
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOuter-sphere effects on the O2 sensitivity, catalytic bias and catalytic reversibility of hydrogenases†
Andrea
Fasano
 ,
Vincent
Fourmond
and
Christophe
Léger
*
,
Vincent
Fourmond
and
Christophe
Léger
*
Laboratoire de Bioénergétique et Ingénierie des Protéines, CNRS, Aix Marseille Université, UMR 7281, Marseille, France. E-mail: leger@imm.cnrs.fr
First published on 15th March 2024
Abstract
The comparison of homologous metalloenzymes, in which the same inorganic active site is surrounded by a variable protein matrix, has demonstrated that residues that are remote from the active site may have a great influence on catalytic properties. In this review, we summarise recent findings on the diverse molecular mechanisms by which the protein matrix may define the oxygen tolerance, catalytic directionality and catalytic reversibility of hydrogenases, enzymes that catalyse the oxidation and evolution of H2. These mechanisms involve residues in the second coordination sphere of the active site metal ion, more distant residues affecting protein flexibility through their side chains, residues lining the gas channel and even accessory subunits. Such long-distance effects, which contribute to making enzymes efficient, robust and different from one another, are a source of wonder for biochemists and a challenge for synthetic bioinorganic chemists.
 Christophe Léger (left), Vincent Fourmond (middle) and Andrea Fasano (right) | Dr Christophe Léger (left) obtained his PhD in physical chemistry from the University of Bordeaux and was a postdoc in the group of Fraser Armstrong. Dr Vincent Fourmond (middle) obtained his PhD in the interface of physics and biology from Université Paris Diderot in 2007. He held postdoctoral positions first in the group of Christophe Léger in Marseille and then Vincent Artero in Grenoble, before coming back to Marseille as a permanent CNRS researcher. CL and VF are both Directeurs de Recherche at CNRS. Dr Andrea Fasano (right) obtained a BSc in 2017 in Biotechnology at the Alma Mater Studiorum university of Bologna, an MSc in Industrial Biotechnology at the University of Padua in 2019, and a PhD at the University of Aix Marseille in 2023. In 2024, he was awarded the PhD prizes of the regional section of the French Chemical Society and of the national CNRS research group on bioinorganic chemistry (FrenchBIC). |
Introduction
Metalloenzymes that use active sites based on transition metals to catalyse the production or consumption of small molecules such as H2, O2, CO2 and N2 have attracted much interest over the last few decades because of the need for cheap and efficient synthetic catalysts for the production and use of solar fuels. There is much hope that the knowledge acquired by characterising these enzymes will be useful to design efficient catalysts. Independently of these technological challenges, metalloenzymes are also useful as model systems, to observe the limits of what chemistry can achieve, and learn about how Nature does it.Hydrogenases are enzymes that use sophisticated inorganic active sites made of transition metals, such as iron and nickel, to catalyse the conversion between molecular hydrogen and protons. Two main classes of hydrogenases exist, named after the metal content of their inorganic active sites: the so-called FeFe hydrogenases have an active site, called the H-cluster, that consists of a dinuclear cluster of Fe covalently attached by a cysteine sulphur to a [4Fe4S] cluster (Fig. 1A); NiFe hydrogenases use a dinuclear cluster of Ni and Fe that is attached to the protein by four cysteine residues (Fig. 1B). These active sites are produced and inserted into the apo-enzyme by dedicated enzymes that are part of a complex maturation machinery. The active site of most hydrogenases is buried in the protein matrix and connected to the solvent by a chain of accessory FeS clusters and a network of acidic residues and water molecules, which mediate long range electron and proton transfers. Preferred pathways (sometimes called “gas channels”) for the diffusion of the substrate H2 and inhibitors (CO, O2, H2S, and Cl−) to and from the active site have also been identified with varying degrees of confidence.
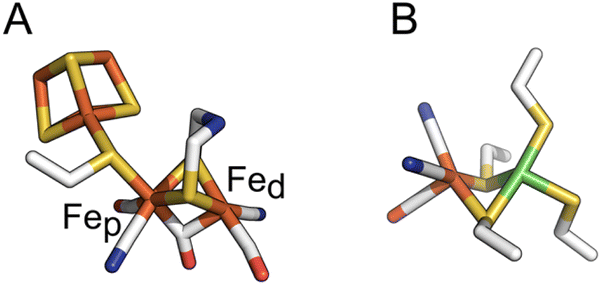 | ||
| Fig. 1 The active sites of FeFe and NiFe hydrogenases, in panels (A) and (B) respectively. In panel (A), the proximal and distal Fe ions are labelled. Colour code: S (yellow), Fe (orange), C (white), O (red), N (blue) and Ni (green). PDB: 4XDC for the FeFe active site and 3UQY for the NiFe active site. | ||
Dihydrogen is a very important energy vector in the microbial world: it is produced as a by-product of fermentation and nitrogen fixation,1,2 and reoxidized in various metabolic pathways, related to e.g. respiration (ATP production)3 or the non-photosynthetic reduction of CO2. As a consequence, most microorganisms produce at least one hydrogenase, but many produce several: Escherichia coli has 4 NiFe hydrogenases,4 and Solidesulfovibrio fructosivorans (Sf, previously known as Desulfovibrio fructosovorans, a model sulfate reducing bacterium) expresses 2 NiFe and 4 FeFe hydrogenases.5 The enzymes in each of the two families are called “homologous”: they embed the same active site (either FeFe or NiFe), but they have similar but distinct amino acid sequences, and therefore distinct structures and a variable number of accessory FeS clusters. They have been classified into groups based on phylogenetic and functional analyses.6,7 Some are monomeric, others may be part of membrane bound or multifunctional protein complexes from which they can be dissociated.
Hydrogenases were discovered nearly a century ago8 and extensively studied using biophysical methods over the last fifty years,9–11 but their biodiversity has been explored only recently.12,13 At the turn of the XXIst century, only about half a dozen of very similar hydrogenases had been used as model enzymes for mechanistic studies. Over the last twenty years, their number has rapidly increased. This work was probably mostly driven by the desire to use hydrogenases for H2 oxidation (in fuel cells) or H2 production (e.g. in photoelectrochemical cells), and it was made possible by a better understanding of their biosynthesis. Tables 1 and 2 list the hydrogenases that we discuss in this review and summarise their main structural features and catalytic properties. The hydrogenases that have been isolated most recently are still not representative of the entire biodiversity, because one tends to focus on small and soluble enzymes that are more easily produced, and yet their characterization studies have revealed an unexpected functional diversity.
| Group6 | Source | Short name | Structure | Properties |
|---|---|---|---|---|
| A | Chlamydomonas reinhardtii | Cr HydA1 | Group M1 (ref. 7) (no accessory FeS cluster), Fig. 3A, pdb 3LX4 (apo form) | Bidirectional (Fig. 2A), O2-sensitive, damaged by UV B19, readily inhibited by HS−,16,82 Cl−17 |
| Desulfovibrio desulfuricans | Dd | Group M2 (ref. 7) (two accessory clusters), dimeric, Fig. 3A, pdb 1HFE | Bidirectional, damaged by visible light,18,125 readily inhibited by HS−82 | |
| C. pasteurianum I | CpI | Group M3 (ref. 7) (four accessory clusters forming a Y-shaped electron transfer conduit126), Fig. 3A, pdb 4XDC | Bidirectional | |
| Clostridium acetobutylicum | CaI | Group M3 | Bidirectional (Fig. 2B), damaged by UV B19, reacts slowly with inhibitors (HS−, Cl−, CO and O2) | |
| Megasphaera elsdenii I | MeI | Group M2, monomeric | Bidirectional, damaged by UV B19, reacts slowly with inhibitors (CO and O2)62 | |
| Acetobacterium woodii | M2-type hydrogenase associated with a formate dehydrogenase | Bidirectional, reacts slowly with O2 (ref. 63) | ||
| C. pasteurianum II | CpII | Group M2 | Catalyses H2 oxidation and evolution irreversibly (Fig. 2D)24 | |
| C. beijerinckii | Cb | 3 accessory clusters (incl. one in a SLBB domain), Fig. 3A, pdb 6TTL | Bidirectional (Fig. 2E), oxygen-stable25,26,80 | |
| B2 | C. pasteurianum III | CpIII | M2, subgroup B2 because they bear a TSCCCP motif27 | Bidirectional24 (Fig. 2G), O2-sensitive27 |
| Megasphaera elsdenii II | MeII | Bidirectional (Fig. 2G)27 | ||
| C | Thermotoga maritima | Tm HydS | Proton transfer pathway different from that in group A111,127 | Bidirectional, very low activity, catalyses H2 oxidation and evolution irreversibly (Fig. 2C)127 |
| D | Thermoanaerobacter mathranii | Tam HydS | Proton transfer pathway different from that in group A (Fig. 3D)36,128 | Bidirectional, very low activity, catalyses H2 oxidation and evolution irreversibly23,33 |
| Group | Structure | pdb | |||
|---|---|---|---|---|---|
| 1b | Desulfovibrio gigas | Dg | Dimeric form (Fig. 4A), large subunit with the NiFe active site (Fig. 1B), small subunit with three FeS clusters: a 4-cysteinyl [4Fe4S] cluster proximal to the active site (Fig. 4C); medial, high redox potential [3Fe4S] cluster; distal [4Fe4S] cluster coordinated by 3 cysteines and one histidine (Fig. 4F) | 1FRV | Bidirectional (see e.g. green CV in Fig. 6A,70 red in Fig. 6B,21,47 although Av is strongly biassed towards H2 oxidation129) and O2-sensitive (grey in Fig. 5B): inhibition by O2 produces a mixture of inactive states, two of which are EPR active (the signatures are called NiA and NiB, the former reactivates more slowly than the latter51,52) |
| D. vulgaris Miyazaki F | 4U9H | ||||
| Solidesulfovibrio fructosivorans, (formerly Desulfovibrio fructosovorans) | Sf Hyn | 1FRF | |||
| 1c | Escherichia coli hydrogenase 2 | Hyd 2 | 6EN9 | ||
| 1e | Allochromatium vinosum | Av | 3MYR | ||
| 1a | Desulfovibrio vulgaris Hildenborough | DvH | Dimer. three cysteine and one selenocysteine ligands130 in the 1st coordination sphere of the Ni ion, instead of four cysteines. A medial [4Fe4S] cluster in the small subunit | 5JSH | Bidirectional. React with O2 to produce EPR-silent inactive states131,132 |
| D. baculatum | 1CC1 | ||||
| 1d | Cupriavidus necator (formerly R. eutropha) membrane bound hydrogenase | Cn MBH | Small subunit with three FeS clusters, incl. a unique [3Fe4S] proximal cluster, coordinated by 6 cysteine residues43–45 (Fig. 4D) | 3RGW | Unidirectional (blue in Fig. 6A), O2-tolerant: they sustain H2 oxidation in the presence of O2 (dark blue in Fig. 5A and B). Inhibition by O2 is reversible and produces mainly the NiB state |
| E. coli 14,70 | Ec Hyd 1 | 3UQY | Cm has high affinity for H2 (ref. 133 and 134) | ||
| Aquifex aeolicus hydrogenase I135 | Aa h2ase I | Aa H2ase I is thermostable, and reactivated upon irradiation by violet light20 | |||
| Cupriavidus metallidurans (formerly Ralstonia metallidurans) | Cm | ||||
| Salmonella enterica hydrogenase 5 | Se Hyd 5 (ref. 42) | 4C3O | |||
| 1h | C. necator actinobacterial-type hydrogenase | Cn AH | Small subunit with three FeS clusters, incl. the proximal [4Fe4S] cluster coordinated by three cysteines and one aspartate (Fig. 4E)46 | 5AA5 | O2-insensitive, low activity (<0.5 μmol of H2 per min per mg), low affinity for H2 (KM ≈ 4 μM)74 |
| 2a | Mycobacterium smegmatis | HucSL | Embeds three [3Fe4S] clusters in each HucSL heterodimer | 8DQV | O2-insensitive, low activity (≈4 μmol of H2 per mg) and high affinity for H2 (Km in the 0.1 μM range, compared to ≈10 μM for Sf Hyn55)56 |
| 2c | C. necator regulatory hydrogenase RH40 | Cn RH | Low activity (≈2 μmol of H2 per mg (ref. 40)) and do not oxidatively inactivate upon exposure to O2 (ref. 40 and 41) | ||
| Rhodobacter capsulatus regulatory hydrogenase41 | |||||
| 3d | C. necator soluble hydrogenases | Cn SH | The initial hypothesis that the active site of Cn SH is coordinated to four (rather than 2) cyanides136 was ruled out137 | Bidirectional. Couples H2 oxidation to NAD+ reduction.138 O2-tolerant (is inactivated by O2 (ref. 137)). In a series of homologous SH hydrogenases, Cn SH is the enzyme that has the largest activity (200 μmol of H2 per min per mg (ref. 139)) and retains the greatest activity in the presence of O2 (ref. 86) | |
| Hydrogenophilus thermoluteolus soluble hydrogenase | Ht SH | These soluble hydrogenases embed only low-potential accessory clusters ([2Fe2S] and [4Fe4S]) | 5XFA | Thermophilic. Oxidatively inactivated to a peculiar inactive state, initially called Ni–B-like86 and then demonstrated to involve an overoxidized Ni(IV) ion39,85 (pdb 5XF9, orange in Fig. 4B) | |
| Synechocystis sp. PCC 6803 | HoxEFUYH | Bidirectional92 | |||
| 3b | Pyrococcus furiosus hydrogenase I | PfSHI | Bidirectional. Distinct aerobic inactivation kinetics140 |
Some hydrogenases oxidise a few hydrogen molecules per second, while others exhibit turnover frequencies in excess of thousands per second. Some are tolerant to O2,14 whereas the active site of others is destroyed upon exposure to oxygen.15 Much variability has also been observed regarding the reactivity with other inhibitors such as sulfide,16 chloride17 or CO. Some FeFe hydrogenases are damaged by light in the visible range18 whereas others are only affected by UV B,19 and one NiFe hydrogenase was reported to be activated by light.20 All hydrogenases inactivate, more or less reversibly, under very oxidising or reducing conditions, but again this varies from one hydrogenase to another. Some hydrogenases are unidirectional (they are only active in one direction of the reaction, H2 oxidation or evolution) whereas others are bidirectional.21 And some are active in response to a very small departure from equilibrium, whereas others catalyse H2 oxidation or evolution only in response to a large overpotential (these behaviours have been termed reversible and irreversible catalysis, respectively22).
