers. According to a report by Ghosh and Hart,66 the radical cyclization of 172 afforded the desired tricyclic products 173 in 54–75% yield. Although the partial reduction of the p-chlorophenyl moiety of 173b occurred during the reductive desulfonylation step, p-tolyl analog 173a was successfully converted into the constrained cocaine analog 174a. An alternative tricyclic compound was also synthesized via the intramolecular (5 + 2) cycloaddition of betaine 175. The resultant 176 was converted into allylic acetate 177, which was subjected to SN2′ alkylation to afford 178 and 179 in 42% and 20% yields, respectively, along with C2 epimers. Finally, the hydrogenation of the major product 178 afforded 180 and 181 in a 3:1 ratio. Preliminary biological experiments showed that 174a and 180 exhibited substantial affinity for the dopamine transporter. Therefore, the orientation of the N-lone pairs has no impact on the binding affinity. Moreover, the same group synthesized the benzo-fused constrained cocaine analog 183 from pyridinium salt 182·HBr, as well as other analogs 184 and 185 from 175.67 The binding affinities of several monoamine transporters were investigated using the analogs described above. The binding affinities of the constrained analogs for the dopamine transporter were found to be 2.5- to 104-fold higher than that for cocaine.
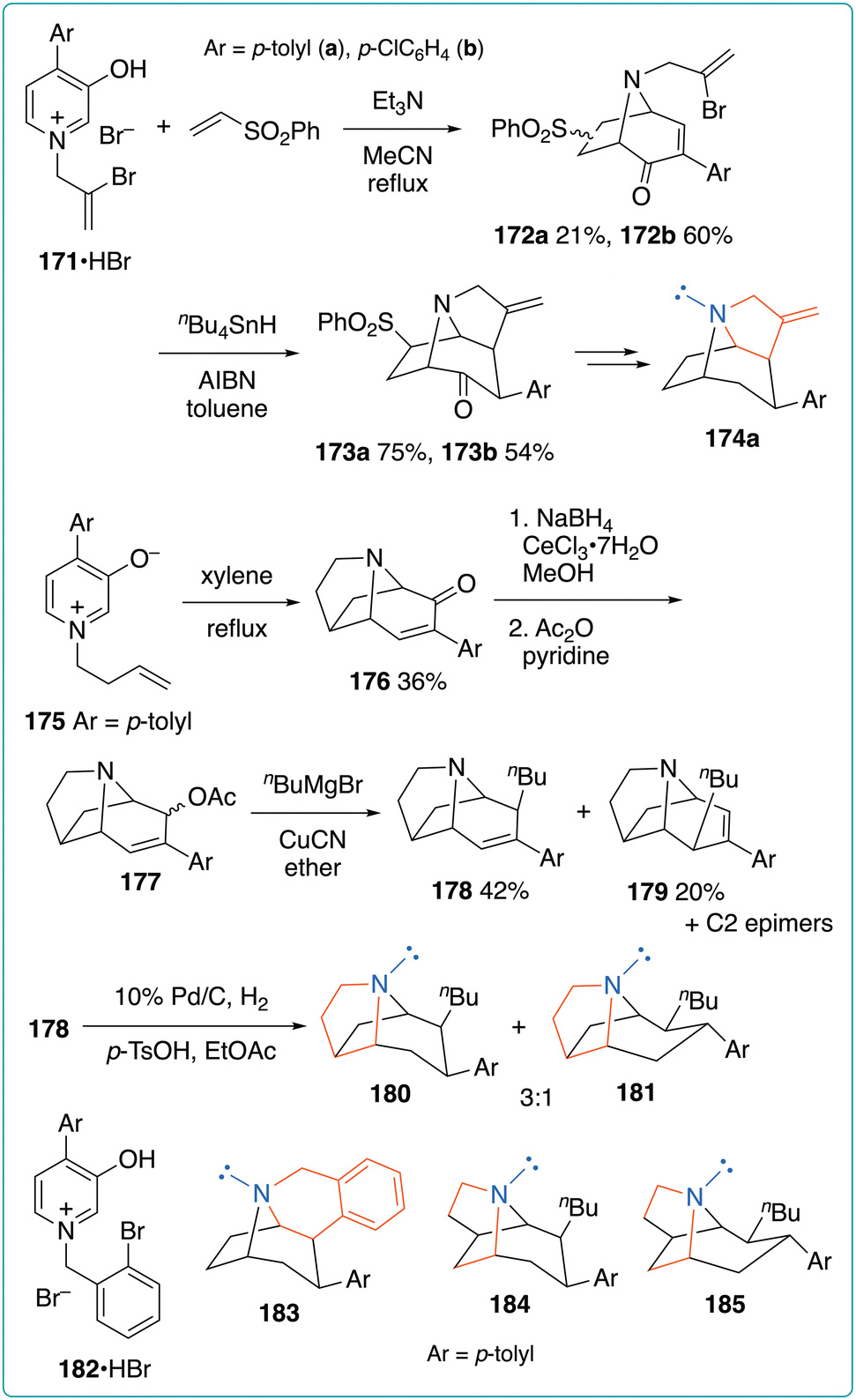 | ||
| Fig. 26 Synthesis of constrained cocaine analogs via the (5 + 2) cycloaddition of 4-aryl-2-oxidopyridinium betaines. | ||
 | ||
| Fig. 27 (A) Kinetic resolution of oxidopyridinium (5 + 2) cycloadducts. (B) Enantiodivergent synthesis of pyrrolidine-fused tropanes. | ||
The (5 + 2) cycloadducts derived from oxidopyridinium betaines and vinyl sulfones are highly versatile platforms for the synthesis of tropanone derivatives.69 Marsden et al. investigated the divergent transformations of vinyl-sulfone-derived (5 + 2) cycloadducts to construct a tropane-based molecular library (Fig. 28A).70 To this end, they transformed 188 into 189via hydrogenation, 190via intramolecular Michael addition, and 191via Rh-catalyzed conjugate arylation. In addition, the reductive Heck cyclization of N-(o-bromophenyl)methyl derivative 192, leading to tetracyclic derivative 193, was performed under conditions similar to those reported by the Grigg group.71 Furthermore, diverse fused tropane derivatives were prepared by converting saturated and unsaturated tropanones, such as 188 and 189. Selected examples are shown in Fig. 28B.
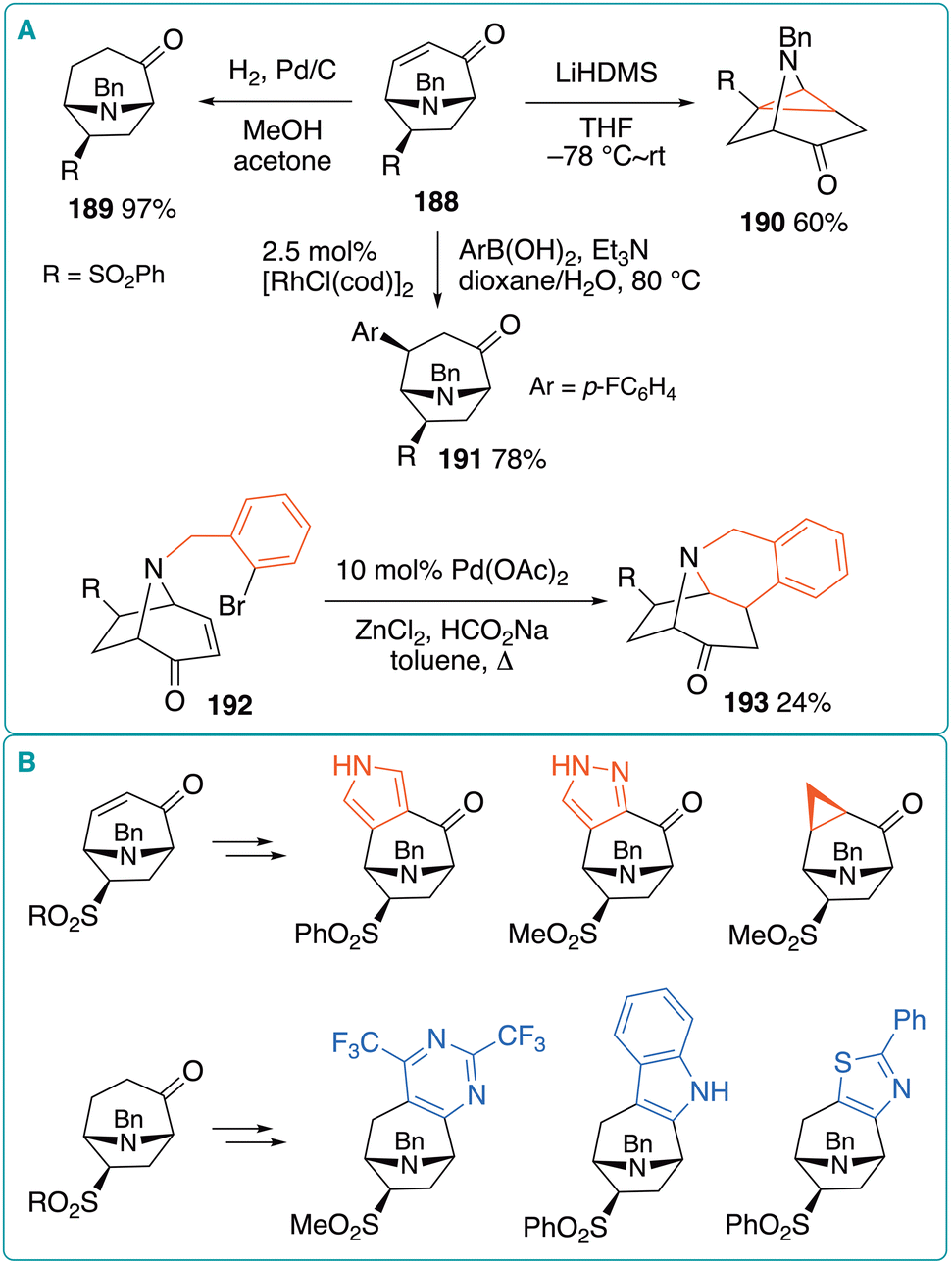 | ||
| Fig. 28 (A) Transformations of oxidopyridinium (5 + 2) cycloadducts derived from phenyl vinyl sulfone. (B) Divergent transformations of saturated and unsaturated tropanones into fused tropane derivatives. | ||
The trifluoromethyl (CF3) group is one of the most common fluoroalkyl moieties, and its introduction into bioactive compounds can modify biological properties such as lipophilicity, metabolic stability, and binding affinity to target receptors.72 Thus, CF3-substituted tropanes are promising candidates for drug discovery. Yamamoto et al. reported the dearomative transformation of CF3-substituted pyridine-3-ols via oxidopyridinium cycloaddition.73 The authors investigated the transformation of pyridin-3-ols bearing a CF3 group at different positions and found that the regio- and stereoselectivity changed depending on the substitution positions (Fig. 29). The N-methylation of 2-(trifluoromethyl)pyridin-3-ol 194 was conducted using MeOTf in toluene at 90 °C for 1 h, and the resultant pyridinium and N-methylmaleimide (1 equiv.) were treated with triethylamine (2 equiv.) in toluene at 110 °C for 16 h in the same pot, affording a mixture of endo- and exo-195 in 87% yield with a 4.8:1 ratio. Similarly, 6-(trifluoromethyl)pyridin-3-ol 196 was subjected to one-pot N-methylation/(5 + 2) cycloaddition to afford a mixture of endo- and exo-197 in 95% yield with a lower stereoselectivity of 2.1:1. In contrast, the transformation of 5-(trifluoromethyl)pyridin-3-ol 198 exclusively produced exo-199 in 68% yield. Thus, the position of the CF3 group has a notable impact on the stereoselectivity. Furthermore, a similar reaction of 194 with phenyl vinyl sulfone exclusively afforded exo-200 in 84% yield, whereas an 8:1 mixture of exo-201 and endo-202 was obtained in 78% yield from 196. The latter result suggests that the bulky CF3 group altered the regioselectivity of the cycloaddition, such that the sulfonyl group adopted a position distal to the CF3 group. A similar stereocontrolling effect of the CF3 group was observed in the reaction with dimethyl fumarate: in this case, products 203 and 204, in which the CF3 and adjacent methoxycarbonyl groups were mutually trans, were selectively obtained from 194 and 196, respectively.
