
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFulleride superconductivity tuned by elastic strain due to cation compositional disorder†
H. Esma
Okur
 a,
Ross H.
Colman
b,
Yasuhiro
Takabayashi
c,
Peter
Jeglič
d,
Yasuo
Ohishi
e,
Kenichi
Kato
f,
Denis
Arčon
dg,
Yoshiki
Kubota
h and
Kosmas
Prassides
*dhi
a,
Ross H.
Colman
b,
Yasuhiro
Takabayashi
c,
Peter
Jeglič
d,
Yasuo
Ohishi
e,
Kenichi
Kato
f,
Denis
Arčon
dg,
Yoshiki
Kubota
h and
Kosmas
Prassides
*dhi
aDepartment of Chemistry, Faculty of Engineering and Natural Sciences, Bursa Technical University, TR-16310 Bursa, Turkey
bDepartment of Condensed Matter Physics, Faculty of Mathematics and Physics, Charles University, 121 16 Prague, Czech Republic
cDepartment of Physical Science and Engineering, Nagoya Institute of Technology, Nagoya 466-8555, Japan
dJozef Stefan Institute, Jamova c. 39, SI-1000 Ljubljana, Slovenia
eJapan Synchrotron Radiation Facility, SPring-8, Hyogo 679-5198, Japan
fRIKEN SPring-8 Center, Hyogo 679-5148, Japan
gFaculty of Mathematics and Physics, University of Ljubljana, Jadranska c. 19, SI-1000 Ljubljana, Slovenia
hDepartment of Physics, Graduate School of Science, Osaka Metropolitan University, Osaka 599-8531, Japan. E-mail: k.prassides@omu.ac.jp
iFaculty of Engineering, Kyoto University of Advanced Science, Kameoka 621-8555, Japan
First published on 3rd September 2024
Abstract
Dynamical fluctuations of the elastic strain in strongly correlated systems are known to affect the onset of metal-to-insulator or superconducting transitions. Here we report their effect on the properties of a family of bandwidth-controlled alkali-intercalated fullerene superconductors. We introduce elastic strain through static local structural disorder in a systematic and controllable way in the fcc-structured KxCs3−xC60 (with potassium content, 0.22 ≤ xK ≤ 2) series of compositions by utilizing the difference in size between the K+ and Cs+ co-dopants. The occurrence of the crossover from the Mott–Jahn–Teller insulating (MJTI) state into the strongly correlated Jahn–Teller metal (JTM) on cooling is evidenced for the compositions with xK < 1.28 by both synchrotron X-ray powder diffraction (SXRPD) – anomalous reduction of the unit cell volume – and 133Cs NMR spectroscopy – sudden suppression in the 133Cs spin-lattice relaxation rates. The emerging superconducting state with a maximum critical temperature, Tc = 30.9 K shows a characteristic dome-like dependence on the unit-cell volume or equivalently, on the ratio between the on-site Coulomb repulsion, U, and the bandwidth, W. However, compared to the parent Cs3C60 composition in which cation disorder effects are completely absent, the maximum Tc is lower by ∼12%. The reduction in Tc displays a linear dependence on the variance of the tetrahedral-site cation size, σT2, thus establishing a clear link between structural-disorder–induced attenuation of critical elastic strain fluctuations and the electronic ground state.
1 Introduction
Superconductors in which the electronically active units are molecules rather than atoms typically incorporate π-electron open-shell units. Among the members of the family of molecular superconductors, the trivalent alkali fullerides, A3C60 (A = alkali metal) are best-in-class representatives exhibiting the highest superconducting transition temperature, Tc (at 38 K under applied pressure)1 and with their zero-resistance state surviving to extremely high magnetic fields (Hc2 > 90 T).2 Very importantly, it has been also established that electron correlations play a dominant role in the superconducting pairing mechanism, thereby rendering these materials members of the family of unconventional high-Tc superconductors.3–7All studies reported so far attest that the transitions between the various electronic states of the A3C60 fullerides are achieved principally via altering the interfullerene separation, which modifies the size of the bandwidth, W, and therefore tunes the ratio (U/W), where U is the on-site Coulomb repulsion.8 For molecular systems like the A3C60 fullerides, U is on the order of 1 eV and varies little across the various compositions.5–7 The parent hyperexpanded compound Cs3C60, in which (U/W) is maximal, is a magnetic insulator at ambient pressure and crystallizes as either the A15-structured (primitive cubic phase comprising orientationally ordered C603− units) or the fcc-structured (face-centred cubic phase comprising merohedrally disordered C603− units) polymorph.3,4 Application of physical pressure to both polymorphs suppresses the magnetic state leading to a Mott-insulator-to-metal transition as the interfullerene separation is reduced, W increases, and (U/W) shifts below a critical value. Upon cooling, superconductivity emerges from the metallic state with a dome-shaped dependence of Tc with volume, V, occupied per C603− anion.1,3,4,8–10 The maximum Tc is somewhat higher for the A15 Cs3C60 phase at 38 K compared to the value measured for the fcc Cs3C60 phase (35 K).
However, while the A15 structure is encountered only at the high-packing densities of the parent Cs3C60 phase, the fcc-structured A3C60 phases are stable over a broad range of packing densities, and therefore (U/W) ratios. As a result, application of physical and/or chemical pressure to the most expanded fcc-structured Cs3C60 phase has allowed the exploration of the electronic phase diagram of the fullerides in great detail.11–18 Firstly, as the orbital overlap of neighbouring C603− anions increases upon pressurisation, the MJTI state is destroyed. The metallic state, which emerges from the insulator is unconventional and is characterized by fluctuating microscopically heterogeneous co-existence of both localized Jahn–Teller-active and itinerant electrons (JTM). Further pressurization, leads to the gradual disappearance of localised Jahn–Teller features and the evolution of the unconventional JTM to a conventional Fermi liquid. At the same time, the s-wave superconducting state evolves from an unconventional correlated superconductor in the strong-coupling limit to a weak-coupling BCS-like superconductor. Notably, the JTM is the parent state of the unconventional fulleride superconductor with the highest Tc encountered at the crossover between JTM and Fermi liquid behaviour.11
Manipulation of the fcc-structured Cs3C60 phase by chemical pressure was successfully achieved by altering the cation occupation of the tetrahedral (T) interstitial sites, which are smaller (rT = 1.12 Å) than the ionic size of the heavier alkali dopants (K+, Rb+, and Cs+). As a result, fulleride packing density can be sensitively controlled by the occupation of the tetrahedral sites as the proportions of isovalent K+, Rb+, and Cs+ ions vary. Increased amounts of the smaller size ions lead to lattice contraction, increased values of W, and smaller values of (U/W).11 On the other hand, the occupancy of the octahedral (O) sites by different alkali ions plays a minor role as the hole size (rO = 2.06 Å) is larger than the ionic radius of all alkali ions. The detailed investigation of the chemical pressure effect across the fcc-structured members of the RbxCs3−xC60 (0.35 ≤ x ≤ 2) series revealed the existence of an electronic phase diagram entirely comparable to that established upon physical pressurization of the parent Cs3C60 phase.11 This is consistent with the dominant role of the intermolecular overlaps in controlling the magnetic and metallic/superconducting states of the materials.3,4 Notably, however, these studies revealed a small but significant (∼5%) reduction of the maximum Tc, which appears smaller (33.2 K) in the chemically pressurized analogues when compared to physically pressurized Cs3C60 (35.1 K). This Tc reduction is evident at all packing densities, V despite the expected identical values of the bandwidth, W and the corresponding (U/W) ratios.
