
Targeted liposomal drug delivery: a nanoscience and biophysical perspective
Yibo
Liu
ab,
Karla M.
Castro Bravo
a and
Juewen
Liu
 *ab
*ab
aDepartment of Chemistry, Waterloo Institute for Nanotechnology, University of Waterloo, Waterloo, Ontario N2L 3G1, Canada. E-mail: liujw@uwaterloo.ca
bCentre for Eye and Vision Research, 17W Hong Kong Science Park, Hong Kong
First published on 21st December 2020
Abstract
Liposomes are a unique platform for drug delivery, and a number of liposomal formulations have already been commercialized. Doxil is a representative example, which uses PEGylated liposomes to load doxorubicin for cancer therapy. Its delivery relies on the enhanced permeability and retention (EPR) effect or passive targeting. Drug loading can be achieved using both standard liposomes and also those containing a solid core such as mesoporous silica and poly(lactide-co-glycolide) (PLGA). Developments have also been made on active targeted delivery using bioaffinity ligands such as small molecules, antibodies, peptides and aptamers. Compared to other types of nanoparticles, the surface of liposomes is fluid, allowing dynamic organization of targeting ligands to achieve optimal binding to cell surface receptors. This review article summarizes development of liposomal targeted drug delivery systems, with an emphasis on the biophysical properties of lipids. In both passive and active targeting, the effects of liposome size, charge, fluidity, rigidity, head-group chemistry and PEGylation are discussed along with recent examples. Most of the examples are focused on targeting tumors or cancer cells. Finally, a few examples of commercialized formulations are described, and some future research opportunities are discussed.
 Yibo Liu | Yibo Liu received her BSc from Heilongjiang University in China in 2009 and MSc from the University of Waterloo in 2013. After working as a research assistant with Prof. Michael Tam at the University of Waterloo, she joined Prof. Juewen Liu's group in 2016, received her PhD in 2020, and continued as a Postdoctoral Fellow. Her PhD research involves fundamental understanding of biointerface interactions of metal ions and nanomaterials with liposomes and their biomedical applications. |
 Juewen Liu | Prof. Juewen Liu received his PhD degree from the University of Illinois at Urbana-Champaign in 2005. He is currently a professor of chemistry at the University of Waterloo, and holds a University Research Chair position. He received a Fred Beamish Award (2014) and a W. A. E. McBryde Medal (2018) from the Canadian Society for Chemistry. He is a College member of the Royal Society of Canada (RSC). He serves as a Section Editor for Biosensors & Bioelectronics, an Associate Editor for Chinese Chemical Letters, and a Contributing Editor for Trends in Analytical Chemistry. He is on the editorial board of FACETS and advisory board of Langmuir. He is interested in metal-dependent DNAzymes, aptamers, biointerface sciences, and nanozymes. Since 2002, he has published over 300 papers, receiving over 25 |
1. Introduction
Targeted drug delivery offers tremendous advantages by improving therapeutic efficacy and reducing side effects.1 To achieve this goal, a diverse range of nanoscale delivery vehicles have been developed.2–4 Lipid vesicles such as liposomes are particularly attractive for drug delivery. For one, lipids are the basic component of the cell membrane and many lipids are highly biocompatible, omitting the problem of biodegradation.5,6 In addition, many types of drug molecules, both hydrophilic and hydrophobic ones can be loaded. Lipids can also wrap around inorganic and polymeric nanoparticles to form supported lipid monolayers or bilayers,7–9 where fluid lipid surfaces then allow for attaching targeting ligands.10 The fundamental biophysics of lipid membranes is very intriguing and interesting, which in turn may affect drug delivery.11–15 Properties like lipid domain formation, fluidity, polyvalent binding, leakage and fusion have all been used in drug delivery. Compared to inorganic delivery vehicles, liposomes are softer and can be more easily deformed. Fluidic membranes allow dynamic ligand reorganization. Therefore, fundamental studies on the biophysical front can also guide rational design of drug formulations.Liposomal drug delivery has already been extensively reviewed.8,16–20 Fundamental physical studies of lipid membranes are also advanced.21–23 However, connections between these two communities exist only in limited and scattered examples. The goal of this review is to emphasize on the biophysical aspects of lipids and connect them to the performance of drug delivery formulations. Most examples covered in this review were published within the past decade.
2. Some common and novel lipids
A typical lipid molecule contains a hydrophilic headgroup and two hydrophobic tails (Fig. 1a). The charge of lipids and their chemical properties can be varied by changing the headgroup, while the hydrophobic tails mainly govern the packing in membranes. A vast number of lipids are found in nature and more are available via chemical synthesis. A few commonly used lipids are listed in Fig. 1. Phosphatidylcholine (PC) lipids are the main constituent of the outer membrane of eukaryotic cells and they are highly biocompatible. The structure of 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC) is shown in Fig. 1c. A PC headgroup contains a negative-charged phosphate and a positive-charged choline. Since phosphate has a pKa lower than 2 and choline is a quaternary ammonium cation,24,25 PC is zwitterionic and overall charge neutral over a wide pH range.24,25 PC is highly hydrated possessing strong anti-fouling properties (e.g. resisting protein adsorption).26,27 The tail structures can be changed rendering different phase transition temperatures (Tc). Tc is an important parameter that governs the fluidity of lipid bilayers. Above Tc, lipid tails have gauche conformation and can diffuse more freely, and the membrane exist in a liquid crystalline phase.28 Below Tc, lipid tails are extended and diffuse slowly, and the membrane is in a gel-like state. It needs to be noted though, the diffusion coefficient of lipids within membranes differ only by a few folds for the fluid and gel phased membranes.29 Generally, Tc increases with the length of carbon chain, but decreases significantly when a double bond is present. For example, DOPC containing two double bonds in the tail has a Tc of −20 °C. 1,2-Dipalmitoyl-sn-glycero-3-phosphocholine (DPPC) (Fig. 1d) and 1,2-dimyristoyl-sn-glycero-3-phosphocholine (DMPC) (Fig. 1e) both have saturated tails, and their Tc's are 41 °C and 23 °C, respectively.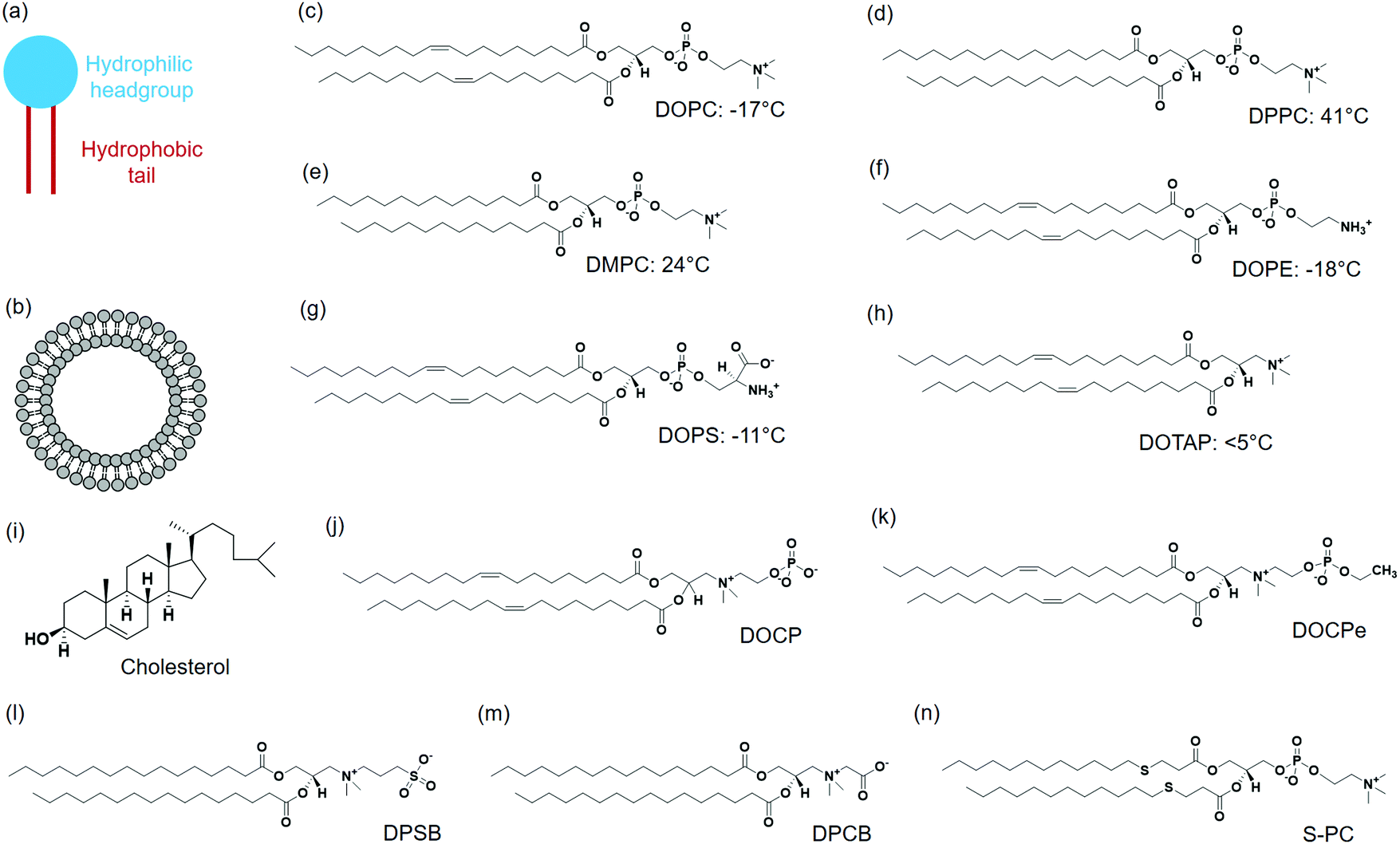 | ||
| Fig. 1 Schematic illustration of (a) a lipid and (b) a liposome. Chemical structures of a few representative lipids and their phase transition temperatures, (c) DOPC, (d) DPPC, (e) DMPC, (f) DOPE, (g) DOPS, (h) DOTAP, (i) cholesterol, (j) DOCP, (k) DOCPe, (l) DPSB, (m) DPCB and (n) S-PC. | ||
Phosphatidylethanolamine (PE) lipid (Fig. 1f) is the second most abundant headgroup in mammalian cell membranes (∼25% of total lipids), and it is especially rich in brain membranes reaching around 45%.30 Compared to PC lipids, PE lipids have a smaller headgroup and prefer negative curvature. As a result, they tend to form nonlamellar structures and pure PE lipids cannot form stable bilayers.31,32 Other lipids need to be mixed with PE lipids to form liposomes.33,34 PE lipids are usually incorporated in liposomes to induce membrane fusion.30
Phosphatidylserine (PS) lipids are another important component in the cell membrane. Although only at around 3%, PS lipids play important biological functions.35,36 The PS headgroup is negatively charged at physiological pH and contains a few metal binding ligands such as amine and carboxyl groups (Fig. 1g).37 PS lipids are enrich in the inner leaflet of the cell membrane, and flipping of PS lipids to the cell surface initiates cell apoptosis.38 A number of proteins bind to PS specifically, and a well-known example is annexin V, which is a Ca2+ dependent PS-binding protein. Annexin V binding with cell membrane can signal the translocation of PS to the external cell surface, an early indication of cell apoptosis.39
Although the majority of natural lipids are negative or neutral, positively charged lipids can be obtained synthetically. 1,2-Dioleoyl-3-trimethylammonium-propane (DOTAP) is a positive-charged synthetic lipid (Fig. 1h). Since the cell membrane is negatively charged, positive charged DOTAP could interact with cells via electrostatic interactions. Cationic liposomes are effective to load negatively charged nucleic acids, and they were widely used as transfection agents.40,41 However, positively charged liposomes can elicit immune responses and affect signaling pathways, and are known to be toxic to cells.42
Cholesterol is another important lipid, representing ∼30–40 mol% of the total lipids of erythrocyte membranes. Cholesterol consists of four fused hydrocarbon rings with one end linked to a small polar hydroxyl group (Fig. 1i). With a planar and rigid structure, cholesterol is a key regulator of membrane fluidity and plays an important role in lipid lateral organization. Cholesterol can make fluid phase membranes more rigid, but broaden the transition of gel phase membranes. In addition, cholesterol can decrease the permeability of lipid membranes.43–45
2-((2,3-Bis(oleoyloxy)propyl)dimethylammonio)ethyl hydrogen phosphate (DOCP) (Fig. 1j) and 2-((2,3-bis(oleoyloxy)propyl)dimethylammonio)ethyl ethyl phosphate (DOCPe) (Fig. 1k) are two synthetic lipids.46 In these lipids, the orientation of headgroup dipole is inversed compared to that in PC lipids.47 DOCP is negatively charged, and DOCPe is charge neutral showing antifouling properties.48 A few CPe dendrimers and polymers were reported to strongly adhere to PC membranes based on dipole mediated interactions,49,50 although we did not observe such interactions using CPe liposomes.48
1,2-Dipalmitoyl-sn-glycero-3-sulfobetaine (DPSB) (Fig. 1l) and 1,2-dipalmitoyl-sn-glycero-3-carboxybetaine (DPCB) (Fig. 1m) are newly emerged zwitterionic headgroup-inversed lipids without a phosphate group. Both show intermolecular interactions with PC lipids that arise from the oppositely charged headgroups.51,52 Some lipids have modifications at lipid tails, for example, thioether phosphatidylcholines (S-PCs) (Fig. 1n).53 Such a modification is sensitive to the environment, providing opportunity for stimuli-responsive drug release.