 | ||
| Fig. 29 Dearomative transformation of trifluoromethyl-substituted pyridin-3-ols into the corresponding tropane derivatives. | ||
The use of 3-oxidopyridinium betaines as bioorthogonal dipoles was reported by Pezacki et al. (Fig. 30).74 They performed the (5 + 2) cycloaddition of betaines 205, bearing a substituent R at the 5-position with 4-dibenzocyclooctynol 206 in acetonitrile at 25 °C, and bimolecular rate constants k2 were determined through UV/Vis absorption spectroscopy under pseudo-first-order kinetic conditions. This study demonstrated that the introduction of electron-donating groups at the 5-position led to significant rate increases. The stability of the most reactive derivative (205, R = OMe) in an aqueous medium relevant to chemical biology was quantitatively investigated by UV/Vis spectroscopy in 9:1 phosphate-buffered saline (pH 7.4)/DMSO with equimolar concentrations of dithiothreitol: under these conditions, less than 5% decrease in absorbance was observed after 18 h, demonstrating the stability of 205 under biological conditions.
 | ||
| Fig. 30 Kinetic study of the (5 + 2) cycloaddition of oxidopyridinium betaines with 4-dibenzocyclooctynol. | ||
3. Advances in (5 + 4) and (5 + 6) cycloaddition
3.1 (5 + 4) cycloaddition with 1,3-dienes
The (5 + 4) cycloaddition of 3-oxidopyridinium betaines with 1,3-dienes has been underdeveloped in relation to the (5 + 2) cycloadditions discussed in the previous sections although the expected products are fascinating bridged nitrogen heterocycles. As a synthetic application, Cha et al. reported the construction of the tricyclic core of sarain A via (5 + 4) cycloaddition (Fig. 31A).75 The key reaction was performed upon treatment of 20·HCl and cyclopentadiene with triethylamine, affording (5 + 4) cycloadducts 208a and 208b in 58% and 4% yields, respectively, along with (5 + 2) cycloadduct 209 (27% yield). The enamine moiety of 208a was reduced using NaBH3CN/TFA to produce 210 in 70% yield. Because the 5-nitropyridin-2-yl-substituted products were less soluble in common organic solvents, the nitro group of 210 was reduced to an amine, and 211 was obtained in 85% yield after Boc protection of the resultant amino group. After many steps, including the oxidative ring opening of the cyclopentene moiety, 211 was transformed into the tricyclic core model of sarain A (212). However, the removal of the N-Boc-5-aminopyridin-2-yl group was not achieved. Therefore, the Cha group examined the use of a 3-oxidopyridinium with the removable N-group (Fig. 31B). After screening several (E)-β-(benzoyl)vinyl-substituted betaines for the (5 + 4) cycloaddition with cyclopentadiene, p-fluorophenyl analog 213 was found to be optimal. The desired (5 + 4) cycloadduct 214 was obtained in 60% yield along with the undesired (5 + 2) cycloadduct 215 (15% yield). After the reduction of the enamine moiety of 214, the benzoylvinyl group of the resultant 216 was replaced with a readily removable Boc group in high yield. Thus, 217 was transformed into a tricyclic core model of sarain A in a manner similar to the conversion of 211 into 212. | ||
| Fig. 31 (A) Construction of the tricyclic core of sarain A via the (5 + 4) cycloaddition of 1-(5-nitropyridin-2-yl)-3-oxidopyridinium with cyclopentadiene. (B) (5 + 4) cycloaddition of 3-oxidopyridinium bearing an (E)-β-(4′-fluorobenzoyl)vinyl group with cyclopentadiene and subsequent transformations. | ||
Electron-withdrawing N-substituents on 3-oxidopyridinium betaines are necessary for efficient (5 + 4) cycloaddition with 1,3-dienes. This requirement limits the application of (5 + 4) cycloaddition products in natural product synthesis, as illustrated above (Fig. 31). To address this limitation, Harmata et al. developed the (5 + 4) cycloaddition of 1-methyl-3-oxidopyridinium bearing an ester substituent at the 5-position, thereby eliminating the need for electron-withdrawing groups on the nitrogen atom (Fig. 32A).76 They conducted the N-methylation of methyl nicotinate (218) with methyl triflate in dichloromethane at ambient temperature, and the resultant pyridinium was directly used for the subsequent (5 + 4) cycloaddition with 2,3-dimethylbuta-1,3-diene (10 equiv.) in the presence of triethylamine (3 equiv.) in acetonitrile at 85 °C (sealed tube). Using this approach, the target product 219 was quantitatively obtained. DFT calculations were performed to identify the most stable transition state (also see below). A representative range of 1,3-dienes is shown in Fig. 32A. The parent 1,3-butadiene and exocyclic dienes were used to obtain the corresponding products in high yield. The use of penta-1,3-diene afforded 220 as a 1:1 diastereomeric mixture in 70% yield. In contrast, the reaction with 1-phenylbuta-1,3-diene produced 221 in 86% yield with a regioisomeric ratio of 10:1. The use of 2-methylbuta-1,3-diene led to the formation of the regioisomers of 222 in 80% yield with a ratio of 1.2:1. To realize an intramolecular (5 + 4) cycloaddition, the same authors prepared a pyridinium salt bearing a pendant 1,3-diene on the nitrogen atom from ethyl nicotinate (223) and triflate 224 (Fig. 32B). The subsequent reaction was conducted in the same manner as the intermolecular reaction, affording the desired tricyclic product 226 in 84% yield, albeit with low diastereoselectivity.
 | ||
| Fig. 32 (A) Tandem N-methylation/(5 + 4) cycloaddition of methyl nicotinate with 1,3-dienes. (B) Intramolecular (5 + 4) cycloaddition of a pyridinium salt derived from ethyl nicotinate. | ||
Given that the (5 + 4) cycloaddition of 1-alkyl-substituted 1,3-butadienes regioselectively proceeded, further scope studies were conducted by the Harmata group.77 1-Alkyl-substituted 1,3-butadienes were subjected to (5 + 4) cycloaddition with 3-oxidopyridinium 227 to regioselectively obtain the corresponding products 228 in 61–97% yields with diastereomeric ratios ranging from 38:62 to 56:44 (Fig. 33A). In addition to alkyl substituents, a benzoate substituent was also compatible. When vinylcyclohexene derivative 229 was used as the 1,3-diene component, tetracyclic product 230 was obtained in 94% yield with a 37:63 diastereomeric ratio. The use of sorbyl acetate as the diene component produced a mixture of four isomeric cycloadducts. In contrast, the reaction of butadienyl phenyl sulfide 231 afforded endo-cycloadduct 232 in 41% yield with a 75:25 regioisomeric ratio.
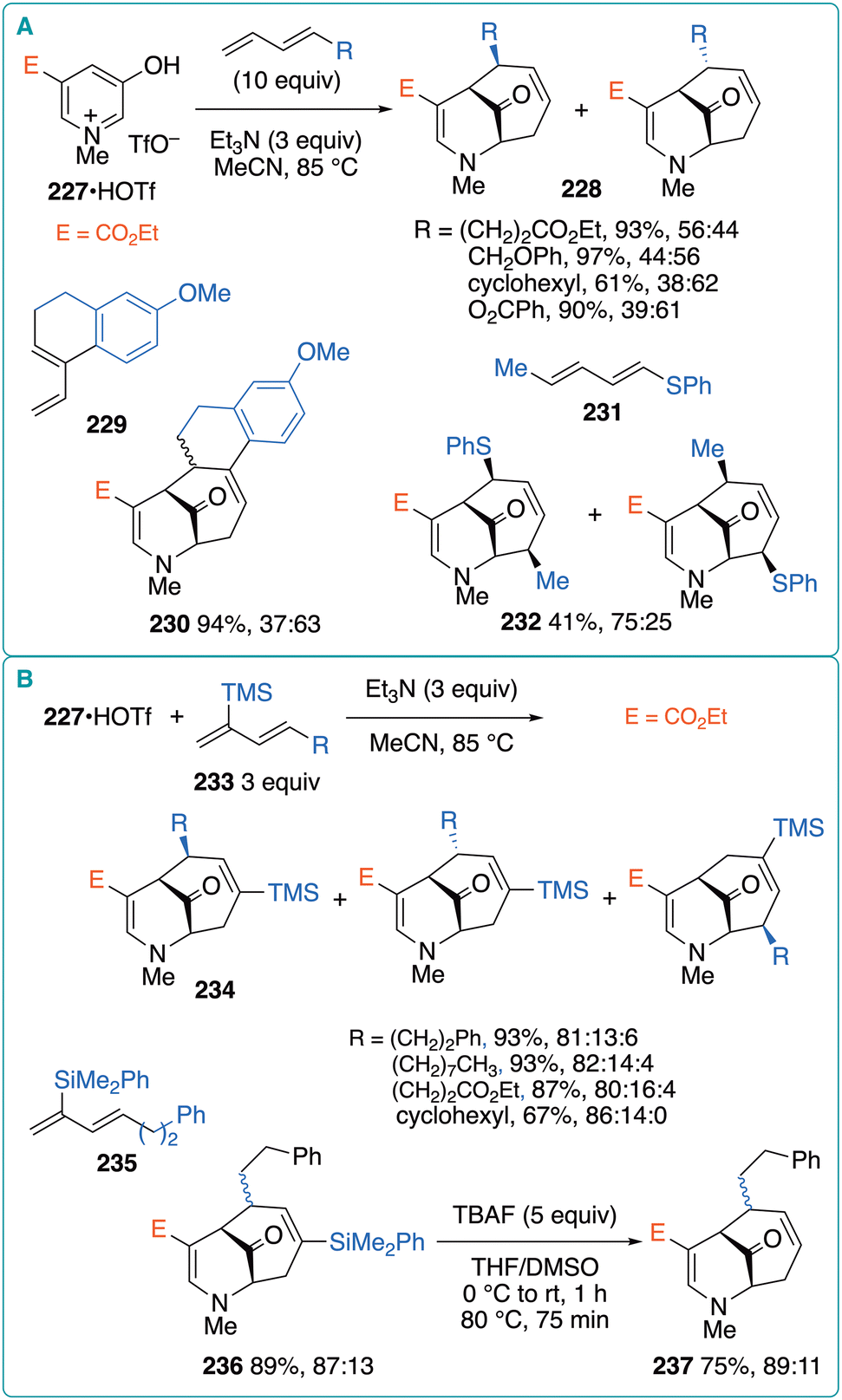 | ||
| Fig. 33 (A) (5 + 4) cycloaddition of ester-substituted oxidopyridinium with unsymmetrical 1,3-dienes. (B) (5 + 4) cycloaddition of ester-substituted oxiopyridinium with dienylsilanes. | ||
Moreover, dienylsilanes 233 were used as the diene component with the expectation that steric interactions between the bulky TMS group and ester substituent on the betaine would lead to improved diastereoselectivity (Fig. 33B).78 Actually, the reaction using 227·HOTf and 233 produced 234 as the major isomer. The use of diene 235 with a much bulkier dimethylphenylsilyl group resulted in the formation of 236 as the major isomer in 89% yield with an 87:13 diastereomeric ratio. The treatment of 236 with tetra(n-butyl)ammonium fluoride (TBAF) afforded desilylation product 237 in 75% yield with an 89:11 diastereomeric ratio.