This observation necessitates a revisit of the role of the alkali ions in determining the superconducting properties of the fulleride phases. In particular, there is a well-defined difference in the occupation of the tetrahedral interstices in the RbxCs3−xC60 (0 ≤ x ≤ 3) phase field: when x = 0, 2, and 3, the tetrahedral holes are occupied by either Cs+ or Rb+ alone, while for all other values of x, they are occupied by a disordered mixture of Rb+ and Cs+ cations. Such disorder of cation occupation of available structural cavities and the associated cooperative fluctuations of the elastic strain generated have been shown before to influence sensitively a variety of electronic responses19 including metal-insulator and superconducting transitions in strongly correlated transition metal oxide phases such as the magnetoresistive manganites20 and the high-Tc cuprates,21 respectively.
This raises the issue whether the generic nature of such elastic interactions is also relevant to the electronic properties of fulleride superconductors and could be responsible for the small, but significant, suppression in the magnitude of their Tc encountered when cation disorder is present. Here, we undertake a systematic study of the structural and electronic properties of the series of fcc-structured KxCs3−xC60 (with nominal content, 0.12 ≤ x ≤ 2) fullerides at both ambient and elevated pressures. Due to the increased difference in size between the K+ and Cs+ constituent ions, we are able to enhance further the ionic size effects, culminating in even larger suppression (on the order of 12%) of the values of Tc observed in the absence of disorder at the same fulleride packing density. The high quality of the experimental data allowed us to establish quantitatively the effects of cation disorder and the associated cooperative elastic interactions on Tc.
2 Materials and methods
2.1 Synthesis and characterisation
Polycrystalline KxCs3−xC60 (nominal K content, 0.12 ≤ x ≤ 2) compounds were synthesized by high-temperature reactions of stoichiometric quantities of sublimed C60 and phase-pure K6C60 and Cs6C60 precursors (Fig. S1†). The exact experimental protocol is detailed in the ESI.† As small amounts of reactants are employed in these preparations, the use of free-flowing A6C60 (A = K, Cs) powders instead of K and Cs metals allows better stoichiometry control of the target compositions. Due to the extreme air- and moisture-sensitivity, all sample manipulations were carried out in an Ar-filled glove box. Laboratory X-ray powder diffraction (Bruker D8 Advance, Debye–Scherrer geometry, CuKα1 radiation, λ = 1.54056 Å) was used to monitor the quality of all intermediate and the final polycrystalline products. Samples were obtained as free-flowing black-coloured powders.2.2 Structural measurements at ambient and elevated pressures
High-resolution synchrotron X-ray powder diffraction (SXRPD) measurements of the samples at ambient pressure were performed using a helium-flow cryostat between 10 and 300 K with the multidetector diffractometer on beamline ID31 and ID22 (λ ∼ in the range 0.35 to 0.4 Å) at the European Synchrotron Radiation Facility (ESRF), Grenoble, France. Data for the x = 1 sample were obtained between 112 and 300 K with the diffractometer on beamline BL44B2 (λ = 0.81887 Å) at SPring-8, Japan.22 Pressure-dependent high-resolution SXRPD data (λ ∼ 0.41 Å) were collected at 7 K for samples of nominal composition KxCs3−xC60 (x = 0.25, 0.75, 1) at pressures between 0.2 and 10 GPa on beamline BL10XU, SPring-8, Japan. Data analysis of the SXRPD profiles at both ambient and elevated pressures was performed with the GSAS suite of Rietveld analysis programs.232.3 Magnetic measurements at ambient and elevated pressures
Ambient-pressure magnetic measurements were carried out with a Quantum Design SQUID MPMS magnetometer on ∼20–25 mg powder samples loaded in thin-walled quartz ampoules and sealed under a He gas partial pressure (∼300 mbar). The magnetization, M, of all samples was measured at an applied field of either 10 or 20 Oe between 1.8 and 40 K under both zero-field-cooled (ZFC) and field-cooled (FC) protocols. Temperature-dependent magnetic susceptibility, χ, data were also collected at 3 and 5 T under FC protocols between 1.8 and 300 K. High-pressure magnetization measurements at 20 Oe under ZFC protocols were carried out under applied pressures of up to 10.3 kbar with an easyLab Technologies piston-cylinder high-pressure cell (Mcell 10). High-purity Sn was used as an in situ manometer with Daphne mineral oil as the pressure transmitting medium.2.4 NMR spectroscopy
133Cs (spin I = 7/2) and 39K NMR (spin I = 3/2) spectra and spin-lattice relaxation, T1, times were measured for KxCs3−xC60 [nominal content, x = 0.35, 0.5, 2; refined content, xK = 0.53, 0.64, 2 (vide infra)] powders at a magnetic field of 9.39 T. Samples were sealed into Pyrex glass tubes under a low-pressure He atmosphere. Measurements were conducted between 4 and 420 K in a liquid He flow cryostat with a temperature stability better than ±0.1 K over the entire temperature range. As a 133Cs NMR reference, a CsNO3 standard was used with a corresponding reference frequency, νref(133Cs) = 52.4609 MHz. In the 133Cs NMR experiments, a solid-echo pulse sequence (β)–τ–(β)–τ-echo was used with a pulse length, τ(β) = 4 μs, an interpulse delay, τ = 50 μs and appropriate phase cycling. The same pulse sequence was also used to detect the 39K NMR signal (KCl standard, νref(39K) = 18.6633 MHz) but with a larger pulse width of 14 μs and an interpulse delay, τ of 200 μs. For spin-lattice relaxation measurements, the inversion recovery technique was applied in both cases.As the KxCs3−xC60 phase assemblages might contain Cs4C60 and/or CsC60 impurity phases (vide infra), we employed a similar strategy as in our earlier work on overexpanded Cs3C60 samples3,4 in order to relatively enhance the 133Cs NMR signal arising from the fcc phases. The symmetry of the Cs+ sites of the Cs4C60 phase is lower than axial (point group C2v), leading to broadening of the respective NMR spectral lines by quadrupole interactions. As a result, such lineshape broadening together with the low phase concentration, and the experimental pulse-length optimization for the fcc Cs3C60 phase suppress the Cs4C60 contribution to the NMR signal amplitude.3,4 In addition, the 133Cs NMR signal of the CsC60 impurity is shifted by ∼800 ppm relative to the reference and is thus well separated from the main O- and T-site signals. Indeed, while the 133Cs NMR signal of the CsC60 impurity is detected in Cs3C60 samples,3,4 it is too low in intensity to be observed in the KxCs3−xC60 series (Fig. S2†).
