
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNanoconfined polymerization: advantages of lyotropic liquid crystals as soft templates
Seyed Mostafa
Tabatabaei
 and
Reza
Foudazi
*
and
Reza
Foudazi
*
School of Sustainable Chemical, Biological and Materials Engineering, The University of Oklahoma, Norman, OK 73019, USA. E-mail: rfoudazi@ou.edu
First published on 27th February 2025
Abstract
Polymerization within nanoconfinement offers a versatile approach to creating nanostructured materials with unique properties and a wide range of applications. Therefore, it is important to understand the nature of polymerization in both hard and soft nanoconfinements, which have been classified based on their mechanical modulus in this perspective. We also evaluate factors affecting the kinetics of polymerization within different templates. Template walls, mainly in hard nanoconfinement, may have a catalytic effect and enhance initial polymerization rates. Additionally, increased termination rates as well as lower limiting conversion are observed in those templates. On the other hand, we discuss the self-assembled amphiphilic molecules in selective solvents, known as lyotropic liquid crystals (LLCs), as a common class of soft templates inducing nanoconfinement during polymerization. Key factors such as initiator type, monomer chemistry, crosslinking density, and arrangement of the micelles in LLC templates are brought into a framework in this perspective to analyze their impact on polymerization rates and structural retention in LLCs.
 Seyed Mostafa Tabatabaei | Mostafa Tabatabaei is a Ph.D. student in the School of Sustainable Chemical, Biological and Materials Engineering at the University of Oklahoma, working in the SMaRT Lab under the supervision of Dr. Reza Foudazi. His research focuses on the self-assembly of block copolymers and the rheology of lyotropic liquid crystals (LLCs). He employs the LLC templating method to develop advanced bioseparation membranes designed for viral clearance in monoclonal antibody (mAb) production. |
 Reza Foudazi | Dr. Reza Foudazi is an Associate Professor in the School of Sustainable Chemical, Biological and Materials Engineering at the University of Oklahoma. The current research activities in his group are self-assembly of amphiphilic molecules, templating approach for synthesis of porous polymers, and rheology of soft matter, with the long-term goal of producing responsive multifunctional materials for sustainability and environmental applications. He is the recipient of Polymer Processing Society Early Career Award in 2019 and ACS PMSE Young Investigator Award in 2020. |
Introduction
Nanoconfinement in polymer science in the form of thin films, pores, or nanodroplets refers to polymeric materials restricted in domain sizes on the order of radius of gyration of polymer chains, Rg. The confined geometries induce stronger deviation in polymer properties from bulk properties as the confinement size decreases, typically in the 1–100 nm range. Interfacial effects and interactions become more dominant in such length scales.1 Depending on the nature of the confinement wall, soft and hard nanoconfinements can be envisioned. Soft nanoconfinement (i.e., nanoconfinement in soft matter domains) occurs in a flexible and dynamic environment such as micelles, vesicles, or thin films on liquid interfaces allowing for some degree of polymer chain mobility and dynamic change in confinement size. On the other hand, hard nanoconfinement originates from rigid and static environments like nanopores in metal oxides and mesoporous inorganic materials imposing significant spatial restrictions on polymer chains. We suggest that if the modulus of template is >3 orders of magnitude higher than the final polymer, it can be considered as a hard template. This suggestion is based on modulus change upon glass transition in amorphous polymers.As summarized in Fig. 1, nanoconfinement has demonstrated significant potential across various applications, utilizing both soft and hard templates. Membranes fabricated by polymerization of or in nanoconfined templates showed improved separation efficiency and fouling behavior compared to commercially available membranes. The obtained membranes offer narrow pore size distribution for nanofiltration and ultrafiltration processes.2–5 In the case of developing hydrogels using templates,6–8 some works show not only faster nanoconfined polymerization rates compared to the isotropic solutions but also enhanced network swelling and diffusivity.9 Many other applications such as catalysis, energy storage, biomedical devices, and optoelectronics are reported based on templates inducing soft nanoconfinement, thanks to their tunable nanostructures and enhanced mechanical, thermal, and chemical properties.10 Polymeric nanomaterials such as nanorods, nanofibers, and nanotubes can also be produced using hard templates.11 Nanoconfined polymerization in hard templates enables applications in energy storage (supercapacitors, conducting polymer electrodes), sensing (biosensors, electrochemical sensors), functional surfaces (self-cleaning coatings, cell adhesion for tissue engineering), biomimetic synthesis (bone tissue formation), advanced smart materials (stimuli-responsive polymers for biomedicine and electronics), and high-performance polymer nanostructures with tailored mechanical and physicochemical properties.12
 | ||
| Fig. 1 Summary of nanoconfined polymerization applications. | ||
Polymer chain mobility is altered under nanoconfinement which imposes nanoscale spatial restrictions on polymers leading to significant changes in their physical and chemical properties. A major consequence of nanoconfinement is the deviation from Gaussian distribution of end-to-end distance of polymer chains as the chain stiffness increases or chain mobility becomes restricted to one or two spatial directions.13 Glass transition temperature,14 crystallinity and morphology,15 diffusion and transport properties,16 and mechanical properties17 are examples of properties which are affected by nanoconfinement as have been reported in many reviews. In addition, there has recently been interest in the nanoconfinement role in polymerization kinetics and final product properties. Nanoconfinement may enable polymerization at operational conditions like temperature that are not possible for bulk polymerization, resulting in higher reactivity and efficiency.18
Various parameters may affect polymerization in nanoconfinement. The degree of confinement, which represents the characteristic size of the nanoconfined medium, is one of the most important parameters. Higher degree of confinement (smaller characteristic size) increases the local monomer concentration and decreases the diffusion rate. While the former enhances polymerization rate, the latter can reduce it.19 In addition, the segregation of the monomers in specific confined spaces limits their diffusion to fewer directions. The presence of interfaces in nanoconfined polymerization can act as a nucleation site or catalyst for polymerization, influencing the rate and growth pattern of the polymer.11 Nanoconfinement can also influence the activation energy required for polymerization. Smaller pore sizes and specific surface interactions can reduce activation energy, thereby, affecting the thermal dynamics of the polymerization process. This is particularly evident when comparing hydrophilic and hydrophobic pore effects, where the former tends to lower activation energy more significantly.20 The stiffness of the template also plays a crucial role in determining the degree of confinement during polymerization, which in turn affects the polymerization rate and the final properties of the polymer. In other words, the natural tendency of growing chains towards random coil conformation may induce mechanical stress on the nanoconfinement boundaries and alter the confinement size. A stiffer template can better maintain the confined environment, leading to a more controlled polymerization process. In contrast, soft nanoconfinements can undergo an increase or decrease in domain size during polymerization due to the competition between density change, chain conformation, phase separation, and solidification during polymerization (see Fig. 2).20
 | ||
| Fig. 2 Illustration of changes in hard and soft nanoconfinement domain size upon polymerization due to natural tendency of random coil formation. | ||
Qavi et al.20 hypothesized a relationship between ratio of change in the domain size of the nanoconfinement and its elastic modulus as follows:
 | (1) |
Free radical polymerization is the most commonly used method for nanoconfinement studies. The main question is how initiation, propagation, termination, and transfer rates are influenced by nanoconfinement. Fig. 3 summarizes the effects that nanoconfinement has on geometrical properties of the template, physicochemical aspects and consequently free radical polymerization steps that are affected. This perspective investigates the role of different parameters on polymerization in hard and soft nanoconfinement and their applications.