3. Liposome preparation and drug loading
Liposomes are vesicles made of lipid bilayers (Fig. 1b). We first briefly introduce a few common methods for preparing liposome and related drug loading. The most common Bangham method involves the formation of a lipid film by evaporating the organic solvent used to dissolve lipids.54 After hydration with an aqueous solution, multilamellar vesicles with a heterogeneous size distribution are formed. By extrusion through a polycarbonate membrane, small unilamellar vesicles (SUVs) with a narrow size distribution are obtained (Fig. 2a). The extrusion temperature needs to be higher than the Tc of the lipids. This method can produce liposomes from ∼50 nm to ∼200 nm. The larger the membrane pores, the more likely to form multilaminar vesicles. Other methods to prepare liposomes include sonication, reverse phase evaporation, solvent injection technique, and detergent dialysis.20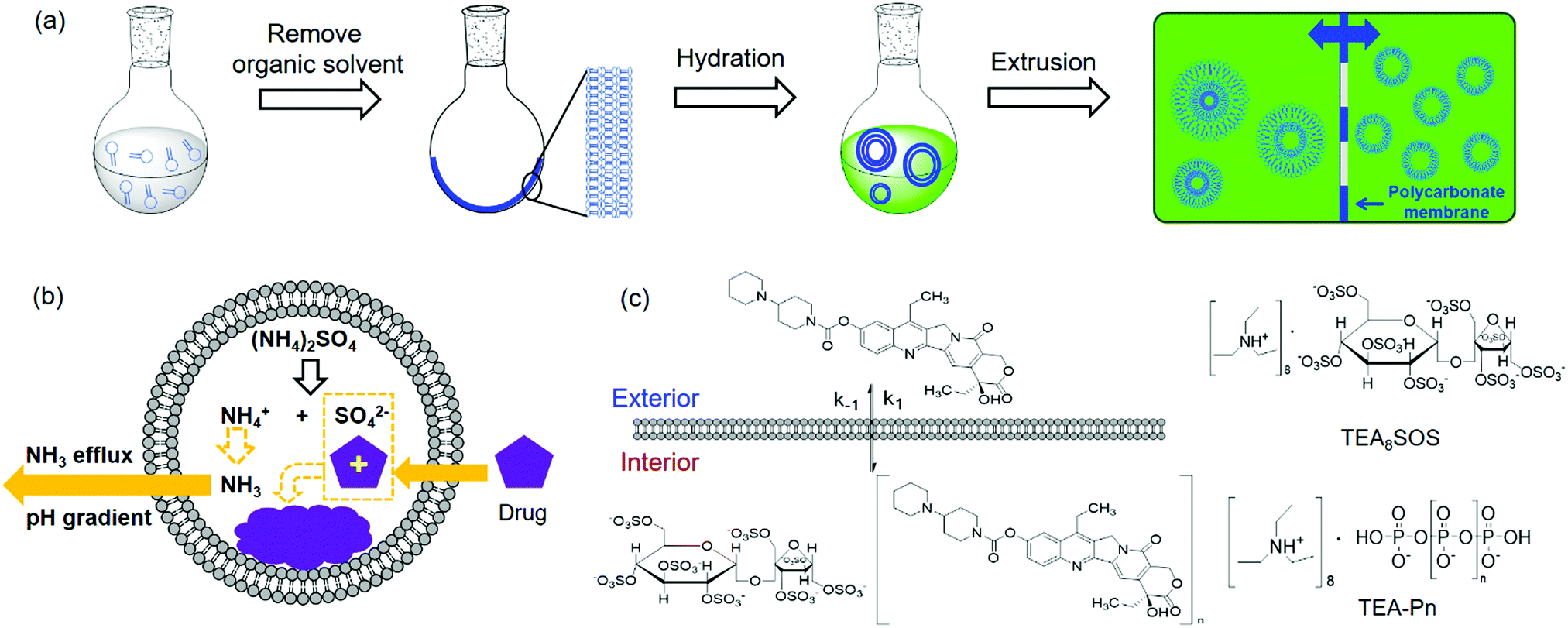 | ||
| Fig. 2 (a) Schematic illustration of liposome preparation via hydration of dried lipid films followed by extrusion. (b) Drug loading using the ammonium gradient method. (c) Schematic illustration of the loading of CPT-11 using polyanionic trapping agents. The structures of polyanionic trapping agents of TEA8SOS and TEA-Pn are shown. | ||
For hydrophobic drugs, they are often loaded in the lipid bilayer region. This requires co-dissolving lipids and drugs in an organic solvent. After evaporating the solvent, the drug molecules are trapped with a high efficiency. Hydrophilic drugs are typically loaded during hydration of lipid films by using a drug containing solution for hydration, which is called passive loading.55–57 Since the volume fraction of liposomes is quite small compared to the volume of the whole solution, the loading efficiency is low for passive loading. Efforts have been made to increase the loading efficiency by varying lipid composition, preparation methods, and lipid–drug interactions.58–61
Active drug loading, also known as remote loading, refers to loading drugs into preformed liposomes. Active loading usually takes advantages of diffusion properties when a pH gradient is established across lipid bilayers.55 This method requires drugs to have both an uncharged form and a charged form, where only the uncharged drugs can cross liposome membranes. Once diffused into liposomes, they become charged and membrane-impermeable and entrapped inside. The remote loading method has led to the successful development of many commercial formulations. For Myocet®, a pH gradient was created by preparing liposomes in acidic citrate solution followed by adding a basic solution to raise the pH of the external solution, driving doxorubicin (DOX) inside the liposomes. For Doxil®, a pH gradient was generated by a transmembrane ammonium sulphate gradient. Ammonium salts could dissociate into ammonia and protons. Since ammonia has a high membrane diffusivity, a pH gradient can also be created. Thus, DOX can influx into liposomes and precipitate with ammonium counter ions remained inside to form membrane-impermeable drug complexes (Fig. 2b).55 Kirpotin and coworkers improved the loading efficiency by using a sterically hindered amine with a highly-charged multivalent anionic trapping agent, triethylammonium salts of poly(phosphate) (TEA-Pn) or sucrose octasulfate (TEA8SOS) (Fig. 2c).62 Each TEA8SOS releases eight protons and forms an octavalent SOS8−. SOS8− could facilitate the formation of a stable intraliposomal drug–polyanion complex. At the same time the displaced triethylammonium could dissociate and traverse the lipid bilayer as triethylamine, generating a pH gradient. With this method, a water-soluble cancer drug, CPT-11 (irinotecan), was encapsulated in liposomes up to 800 g CPT-11 per mol phospholipid (a drug-to-phospholipid molar ratio of 1.36![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1).
:1).
In addition, a concentration gradient can be used to improve drug loading efficiency when the pH gradient method is not suitable. For example, gemcitabine (Gem) has a pKa of 3.6, which cannot be extensively ionized in the acidic compartment of liposomes. Yeo et al. reported loading of Gem into liposomes using combined ammonium and concentration gradients, in which osmosis pressure provided an additional driving force pushing Gem inside the liposomes.63 For example, the liposomes were dispersed in a saturated Gem solution thus generating the maximum concentration gradient across the liposomal membrane. Alternatively, liposomes were first filled with a hypertonic sodium chloride solution (462 mM) and suspended in a Gem solution. A high osmotic pressure created by the different ionic strengths drove the diffusion of Gem along with water into the liposomes. By combining a transmembrane pH gradient and a concentration gradient, the loading efficiency reached about 10 wt%.
4. Biophysical properties of liposomes for drug delivery
4.1 Liposome size
The size of liposomes is very important for drug delivery.64 On one hand, it affects the blood circulating time of liposomes. The clearance of nanoparticles and macromolecules in the bloodstream is mediated by the renal system, mononuclear phagocytic system (MPS) or reticuloendothelial system (RES). Since the estimated threshold for first-pass elimination by the kidneys is 10 nm, molecules and nanoparticles below 10 nm are rapidly eliminated by the renal system, while larger nanoparticles are mainly cleared by MPS.65 On the other hand, vasculature in tumors is leaky due to enlarged endothelial pores, allowing nanoparticles with proper size to escape from the bloodstream and accumulate at tumor tissues rather than healthy organs. This is known as the enhanced permeability and retention (EPR) effect, although some recent work suggested that the EPR effect might not be as important in real tumors.66,67 Dysfunctional lymphatic drainage in tumors also helps to retain accumulated nanocarriers and allows them to release drugs into the vicinity of tumor cells. Experiments using liposomes of different sizes suggested that the upper threshold for extravasation into tumors was ∼400 nm,68 and other studies have shown that particles smaller than 200 nm were more effective.19 In comparison, penetration through regular healthy vasculature is limited to 1–2 nm.694.2 Liposome charge
To achieve a long blood-circulation time, charge neutral zwitterionic PC liposomes are the most frequently used to reduce protein binding.26,27 To further increase the circulation time, liposomes were modified with hydrophilic polymers to evade MPS detection (i.e. stealth liposomes). Some early research coated monosialotetrahexosylganglioside (GM1) or hydrated phosphatidylinositol (HPI) to mimic cellular polysaccharide coating.70,71 However, they are too costly for large scale preparation. PEG is currently used in clinics for this purpose. PEGylation increases liposome surface hydration, inhibits liposome aggregation, and further discourages adsorption of proteins.72,73Incorporation of negatively charged lipids accelerates clearance of liposomes due to adsorption of proteins.17,74,75 Cationic lipids are mostly used for nucleic acids delivery.76 Since nucleic acids are negatively charged, they can be condensed by cationic lipids facilitating cellular uptake. Cationic lipids are in general toxic to cells and are rapidly cleared from circulation.77 Systemic delivery using cationic liposomes was rarely explored and we will not cover cationic lipids further in this review.
