
Kinetics of tautomerisation of thiouracils and cognate species at low temperatures: theory versus experiment†
Judith
Wurmel‡
 *a and
John M.
Simmie‡
*b
*a and
John M.
Simmie‡
*b
aDepartment of Analytical, Biopharmaceutical and Medical Sciences, Atlantic Technological University, Galway, Ireland. E-mail: judith.wurmel@atu.ie
bSchool of Biological and Chemical Sciences, University of Galway, Galway, H91 TK33, Ireland
First published on 20th November 2024
Abstract
Hydrogen-atom tunnelling is an important component in some chemical reactions particularly at low temperatures ≤300 K. Recent experiments by Rostkowska et al. [H. Rostkowska, L. Lapinski and M. J. Nowak, Intramolecular Hydrom-Atom Tunnelling in Matrix-Isolated Heterocyclic Compounds: 2-Thiouracil and Its Analogues, Phys. Chem. Chem. Phys., 2024, 26, 23944–23950.] showed that higher energy monomeric conformers of thiouracil and cognate species (thiols) prepared on neon and argon matrices at 3.5 K spontaneously reverted to the lower energy conformer (thiones) presumably by hydrogen-atom quantum mechanical tunnelling. We have shown that these observations can be rationalised by carrying out chemical kinetic calculations employing canonical variational transition state theory with tunnelling effects on these systems in the gas-phase. We show that tunnelling is totally dominant in these systems from 300 K down and discount the possibility of adventitious water contaminating the experimental observations.
Tautomerism is a well known feature in organic chemistry where easily interchangeable isomers may possess different chemical properties. This is best exemplified by the enols, compounds which are part alkenes and part alcohols, which can convert to ketones via a hydrogen-atom transfer reaction, resulting in the well-known keto ⇌ enol tautomerisation such as vinyl alcohol and acetaldehyde, H2C
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH(OH) ⇌ H3C–CH(O). Normally the ketonic form is the more stable but there are numerous counterexamples as for example phenol.
CH(OH) ⇌ H3C–CH(O). Normally the ketonic form is the more stable but there are numerous counterexamples as for example phenol.
Here we are interested in those reactions where the sulfur analogue of an enol, or thiol, tautomerises to form a thione via the transfer of an hydrogen atom from sulfur to nitrogen, NC–S–H → H–N–CS. This focus is as a result of our previous work in which we have reported on the kinetics and thermochemistry of the tautomerisation of imidic and thioimidic acids,1,2 four and five-membered cyclic lactims to lactams3,4 and more pertinently the kinetics of both the direct and water-mediated tautomerisation of nucleobases including uracil.5
Sulfur-substituted nucleobases or thiobases have been widely used compounds for chemotherapeutic and immunosuppression purposes since the Nobel work of Hitchings and Elion.6 Their medical potential arises because they are efficient absorbers of ultraviolet-B and A regions, generating long-lived reactive triplet electronic states useful for photo-therapeutic applications, but here we are dealing with reactions of their ground electronic states.
The primary focus of this work is to provide a theoretical framework for the series of elegant experiments which Rostkowska et al.7 carried out in which they generated the higher energy conformers of a number of thiouracils and related species by the UV-irradiation at a wavelength of 305 nm of thiones in low temperature, 3.5 K, neon and argon matrices. They were then able to follow the spontaneous reversion of said thiols back to the more stable thiones by IR-spectroscopy. Two examples are shown in Fig. 1 of thiols reverting to the thiouracil. They ascribe the results that they obtained by postulating that the observed reactivities are the consequence of hydrogen atom quantum mechanical tunnelling in essence by virtue of the high barriers to reaction and the extremely low temperature of their experiments.
 | ||
| Fig. 1 Reactions of thiouracils; reaction barriers/kJ mol−1. | ||
The objective of this work then is to model their experiments by carrying out quantum chemistry calculations of the various reactions in the gas-phase in alliance with canonical variational transition state computations incorporating small-curvature and quantised-reaction-states tunnelling effects. We also consider whether the extraneous presence of water on the matrix or whether bimolecular interactions may play roles in the overall reaction.