In Marseille, we have been particularly active in using direct electrochemistry to study and compare hydrogenases from various sources.28 In this technique the enzyme is adsorbed onto and undergoes direct electron transfer with an electrode, which is spun to avoid H2 depletion or accumulation near the electrode surface.28–31 The current is proportional to the activity, which can be monitored either as a function of time at a constant electrode potential (E), or as a function of electrode potential in experiments called cyclic voltammetry where the electrode potential is repeatedly swept up and down at a certain scan rate (Fig. 2). The sign of the current indicates the direction of electron flow (we count positive and negative the H2 oxidation and evolution currents, respectively), and the magnitude of the current is proportional to turnover frequency (TOF) times the amount of enzyme that contributes:
| i α TOF × (surface coverage of the active enzyme) | (1) |
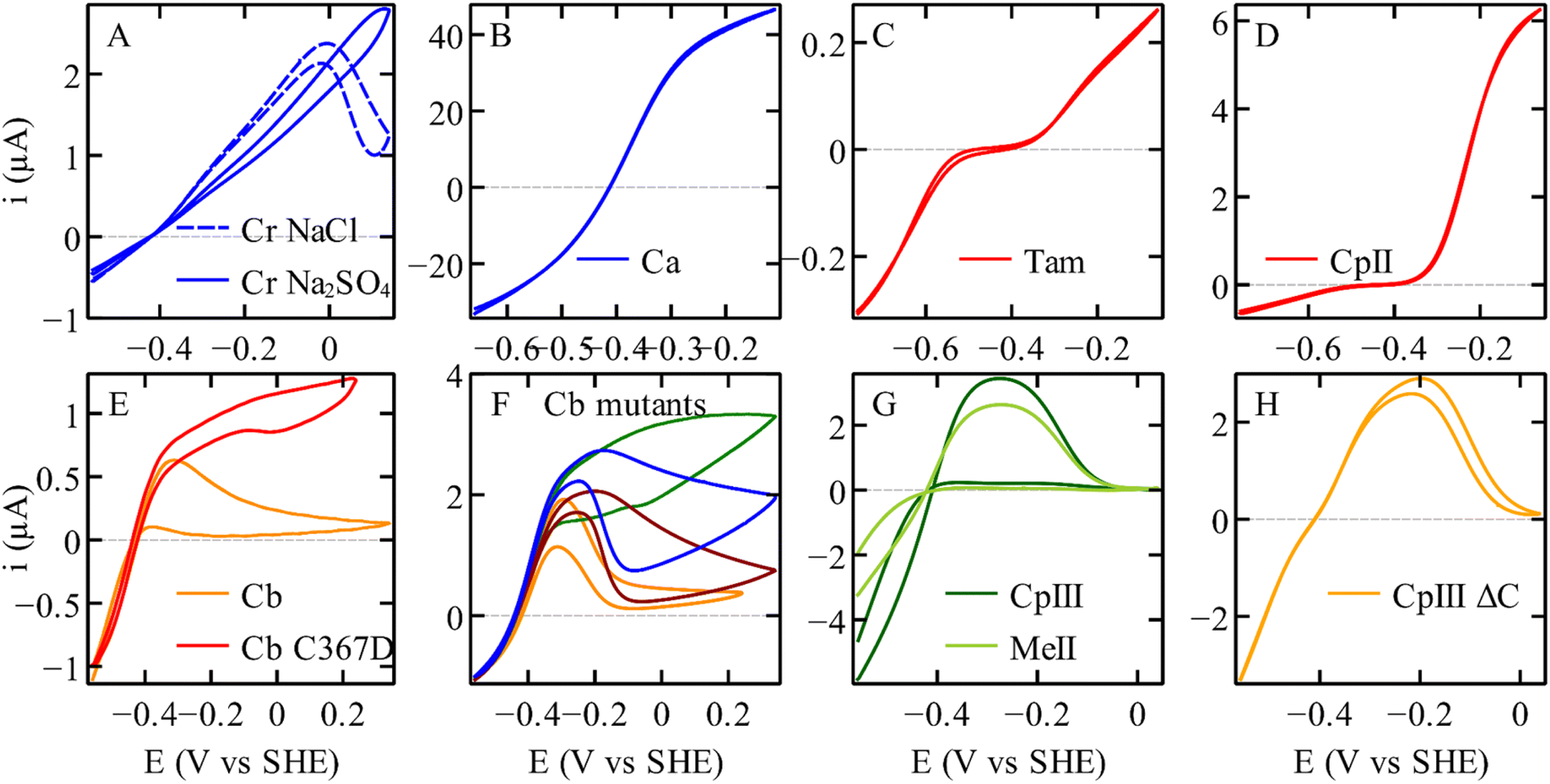 | ||
| Fig. 2 The electrochemical diversity of FeFe hydrogenases, illustrated by catalytic voltammograms recorded under conditions of direct electron transfer and using a rotating disc electrode, all under one atm. of H2. (A) C. reinhardtii HydA1.17 (B) C. acetobutylicum hydrogenase I. (C) Thermoanaerobacter mathranii HydS.23 (D) C. pasteurianum hydrogenase II.24 (E) C. beijerinckii hydrogenase, and the C367D variant. (F) Variants of Cb, A561F (green); P386L (blue); L364F (dark red); M382E (orange).25,26 (G) C. pasteurianum hydrogenase III and M. elsdenii hydrogenase II.24,27 (H) The variant of C. pasteurianum hydrogenase III where the supernumerary cysteine (yellow in Fig. 3C) is deleted.27 All the voltammograms were recorded in a mixed buffer (MES, CHES, HEPES, TAPS, and Na acetate all [5 mM], and Na2SO4 (0.1 M), or NaCl (0.1 M) if specified). Panels (A), (B), (C), (D), (G), and (H) conditions: pH 7; 30 °C; 20 mV s−1; 3000 rpm. Panels (E) and (F) conditions: pH 7; 5 °C; 20 mV s−1; 3000 rpm, currents normalised at −559 mV. | ||
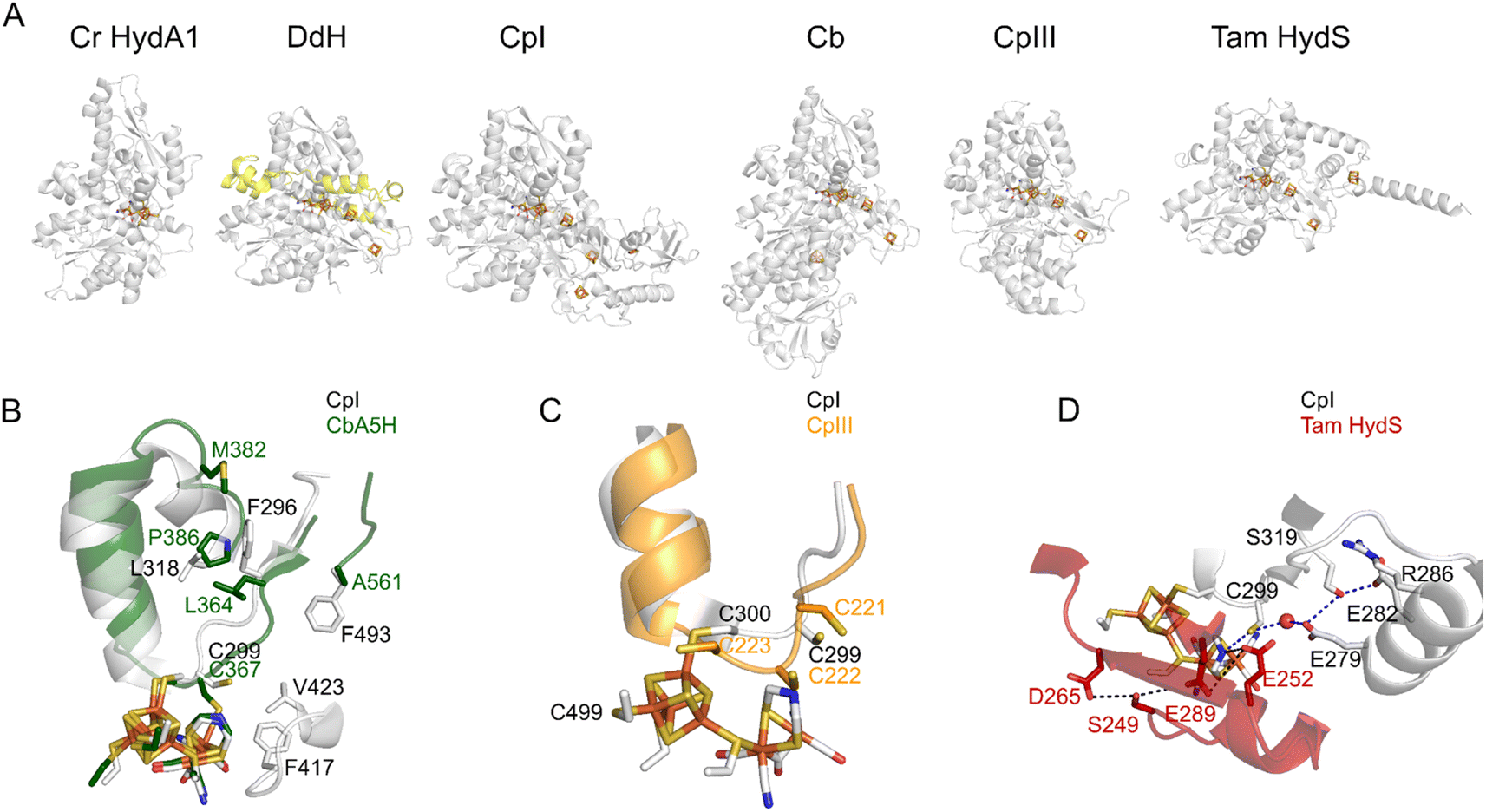 | ||
| Fig. 3 The structures of various FeFe hydrogenases. (A) The overall structures of the enzymes from C. reinhardtii (pdb 3LX4), D. desulfuricans (the two subunits are shown in grey and yellow, pdb 1HFE), C. pasteurianum (CpI, pdb 6N59), C. beijerinckii (Cb, AlphaFold model, since the structure pdb 6TTL is incomplete), CpIII (AlphaFold model), and T. mathranii HydS (AlphaFold model). (B) The residues that define the conformational change in Cb25,26 (P386; L364; A561; M382, Cb numbering) and CO access to the active site in Cr HydA1 (ref. 35) (F417 and V423, CpI numbering). (C) The supernumerary cysteine of CpIII (C221, CpIII numbering). (D) The proton transfer pathways in CpI (gray) and Tam HydS (red).36 Regarding the AlphaFold predictions shown in this figure, ESI Fig. 1† shows the confidence values along the peptide chain. | ||
The turnover frequency depends on the electrode potential (and other experimental parameters such as T, pH, and [H2]) and this dependence informs on the properties of the intermediates in the catalytic cycle.32,33 The second term in the right-hand side of eqn (1), the surface concentration of the active enzyme, is smaller than the total amount of enzyme adsorbed on the electrode, first because some enzyme molecules may be adsorbed in a configuration that does not allow electrical communication with the electrode (and hence do not contribute to the current), and second because some enzyme molecules may (in)activate in a certain range of electrode potentials. If such a redox-driven (in)activation process is slow on the time scale of the experiment, it results in a hysteresis in the voltammetric signature.34
The cyclic voltammograms recorded with various homologous FeFe hydrogenases, shown in Fig. 2, display a striking diversity of shapes. All enzymes appear to catalyse H2 oxidation and reduction, although to various extents. Panel (D) illustrates the case of an enzyme whose response is nearly unidirectional (it is mostly active for H2 oxidation, see also Fig. 6 below). Panel (C) shows the irreversible response of a bidirectional enzyme. Very strong hystereses are seen in panels (A), (E)–(G); they illustrate the effects of slow, reversible, redox-driven transformations between active and inactive forms of the enzyme.34
The immediate implication of the observation that distinct hydrogenases exhibit distinct catalytic properties is that in designing a device that would use a hydrogenase for a particular purpose, care must be taken to choose the right enzyme: not an O2-sensitive enzyme if the system operates in air, or not a photosensitive enzyme if the goal is to photo-produce H2. There is still much hope that by exploring the biodiversity of these enzymes further, one will identify “new” hydrogenases that are even more robust or better suited for a given application.