4.3 Liposome fluidity
The structure of lipid tails strongly influences the Tc of lipids, and controls their mechanical strength, lateral diffusion and permeability. The membrane permeability is the largest at Tc because of the coexistence and interconversion of the two phases, creating leaky phase boundaries (Fig. 3a).78,79 Many thermal-responsive liposomal drug delivery systems have been developed and reviewed.80 A distinct feature of lipid membranes compared to inorganic surfaces is surface fluidity, allowing dynamic organization of the anchored ligands.81–83 Rearrangement of immobilized ligands allows for optimal polyvalent binding, increasing binding affinity (so called avidity for describing polyvalent interactions) (Fig. 3b).84,85 Tumor targeting by manipulating membrane fluidity was demonstrated in a recent work, with fluid liposomes preferentially targeting the tumor cells and gel-phase liposomes targeting the healthy cells.86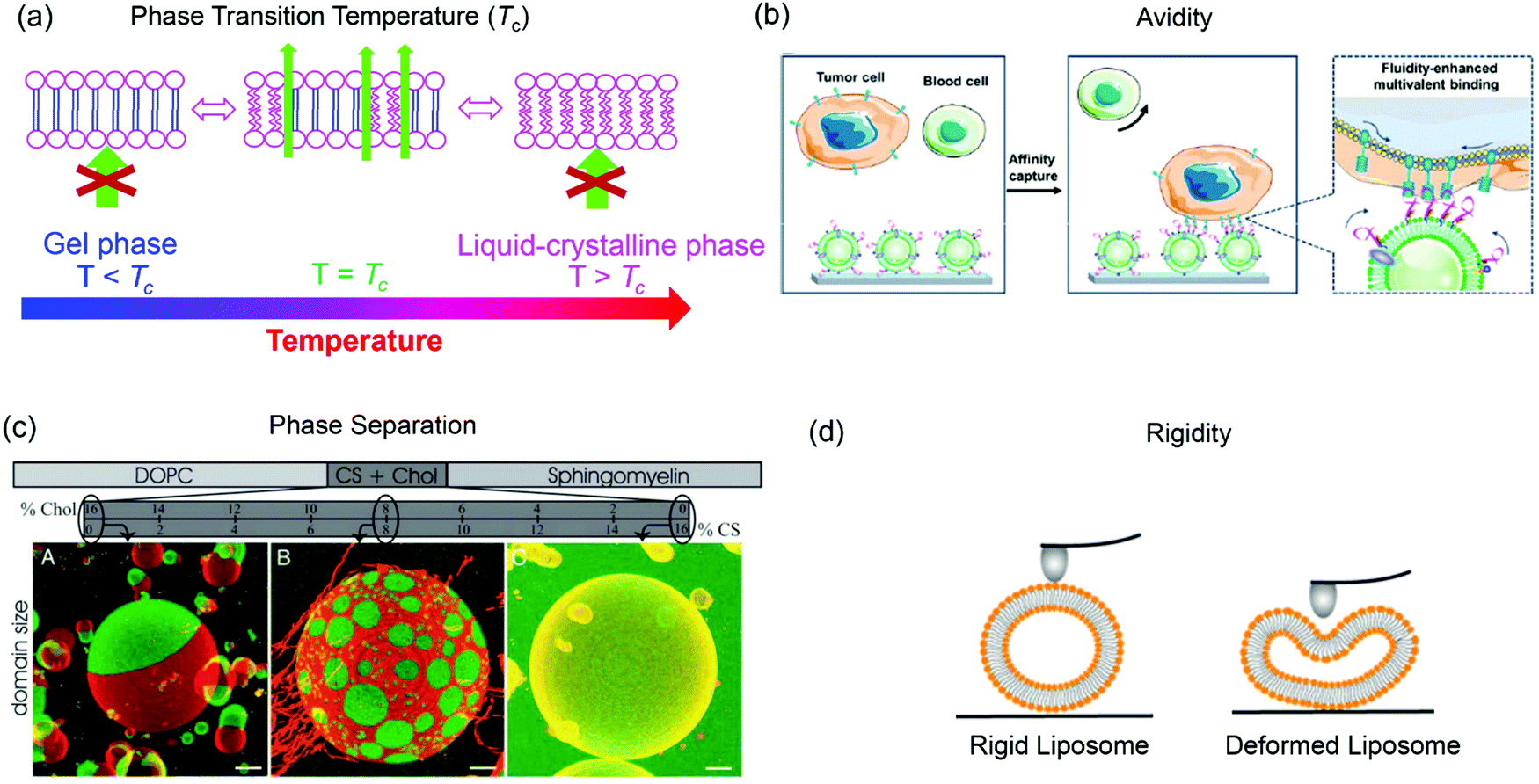 | ||
| Fig. 3 Some properties of liposomes provided by lipid bilayer fluidity. (a) Lipid phase transition temperature, (b) binding avidity, (c) Ld/Lo phase separation, and (d) liposome rigidity. Panel (b) reproduced with permission from ref. 85. Copyright © 2020 American Chemical Society. Panel (c) reproduced with permission from ref. 87. Copyright © 2005 The National Academy of Sciences. Panel (d) adapted with permission from ref. 89. Copyright © 2019 American Chemical Society. | ||
Another interesting property of lipid bilayers is their lateral organization. In addition to the liquid-crystalline phase and gel phases, phospholipids can form a liquid crystalline ordered phase (Lo) in the presence of cholesterol. Some ternary or four-component lipid mixtures can separate into distinct phases, a liquid-disordered (Ld) phase comprising mainly unsaturated lipid species, and a more packed liquid-ordered (Lo) phase enriched in saturated lipid and cholesterol.87,88 By incorporating fluorescent probes that selectively partition in the Ld or Lo phase, phase separation can be observed by confocal fluorescence microscopy (Fig. 3c).87
Another related factor that can be manipulated to facilitate drug delivery is lipid membrane rigidity or mechanical properties (Fig. 3d).89 Lipids with a higher Tc afford a more rigid structure with less deformability, while lipids with lower Tc are more flexible.89,90 Membrane rigidity influences both cellular uptake and liposome penetration in extracellular matrix (ECM) environment.89,91,92
4.4 Liposome fusion
Lipid membrane fusion is critical in many biological processes, but it does not happen spontaneously.93 Membranes need to come close enough and overcome repulsion and hydration. Liposome fusion can be induced by metal ions and fusogenic lipids (e.g. PE lipids). In addition, the recognition of ligands pair, including DNA hybridization,94–96 peptide binding,97–99 and small molecules can also facilitate liposome fusion.100–102Intracellular lipid bilayer fusion is mediated by the SNARE protein family (soluble N-ethyl-maleimide-sensitive fusion protein attachment protein receptor).103 With v-SNARE (vesicle-SNARE) and t-SNARE (target-SNARE) located at the transport vesicle and the target membrane, respectively, the formation of coiled coil complementary SNARE protein complex brings membranes closer, induces lipid rearrangements, and consequently merges the lipids. Drug delivery directly via liposome fusion with cell membrane is possible.104,105 In comparison with cellular uptake via endocytosis, the membrane fusion strategy delivers drug into the cytosol directly, avoiding lysosomal degradation.
5. Passive targeting
5.1 Effect of liposome size
Passive targeting refers to the accumulation of drug-loaded liposomes or other nanomaterials in tumors based on the EPR effect, while active targeting relies on specific ligand binding.106 Under typical tumor conditions such as inflammation and hypoxia, endothelium of blood vessels are permeable to accumulate nanoparticles. At the same time, nanoparticles are better retained due to a lack of the normal lymphatic drainage.107,108 The size of liposomes, which is an important parameter related to blood circulation time, extravasation through leaky vasculature, and macrophage uptake, has long been studied for their effect on drug delivery. Generally, a decrease in liposome size reduces their uptake by the RES system, increasing blood circulation time. Liposomes of ∼100 nm are most commonly used for long blood circulation and EPR effect.109,110 A recent research studying rheumatoid arthritis (RA) targeting also showed egg yolk lecithin (EPC) liposomes with a size of 100 nm had a longer blood circulation time of 12.85 h than liposomes with a size of 70 nm, 200 nm or 350 nm in healthy mice.111 The longer circulation time also resulted in a higher accumulation in inflamed joints. Although size is an important parameter, liposome circulation and accumulation are also affected by lipid composition and surface modifications.5.2 Polarity of lipid headgroup
While the majority of drug delivery work used PC-based lipids, we herein give an example of its headgroup inversed CPe liposomes (see Fig. 1k for structure). Based on the fundamental studies that DOCPe liposomes did not adhere to DOPC liposomes, cells or proteins, Li et al. studied the circulation time of DOCPe liposomes compared to DOPC liposomes in vivo.48 Near-infrared (NIR) fluorophore-labeled liposomes were injected in the tail vein of mice and their concentrations in blood were followed by measuring the fluorescence in the plasma at different time points. The DOCPe liposomes had a longer circulation time than the DOPC liposomes (Fig. 4a), which was attributed to the greater resistance of DOCPe uptake by macrophages. Such longer circulation allowed better accumulation of the DOCPe liposomes in tumors based on the EPR effect (Fig. 4b).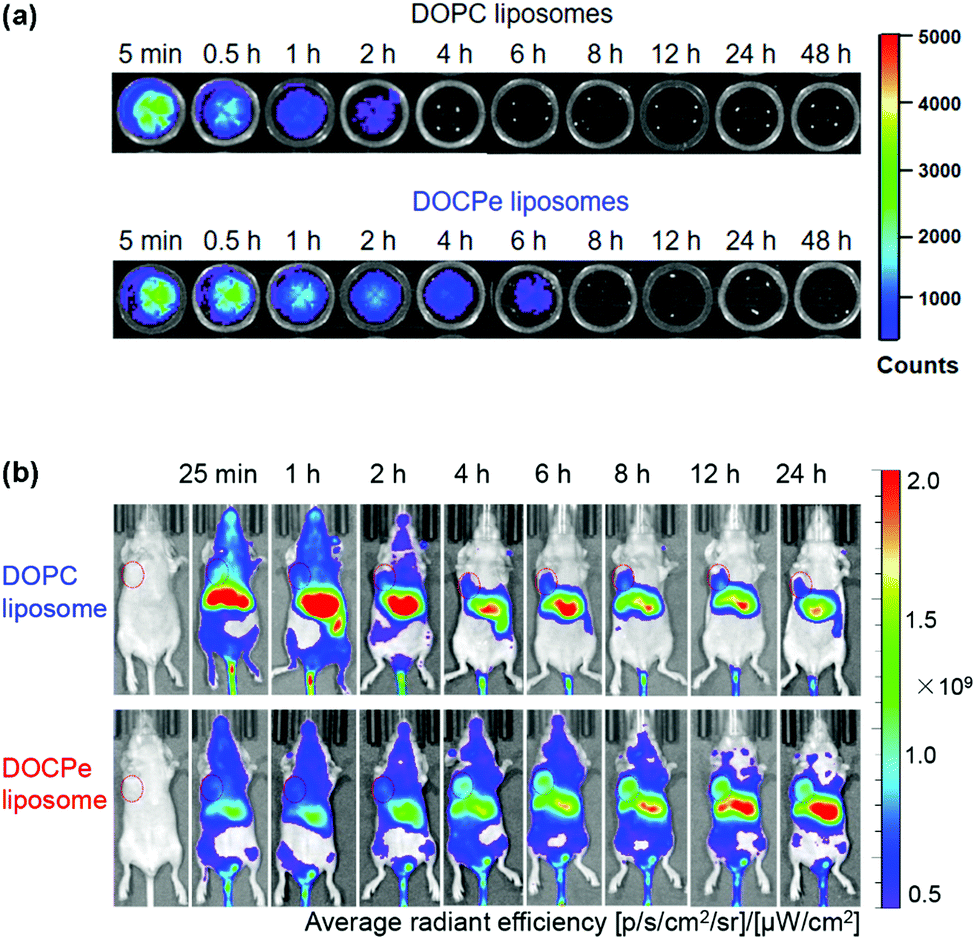 | ||
| Fig. 4 (a) In vitro fluorescence images of the mice plasma extracted at different points after injecting DiR- or DiO-loaded DOPC and DOCPe liposomes. (b) Time-dependent imaging of mice with MDA-MB-231 breast cancer xenografts injected with DiR-loaded DOPC and DOCPe liposomes. The tumors are indicated by the red circles. Reproduced with permission from ref. 48. Copyright © 2017 American Chemical Society. | ||
Taking advantage of the longer circulation time and enhanced tumor site accumulation, the simultaneous encapsulation of a radiotherapy sensitizer, voinostat (a histone deacetylase inhibitor), and hypoxia-activated banoxantrone dihydrochloride (AQ4N) in DOCPe liposomes for cascaded chemo-radiotherapy was developed.112 On one hand, voinostate is an effective radiosensitizer that overcame the intrinsic radioresistance by inhibiting repair of damaged DNA caused by radiation. On the other hand, radiation consumed O2 and generated a hypoxia status in cells, which activated the cytotoxicity of AQ4N to kill the hypoxia tumor cells. This DOCPe-based formulation exhibited 2.9-fold higher tumoral accumulation than the DOPC liposomes in BALB/c mice, and the radiation therapy with the DOCPe formulation showed ∼75% tumor suppression after 20 days.