We employ a highly abbreviated notation to label each species under consideration largely because the full proper IUPAC names, even for the better known thione conformers, are extremely cumbersome and partly because it accords with the naming conventions that we5 and Piacenza and Grimme8 have used in previous work on nucleobases—thus uracil conformer U2 becomes thiouracil conformer SU2etc. The other species under consideration are shown in Fig. 2 and include, in their thione forms, a methyl-substituted thiouracil MeSU3, a triazine NSU3 3,4-dihydro-3-thioxo-1,2,4-triazin-5(2H)-one, a diazine NNSH 3(2H) pyridazinethione and a five-membered ring compound 5SH 2-mercaptoimidazole.
 | ||
| Fig. 2 Thiols: MeSU3, NSU3, NNSH and 5SH. | ||
1 Computational methods
Energy minimisation and frequency determinations of all the species were computed by density functional theory embodied by the functional M06-2X9 with the Def2TZVP valence triple-zeta polarization basis set10 using the finest integration grid possible. The same combination of functional and basis set was used for intrinsic reaction coordinate (IRC) calculations11 which ensured that reliable connections existed between reactant, transition state and product for each individual reaction. Water-mediated reactions at B3LYP12 and the basis Def2TZVP included an additional dispersion term D3BJ to better capture the weak intermolecular interactions between reactant and water forming pre- and post-reaction complexes.13 All of these computations utilised quantum chemistry code GAUSSIAN14 with imaging and manipulation of the results used the application CHEMCRAFT.15Improvements to the zero-point corrected electronic energies were made by employing a composite method, Wuhan–Minnesota scaling (WMS), which combines low-cost coupled cluster energies CCSD(T)-F12b together with a variety of corrections to achieve a CCSD(T)/CBS valence correlation energy.16 The method uses pre-determined geometries obtained typically at lower levels of theory before embarking on the more exacting calculations. These coupled cluster results were obtained with Molpro (version 2024.1).17
As regards the determination of rate constants from 15 K to 300 K, these were calculated with the application Pilgrim which employs canonical variational transition state theory and incorporates multi-dimensional quantum effects with zero- and small-curvature and quantised-reaction-states tunnelling corrections.18–21 The Pilgrim work, which in essence tracks the reaction path via IRC calculations, was all performed at the M06-2X/Def2TZVP level of theory but additionally we also tested the functionals ω B97XD,22 B3LYP,12 APF,23 and, B2PLYP24 for the reaction MeSU3 → MeSU1 in order to establish just how much or how little variability can result. Although there are number of recommended25,26 scale factors for these functionals with the Def2TZVP basis set, for both frequencies and zero-point energies, we have used one of 0.985 throughout for reasons of consistency.
2 Results and discussion
2.1 Structures
The majority of the species have closed-shells ground electronic states 1A′, from which the thiol to thione reaction is assumed to proceed, and adopt a planar Cs point group with a′ and a′′ vibrational modes; the water-mediated pre- and post-reaction complexes as well as their transition states naturally exhibit lesser symmetry.2.2 Energetics of the direct reaction
Our computed barriers to reaction, E‡, and reaction enthalpies are presented in Table 1 for the direct reactions; the zero-point corrected electronic energies derived from WMS calculations are compared to those reported by Rostkowska et al.7 from MP2/6-311++G(2d,p) calculations which are shown in round brackets ().| Reaction | E ‡ | ΔrH (0 K) | |
|---|---|---|---|
| SU2 → SU1 | 102.5 | (107) | −32.3 |
| SU3 → SU1 | 90.6 | (93) | −65.8 |
| MeSU3 → MeSU1 | 84.0 | (88) | −68.0 |
| NSU3 → NSU1 | 95.5 | (99) | −53.6 |
| NNSH → NNHS | 103.8 | (102) | −17.0 |
| 5SH → 5HS | 129.2 | (137) | −23.1 |
Given the different approaches employed, there is reasonable qualitative agreement between the two sets of results. Our calculated WMS barriers show very little variation with the geometry of the reactants and transition states; e.g., for MeSU3 → MeSU1E‡ = 83.71 ± 0.53 kJ mol−1 for B3LYP, M062-X, APF, ω B97XD and B2PLYP-based structures.
Comparison with our previous results for the tautomerisation of uracils are instructive. For U2 → U1 we reported a barrier of 133.9 kJ mol−1 and for U3 → U1 120.2 kJ mol−1 at effectively the same level of theory.