From a more fundamental perspective, this functional diversity clearly demonstrates that the properties of each enzyme are not dictated by the active site itself. Although inorganic chemists are used to the idea that ligands that do not directly coordinate the metal may have an influence on catalysis, these effects are usually not considered beyond the so-called second coordination sphere.49 In contrast, research conducted over the past two decadesdescribing the comparison of distinct hydrogenases and their modification by protein engineering has demonstrated that the molecular factors influencing their reaction with inhibitors, catalytic bias and catalytic reversibility are very diverse. These factors include structural features that may be distant from the active site and may have an influence even across different subunits in the same enzyme complex.
O2-tolerance
Most of the hydrogenases isolated so far – and all the highly active ones – are inhibited by O2, which is recognized as a main obstacle to using these enzymes. The reactions of hydrogenases with O2 are complex, and the actual effect of O2 varies greatly between FeFe and NiFe hydrogenases, and from one enzyme to another in the same family.Dioxygen irreversibly damages the active site of many FeFe hydrogenases (in a complex, multistep reaction15,35,50) and it oxidises the active site of NiFe hydrogenases into one or a mixture of inactive species, two of which (called “ready” and “unready”) can be distinguished by the rates at which they reactivate under reductive conditions.51,52 These two NiFe hydrogenase inactive states can also be obtained upon anaerobic oxidation of the enzyme.53 In addition to this, in some hydrogenases, dioxygen may damage the accessory FeS clusters involved in mediating long range ET. Comparing the O2 sensitivity of hydrogenases is therefore not trivial because there is no such thing as an “overall” sensitivity that could be quantified by a single parameter, such as the value of just one rate constant or one inhibition constant. The expression “O2-tolerance” refers to the observation that a hydrogenase can oxidise H2 in the presence of O2 for a significant amount of time. This implies that dioxygen acts as a reversible inhibitor (the ratio of reversible inactivation over reactivation rate constants defines an apparent inhibition constant, the magnitude of which is related to the extent of inhibition) and that if any irreversible inactivation occurs, it is slow on the time scale of the particular experiment where the aerobic H2 oxidation activity is monitored.
Blocking the gas channel of an O2-sensitive NiFe hydrogenase may hinder O2 access, but does not make the enzyme O2-tolerant
Hypotheses regarding the reason why the NiFe hydrogenase sensors (RH) from Rhodobacter capsulatus and Cupriavidus necator (previously known as Ralstonia eutropha) resist O2 came from the structural description of the gas channels that connect the active site to the solvent. Volbeda and coworkers observed that in many oxygen sensitive NiFe hydrogenases, a bottleneck at the end of this channel is shaped by the side chains of two conserved residues, a leucine and a valine (grey in Fig. 4B), whereas bulkier phenylalanine and isoleucine residues are present at the same positions in the O2-resistant NiFe H2 sensors (yellow in Fig. 4B). Volbeda's hypothesis, that a narrow pathway may hamper O2 access to the active site and make the enzyme resistant to O2,54 was supported by two site-directed mutagenesis (SDM) studies of these H2 sensors (group 2c): the double replacement of the phenylalanine and isoleucine with leucine and valine makes the enzymes susceptible to oxidative inhibition.40,41 However, the rates of aerobic inactivation of the double mutants remain orders of magnitude slower than that of prototypical hydrogenases (about 10−3 s−1 (atm O2)−1 based on the data in Fig. 4 of ref. 40, 10−5 s−1 (atm O2)−1 from Fig. 2B of ref. 41, compared to ≈10 s−1 (atm O2)−1 for Sf Hyn55), which shows that these two residues are not the only key.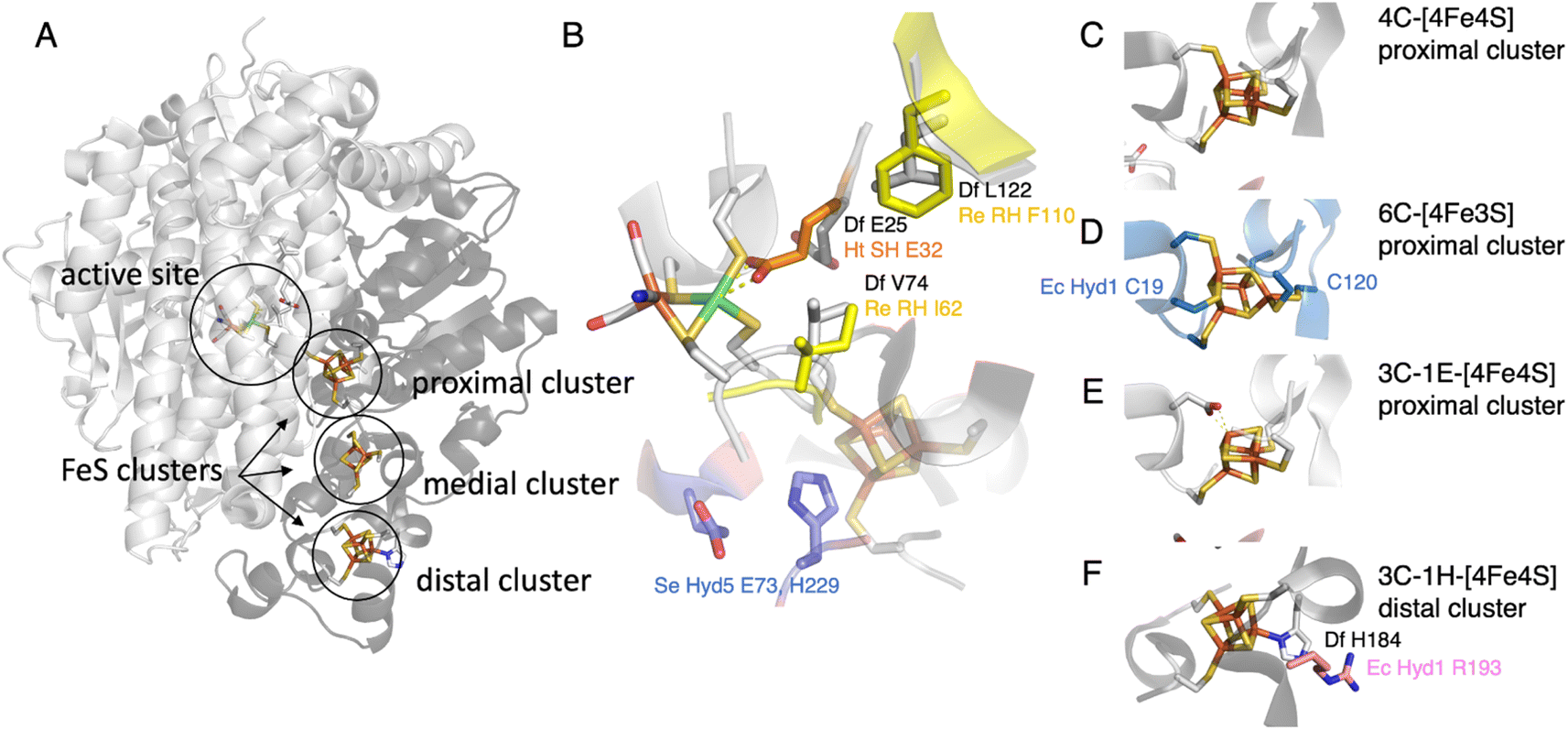 | ||
| Fig. 4 The NiFe hydrogenase dimer. (A) Overview of the structure of prototypical hydrogenases, the large subunit (light grey) embeds the active site, and the small subunit (dark grey) a chain of FeS clusters. (B) The environment of the active site, showing the V74/L122/E25 (Sf Hyn numbering) residues in prototypical hydrogenases,37,38 the residue E32 binding the Ni in the oxidised form of Ht SH,39 the I62/F110 residues in Cupriavidus necator (Cn, previously known as Ralstonia eutropha) MBH40,41 (based on an AlphaFold model), and the E73 and H229 residues in S. enterica (Se) Hyd 5.42 (C) The proximal [4Fe4S] cluster in prototypical NiFe hydrogenases. (D) The proximal [4Fe3S] cluster in group 1d hydrogenases, coordinated by two supernumerary cysteines, C19 and C120 (E. coli Hyd 1 numbering).43–45 (E) The proximal cluster coordinated by 3 cysteines and one aspartate in the AH enzyme from C. necator.46 (F) The histidine ligated distal cluster in prototypical NiFe hydrogenases,47 and the arginine residue substituted in E. coli Hyd 1.48 | ||
The gas channels of the recently characterised Mycobacterium smegmatis NiFe hydrogenases Huc (group 2a)56 and of the actinobacterial-type hydrogenase from C. necator (AH, group 1h)46 are actually reported to be particularly narrow; it has been suggested that this may play a role in ref. 46 (or result in ref. 56) O2 tolerance, but these hypotheses have not been tested by SDM. A computational comparison of NiFe hydrogenases from group 1 led to the hypothesis that O2 sensitivity correlates with a more complex tunnel matrix into the protein, with an increased number of openings toward the solvent compared to the O2-tolerant hydrogenases.57 The calculated O2-diffusion pathways are also different in NiFe and NiFeSe hydrogenases,58 and include a hydrophilic channel in the latter.59
In attempts to increase the O2 resistance of the O2-sensitive NiFe hydrogenase from S. fructosivorans (Sf Hyn) by blocking the gas channel, we designed many site-directed variants where we replaced the valine (at position 74) or the leucine (122, grey in Fig. 4B) with bulkier residues, and we used two kinetic methods to experimentally evaluate the rates of diffusion along these modified channels. One method consists in using mass spectrometry to monitor the “isotope exchange reaction” (D2 + 2H+ → H2 + 2D+), whose kinetics depends on the rate of diffusion in the channel; the other consists in using electrochemistry to measure the bimolecular rate constant of inhibition by CO, a competitive inhibitor that uses the same gas channel as O2 to access the active site.60 Single-point mutations (especially at position 74) alter the rate of intramolecular diffusion by more than three orders of magnitude, and both the charge and the size of the side chains at positions 74 and 122 matter.60,61 However, the effect of the mutations on the rate of inhibition by O2 is rather small (only up to ten-fold), because in the WT enzyme and in most variants, the diffusion of O2 along the gas channel is much faster than the rate of reaction of O2 at the active site, and it is not the rate limiting step of the inhibition reaction.61 Mutations that have the most severe effect on the diffusion rate do slow down O2 inhibition, but this effect alone only delays the loss of enzyme activity upon exposure to O2,61 because the formation of the oxidised inactive states of the active site remains irreversible under the experimental conditions. We concluded that, at least in the particular case of this hydrogenase, slowing diffusion along the gas channel is not a strategy that makes the enzyme O2-tolerant.