3 Results
3.1 Structural evolution of KxCs3−xC60 at ambient and elevated pressures
A series of eight KxCs3−xC60 samples with nominal x varying between 0.12 and 2 were synthesized. Inspection of their high-resolution SXRPD profiles (Fig. S3†) collected at ambient temperature reveals in all cases the formation of cubic fulleride phases with fcc symmetry (Fig. 1a) and with an increasing lattice size, a as the content of the smaller K+ cation, x decreases. At the same time, the purity of the samples degrades with increasing Cs+ content – decreasing x – as the fractions of co-existing Cs4C60 and CsC60 phases grow at the expense of that of the fcc-structured phases. As established before, both fcc Cs3C60 (ref. 4) and the expanded fcc fullerides RbxCs3−xC60 (ref. 11) are not accessible in phase-pure form due to the competing disproportionation reaction into CsC60 and Cs4C60 – driven by the size mismatch between the large Cs+ and Rb+ cations and the small tetrahedral fcc interstitial sites – that leads to the isolation of three-phase assemblages. Rietveld analysis of the SXRPD data was undertaken by employing the established merohedrally disordered fulleride model (space group Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m) for the fcc-structured components.
m) for the fcc-structured components.
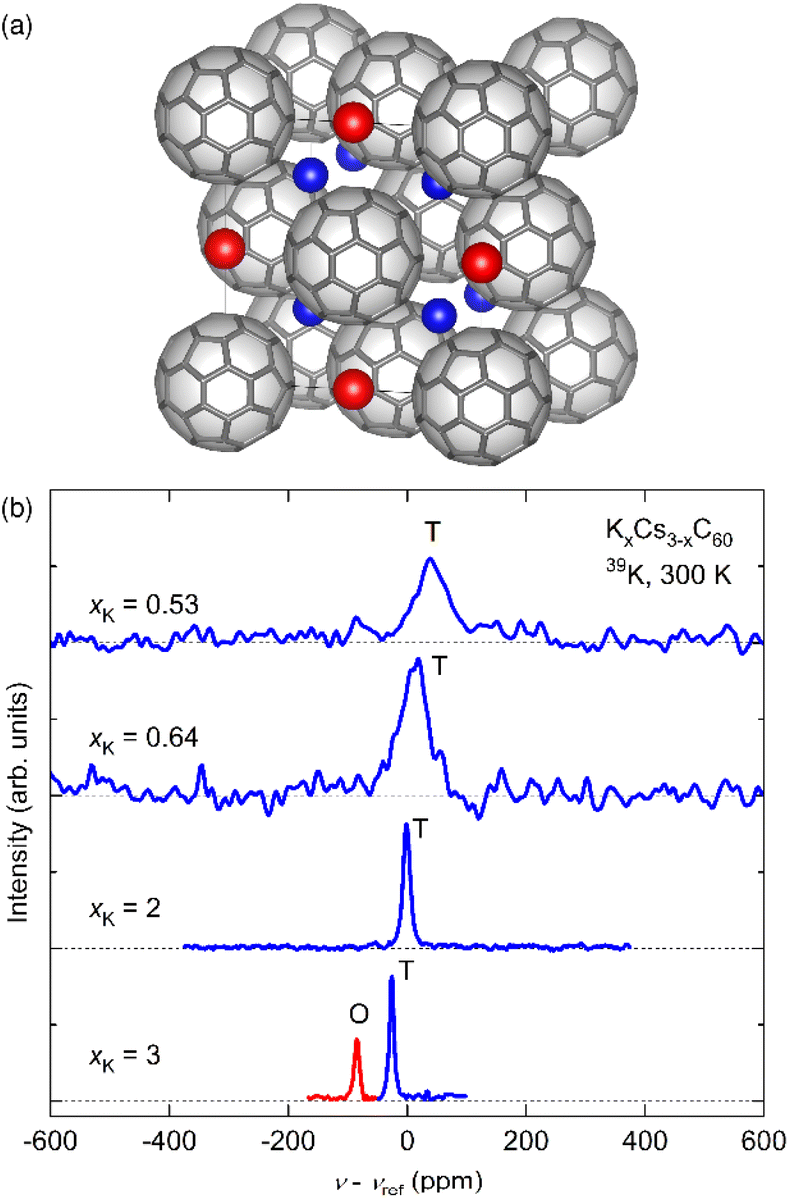 | ||
| Fig. 1 (a) Crystal structure of fcc fullerides (space group Fmm) with stoichiometry A3C60 (A is an alkali metal cation; blue spheres depict cations residing in tetrahedral (T) and red in octahedral (O) interstices, respectively). The C603− units are merohedrally disordered; here only one of the two possible orientations is depicted at each lattice position. (b) 39K NMR spectra measured for K3−xCsxC60 (refined xK = 0.53, 0.64, 2, 3) powders at 300 K. The T and O peaks are labelled in blue and red colour, respectively. The spectrum of K3−xCsxC60 (x = 3) is been reproduced from ref. 24. | ||
An important issue which arises from the different size of the octahedral (rO = 2.06 Å) and tetrahedral (rT = 1.12 Å) cavities in the fcc unit cell relates to their possible preferred occupation by the larger Cs+ (rCs+ = 1.67 Å) and smaller K+ (rK+ = 1.38 Å) ions, respectively. This can be addressed experimentally by an element-specific experimental technique such as NMR spectroscopy. Fig. 1b shows the 39K NMR spectra for the KxCs3−xC60 series (with refined xK varying between 0.53 and 3) at ambient temperature. For disorder-free K3C60 (x = 3), sharp peaks of both O- and T-lattice-sites can be clearly recognized.24 On the other hand, the O-site peak is completely suppressed for K2CsC60 (xK = 2), implying preferential occupation of the O site by the larger Cs+ ion with K+ residing in the T cavities. This trend persists as x becomes smaller, albeit with the observation of a significantly broadened T-site 39K NMR peak (Fig. S4†). This is the signature of increased cation disorder due to the random distribution of the differently sized K+ and Cs+ cations amongst the T-sites of the fcc lattice (vide infra).