Harmata et al. envisioned that the intramolecular (5 + 4) cycloaddition (cf.Fig. 32B) would provide a straightforward route to the ABC-ring system of natural alkaloid daphnicyclidin A. However, the reaction of 225·HOTf afforded the desired (5 + 4) cycloadduct with unsatisfactory diastereoselectivity. Therefore, they reinvestigated the influence of the substituted dienes on the intramolecular (5 + 4) cycloaddition (Fig. 34A).79 The alkylation of 223 with triflate 239 was conducted under neat conditions (no solvent, 80 °C, 18 h) to quantitatively afford pyridinium salt 240·HOTf, which was treated with triethylamine (3 equiv.) in acetonitrile at 85 °C for 9 h. Under these conditions, cycloadduct 241 was obtained in 80% yield as the sole product. Thus, the methyl substituent at the 5′-position is beneficial for diastereocontrol. The terminal methyl substituent of 242·HOTf was also found to be effective: cycloadduct 243 was obtained with high diastereoselectivity, albeit with moderate yield (51%). The reaction of pyridinium 244·HOTf, which has a TMS substituent at the 6′-position, produced cycloadduct 245 in 66% yield as the sole diastereomer. A higher reaction temperature was required for the reaction of pyridinium salts with shorter two-carbon tethers between the nitrogen and diene moieties. The reaction of 246·HOTf was conducted using sodium benzoate as a base in benzonitrile at 220 °C for 0.5 h, affording the desired ABC-ring unit 247 in 74% yield with complete diastereoselectivity. Moreover, the same group prepared pyridinium salt 249, possessing a pendant 3-sulfolene, from 223 and triflate 248 (Fig. 34B).80 The treatment of 249 with sodium benzoate in benzonitrile at 180 °C for 1 h successfully produced the ABCE-ring unit 251 in 70% yield, via the formation of exocyclic diene 250 with concomitant extrusion of SO2.
 | ||
| Fig. 34 (A) Intramolecular (5 + 4) cycloaddition of ester-substituted oxidopyridiniums with pendant 1,3-dienes. (B) Intramolecular (5 + 4) cycloaddition of ester-substituted oxidopyridinium with a pendant 3-sulfolene. | ||
Burns and Boittier performed DFT calculations to investigate the reaction of 5-methoxycarbonyl-substituted oxidopyridinium 86 with 1,3-butadiene (Fig. 35).81 In accordance with the DFT analysis by Harmata et al.,76 two TSs were found for the (5 + 4) cycloaddition leading to 252. Among these, TS1 for the exo-approach was slightly more efficient than TS2 for the endo-approach. TS3 for the (5 + 2) cycloaddition can be located 4.1 kcal mol−1 above TS1, and (5 + 2) cycloadduct 253 is 12.8 kcal mol−1 less stable than (5 + 4) cycloadduct 252. Moreover, the [3,3]-sigmatropic rearrangement of 253 led to the formation of 252via TS4. Accordingly, (5 + 4) cycloadduct 252 should be produced selectively.
 | ||
| Fig. 35 DFT calculations of the (5 + 4) and (5 + 2) cycloaddition of 1-methyl-3-methoxycarbonyl-3-oxidopyridinium with 1,3-butadiene [Gibbs energies (kcal mol−1) are shown in parentheses]. | ||
Jørgensen et al. achieved the enantioselective (5 + 2) cycloaddition of oxidopyridinium betaine 22 with acrolein derivatives using proline-derived organocatalyst 69 (Fig. 10).35 Although dienamine 71 is involved in the enantioselective cycloaddition, no (5 + 4) cycloadduct was observed. It was considered that 71 attacks betaine 22 to produce iminium intermediate 72, which reversibly generates (5 + 2) intermediate 73 or (5 + 4) intermediate 254 (Fig. 36A). Because catalyst restoration from 254 is impossible, a (5 + 2) cycloadduct (endo-70) was selectively obtained upon catalyst release from 73. To achieve the enantioselective (5 + 4) cycloaddition, the same group investigated the reaction of dienal 255 (Fig. 36B).82 In the presence of catalyst 256 (20 mol%) and pivalic acid (1 equiv.), 255 and the oxidopyridinium dimer of 23 were allowed to react in o-dichlorobenzene (oDCB) at room temperature for one day. However, rather than the expected (5 + 4) product 258, cyclopentene-fused cycloheptenone 261 was obtained in 52% yield with 59% ee. This result was rationalized as the intramolecular attack of the enamine moiety on the formyl group generating iminium intermediate 259, which is hydrolyzed to afford aminoaldehyde 260. The subsequent E1cB elimination of H2O from 260 ultimately produces 261. Although the product yields were low to moderate, several alkyl substituents on the dienal component were compatible; however, tricyclic product 262 was obtained in low yield with low enantioselectivity. The enantioselectivity was improved when (hetero)aromatic substituents were introduced into the dienal components (i.e., 263–265).
 | ||
| Fig. 36 (A) Mechanistic rationale explaining why no (5 + 4) cycloadduct is obtained from dienamine intermediate and oxiopyridinium. (B) Enantioselective formation of cyclopentene-fused cycloheptenones via (5 + 4) cycloaddition using dienals. | ||
In the above examples, oxidopyridinium betaines possessing N-heteroaryl or 5-alkoxycarbonyl substituents were essential for efficient (5 + 4) cycloadditions. In contrast, Yamamoto et al. reported that highly reactive o-quinodimethane could be used as the 1,3-diene component for the (5 + 4) cycloaddition of various oxidopyridinium betaines.83 As the o-quinodimethane precursor, [(trimethylsilyl)methyl]benzyl acetate (267) was used with 1-(pyrimidin-2-yl)-3-oxidopyridinium dimer 266 (Fig. 37A). These substrates were treated with KF (3.6 equiv.) in DMF at 100 °C for 3 h, affording the expected (5 + 4) cycloadduct 268 in 80% yield. More importantly, the N-pyrimidyl group was not necessary. N-Methylated pyrimidinium 269·HI and 267 were treated with KF (4 equiv.) in DMF at 100 °C for 5 h, affording 270 in 72% yield from 5-chloropyridin-3-ol. In this method, KF was used as the activator of 267 and as the base for the deprotonation of 269·HI. Similarly, 5-phenyl, 5-methoxycarbonyl, and 6-trifluoromethyl derivatives were obtained in 52–65% yield. The same authors also showed that sultine 271 could be used as a precursor of o-quinodimethane (Fig. 37B). Oxidopyridinium dimer 266 and sultine 271 were simply heated in 1,2-dichloroethane (DCE) under reflux to afford 268 in 73% yield. Symmetrically substituted sultines were used to obtain the corresponding (5 + 4) cycloadducts in high yields. Moreover, 5-halogenated pyridinium salts 272·HCl and 271 were treated with AcONa as the base in refluxing DCE to obtain the corresponding cycloadducts 273 in 63–94% yield. The stereoselective transformations of (5 + 4) cycloadduct 268 at its enamine and carbonyl moieties were also achieved as shown in Fig. 37C.