Inhibitor access to the active site of FeFe hydrogenases depends on unidentified details of the protein matrix
Regarding FeFe hydrogenases, the rate of inhibition by CO and O2 is three orders of magnitude slower for MeI hydrogenase than for Dd62 (cf. ESI Table S3 in ref. 62). The trend is the same regarding the rate of inhibition by sulfide16 and chloride,17 with the bimolecular rates of inhibition increasing in the order CpI < Cr HydA1 < Dd. The FeFe hydrogenase from Acetobacterium woodii also reacts slowly with O2.63 We believe that these differences are related to the rates of diffusion through the protein matrix, but which residues are responsible for this is unknown. The mutation of a phenylalanine residue that is in the gas channel (F417 in Fig. 3B) to tyrosine in Cr HydA1 slows both the inhibition by O2 (two-fold) and CO (ten-fold).35 Other mutations of FeFe hydrogenases have been shown to moderately slow down O2 inhibition,64–66 but the link between O2 diffusion kinetics and protection has not been demonstrated. When random mutagenesis was used to identify O2-resistant variants of CpI, the hot-spots were found close to the accessory clusters, rather than close to the putative gas channel.65,66A non-natural O2-protection mechanism of NiFe hydrogenases: the substitution of a conserved valine in the second coordination sphere of the Ni ion affects the rates of reactivation after O2 inhibition
Modifying the V74 and L122 residues in the large subunit of Sf Hyn (grey in Fig. 4B), with the initial aim of blocking the gas channel, had two unexpected effects, one of which is an improvement of O2 tolerance that is not related to the rate of O2 access: many substitutions of the position 74 valine increase the rate at which the enzyme reactivates after inhibition by O2. The V74C67 and V74H38 replacements are particularly effective (Fig. 5A). The most significant enhancements of the reactivation rates are observed when the side chain of the position-74 residue is hydrophilic38 (although this rule is not strict68), but the reason for this is unknown. Replacing the equivalent valine in the oxygen-tolerant hydrogenase E. coli Hyd 1 also increases oxygen tolerance further (although at the expense of decreasing activity).69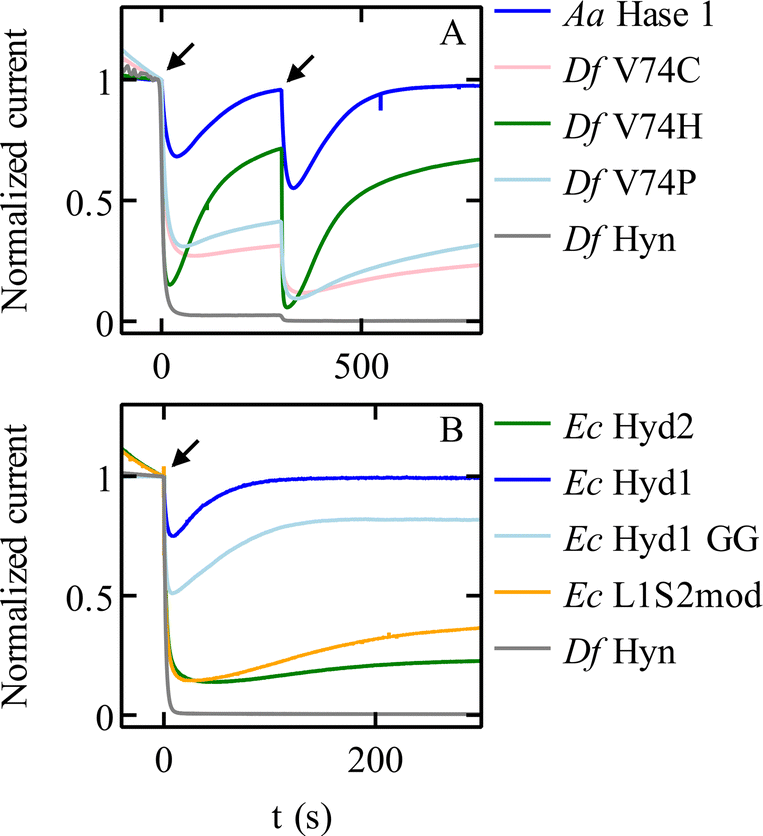 | ||
| Fig. 5 Chronoamperometric responses of NiFe hydrogenases to transient exposures to O2. (A) Various mutants of Sf Hyn, obtained by replacing the position 74 valine, compared to the O2-tolerant enzyme from Aquifex aeolicus (Fig. 4B).38 (B) E. coli Hyd 1 and Hyd 2, the C19G/C120G (called GG) variant of E. coli Hyd 1 (Fig. 4D) where the proximal cluster is a standard cubane cluster, the chimeric “L1S2mod” dimer made of the large subunit of Ec Hyd 1 and the small subunit of Ec Hyd 2,70 and Sf Hyn.55 Arrows show the time of injection of O2 saturated buffer, to reach a final concentration of 4 μM and 8 μM at time t = 0 s and t = 300 s in panel (A), and 8 μM at time = 0 s in panel (B). Conditions: in panel (A) E = 140 mV; 40 °C; pH 5.5; 3000 rpm; 1 atm H2; in panel (B) E = 140 mV; 40 °C; pH 6; 3000 rpm; 1 atm H2; 8 μM O2 injection. In all cases, the concentration of O2 increases suddenly after the injection of an aliquot of O2-saturated solution, and then decreases exponentially as the solution is flushed with H2.55 | ||
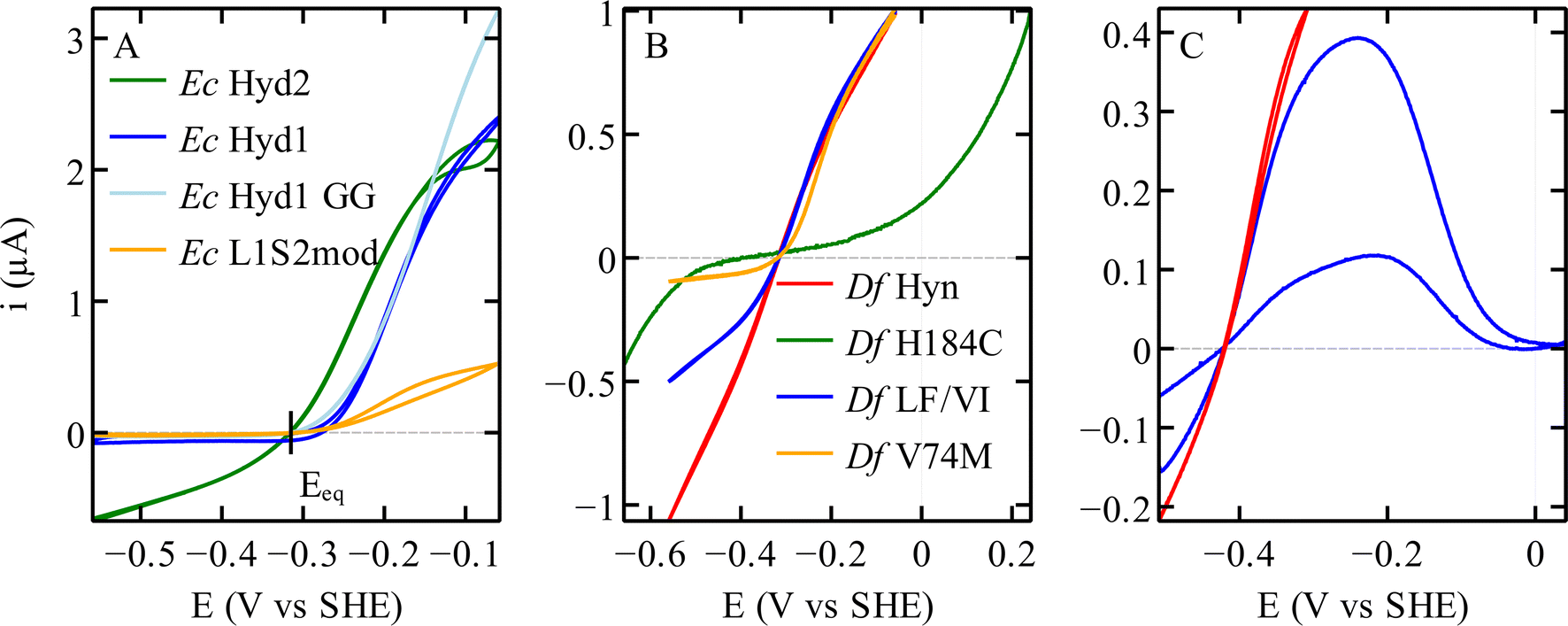 | ||
| Fig. 6 Catalytic bias in voltammetry for different hydrogenases. (A) NiFe hydrogenases from E. coli Hyd 1 and Hyd 2, the variant C19G/C120G (called GG) and the chimeric dimer L1S2mod.70 Conditions: pH 6; 40 °C; 3% H2. (B) Variants of the NiFe hydrogenase Hyn from S. fructosivorans. The curves in this panel were scaled for normalisation. Conditions: pH 5.5; 40 °C; 10% H2 for all21 except H184C,47 pH 6; 40 °C; 100% H2. (C) FeFe hydrogenase CpIII WT,27 in red a voltammogram where the potential is reversed before reaching a high value, showing the steady-state catalytic response and no inactivation. In blue a voltammogram recorded over a large potential range showing the oxidative inactivation; pH 7; 5 °C; 100% H2. | ||
It is somewhat surprising that this valine is very conserved, whereas many of its substitutions improve the resistance to O2.38 The effects of these substitutions on the catalytic bias (see below) may have put a selective pressure on the conservation of this residue.
O2-tolerant NiFe hydrogenases from group 1d: the whole small subunit is crucial, not just the proximal cluster
O2-tolerant NiFe hydrogenases in group 1d have attracted much interest when various investigations showed that they differ from prototypical hydrogenases by the presence of a peculiar [4Fe3S] cluster, attached to the protein by 6 cysteine residues (including C19 and C120, E. coli Hyd 1 numbering, Fig. 4D). This cluster is proximal to the active site but embedded in the small accessory subunit, and replaces the standard 4-Cys [4Fe4S] cluster found in many other NiFe hydrogenases and shown in Fig. 4C.The most familiar hypothesis is that the proximal cluster makes the enzyme O2-tolerant by providing two electrons to reduce the attacking O2, favouring the formation of the inactive state NiB, which reactivates quickly, over the more inert inactive state NiA.43,71 However, this hypothesis has been challenged by recent investigations based on the engineering of this proximal cluster.
Very recently, Lenz and coworkers showed that replacing with glycine either one or two of the supernumerary cysteines around the proximal cluster of the O2-tolerant NiFe hydrogenase from C. necator (MBH) does make the enzyme more O2-sensitive (as observed before in E. coli Hyd 1 (ref. 72)), but without favouring the formation of the NiA inactive state over NiB.73 According to the authors, the observation that group 1d NiFe hydrogenases react with O2 to form only the NiB state is therefore not related to the two-electron transfer capacity of the proximal cluster.73
In our study of the O2-tolerant NiFe hydrogenase from E. coli (Hyd 1), by comparing variants where we replaced either the proximal [4Fe3S] cluster with a standard [4Fe4S] cluster (double substitution C19G/C120G, Fig. 4D), or the entire accessory subunit with that of an O2-sensitive NiFe hydrogenase (by designing a chimeric dimer, called L1S2mod, made of the large subunit of E. coli Hyd 1 and the small subunit of Hyd 2), we concluded that the kinetics of reactivation after oxidative anaerobic inactivation of the enzyme is determined by the large subunit, which bears the NiFe active site, whereas the accessory subunit that houses the electron transfer chain is mostly responsible for O2 tolerance (pale blue and orange in Fig. 5B).70 Most importantly, the variant of E. coli Hyd 1 that bears a standard proximal cluster is much less O2-sensitive than standard hydrogenases (pale blue and green in Fig. 5B), showing that this cluster is not the unique determinant of O2-resistance.70
A SDM study of S. enterica Hyd5 (also from group 1d) showed that the substitutions of two residues (E73 and H229, blue in Fig. 4B) negatively impact O2-tolerance. They are in the large subunit, but H229 is close to the proximal cluster of the small subunit. However these residues are not markers of O2-tolerance: the histidine is very conserved (including in O2-sensitive NiFe hydrogenases) and some enzymes from group 1d (e.g. Aquifex aeolicus hydrogenase I) have a glutamine instead of the glutamate, as in O2-sensitive enzymes.42
Other NiFe hydrogenases (those in groups 1h74,75 and 2a76) appear to be O2-tolerant despite lacking the proximal [4Fe3S] cluster, but their catalytic activity is very low.77 The narrowness of gas channels has been hypothesised to play a role, as mentioned above. However, the O2-insensitivity of the AH enzyme from C. necator (group 1h, formerly group 5) is lost upon substitution of the aspartate residue that coordinates (in an unusual fashion) the proximal FeS cluster of its electron transfer chain (Fig. 4E); this observation led the authors to suggest again an O2 tolerance mechanism involving the reductive removal of the attacking O2.46
Protecting the hydrogenase active site by saturating the coordination sphere of the metal ions in FeFe and NiFe hydrogenases
Most FeFe hydrogenases are irreversibly damaged by O2. Some of them partially recover activity after a short exposure to O2,35,78,79 but this does not prevent irreversible degradation upon continuous exposure. However, Morra and co-workers recently discovered that the FeFe hydrogenase from C. beijerinckii (Cb) is “O2-stable”: it can be exposed to air with minimal damage.80,81Various observations led to the conclusion that this peculiar protection mechanism results from the binding of the thiolate of a conserved cysteine residue to the distal Fe of the active site, under oxidising conditions. First, the X-ray structure of the Cb enzyme in the Hinact state (pdb 6TTL) is consistent with the formation of a bond between the cysteine and the distal Fe (Fig. 3B).25 Second, the FTIR signature of the oxidised active site of Cb hydrogenase is similar to the so-called “Hinact state”, obtained by oxidising the FeFe hydrogenases from Dd or Cr in the presence of hydrogen sulfide;16,82,83 in this state, a sulfido ligand binds the distal iron (“Fed” in Fig. 1A) and prevents the binding of H2 or O2, hence the enzyme is inactive but protected from O2.84 Third, the replacement of the cysteine with an aspartate suppresses both the stability in air and the formation of the Hinact state under oxidising conditions.25
That the cysteine binds to the oxidized H-cluster in Cb but not in other FeFe hydrogenases is due to a conformational change that involves residues very far away from the active site (up to 20 Å, see below the section on Redox-driven changes in active site structure).25,26 It is a clear illustration of how the protein matrix may have a long distance effect on active site chemistry.