 | ||
| Fig. 3 Physiochemical consequences of confinement effect altering free radical polymerization steps. | ||
Polymerization in hard nanoconfinement
Zhao et al.21 reported that hard nanoconfinement significantly accelerates the kinetics of free radical polymerization of methyl methacrylate (MMA). Their results show that although initial reaction rate is the same as bulk polymerization, autoaceleration takes place much earlier leading to faster completion of the reaction in nanoconfined spaces compared to the bulk. A total higher reaction rate can be due to the reduced diffusivity of polymer chains and suppression of chain transfer, which decrease the rate of termination relative to propagation. This effect results in higher molecular weights and lower polydispersity index at full conversion.Tian et al.22 reported the same behavior in polymerization of alkyl methacrylate in controlled pore glass (CPG). Hard nanoconfinement in this case accelerates depropagation relative to propagation at higher temperatures compared to bulk polymerization and also decreases the apparent activation energy as a strong function of pore size. It is worth mentioning that not all hard confinement mediums have the same effect. In a study of polymerization of benzyl methacrylate by Zhai et al.,23 polymerization rate in CPG and mesoporous silica is reported to be higher than that of bulk polymerization and inversely proportional to pore diameter. However, reaction rates in mesoporous carbon are lower than bulk, decreasing linearly with reciprocal of pore diameter. This observation is attributed to a retardation effect likely caused by the functional groups present on the carbon pore surface. Depending on the monomer, autoacceleration is not always improved in hard nanoconfinement. Fig. 4 shows the most common polymerizable functional groups used for nanoconfined polymerization. For example, in the case of dodecyl methacrylate (DMA), suppression of autoacceleration is observed in contrast to other methacrylates. In bulk polymerization, the long alkyl side chain promotes chain transfer reactions leading to branching and cross-linking, improving propagation and decreasing termination rates. In nanoconfined environments, the high flexibility of polyDMA chain delays the onset of autoacceleration, which can suppress the autoacceleration alongside with chain transfer from the propagating radical to the alkyl group of the ester.24
 | ||
| Fig. 4 Structure of the most common polymerizable groups and monomers used for polymerization in nanoconfinement: (a) acrylate monomers,20,25–29 (b) diacrylate monomers,27,30–34 (c) methacrylate monomers,21,24,35–37 (d) dimethacrylate monomers,38–40 (e) vinyl acetate,41 (f) styrene,42–44 (g) 1,4-diphenylbutadiyne,45 (h) divinyl benzene,46 (i) bisphenol A diglycidyl ether crosslinked with (j) 1-ethyl-3-methylimidazolium dicyanamide,47 (k) 3-hexylthiophene,48 (l) 2,3-dihydrothieno[3,4-b]-1,4-dioxin,49–51 (m) dimethyldimethoxysilane crosslinked with (n) methyltrimethoxysilane,52 (o) aniline,53 (p) phenol and (q) formaldehyde,54,55 (r) phloroglucinol crosslinked with (q) formaldehyde,56,57 (s) glyoxal,58,59 or (t) 1,4-bis(chloromethyl)benzene,60 (u) resorcinol crosslinked with (q) formaldehyde,57 and (v) 3,9-dioxidophenanthrene-1,5-dicarboxylate.61 | ||
To understand the thermodynamics of polymerization, the changes in Gibbs free energy should be considered:
| ΔG = ΔH − TΔS | (2) |
In this equation, ΔG, ΔH, and ΔS show changes in Gibbs free energy, enthalpy, and entropy, respectively; and T shows the temperature. As a result of higher entanglement and lower flexibility of the chains, the polymerization is entropically unfavorable with negative ΔS. On the other hand, the negative ΔH makes polymerization favorable from an enthalpy perspective, for example in radical polymerization, due to the conversion of π-bonds in monomers to σ-bonds in the polymer, energy is released. Considering eqn (2), we can discuss the hard nanoconfinement effect on the thermodynamics of polymerization. ΔH contribution to free energy can be enhanced or diminished under confinement depending on the interaction of confinement wall with monomer and polymer. The same trend may be expected for entropic contribution, because the confinement decreases the entropy of both monomer molecules and polymer chains. However, we hypothesize that nanoconfinement effect on polymer is more significant, thus, the |ΔS| of confined polymerization is higher than that of bulk polymerization and the confined polymerization is less entropically favorable.
Although hard nanoconfinement offers some improvements towards polymerization rate and final product properties, there are some problems due to the nature of hard confinements. For example, removal of the template from the resulting polymer may be difficult in some cases and need harsh conditions like using organic solvents to extract the template.21 In addition, by removing the template, the confined polymers will be disintegrated. Therefore, it will be challenging to produce a nano-heterogeneous monolith that is entirely composed of soft matter. With crystalline-like ordered structures while having flowability, lyotropic liquid crystals (LLCs) have shown great potential as highly ordered mesostructures for polymerization. Polymerized LLCs (polyLLC) have been shown to be excellent candidates for separation membranes4 and hydrogels62 with significantly improved performance.