 | ||
| Fig. 37 (A) (5 + 4) cycloaddition using [(trimethylsilyl)methyl]benzyl acetate as the o-quinodimethane precursor. (B) (5 + 4) cycloaddition using sultines as the o-quinodimethane precursors. (C) Stereoselective transformation of the representative cycloadduct. | ||
3.2 (5 + 6) Cycloaddition with fulvenes
Katritzky et al. reported that the reaction of N-(5-nitropyridin-2-yl)-substituted oxidopyridinium betaine with 6,6-dimethylfulvene produced (5 + 6) cycloadduct 26, instead of the corresponding (5 + 4) or (5 + 2) cycloadduct involving the cyclopentadienyl moiety of the fulvene (Fig. 4B).17b The generality of this (5 + 6) cycloaddition was later investigated by Radhakrishnan and coworkers (Fig. 38A).84N-(Pyrimidin-2-yl)-substituted pyridinium salt 274·HBr and 6,6-diphenylfulvene (275) were treated with triethylamine in THF at 0 °C to room temperature, affording (5 + 6) cycloadduct 277 in 68% yield via a 1,5-H shift of the initially generated intermediate 276. The reactions using 6,6-dialkylfulvenes also afforded the corresponding products in similar yields. In addition, 5-nitropyridin-2-yl-substituted pyridinium salts were used to obtain the corresponding (5 + 6) cycloadducts in similar yields. Interestingly, the sterically demanding adamantane-derived fulvene 279 was allowed to react with oxidopyridinium betaine 278 under the same conditions to produce cycloadduct 280 in 36% yield, without a subsequent 1,5-H shift (Fig. 38B). The structure of 280 was unambiguously confirmed by X-ray crystallography. In addition, (5 + 2) cycloadduct 281 was also obtained in 26% yield.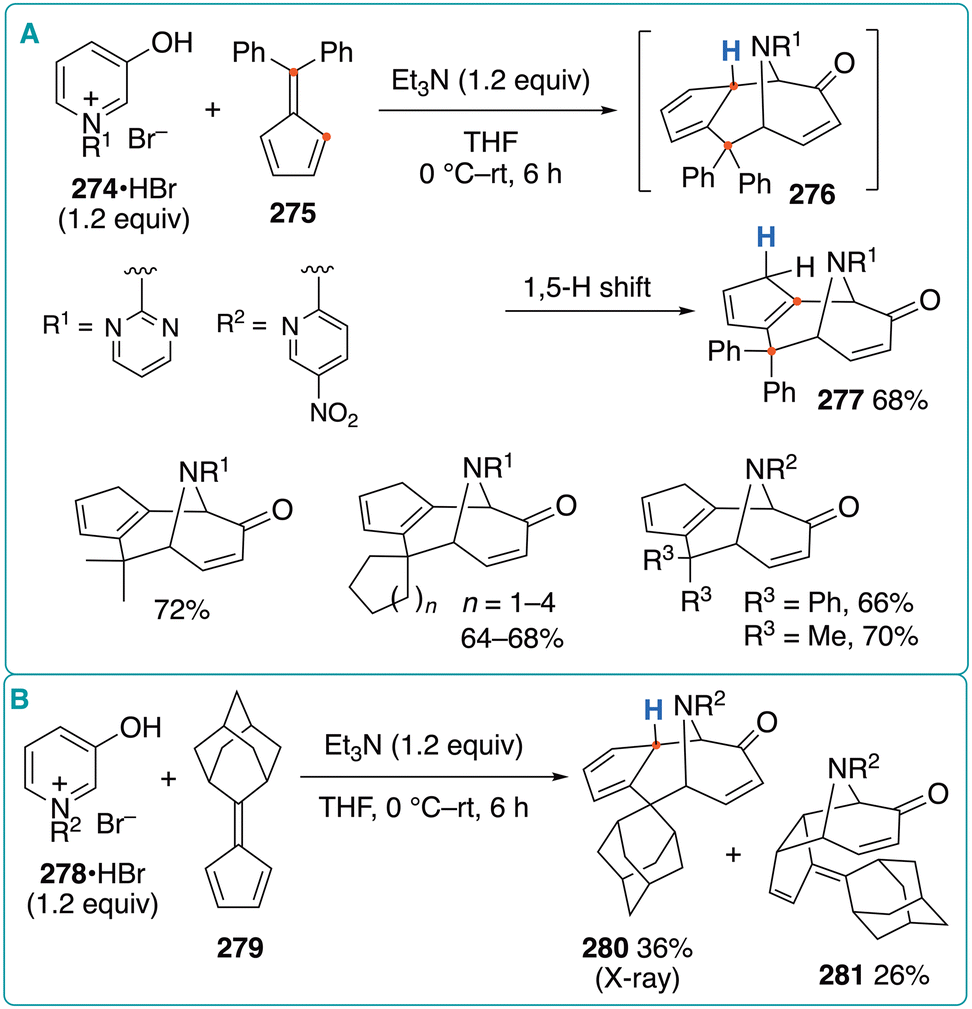 | ||
| Fig. 38 (A) (5 + 6) cycloaddition of N-heterocycle-substituted oxidopyridinium betaines with fulvenes. (B) Reaction of N-(5-nitropyridin-2-yl)-substituted oxidopyridinium with adamantane-derived fulvene. | ||
Yamamoto et al. also revisited the (5 + 6) cycloaddition of oxidopyridium 274 with 6,6-dimethylfulvene (282) and observed a different outcome (Fig. 39A).20d In the presence of triethylamine, 274·HCl and 282 (2 equiv.) were heated in THF under reflux for 23 h, affording a 1.5:1 mixture of the known (5 + 6) cycloadduct 283 and minor product 284 in 60% combined yield. Because the similar peak patterns of cyclopentadienyl methylene protons were observed in their 1H NMR spectra, it was assumed that 283 and 284 are mutually isomeric. DFT calculations suggest that the cycloaddition of 274 and 282 proceeds via an ambimodal TS, from which post-TS bifurcation occurs to generate both (5 + 4) cycloadduct 285 and initial (5 + 6) product 286 (Fig. 39B). The former is 12.9 kcal mol−1 less stable than the latter, and 285 can be converted into 286via a [3,3]-sigmatropic rearrangement. The subsequent 1,5-H shift from 286 produces the experimentally observed major product 283, which is 2.3 kcal mol−1 more stable than 286. Moreover, two additional 1,5-H shifts from 283 would produce isomeric product 284, which is only 0.5 kcal mol−1 less stable than 283. According to these results, the experimentally observed minor product can be assigned to 284.
 | ||
| Fig. 39 (A) (5 + 6) cycloaddition of N-(pyrimidin-2-yl)-substituted oxidopyridinium betaine with 6,6-dimethylfulvene. (B) DFT analysis of the formation of isomeric (5 + 6) products from N-(pyrimidin-2-yl)-substituted oxidopyridinium betaine and 6,6-dimethylfulvene [Gibbs energies (kcal mol−1) are indicated in parentheses]. | ||
4. Cycloadditions of related six-membered N-heterocyclic betaines
In this section, the reactions of six-membered N-heterocyclic betaines relevant to 3-oxidopyridiniums are briefly discussed. Early examples of cycloadditions involving benzodiazine-derived betaines are shown in Fig. 40. In 1969, Ames and Novitt reported that the reaction of 2-methyl-4-oxidocinnolinium 287 with DMAD afforded (5 + 2) cycloadduct 288 in 32% yield (Fig. 40A).85 In 1975, Katritzky et al. investigated the (5 + 2) cycloaddition of 6-chloro-2-methyl-4-oxidocinnolinium with alkynes.86 The reaction with DMAD afforded 289a in 66% yield, which was higher than the yield of 288. Although the reaction with less reactive diphenylacetylene required a higher reaction temperature (180 °C), 289b was obtained, albeit in a significantly lower yield (30%). The use of phenylacetylene as the unsymmetrical alkyne led to the regioselective formation of 289c in 60% yield. The Katritzky group also reported the reaction of 3-methyl-1-oxidophthalazinium 290 with diphenylacetylene,86 which afforded the corresponding (5 + 2) cycloadduct 291 in 80% yield (Fig. 40B). Moreover, the reaction of the parent phthalazine-1(2H)-one (292) with benzyne, derived from diazonium 293, produced 294 in 75% yield. One possibility is that 292 reacts with one benzyne molecule to generate betaine 295, which then reacts with a second benzyne molecule to produce 294. Similarly, the reaction of 292 with DMAD afforded 1:2 product 296 in 58% yield.
 | ||
| Fig. 40 (A) (5 + 2) cycloaddition of 2-methyl-4-oxidocinnolinium betaines with alkynes. (B) (5 + 2) cycloaddition of 1-oxidophthalazinium betaines with alkynes and benzyne. | ||
Joule et al. found that the reaction of oxidopyrazinium 297 with dipolarophiles afforded cyclic enamides 299 through the tautomerization of the initial (5 + 2) cycloadduct 298 (Fig. 41A).87 The reaction with methyl acrylate selectively afforded exo-299a in 85% yield, whereas exo- and endo-299b were obtained in 25% and 29% yields, respectively, from the reaction with acrylonitrile. Cycloaddition with methyl propiolate or indene selectively produced 299c or endo-299d in 75% and 40% yields, respectively. As a model reaction for the synthesis of quinocarcin, the reaction of betaine 300 with methyl acrylate was conducted to afford exo-301, bearing a benzylidene moiety, in moderate yield. The Joule group further investigated the reactivity of more sterically hindered oxidopyraziniums (Fig. 41B). The reaction of tetramethyl oxidopyrazinium 302 with methyl acrylate proceeded with opposite regioselectivity to that observed in the reaction of 297.88 As a result, exo-cycloadduct 303 was obtained in 58% yield, and its structure was unambiguously confirmed by X-ray crystallography. The reaction of trimethyl analog 304 with methyl methacrylate produced tricyclic product 307 in 59% yield.89 A follow-up theoretical study suggested that the initial (5 + 2) cycloadduct 305 undergoes skeletal rearrangement to generate betaine intermediate 306, from which lactonization proceeds to afford 307.90 Moreover, N-PMB derivative 308 underwent dimerization to produce tetracyclic product 310via an intramolecular reaction between the enamine and iminium moieties in 309.
 | ||
| Fig. 41 (A) (5 + 2) cycloaddition of oxidopyrazinium betaines with dipolarophiles. (B) Reactions of highly hindered oxidopyrazinium betaines. | ||
In relation to oxidopyrazinium betaines, cyclic azomethine ylides 312 were also used for natural product syntheses. The photolysis of bicyclic aziridines 311 generated ylides 312, which were trapped by dipolarophiles to afford (5 + 2) cycloadducts 313 (Fig. 42A).91 This method has been applied to the asymmetric total synthesis of natural products, such as (–)-quinocarcin, (–)-tetrazomine, and (–)-lemonomycin.92 Recently, new pyridinium betaines derived from 1-aryl-3,4-dialkylpyridiniums were reported by Hansmann et al. (Fig. 42B).93 For example, pyridinium 314 was treated with potassium hexamethyldisilazide (KHMDS) at −40 °C to generate betaine 315 in 77% yield. In contrast to oxidopyridinium betaines, 315 underwent the (3 + 2) cycloaddition with activated alkenes to afford 316a–c in high yields with excellent regio- and diastereoselectivities.
 | ||
| Fig. 42 (A) (5 + 2) cycloaddition of azomethine ylides derived from imide-fused aziridines. (B) (3 + 2) cycloaddition of pyridinium betaines generated from deprotonation of 1-aryl-3,4-dialkylpyridiniums. | ||
5. Conclusions
Katritzky's pioneering studies revealed the multifaceted cycloaddition reactivities of oxidopyridinium betaines. The most investigated reaction of oxidopyridinium betaines is the (5 + 2) cycloaddition, in which acrylates, acrylonitrile, vinyl sulfones, and styrene are commonly used as the dipolarophiles. Although 1-alkyl-3-oxidopyridinium betaines showed limited scope, 1-(heteroaryl)-3-oxidopyridiniums were found to exhibit significantly improved reactivity toward various alkenes. Moreover, the latter class of betaines was used for the (5 + 4) cycloaddition with 1,3-butadienes and (5 + 6) cycloaddition with fulvenes. The (5 + 2) cycloaddition has been continuously studied to improve its scope and selectivity, leading to the development of enantioselective methods, intramolecular cycloadditions, and some modern methods involving solid-phase synthesis, ultrasound irradiation, and mechanochemical techniques. Consequently, oxidopyridinium (5 + 2) cycloadditions have been successfully applied to the total synthesis of natural products and the creation of medicinally important molecules. In contrast, the development of (5 + 4) and other cycloadditions has been slower than that of the (5 + 2) cycloaddition. Nevertheless, Harmata et al. reported a breakthrough discovery that 1-methyl-5-(alkoxycarbonyl)-3-oxidopyridinium betaines exhibit high reactivity toward the (5 + 4) cycloaddition with 1,3-butadienes. They also demonstrated the synthetic utility of their method by applying it to the synthesis of natural product subunits.A potential limitation of this methodology is the limited availability of oxidopyridinium betaine precursors (i.e., pyridine-3-ol and its derivatives). Therefore, new methods to generate betaines from readily available precursors should be developed. The transition-metal-mediated generation of 4-oxidoisoquinoliniums from alkynes is one viable approach.