Rietveld analysis of the ambient-temperature data (Fig. 2, S5–S11 and Table S1†) was therefore undertaken by incorporating the information from NMR spectroscopy – K+ ions were only included in the T sites while, for x < 2, Cs+ ions were allowed to occupy both T and O sites. Across the KxCs3−xC60 series, we find that the lattice constants of the cubic phases with fcc symmetry decrease monotonically as the K content increases. At the same time, the refined stoichiometry of the fcc phase in each sample and the composition of the phase assemblage were determined. The refined stoichiometries differ from the nominal ones, except for the x = 2 sample, with the refined K-content, xK being consistently higher than the nominal value. At the same time, the refined fcc phase fractions increase from 31.5(2)% (the remaining comprising Cs4C60 and CsC60) for the most expanded phase, xK = 0.22(1) to 94.05(1)% for xK = 1.626(4) and 100% for xK = 1.996(6). The results are summarized in Tables S2–S9.† Fig. S12† shows that the fcc lattice parameter, a varies linearly with the refined K content, xK across the KxCs3−xC60 series in agreement with Vegard's law and confirming the random distribution of the K+ and Cs+ alkali ions amongst the lattice T-sites. Notably the slope da/dxK = −0.234(3) Å is significantly larger than that determined for the RbxCs3−xC60 analogues (da/dxRb = −0.150(4) Å (ref. 11)), as expected by the smaller ionic size of K+ than that of the Rb+ intercalant.
 | ||
| Fig. 2 Rietveld refinement of the synchrotron XRPD data collected at 300 K for fcc-structured KxCs3−xC60 with refined K-content, xK = 0.87(1) (λ = 0.39984 Å). Red circles, blue lines and green lines represent the observed, calculated and difference profiles, respectively. Ticks mark the reflection positions of the fcc (85.18(7)%, red) and the body-centred-orthorhombic (bco) Cs4C60 (14.8(1)%, black) phases. Inset: expanded view in the range 4.7° to 5.7° with reflections labelled by their hkl Miller indices. | ||
The temperature dependence of the fcc crystal structures of the members of the KxCs3−xC60 series with xK = 0.35, 0.64, 0.87, and 1.28, as studied by SXRPD, shows that the cubic structure is robust and survives to the lowest temperature of the present experiments. In particular, the thermal behaviour of the unit cell volume of the KxCs3−xC60 composition with a high K-content, xK = 1.28 mimics that of the parent Cs3C60 (ref. 4) showing a smooth lattice contraction on cooling that can be accounted for with a Debye–Grüneisen model (see the corresponding text in the ESI† for details of the Debye–Grüneisen analysis).25 On the other hand, the contraction of the unit cell volume of the compositions with xK = 0.35, 0.64, and 0.87 with decreasing temperature is not smooth and exhibits an anomalous reduction, ΔV from the value derived from the Debye–Grüneisen fits below a characteristic temperature, T′ ∼ 97, 170, and 220 K, respectively (Fig. 3 and Table S10†). The normalised volume changes, ΔV/VT′ range between 0.2 and 0.4% (inset Fig. 3). Rietveld refinements (Fig. S13–S16, Tables S4–S6 and S8†) establish the isosymmetric nature of the structure across the observed anomalies in the lattice metrics. In a similar fashion to the V(T) response of the RbxCs3−xC60 (0.25 ≤ x ≤ 1.5) analogues,11,14T′ shifts to higher temperatures and the anomalous behaviour extends over a larger temperature range as the lattice contracts with increasing K content, xK.
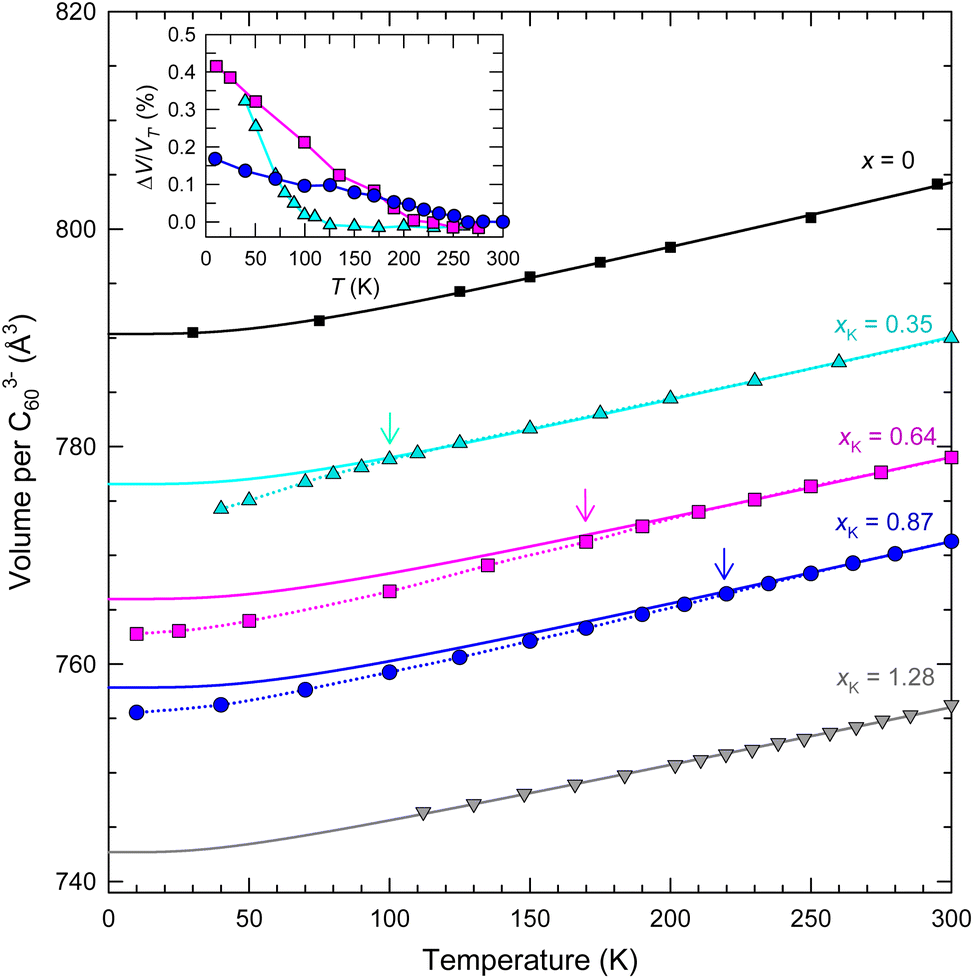 | ||
| Fig. 3 Temperature evolution of volume, V, occupied per C603− anion for fcc-structured KxCs3−xC60 samples with xK = 0.35, 0.64, 0.87 and 1.28 and for Cs3C60.4 The arrows mark the onset temperatures of the change in lattice response, denoted T′, where present. The solid lines through the data are Debye–Grüneisen fits25 for T > T′ (or over all T for x = 0 and xK = 1.28) (Table S10†). The dotted lines through the data at T < T′ are guides to the eye. Inset: temperature dependence of the normalised volume change, ΔV/VT′ for xK = 0.35, 0.64 and 0.87 – ΔV is the difference in volume between that predicted by the Debye–Grüneisen fits and that derived experimentally. The errors in V per C603− from Rietveld analysis are smaller than the size of the data points. | ||
The pressure dependence of the low-temperature crystal structures of the three KxCs3−xC60 samples with xK = 0.35, 0.87 and 1.28 was also followed by SXRPD to 10 GPa at 7 K. Rietveld analysis of the diffraction profiles confirms the robustness of the fcc structure under the high-pressure conditions too (Fig. S17–S19 and Tables S11–S13†). Fits of the non-linear pressure dependence of the 7 K unit cell volume to the semi-empirical second-order Murnaghan equation-of-state (EoS)26 for each composition (Fig. S20†) allow us to extract the values of the zero-pressure isothermal bulk moduli, K0 (∼18–19 GPa) and their pressure derivatives,  These, and the resulting low-temperature volume compressibilities, κ (∼0.05 GPa−1) are comparable to those derived before for fcc-structured Cs3C60 (ref. 4) and the RbxCs3−xC60 analogues11 (Table S14†).