Polymerization in LLC templates
The self-assembly of amphiphilic molecules in selective solvent(s) yields different meso-ordered structures with sizes in the 2–50 nm range.3 Presence of surfactants as building blocks of LLCs forms both hydrophilic and hydrophobic phases. In other words, microphase separation of these domains leads to LLC formation. Based on the concentration of the surfactant and interaction of the surfactant with selective solvent(s), different micellar shapes can be formed, including spherical, cylindrical, and planar micelles, which give rise to different mesostructures, such as normal (oil-in-water) micelles (L1), normal micellar cubic (I1, e.g., body-centered cubic and face-centered cubic), normal hexagonal (H1), lamellar (Lα), normal bicontinuous cubic (V1), reverse (water-in-oil) bicontinuous cubic (V2), reverse hexagonal (H2), reverse micellar cubic (I2), and reverse micelles (L2).10Fig. 5 shows a typical phase diagram of an LLC system containing Pluronic P84 as the surfactant, and mixture of water and p-xylene.63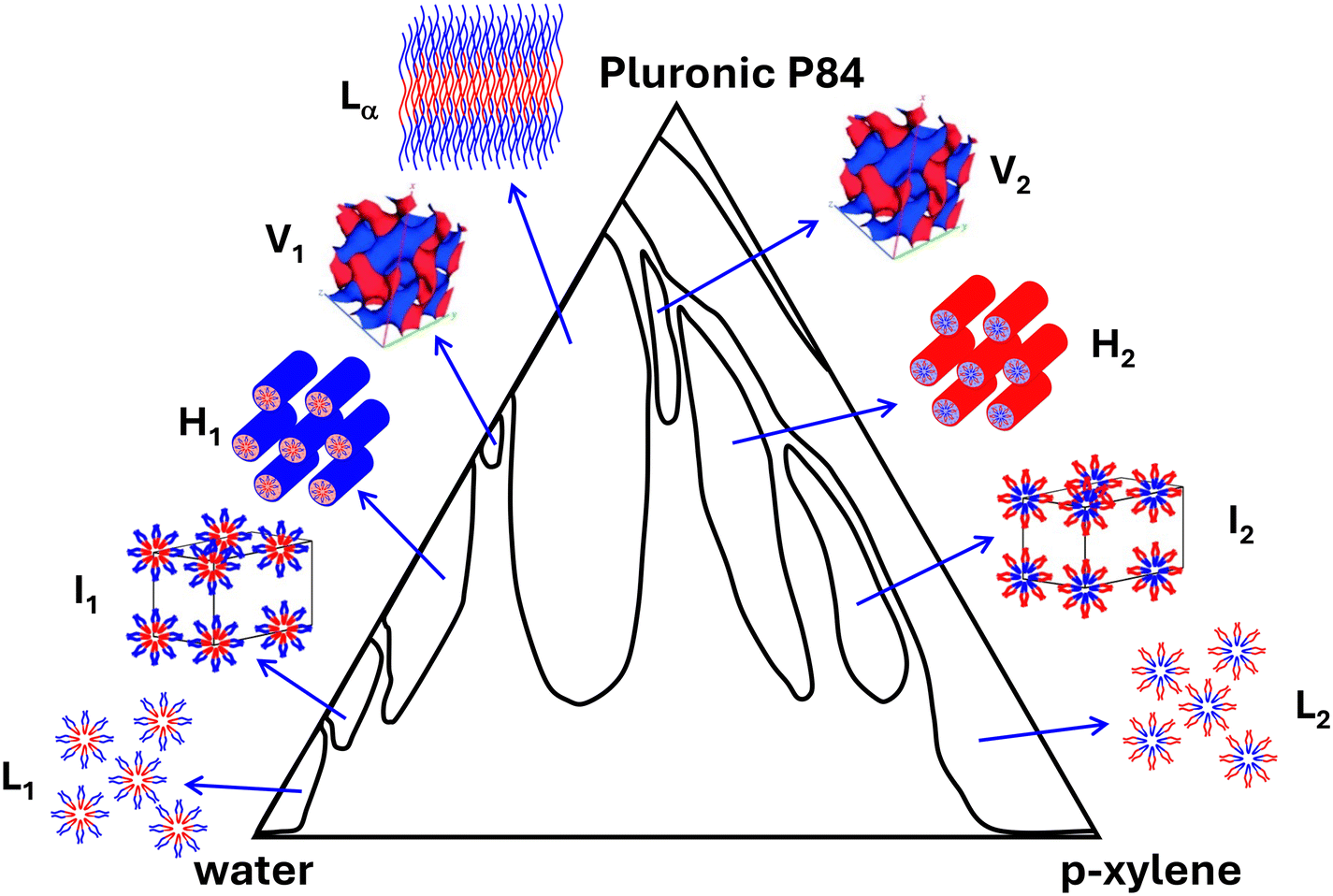 | ||
| Fig. 5 Typical phase diagram of water/oil/surfactant mixtures. Reproduced from ref. 63 with permission from American Chemical Society, copyright ©1998. | ||
LLCs have various applications in developing membranes, drug delivery, hydrogels, and biosensors to name a few due to their tunable hierarchical nanostructure and possibility to incorporate stimuli-responsiveness.10 However, LLCs are in the form of gels (i.e., have solid-like rheological behavior)64,65 and lack thermal and/or chemical stability, which hinder their application in fields such as membrane science or biosensors. One way to overcome this issue is to use LLCs as a template and polymerize one of the phases or the surfactant at the interface, producing polyLLCs. In this case, if the mesostructure is retained, the mechanical and thermal stability of the system is significantly improved.42 Therefore, many studies have focused on polymerization of LLCs and understanding the mechanisms involved in this process and methods to improve the final product properties. Templating can be broadly categorized into synergistic and transcriptive ones, which will be reviewed in the next sub-sections.
Transcriptive templating
Transcriptive templating forms the desired material through polymerization and/or cross-linking of monomers in the nano-confined spaces of the LLC template, aiming to replicate the LLC structure precisely. This method is used to fabricate nanostructured materials like hydrogels,62 membranes,29 and catalysts,66 benefiting from precise nanostructure control. Transcriptive templating involves non-reactive surfactants forming the LLC (and thus, the nanoconfinement), in which monomers are polymerized inside or outside of the micelles, ideally forming the polymer with the same shape.5,67 Preserving the structure during polymerization is a challenge in transcriptive templating since surfactants are prone to reorganize during polymerization as the surface energy of polymerizing phase changes by degree of polymerization, as shown in eqn (3) based on an empirical correlation.68| γ = γ∞(1 − CMn−z) | (3) |
In this equation, γ represents the interfacial tension, γ∞ represents the infinite molecular weight limit, Mn is the number average molecular weight, and C and z are constants. This equation shows that as the degree of polymerization increases, the interfacial tension increases too. As the polymerization takes place, longer chain length leads to increased chain entanglement, which increases the total energy required to create a new interface.68
To analyze the effect of soft nanoconfinement on polymerization thermodynamics, we can use a modified version of Flory–Huggins theory by including the degree of polymerization and choose an isotropic solution as a reference state. Eqn (4)–(6) show dependence on degree of polymerization.69
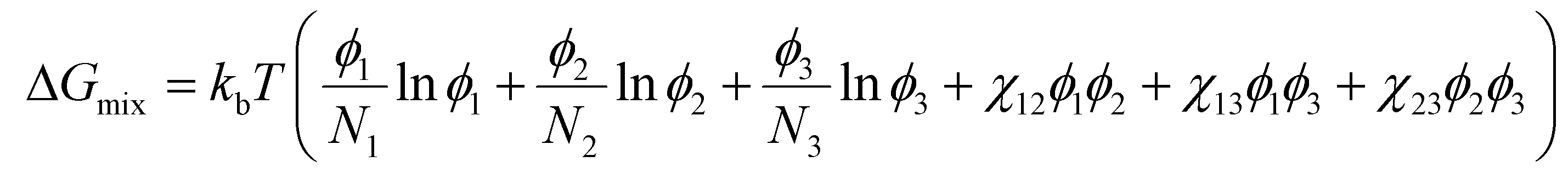 | (4) |
| ΔHmix = kbT(χ12ϕ1ϕ2 + χ13ϕ1ϕ3 + χ23ϕ2ϕ3) | (5) |
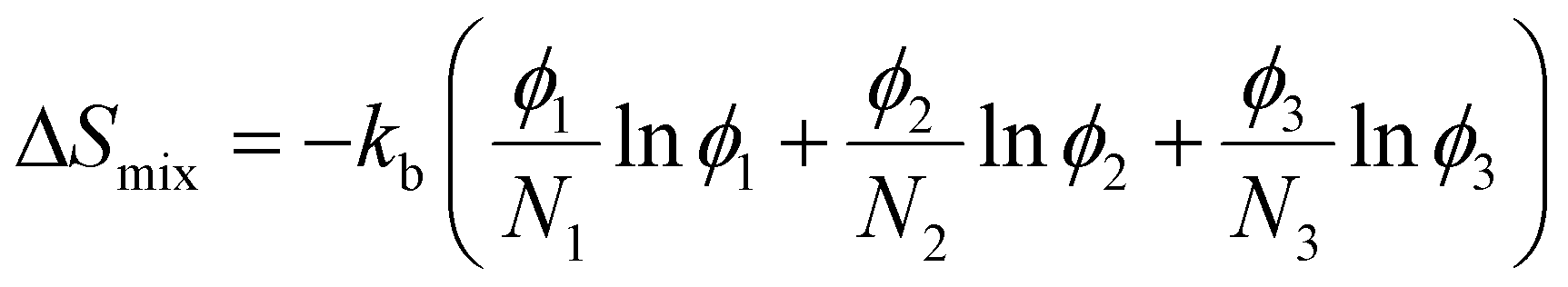 | (6) |
In these equations, N shows the number of statistical repeating units (related to degree of polymerization), ϕ shows the volume fraction, kb is the Boltzmann constant, and χ shows the Flory–Huggins interaction parameter. It should be noted that the interaction parameter may need to be adjusted for confinement effect, but Flory–Huggins model remains a good approximation for phase separation in confined spaces.70 The subscript 1 refers to the monomer in the system, 2 refers to the polymer, and 3 refers to the surfactant segment (head or tail) that is in contact with the polymerizing phase. It should be noted that the surfactant has usually a short oligomer chain, but in some cases, it can be an amphiphilic block copolymer. The solvent in LLC formulation, which segregates from the monomer and polymer (e.g., water), is excluded from the total free energy of the polymerizing phase. It is worth mentioning that before polymerization, there is no polymer in the system. Upon polymerization, ϕ3 remains constant and ϕ1 decreases while ϕ2 increases. Furthermore, N1, which is equal to 1 for monomers and higher than 1 if macromers are used, remains constant while N2 increases during polymerization.