5.3 Effect of liposome rigidity
Membrane rigidity and mechanical properties not only affect cellular uptake but also extracellular diffusion.89,91,92 The rigidity of lipid membranes can be adjusted by varying lipid tail chemistry or incorporation of cholesterol. Generally, membrane rigidity increases with the increase of tail length or incorporation of cholesterol into fluid phase lipids.25,113–115 The Schroeder's group studied the effect of lipid composition on cellular uptake of PC liposomes by triple negative 4T1 breast cancer cells. By using flow cytometry, the authors demonstrated that the cellular uptake increased with the increase of acyl chain length, with DSPC (18:0) > DPPC (16:0) > DMPC (14:0). They further showed that the DMPC (14:0) and DLPC (12:0) liposomes could destabilize cell membranes leading to decreased cell viability. In contrast, liposomes with longer acyl chains, e.g., DSPC (18:0) and DPPC (16:0) enhanced cancer cell proliferation due to the integration of the liposomal lipids into cancer cell membranes. Rigidification of DMPC (14:0) membrane by incorporation cholesterol also improved cellular uptake.91
Tumors are surrounded by an extracellular matrix that provides a barrier for vesicles to reach the cells. Membrane rigidity and deformability of liposomes affect their diffusion through extracellular matrix and multicellular spheroid (MCS) penetration. Dai et al. prepared liposomes with the same surface properties but different rigidity by varying lipid chain length and degree of saturation.89In vitro cellular uptake studies showed that liposomes with a stiff membrane (Lip4, DSPC 18:0) had a greater cellular uptake and cytotoxicity. However, in MCS rich in fibrous matrix mimicking the microenvironment of solid tumors, the liposomes with a moderate rigidity (Lip3, DPPC, 16:0) exhibited the best penetration and retention in tumor interstitial space. In addition, hydroxycamptothecin (HCPT) loaded Lip3 showed significantly enhanced tumor toxicity compared to the stiffer or softer liposomes (e.g., Lip2, DMPC). The improved tumor suppression effect of Lip3 was explained by its transformation to a rod-like shape as observed by stimulated emission depletion (STED) microscopy, while the stiff liposomes hardly deformed and softer liposome (with unsaturated acyl chains) changed to irregular shapes (Fig. 5), both slowed the MCS penetration.
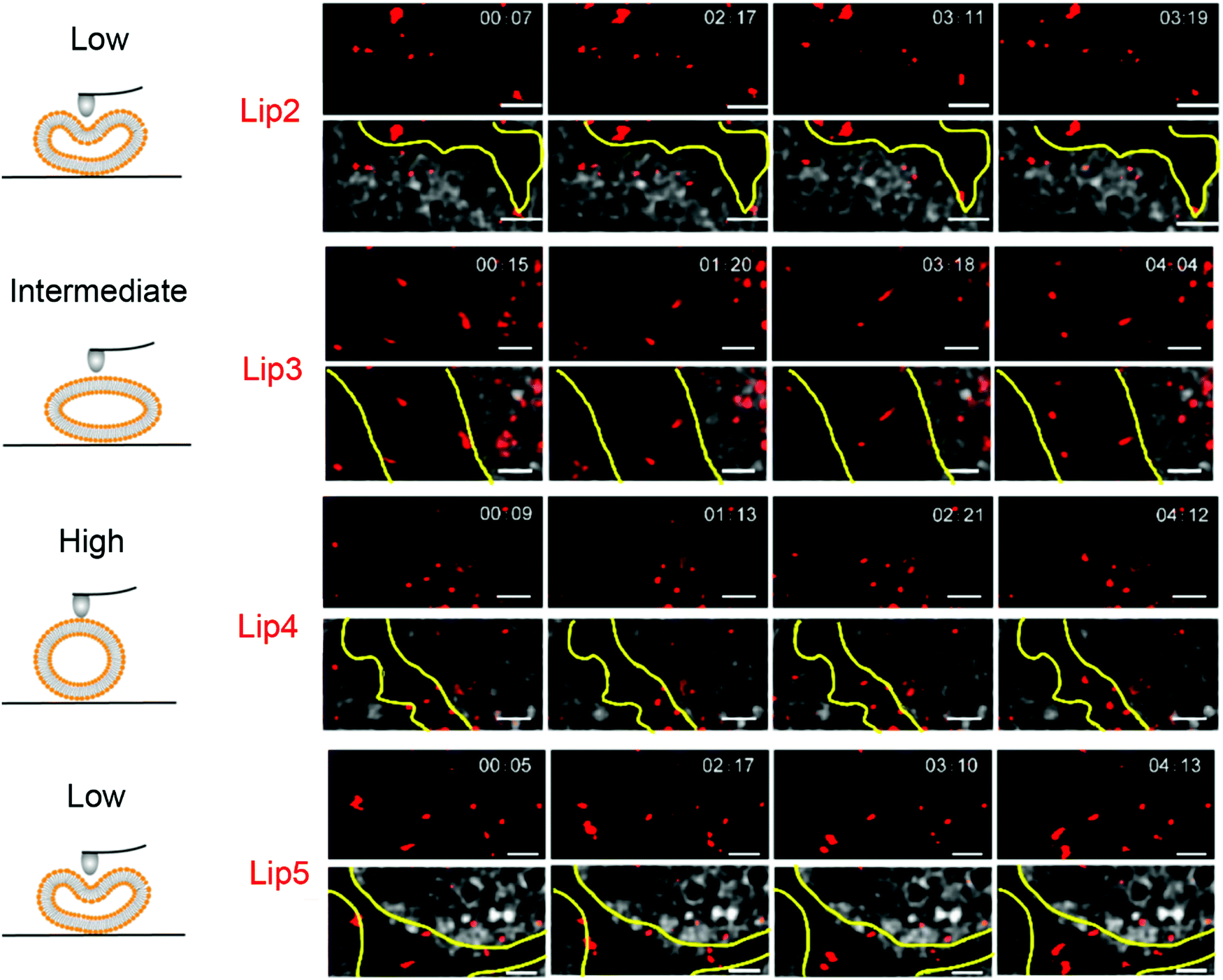 | ||
| Fig. 5 Snapshots and trajectories of liposomes in BxPC3-HPSC MCSs imaged by STED microscopy at different time points. The schemes on the left indicate the deformation of liposomes of different rigidity. On the right, the red spots are from the DiI-labeled liposomes. The areas between the yellow lines indicate the tumor stroma. The soft liposomes, Lip2 and Lip5, were deformed irregularly and diffused in a small area. Lip4 with high rigidity showed little deformation during movement. Lip3 with intermediate rigidity changed from spherical to ellipsoidal with high-speed movement. Scale bars: 1 μm. Adapted with permission from ref. 89. Copyright © 2019 American Chemical Society. | ||
Sakai-Kato and coworkers also reported that liposome membrane rigidity (characterized by bend moduli Kc) affected their penetration into three-dimensional tumor spheroids of HeLa cells.92 In their work, the liposomes were prepared using HSPC/HSPC–PEG (94.7/5.3), and the Kc was increased by including cholesterol (HSPC/chol/PEG–HSPC, 56.3/38.4/5.3). Penetration in the tumor spheroids was governed by both diffusion through the intercellular space and cellular uptake. They found the penetration efficiency of liposomes increased as Kc increased, while the cellular take efficiency was saturated when Kc was greater than 3.3 × 10−19 J, indicating the penetration of liposomes into spheroids was not solely controlled by cellular uptake. Additional contributions may be from less deformation of the liposomes with a larger bending modulus resulting in a weak adsorption on the extracellular matrix (and thus less impeded by the matrix).