In addition to experimental studies, modern theoretical investigations using DFT methods have been reported that provide insights into the reactivity and selectivity of oxidopyridinium betaines. It is expected that combined experimental and computational studies will accelerate the development of new methods and applications in a wide range of research areas such as drug discovery, chemical biology, and materials science, to which this exciting methodology can add so much.
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research is partially supported by the Platform Project for Supporting Drug Discovery and Life Science Research (Basis for Supporting Innovative Drug Discovery and Life Science Research (BINDS) from AMED under Grant Number JP24ama121044).References
- (a) F. Lovering, J. Bikker and C. Humblet, Escape from Flatland: Increasing Saturation as an Approach to Improving Clinical Success, J. Med. Chem., 2009, 52, 6752 CrossRef CAS PubMed; (b) F. Lovering, Escape from Flatland 2: complexity and promiscuity, Med. Chem. Commun., 2013, 4, 515 RSC.
- (a) J. Jia, F. Hu and Y. Xia, Transition-Metal-Catalyzed Nucleophilic Dearomatization of Electron-Deficient Heteroarenes, Synthesis, 2022, 92 Search PubMed; (b) M. Escolano, D. Gaviña, G. Alzuet-Piña, S. Díaz-Oltra, M. Sánchez-Roselló and C. del Pozo, Recent Strategies in the Nucleophilic Dearomatization of Pyridines, Quinolines, and Isoquinolines, Chem. Rev., 2024, 124, 1122 CrossRef CAS; (c) H. Jia, Z. Tan and M. Zhang, Reductive Functionalization of Pyridine-Fused N-Heteroarenes, Acc. Chem. Res., 2024, 57, 795 CrossRef CAS PubMed.
- (a) N. Dennis, A. R. Katritzky and Y. Takeuchi, Synthetic Applications of Heteroaromatic Betaines with Six-Membered Rings, Angew. Chem., Int. Ed. Engl., 1976, 15, 1 CrossRef CAS PubMed; (b) A. R. Katritzky and N. Dennis, Cycloaddition Reactions of Heteroaromatic Six-Membered Rings, Chem. Rev., 1989, 89, 827 CrossRef CAS; (c) W. Śliwa, Cycloaddition Reactions of Pyridinium Ylides and Oxidopyridiniums, Heterocycles, 1996, 43, 2005 CrossRef; (d) K. V. Radhakrishnan, Heterocycles via Pyrylium and Pyridinium Betaines, Top. Heterocycl. Chem., 2008, 13, 71 CAS.
- Selected reviews of related cycloaddition chemistry: (a) M. Harmata, J. Tu and M. M. Clark, (4+3) Cycloadditions of Allylic and Related Cations, Org. React., 2024, 115, 1 Search PubMed; (b) K. Gao, Y.-G. Zhang, Z. Wang and H. Ding, Recent Development on the [5+2] Cycloadditions and Their Application in Natural Product Synthesis, Chem. Commun., 2019, 55, 1859 RSC; (c) J. Huang, L. Zhang and X. Meng, Recent Advances in the Cyclization Reactions of Pyridinium 1,n-Zwitterions (n = 4 and 5): Scope and Mechanism, Org. Chem. Front., 2023, 10, 2813 RSC.
- A. R. Katritzky and J. M. Lagowski, Prototropic Tautomerism of Heteroaromatic Compounds: II. Six-Membered Rings, Adv. Heterocycl. Chem., 1963, 1, 339 CrossRef CAS.
- Reviews of oxidopyrylium cycloadditions: (a) V. Singh, U. M. Krishna, Vikrant and G. K. Trivedi, Cycloaddition of Oxidopyrylium Species in Organic Synthesis, Tetrahedron, 2008, 64, 3405 Search PubMed; (b) L. P. Bejcek and R. P. Murelli, Oxidopyrylium [5+2] Cycloaddition Chemistry: Historical Perspective and Recent Advances (2008–2018), Tetrahedron, 2018, 74, 2501 CrossRef CAS PubMed.
- J. Banerji, N. Dennis, J. Frank, A. R. Katritzky and T. Matsuo, 1,3-Dipolar Character of Six-membered Aromatic Rings. Part XXVI. 3-Hydroxypyridine and 1-Benzyl-3-oxidopyridinium, J. Chem. Soc., Perkin Trans. 1, 1976, 2334 RSC.
- A symbolism (m + n) is used to identify a cycloaddition between precursors involving m and n linearly connected atoms. The symbolism “(5 + 2)” has been conventionally used for cycloadditions of oxidopyryliums and oxidopyridiniums with 2π components (ref. 3d and 6), whereas a different symbolism “(3 + 2)” has also been used for these cycloadditions. In this review, “(5 + 2)” is used because oxidopyridiniums have delocalized betaine structures, which make a significant contribution to the HOMO and LUMO levels involved in their cycloadditions. Similarly, “(5 + 4)” and “(5 + 6)” are used for the cycloadditions of oxidopyridiniums with 1,3-dienes and fulvenes, respectively, though some have referred to the former as (4 + 3) cycloadditions.
- S. A. El-Abbady, M. S. Abdulhalim, M. K. Awad and A. H. Moustafa, Enhanced Reactivity of Pyridin-3-ol Towards 4-Phenyl-1,2,4-triazoline-3,5-dione: FMO Treatment of the Cycloaddition Process by ASED-MO Calculations Method, Indian J. Heterocycl. Chem., 1995, 5, 55 CAS.
- D. Samuel, K. Norrell and D. G. Hilmey, Novel Ring Chemistry of Vitamine B6 with Singlet Oxygen and an Activated Ene: Isolated Products and Identified Intermediates Suggesting an Operable [3 + 2] Cycloaddition, Org. Biomol. Chem., 2012, 10, 7278 CAS.
- (a) A. R. Katritzky and Y. Takeuchi, A New Route to Tropones Based on the 1,3-Dipolar Reactivity of Heteroaromatic Betaines, J. Am. Chem. Soc., 1970, 92, 4134 CAS; (b) A. R. Katritzky and Y. Takeuchi, 1,3-Dipolar Character of Six-membered Aromatic Rings. Part I. 1-Methyl-3-oxidopyridinium, J. Chem. Soc. C, 1971, 874 CAS; (c) A. R. Katritzky and Y. Takeuchi, 1,3-Dipolar Character of Six-membered Aromatic Rings. Part II. A Novel Route to Tropolones, J. Chem. Soc. C, 1971, 878 CAS.
- T. Sasaki, K. Kanematsu, K. Hayakawa and M. Uchida, Studies of Bridged Benzoheterocycles. Part III. Cycloadditions of 1,4-Epoxy-1,4-dihydronaphthalenne to Some Dipolar Compounds and Dienes, J. Chem. Soc., Perkin Trans. 1, 1972, 2750 RSC.
- Y. Tamura, M. Akita, H. Kiyokawa, L.-C. Chen and H. Ishibashi, 1,3-Dipolar Cycloaddition of 5-Methoxy-1-methyl-3-oxidopyridinium, Tetrahedron Lett., 1978, 1751 CrossRef CAS.
- (a) N. Dennis, A. R. Katritzky, T. Matsuo, S. K. Parton and Y. Takeuchi, 1,3-Dipolar Character of Six-membered Aromatic Rings. Part VII. 1-Phenyl-3-oxidopyridinium, J. Chem. Soc., Perkin Trans. 1, 1974, 746 RSC; (b) N. Dennis, A. R. Katritzky, S. K. Parton, Y. Nomura, Y. Takahashi and Y. Takeuchi, 1,3-Dipolar Character of Six-membered Aromatic Rings. Part XVIII. Adducts from 3-Oxido-1-phenylpyridinium and their Quaternisation and Conversion into Tropone Derivatives, J. Chem. Soc., Perkin Trans. 1, 1976, 2289 RSC.
- (a) S. A. El-Abbady, A. A. Al-Ahmady and A. H. Moustafa, Regio- and Stereochemistry of 1,3-Dipolar Cycloadditions of 3-Pyridinol and 3-Oxidopyridinium Betaines with β-Nitrostyrenes, Indian J. Chem., 1992, 31B, 24 CAS; (b) A. Banerji, S. Haldar and J. Banerji, 1,3-Dipolar Cycloadditions: Part IV—Cycloaddition of 1-Phenyl-3-oxidopyridinium Betaines to Electron-deficient Alkenes, Indian J. Chem., 1999, 38B, 641 CAS.
- (a) N. Dennis, B. Ibrahim and A. R. Katritzky, Rationalisation of the Variation of Reactivity towards Cycloaddition with Structure in 3-Oxido-pyridinium and -azinium Betaines and a Study of 1-(4,6-Dimethyl-2-pyrimidyl)-3-oxidopyridinium, J. Chem. Soc., Chem. Commun., 1975, 425 CAS; (b) N. Dennis, B. Ibrahim and A. R. Katritzky, 1,3-Dipolar Character of Six-membered Aromatic Rings. Part XX. Preparation and Dimerisation of 1-(5-Nitro-2-pyridyl)- and 1-(4,6-Dimethylpyrimiidin-2-yl)-3-oxidopyridinium, J. Chem. Soc., Perkin Trans. 1, 1976, 2296 CAS.
- (a) N. Dennis, B. Ibrahim and A. R. Katritzky, The Dimer of 1-(5-Nitro-2-pyridyl)-3-oxidopyridinium and its Reactions, J. Chem. Soc., Chem. Commun., 1974, 500 RSC; (b) N. Dennis, B. Ibrahim and A. R. Katritzky, 1,3-Dipolar Character of Six-membered Aromatic Rings. Part XXI. Thermal Cycloadditions of 1-(5-Nitro-2-pyridyl)- and 1-(4,6-Dimethylpyrimidyl-2-yl)-3-oxidopyridinium with 2, 4, and 6 π-Electron Components, J. Chem. Soc., Perkin Trans. 1, 1976, 2307 RSC.