Although the inactive state itself (Hinact, cysteine-bound) is protected from O2, whether or not the formation of this state protects the enzyme depends on the relative kinetics of formation of Hinact and O2-induced degradation. In the third FeFe hydrogenase from C. pasteurianum, CpIII (Fig. 3C), the inactivation (Fig. 2G) that results from the formation of the Hinact state upon binding to Fed of a cysteine side chain is too slow to overcome O2 attack.27
The protection of the H-cluster of Cb and CpIII hydrogenases by the binding under oxidising conditions of a nearby cysteine side chain is reminiscent of the protection mechanism evidenced in the NAD+-reducing soluble [NiFe]-hydrogenase (SH) from Hydrogenophilus thermoluteolus (Ht). The oxidised active site of this enzyme includes a six-coordinate Ni, with a conserved glutamic acid previously shown to be involved in proton transfer37 bound to the Ni (orange in Fig. 4B). Like in the case of Cb, the formation of this bond inactivates the enzyme and prevents O2 binding.39,85 It is proposed that the reversible formation of this inactive state under oxidising conditions protects Ht SH from irreversible damage related to metal-assisted ROS production,85 but it is unknown which other mechanism allows this enzyme to perform H2 oxidation (although at a reduced rate) in the presence of O2.86 Replacement of the glutamate residue with alanine or glutamine suppresses the IR signature of the Ni–carboxylate bond in the oxidised state, and greatly decreases the activity (as expected from the substitution of a residue involved in proton transfer),85 but the impact of the substitution of this glutamate on O2-damage has not been evaluated. The reason why this protection mechanism is operational in Ht SH and not in other, homologous NiFe hydrogenases is also unknown.
Catalytic bias
In voltammetry experiments such as those in Fig. 2 and 6, the H2 oxidation and production activities of hydrogenases are assessed successively as the electrode potential is swept up and down across a large potential window. It immediately appears that some enzymes are more active in one direction of the reaction than the other, or even only active in one direction (see e.g.Fig. 2D and 6A). This property is referred to as the “catalytic bias”, or “catalytic preference”, or “catalytic directionality”,22 although there is no consensus in the hydrogenase literature as to how exactly it should be quantified.That a particular hydrogenase (or any other catalyst) may be a unidirectional catalyst does not violate the principles of thermodynamics.87 Thermodynamics imposes that the current is zero when the electrode potential (E) matches the equilibrium potential (Eeq, or “Nernst potential”) of the H+/H2 couple (as indeed observed in all experiments88,89), and it relates the direction of the reaction (H2 oxidation or production) to the sign of the overpotential (the difference between E and Eeq), but it does not constrain the magnitudes of the currents observed under oxidising or reducing conditions.
Elucidating why hydrogenases may be biassed to catalyse the reaction in a certain direction may help the design of synthetic catalysts, very few of which are active in H2 oxidation.90 This question also has implications in terms of physiology. Indeed, some bacteria produce a number of homologous hydrogenases, which are involved in various metabolic functions: for example, as part of respiration and fermentation pathways, these enzymes are used to either oxidise or produce H2, respectively. It is tempting to think that a particular function matches the intrinsic property of each isoform, and that in addition to genetic regulation and cellular localisation, whether a hydrogenase is a better catalyst in one of the two directions of the reaction contributes to defining its metabolic contribution.
Evidence that the standard redox potentials of the FeS of the electron transfer chain determine the catalytic bias of hydrogenase is lacking
Twenty five years ago, P. L. Dutton and coworkers proposed that electron transfer chains in redox enzymes are only optimised in terms of distance between the cofactors (allegedly the most important determinant of the ET rate), whereas the standard redox potentials of the cofactors do not matter. The reasoning was that intramolecular electron transfer was faster than active site chemistry and thus not rate limiting in the catalytic cycle, “endergonic electron transfer steps can still support rapid electron transfer”, and the details of the electron transfer chain (incl. the potentials of the relays) are not important.91 This paradigm has shifted since, at least in the hydrogenase field, where the potential of the accessory clusters is now often considered a determinant of the catalytic bias (higher potential clusters supposedly accelerating catalysis in the direction of H2 oxidation, and lower potential clusters favouring H2 production).Indeed, recently, the bias in the direction of H2 oxidation of the group 2a Huc NiFe hydrogenase from Mycobacterium smegmatis was tentatively ascribed to the presence of three high redox potential [3Fe4S] clusters in the electron transfer chain of this enzyme,56 and the lack of [3Fe4S] clusters in the NiFe-hydrogenase of the Cyanobacterium Synechocystis sp. PCC 6803 was tentatively related to the catalytic bias toward proton reduction.92 Support for this hypothesis from site-directed mutagenesis experiments has not been provided yet. By explicitly considering the redox relay in the kinetic modelling of the catalytic response, we showed that the rates of intramolecular electron transfer matter, not just the potential of the relay,93 but these rate constants are difficult to measure.94
The above-mentioned hydrogenases E. coli Hyd 1 (group 1d) and Hyd 2 (group 1c) have also been studied in this context,14,70 because, despite their strong homology (43% sequence identity for both the large and the small subunits), the former is strongly biassed in the direction of H2 oxidation whereas the latter is bidirectional (dark blue and green, respectively, in Fig. 6A). In NiFe hydrogenases from group 1d, O2-tolerance and the presence of an atypical [4Fe3S] proximal cluster (Fig. 4D) seem to correlate with unidirectionality. Another difference between Hyd 1 and Hyd 2 is that the distal cluster of the former has not been detected by EPR,95 despite its structure and environment being the same as in Hyd 2. Parkin et al. hypothesised that this cluster has higher potential in Hyd 1 than in Hyd 2 and that this may be one of the factors that bias Hyd 1 for H2 oxidation.48
However, investigations of the relation between the redox potential of the clusters of the ET chain and catalytic bias were inconclusive. Decreasing the potential of the distal cluster in Hyd 1 (by substituting a conserved residue, R193, Fig. 4F) has only a small influence on the catalytic bias.48 Replacing either the high potential [3Fe4S] middle cluster or the high potential [4Fe3S] proximal cluster with a low potential [4Fe4S] has no impact.70,72
Regarding the structural determinants of the difference in catalytic bias between Hyd 1 and Hyd 2, the effect of assembling the large subunit of the unidirectional enzyme Hyd 1 with the entire electron transfer subunit of the bidirectional enzyme Hyd 2 is unambiguous: this chimeric dimer has no H2 evolution activity, showing that this catalytic bias is not defined by the electron transfer chain.70
Regarding FeFe hydrogenases, attempts to identify the determinants of the catalytic bias by site-directed mutagenesis have mostly focused on the immediate environment of the H-cluster and the proton transfer chain. Replacing a cysteine ligand of the cubane of the H-cluster (C362 in Cr HydA1, C499 in CpI, see Fig. 3C) with histidine increases the redox potential of the active site and selectively suppressed proton reduction.96 The replacement of the same cysteine with aspartate97 and certain modifications of residues involved in long range proton transfer also suppress98 or favor36 proton reduction. Changing the environment of the CN− ligand of the proximal Fe ion (of the dinuclear fragment of the H-cluster) has a mild effect on the catalytic bias.99 Regarding the manipulation of the accessory clusters, it was shown that replacing the histidine that binds the surface exposed accessory [4Fe4S] cluster in CpI with a cysteine decreases the redox potential of the cluster (≈65 mV), and the bias in solution assays (about two-fold) in the reductive direction.100
The catalytic bias depends on the rate constants of the steps in the catalytic cycle
The rates of catalysis in either direction depend on the rate constants of the steps in the catalytic cycle, and the discussion of the catalytic bias should be based on the measurement of the rate constants of the steps and on the understanding of which steps define the overall TOF. However, the catalytic cycle of complex metalloenzymes involves steps of very different types (active site chemistry, long range proton and electron transfers, diffusion along substrate channels101) and whose individual rate constants are often impossible to measure. Moreover, the overall turnover frequency may be a very complex function of these rate constants, in particular when all steps in the catalytic cycle are reversible32 (indeed, only when a reaction consists of a sequence of irreversible steps is the rate limiting step easily defined as the slowest of these steps102).Modifying the coordination of the distal cluster of Sf NiFe hydrogenase (Fig. 4F) changes the catalytic bias by impacting the rate of H2 oxidation in solution assays more than the rate of H2 evolution; this suggests that the intermolecular ET step contributes to defining the rate of H2 oxidation.47 Furthermore, obstructing the gas channel of the same enzyme by substituting V74 or L122, the residues whose side chains line the gas channel in the large subunit of the enzyme (grey in Fig. 4B), slows H2 evolution much more than H2 oxidation, suggesting that H2 egress is the rate limiting step of the reaction of H2-production in this series of mutants: indeed, the rates of that diffusion step, measured by interpreting the results of isotope exchange assays, exactly match the overall rates of H2 evolution measured in solution assays.21 The effect of these mutations in the large subunit of NiFe hydrogenase on the catalytic bias is also clear from the voltammetric signatures of this series of variants (e.g. red and yellow in Fig. 6B). These are clear examples where the catalytic bias is defined by steps in the catalytic cycle other than active site chemistry.
The discussion of the impact of the mutations on the redox properties of the active site and the catalytic bias (as observed with the C170H mutant of Cr HydA1 (ref. 96)) focussed on how the mutations may affect the standard redox potential of the active site. A complication in catalytic cycles that are bidirectional is that a thermodynamic constraint applies to the series of steps in the catalytic cycle,32,103 and a mutation that tends to accelerate a chemical step in one particular direction of the catalytic cycle must impact the thermodynamics of other steps, possibly redox transitions. There may therefore be cases where the discussion of the rate constants of the chemical steps is intrinsically linked by thermodynamics to the redox potential of some other steps in the catalytic cycle.
Redox-driven changes in the structure of the active site may influence the catalytic bias
A completely distinct discussion of why a catalyst may be better in one particular direction considers the possibility that the structure of the catalyst may change depending on the driving force.A very classical example of this was provided by John Bockris in his studies of oxygen reduction and evolution on metals: the metal surface that is present at low electrode potential under conditions of oxygen reduction is completely distinct from the metal-oxide or hydroxide surface that is present when a high electrode potential is set to drive O2-evolution from water.104 The structure of the active site of many hydrogenases also changes depending on the redox conditions.
The active site of NiFe hydrogenases, for example, is oxidised at high electrode potential (above E ≈ −100 mV, pH 7 (ref. 105)) to either one or a mixture of inactive states in which hydroxo (or maybe peroxo) ligands bind the active site metal ions, which prevents oxidative turnover.53,105,106 Sulfoxygenation of the active site107 or saturation of the Ni coordination sphere with a nearby side chain39,85 may also result in oxidative inactivation of NiFe hydrogenases.
Anaerobic oxidative inactivation does not occur in most FeFe hydrogenases that have been characterized to date (unless chloride17 or sulfide16,82 is present), but the group A FeFe hydrogenase from Clostridium beijerinckii is unusual in that respect. Under mildly oxidising conditions, the side chain of the conserved cysteine that is near the amine of the dithiolate ligand binds the distal Fe of the dinuclear cluster, which inactivates the enzyme by preventing H2-binding (resulting in the formation of the above-mentioned Hinact state); this is detected in voltammetry as a decrease of the H2-oxidation current above ≈−300 mV (orange in Fig. 2E). This oxidative inactivation disappears when this cysteine (C367) is replaced with an aspartate (red in Fig. 2E, the residual hysteresis in this voltammogram is due to the presence of chloride in the buffer).25 The consequence of this inactivation is that the WT FeFe hydrogenase from Cb is protected from O2 damage (as discussed above, the saturation of the coordination sphere of the distal Fe prevents O2 binding) but there is a trade-off: this enzyme is a very poor H2-oxidation catalyst. This is not because the enzyme is intrinsically unable to oxidise H2, but because it cannot stay active for long under conditions where H2 oxidation occurs.
The question remains as to why this conserved cysteine binds the cluster in the FeFe hydrogenase from Cb, and not in prototypical FeFe hydrogenases from group A. The reason is that the formation of the bond between the distal Fe and the cysteine sulphur requires a significant conformational change, which depends on the side chain of non-conserved residues that are remote from the active site. Winkler et al. identified 3 remote residues (at positions 364, 368, and 561, Cb numbering, Fig. 3B), whose bulkier side chains in prototypical FeFe hydrogenase (such as CpI) prevent the conformational change.25 Replacing the amino acids of Cb with those present in CpI slows down the conformational change and prevents oxidative inactivation (Fig. 2F).25 The replacement of the position 382 residue, which is even more distant (18 Å away from the nitrogen of the amine bridge, Fig. 3B), has the opposite effects: it increases the flexibility of the protein and the rate and extent of oxidative inactivation.26 The voltammograms of these variants disclose the intrinsic capability of the active site to oxidise H2.