5.4 Effect of surface PEG
PEGylation is an efficient and popular strategy to prevent the liposome clearance via phagocytosis by reducing plasma protein adsorption. PEG chains are usually attached to liposomes via PEGylated lipids such as PEG–DSPE. Huang and coworkers reported PEG-modified large unilamellar egg PC/chol liposomes (∼200 nm) showing a long blood circulation half-life (t1/2) of 5 h compared to those without PEG (less than 30 min).116 In another work, PEGylation of egg PC/chol liposomes (100 nm) showed a 5-fold increased blood circulation time up to 15.3 h, with significantly reduced liver and spleen uptake and increased accumulation in the implanted tumors.117 The effect of PEG depends on both its density and length, and 5 mol% PEG2000–DSPE is a good starting point for futher optimization.19,118,119In a work studying liposome circulation in healthy ICR mice, 5 kDa PEG showed a longer t1/2 of 15.83 h than 1 kDa (11.21 h), 2 kDa (12.85 h) or 10 kDa (11.41 h). In addition, the liposomes with 10% PEG (5 kDa) had a longer t1/2 of 15.83 h compared to the liposomes with 5% (10.52 h) or 20% PEG (13.6 h). The passive targeting efficiency, however, was similar for the formulations with 5, 10, and 20% PEG (5 kDa), which indicated that the PEG content (within a limited range) might not be crucial for the targeting.111 In addition to the effect on blood circulation time, PEGylation was reported to affect tumor spheroid penetration. The efficiency of penetration into HeLa spheroids was greater for HSPC/chol than for HSPC/Cho/PEG–DSPE, although they had a similar bending module.92
Although the general understanding is that PEGylation enhances the blood circulation, extensive PEGylation can result in negative effects on liposomal drug delivery, including reduced cellular uptake and inhibited endosomal release.120 Moreover, accumulated evidence suggested that PEGylated liposomes are prone to rapid clearance by the accelerated blood clearance (ABC) phenomenon, which is stemmed from the incomplete blockage of liposome surface from proteins adsorption in biological milieu.121,122
5.5 Effect of lipid phase separation
Stachowiak and coworkers studied phase-separated liposomes for delivery to cytoplasm.123 The Ld/Lo phase separation of the ternary DOPC/DPPC/chol system has been well studied. In their work, cationic and fusogenic DOTAP lipid with a low Tc was used in lieu of DOPC. Based on the phase diagram of DOTAP/DPPC/chol (Fig. 6a), phase separated liposomes with 1:1 DPPC/chol containing up to 24 mol% DOTAP were prepared. In addition, DOTAP/DOPC liposomes were prepared as non-phase separated controls. The phase separated liposomes showed 8–10 times higher lipid transfer rates to DOPC GUVs compared to the non-phase-separated ones. In addition, the shielding effect of PEG was less obvious on the phase separated liposomes, which was attributed to the saturated PEGylated lipid palmitoyl tails preferring to reside in the Lo phase while the DOTAP lipids in the Ld phase being less shielded (Fig. 6b). For the cell uptake studies, the 5 mol% DOTAP phase separated liposomes enhanced liposome fusion with cells by 4–5 fold compared to the homogeneous liposomes, which was attributed to the phase separation concentrating the DOTAP lipids in specific regions. Flow cytometry analysis showed the dextran loaded phase-separated GUVs (Fig. 6c) containing 24 mol% DOTAP could deliver dextran 4 times more efficiently than the homogenous vesicles (Fig. 6d). Membrane phase separation reduced the total DOTAP required for fusion to as low as 5 mol%, which was significantly less than typical DOTAP-containing delivery systems with 50–100% DOTAP.
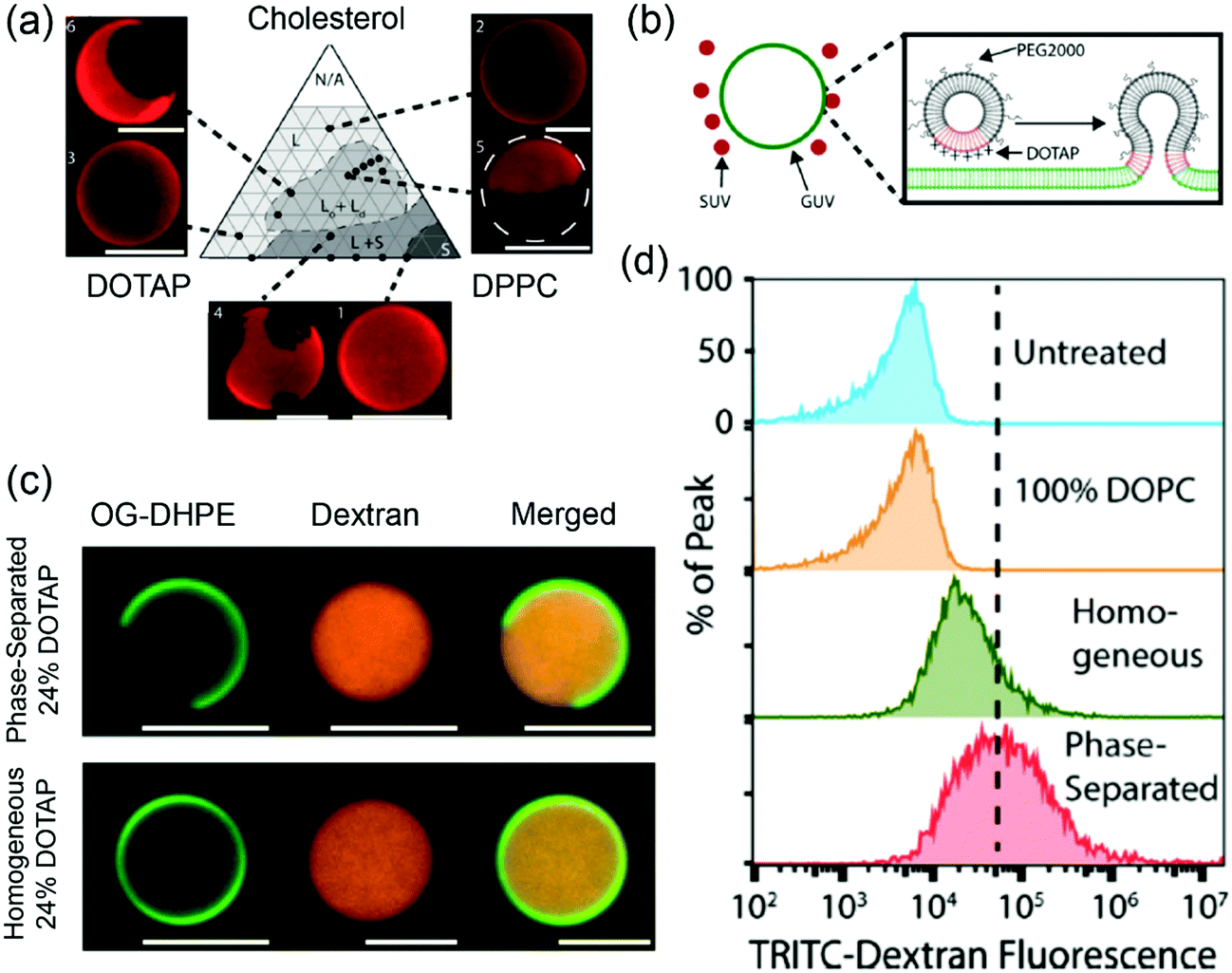 | ||
| Fig. 6 (a) A DOTAP/DPPC/cholesterol phase diagram at 25 °C. Texas red–DPPE (0.3 mol%) was used to visualize different phases. (b) Schematic illustration of phase-separated DOTAP containing SUVs fusing to a GUV. (c) Confocal fluorescence micrographs of phase separated (24 mol% DOTAP, 38 mol% cholesterol, 36 mol% DPPC, 2 mol% PEG2000–DPPE) and homogeneous (24 mol% DOTAP, 74 mol% DOPC, 2 mol% PEG2000–DPPE) liposomes loaded with TRITC–dextran (red). The liposomes were labeled with 1 mol% Oregon green–DPPE (green). (d) Flow cytometry histograms of cells incubated with GUVs loaded with TRITC–dextran. Adapted with permission from ref. 123. Copyright © 2017 Biomedical Engineering Society. | ||
6. Active targeting
6.1 Effect of PEG
Active targeting relies on the functionalization of liposome with affinity ligands such as antibodies, aptamers or small molecules that can bind to surface receptors of target cells. In active targeting, PEG was used as a stealth coating to increase blood circulation and as a spacer to link targeting ligands to liposomes. Although PEG2000 (∼45 repeating units of ethylene glycol: EG 45) is widely used as a clinical standard, Bilgicer, Kiziltepe and coworkers reported enhanced cellular uptake by a shorter PEG on liposomes with a targeting peptide.118 In their work, a short cyclic peptide antagonist of the human epidermal growth factor receptor 2 (HER2) (YCDGFYACYMDV, HER2-pep) was functionalized on liposomes (95:10:3:2 HSPC:Chol:PEG:HER2-pep), and the cellular uptake of the liposomes of different lengths of PEG was compared (Fig. 7a). The HER2-pep targeting liposomes showed no difference in uptake by the HER2 overexpressing SK-BR-3 and BT-474 cells compared to the non-targeting liposomes when PEG2000 coating was used. In addition, the EG45 peptide linker itself limited the availability of the peptide to bind to its receptor. The long PEG chains might fold into globular mushroom like structures that blocked the accessibility of the targeting ligand. When a short PEG350 (∼12 repeating units) was used in combination with an EG12 linker, a ∼9-fold greater uptake was observed than the control, whereas the uptake reduced when the linker was longer than EG24 and completely diminished with EG45 and EG72 (Fig. 7b). Confocal fluorescence images also revealed significant increased cellular uptake of HER2-liposomes with PEG350 and EG12 linker (Fig. 7d). The enhancement observed with shorter linker was attributed to several reasons, including reduced overall entropic loss when cells bind to a shorter linker with reduced translational and conformational freedom, decreased nonspecific interactions of peptides with lipid bilayers and increased avidity.
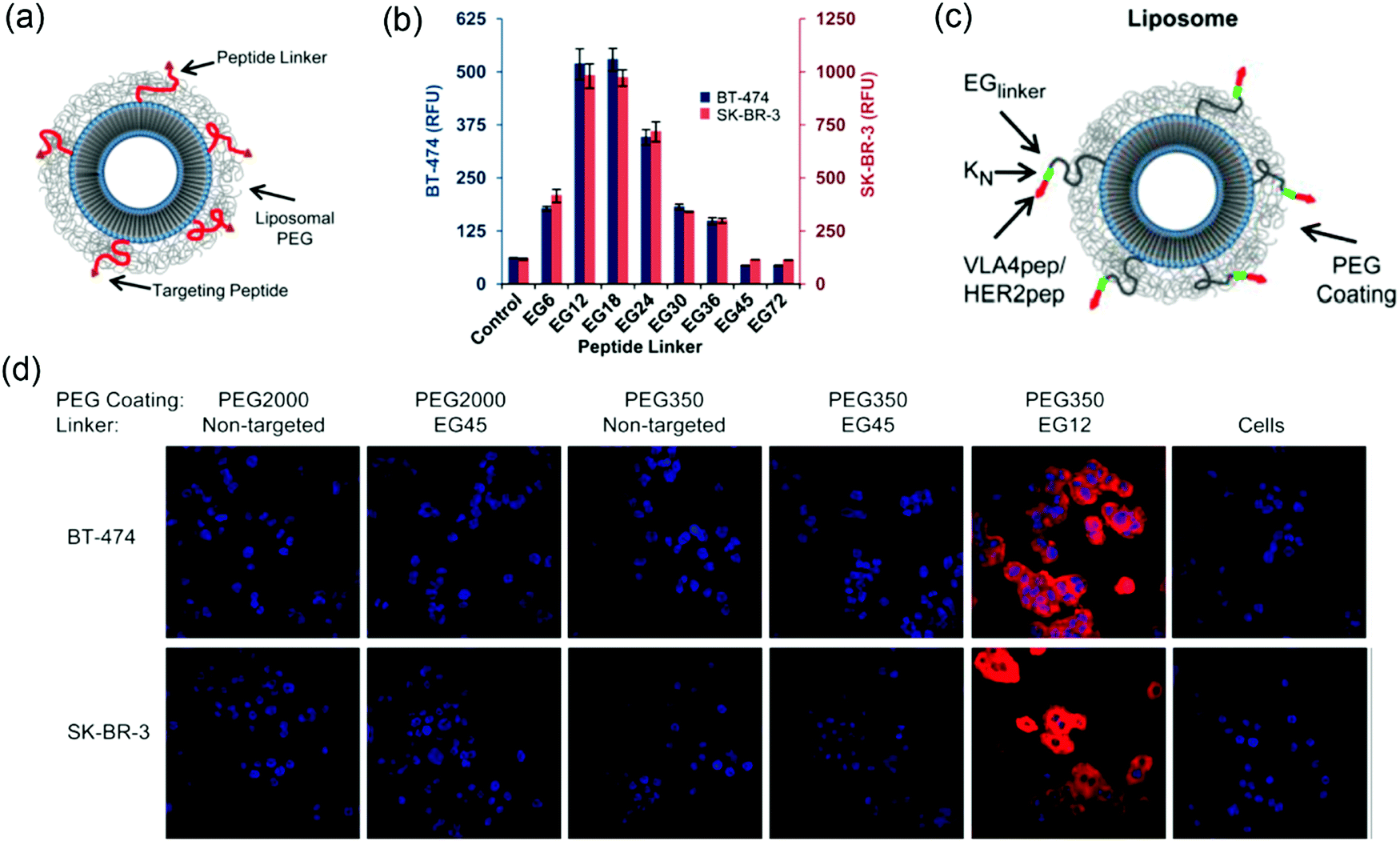 | ||
| Fig. 7 (a) Scheme of ligand-targeted liposomes with PEG coating. (b) Flow cytometry data showing the uptake of liposomes with different linker lengths and PEG350 coating by both BT474 cells (left y-axis; blue columns) and SK-BR-3 cells (right y-axis; red columns). The most significant cellular uptake was observed with the EG12 linker. (c) Scheme of ligand-targeted PEGylated-liposomes with hydrophilic modification adjacent to the targeting peptide. (d) Confocal microscopy of rhodamine-labeled liposomes with the indicated PEG coating and EG-linker incubated with BT-474 (top) and SK-BR-3 (bottom) cell lines for 3 h at 3 °C. The non-targeted liposomes were used as controls. Panels (a, b and d) reproduced with permission from ref. 118. Copyright © 2013 American Chemical Society. Panel (c) reproduced with permission from ref. 124. Copyright @ 2013 American Chemical Society. | ||
In another system, the cellular uptake of VLA-4 pep (YCDPC) labeled liposomes with PEG350 coating and EG12 linker showed a ∼100-fold enhancement by VLA-4 overexpressing myeloma cells. Interestingly, the cellular uptake of the peptide targeted liposomes can be significantly increased by including a short oligolysine (K3, lysine with 3 repeat units) chain adjacent to the targeting peptide (Fig. 7c). A dramatic increase in cellular uptake up to ∼80-fold for the VLA-4/multiple myeloma system was observed using an EG6 peptide-linker and PEG2000 coating.124 The hydrophilic oligolysine chain might increase the display of the peptide, thus enhancing the availability of peptide to bind to its target receptor.