- (a) A. R. Katritzky, J. Banerji, A. Boonyarakvanich, A. T. Cutler, N. Dennis, S. O. Abbas Rizvi, G. J. Sabongi and H. Wilde, 1,3-Dipolar Character of Six-membered Aromatic Rings. Part 34. The Search for Superior 1-Substituents to Facilitate Cycloadditions of 3-Oxidopyridinium, J. Chem. Soc., Perkin Trans. 1, 1979, 399 CAS; (b) A. R. Katritzky, J. Banerji, N. Dennis, J. Ellison, G. J. Sabongi and E.-U. Würthwein, 1,3-Dipolar Character of Six-membered Aromatic Rings. Part 44. Further Examples of Troponoid Synthesis, J. Chem. Soc., Perkin Trans. 1, 1979, 2528 CAS; (c) A. R. Katritzky, A. Boonyarakvanich and N. Dennis, 1,3-Dipolar Character of Six-membered Aromatic Rings. Part 49. 3-Oxido-1-(4-pyridyl)pyridinium, 3-Oxido-1-(2-pyridyl)pyridinium, 3-Oxido-1-(quinoxolin-2-yl)pyridinium, 3-Oxido-1-(5,6-diphenyl-1,2,4-triazin-3-yl)pyridinium, and 3-Oxido-1-(5-phenyl-1,2,4-triazin-3-yl)pyridinium, J. Chem. Soc., Perkin Trans. 1, 1980, 343 CAS; (d) A. H. Moustafa, S. A. El-Abbady and R. A. Jones, Dipolar Additions with 1-(4,6-Dimethylpyrimidin-2-yl)- and 1-(4,6-Dimethoxy-s-triazin-2-yl)-3-oxidopyridinium Betaines, J. Heterocycl. Chem., 1981, 18, 1461 CAS; (e) S. A. El-Abbady, M. G. M. Ahmed, H. A. Abdulhamid, A. A. Shalaby and A. H. Moustafa, Regio- and Stereoselectivity of 1-(Pyridazin-3-yl)-3-oxidopyridinium Betaines towards 2π- and 4π-Conjugated Olefins, J. Prakt. Chem., 1989, 331, 105 CAS; (f) S. A. El-Abbady, H. A. Abdul-Hamid, A. A. Shalaby, M. G. Ahmed and A. H. Moustafa, [4n + 2]π-Cycloadditions of 1-(6-arylpyridazin-3-yl)-3-oxidopyridinium betaines with 2π-1,3-dipolarophiles, Indian J. Chem., 1989, 29B, 923 Search PubMed; (g) S. A. El-Abbady and S. M. Agami, 1-(4-Phenylphthalazin-1-yl)-3-oxidopyridinium Betaine: Rearrangement and Dipolar additions with 2π-1,3-Dipolarophiles, Indian J. Chem., 1995, 34B, 504 CAS; (h) S. A. El-Abbady, S. M. Agami and W. A. M. Mokbel, [π2s + π4s] Dimerisation of 1-(Saccharin-1-yl)pyridinium-3-oxide and Its Dipolar Additions with 2π-1,3-Dipolarophiles, Heterocycl. Commun., 1996, 2, 169 CAS.
- A. R. Katritzky, N. Dennis, M. Chaillet, C. Larrieu and M. El Mouhtadi, 1,3-Dipolar Character of Six-membered Aromatic Rings. Part 38. FMO Treatment of Peri-, Site, Regio, and Stereo-selectivity, and Relative Reaction Rates of Cycloaddition Reactions, J. Chem. Soc., Perkin Trans. 1, 1979, 408 RSC.
- (a) L. Rhyman, H. H. Abdallah, S. Jhaumeer-Laulloo, L. R. Domingo, J. A. Joule and P. Ramasami, The 1,3-Dipolar Cycloaddition of 1H-Pyridinium-3-olate and 1-Methylpyridinium-3-olate with Methyl Acrylate: a Density Functional Theory Study, Tetrahedron, 2010, 66, 9187 CrossRef CAS; (b) L. Rhyman, H. H. Abdallah, S. Jhaumeer-Laulloo, L. R. Domingo, J. A. Joule and P. Ramasami, Regio- and Stereoselectivity of the 1,3-Dipolar Cycloaddition of Pyridinium-3-olates and Pyrazinium-3-olates with Methyl Methacrylate: A Density Functional Theory Exploration, Curr. Org. Chem., 2012, 16, 1711 CrossRef CAS; (c) Y. Lu and D. J. Tantillo, Comparison of (5 + 2) Cycloadditions Involving Oxidopyrylium and Oxidopyridinium Ions: Relative Reactivities, J. Org. Chem., 2021, 86, 8652 CrossRef CAS PubMed; (d) Y. Yamamoto, Y. Shizume, S. Tazawa and T. Yasui, Oxidopyridinium Cycloadditions Revisited: A Combined Computational and Experimental Study on the Reactivity of 1-(2-Pyrimidyl)-3-oxidopyridinium Betaine, J. Org. Chem., 2023, 88, 3193 CrossRef CAS PubMed.
- L. Türker and S. Gümüş, 1,3-Dipolar Cycloaddition Reaction of 1-Methyl-3-Oxidopyridinium Betaine with C70—A Theoretical Study, J. Comput. Theor. Nanosci., 2009, 6, 873 CrossRef.
- (a) N. Dennis, A. R. Katritzky and G. J. Sabounji, 2π + 8π Cycloaddition Reactions of 3-Oxidopyridinium Betaines, Tetrahedron Lett., 1976, 2959 CrossRef CAS; (b) A. R. Katritzky, A. T. Cutler, N. Dennis, G. J. Sabongi, S. Rahimi-Rastgoo, G. W. Fischer and I. J. Fletcher, 1,3-Dipolar Character of Six-membered Aromatic Rings. Part 52. 2π + 8π Cycloaddition Reactions of 1-Substituted 3-Oxidopyridinium Betaines, J. Chem. Soc., Perkin Trans. 1, 1980, 1176 RSC.
- N. Dennis, A. R. Katritzky and Y. Takeuchi, 1,3-Dipolar Character of Six-membered Aromatic Rings. Part III. 2-Methyl-3-oxidoisoquinolinium. A Novel Route to Benzotropones, J. Chem. Soc., Perkin Trans. 1, 1972, 2054 RSC.
- N. Dennis, A. R. Katritzky and S. K. Parton, 1,3-Dipolar Character of Six-membered Aromatic Rings. Part XIII. 2-(2′,4′-Dinitrophenyl)-4-oxidoisoquinolinium, Chem. Pharm. Bull., 1975, 23, 2899 Search PubMed.
- A. R. Katritzky, N. Dennis and H. A. Dowlatshahi, Novel Conversion of a 3-Oxidopyridinium into a 4-Oxidoisoquinolinium, J. Chem. Soc., Chem. Commun., 1978, 316 Search PubMed.
- K.-L. Mok and M. J. Nye, 1,3-Cycloaddition of Heterocyclic Zwitterions to Dienes, J. Chem. Soc., Chem. Commun., 1974, 608 RSC.
- (a) P. G. Sammes and R. A. Watt, Novel Heterocyclic Systems by Intramolecular Cycloadditions with Unactivated Olefins, J. Chem. Soc., Chem. Commun., 1976, 367 RSC; (b) S. M. Bromindge, D. A. Archer and P. G. Sammes, Studies on Intramolecular Cycloadditions Involving 3-Oxidopyridinium, J. Chem. Soc., Perkin Trans. 1, 1990, 353 Search PubMed.
- (a) B. S. Oriek, P. G. Sammes and D. J. Weller, Use of Steric Buttresses to Enhance Intramolecular Cycloadditions, J. Chem. Soc., Chem. Commun., 1993, 1412 Search PubMed; (b) B. S. Oriek, P. G. Sammes and D. J. Weller, Steric Acceleration of Intramolecular Cycloaddition Reactions, Tetrahedron, 1993, 49, 8179 Search PubMed.
- R. A. Joshi and T. Ravindranathan, Intramolecular Cycloadditions of Oxopyridinium Betaines, Indian J. Chem., 1984, 23B, 300 CAS.
- A. Rumbo, A. Mouriño, L. Castedo and J. L. Mascareñas, 3-Hydroxy-4-pyrones as Precursors of 4-Methoxy-3-oxidoptridinium Ylides. An Expeditious Entry to Highly Substituted 8-Azabicyclo[3.2.1]octanes, J. Org. Chem., 1996, 61, 6114 CrossRef CAS PubMed.
- T. Takahashi, T. Hagi, K. Kitano, Y. Takeuchi and T. Koizumi, 1,3-Dipolar Cycloaddition of 1-Methyl-3-oxidopyridinium and Sulfonylethenes. A Synthesis of 2-Tropanols and Monofluorinated 2-Tropanol, Chem. Lett., 1989, 593 CrossRef CAS.
- (a) T. Takahashi, K. Kitano, T. Hagi, H. Nihonmatsu and T. Koizumi, The First Asymmetric 1,3-Dipolar Cycloaddition of 1-Methyl-3-oxidopyridinium and (R)s-p-Tolyl Vinyl Sulfoxide. An Enantioselective Synthesis of (1S)-(–)-2α-Tropanol, Chem. Lett., 1989, 597 CrossRef CAS; (b) T. Takahashi, A. Fujii, J. Sugita, T. Hagi, K. Kitano, Y. Arai, T. Koizumi and M. Shiro, Asymmetric 1,3-Dipolar Cycloaddition of Chiral Sulphinylethenes with 1-Methyl-3-oxidopyridinium and Some Nitrones, Tetrahedron: Asymmetry, 1991, 2, 1379 CrossRef CAS.
- V. K. Aggarwal, R. S. Grainger, G. K. Newton, P. L. Spargo, A. D. Hobson and H. Adams, Highly Diastereoselective 1,3-Dipolar Cycloaddition Reactions of trans-2-Methylene-1,3-dithiolane 1,3-Dioxide with 2-Oxidopyridinium and 3-Oxidopyrylium Betaines: a Route to the Tropane Skeleton, Org. Biomol. Chem., 2003, 1, 1884 RSC.
- N. R. Curtis, R. G. Ball and J. J. Kulagowski, 8-Azabicyclo[3.2.1]oct-3-en-2-ones via Asymmetric 1,3-Dipolar Cycloaddition of a Homochiral 3-Oxidopyridinium Betaine, Tetrahedron Lett., 2006, 47, 2635 CrossRef CAS.
- J. N. Lamhauge, D. A. McLeod, C. L. Barløse, G. A. Oliver, L. Viborg, T. Warburg and K. A. Jørgensen, Enantioselective Synthesis of Tropane Scaffolds by an Organocatalyzed 1,3-Dipolar Cycloaddition of 3-Oxidopyridinium Betaines and Dienamines, Chem. – Eur. J., 2023, 29, e202301830 CrossRef CAS PubMed.
- L.-C. Chen and S.-C. Yang, Synthesis and 1,3-Dipolar Cycloaddition of 3-Hyfroxy-5-methoxypyridinium N-Imine, Heterocycles, 1986, 24, 3411 CrossRef CAS.
- F. Estour, S. Rézel, D. Fraisse, J. Métin, V. Gaumet, C. Lartigue, G. Miscoria, A. Gueiffier, Y. Blache, J. C. Teulade and O. Chavignon, Regioselectivity of 1,3-Dipolar Cycloaddition of 3-Oxidopyridinium Betaines to Olefins and Stereoselective Synthesis of 6-Alkyloxy-5-oxa-9-azatricyclo[5.2.1.04,8]decane-2-one Derivatives, Heterocycles, 1999, 50, 929 CrossRef CAS PubMed.
- H. Ren, C. Wu, X. Ding, X. Chen and F. Shi, Aryne Cycloaddition with 3-Oxidopyridinium Species, Org. Biomol. Chem., 2012, 10, 8975 RSC.
- C. Yuan, S. Sun, G. Wang, S. Sun and J. Wang, Access to 8-Azabicyclo[3,2,1]octanes via the [3 + 2] Cycloaddition of Oxidopyridinium Ions to Maleimides, J. Org. Chem., 2023, 88, 9432 CrossRef CAS PubMed.