As eqn (5) shows, the degree of polymerization does not directly affect the enthalpy of the system. However, as polymerization proceeds and the chain length increases, the interaction of polymer chains and LLC template becomes less favorable, which increases the interfacial tension (see eqn (1)), thus, the χ increases according to Helfand and Sapse model for asymmetric systems:71
 | (7) |
Mours and Winter73 defined mutation time, λmu, the time needed for 1/e change in the material rheological property g by time-resolved rheometry (TSR) according to eqn (8), which enables characterization of the dynamic properties of transient systems. Storage (G′) or loss (G′′) modulus can be used instead of g in this equation.
 | (8) |
By applying TSR on LLCs in the absence of polymerization (e.g., by excluding the initiator), evolution of G′ or G′′ can be used to estimate the mutation time required for structure reorganization. To determine characteristic time for polymerization, chemorheology can be used to analyze polymerization of LLC templates.20,74 If polymerization in LLC templates is considered as a first order reaction, which is true in many cases, the overall kinetic constant rate, k, can be determined from chemorheology or calorimetry techniques.20 1/k shows the time required for the monomer concentration to decrease by a factor of e. Theoretically, λmu should be equal or larger than 1/k to avoid any phase separation during polymerization. Based on this hypothesis, we propose a dimensionless number called Artemis† number,  , according to eqn (9).
, according to eqn (9).  values larger than 1 show that potentially no phase separation takes place during polymerization of the LLC (Fig. 6). It should be noted that k only applies to low conversion regime before autoacceleration kicks in.20 Nevertheless, the low conversion regime seems to be the most critical stage since the changes in ΔG of mixture is more significant. It is worth mentioning that since an essential part of the Artemis number is measuring changes in rheological properties, instrumental limitations might be a challenge to enable polymerization while performing rheological tests.
values larger than 1 show that potentially no phase separation takes place during polymerization of the LLC (Fig. 6). It should be noted that k only applies to low conversion regime before autoacceleration kicks in.20 Nevertheless, the low conversion regime seems to be the most critical stage since the changes in ΔG of mixture is more significant. It is worth mentioning that since an essential part of the Artemis number is measuring changes in rheological properties, instrumental limitations might be a challenge to enable polymerization while performing rheological tests.
 | (9) |
 | ||
Fig. 6 The role of Artemis number, 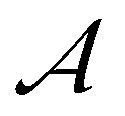 , in successful polymerization in nanoconfinements. , in successful polymerization in nanoconfinements. | ||
There are some strategies to enhance the structural retention upon polymerization: (1) design formulation with favorable thermodynamic interactions between the template and polymer (i.e., minimal change in the surface energy of polymerizing phase),75 (2) increase the system viscosity to limit surfactant diffusion (which may have an unwanted effect on polymerization rate),75 (3) choose surfactant systems with strong interfacial rheology to enhance resistance against coarsening,76 and (4) suppress the conformational rearrangement of chains through the formation of network structure.77 In the latter, cross-linkers are often used, which kinetically trap polymer chains, preventing phase separation or inversion. Higher crosslinking density decreases the characteristic time of network formation to occur before structure reorganization. In other words, below a threshold of crosslinking density, the structure cannot be retained. Controlling local crosslinking density (network heterogeneity) in domains can also affect the polymerization efficiency and final properties.78 An example for implementing the second strategy is to use ionic liquids, which have high viscosity and help retain the structure after polymerization.79
Synergistic templating
In the synergistic templating method, reactive surfactants (i.e., surfactant monomers or surfmers) are polymerized at the interface between hydrophobic and hydrophilic domains. Depending on the structure of the surfactant, the polymerizable group can be in the hydrophilic or hydrophobic moiety of the surfactant.5 This method often results in materials with enhanced mechanical, thermal, and chemical properties, such as membranes with improved permeability, selectivity, and fouling resistance.4In transcriptive templating, as the polymerization takes place and the free energy of the system tends to increase according to eqn (3), surfactants undergo reorganization to minimize the free energy.75 However, the movement of surfactants becomes restricted in synergistic templating as they get crosslinked and covalent bonding between the surfactants reduces the probability of phase separation. In addition, considering that we only have the first two terms for the monomer and polymerizing chain in eqn (3), the unfavorable demixing of monomer and polymer is unlikely. It is important to note that the kinetics of polymerization still play an important role, and  should remain greater than 1 to ensure successful retention of the structure. If the polymerization is slower than characteristic time for self-diffusion of unpolymerized surfactants, their diffusion can disturb the mesostructure during polymerization.
should remain greater than 1 to ensure successful retention of the structure. If the polymerization is slower than characteristic time for self-diffusion of unpolymerized surfactants, their diffusion can disturb the mesostructure during polymerization.
Although synergistic templating method shows great advantages over transcriptive process, the current challenge is the lack of commercially available surfmers that form LLCs. In contrast, there are many widely available monomers, both hydrophilic and hydrophobic ones, that can be implemented in LLCs obtained from conventional surfactants for transcriptive templating. Fig. 7 schematically depicts transcriptive and synergistic templating methods in Lα structure.
 | ||
| Fig. 7 Schematic of LLC polymerization in transcriptive and synergistic templating methods. | ||
Effective parameters in kinetics of LLC polymerization
Both thermal- and photo-polymerization methods have been utilized in LLC templating. As mentioned before, preserving the structure is crucial in this process and shorter reaction time can improve the process and help preserve the structure. The photo-polymerization process within LLC templates is crucial for minimizing polymerization-induced phase separation and/or thermal induced phase separation between the growing polymer network and its template.38 Photo-polymerization of LLC templates can be done in less than 1 min due to fast polymerization rate, which can kinetically trap the LLC structure and preserve the structure with optimum crosslinking density.72 Compared to thermal polymerization, another advantage of photo-polymerization is the mild reaction conditions. Thermal polymerization of LLCs has usually been reported to take place at elevated temperatures at which the LLC may not have the desired structure due to the order-to-order or order-to-disorder transition. The temperature increase is also inevitable during photo-polymerization, although it is usually modest. Therefore, the choice of a proper surfactant that provides a broad thermal stability range is essential in successful LLC templating.The initiator can have a significant effect on both kinetics of polymerization and final product properties. Saadat et al.74 studied the effect of both water-soluble and oil-soluble initiators on the polymerization kinetics of hydrophobic monomers in LLCs. Although the monomers are in the oil phase in this system, surprisingly the polymerization rate and conversion in the presence of water-soluble initiators are higher, which is not due to higher concentration of active free radicals. This superior performance is attributed to the initiation of polymer chains at the water/oil interface and subsequent migration of propagating chains into the monomer phase, which reduces termination rates and enhances overall polymerization rates. In this case, the formation of polymer chains at the interface of two phases leads to improved mechanical properties of polyLLC. Initiator with the larger molecular size shows a lower polymerization rate due to the caging effect and reduced mobility of free radicals, which increases radical recombination under confinement.74 Thus, the initiation efficiency may be reduced under confinement.