Thus, the sulfur analogues face barriers to reaction which are some 30 kJ mol−1 lower; a finding which corresponds to the decrease in barrier heights of approximately 24 kJ mol−1 that we found in going from imidic acids to amides RC(OH) = NH → RC(O)NH2 in comparison to thioimidic acids going to thioamides RC(SH) = NH → RC(S)NH2.1,2
2.3 Kinetics
Computed rate constants, ki, for all six reactions are displayed graphically in Fig. 3 from 300 K down to 15 K and a more complete set tabulated in the ESI† documentation. Not unexpectedly the rate constants follow the same trend as the barrier heights, increasing barriers reduces rate constants, the normal inverse relationship encapsulated by the Arrhenius equation for thermal reactions. Note that the reactions under consideration here are not thermal in origin but are almost exclusively driven by quantum mechanical tunnelling even at temperatures up to 300 K. Tunnelling rates do depend, inter alia, on barrier heights.27 | ||
| Fig. 3 k i /s−1vs. T/K. MeSU3, SU3, NSU3, SU2, NNSH, 5SH. Solid symbols: experiment. | ||
These calculated rate constants are based on WMS computed high-level energies which are not that dissimilar to those from M06-2X/Def2TZVP computations, Table 1.
Rostkowska et al.7 reported time constants, τi, from their experiments (equivalent to reciprocal rate constants, ki = 1/τi) with MeSU3 rated at the fastest with a τ = 1.6 hours or k = 1.7 × 10−4 s−1 and the slowest that was measurable, NSU3 rated at 1160 hours or k = 2.4 × 10−7 s−1. SU3 lies in between with time constants of 68–76 hours depending on whether a neon or argon matrix was being used, or k = 3.7–4.1 × 10−6 s−1.
The order of reactivity is correctly predicted by our theoretical calculations, that is, MeSU3 is more reactive than SU3 is more reactive than NSU3 and the remainder, SU2, NNSH and 5SH are totally unreactive within the timespan of the laboratory experiments. So qualitative agreement has been achieved—as also quantitative, this can be seen in Fig. 3 which illustrates the agreement between experiments and theory. At 3.5 K, the time constants predicted are MeSU3 1.5 (1.6), SU3 132 (72) and NSU3 3960 (1160) h compared to the experimental values.
It is clear that theory and experiments are in quite good agreement; the question naturally arises, is this fortuitous or should other factors be considered?
The experiments demonstrate a small but not insignificant impact of the environment on the reactivity of SU3 → SU1 which the theory entirely neglects. The surface is not therefore totally inert but plays a role in the overall reaction.
S, have time constants of 81 for SCT and 45 for LAT as against the experimental values30 of 52, all in units of hours. They performed their work at the MPWB1K/6-31+G(d,p) level buttressed by higher level single-point calculations at CCSD(T)-F12b/cc-pVTZ-F12. Note that in their study SCT underestimates the reactivity of the thiol → thione tautomerisation; however, a factor of less than 2 difference between SCT and experiment, if maintained in this work, is certainly acceptable.
Indeed that is the case here too; for example, SU3 forms strong complexes with water, M⋯H2O, a pre-reaction complex which lies 33.3 kJ mol−1 below the separate reactants to which all energies are referenced; similar complexes have been previously reported.31 In turn the transition state is at −11.0 kJ mol−1 leading to a post-reaction complex, P⋯H2O, at −94.3 kJ mol−1 which eventually results in the final products P or in this case SU1 and H2O at −65.8 kJ mol−1:

In summary the forward barrier to reaction from pre-complex to transition state is 22.3 kJ mol−1 and the reverse barrier from post-complex is 83.3 kJ mol−1. This is a very similar situation to that that we encountered for the oxygen-nucleobases,5 the forward barrier for U3⋯H2O TS was 26.9 kJ mol−1 and the reverse 80.9 kJ mol−1.
All of which impacts on the overall kinetics depending upon whether the pre-complex formation is faster or slower than the subsequent step. If we assume that complex formation is slow and dominates the overall water-mediated reaction then there is no tunnelling effect as such—the overall rate is controlled by the formation of complex governed by long-range transition state theory32 which given the bidentate interactions between the thiol bases SU2, SU3, NSU3 and MeSU3 and water are essentially identical means that these all take place at the same rate independent of the exact nature of the thiol. Pre-complex formation is the bottleneck governing the overall rate of reaction.