- S. Caix-Haumesser, I. Hanna, J.-Y. Lallemand and J.-F. Peyronel, Solid-phase Synthesis of Functionalized Tropane Derivatives via 1,3-Dipolar Cycloaddition, Tetrahedron Lett., 2001, 42, 3721 CrossRef CAS.
- A. Aboelnaga, M. Hagar and S. M. Soliman, Ultrasonic Synthesis, Molecular Structure and Mechanistic Study of 1,3-Dipolar Cycloaddition Reaction of 1-Alkynylpyridinium-3-olate and Acetylene Derivatives, Molecules, 2016, 21, 848 CrossRef PubMed.
- A. Aboelnaga and S. Abbady, Ball Milling Assisted 1,3-Dipolar Cycloaddition Reaction of 1-(4-Nitrobenzyl)-pyridinium-3-olate, Indian J. Heterocycl. Chem., 2017, 27, 423 Search PubMed.
- A. Padwa, D. C. Dean, M. H. Osterhout, L. Precedo and M. A. Semone, Synthesis of Functionalized Azomethine Ylides via the Rh(II)-Catalyzed Cyclization of α-Diazo Carbonyls onto Imino π-Bonds, J. Org. Chem., 1994, 59, 5347 CrossRef CAS.
- H.-S. Yeon, J.-E. Lee and S. Shin, Gold-Catalyzed Waste-Free Generation and Reaction of Azomethine Ylides: Internal Redox/Dipolar Cycloaddition Cascade, Angew. Chem., Int. Ed., 2008, 47, 7040 CrossRef PubMed.
- J. Jeong, H.-S. Yeom, O. Kwon and S. Shin, Auxiliary-Controlled Asymmetric [3+2]-Dipolar Cycloaddition of Azomethine Ylides Generated from Au-Catalyzed Intramolecular Redox Reaction of Nitronyl Alkynes, Chem. – Asian J., 2011, 6, 1977 CrossRef CAS PubMed.
- G. Song, D. Chen, Y. Su, K. Han, C.-L. Pan, A. Jia and X. Li, Isolation of Azomethine Ylides and Their Complexes: Iridium(III) Mediated Cyclization of Nitrone Substrates Containing Alkynes, Angew. Chem., Int. Ed., 2011, 50, 7791 CrossRef CAS PubMed.
- Y.-L. Du, Y.-F. Zhu and Z.-Y. Han, Pd(II)-Catalyzed Cycloisomerization/Dipolar Cycloaddition Cascade of N-Arylnitrone Alkyne with Olefins, J. Org. Chem., 2015, 80, 7732 CrossRef CAS PubMed.
- B. Hu, X. Zhang, Y. Mo, J. Li, L. Lin, X. Liu and X. Feng, Catalytic Asmmetric Tandem Cycloisomerization/[5+2] Cycloaddition Reaction of N-Aryl Nitrone Alkynes with Methyleneindolinones, Org. Lett., 2020, 22, 1034 CrossRef CAS PubMed.
- Y. Tamura, T. Saito, H. Kiyokawa, L.-C. Chen and H. Ishibashi, Syntheses of Stipitatic Acid and Hinokitiol, Tetrahedron Lett., 1977, 4075 CrossRef CAS.
- M. E. Jung, Z. Longmei, P. Tangsheng, Z. Huiyan, L. Yan and S. Jingyu, Total Synthesis of Bao Gong Teng A, a Natural Antiglaucoma Compound, J. Org. Chem., 1992, 57, 3528 CrossRef CAS.
- X.-F. Pei and J.-X. Shen, Synthesis of (±)-2β-Hydroxy-6α-Acetoxynortropane, Heterocycles, 1993, 36, 2549 CrossRef CAS.
- V. C. Pham and J. L. Charlton, Methyl (Σ)-Lactate as a Chiral Auxiliary in the Asymmetric Synthesis of Bao Gong Teng A, J. Org. Chem., 1995, 60, 8051 CrossRef CAS.
- (a) K. M. Peese and D. Y. Gin, Efficient Synthetic Access to the Hetisine C20-Diterpenoid Alkaloids. A Concise Synthesis of Nominine via Oxidoisoquinolinium-1,3-Dipolar and Dienamine-Diels−Alder Cycloadditions, J. Am. Chem. Soc., 2006, 128, 8734 Search PubMed; (b) K. M. Peese and D. Y. Gin, Enantioselective Approach to the Hetisine Alkaloids. Synthesis of the 3-Methyl-1-aza-tricyclo[5.2.1.03,8]decane Core via Intramolecular Dipolar Cycloaddition, Org. Lett., 2005, 7, 3323 Search PubMed.
- K. M. Peese and D. Y. Gin, Asymmetric Synthetic Access to the Hetisine Alkaloids: Total Synthesis of (+)-Nominine, Chem. – Eur. J., 2008, 14, 1654 Search PubMed.
- S. Krüger and T. Gaich, Enantioselective, Protecting-Group-Free Total Synthesis of Sarpagine Alkaloids—A Generalized Approach, Angew. Chem., Int. Ed., 2015, 54, 315 CrossRef PubMed.
- S. Krüger and T. Gaich, Total Syntheses of Vellosimine, N-Methylvellosimine, and 10-Methoxyvellosimine and Formal Synthesis of 16-Epinormacusine B through a [5+2] Cycloaddition, Eur. J. Org. Chem., 2016, 4893 CrossRef.
- H. Rebmann, C. K. G. Gerlinger and T. Gaich, Gram-Scale Total Synthesis of Spargine Alkaloids and Non-Natural Derivatives, Chem. – Eur. J., 2019, 25, 2704 Search PubMed.
- C. K. G. Gerlinger, S. Krüger and T. Gaich, Total Synthesis of Parvinestemonine by Structure Pattern Recognition: A Unified Approach to Stemona and Sarpagine Alkaloids, Chem. – Eur. J., 2018, 24, 3994 CrossRef CAS PubMed.
- J. C. DiCesare, J. P. Burgess, S. W. Mascarella, F. I. Carroll and R. B. Rothman, Synthesis and Structural Determination of 5H-Benzocyclohepten-5,8-imines, J. Heterocycl. Chem., 1994, 31, 187 CrossRef CAS.
- K. P. Constable, B. E. Blough and F. I. Carroll, Benzyne Addition to N-Alkyl-4-hydroxy-1-methylisoquinolinium Salts; a New and Convenient Synthesis of (±)-5-Methyl-10,11-dihydro-5H-dibenzo-[a,d]cycloheptene-5,10-imine (MK801), Chem. Commun., 1996, 717 Search PubMed.
- (a) R. L. Clarke, S. J. Daum, A. J. Gambino, M. D. Aceto, J. Pearl, M. Levitt, W. R. Cumiskey and E. F. Bogado, Compounds Affecting the Cenral Nervous System. 4. 3β-Phenyltropane-2-carboxylic Esters and Analogs, J. Med. Chem., 1973, 16, 1260 CAS; (b) F. I. Carroll, Y. Gao, M. A. Rahman, F. Abraham, K. Oarham, A. H. Lewin, J. W. Boja and M. J. Kuhar, Synthesis, Ligand Binding, QSAR, and CoMFA Study of 3β-(p-Substituted phenyl)tropane-2β-carboxylic Acid Methyl Esters, J. Med. Chem., 1991, 34, 2719 Search PubMed.
- G. L. Araldi, K. R. C. Prakash, C. George and A. P. Kozikowski, An Enantioselective Synthesis of 2-Alkyl-3-phenyltropanes by an Asymmetric 1,3-Dipolar Cycloaddition Reaction, Chem. Commun., 1997, 1875 Search PubMed.
- (a) A. P. Kozikowski, G. L. Araldi, K. R. C. Prakash, M. Zhang and K. M. Johnson, Synthesis and Biological Properties of New 2β-Alkyl- and 2β-Aryl-3-(substituted phenyl)tropane Derivatives: Stereochemical Effect of C-3 on Affinity and Selectivity for Neuronal Dopamine and Serotonin Transporters, J. Med. Chem., 1998, 41, 4973 CAS; (b) K. R. C. Prakash, M. Trzcinska, K. M. Johnson and A. P. Kozikowski, An Enantioselective Synthesis and Biobehavioral Evaluation of 7-Fluoro-3-(p-fluorophenyl)-2-propyltropanes, Bioorg. Med. Chem. Lett., 2000, 10, 1443 Search PubMed.
- (a) A. P. Kozikowski, G. L. Araldi and R. G. Ball, Dipolar Cycloaddition Route to Diverse Analogues of Cocaine: The 6- and 7-Substituted 3-Phenyltropanes, J. Org. Chem., 1997, 62, 503 Search PubMed; (b) X.-F. Pei, T. H. Gupta, B. Badio, W. L. Padgett and J. W. Daly, 6β-Acetoxynortropane: A Potent Muscarinic Agonist with Apparent Selectivity toward M2-Receptors, J. Med. Chem., 1998, 41, 2047 Search PubMed; (c) A. J. Airaksinen, J. Lipsonnen, M. Ahlgren, P. Vainiotalo, K. A. Bergström, R. Laatikainen and J. Vepsäläinen, Reduction of 6/7-Substituted 3-Phenyltrop-3-en-2-ones: Stereoselectivity and Conformational Analysis of the Products, Tetrahedron, 2003, 59, 377 Search PubMed.
- M. P. Smith, C. George and A. P. Kozikowski, The Synthesis of Tricyclic Cocaine Analogs via the 1,3-Dipolar Cycloaddition of Oxidopyridinium Betaines, Tetrahedron Lett., 1998, 39, 197 Search PubMed.
- T. Ghosh and H. Hart, Scope of Tandem Cycloaddition/Radical Cyclization Methodology, J. Org. Chem., 1989, 54, 5073 CrossRef CAS.
- M. P. Smith, K. M. Johnson, M. Zhang, J. L. Plippen-Anderson and A. P. Kozikowski, Tuning the Selectivity of Monoamine Transporter Inhibitors by the Stereochemistry of the Nitrogen Lone Pair, J. Am. Chem. Soc., 1998, 120, 9072 Search PubMed.
- H. Xu, C. Golz, C. Strohmann, A. P. Antonchick and H. Waldmann, Enantiodivergent Combination of Natural Product Scaffolds Enabled by Catalytic Enantioselective Cycloaddition, Angew. Chem., Int. Ed., 2016, 55, 7761 Search PubMed.
- (a) P.-H. Ducrot and J. Y. Lallemand, Structure of the Calystegines: new alkaloids of the norytopane family, Tetrahedron Lett., 1990, 31, 3879 CrossRef CAS; (b) S. A. Lomenzo, J. L. Enmon, M. C. Troyer and M. L. Trudell, A Facile and Efficient Synthesis of (±)-Tropan-2-one, Synth. Commun., 1995, 25, 3681 CrossRef CAS.