The FeFe hydrogenase CpIII is from the phylogenetic group B, which has barely been investigated. Like Cb, CpIII inactivates under mildly oxidising conditions (Fig. 2G)27 and is observed by IR in a state that resembles Hinact.24 The loop that bears the above-mentioned proton transfer cysteine is characterised in CpIII by an unusual motif with three vicinal cysteine residues. According to the alpha-fold structure of CpIII (Fig. 3C),27 we hypothesised that one cysteine binds the [4Fe4S] cluster of the active site (as in prototypical FeFe hydrogenases), another one takes the position of the above mentioned proton transfer cysteine and may bind the distal Fe (to produce the Hinact-like state), and the third one may also be involved in proton transfer.108
Like the case of Cb, the analysis of the voltammetric responses of CpIII shows that the environment of its active site is not particularly tuned in a way that makes it better at evolving H2 than oxidising H2; the reason this enzyme has low H2 oxidation activity in solution assays is that it inactivates under oxidising conditions.27
Reversibility
We22 and others before us90,109 have proposed to call “reversible catalysis” the action of a “reversible catalyst”, which becomes active in one direction or the other in response to a small departure from equilibrium. In contrast, an irreversible redox catalyst is only active at high overpotential.Irreversibility may be the mere consequence of slow interfacial electron transfer (between the electrode and the enzyme), as observed in the H184C mutant of Sf Hyn (Fig. 4F, and green in Fig. 6B).47 In contrast, in the case of the sensory FeFe hydrogenase Tam HydS discussed below, we concluded from the analysis of the waveshape that slow electron transfer is not the reason the catalytic response is irreversible, this is an intrinsic property of the enzyme.23
Fig. 2 shows reversible and irreversible responses obtained with homologous FeFe hydrogenases. The catalytic response in panel C is from the putative sensory hydrogenase from Thermoanaerobacter mathranii (Tam); it is classified as group D, but it resembles the sensory hydrogenase HydS from Thermotoga maritima (Tm) which is in group C. In both enzymes, the proton transfer chain and the environment of the H-cluster differ from that in group A hydrogenases (Fig. 3D).108 Irreversibility is not specific to hydrogen sensors: the irreversible response in Fig. 2D is that of the 2nd hydrogenase from C. pasteurianum (CpII, from group A, prototypical FeFe hydrogenases).24
The separation between the two catalytic waves (the difference, for a given enzyme, between the values of the two catalytic potentials, the midpoint potentials of the H2 oxidation and H+ reduction catalytic waves) is related to the difference between the redox potentials of the two transitions of the active site, although the catalytic potentials are shifted from the thermodynamic values, just like Michaelis constants depart from the true dissociation constants.22,32,110 This implies that the more stable the half reduced state of the active site (the so-called Hred state), the more irreversible the response; this appears to be the main reason why the enzyme from Tam behaves very irreversibly,33 although it is still unknown which residues in the environment of the active site make the half reduced state very stable.111 That the proton relay near the H-cluster has a lower pKa in Tam than in Cr also contributes to making the response of the former less reversible.33
Implications for the performance of hydrogenase-based catalytic devices
The functional variations that are observed among homologous hydrogenases that share the same active site are clear illustrations of very long range effects in these biological inorganic catalysts. They also have immediate implications regarding the design of any device that uses a hydrogenase as a catalyst for oxidation or production. Care must be taken that the properties of the particular hydrogenase that is chosen match the needs and the operational conditions of the device, in terms of activity, directionality, reversibility, resistance to light and inhibitors (including O2), stability, cost of production, etc. Since it appears that no hydrogenase checks all the boxes, the right choice is necessarily a compromise.Any attempt to evolve H2 by coupling hydrogenase with a photosensitizer would fail if the enzyme from D. desulfuricans is used since that enzyme is destroyed by white light.112,113 Other hydrogenases appear more robust in that respect, but the possibility of photodamage19 should be considered in all sunlight-dependent H2-evolution systems.114
Since long term stability is required, enzymes produced by hyperthermophilic organisms may be particularly useful.115,116 However, some hydrogenases from mesophilic organisms were shown to be very stable: the NiFe hydrogenase from Desulfovibrio gigas could be used for oxidising H2 at 40 °C for weeks without any loss of activity.117
The most important concern regarding the use of hydrogenases is their sensitivity to oxygen. The O2-tolerant NiFe hydrogenases from group 1d have been extensively used for that reason because all other O2-tolerant hydrogenases have very low H2-oxidation activity. The group-1d hydrogenase from Cupriavidus metallidurans, which combines O2-tolerance and high affinity for H2, was used as the anode catalyst in a membraneless fuel cell operating on 3% H2 in air.118
But even oxygen sensitive hydrogenases can be used as electrocatalysts of H2-oxidation in the presence of O2, under the condition that the matrix that supports the enzyme blocks O2. This is the basis of the strategy used by Plumeré and coworkers, who have shown that when a hydrogenase is embedded in the thick layer of a redox polymer, a fraction of the incoming H2 can be redirected to catalytically produce electrons that reduce any molecule of O2 that may penetrate the film. Long term stability can be achieved under the harsh conditions of a fuel cell,119 even with the most O2-sensitive FeFe hydrogenase.120 The protective effect of the matrix and the intrinsic O2-resistance of the enzyme can be combined to make the system particularly robust.121
Reversibility22 is also desirable when a hydrogenase is used as an electrocatalyst because any overpotential needed to trigger catalysis is a waste of energy. Care must also be taken that this useful property is not lost when the electron transfer between the enzyme and an electrode is mediated.122
The active sites of hydrogenases are based on cheap metals, Ni and Fe, but the cost of producing the enzyme in large amounts is rarely discussed in the literature. The heavy biological machinery that the living cells must use to produce the complex active sites shown in Fig. 1 hampers the biological synthesis of these enzymes, and upscaling their production is a challenge. A huge step forward was made by an international collaboration ten years ago when it was demonstrated that some FeFe hydrogenases can be produced in an “apo” form (that lacks the dinuclear fragment of the H-cluster), and then activated by the spontaneous insertion of a synthetic dinuclear cluster.123 This opened the way for the cheap, large-scale production of simple FeFe hydrogenases, and recent results regarding the biological maturation of the NiFe active site124 makes us hope that a similar process will one day be available to produce NiFe hydrogenases.
Data availability
The text files of the electrochemical data shown in this paper can be downloaded from the Zenodo repository at https://zenodo.org/records/10849830.Author contributions
AF, VF and CL co-wrote the manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
We are very grateful to many colleagues and former students who contributed to our work on hydrogenase in Marseille: Abbas Abou Hamdan, Carole Baffert, Aurore Bailly, Melisa del Barrio, Myriam Brugna, Pierre Ceccaldi, Sebastien Dementin, Christina Felbek, Marie-Thérèse Giudici-Orticoni, Chloé Guendon, Pascale Infossi, Arlette Kpebbe, Fanny Leroux, Pierre-Pol Liebgott, Matteo Sensi and Jeremy Wozniak. And to our colleagues Gustav Berggren, Luca Bertini, James Birrell, Jochen Blumberger, Maurizio Bruschi, Marc Fontecave, Luca de Gioia, Claudio Greco, Thomas Happe, Adam Kubas, Ines Perreira, Nicolas Plumeré, Sven Stripp, Francesca Valletti, Martin Winkler and their students and colleagues. We also thank Oliver Lenz, Stefan Frielingsdorf and Marius Horch for helpful discussions. This research was funded by the Centre National de la Recherche Scientifique, Aix Marseille Université, Agence Nationale de la Recherche (ANR-12-BS08-0014, ANR-14-CE05-0010, ANR-15-CE05-0020, ANR-21-CE50-0041, and ANR-23-CE50-0016), Région Sud, and the French Government under the France 2030 investment plan, as part of the Initiative d'Excellence d'Aix-Marseille Université – A*MIDEX, AMX-22-RE-AB-097 and ANR-11-IDEX-0001-02. The authors are part of the FrenchBIC CNRS network (https://www.frenchbic.cnrs.fr/).References
- O. Einsle and D. C. Rees, Chem. Rev., 2020, 120, 4969–5004 CrossRef CAS PubMed.
- B. M. Hoffman, D. Lukoyanov, Z.-Y. Yang, D. R. Dean and L. C. Seefeldt, Chem. Rev., 2014, 114, 4041–4062 CrossRef CAS.
- D. G. Nicholls, Bioenergetics, Academic Press, 2013 Search PubMed.
- F. Sargent, in Advances in Microbial Physiology, ed. R. K. Poole, Academic Press, 2016, vol. 68, pp. 433–507 Search PubMed.
- C. Baffert, A. Kpebe, L. Avilan and M. Brugna, in Advances in Microbial Physiology, ed. R. K. Poole, Academic Press, 2019, vol. 74, pp. 143–189 Search PubMed.
- C. Greening, A. Biswas, C. R. Carere, C. J. Jackson, M. C. Taylor, M. B. Stott, G. M. Cook and S. E. Morales, ISME J., 2016, 10, 761–777 CrossRef CAS PubMed.
- J. Meyer, Cell. Mol. Life Sci., 2007, 64, 1063–1084 CrossRef CAS.
- M. Stephenson and L. H. Stickland, Biochem. J., 1931, 25, 205–214 CrossRef CAS.
- S. T. Stripp, B. R. Duffus, V. Fourmond, C. Léger, S. Leimkühler, S. Hirota, Y. Hu, A. Jasniewski, H. Ogata and M. W. Ribbe, Chem. Rev., 2022, 122, 11900–11973 CrossRef CAS PubMed.
- J. A. Birrell, P. Rodríguez-Maciá, E. J. Reijerse, M. A. Martini and W. Lubitz, Coord. Chem. Rev., 2021, 449, 214191 CrossRef CAS.
- W. Lubitz, H. Ogata, O. Rüdiger and E. Reijerse, Chem. Rev., 2014, 114, 4081–4148 CrossRef CAS PubMed.
- S. Morra, Front. Microbiol., 2022, 13, 853626 CrossRef.
- J. W. Peters, G. J. Schut, E. S. Boyd, D. W. Mulder, E. M. Shepard, J. B. Broderick, P. W. King and M. W. W. Adams, Biochim. Biophys. Acta, 2015, 1853, 1350–1369 CrossRef CAS.
- M. J. Lukey, A. Parkin, M. M. Roessler, B. J. Murphy, J. Harmer, T. Palmer, F. Sargent and F. A. Armstrong, J. Biol. Chem., 2010, 285, 3928–3938 CrossRef CAS.
- J. Esselborn, L. Kertess, U.-P. Apfel, E. Hofmann and T. Happe, J. Am. Chem. Soc., 2019, 141, 17721–17728 CrossRef CAS PubMed.
- C. Felbek, F. Arrigoni, D. de Sancho, A. Jacq-Bailly, R. B. Best, V. Fourmond, L. Bertini and C. Léger, ACS Catal., 2021, 11, 15162–15176 CrossRef CAS.
- M. Del Barrio, M. Sensi, L. Fradale, M. Bruschi, C. Greco, L. de Gioia, L. Bertini, V. Fourmond and C. Léger, J. Am. Chem. Soc., 2018, 140, 5485–5492 CrossRef CAS.
- S. P. J. Albracht, W. Roseboom and E. C. Hatchikian, J. Biol. Inorg. Chem., 2006, 11, 88–101 CrossRef CAS PubMed.
- M. Sensi, C. Baffert, L. Fradale, C. Gauquelin, P. Soucaille, I. Meynial-Salles, H. Bottin, L. de Gioia, M. Bruschi, V. Fourmond, C. Léger and L. Bertini, ACS Catal., 2017, 7, 7378–7387 CrossRef CAS.
- A. Ciaccafava, C. Hamon, P. Infossi, V. Marchi, M.-T. Giudici-Orticoni and E. Lojou, Phys. Chem. Chem. Phys., 2013, 15, 16463–16467 RSC.
- A. Abou Hamdan, S. Dementin, P.-P. Liebgott, O. Gutierrez-Sanz, P. Richaud, A. L. De Lacey, M. Rousset, P. Bertrand, L. Cournac and C. Léger, J. Appl. Chem. Sci., 2012, 134, 8368–8371 CAS.
- V. Fourmond, N. Plumeré and C. Léger, Nat. Rev. Chem., 2021, 5, 348–360 CrossRef CAS PubMed.
- A. Fasano, H. Land, V. Fourmond, G. Berggren and C. Léger, J. Am. Chem. Soc., 2021, 143, 20320–20325 CrossRef CAS PubMed.
- J. H. Artz, O. A. Zadvornyy, D. W. Mulder, S. M. Keable, A. E. Cohen, M. W. Ratzloff, S. G. Williams, B. Ginovska, N. Kumar, J. Song, S. E. McPhillips, C. M. Davidson, A. Y. Lyubimov, N. Pence, G. J. Schut, A. K. Jones, S. M. Soltis, M. W. W. Adams, S. Raugei, P. W. King and J. W. Peters, J. Am. Chem. Soc., 2020, 142, 1227–1235 CrossRef CAS PubMed.
- M. Winkler, J. Duan, A. Rutz, C. Felbek, L. Scholtysek, O. Lampret, J. Jaenecke, U.-P. Apfel, G. Gilardi, F. Valetti, V. Fourmond, E. Hofmann, C. Léger and T. Happe, Nat. Commun., 2021, 12, 756 CrossRef CAS PubMed.
- A. Rutz, C. K. Das, A. Fasano, J. Jaenecke, S. Yadav, U.-P. Apfel, V. Engelbrecht, V. Fourmond, C. Léger, L. V. Schäfer and T. Happe, ACS Catal., 2023, 13, 856–865 CrossRef CAS PubMed.
- A. Fasano, A. Bailly, J. Wozniak, V. Fourmond and C. Léger, bioRxiv, 2023, preprint, DOI:10.1101/2023.06.23.541094, ACS Cat. (2024), in press.
- M. Sensi, M. del Barrio, C. Baffert, V. Fourmond and C. Léger, Curr. Opin. Electrochem., 2017, 5, 135–145 CrossRef CAS.