If we assume that the pre-complex formation is fast then reaction over a barrier of only 22.3 kJ mol−1 for SU3⋯H2O → TS is both thermally and quantum mechanically also faster, approaching 1.6 × 102 s−1 at 15 K for SU3, that is, tautomerisation of SU3 would occur with a time constant of 6 ms. From all of which we conclude that that it is highly unlikely that water–ice played any significant role in the experiments; a view with which the experimentalists7 concur.
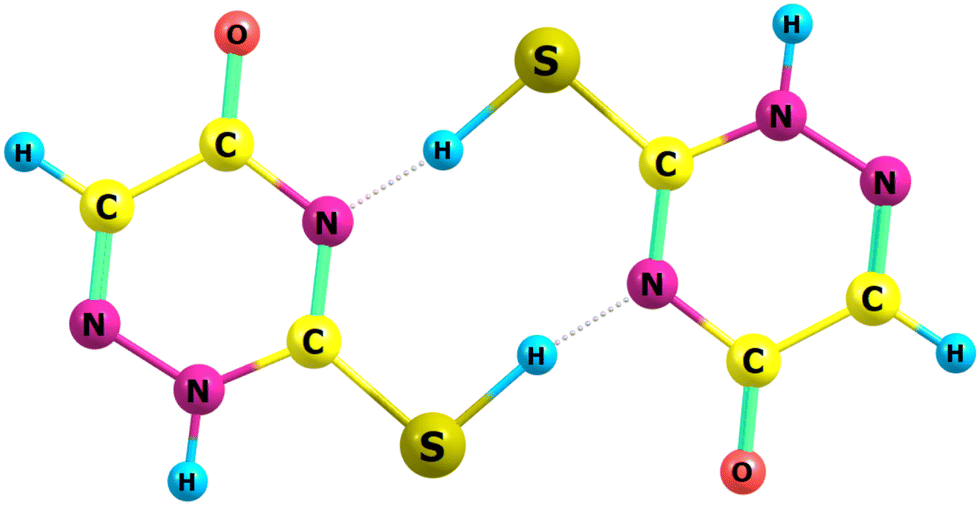 | ||
| Fig. 4 Transition state for dimeric H-atom transfer in NSU3. | ||
The reaction follows a well-trodden route, formation of a complex between two thiols, transition state, post-reaction complex then through to final thione products. Complex formation is at −67 kJ mol−1 with the subsequent barrier only 3.8 kJ mol−1 at the CBS-QB333 level of theory.
But just how important or relevant such considerations are and whether they can be applied to the experimental solid-state reactions is a matter for another day; it would depend upon the exact experimental procedures employed in preparing the matrix itself, deposition rates, etc. Dimerisation in the gas-phase is certainly feasible but depends upon external factors such as species concentration or pressure.
A very recent study by Góbi et al. on the hydrogen-atom assisted tautomerisation of thioacetamide CH3C(S)NH2 ⇌ CH3C(SH) = NH on a low temperature substrate explores these very issues in great detail including the participation of a double H-atom transfer between a thione dimer and a thiol dimer.34 However, their work was not carried out in a noble-gas matrix but rather in amorphous thioacetamide at 10 K where dimer formation would be heavily favoured.
3 Conclusion
We have computed the energy barriers to the tautomerisation reaction where hydrogen atom is transferred from sulfur to nitrogen, –C(SH)N– → –C(S)NH–, in a variety of cyclic and an acyclic system. These results are in relatively good agreement with the literature.
Chemical kinetic calculations then predict correctly the relative velocities with which these reactions proceed at very low temperatures clearly pointing to H-atom tunnelling as the primary contributor to the laboratory observations. Although the experiments demonstrate enhanced reactivity vis-à-vis our results.
The possibility of water molecules inadvertently making any contribution to the experimental findings can be ruled out but whether other solid-state effects such as dimerisation in or on the matrix surface can take place is open to further study.
Data availability
Summaries of all Gaussian output files which include optimised structures in Cartesian coordinates, frequencies, etc.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Computational resources were provided by the Irish Center for High-End Computing (ICHEC) under project p200357. JMS thanks Professors Hans-Joachim Werner (Stuttgart) and Peter J. Knowles (Cardiff) for a Molpro license.References
- J. Wurmel and J. M. Simmie, Substituent effects in the tautomerization of imidic acids R–C(OH)NH → R–C(O)NH2: Kinetic implications for the formation of peptide bonds in the interstellar medium, Int. J. Chem. Kinet., 2023, 55(7), 381–391 CrossRef.