These, and the resulting low-temperature volume compressibilities, κ (∼0.05 GPa−1) are comparable to those derived before for fcc-structured Cs3C60 (ref. 4) and the RbxCs3−xC60 analogues11 (Table S14†).
3.2 Electronic properties of KxCs3−xC60 at ambient and elevated pressures
The superconducting properties of the KxCs3−xC60 samples (xK content in the range between 0.22 and 2) at ambient pressure were investigated by low-field magnetization measurements. Bulk superconductivity is observed with Tc initially increasing from 26.5 K in overexpanded K0.22Cs2.78C60 (shielding fraction 16%) to 30.9 K in optimally expanded K0.87Cs2.13C60 (40%) and then decreasing to 22.8 K in underexpanded K2CsC60 (52%) (Fig. 4a, b and S21†). The variation of Tc with K content in fcc KxCs3−xC60 mimics the Tc(V) behaviour of fcc Cs3C60 (ref. 4) and the Tc(V) and Tc(x) behaviour of isostructural RbxCs3−xC60,11 displaying well-defined dome shape (Fig. 4c). At large unit cell volumes – i.e. at low co-doping levels of K+ or Rb+ – there is little change in Tc at fixed lattice sizes between the KxCs3−xC60 and RbxCs3−xC60 systems. However, at small unit cell volumes as the amount of the K+ or Rb+ co-dopants in the tetrahedral interstices increases, Tc is markedly suppressed in the KxCs3−xC60 family relative to those in RbxCs3−xC60 (maximum Tc at 33.2 K) and pressurized Cs3C60 (maximum Tc at 35.1 K). | ||
| Fig. 4 Temperature dependence of the ZFC magnetisation, M, of the KxCs3−xC60 samples divided by the applied magnetic field. (a) Samples with K-content, 0.22 ≤ xK ≤ 0.87. (b) Samples with K-content, 1.28 ≤ xK ≤ 2. The insets show expanded regions of the respective M(T) data near the superconducting Tc. (c) Variation of ambient pressure superconducting Tc with volume, V occupied per C603− anion at T = Tc for KxCs3−xC60 (0.22 ≤ xK ≤ 2) and for RbxCs3−xC60 (0.22(1) ≤ xRb ≤ 2)11,14 together with that for fcc Cs3C60 under pressure.4 Volumes per C603− for KxCs3−xC60 at Tc are estimated by extrapolation of the unit cell volumes determined from low-temperature structural data. The inset shows the dependence of Tc on xK (red squares) and xRb (blue circles). All solid lines through the data points are guides-to-the-eye. | ||
The superconducting properties of the materials were also investigated by magnetization measurements, M(T) as a function of both chemical and physical pressure (to ∼10.3 kbar) (Fig. S22†). For the most expanded sample studied, K0.35Cs2.65C60, Tc increases with increasing pressure with an initial pressure coefficient, (dTc/dP)P=0 = +1.5(2) K kbar−1. As xK increases, the rate of change rapidly decreases, approaching zero for the K0.87Cs2.13C60 composition, which shows the highest Tc at ambient pressure. (dTc/dP)P=0 then becomes negative upon further chemical pressurization (−0.88(1) K kbar−1 for K1.28Cs1.72C60, −0.99(3) K kbar−1 for K2CsC60). Further increase in pressure leads to superconductivity domes for the 0.35 ≤ xK ≤ 0.87 compositions while Tc of the 1.28 ≤ xK ≤ 2 compositions decreases smoothly (Fig. 5a). The variation of the superconducting Tc of KxCs3−xC60 (0.35 ≤ x ≤ 2) with unit cell volume, V, at low temperature extracted using the Tc(P) data together with 7 K V(P) data (Fig. S20†) is shown in Fig. 5b – the Tc(V) domes show maxima in the vicinity of 760 Å3 per C603− in a comparable fashion to the behaviour of pressurized fcc-structured Cs3C60 and RbxCs3−xC60.
 | ||
| Fig. 5 (a) Variation of the superconducting critical temperature, Tc with pressure for fcc-structured KxCs3−xC60 (0.35 ≤ xK ≤ 2). Solid lines through data points are guides to the eye except those for xK = 1.28 and 2 that represent linear fits to the data. (b) Variation of Tc with volume, V, occupied per C603− anion at low temperature for fcc-structured KxCs3−xC60 (0.35 ≤ x ≤ 2), RbxCs3−xC60 (0.35 ≤ x ≤ 2) (grey solid lines) and Cs3C60 (dashed black line). Solid lines through xK data points are guides-to-the-eye. The inset shows an expanded view of dome-shaped Tc (V) data. | ||
Paramagnetic susceptibility measurements, χ(T), as a function of temperature at ambient pressure in the normal state have proven before3,4,11 to be an excellent probe of the electronic properties of fullerides, sensitively tracking the insulator-to-metal crossover associated with the lattice anomalies evident in diffraction for selected compositions (vide supra). However, χ(T) data for the present KxCs3−xC60 phase assemblages at small xK suffer from also incorporating significant impurity-phase contributions to the measured susceptibility. As a result, well-defined cusps on cooling that provide evidence for insulator-to-metal crossover are hard to ascertain – there is only some weak evidence of maxima in χ(T) at ∼68 and 97 K for K0.22Cs2.78C60 and K0.35Cs2.65C60, respectively (Fig. S23†). The χ(T) data for moderately expanded compositions with xK = 0.53 and 0.64 no longer display any cusps but they do not exhibit a temperature-independent Pauli susceptibility term either. Such a term, which is consistent with a metallic ground state from which superconductivity emerges on cooling, appears to be present for underexpanded compositions with xK = 1.28 and 2.