Liu and coworkers studied hyaluronan (HA)-grafted liposomes targeting tumor cells overexpressing CD44 via receptor-mediated endocytosis.75 The incorporation of negatively charged HA on the liposome surface led to decreased blood circulation and tumor accumulation in CD44+ human breast cancer MDA-MB-231 xenografts compared to the HA-free PEGylated liposomes. Coating PEG on the HA-liposomes reduced the cellular uptake in vitro, but resumed the long circulation in vivo. The PEG–HA-liposomes and PEG-liposomes displayed similar tumor accumulation, but the PEG–HA-liposomes had better cellular internalization due to the active targeting effect. In this work, the tumor-targeting ligands did not increase the total tumor accumulation at the targeted site, but the passive targeting EPR effect determined the blood circulation and accumulation of the HA-liposome. However, ligand–receptor interactions increased cellular internalization after delivered to the target site.
6.2 Effect of surface protein adsorption
Liposomes, like other nanoparticles, adsorb proteins in blood and form a protein corona.125 Such protein corona has significant effects on their blood circulation, cellular uptake and biodistribution.125–129 Even with PEG modification, plasma protein adsorption on liposomes is inevitable.120 By rational design of peptide structure, Zhan and coworkers reported enhanced immunocompatibilty of brain-targeting liposomes via manipulating IgM adsorption.130 A retro-inverso analog of a brain-targeting peptide DCDX was selected and conjugated to the surface of stealth liposomes (HSPC/cholesterol/PEG) due to their stability towards proteolysis compared to the natural LCDX. The DCDX-modified stealth liposomes adsorbed a significant amount of IgM leading to an immune response and rapid liposome clearance. By analysis of peptide binding with the brain target receptor, nicotinic acethylcholine receptor (nAChRs), a short peptidomimetic D8 peptide was obtained by computational peptide design. D8-sLip had comparable stability, binding affinity, and brain targeting property to DCDX-sLip, but the protein corona formed in vivo showed that D8-sLip adsorbed much less IgM than DCDX-sLip. The pharmacokinetics and biodistribution of D8-sLip in BALB/c mice showed prolonged circulation and decreased liver and spleen distribution. As expected, D8-Lipd generated significantly less immunogenicity than DCDX-sLip. By intravenously injecting DOX-loaded liposomes in ICR mice, both DCDX-sLip and D8-sLip/DOX promoted the liposomes to cross the blood–brain barrier (BBB) in vivo compared to sLip, and D8-sLip/DOX showed slightly longer duration than DCDX-sLip/DOX.131In another study, the group showed that a short nontoxic peptide Aβ25-35 (derived from β-amyloid (Aβ)) modified liposomes (SP-sLip) specifically adsorbed brain-targeting plasma apolipoproteins, ApoA1, ApoE and ApoJ.132 The SP-sLip interacted with the lipid-binding domain of apolipoproteins, and their receptor-binding domains exposed on the liposomal surface were still active for recognition of multiple receptors (LRP1/ApoE, LRP2/ApoJ, and SR-B1/ApoA1). The direct interactions between SP-sLip and receptor LRP1 were too weak to induce efficient uptake by brain capillary endothelial cells (bEnd.3), and protein adsorption from rhApoE or mouse plasma was required to boost endocytosis of SP-sLip. Intravenous injection of SP-sLip through the tail vein of healthy BALB/c mice showed a significant distribution in hippocampus and cortex in comparison to sLip, indicating the penetration of BBB. Using DOX-loaded liposomes, SP-sLip/DOX showed a significantly higher brain distribution of DOX than sLip/DOX. The therapeutic efficacy of DOX-liposomes in nude mice bearing intracranial human glioma cells (U87) was studied for the potential therapeutic value of SP-sLip for glioma. Intravenously injecting free or liposome-formulated DOX at 10 mg kg−1 body weight did little in improving mouse survival, while SP-sLip/DOX significantly prolonged the median survival time of nude mice from 27 days to 50 days.
6.3 Effect of fusogenic liposomes
Kro and coworkers delivered drugs by liposome fusion using coiled coil forming peptides E4 (EIAALEK)4 and K4 (KIAALKE)4 (Fig. 8).133 E4 was conjugated to a PEG4 (PEG with 4 repeat units) spacer with a cholesterol anchor inserted in the liposomes. In vitro experiments showed that DOX-loaded E4-liposomes (E4-Lipo-DOX) delivered DOX into K4-encoded HeLa cells (HeLa-K) upon coiled coil formation with enhanced cellular uptake compared to free DOX or liposomal DOX without E4 (Lipo-DOX). Using several endocytosis inhibitors, it was shown that endocytosis was a minor pathway, suggesting that direct fusion with the cell membrane might be the major pathway. In the in vivo experiment, posterior cardinal vein (CV) injection of E4-Lipo-DOX at 1 μmol per kgBW (kilogram per body weight) into HeLa-K cell implanted zebrafish significantly reduced tumor proliferation compared to Lipo-DOX or free DOX treatment (Fig. 8b). The administrated amount of DOX using E4-Lipo was 5-fold lower than that typically used in clinical settings. Although this system required pre-labeling target cells with the K4 peptide, it demonstrated that the coiled coil targeting mechanism enabled direct liposome fusion for drug delivery. | ||
| Fig. 8 (a) Schematic illustration of liposome fusion with cells by E4/K4 coiled coil formation. (b) E4/K4 coiled coil formation enhanced anticancer efficacy of DOX in xenograft zebrafish. E4-Lipo-DOX, Lipo-DOX, or 0.25 mM free-DOX (1 nL of 1 mM) was injected and imaged at 72 h postfertilization. HeLa-K xenograft with E4-Lipo-Dox injection (second panel) shows the decrease of red fluorescence. Green and red fluorescence indicate vasculatures and cancer cells, respectively. Reproduced with permission from ref. 133. Copyright © 2016 American Chemical Society. | ||
Qin and coworkers prepared membrane fusogenic liposomes (MFLp) with a cell penetrating arginine fragment (DSPE-4A) and PEG coating having a pH-sensitive linker of benzaldhyde (DSPE-Hy-PEG2k). They were used to co-deliver anti-S100A4 antibody (S-mAb) and DOX for synergistic treatment of metastatic tumors. In tumor's acidic microenvironment, the PEG chains were detached from the liposomes and the cell-penetrating 4A peptides were exposed, inducing liposome fusion with cell membranes. Liposome fusion bypassing endocytosis resulted in a quick release of the payload into the cytoplasm in 30 s. S100A4 is a key protein in promoting metastasis. In breast cancer 4T1 cells, MFLp/S-mAb effectively downregulated S100A4 expression, increased the expression of metastasis suppressor proteins, and subsequently resulted in the suppression of cell motility and migration. In vitro experiments showed that MFLp/DOX plus S-mAb induced more 4T1 cell apoptosis of ∼32% compared to MFLp/DOX of 24.4% due to the additional effect of S-mAb. For in vivo experiment, 4T1 tumor-bearing BALB/c mice treated with MFLp/DOX + S-mAb showed a significant tumor volume reduction of 88.57% without spleen swelling or liver metastasis, indicating the inhibition of both local growth and metastasis by the anti-S100A4 antibody.134
Szoka's group prepared phosphatase-triggered lipid-based particles (PTPs) by coating liposomes with a phosphate-modified fusion peptide for cytoplasmic delivery of cell-impermeable compounds.135 The phosphate modified HIV gp41 N-terminus fusion peptide with a cholesterol end (FP-2PT-Chems) was inserted in POPC liposomes. The phosphate modification shielded the hydrophobic character of the peptide, inactivating the peptide for the fusogenic property. Once inside the tumor microenvironment with overexpressed phosphatase, dephosphorylation occurred and the fusogenic activity of the peptide was restored. Cytoplasmic delivery small molecules such as carboxyfluorescein and propidium iodide, or macromolecules such as FITC–dextran encapsulated in the PTPs were demonstrated.