- J. Wurmel and J. M. Simmie, Kinetics of the tautomerization of thioimidic acids R–C(SH)NH → R–C(S)NH2: where R is H, F, HO, CN, NC, H2N, HC(O), HC(S), HC#C, CH3, CF3, H2C = CH, HOCH2, H2NCH2, CH3C(O), C2H5, (CH3)2CH and C6H5, Int. J. Chem. Kinet., 2023, 55(11), 731–742 CrossRef.
- J. Wurmel and J. M. Simmie, Kinetics of direct and water-mediated tautomerization reactions of four-membered cyclic lactims to amides lactams, Int. J. Chem. Kinet., 2024, 56(11), 649–660 CAS.
- J. Wurmel and J. M. Simmie, Kinetics of direct and water-mediated tautomerization reactions of five-membered cyclic amides or lactams, Int. J. Chem. Kinet., 2024, 56(8), 458–468 CrossRef CAS.
- J. Wurmel and J. M. Simmie, Kinetics of direct and water-mediated tautomerisation reaction of nucleobases at low temperatures ≤200 K, Int. J. Chem. Kinet., 2024, 56, 105–116 CrossRef CAS.
- B. Ashwood, M. Pollum and C. E. Crespo-Hernández, Photochemical and Photodynamical Properties of Sulfur-Substituted Nucleic Acid Bases, Photochem. Photobio., 2019, 95(1), 33–58 CrossRef CAS PubMed.
- H. Rostkowska, L. Lapinski and M. J. Nowak, Intramolecular Hydrom-Atom Tunnelling in Matrix-Isolated Heterocyclic Compounds: 2-Thiouracil and Its Analogues, Phys. Chem. Chem. Phys., 2024, 26, 23944–23950 RSC.
- M. Piacenza and S. Grimme, Systematic quantum chemical study of DNA-base tautomers, J. Comput. Chem., 2004, 25, 83–98 CrossRef CAS PubMed.
- Y. Zhao and D. G. Truhlar, The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- A. Hellweg and D. Rappoport, Development of new auxiliary basis functions of the Karlsruhe segmented contracted basis sets including diffuse basis functions (def2-SVPD, def2-TZVPPD, and def2-QVPPD) for RI-MP2 and RI-CC calculations, Phys. Chem. Chem. Phys., 2014, 17(2), 1010–1017 RSC.
- K. Fukui, The path of chemical-reactions - the IRC approach, Acc. Chem. Res., 1981, 14(12), 363–368 CrossRef CAS.
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, Ab Initio calculation of vibrational absorption and circular dichroism spectra using density functional force fields, J. Phys. Chem., 1994, 98(45), 11623–11627 CrossRef CAS.
- S. Grimme, S. Ehrlich and L. Goerigk, Effect of the damping function in dispersion corrected density functional theory, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson and H. Nakatsuji, et al., Gaussian 16 Revision C.02, 2016, https://gaussian.com/gaussian16/ Search PubMed.
- G. A. Zhurko, Chemcraft - graphical program for visualization of quantum chemistry computations. Ivanovo, Russia, 2005. Version 1.8, build 688b. https://chemcraftprog.com Search PubMed.
- Y. Zhao, L. Xia, X. Liao, Q. He, M. X. Zhao and D. G. Truhlar, Extrapolation of high-order correlation energies: the WMS model, Phys. Chem. Chem. Phys., 2018, 20, 27375–27384 RSC.
- H. J. Werner, P. J. Knowles, G. Knizia, F. R. Manby and M. Schütz, Molpro: A general-purpose quantum chemistry program package, Wiley Interdiscip. Rev.:Comput. Mol. Sci., 2012, 2(2), 242–253, DOI:10.1002/wcms.82 . Accessed 20-March-2023.
- D. Ferro-Costas, D. G. Truhlar and A. Fernández-Ramos, Pilgrim-version 2021.5 (University of Minneapolis, Minnesota, MN, and Universidade de Santiago de Compostela, Spain 2021), Comput. Phys. Commun., 2020, 256, 107457 CrossRef CAS Pilgrim: A thermal rate constant calculator and a chemical kinetics simulator.