- J. N. Butt, L. J. C. Jeuken, H. Zhang, J. A. J. Burton and A. L. Sutton-Cook, Nat. Rev. Methods Primers, 2023, 3, 1–19 CrossRef.
- C. Léger and P. Bertrand, Chem. Rev., 2008, 108, 2379–2438 CrossRef PubMed.
- M. Del Barrio, M. Sensi, C. Orain, C. Baffert, S. Dementin, V. Fourmond and C. Léger, Acc. Chem. Res., 2018, 51, 769–777 CrossRef CAS PubMed.
- V. Fourmond, E. S. Wiedner, W. J. Shaw and C. Léger, J. Am. Chem. Soc., 2019, 141, 11269–11285 CrossRef CAS PubMed.
- A. Fasano, C. Baffert, C. Schumann, G. Berggren, J. A. Birrell, V. Fourmond and C. Léger, J. Am. Chem. Soc., 2024, 146(2), 1455–1466 CrossRef CAS PubMed.
- M. Barrio and V. Fourmond, ChemElectroChem, 2019, 6, 4949–4962 CrossRef.
- A. Kubas, C. Orain, D. De Sancho, L. Saujet, M. Sensi, C. Gauquelin, I. Meynial-Salles, P. Soucaille, H. Bottin, C. Baffert, V. Fourmond, R. B. Best, J. Blumberger and C. Léger, Nat. Chem., 2017, 9, 88–95 CrossRef CAS.
- P. R. Cabotaje, K. Walter, A. Zamader, P. Huang, F. Ho, H. Land, M. Senger and G. Berggren, ACS Catal., 2023, 13, 10435–10446 CrossRef CAS PubMed.
- S. Dementin, B. Burlat, A. L. De Lacey, A. Pardo, G. Adryanczyk-Perrier, B. Guigliarelli, V. M. Fernandez and M. Rousset, J. Biol. Chem., 2004, 279, 10508–10513 CrossRef CAS PubMed.
- A. Abou Hamdan, P.-P. Liebgott, V. Fourmond, O. Gutiérrez-Sanz, A. L. De Lacey, P. Infossi, M. Rousset, S. Dementin and C. Léger, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 19916–19921 CrossRef CAS PubMed.
- Y. Shomura, M. Taketa, H. Nakashima, H. Tai, H. Nakagawa, Y. Ikeda, M. Ishii, Y. Igarashi, H. Nishihara, K.-S. Yoon, S. Ogo, S. Hirota and Y. Higuchi, Science, 2017, 357, 928–932 CrossRef CAS.
- T. Buhrke, O. Lenz, N. Krauss and B. Friedrich, J. Biol. Chem., 2005, 280, 23791–23796 CrossRef CAS PubMed.
- O. Duché, S. Elsen, L. Cournac and A. Colbeau, FEBS J., 2005, 272, 3899–3908 CrossRef.
- L. Bowman, L. Flanagan, P. K. Fyfe, A. Parkin, W. N. Hunter and F. Sargent, Biochem. J., 2014, 458, 449–458 CrossRef CAS.
- J. Fritsch, P. Scheerer, S. Frielingsdorf, S. Kroschinsky, B. Friedrich, O. Lenz and C. M. T. Spahn, Nature, 2011, 479, 249–252 CrossRef CAS PubMed.
- Y. Shomura, K.-S. Yoon, H. Nishihara and Y. Higuchi, Nature, 2011, 479, 253–256 CrossRef CAS PubMed.
- A. Volbeda, P. Amara, C. Darnault, J.-M. Mouesca, A. Parkin, M. M. Roessler, F. A. Armstrong and J. C. Fontecilla-Camps, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 5305–5310 CrossRef CAS.
- C. Schäfer, M. Bommer, S. E. Hennig, J.-H. Jeoung, H. Dobbek and O. Lenz, Structure, 2016, 24, 285–292 CrossRef PubMed.
- S. Dementin, V. Belle, P. Bertrand, B. Guigliarelli, G. Adryanczyk-Perrier, A. L. De Lacey, V. M. Fernandez, M. Rousset and C. Léger, J. Am. Chem. Soc., 2006, 128, 5209–5218 CrossRef CAS PubMed.
- H. Adamson, M. Robinson, J. J. Wright, L. A. Flanagan, J. Walton, D. Elton, D. J. Gavaghan, A. M. Bond, M. M. Roessler and A. Parkin, J. Am. Chem. Soc., 2017, 139, 10677–10686 CrossRef CAS PubMed.
- R. M. Bullock and A. Dey, Chem. Rev., 2022, 122, 11897–11899 CrossRef CAS PubMed.
- K. D. Swanson, M. W. Ratzloff, D. W. Mulder, J. H. Artz, S. Ghose, A. Hoffman, S. White, O. A. Zadvornyy, J. B. Broderick, B. Bothner, P. W. King and J. W. Peters, J. Am. Chem. Soc., 2015, 137, 1809–1816 CrossRef CAS PubMed.
- R. Cammack, V. M. Fernandez and K. Schneider, Biochimie, 1986, 68, 85–91 CrossRef CAS.
- V. M. Fernandez, E. C. Hatchikian and R. Cammack, Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol., 1985, 832, 69–79 CrossRef CAS.
- A. Abou Hamdan, B. Burlat, O. Gutiérrez-Sanz, P.-P. Liebgott, C. Baffert, A. L. De Lacey, M. Rousset, B. Guigliarelli, C. Léger and S. Dementin, Nat. Chem. Biol., 2013, 9, 15–17 CrossRef PubMed.
- A. Volbeda, Y. Montet, X. Vernède, E. C. Hatchikian and J. C. Fontecilla-Camps, Int. J. Hydrogen Energy, 2002, 27, 1449–1461 CrossRef CAS.
- C. Léger, S. Dementin, P. Bertrand, M. Rousset and B. Guigliarelli, J. Appl. Chem. Sci., 2004, 126, 12162 Search PubMed.
- R. Grinter, A. Kropp, H. Venugopal, M. Senger, J. Badley, P. R. Cabotaje, R. Jia, Z. Duan, P. Huang, S. T. Stripp, C. K. Barlow, M. Belousoff, H. S. Shafaat, G. M. Cook, R. B. Schittenhelm, K. A. Vincent, S. Khalid, G. Berggren and C. Greening, Nature, 2023, 615, 541–554 CrossRef CAS PubMed.
- J. Kalms, A. Schmidt, S. Frielingsdorf, P. van der Linden, D. von Stetten, O. Lenz, P. Carpentier and P. Scheerer, Angew Chem. Int. Ed. Engl., 2016, 55, 5586–5590 CrossRef CAS PubMed.
- T. M. Barbosa, C. S. A. Baltazar, D. R. Cruz, D. Lousa and C. M. Soares, Sci. Rep., 2020, 10, 10540 CrossRef CAS PubMed.
- S. Zacarias, A. Temporão, M. del Barrio, V. Fourmond, C. Léger, P. M. Matias and I. A. C. Pereira, ACS Catal., 2019, 9, 8509–8519 CrossRef CAS.
- F. Leroux, S. Dementin, B. Burlat, L. Cournac, A. Volbeda, S. Champ, L. Martin, B. Guigliarelli, P. Bertrand, J. Fontecilla-Camps, M. Rousset and C. Léger, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 11188–11193 CrossRef CAS PubMed.
- P.-P. Liebgott, F. Leroux, B. Burlat, S. Dementin, C. Baffert, T. Lautier, V. Fourmond, P. Ceccaldi, C. Cavazza, I. Meynial-Salles, P. Soucaille, J. C. Fontecilla-Camps, B. Guigliarelli, P. Bertrand, M. Rousset and C. Léger, Nat. Chem. Biol., 2010, 6, 63–70 CrossRef CAS PubMed.
- G. Caserta, C. Papini, A. Adamska-Venkatesh, L. Pecqueur, C. Sommer, E. Reijerse, W. Lubitz, C. Gauquelin, I. Meynial-Salles, D. Pramanik, V. Artero, M. Atta, M. Del Barrio, B. Faivre, V. Fourmond, C. Léger and M. Fontecave, J. Am. Chem. Soc., 2018, 140, 5516–5526 CrossRef CAS PubMed.
- P. Ceccaldi, K. Schuchmann, V. Müller and S. J. Elliott, Energy Environ. Sci., 2017, 10, 503–508 RSC.
- C. Brocks, C. K. Das, J. Duan, S. Yadav, U.-P. Apfel, S. Ghosh, E. Hofmann, M. Winkler, V. Engelbrecht, L. V. Schäfer and T. Happe, ChemSusChem, 2023, e202301365 Search PubMed.
- J. Koo and J. R. Swartz, Metab. Eng., 2018, 49, 21–27 CrossRef CAS PubMed.
- A. S. Bingham, P. R. Smith and J. R. Swartz, Int. J. Hydrogen Energy, 2012, 37, 2965–2976 CrossRef CAS.
- P.-P. Liebgott, A. L. de Lacey, B. Burlat, L. Cournac, P. Richaud, M. Brugna, V. M. Fernandez, B. Guigliarelli, M. Rousset, C. Léger and S. Dementin, J. Am. Chem. Soc., 2011, 133, 986–997 CrossRef CAS PubMed.
- A. Volbeda, L. Martin, P.-P. Liebgott, A. L. De Lacey and J. C. Fontecilla-Camps, Metallomics, 2015, 7, 710–718 CrossRef CAS PubMed.
- M. del Barrio, C. Guendon, A. Kpebe, C. Baffert, V. Fourmond, M. Brugna and C. Léger, ACS Catal., 2019, 9, 4084–4088 CrossRef CAS.
- A. Fasano, C. Guendon, A. Jacq-Bailly, A. Kpebe, J. Wozniak, C. Baffert, M. D. Barrio, V. Fourmond, M. Brugna and C. Léger, J. Am. Chem. Soc., 2023, 145, 20021–20030 CrossRef CAS PubMed.
- J. A. Cracknell, A. F. Wait, O. Lenz, B. Friedrich and F. A. Armstrong, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 20681–20686 CrossRef PubMed.
- M. J. Lukey, M. M. Roessler, A. Parkin, R. M. Evans, R. A. Davies, O. Lenz, B. Friedrich, F. Sargent and F. A. Armstrong, J. Am. Chem. Soc., 2011, 133, 16881–16892 CrossRef CAS PubMed.
- A. Schmidt, J. Kalms, C. Lorent, S. Katz, S. Frielingsdorf, R. M. Evans, J. Fritsch, E. Siebert, C. Teutloff, F. A. Armstrong, I. Zebger, O. Lenz and P. Scheerer, Chem. Sci., 2023, 14, 11105–11120 RSC.
- C. Schäfer, B. Friedrich and O. Lenz, Appl. Environ. Microbiol., 2013, 79, 5137–5145 CrossRef.
- R. A. Schmitz, A. Pol, S. S. Mohammadi, C. Hogendoorn, A. H. van Gelder, M. S. M. Jetten, L. J. Daumann and H. J. M. Op den Camp, ISME J., 2020, 14, 1223–1232 CrossRef CAS PubMed.
- H. Koch, A. Galushko, M. Albertsen, A. Schintlmeister, C. Gruber-Dorninger, S. Lücker, E. Pelletier, D. Le Paslier, E. Spieck, A. Richter, P. H. Nielsen, M. Wagner and H. Daims, Science, 2014, 345, 1052–1054 CrossRef CAS.
- C. Greening and R. Grinter, Nat. Rev. Microbiol., 2022, 20, 513–528 CrossRef CAS.
- C. Baffert, M. Demuez, L. Cournac, B. Burlat, B. Guigliarelli, P. Bertrand, L. Girbal and C. Léger, Angew Chem. Int. Ed. Engl., 2008, 47, 2052–2054 CrossRef CAS PubMed.
- C. Orain, L. Saujet, C. Gauquelin, P. Soucaille, I. Meynial-Salles, C. Baffert, V. Fourmond, H. Bottin and C. Léger, J. Am. Chem. Soc., 2015, 137, 12580–12587 CrossRef CAS PubMed.
- S. Morra, M. Arizzi, F. Valetti and G. Gilardi, Biochemistry, 2016, 55, 5897–5900 CrossRef CAS PubMed.
- F. Valetti, S. Morra, L. Barbieri, S. Dezzani, A. Ratto, G. Catucci, S. J. Sadeghi and G. Gilardi, Faraday Discuss., 2024 10.1039/D4FD00010B.
- P. Rodríguez-Maciá, E. J. Reijerse, M. van Gastel, S. DeBeer, W. Lubitz, O. Rüdiger and J. A. Birrell, J. Am. Chem. Soc., 2018, 140, 9346–9350 CrossRef PubMed.
- P. Rodríguez-Maciá, L. M. Galle, R. Bjornsson, C. Lorent, I. Zebger, Y. Yoda, S. P. Cramer, S. DeBeer, I. Span and J. A. Birrell, Angew Chem. Int. Ed. Engl., 2020, 59, 16786–16794 CrossRef PubMed.
- A. A. Oughli, S. Hardt, O. Rüdiger, J. A. Birrell and N. Plumeré, Chem. Commun., 2020, 56, 9958–9961 RSC.