Chemistry and properties of monomers and crosslinkers are other key factors influencing the reaction rate in transcriptive templating. However, their effect depends on the type of structure which is affected by the concentration of the surfactant. A general trend in nanoconfinement is that higher local monomer concentration in more confined systems results in a higher termination rate due to increased proximity of propagating chains.11 However, as discussed by Worthington et al.,80 the polymerization rate of hydrophilic monomers in LLC templates is significantly enhanced by increasing the surfactant concentration forming smaller confinement medium by changing the structure from I1 to Lα. This enhancement is primarily due to the segregation of monomers into polar domains, which increases local monomer concentration leading to higher free radical propagation and limits the propagating polymer chain diffusion, leading to reduced termination rate. Although the propagation rate is less sensitive to the type of structure and surfactant concentration, the overall polymerization rate is substantially accelerated, enabling the creation of polymers with well-defined nanostructures and desirable properties.
Goujon et al.81 showed that slight change in the monomer chemistry from 2-hydroxyethyl acrylate (HEA) to hydroxyethyl methacrylate (HEMA) changes the structure of LLC precursor from I1 to H1 structure and decreases the polymerization rate. Furthermore, polyethylene glycol diacrylate (PEGDA, 575 g mol−1) and polyethylene glycol dimethacrylate (PEGDMA, 400 g mol−1) both show high reaction rates due to flexibility of ethylene glycol units. However, PEGDMA-derived polymers show higher order and thermal stability. In addition, it is found that within the same phase, decreasing PEGDA concentration results in larger pre-polymerization domain sizes and slower polymerization rates. This result can be attributed to the presence of PEGDA at the headgroup interface due to its hydrophilic nature. However, the smaller molecular weight of PEGDMA enables its slightly deeper penetration into the hydrophobic region and influences both headgroup interface and hydrophobic region.
Hydrophobic monomers show the opposite trend, where the polymerization rate declines as the LLC structure transitions from I1 (FCC or BCC) to H1 to Lα. This shift in structure is generally obtained by an increase in surfactant concentration, which expands the volume fraction of the apolar domains. Consequently, as the size of the apolar domains increases, the local monomer concentration decreases, resulting in reduced polymerization rates.10 More specifically, the propagation rate of hydrophobic monomers decreases as the concentration of surfactant is increased, while it may be constant or have a slight increase for hydrophilic monomers. However, the termination rate for both hydrophilic and hydrophobic monomers in LLC templates has the same declining trend with increasing surfactant concentration.27
When it comes to reverse micelles, the trend is the same based on packing of the micelles, but not the concentration of the surfactants. In the study of Qavi et al.,20 in which the apolar domain size of the H2 is smaller than the Lα structure, higher degree of confinement in Lα structure results in higher termination rate, which leads to lower polymerization rate and overall conversion. In addition to the confinement size effect, which is smaller in Lα structure, it is hypothesized that as the elastic modulus of the soft template increases, the confinement effect is enhanced. Higher stiffness restricts the flexibility of domains containing monomers and radicals, increasing the probability of termination reactions. However, this might not be the general trend, as there is a possibility of the dynamic changes of self-assembly due to the change in interfacial properties of the polymerizing phase. Overall, there may be some exceptions too, as Saadat et al.74 found that in the case of LLCs with hydrophilic initiator and hydrophobic monomers, H2 structure shows lower polymerization rate than Lα due to a pull-push effect of gradual increase in interfacial concentration of radicals and improved termination reactions at the interface of Lα structure.
Although all the discussions mentioned about the polymerization of hydrophilic and hydrophobic monomers in confinement seem to be correct, they might look counterintuitive and confusing. Thus, there is a need for a general explanation. Change in the structure of LLCs is a result of change in the critical packing factor (CPP) of the surfactant which can be calculated as follows:10
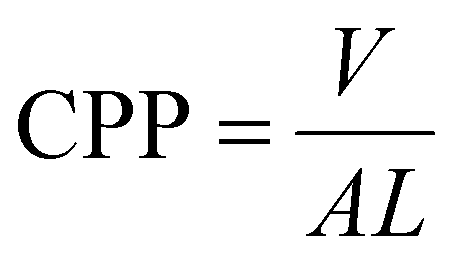 | (10) |
In this equation, V, A, and L represent the hydrophobic tail volume, cross-sectional area of the hydrophilic head group, and hydrophobic tail chain length, respectively. As the shape of the micelle evolves from spheres to planar bilayer and consequently the LLC structure evolves from normal micelles to Lα, CPP increases to the value of 1. When the CPP exceeds the value of 1, the micelles will be inverted and form structures like H2 and I2 in which the hydrophobic part is outside of the micelle. Therefore, at CPPs lower than 1, the hydrophilic domain is the continuous phase and at CPPs higher than 1, the hydrophobic domain forms the continuous phase.
Since polymerization in transcriptive templating usually takes place in the continuous phase, to maintain the structure after template removal, the choice of the monomer depends on the continuous phase. Evaluating the results in the literature suggests that CPP influences the polymerization rate, as schematically shown in Fig. 8. When polymerization takes place inside the micelles, transcriptive polymerization rate decreases with increasing CPP, while the trend is opposite for polymerization outside the micelles. It is worth mentioning that Fig. 8 only shows the overall trend and should not be used to conclude that the polymerization rate of hydrophobic monomers are higher than hydrophilic ones or vice versa. Another benefit of using CPP over surfactant concentration or the order (a terminology used by Worthington et al.80 to contrast structures with Lα having the highest order, which may be confusing as all LLC structures are ordered and periodic) is that not always increasing the concentration leads to higher CPP. For example, in the study by Qavi et al.,20 in which the monomer fraction was fixed, H2 system had lower surfactant concentration than Lα structure. However, as a general trend, when polymerization takes place outside the micelles, the confinement size which is the space between micelles, where monomers are present and propagation takes place, increases as the CPP is increased.65
 | ||
| Fig. 8 Proposed relationship between CPP of the structure and transcriptive polymerization rate. | ||
While there are experimental reports on polymerization of hydrophobic monomers in both normal and reverse micelles and hydrophilic monomers in normal micelles, there is no publication on the polymerization of hydrophilic monomers in reverse micelles to the knowledge of the authors. Nevertheless, we hypothesize the trend in Fig. 8.
It is worth mentioning that although polymerization kinetics has been studied in different structures, the role of confinement shape as an independent factor is still unknown and not studied. One can hypothesize that the number of directions in which radicals can propagate may be different based on the shape of the micelles. Considering a cartesian coordinate system (see Fig. 9), the surfactant molecules in a spherical micelle are confined in a 3D space. So, the degree of freedom for monomers inside or outside the micelle becomes 0, and movement of the propagating chain becomes limited. In rod-like micelles, the surfactants are confined in two directions, allowing for monomer chains to propagate just in one direction. Planar micelles in Lα structures have 1D confinement, which allows chains to freely propagate in 2 directions.
 | ||
| Fig. 9 Direction of monomer propagation inside (a) cubic, (b) hexagonal, and (c) planar micelles. | ||
While the above-mentioned hypothesis may be presented for transcriptive templating, synergistic templating is a different case. Since the surfactants are being crosslinked together in synergistic templating, polymerization takes place at the interface (2D space). It could be the surface of a sphere in cubic structures, the surface of a cylinder in hexagonal structures, or the surface of a plane in Lα structures. Thus, propagation can proceed just in two directions in synergistic templating (1D confinement). However, segregation becomes more important in this case. In other words, segregation effect can be described as fraction of total surfactant molecules in the confined space to allow propagation since the propagating chains cannot jump from one confined space to another one. Segregation effect in the case of LLC synergistic templating can be quantified using aggregation number which shows the number of surfactants in a micelle. In transcriptive templating, two parameters control the segregation effect for polymerization of monomers in discontinuous domains: (1) shape: as the structure shifts from spherical to cylindrical to planar, the segregation effect becomes weaker due to the enhanced continuity of polymerizing phase, and (2) size: as the domain size decreases, the segregation effect is enhanced.