- B. C. Garrett and D. G. Truhlar, Criterion of minimum state density in the transition-state theory of bimolecular reactions, J. Chem. Phys., 1979, 70(4), 1593 CrossRef CAS.
- R. T. Skodje, D. G. Truhlar and B. C. Garrett, A general small-curvature approximation for transition-state-theory transmission coefficients, J. Phys. Chem., 1981, 85(21), 3019–3023 CrossRef CAS.
- D. G. Truhlar, in Semiclassical Multidimensional Tunneling Calculations, Tunnelling in Molecules: Nuclear Quantum Effects from Bio to Physical Chemistry, ed. J. Kästner and S. Kozuch, RSC Publishing, Cambridge, 2021, pp. 261–282 Search PubMed.
- J.-D. Chai and M. Head-Gordon, Long-range corrected hybrid density functionals with damped atom-atom dispersion corrections, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC.
- A. Austin, G. Petersson, M. J. Frisch, F. J. Dobek, G. Scalmani and K. Throssell, A density functional with spherical atom dispersion terms, J. Chem. Theory and Comput., 2012, 8, 4989–5007 CrossRef CAS PubMed.
- L. Goerigk and S. Grimme, Efficient and Accurate Double-Hybrid-Meta-GGA Density Functionals-Evaluation with the Extended GMTKN30 Database for General Main Group Thermochemistry, Kinetics, and Noncovalent Interactions, J. Chem. Theory Comput., 2011, 7, 291–309 CrossRef CAS PubMed.
- M. K. Kesharwani, B. Brauer and J. M. L. Martin, Frequency and zero-point vibrational energy scale factors for double-hybrid density functionals (and other selected methods): can anharmonic force fields be avoided?, J. Phys. Chem. A, 2015, 119(9), 1701–1714 CrossRef CAS PubMed.
- I. M. Alecu, J. Zheng, Y. Zhao and D. G. Truhlar, Computational Thermochemistry: Scale Factor Databases and Scale Factors for Vibrational Frequencies Obtained from Electronic Model Chemistries, J. Chem. Theory Comput., 2010, 6, 2872–2887 CrossRef CAS PubMed.
- J. Meisner and J. Kästner, Atom Tunneling in Chemistry, Angew. Chem., Int. Ed., 2016, 55, 5400–5413 CrossRef CAS PubMed.
- I. Mosquera-Lois, D. Ferro-Costas and A. Fernández-Ramos, Chemical reactivity from the vibrational ground-state. The role of the tunnelling path in the tautomerisation of urea and derivatives, Phys. Chem. Chem. Phys., 2020, 22, 24951–24963 RSC.
- R. Meana-Pañeda, D. G. Truhlar and A. Fernández-Ramos, Least-Action Tunneling Transmission Coefficient for Polyatomic Reactions, J. Chem. Theory Comput., 2010, 6, 6–17 CrossRef PubMed.
- H. Rostkowska, L. Lapinski and M. J. Nowak, Proton-transfer process in thiourea: UV-induced thiol to thione reaction and ground state thiol to thione tunnelling, J. Phys. Chem. A, 2003, 107, 6373–6380 CrossRef CAS.
- E. Kryachko, M. T. Nguyen and T. Zeegers-Huyskens, Thiouracils: Acidity, Basicity, and Interaction with Water, J. Phys. Chem. A, 2001, 105, 3379–3387 CrossRef CAS.
- Y. Georgievskii and S. J. Klippenstein, Long-range transition state theory, J. Chem. Phys., 2005, 122, 194103 CrossRef PubMed.
- J. A. Montgomery Jr., M. J. Frisch, J. W. Ochterski and G. A. Petersson, A complete basis set model chemistry. VII. Use of the minimum population localization method, J. Chem. Phys., 2000, 112, 6532–6542 CrossRef.
- S. Góbi, I. Reva, G. Ragupathy, R. Fausto and G. Tarczay, Hydrogen-Atom Assisted Tautomerization on Solid Surfaces—The Case Study of Thioacetamide, J. Phys. Chem. C, 2024 DOI:10.1021/acs.jpcc.4c05817.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cp04038d |
| ‡ Both authors contributed equally. |
| This journal is © the Owner Societies 2024 |