- R. A. Lowe, D. Taylor, K. Chibale, A. Nelson and S. P. Marsden, Synthesis and Evaluation of the Performance of a Small Molecule Library Based on Diverse Tropane-related Scaffolds, Bioorg. Med. Chem., 2020, 28, 115442 Search PubMed.
- M. R. Fielding, R. Grigg, V. Sridharan, M. Thorn-Pett and C. J. Urch, Sequential Oxopyridinium Betaine Cycloaddition−Palladium Catalyzed Cyclisation−Anion Capture Processes, Tetrahedron, 2001, 57, 7737 Search PubMed.
- (a) K. Müller, C. Faeh and F. Diederich, Fluorine in Pharmaceuticals: Looking Beyond Intuition, Science, 2007, 317, 1881 CrossRef PubMed; (b) S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Fluorine in Medicinal Chemistry, Chem. Soc. Rev., 2008, 37, 320 Search PubMed; (c) N. A. Meanwell, Fluorine and Fluorinated Motifs in the Design and Application of Bioisosteres for Drug Design, J. Med. Chem., 2018, 61, 5822 Search PubMed.
- Y. Yamamoto, S. Tazawa, R. Tadano and T. Yasui, Trifluoromethylated Oxidopyridinium Betaines: Unique (5 + 2) Cycloaddition Selectivity Imposed by 2- or 6-Trifluoromethyl Groups, Chem. – Asian J., 2023, 18, e202300211 CrossRef CAS PubMed.
- M. Serhan, J. D. Josephson, S. S. Masoud, M. Nakajima and J. P. Pezacki, 3-Oxidopyridinium Ions Are Versatile Bioorthogonal Dipoles for Use in Cycloadditions with Cyclooctynes, Chem. – Eur. J., 2024, 30, e202303699 CrossRef CAS PubMed.
- (a) M. J. Sung, H. I. Lee, Y. Chong and J. K. Cha, Facile Synthesis of the Tricyclic Core of Sarain A. 3-Oxidopyridinium Betaine Cycloaddition Approach, Org. Lett., 1999, 1, 2017 CrossRef CAS PubMed; (b) H. I. Lee, M. J. Sung, H. B. Lee and J. K. Cha, An Improved Synthesis of the Tricyclic Core of Sarains by a 3-Oxidopyridinium Betaine Cycloaddition, Heterocycles, 2004, 62, 407 CrossRef CAS PubMed.
- C. Fu, N. Lora, P. L. Kirchhoefer, D. R. Lee, E. Altenhofer, C. L. Barnes, N. L. Hungerford, E. H. Krenske and M. Harmata, (4+3) Cycloaddition Reactions of N-Alkyl Oxidopyridinium Ions, Angew. Chem., Int. Ed., 2017, 56, 14682 CrossRef CAS PubMed.
- C. Fu, S. P. Kelley, J. Tu and M. Harmata, Generation of the 7-Azabicyclo[4.3.1]decane Ring System via (4 + 3) Cycloaddition of Oxidopyridinium Ions, J. Org. Chem., 2021, 86, 7028 CrossRef CAS PubMed.
- (a) W. Sungnoi, A. B. Keto, R. B. Roseli, J. Liu, H. Wang, C. Fu, E. L. Regalado, E. H. Krenske and M. Harmata, Endo Selectivity in the (4 + 3) Cycloaddition of Oxidopyridinium Ions, Org. Lett., 2021, 23, 8302 Search PubMed; (b) W. Sungnoi and M. Harmata, Oxidative Bridgehead Functionalization of (4 + 3) Cycloadducts Obtained from Oxidopyridinium Ions, RSC Adv., 2022, 12, 28572 Search PubMed.
- J. Tu, R. A. Ripa, S. P. Kelley and M. Harmata, Intramolecular (4 + 3) Cycloadditions of Oxidopyridinium Ions: Towards Daphnicyclidin A, Chem. – Eur. J., 2022, 28, e202200370 Search PubMed.
- J. Tu, M. M. Clark and M. Harmata, First Synthesis of an ABCE Ring Substructure of Daphnicyclidin A, Org. Biomol. Chem., 2022, 20, 6547 RSC.
- J. M. Burns and E. D. Boittier, Pathway Bifurcation in the (4 + 3)/(5 + 2)-Cycloaddition of Butadiene and Oxidopyrylium Ylides: The Significance of Molecular Orbital Isosymmetry, J. Org. Chem., 2019, 84, 5997 CrossRef CAS PubMed.
- J. Faghtmann, M. Eugui, J. N. Lamhauge, S. S. P. Andresen, A. R. Østergaard, E. B. Svenningsen, T. B. Poulsen and K. A. Jørgensen, An Enantioselective Aminocatalytic Cascade Reaction Affording Bioactive Hexahydroazulene Scaffolds, Chem. – Eur. J., 2024, 30, e202401156 Search PubMed.
- Y. Yamamoto, Y. Ide and T. Yasui, Dearomative Transformation of Pyridin-3-ols into 2,3,6,7-Tetrahydro-1H-2,6-methanobenzo[d]azonin-12-ones via Oxidopyridinium/o-Quinodimethane (5 + 4) Cycloaddition, Org. Lett., 2025, 27, 63 Search PubMed.
- J. M. Kuthanapillil, S. Thulasi, R. Rajan, K. S. Krishnan, E. Suresh and K. V. Radhakrishnan, Expeditious Synthesis of N-Bridged Heterocycles via Dipolar Cycloaddition of Pentafulvenes with 3-Oxidopyridinium Betaines, Tetrahedron, 2011, 67, 1272 CrossRef CAS.
- D. E. Ames and B. Novitt, Cinnolines. Part XIII. Structure and Reactions of the Anhydro-base of 4-Hydroxy-2-methylcinnolinium Hydroxide, J. Chem. Soc. C, 1969, 2355 Search PubMed.
- N. Dennis, A. R. Katritzky and M. Ramaiah, 1,3-Dipolar Character of Six-membered Aromatic Rings. Part X. Pyridazine and Benzopyridazine Betaines, J. Chem. Soc., Perkin Trans. 1, 1975, 1506 Search PubMed.
- (a) M. Kiss, J. Russell-Maynard and J. A. Joule, 1,3-Dipolar Cycloadditions to Oxidopyraziniums, Tetrahedron Lett., 1987, 28, 2187 Search PubMed; (b) P. A. Allway, J. K. Sutherland and J. A. Joule, A Synthesis of (±)-7-Methoxycarbonyl-2-(3-Methoxyphenylmethylidene)-8-methyl-3,8-diazabicyclo[3.2.1]octan-4-one (1b) Using Dipolar Cycloaddition to a 3-Oxidopyrazinium, Tetrahedron Lett., 1990, 31, 4781 CrossRef CAS; (c) N. D. Yates, D. A. Peters, P. A. Allway, R. L. Beddoes, D. I. C. Scopes and J. A. Joule, 1,3-Dipolar Cycloadditions to Oxidopyraziniums, Heterocycles, 1995, 40, 331 Search PubMed; (d) L. Rhyman, H. H. Abdallah, S. Jhaumeer-Laulloo, L. R. Domingo, J. A. Joule and P. Ramasami, 1,3-Dipolar Cycloaddition of 1H-Pyrazinium-3-olate and N1- and C-Methyl Substituted Pyrazinium-3-olates with Methyl Acrylate: a Density Functional Theory Study, Tetrahedron, 2011, 67, 8383 CrossRef CAS.
- M. Helliwell, Y. You and J. A. Joule, The 1,3-Dipolar Cycloaddition of Methyl Actylate to Hindered 3-Oxidopyraziniums, Heterocycles, 2006, 70, 87 CrossRef CAS PubMed.
- (a) Z. Joomun, J. Raftery, K. Delawarally, S. J. Laulloo and J. A. Joule, 3-Oxidopyraziniums—[4+2] versus, [3+2] Cycloadditions, ARKIVOC, 2007, xvi, 51 Search PubMed; (b) G. Riesco-Llach, M. Planas and J. A. Joule, Exploiting 3-Oxidopyraziniums toward Diazabicyclo[3.2.1]octanes and Their Conversion into Diazabicyclo[2.2.2]octanes and Tricyclic Lactone-Lactams, J. Org. Chem., 2024, 89, 2904 CrossRef CAS PubMed.
- L. R. Domingo, J. A. Sáez, J. A. Joule, L. Rhyman and P. Ramasami, A DFT Study of the [3 + 2] versus [4 + 2] Cycloaddition Reactions of 1,5,6-Trimethylpyrazinium-3-olate with Methyl Methactylate, J. Org. Chem., 2013, 78, 1621 CrossRef CAS PubMed.
- (a) S. Oida and E. Ohki, 1,3-Dipolar Cycloaddition Reaction of Aziridinedicarboximide, Chem. Pharm. Bull., 1968, 16, 764 CrossRef CAS; (b) R. Huisgen and H. Mäder, Configuration of a cis-Disubstituted Azomethine Ylide, Angew. Chem., Int. Ed. Engl., 1969, 8, 604 Search PubMed.
- (a) P. Garner, K. Sunitha and T. Shanthilal, An Approach to the 3,8-Diazabicyclo[3.2.1]octane Moiety of Naphthyidinomycin and Quinocarcin via 1,3-Dipolar Cycloaddition of Photochemically Generated Azomethine Ylides, Tetrahedron Lett., 1988, 29, 3525 Search PubMed; (b) P. Garner, K. Sunitha, W.-B. Ho, W. J. Youngs, V. O. Kennedy and A. Djebli, An Asymmetric Approach to Naphthyridinomycin and Quinocarcin via a Remarkably Selective Intramolecular 1,3-Diplar Cycloaddition Reaction, J. Org. Chem., 1989, 54, 2041 CAS; (c) P. Garner, F. Arya and W.-B. Ho, Complex Pyrrolidines via a Tandem Michael Reaction/1,3-Dipolar Cycloaddition Sequence. A Novel Method for the Generation of Unsymmetrical Azomethine Ylides, J. Org. Chem., 1990, 55, 412 CrossRef CAS; (d) P. Garner, W.-B. Ho and H. Shin, The Asymmetric Synthesis of (–)-Quinocarcin via a 1,3-Dipolar Cycloadditive Strategy, J. Am. Chem. Soc., 1993, 115, 10742 CAS; (e) J. D. Scott and R. M. Williams, Total Synthesis of (–)-Tetrazomine. Determination of the Stereochemistry of Tetrazomine and the Synthesis and Biological Activity of Tetrazomine Analogues, J. Am. Chem. Soc., 2002, 124, 2951 CAS; (f) E. R. Ashley, E. G. Cruz and B. M. Stoltz, The Total Synthesis of (–)-Lemonomycin, J. Am. Chem. Soc., 2003, 125, 15000 CAS.
- Q. Sun, A. Eitzinger, R. Esken, P. W. Antoni, R. J. Mayer, A. R. Ofial and M. M. Hansmann, Pyridinium-Derived Mesoionic N-Heterocyclic Olefins (py-mNHOs), Angew. Chem., Int. Ed., 2024, 63, e202318283 CAS.
| This journal is © the Partner Organisations 2025 |