Given the ambiguities associated with the χ(T) datasets due to the multiphase nature of the samples, we used the local-probe NMR technique to follow the temperature evolution of the electronic properties of the K0.64Cs2.36C60 composition. The 133Cs spin-lattice relaxation rate divided by temperature, 1/133T1T, which measures the imaginary part of the dynamic electron spin susceptibility, provides a sensitive insight into the electronic degrees of freedom as they develop across the insulator-to-metal crossover.3,4,11 On cooling from high temperatures, 1/133T1T for both O- and T-sites of K0.64Cs2.36C60 slightly increases with decreasing temperature (see Fig. S24† and the corresponding text in the ESI† for details of the spectral and 133Cs relaxation rate analysis) showing qualitatively comparable behaviour to that previously observed in RbCs2C60 and in Mott-insulating low-spin (S = ½) Cs3C60 (ref. 4, 11 and 14) (Fig. 6). Consistent with the structural results, 1/133T1T of K0.64Cs2.36C60 begins to decrease below T' ∼ 170 K, as expected for a crossover to a Jahn–Teller metal11 and in close similarity with the behaviour of RbCs2C60. Above T′, the 1/133T1T datasets for the two samples with comparable unit-cell volumes show different dependences with increasing temperature with the difference increasing as we move away from the insulator-to-metal crossover line. This implies the existence of another control parameter, namely interstitial site disorder (vide infra), which further controls the local electronic properties.
 | ||
| Fig. 6 Temperature dependence of the 133Cs spin-lattice relaxation rates divided by temperature, 1/133T1T for K0.64Cs2.36C60 (solid symbols) and for similarly expanded RbCs2C60 (open symbols) from ref. 11. In both cases, squares and circles mark the data for T- and O-sites, respectively. The dotted lines mark the temperature, T′ at which maxima in 1/133T1T are observed and the onset of superconductivity at Tc in the NMR magnetic field of 9.39 T. | ||
Below Tc = 27 K (the value of the critical temperature in the NMR magnetic field of 9.39 T), 1/133T1T of K0.64Cs2.36C60 is suddenly suppressed indicating the opening of a superconducting gap, Δ, while at the same time, no Hebel–Slichter coherence peak is observed. The spin-lattice relaxation rate adopts a thermally activated dependence 1/133T1 ∝ exp[−Δ0/kBT], in agreement with an isotropic s-wave superconducting gap (Fig. S25†). The semilog plot allows us to extract an estimate of the superconducting gap, Δ0. The normalized gap value, 2Δ0/kBTc ∼ 5 is significantly enhanced relative to that of a weakly coupled BCS superconductor (2Δ0/kBTc = 3.52) in a similar fashion to the behaviour of the overexpanded members of the RbxCs3−xC60 family.11
4 Discussion
The electronic phase diagram of selected fcc-structured A3C60 fullerides was mapped out in the past both by the application of physical pressure to the highly expanded MJTI Cs3C60 phase4,13 and of chemical pressure in the series of RbxCs3−xC60 (0 ≤ x ≤ 2) compositions in which Cs+ is quasi-continuously replaced by the smaller Rb+ cation.11,14,27 The KxCs3−xC60 (0 ≤ x ≤ 2) phases investigated here are direct analogues of their antecedents in the RbxCs3−xC60 (0 ≤ x ≤ 2) series with the smaller K+ ion being now employed as a direct substituent of Cs+. The fcc unit cell size decreases again linearly with increasing content of the smaller K+ ion (Fig. S12†), thereby permitting the exploration of the electronic properties of the KxCs3−xC60 (0 ≤ x ≤ 2) fullerides across a broad region of the phase diagram from underexpanded to overexpanded via optimally expanded compositions. The existence of a superconductivity dome and the presence of a crossover of the normal metallic state from a correlated JT metal to a Fermi liquid, as the packing density of the fulleride units decreases are essentially identical observations to those established before for the fcc RbxCs3−xC60 compounds.11,14 However, a noticeable difference in behaviour of the KxCs3−xC60 (0 ≤ x ≤ 2) compositions, as evidenced in Fig. 4c and 5b, is the presence of a significant suppression of the superconducting transition temperature, Tc when compared with the values measured in both RbxCs3−xC60 and pressurised Cs3C60 at the same packing densities, V/C603−.In order to understand the origin of the suppression of Tc in the KxCs3−xC60 series, we first examine the variation of the packing density, V/C603− with the average radius of the ions occupying the tetrahedral sites, 〈rA,T〉 (A = K, Rb, Cs). Fig. S26† shows that the identity of the ions themselves is important in determining the packing density. As the amount of the K+ or Rb+ co-intercalants into the tetrahedral sites of the Cs3C60 structure increases by partially replacing the larger Cs+ ions, the packing density smoothly decreases. However, the rate of decrease is distinctly different between the KxCs3−xC60 (dV/d〈rA,T〉 = 250(3) Å2) and the RbxCs3−xC60 (dV/d〈rA,T〉 = 306(9) Å2) series. This leads to an increasing deviation between their corresponding V/C603−vs. 〈rA,T〉 lines at the same average ionic size for large co-intercalant content (Fig. S26†). It is an indication of the potential importance of disorder in the tetrahedral sites – that arises from the difference in size mismatch between the intercalants, namely (rCs+ − rK+) > (rCs+ − rRb+) – on the structural and electronic properties of the materials.
The degree of cation disorder may be quantified by the magnitude of the statistical variance, σT2 in the distribution of the T-site ionic radii, namely σT2 = ∑xA,TrA,T2 − 〈rA,T〉2. The inset in Fig. S26† shows the variation in cation disorder with the average cation radius, 〈rA,T〉 of the tetrahedral sites for both KxCs3−xC60 and RbxCs3−xC60. The magnitude of σT2 gradually increases as we replace Cs+ with K+ or Rb+ ions at the T-sites of fcc-structured Cs3C60. In both cases, it reaches a maximum value and then decreases, producing a dome-shaped scaling of cation size variance with 〈rA,T〉. The degree of cation disorder is significantly larger in the case of the K+ co-intercalant for the KxCs3−xC60 series whereas it is absent in the parent Cs3C60 and A2CsC60 (A = K, Rb) phases for which σT2 = 0.
The static local structural disorder quantified by σT2 is directly associated with the elastic strain whose fluctuations affect critical temperatures. Returning to Fig. 5b, which displays the values of superconducting Tc for fcc Cs3C60 under pressure together with those for RbxCs3−xC60 and KxCs3−xC60 at ambient pressure vs. the packing density of the fulleride units, it is apparent that there are three distinct domes peaking roughly at comparable values of V/C603− but displaced vertically – one for each family. The highest value of the measured Tcs at each V is encountered for pressurized Cs3C60, followed by that for RbxCs3−xC60, and then by that for KxCs3−xC60. For instance, (Tc)max for the KxCs3−xC60 (xK = 0.87, V = 757.9 Å3) composition at ambient pressure at the top of the dome is 30.9 K. For the same lattice size of the fulleride salts, Tc increases first to 33.2 K at ambient pressure for the RbxCs3−xC60 family and then to 35.1 K for pressurized Cs3C60, an overall increase of 4.2 K. This trend is associated with a decrease in the value of the T-site cation size variance, σT2 from 0.021 Å2 (in the K-substituted series) to 0.005 Å2 (in the Rb-substituted series) to 0 Å2 (in pressurized Cs3C60). Fig. 7 displays the variation of the Tc suppression, ΔTc = Tc(Cs3C60) − Tc(KxCs3−xC60, P = 1 atm) with σT2 and with packing density, V/C603− as an implicit parameter for compositions lying at the maximum and on the overexpanded side of the superconductivity dome. ΔTc varies linearly with σT2 with a slope of dΔTc/dσT2 = 210(4) K Å−2, thus demonstrating a clear link between T-site disorder of the electronically inactive alkali ions and suppression of Tc.