For synergistic templating, as the aggregation number increases (with increasing the surfactant concentration), there are fewer discrete domains of surfactants; thus, while the free radical concentration may not change, the number of radicals per discrete domains will be higher. Therefore, the probability of termination increases. This higher local concentration can increase the propagation rate too, which may lead to an increased overall kinetic rate, as will be discussed in the next paragraph. This trend has been confirmed for the cases in which reactive groups are present near the headgroup of the micelles.82,83 However, the same studies have shown an opposite trend for micelles with reactive groups near surfactant tails. It seems that when the reactive groups are inside the micelles, higher curvature of the micelle or lower CPP increases the local free radical concentration, leading to an increased termination and propagation rate in I1 than Lα structure. Similar explanation applies to the segregation effect on termination reaction in transcriptive templating.
As mentioned before, the kinetics of polymerization in transcriptive LLC templates can be considered as a first-order reaction assuming a steady-state hypothesis for free radical concentration. In this case, the overall reaction kinetic rate, k, can be defined using a combination of kinetic rate parameters as follows:
 | (11) |
In which kp shows the rate constant of propagation, f shows the initiator efficiency, kd shows the rate constant of initiator decomposition, kt shows the rate constant of termination, and [I] is the concentration of the initiator. Based on what is discussed throughout this review, it seems that the termination rate is the most affected parameter by the soft nanoconfinement of LLC templates compared to bulk polymerization and determines the difference in overall reaction rate. However, further studies are still required to evaluate all the parameters in (eqn 11) in LLC polymerization against bulk polymerization.
Considering changes of kinetic rates between different LLC structures, comparing the kt in all reviewed studies reveal that in all cases, it decreases as the CPP increases. The other governing parameter which has a determining effect on k is kp. Since kd ∼ kp1 and k ∼ kt−1/2, a decrease in kp leads to lower k despite the increasing effect of lower termination rate. As the CPP increases, hydrophobic volume of the system becomes larger while the hydrophilic part becomes smaller. Thus, the local monomer concentration can be affected depending on the presence of monomers inside or outside of the micelles. As a general trend, higher local monomer concentration results in an increased propagation rate.
Other polymerization techniques in templating
While step-growth polymerization is extensively used in polymer science and industries, most of the studies on polymerization in nanoconfinements have employed free radical chain growth polymerization, except for a few cases. For example, Forney84 used thiol–ene chemistry to photo-polymerize the LLC phase. This reaction involves a step-growth process where thiol and ene monomers alternately react through alternating propagation and chain transfer steps to form a crosslinked polymer network. In a similar study, Kotsiras85 observed exceptional preservation of H1 structures and also much faster normalized rate of polymerization with respect to an unconfined thiol–ene system. The highest normalized maximum rate of polymerization reported in this study is 1.2 s−1 which is considerably high.Classical condensation polymerization has also been used with LLC templating. As an example, phenol-formaldehyde reaction (in the form of water-soluble phenolic resins or resole) was combined with evaporation induced self-assembly of amphiphilic block copolymers to produce polyLLC with highly ordered mesostructure. The polymerization of the resole around the formed template can make a stiff and solid polymer. The resole precursor can be synthesized through the sodium hydroxide-catalyzed condensation of phenol and formaldehyde.86 The polymerization of resole at low temperatures results in a phenolic resin crosslinked by benzene rings through an aliphatic or an ether bridge.55 Uniform and monodispersed mesoporous carbon nanospheres could be obtained by further carbonization of such templates showing small particle sizes and large mesopores with narrow size distribution.87
Conclusion and future prospects
This perspective highlights the significant influence of nanoconfinement via soft and hard templates on the polymerization kinetics and final properties of polymers. Although hard nanoconfinement can provide more controlled polymerization and negligible change in the domain size of the template, template removal in this case can be a challenge. The unique interplay between the confinement environment and polymerization parameters such as initiator type, monomer chemistry, and crosslinking density has been reviewed in this study, revealing critical insights into how these factors contribute to the polymerization process. Physics of nanoconfinement can also affect polymerization steps in terms of template's size, shape, and wall effect.LLCs are in the form of gels with dimensions in range of 2–50 nm offering a versatile and dynamic environment that can affect polymerization rates, improve structural retention, and yield polymers with well-defined nanostructures. In contrast to hard templating, soft matter nature of LLCs enhances template removal after polymerization using mild conditions like extraction with water. Although synergistic templating offers a higher chance of retention of the structure over transcriptive templating, the latter can be done using widely available commercial monomers and crosslinkers.
The general trend obtained from the literature for polymerization in LLC templates shows that for hydrophilic monomers, polymerization rate increases by changing the structure from I1 to H1 to Lα while the trend can be the opposite for hydrophobic monomers. We propose a universal rule of thumb for polymerization rate by using CPP covering both normal and reverse micelles. As the CPP increases, the micelle shape and LLC structure evolve from I1 to Lα (CPP = 0–1) and then to reverse spherical micelles (CPP > 1). Generally, increasing CPP leads to enhanced overall polymerization rates outside of the micelles in transcriptive templating while the trend is the opposite when polymerization takes place inside of the micelles. It has been shown that no matter where the polymerization takes place, the termination rate always decreases by increasing CPP. The propagation, however, is directly influenced by local monomer concentration which determines the overall kinetic rate.
Although the main focus of this perspective is on the most utilized polymerization pathway, i.e., chain growth free radical polymerization, some step-growth mechanisms such as thiol–ene and phenol-formaldehyde polymerization are also reviewed. However, the role of different mentioned parameters on step-growth polymerization kinetics in LLCs is still unknown. Unlike chain growth polymerization in which there is a sudden increase in viscosity, there is a rather gradual increase in viscosity in step-growth polymerization until the gelation point. Considering the soft nature of LLCs and possibility of phase separation during polymerization, it is necessary to understand how the LLC structure changes during step-growth polymerization.
It is worth mentioning that despite various studies investigating the effect of LLC structure on polymerization kinetics, the effect of micelle shape (i.e., confinement shape) on different steps of polymerization is yet to be determined. Also, the effect of confinement shape in hard templating is rarely studied. In addition, there is no study on ionic polymerization or controlled radical polymerization, which could be due to their sensitivity to impurities and/or solvents present in conventional formulations for templating. Nevertheless, since LLCs can also be made with ionic liquids (anhydrous LLCs), there is a possibility to make polyLLCs containing confined block copolymers, living polymers, etc.
There are some important research questions in the field that need to be answered. The most important one from the authors’ perspective is how the confinement shape affects steps of free radical polymerization. Although our hypothesis is presented here, it still needs to be validated. Another research gap in the field is the behavior of hydrophilic monomers in LLC structures with CPP higher than 1 (reverse micelles) to validate the trend in the bottom right part of the Fig. 8.
Based on this study, we present the following as potential future work in this field:
• Evaluate the propagation rate constant and termination rate constant of polymerization in soft and hard nanoconfined templates versus bulk polymerization.
• Explore the role of template surface chemistry such as hydrophilic and hydrophobic surface functionalization on different steps of polymerization.
• Minimize polymer disintegration while removing template by developing innovative template removal methods or using hybrid hard–soft templates.
• Study the changes in polymerization rates in synergistic templating methods in LLC systems for a wide range of CPP values (especially higher than 1).