- C. J. Kulka-Peschke, A.-C. Schulz, C. Lorent, Y. Rippers, S. Wahlefeld, J. Preissler, C. Schulz, C. Wiemann, C. C. M. Bernitzky, C. Karafoulidi-Retsou, S. L. D. Wrathall, B. Procacci, H. Matsuura, G. M. Greetham, C. Teutloff, L. Lauterbach, Y. Higuchi, M. Ishii, N. T. Hunt, O. Lenz, I. Zebger and M. Horch, J. Am. Chem. Soc., 2022, 144, 17022–17032 CrossRef CAS PubMed.
- J. Preissler, S. Wahlefeld, C. Lorent, C. Teutloff, M. Horch, L. Lauterbach, S. P. Cramer, I. Zebger and O. Lenz, Biochim. Biophys. Acta, Bioenerg., 2018, 1859, 8–18 CrossRef CAS PubMed.
- A. Cornish-Bowden, Fundamentals of Enzyme Kinetics (English Edition), Wiley-Blackwell, 4th edn, 2013 Search PubMed.
- J. N. Butt, M. Filipiak and W. R. Hagen, Eur. J. Biochem., 1997, 245, 116–122 CrossRef CAS PubMed.
- H. R. Pershad, J. L. Duff, H. A. Heering, E. C. Duin, S. P. Albracht and F. A. Armstrong, Biochemistry, 1999, 38, 8992–8999 CrossRef CAS PubMed.
- A. Dutta, A. M. Appel and W. J. Shaw, Nat. Rev. Chem, 2018, 2, 244–252 CrossRef CAS.
- C. C. Page, C. C. Moser, X. Chen and P. L. Dutton, Nature, 1999, 402, 47–52 CrossRef CAS PubMed.
- C. L. McIntosh, F. Germer, R. Schulz, J. Appel and A. K. Jones, J. Am. Chem. Soc., 2011, 133, 11308–11319 CrossRef CAS PubMed.
- V. Fourmond, C. Baffert, K. Sybirna, T. Lautier, A. Abou Hamdan, S. Dementin, P. Soucaille, I. Meynial-Salles, H. Bottin and C. Léger, J. Am. Chem. Soc., 2013, 135, 3926–3938 CrossRef CAS PubMed.
- S. Dementin, B. Burlat, V. Fourmond, F. Leroux, P.-P. Liebgott, A. Abou Hamdan, C. Léger, M. Rousset, B. Guigliarelli and P. Bertrand, J. Am. Chem. Soc., 2011, 133, 10211–10221 CrossRef CAS PubMed.
- M. M. Roessler, R. M. Evans, R. A. Davies, J. Harmer and F. A. Armstrong, J. Am. Chem. Soc., 2012, 134, 15581–15594 CrossRef CAS PubMed.
- P. Rodríguez-Maciá, L. Kertess, J. Burnik, J. A. Birrell, E. Hofmann, W. Lubitz, T. Happe and O. Rüdiger, J. Am. Chem. Soc., 2019, 141, 472–481 CrossRef PubMed.
- L. Kertess, A. Adamska-Venkatesh, P. Rodríguez-Maciá, O. Rüdiger, W. Lubitz and T. Happe, Chem. Sci., 2017, 8, 8127–8137 RSC.
- A. J. Cornish, K. Gärtner, H. Yang, J. W. Peters and E. L. Hegg, J. Biol. Chem., 2011, 286, 38341–38347 CrossRef CAS PubMed.
- O. Lampret, A. Adamska-Venkatesh, H. Konegger, F. Wittkamp, U.-P. Apfel, E. J. Reijerse, W. Lubitz, O. Rüdiger, T. Happe and M. Winkler, J. Am. Chem. Soc., 2017, 139, 18222–18230 CrossRef CAS PubMed.
- C. E. Lubner, J. H. Artz, D. W. Mulder, A. Oza, R. J. Ward, S. G. Williams, A. K. Jones, J. W. Peters, I. I. Smalyukh, V. S. Bharadwaj and P. W. King, Chem. Sci., 2022, 13, 4581–4588 RSC.
- S. M. Kim, S. H. Kang, B. W. Jeon and Y. H. Kim, Bioresour. Technol., 2024, 394, 130248 CrossRef CAS PubMed.
- W. J. Ray Jr, Biochemistry, 1983, 22, 4625–4637 CrossRef CAS PubMed.
- A. Fasano, V. Fourmond and C. Léger, The difference bidirectionality makes to the kinetic modeling of molecular catalysis, Curr. Opin. Electrochem., 2024 DOI:10.1016/j.coelec.2024.101489.
- A. Damjanovic, A. Dey and J. O. M. Bockris, J. Electrochem. Soc., 1966, 113, 739 CrossRef CAS.
- A. K. Jones, S. E. Lamle, H. R. Pershad, K. A. Vincent, S. P. J. Albracht and F. A. Armstrong, J. Am. Chem. Soc., 2003, 125, 8505–8514 CrossRef CAS PubMed.
- V. Fourmond, P. Infossi, M.-T. Giudici-Orticoni, P. Bertrand and C. Léger, J. Am. Chem. Soc., 2010, 132, 4848–4857 CrossRef CAS PubMed.
- M. Horch, L. Lauterbach, M. A. Mroginski, P. Hildebrandt, O. Lenz and I. Zebger, J. Am. Chem. Soc., 2015, 137, 2555–2564 CrossRef CAS PubMed.
- J. Duan, M. Senger, J. Esselborn, V. Engelbrecht, F. Wittkamp, U.-P. Apfel, E. Hofmann, S. T. Stripp, T. Happe and M. Winkler, Nat. Commun., 2018, 9, 4726 CrossRef PubMed.
- F. A. Armstrong and J. Hirst, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 14049–14054 CrossRef CAS PubMed.
- A. Fasano, V. Fourmond and C. Léger, Bioelectrochemistry, 2024, 155, 108511 CrossRef CAS PubMed.
- N. Chongdar, P. Rodríguez-Maciá, E. J. Reijerse, W. Lubitz, H. Ogata and J. A. Birrell, Chem. Sci., 2023, 14, 3682–3692 RSC.
- M. Sensi, C. Baffert, V. Fourmond, L. De Gioia, L. Bertini and C. Léger, Sustainable Energy Fuels, 2021, 5, 4248–4260 RSC.
- P. Rodríguez-Maciá, J. A. Birrell, W. Lubitz and O. Rüdiger, ChemPlusChem, 2017, 82, 540–545 CrossRef PubMed.
- D. Adam, L. Bösche, L. Castañeda-Losada, M. Winkler, U.-P. Apfel and T. Happe, ChemSusChem, 2017, 10, 894–902 CrossRef CAS PubMed.
- C. Beaufils, H.-M. Man, A. de Poulpiquet, I. Mazurenko and E. Lojou, Catalysts, 2021, 11, 497 CrossRef CAS.
- A. de Poulpiquet, A. Ciaccafava, R. Gadiou, S. Gounel, M. T. Giudici-Orticoni, N. Mano and E. Lojou, Electrochem. Commun., 2014, 42, 72–74 CrossRef CAS.
- M. A. Alonso-Lomillo, O. Rüdiger, A. Maroto-Valiente, M. Velez, I. Rodríguez-Ramos, F. J. Muñoz, V. M. Fernández and A. L. De Lacey, Nano Lett., 2007, 7, 1603–1608 CrossRef CAS PubMed.
- K. A. Vincent, J. A. Cracknell, J. R. Clark, M. Ludwig, O. Lenz, B. Friedrich and F. A. Armstrong, Chem. Commun., 2006, 5033–5035 RSC.
- V. Fourmond, S. Stapf, H. Li, D. Buesen, J. Birrell, O. Rüdiger, W. Lubitz, W. Schuhmann, N. Plumeré and C. Léger, J. Am. Chem. Soc., 2015, 137, 5494–5505 CrossRef CAS PubMed.
- A. A. Oughli, F. Conzuelo, M. Winkler, T. Happe, W. Lubitz, W. Schuhmann, O. Rüdiger and N. Plumeré, Angew Chem. Int. Ed. Engl., 2015, 54, 12329–12333 CrossRef CAS PubMed.
- H. Li, D. Buesen, S. Dementin, C. Léger, V. Fourmond and N. Plumeré, J. Am. Chem. Soc., 2019, 141, 16734–16742 CrossRef CAS PubMed.
- S. Hardt, S. Stapf, D. T. Filmon, J. A. Birrell, O. Rüdiger, V. Fourmond, C. Léger and N. Plumeré, Nat. Catal., 2021, 4, 251 CrossRef CAS PubMed.
- J. Esselborn, C. Lambertz, A. Adamska-Venkates, T. Simmons, G. Berggren, J. Noth, J. Siebel, A. Hemschemeier, V. Artero, E. Reijerse, M. Fontecave, W. Lubitz and T. Happe, Nat. Chem. Biol., 2013, 9, 607–609 CrossRef CAS PubMed.
- G. Caserta, S. Hartmann, C. Van Stappen, C. Karafoulidi-Retsou, C. Lorent, S. Yelin, M. Keck, J. Schoknecht, I. Sergueev, Y. Yoda, P. Hildebrandt, C. Limberg, S. DeBeer, I. Zebger, S. Frielingsdorf and O. Lenz, Nat. Chem. Biol., 2023, 19, 498–506 CrossRef CAS PubMed.
- W. Roseboom, A. L. De Lacey, V. M. Fernandez, E. C. Hatchikian and S. P. J. Albracht, J. Biol. Inorg. Chem., 2006, 11, 102–118 CrossRef CAS PubMed.
- K. Zuchan, F. Baymann, C. Baffert, M. Brugna and W. Nitschke, Biochim. Biophys. Acta, Bioenerg., 2021, 1862, 148401 CrossRef CAS PubMed.
- N. Chongdar, J. A. Birrell, K. Pawlak, C. Sommer, E. J. Reijerse, O. Rüdiger, W. Lubitz and H. Ogata, J. Am. Chem. Soc., 2018, 140, 1057–1068 CrossRef CAS PubMed.
- H. Land, A. Sekretareva, P. Huang, H. J. Redman, B. Németh, N. Polidori, L. S. Mészáros, M. Senger, S. T. Stripp and G. Berggren, Chem. Sci., 2020, 11, 12789–12801 RSC.
- C. Léger, A. K. Jones, W. Roseboom, S. P. J. Albracht and F. A. Armstrong, Biochemistry, 2002, 41, 15736–15746 CrossRef PubMed.
- L. B. Maia, B. K. Maiti, I. Moura and J. J. G. Moura, Molecules, 2024, 29(1), 120 CrossRef CAS PubMed.
- P. Ceccaldi, M. C. Marques, V. Fourmond, I. C. Pereira and C. Léger, Chem. Commun., 2015, 51, 14223–14226 RSC.
- A. Parkin, G. Goldet, C. Cavazza, J. C. Fontecilla-Camps and F. A. Armstrong, J. Am. Chem. Soc., 2008, 130, 13410–13416 CrossRef CAS PubMed.
- M. Ludwig, J. A. Cracknell, K. A. Vincent, F. A. Armstrong and O. Lenz, J. Biol. Chem., 2009, 284, 465–477 CrossRef CAS PubMed.
- J. A. Cracknell, K. A. Vincent, M. Ludwig, O. Lenz, B. Friedrich and F. A. Armstrong, J. Am. Chem. Soc., 2008, 130, 424–425 CrossRef CAS PubMed.
- M.-E. Pandelia, V. Fourmond, P. Tron-Infossi, E. Lojou, P. Bertrand, C. Léger, M.-T. Giudici-Orticoni and W. Lubitz, J. Am. Chem. Soc., 2010, 132, 6991–7004 CrossRef CAS PubMed.
- E. Van der Linden, T. Burgdorf, M. Bernhard, B. Bleijlevens, B. Friedrich and S. P. J. Albracht, J. Biol. Inorg. Chem., 2004, 9, 616–626 CrossRef CAS PubMed.
- M. Horch, L. Lauterbach, M. Saggu, P. Hildebrandt, F. Lendzian, R. Bittl, O. Lenz and I. Zebger, Angew Chem. Int. Ed. Engl., 2010, 49, 8026–8029 CrossRef CAS PubMed.
- T. Burgdorf, E. van der Linden, M. Bernhard, Q. Y. Yin, J. W. Back, A. F. Hartog, A. O. Muijsers, C. G. de Koster, S. P. J. Albracht and B. Friedrich, J. Bacteriol., 2005, 187, 3122–3132 CrossRef CAS PubMed.
- E. van der Linden, T. Burgdorf, A. L. de Lacey, T. Buhrke, M. Scholte, V. M. Fernandez, B. Friedrich and S. P. J. Albracht, J. Biol. Inorg. Chem., 2006, 11, 247–260 CrossRef CAS PubMed.
- P. Kwan, C. L. McIntosh, D. P. Jennings, R. C. Hopkins, S. K. Chandrayan, C.-H. Wu, M. W. W. Adams and A. K. Jones, J. Am. Chem. Soc., 2015, 137, 13556–13565 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc00691g |
| This journal is © The Royal Society of Chemistry 2024 |