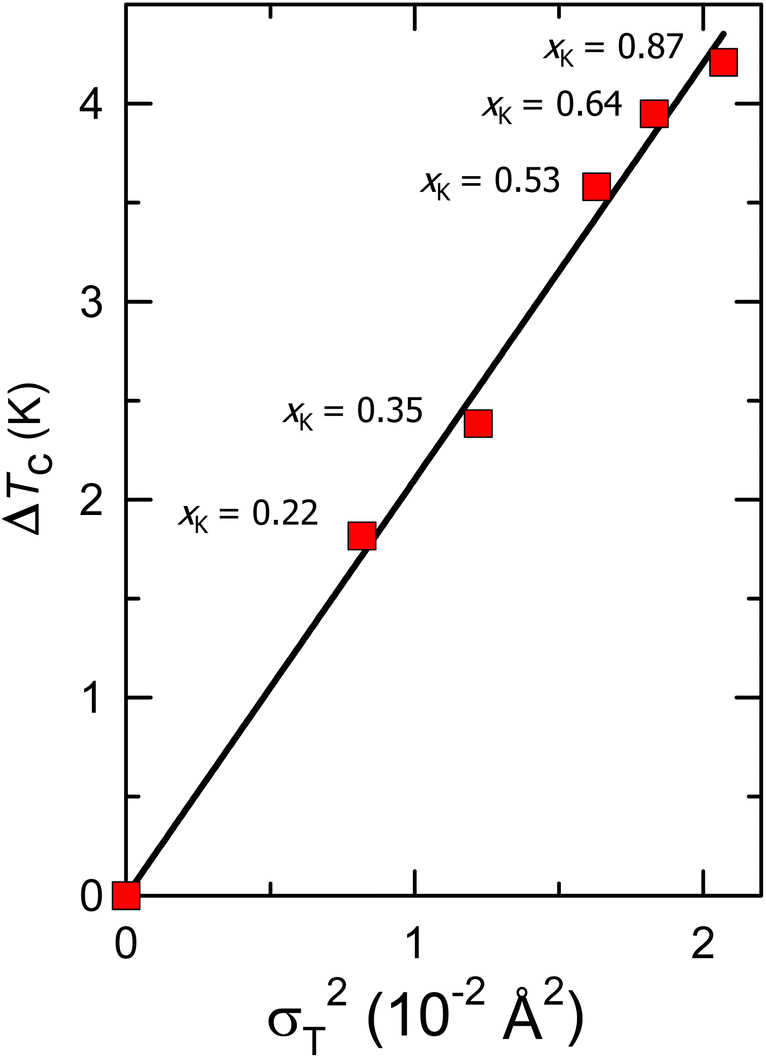 | ||
| Fig. 7 Dependence of the suppression of the ambient-pressure superconducting Tc of the KxCs3−xC60 (0.22 ≤ xK ≤ 0.87) compounds relative to that of the pressurized binary analogue, Cs3C60 at the same C60 packing density on the tetrahedral-site cation size variance. The solid red line is a linear fit to the data. | ||
Such effects have attracted considerable interest before in other classes of strongly correlated materials, namely perovskite transition-metal oxides. For instance, it was demonstrated that the metal-to-insulator transition temperature in colossal magnetoresistive manganites, (RE1−xAEx)MnO3 responds sensitively to changes in lattice strain associated with the variation in ionic size mismatch of the electronically inactive rare earth (RE) and alkaline earth (AE) supporting cations.20,28 In a similar fashion, the superconducting Tc in high-temperature cuprate superconductors, (RE1−xAEx)2CuO4 decreases linearly with increasing disorder of the supporting cation site arising from differences in size between the rare earth and alkaline earth cations.21,29 Similar observations of the importance of ionic size disorder effects in controlling the onset of phase transitions have been also made for nickelates,30 titanates,31 and ferroelectrics.32 Theoretically the large effect of ionic size disorder on Mott-like phase transitions has been understood in terms of dynamic fluctuations of the resulting elastic strain as demonstrated in statistical mechanical models, which incorporate cooperative lattice distortions coupled explicitly to the electronic degrees of freedom.19 Thus it appears that comparable strong elastic interactions are also relevant in strongly correlated molecular systems such as the fcc-structured alkali fullerides, 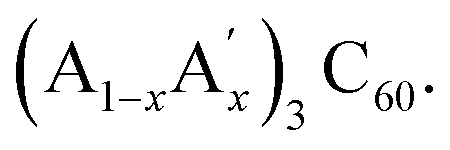 Here the onset of superconductivity is shown to be tunable not only by purely electronic effects associated with the fulleride sublattice – as quantified by the (U/W) ratio – but also by elastic strain effects – as quantified by the variance in the distribution of the radii of the supporting A and A′ cations residing in the T-sites of the fcc structure.
Here the onset of superconductivity is shown to be tunable not only by purely electronic effects associated with the fulleride sublattice – as quantified by the (U/W) ratio – but also by elastic strain effects – as quantified by the variance in the distribution of the radii of the supporting A and A′ cations residing in the T-sites of the fcc structure.
5 Conclusions
In conclusion, we have investigated in detail the structural and electronic properties of the series of fcc-structured KxCs3−xC60 fullerides (0.22 ≤ xK ≤ 2) by means of SXRPD, NMR spectroscopy, and SQUID magnetometry at both ambient and elevated pressures. High-resolution synchrotron XRPD shows that there are no structural phase transitions occurring for any of the compositions studied at all temperatures and pressures investigated. Insulator-to-metal crossovers are encountered on cooling at ambient pressure, evident by both a lattice size decrease without an accompanying crystal symmetry change and a maximum in the 1/133T1T vs. T133Cs NMR spin lattice relaxation data. Composition-dependent Tc measurements at both ambient and elevated pressures reproduce dome-shaped responses to changes in the fulleride packing density.At the same time, the results permit the extension of the electronic phase diagram of the fcc-structured A3C60 fullerides to compositions with significantly increased chemical disorder, as quantified by the statistical variance, σT2 in the distribution of tetrahedral-site ionic radii. We establish a very sensitive dependence of Tc on the lattice strains associated with the cation size variance at the same packing density, V/C603− – the suppression of Tc from its value in the absence of disorder varies linearly with lattice strains at a rate of 210(4) K Å−2.
Data availability
All the necessary data to support the findings of this study can be found within the main text and ESI.†Author contributions
Synthesis, H. E. O. and R. H. C.; synchrotron X-ray diffraction—data collection and analysis, H. E. O., R. H. C., Y. T., K. K., and K. P.; high-pressure synchrotron X-ray diffraction—data collection and analysis, H. E. O., R. H. C., Y. T., Y. O., and K. P.; NMR spectroscopy, P. J. and D. A.; conceptualization and overall conclusions, D. A., Y. K., and K. P.; project supervision, K. P.; writing—review and editing, D. A. and K. P. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the ESRF and SPring-8 for access to synchrotron X-ray facilities. This work was financially supported by Grants-in-Aid for Scientific Research (JSPS KAKENHI Grant Numbers JP21H01907 and JP22K18693), the Japan Atomic Energy Agency (JAEA) (REIMEI Research Program) and the Slovenian Research Agency (Grant Numbers P1-0125 and J1-3007). The experiments at SPring-8 were performed under Proposals No. 2013A1194 and 2017A1363.Notes and references
- A. Y. Ganin, Y. Takabayashi, Y. Z. Khimyak, S. Margadonna, A. Tamai, M. J. Rosseinsky and K. Prassides, Nat. Mater., 2008, 7, 367–371 CrossRef CAS.