• Understand the role of different parameters on step-growth polymerization kinetics in nanoconfined templates. Unlike chain-growth polymerization, step-growth polymerization involves a gradual increase in viscosity, making it crucial to study how the LLC structure can be retained during the process.
• Investigate the effect of confinement shape (i.e., micelle shape) on different steps of polymerization. The shape can influence the degree of freedom for monomers and propagating chains, impacting the polymerization process.
• Study ionic polymerization or controlled radical polymerization in LLCs, especially with anhydrous LLCs made with ionic liquids. This could lead to the creation of polyLLCs containing confined block copolymers or living polymers for electrochemical applications.
• Validate the Artemis number for polymerization processes and finding challenges and viability of measurements.
Author contributions
S. M. T.: conceptualization, writing – original draft, writing – review & editing. R. F.: conceptualization, writing – review & editing, supervision, funding acquisition.Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was prepared using Federal funds under award #08_79_05677 from Economic Development Administration, U.S. Department of Commerce. The statements, findings, conclusions, and recommendations are those of the authors and do not necessarily reflect the views of the Economic Development Administration or the U.S. Department of Commerce.References
- C. B. Roth, Chem. Soc. Rev., 2021, 50, 8050–8066 RSC.
- X. Feng, Q. Imran, Y. Zhang, L. Sixdenier, X. Lu, G. Kaufman, U. Gabinet, K. Kawabata, M. Elimelech and C. O. Osuji, Sci. Adv., 2019, 5, eaav9308 CrossRef CAS PubMed.
- Y. Saadat, S. M. Tabatabaei, K. Kim and R. Foudazi, J. Membr. Sci., 2023, 684, 121861 Search PubMed.
- Y. Saadat, S. M. Tabatabaei, K. Kim and R. Foudazi, ACS ES&T Eng., 2024, 4, 1454–1468 Search PubMed.
- Y. Saadat, K. Kim and R. Foudazi, ACS Appl. Polym. Mater., 2022, 4, 8156–8165 CrossRef CAS.
- R. Foudazi, R. Zowada, I. Manas-Zloczower and D. L. Feke, Langmuir, 2023, 39, 2092–2111 CrossRef CAS PubMed.
- J. R. McLaughlin, N. L. Abbott and C. A. Guymon, Polymer, 2018, 142, 119–126 Search PubMed.
- K. S. Worthington, B. J. Green, M. Rethwisch, L. A. Wiley, B. A. Tucker, C. A. Guymon and A. K. Salem, Biomacromolecules, 2016, 17, 1684–1695 Search PubMed.
- J. D. Clapper, S. L. Iverson and C. A. Guymon, Biomacromolecules, 2007, 8, 2104–2111 CrossRef CAS PubMed.
- Y. Saadat, O. Q. Imran, C. O. Osuji and R. Foudazi, J. Mater. Chem. A., 2021, 9, 21607–21658 RSC.
- B. Sanz, N. Ballard, J. M. Asua and C. Mijangos, Macromolecules, 2017, 50, 811–821 CrossRef CAS.
- C. Mijangos and J. Martin, Polymers, 2023, 15, 525 CrossRef CAS PubMed.
- J. Kim, J. M. Kim and C. Baig, Polymer, 2021, 213, 123308 CrossRef CAS.
- N. Patsalidis, G. Papamokos, G. Floudas and V. Harmandaris, J. Chem. Phys., 2024, 160, 104904 Search PubMed.
- J. Yang, Y. Chen, Z. Yang, L. Dai, H. Choi and Z. Meng, Polymers, 2024, 16, 1155 Search PubMed.
- X. Chen and X. Kong, Nano Lett., 2023, 23, 5194–5200 Search PubMed.
- J. Martín-de León, J. L. Pura, M. L. Rodríguez-Méndez and M. A. Rodríguez-Pérez, Eur. Polym. J., 2024, 214, 113181 Search PubMed.
- P. Züblin, A. Zeller, C. Moulis, M. Remaud-Simeon, Y. Yao and R. Mezzenga, Angew. Chem., Int. Ed., 2024, 63, e202312880 Search PubMed.
- C. Zhai, PhD Dissertation, Texas Tech University, 2021.
- S. Qavi, A. Bandegi, M. Firestone and R. Foudazi, Soft Matter, 2019, 15, 8238–8250 Search PubMed.
- H. Zhao and S. L. Simon, Polymer, 2020, 211, 123112 CrossRef CAS.
- Q. Tian, H. Zhao and S. L. Simon, Polymer, 2020, 205, 122868 Search PubMed.
- C. Zhai, Y. P. Koh, B. D. Vogt and S. L. Simon, J. Polym. Sci., 2024, 62, 1922–1933 CrossRef CAS.
- Q. Tian, Y. P. Koh, S. V. Orski and S. L. Simon, Macromolecules, 2022, 55, 8723–8730 Search PubMed.
- M. A. DePierro, K. G. Carpenter and C. A. Guymon, Radtech Tech. Proc., 2006 Search PubMed https://www.radtech.org/proceedings/2006/papers/069.pdf .
- M. A. DePierro, C. Baguenard and C. A. Guymon, J. Polym. Sci., Part A: Polym. Chem., 2016, 54, 144–154 CrossRef CAS.
- C. L. Lester, C. D. Colson and C. A. Guymon, Macromolecules, 2001, 34, 4430–4438 CrossRef CAS.
- M. E. Tousley, X. Feng, M. Elimelech and C. O. Osuji, ACS Appl. Mater. Interfaces, 2014, 6, 19710–19717 CrossRef CAS PubMed.
- S. Qavi, A. P. Lindsay, M. A. Firestone and R. Foudazi, J. Membr. Sci., 2019, 580, 125–133 Search PubMed.
- M. A. DePierro, A. J. Olson and C. A. Guymon, Polymer, 2005, 46, 335–345 Search PubMed.
- M. A. DePierro and C. A. Guymon, Macromolecules, 2006, 39, 617–626 CrossRef CAS.
- B. S. Forney and C. A. Guymon, Macromolecules, 2010, 43, 8502–8510 Search PubMed.
- D. T. McCormick, K. D. Stovall and C. A. Guymon, Macromolecules, 2003, 36, 6549–6558 Search PubMed.
- J. D. Clapper and C. A. Guymon, Adv. Mater., 2006, 18, 1575–1580 Search PubMed.
- D. M. Anderson and P. StrÖm, in Polymer Association Structures, American Chemical Society, 1989, pp. 204–224 Search PubMed.
- M. Antonietti, R. A. Caruso, C. G. Göltner and M. C. Weissenberger, Macromolecules, 1999, 32, 1383–1389 Search PubMed.
- W. Meier, Macromolecules, 1998, 31, 2212–2217 Search PubMed.
- J. Jennings, B. Green, T. J. Mann, C. A. Guymon and M. K. Mahanthappa, Chem. Mater., 2018, 30, 185–196 Search PubMed.
- J. D. Clapper and C. A. Guymon, Macromolecules, 2007, 40, 7951–7959 CrossRef CAS.
- J. D. Clapper and C. A. Guymon, Mol. Cryst. Liq. Cryst., 2009, 509, 30/[772]–38/[780] Search PubMed.
- J. Hwang, H.-C. Lee, M. Antonietti and B. V. K. J. Schmidt, Polym. Chem., 2017, 8, 6204–6208 Search PubMed.
- A. Bandegi, J. L. Bañuelos and R. Foudazi, Soft Matter, 2020, 16, 6102–6114 Search PubMed.
- M. Jung, A. L. German and H. R. Fischer, Colloid Polym. Sci., 2001, 279, 105–113 Search PubMed.