7. Supported lipid bilayers
Coating a lipid bilayer on nanoparticles can normalize the surface properties of the cores and the lipid bilayer is mainly responsible for interacting with cells.9 Drugs can be adsorbed into or entrapped by the core materials to achieve a high loading efficiency. The use of lipid enveloped nanoparticles for drug delivery has been previously reviewed,7,8 and we focus on the biophysical aspect of it.7.1 Effect of core rigidity
Supported lipid bilayers allow the variation of NP rigidity while using the same lipid composition.136 Jiang and coworkers prepared lipid–polymer NPs (LPNs) of the same size, with PLGA cores covered with either DPPC lipid monolayers (MPs) or bilayers (BPs) (Fig. 9a and b). Atomic force microscopy (AFM) and atomistic simulation revealed that the MPs had a lower flexibility than the BPs. In vitro cellular uptake was carried out by incubating drug-loaded LPNs (DOX loaded in the PLGA core for cancer chemotherapy and combretastatin A4 (CA4) loaded in the bilayer region for treating vasculature damages) with HeLa and HUVEC (blood capillary) cells. This system allowed for simultaneous observation of inhibition of both cancer cells and blood capillary cells that are important for eradication of most types of solid tumors. The dual drug-loaded MPs had more significant cytotoxicity than the BPs (Fig. 9c), which was attributed to the more flexible BPs dissipating more energy at the LNP–cell interface and making it more difficult to be internalized by cancer cells. Both drug-loaded LPNs inhibited tumor growth with the MP being more efficient due to enhanced cellular uptake (Fig. 9d). In addition, in vivo imaging showed that the MPs accumulated in the tumor sites more rapidly than the BPs.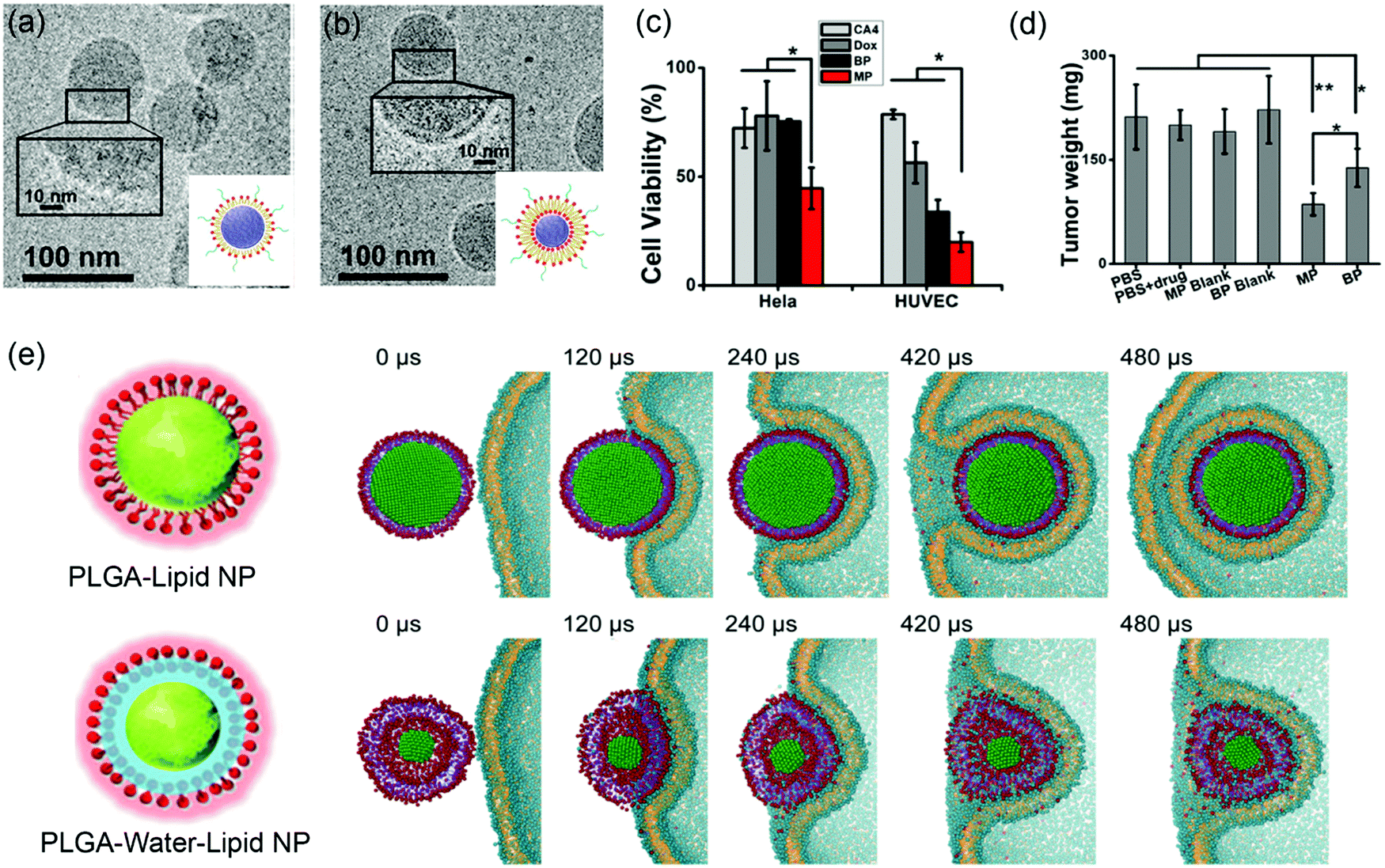 | ||
| Fig. 9 Cryo-TEM micrographs and schematic illustration of (a) lipid-monolayer-shell (MPs) and (b) lipid-bilayer-shell (BPs). (c) Cell viability after being treated by MPs, BPs, and free drugs. (d) Weight of the excised tumors after different treatments. Adapted with permission from ref. 136. Copyright © 2015 American Chemical Society. (e) Rigidity-governed deformation influencing cellular uptake as depicted by MD simulations. Top: The rigid P–L NP is internalized smoothly via wrapping of the cellular membrane, showing a mild shape deformation. Bottom: The less rigid or “soft” P–W–L NP underwent a large deformation during the internalization, and finally it is trapped on the cell surface. Reproduced with permission from ref. 137. Copyright © 2014 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
To further explore the rigidity of NPs on cellular uptake, Jiang and coworkers prepared lipid-covered PLGA NPs by tuning the interfacial water layer.137 For the P–L NPs, the lipid shell was tightly attached to the PLGA core, while the P–W–L NPs contained a thin water layer between the lipid shell and PLGA core. AFM showed that the 40 nm P–L and P–W–L had Yong's modulus of 1.2 ± 0.11 GPa and 0.76 ± 0.07 GPa, respectively. A significantly higher cellular uptake was observed from the more rigid P–L NPs in HeLa Cells. To understand the mechanism, MD simulation was performed to reproduce the internalization process (Fig. 9e). The simulation showed that the rigid P–L NPs were fully wrapped by the cell membrane and internalized smoothly (Fig. 9e top), whereas the P–W–L NPs significant deformed during the internalization before trapped within the cell membrane (Fig. 9e bottom). They analyzed the elastic deformation energy as a function of the wrapping fraction for spherical and ellipsoidal NPs, and revealed that complete wrapping of the ellipsoidal NPs required about 30% more energy than the spherical ones. This may partially explain the less rigid and more flexible NPs being relatively more difficult for cellular uptake.
Moses and Auguste and coworkers synthesized a hybrid nanolipogel (NLG), an alginate encapsulated liposome with defined size, surface charge and tunable elasticity.138 The elasticity was modulated by the extent of crosslinking of the hydrogel core controlled by calcium concentration (Fig. 10a). As determined by AFM, the free DOPC liposomes showed the lowest Young's modules of 45 ± 9 kPa (MP-45 kPa), the uncrosslinked NLGs were stiffer with a Young's module of 1.6 MPa (NLG-1.6 MPa), and the crosslinked NLGs were even stiffer of 5.3 MPa (NLG-5.3 MPa), 13.8 MPa (NLG-13.8 MPa), and 19 MPa (NLG-19 MPa). In vitro cellular uptake experiments revealed decreased cellular uptake with increasing of the modulus independent of cell type. The maximal uptake occurred with MP-45 kPa, 80% greater than with NLG-19 MPa. Interestingly, particles with differently elasticity were taken by cells via distinct internalization pathways, in which the MLP-45 kPa entered cells predominately via fusion, while the NLG-19 MPa internalized strictly via endocytosis (Fig. 10b). Ex vivo tumor uptake by tail vein injection in a 4T1 breast tumor model showed that the uptake was significantly increased with the NLP-45 kPa and NLG-1.6 MPa compared to NLG-19 MPa at 48 h, suggesting that decreasing elastic modules enhanced both cellular internalization and permeation in tumors. In vivo tumor uptake indicated the NLG with higher Young's moduli (above 13.8 MPa) showed higher liver uptake, while NLP-45 kPa and NLG-1.6 MPa had significantly increased tumor uptake. This also suggested that decreasing the elastic modulus preferentially enhanced both cellular internalization and permeation of tumors. It is noteworthy that the conclusion of NPs with lower elastic modules showed enhanced cellular uptake does not agree with the abovementioned rigid lipid-coated PLGA entering cells more efficiently (Fig. 9). This may be due to that the PLGA cores were much more rigid, so that endocytosis was the dominant internalization pathway and no liposome fusion was involved.