- Y. Kasahara, Y. Takeuchi, R. H. Zadik, Y. Takabayashi, R. H. Colman, R. D. McDonald, M. J. Rosseinsky, K. Prassides and Y. Iwasa, Nat. Commun., 2017, 8, 14467 CrossRef CAS.
- Y. Takabayashi, A. Y. Ganin, P. Jeglic, D. Arcon, T. Takano, Y. Iwasa, Y. Ohishi, M. Takata, N. Takeshita, K. Prassides and M. J. Rosseinsky, Science, 2009, 323, 1585–1590 CrossRef CAS.
- A. Y. Ganin, Y. Takabayashi, P. Jeglič, D. Arčon, A. Potočnik, P. J. Baker, Y. Ohishi, M. T. McDonald, M. D. Tzirakis, A. McLennan, G. R. Darling, M. Takata, M. J. Rosseinsky and K. Prassides, Nature, 2010, 466, 221–227 CrossRef CAS.
- M. Capone, M. Fabrizio, C. Castellani and E. Tosatti, Rev. Mod. Phys., 2009, 81, 943–958 CrossRef.
- Y. Nomura, S. Sakai, M. Capone and R. Arita, Sci. Adv., 2015, 1, e1500568 CrossRef.
- S. Hoshino and P. Werner, Phys. Rev. Lett., 2017, 118, 177002 CrossRef.
- Y. Takabayashi and K. Prassides, Philos. Trans. R. Soc., A, 2016, 374, 20150320 CrossRef.
- H. Alloul, P. Wzietek, T. Mito, D. Pontiroli, M. Aramini, M. Riccò, J. P. Itie and E. Elkaim, Phys. Rev. Lett., 2017, 118, 237601 CrossRef CAS.
- Y. Ihara, H. Alloul, P. Wzietek, D. Pontiroli, M. Mazzani and M. Ricco, Phys. Rev. Lett., 2010, 104, 256402 CrossRef CAS.
- R. H. Zadik, Y. Takabayashi, G. Klupp, R. H. Colman, A. Y. Ganin, A. Potocnik, P. Jeglic, D. Arcon, P. Matus, K. Kamaras, Y. Kasahara, Y. Iwasa, A. N. Fitch, Y. Ohishi, G. Garbarino, K. Kato, M. J. Rosseinsky and K. Prassides, Sci. Adv., 2015, 1, e1500059 CrossRef.
- Y. Takabayashi and K. Prassides, Struct. Bonding, 2016, 172, 119–138 CrossRef.
- R. H. Zadik, Y. Takabayashi, R. H. Colman, G. Garbarino and K. Prassides, Mater. Chem. Front., 2018, 2, 993–998 RSC.
- M. Menelaou, Y. Takabayashi, H. E. Okur, R. H. Zadik and K. Prassides, Int. J. Mod. Phys. B, 2018, 32, 1840020 CrossRef CAS.
- G. Klupp, P. Matus, K. Kamarás, A. Y. Ganin, A. McLennan, M. J. Rosseinsky, Y. Takabayashi, M. T. McDonald and K. Prassides, Nat. Commun., 2012, 3, 912 CrossRef.
- P. Wzietek, T. Mito, H. Alloul, D. Pontiroli, M. Aramini and M. Ricco, Phys. Rev. Lett., 2014, 112, 066401 CrossRef CAS.
- A. Potočnik, A. Krajnc, P. Jeglič, Y. Takabayashi, A. Y. Ganin, K. Prassides, M. J. Rosseinsky and D. Arčon, Sci. Rep., 2014, 4, 4265 CrossRef.
- A. Potocnik, A. Y. Ganin, Y. Takabayashi, M. T. McDonald, I. Heinmaa, P. Jeglic, R. Stern, M. J. Rosseinsky, K. Prassides and D. Arcon, Chem. Sci., 2014, 5, 3008–3017 RSC.
- G. G. Guzman-Verri, R. T. Brierley and P. B. Littlewood, Nature, 2019, 576, 429–432 CrossRef CAS.
- L. M. Rodriguez-Martinez and J. P. Attfield, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, R15622–R15625 CrossRef CAS.
- J. P. Attfield, A. L. Kharlanov and J. A. McAllister, Nature, 1998, 394, 157–159 CrossRef CAS.
- K. Kato and H. Tanaka, Adv. Phys.: X, 2016, 1, 55–80 Search PubMed.
- A. C. Larson and R. B. Von Dreele, General Structure Analysis System (GSAS) Report LAUR 86-748, Los Alamos National Laboratory, 2004 Search PubMed.
- Y. Yoshinari, H. Alloul, V. Brouet, G. Kriza, K. Holczer and L. Forro, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 6155–6166 CrossRef CAS.
- F. Sayetat, P. Fertey and M. Kessler, J. Appl. Crystallogr., 1998, 31, 121–127 CrossRef.
- F. D. Murnaghan, Am. J. Math., 1937, 59, 235–260 CrossRef.
- H. E. Okur and K. Prassides, J. Phys. Chem. Solids, 2019, 131, 44–49 CrossRef CAS.
- Y. L. Wang, M. F. Liu, R. Liu, Y. L. Xie, X. Li, Z. B. Yan and J.-M. Liu, Sci. Rep., 2016, 6, 27840 CrossRef CAS.
- J. A. McAllister and J. P. Attfield, Phys. Rev. B: Condens. Matter Mater. Phys., 2002, 66, 014514 CrossRef.
- S. Catalano, M. Gibert, J. Fowlie, J. Iniguez, J.-M. Triscone and J. Kreisel, Rep. Prog. Phys., 2018, 81, 046501 CrossRef CAS.
- T. Katsufuji, Y. Taguchi and Y. Tokura, Phys. Rev. B: Condens. Matter Mater. Phys., 1997, 56, 10145–10153 CrossRef CAS.
- P. V. Balachandran, S. R. Broderick and K. Rajan, Proc. R. Soc. A, 2011, 467, 2271–2290 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional details of the experimental methods; tables of the refined parameters of the Rietveld analysis results of the synchrotron X-ray diffraction data at ambient and elevated pressures (Tables S1–S14); figures of the Rietveld fits of the diffraction data, of additional NMR spectral information, and of magnetic susceptibility data (Fig. S1–S26). See DOI: https://doi.org/10.1039/d4sc03399j |
| This journal is © The Royal Society of Chemistry 2024 |