- J. M. Giussi, I. Blaszczyk-Lezak, M. S. Cortizo and C. Mijangos, Polymer, 2013, 54, 6886–6893 Search PubMed.
- S. Ghosh, L. Ramos, S. Remita, A. Dazzi, A. Deniset-Besseau, P. Beaunier, F. Goubard, P.-H. Aubert and H. Remita, New J. Chem., 2015, 39, 8311–8320 RSC.
- T. Uemura, D. Hiramatsu, Y. Kubota, M. Takata and S. Kitagawa, Angew. Chem., Int. Ed., 2007, 46, 4987–4990 CrossRef CAS PubMed.
- D. Y. Liu and D. V. Krogstad, Macromolecules, 2021, 54, 988–994 CrossRef CAS.
- D. Floresyona, F. Goubard, P.-H. Aubert, I. Lampre, J. Mathurin, A. Dazzi, S. Ghosh, P. Beaunier, F. Brisset, S. Remita, L. Ramos and H. Remita, Appl. Catal., B, 2017, 209, 23–32 CrossRef CAS.
- J. F. Hulvat and S. I. Stupp, Angew. Chem., Int. Ed., 2003, 42, 778–781 Search PubMed.
- J. F. Hulvat and S. I. Stupp, Adv. Mater., 2004, 16, 589–592 Search PubMed.
- S. Ghosh, H. Remita, L. Ramos, A. Dazzi, A. Deniset-Besseau, P. Beaunier, F. Goubard, P.-H. Aubert, F. Brisset and S. Remita, New J. Chem., 2014, 38, 1106–1115 RSC.
- M. N. Wadekar, R. Pasricha, A. B. Gaikwad and G. Kumaraswamy, Chem. Mater., 2005, 17, 2460–2465 CrossRef CAS.
- S. Dutt and P. F. Siril, J. Appl. Polym. Sci., 2014, 131, 40800 CrossRef.
- I. Moriguchi, A. Ozono, K. Mikuriya, Y. Teraoka, S. Kagawa and M. Kodama, Chem. Lett., 1999, 1171–1172 Search PubMed.
- Y. Meng, D. Gu, F. Zhang, Y. Shi, L. Cheng, D. Feng, Z. Wu, Z. Chen, Y. Wan, A. Stein and D. Zhao, Chem. Mater., 2006, 18, 4447–4464 Search PubMed.
- C. Liang and S. Dai, J. Am. Chem. Soc., 2006, 128, 5316–5317 Search PubMed.
- X. Wang, C. Liang and S. Dai, Langmuir, 2008, 24, 7500–7505 CrossRef CAS PubMed.
- C. M. Ghimbeu, M. Sopronyi, F. Sima, C. Vaulot, L. Vidal, J. M. Le Meins and L. Delmotte, RSC Adv., 2015, 5, 2861–2868 Search PubMed.
- S. Herou, M. C. Ribadeneyra, R. Madhu, V. Araullo-Peters, A. Jensen, P. Schlee and M. Titirici, Green Chem., 2019, 21, 550–559 RSC.
- J. Zhang, Z. A. Qiao, S. M. Mahurin, X. Jiang, S. H. Chai, H. Lu, K. Nelson and S. Dai, Angew. Chem., Int. Ed., 2015, 54, 4582–4586 CrossRef CAS PubMed.
- Z. Li, W. Yan and S. Dai, Carbon, 2004, 42, 767–770 CrossRef CAS.
- S. Wang, D. P. Maruri, J. M. Boothby, X. Lu, L. K. Rivera-Tarazona, V. D. Varner and T. H. Ware, J. Mater. Chem. B, 2020, 8, 6988–6998 RSC.
- P. Alexandridis, U. Olsson and B. Lindman, Langmuir, 1998, 14, 2627–2638 Search PubMed.
- S. Qavi, M. A. Firestone and R. Foudazi, Soft Matter, 2019, 15, 5626–5637 Search PubMed.
- S. Qavi and R. Foudazi, Rheol. Acta, 2019, 58, 483–498 Search PubMed.
- K. E. Culley, C. Johnson and D. L. Gin, Chem. Commun., 2023, 59, 11105–11108 Search PubMed.
- R. Ni, A. P. Gantapara, J. De Graaf, R. Van Roij and M. Dijkstra, Soft Matter, 2012, 8, 8826–8834 Search PubMed.
- S. H. Anastasiadis, in Polymer Thermodynamics, ed. B. A. Wolf and S. Enders, Springer Berlin Heidelberg, Berlin, Heidelberg, 2011, pp. 179–269 Search PubMed.
- M. Rubinstein and R. H. Colby, Polymer Physics, Oxford University Press, Oxford, 2003, pp. 137–190 Search PubMed.
- M. Bažec and S. Žumer, Phys. Rev. E:Stat., Nonlinear, Soft Matter Phys., 2006, 73, 021703 CrossRef PubMed.
- E. Helfand and A. M. Sapse, J. Chem. Phys., 1975, 62, 1327–1331 Search PubMed.
- A. Kotsiras, J. Whitley, E. Orozco and C. A. Guymon, ACS Appl. Polym. Mater., 2024, 6, 2442–2452 CrossRef CAS PubMed.
- M. Mours and H. H. Winter, Rheol. Acta., 1994, 33, 385–397 Search PubMed.
- Y. Saadat, K. Kim and R. Foudazi, Polym. Chem., 2021, 12, 2236–2252 Search PubMed.
- S. Gu, L. Zhang, L. de Campo, L. A. O'Dell, D. Wang, G. Wang and L. Kong, Membranes, 2023, 13, 549 Search PubMed.
- M. Zhou and R. Foudazi, Langmuir, 2021, 37, 7907–7918 Search PubMed.
- D. L. Gin, X. Lu, P. R. Nemade, C. S. Pecinovsky, Y. Xu and M. Zhou, Adv. Funct. Mater., 2006, 16, 865–878 CrossRef CAS.
- L. N. Bodkin, C. W. Johnson, K. E. Culley, Z. A. Krajnak, J. R. Hage, N. K. Kim, C. O. Osuji and D. L. Gin, Polymer, 2024, 291, 126615 Search PubMed.
- S. Cao, M. Yoshio and A. Seki, Crystals, 2020, 10, 276 Search PubMed.
- K. S. Worthington, C. Baguenard, B. S. Forney and C. A. Guymon, J. Polym. Sci., Part B:Polym. Phys., 2017, 55, 471–489 CrossRef CAS.
- N. Goujon, L. F. Dumée, N. Byrne, G. Bryant and M. Forsyth, Macromol. Chem. Phys., 2018, 219, 1800307 Search PubMed.
- C. L. Lester and C. A. Guymon, Polymer, 2002, 43, 3707–3715 CrossRef CAS.
- L. Sievens-Figueroa and C. A. Guymon, Macromolecules, 2009, 42, 9243–9250 CrossRef CAS.
- B. S. Forney, PhD thesis, The University of Iowa, 2013 Search PubMed.
- A. Kotsiras, PhD thesis, The University of Iowa, 2023 Search PubMed.
- L. Song, D. Feng, N. J. Fredin, K. G. Yager, R. L. Jones, Q. Wu, D. Zhao and B. D. Vogt, ACS Nano, 2010, 4, 189–198 CrossRef CAS PubMed.
- L. Hu, Z. Qian, W. Gao, X. Wang and Y. Tian, J. Mater. Sci., 2020, 55, 2052–2067 CrossRef CAS.
Footnote |
| † In greek mythology, Artemis symbolizes the balance between the natural order and the unpredictable aspects of nature. |
| This journal is © The Royal Society of Chemistry 2025 |