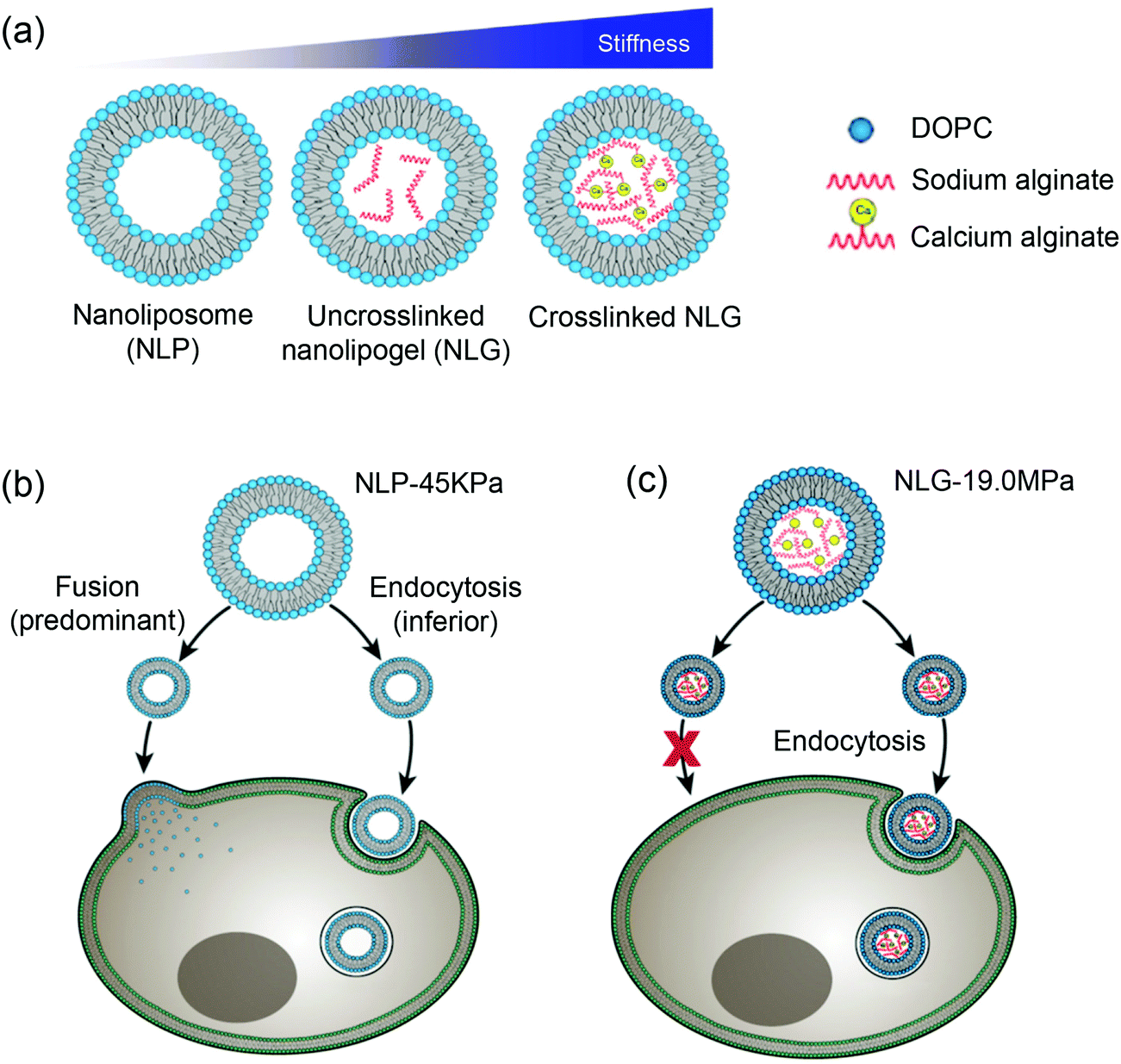 | ||
| Fig. 10 (a) Schematic illustration of nanoliposome–hydrogel complex systems. Cell internalization pathways shifted by varying particle elasticity: (b) soft NLP-45 kPa enters the cells via two pathways: fusion (predominant) and endocytosis (inferior), while (c) hard NLG-19 MPa enters the cells only via clathrin-mediated endocytosis. Reproduced with permission from ref. 138. Copyright © 2018 Springer Nature. | ||
7.2 Effect of lipid fluidity
The Brinker's group has pioneered the use of mesoporous silica nanoparticle supported lipid bilayers for targeted drug delivery.10 They fused liposomes onto mesoporous silica cores (Fig. 11a) followed by modification the supported bilayers with targeting peptides. The peptides on supported DOPC liposomes had 100-fold lower Kdversus the supported DPPC. This higher avidity was contributed by the enhanced lipid fluidity allowing dynamic organization of targeting peptides and a minimal number of targeting peptides can be used to achieve highly efficient binding (Fig. 11b). Such supported bilayers were used to deliver multiple classes of cargos, including small molecule drugs, siRNA, and toxin proteins to cells.139,140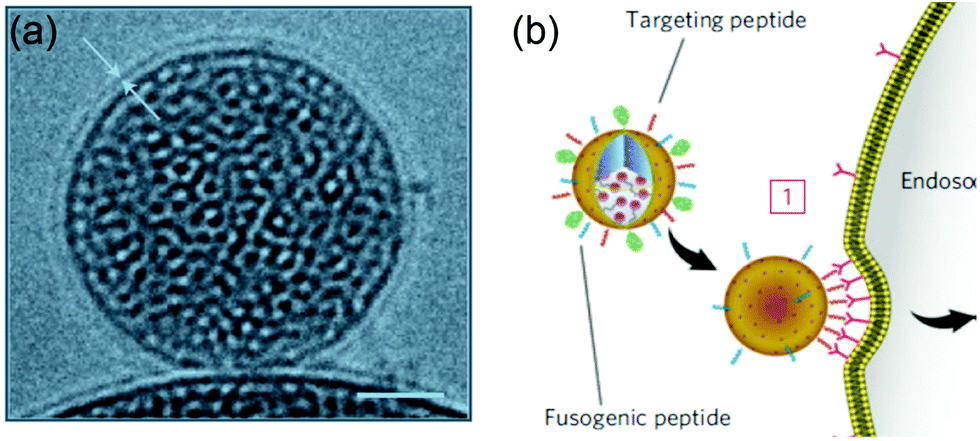 | ||
| Fig. 11 (a) Cryogenic TEM image of a lipid bilayer (∼4 nm thick) coated on a mesoporous silica nanoparticle. Scale bar = 25 nm. (b) Schematic illustration of a supported bilayer with targeting peptides binding to cells with high affinity owing to recruitment of targeting peptides to the cell surface. Adapted with permission from ref. 10. Copyright © 2011 Macmillan Publishers Limited. | ||
In addition to increasing binding affinity, the fluidity of lipid bilayers was also demonstrated to affect the colloidal stability of nanoparticles, which could be critical in achieving effective targeting. In another work from Brinker and coworkers, monodispersed supported bilayers were prepared that remained stable even in a complex media.141 The lipid fluidity effected colloidal stability along with other synthetic parameters including lipid:silica ratio, ionic strength, mesoporous silica nanoparticle size, shape and pore size, extent of PEGylation and surface display of targeting ligands. The size of both PEGylated and non-PEGylated DOPC-based supported bilayers increased progressively from 24 to 72 h in PBS buffer, while the DSPC-based formulation remained stable for >72 h under the same condition. This instability of the DOPC shell was attributed to the oxidation of double bond in the acyl chain in the unsaturated lipid as the DOPC-based formulation had long-term stability in an oxygen reduced buffer. Interestingly, the DOPC and DSPC based supported bilayers had identical colloidal stability in a complete cell medium, suggesting the adsorption of protein provided additional barriers against aggregation.
8. Some successful examples
The above sections have summarized the fundamental biophysical and nanoscience aspects in the field. Many liposomal formulations have been successfully commercialized and currently, thirteen Food and Drug Administration (FDA) approved liposome and lipid-based therapeutics are available, with more under clinical investigation.142 Among them, DOXIL is the first FDA approved nanomedicine. DOXIL has a size of 100 nm, a PEG surface that creates a “stealth” liposome, and uses remote loading that improves the drug loading efficiency.143 In addition, compared to free DOX, DOXIL results in 4–16 fold higher drug concentration in malignant effusions, demonstrating the EPR effect.142 DOXIL has been approved to treat AIDS-related Kaposi's Sarcoma (KS), as well as ovarian cancer, breast cancer, and multiple myeloma. Myocet is another DOX-based liposomal drug, a nonpegylated liposomal formulation with a size of 190 nm. Myocet has a t1/2 of 2.5 h compared to DOXIL's t1/2 of 55 h,144 possibly due to the increased liposome size. Myocet has been approved for the treatment of metastatic breast cancer in the EU and Canada.Besides DOX, other anticancer drugs have also been formulated for liposomal delivery. DaunoXome, which encapsulates daunorubicin, was developed using a nonpegylated liposome platform.145 It has been granted accelerated approval for KS treatment, although DaunoXome appeared to be less effective than DOXIL. Another nonpegylated liposomal vincristine formulated with egg sphingomyelin and cholesterol, Marqibo, has been approved for adult patients with Philadelphia chromosome-negative (Ph−) acute lymphoblastic leukemia (ALL) relapse.144 Lastly, DepoCyt, a multivesicular liposome platform encapsulating cytarabine, larger in size and with a slow release, has received accelerated approval for lymphomatous meningitis.146 In addition to the approved liposomal formulations, several are under clinical investigation, including liposomal cisplatin, liposomal irinotecan, and liposomal docetaxel.142
9. Summary and future perspectives
In summary, liposome-based drug delivery with an emphasis on the biophysical properties of lipids was reviewed. The self-assembly, soft and fluid nature of liposomes together with the variety of lipid composition makes it interesting and feasible to systematically tune their biophysical properties. This includes vesicle size, surface charge, membrane rigidity, headgroup dipole and lipid phase separation. Although fundamental biophysical studies of lipids and applied drug delivery studies are different research directions, as seen in this review, many papers have established connections between them. Both passive and active targeted delivery have been discussed. For active targeted delivery, the unique property of lipid fluidity allows lateral reorganization of targeting ligands to enhance binding affinity with cells. In addition, the membrane fusion property provides unique opportunities for drug delivery. Given the complexity of both liposomal formulation, as well as in vitro and in vivo biological environments, continued development of liposomal nanomedicine is expected.First, biophysical measurements such as surface charge, lateral diffusion and mechanical properties are typically done in simple buffers, while drug delivery has to experience much more complex environment and protein adsorption is a key difference. Often times if fluorescence microscopy is used (e.g. looking at phase separation and lipid diffusion), giant liposomes are needed. This is also different from the materials used for drug delivery. Such gaps exist in measurement and their influence needs to be better addressed. A more challenging direction is to use synthetic chemistry to systematically vary lipid properties beyond the current available ones and to examine their drug delivery properties along with measurement of biophysical properties. The study of the PC and CP (or CPe) liposomes is an interesting example. Further comparisons with SB and CB liposomes (Fig. 1l and m) can offer insights into the effect of both headgroup chemistry and polarity.51,52 Furthermore, an interesting research direction is to harness natural cell membranes to coat on inorganic or polymeric nanoparticle cores.8,147–150 Probing and tuning the biophysical properties of such complex system would be interesting to do. For example, adding new lipids to such membranes could be a useful way to perturb the system. Finally, understanding of the nanomedicine field is still rapidly involving. For example, the amount of drug accumulation at tumor sites is under debate.151,152 Even the EPR effect is currently being questioned. Since liposomes are already commercially used and they are highly tunable, we can expect systematic studies using liposomes to give more solid answers to these questions.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Funding for this work was from the Natural Sciences and Engineering Research Council of Canada (NSERC) and the Centre for Eye and Vision Research (CEVR).References
- R. Langer, Nature, 1998, 392, 5–10 CAS.
- O. C. Farokhzad and R. Langer, ACS Nano, 2009, 3, 16–20 CrossRef CAS.
- J. Liu, Q. Chen, L. Feng and Z. Liu, Nano Today, 2018, 21, 55–73 CrossRef CAS.
- W. H. D. Jong and P. J. Borm, Int. J. Nanomed., 2008, 3, 133–149 CrossRef.
- N. Feliu, D. Docter, M. Heine, P. d. Pino, S. Ashraf, J. Kolosnjaj-Tabi, P. Macchiarini, P. Nielsen, D. Alloyeau, F. Gazeau, R. H. Stauber and W. J. Parak, Chem. Soc. Rev., 2016, 45, 2440–2457 RSC.
- J. M. Anderson and M. S. Shive, Adv. Drug Delivery Rev., 2012, 64, 72–82 CrossRef.
- S. Tan, X. Li, Y. Guo and Z. Zhang, Nanoscale, 2013, 5, 860–872 RSC.
- W. Gao, C.-M. J. Hu, R. H. Fang and L. Zhang, J. Mater. Chem. B, 2013, 1, 6569–6585 RSC.
- J. Liu, Langmuir, 2016, 32, 4393–4404 CrossRef CAS.
- C. E. Ashley, E. C. Carnes, G. K. Phillips, D. Padilla, P. N. Durfee, P. A. Brown, T. N. Hanna, J. Liu1, B. Phillips, M. B. Carter, N. J. Carroll, X. Jiang, D. R. Dunphy, C. L. Willman, D. N. Petsev, D. G. Evans, A. N. Parikh, B. Chackerian, W. Wharton, D. S. Peabody and C. J. Brinker, Nat. Mater., 2011, 10, 389–397 CrossRef CAS.
- C. Peetla, S. Vigayaraghavalu and V. Labhasetwar, Adv. Drug Delivery Rev., 2013, 65, 1686–1698 CrossRef CAS.
- C. Peetla, A. Stine and V. Labhasetwar, Mol. Pharmaceutics, 2009, 6, 1264–1276 CrossRef CAS.
- M. Deleu, J.-M. Crowet, M. N. Nasir and L. Lins, Biochim. Biophys. Acta, Biomembr., 2014, 1838, 3171–3190 CrossRef CAS.
- R. Friedman, S. Khalid, C. Aponte-Santamaría, E. Arutyunova, M. Becker, K. J. Boyd, M. Christensen, J. T. S. Coimbra, S. Concilio, C. Daday, F. J. v. Eerden, P. A. Fernandes, F. Gräter, D. Hakobyan, A. Heuer, K. Karathanou, F. Keller, M. J. Lemieux, S. J. Marrink, E. R. May, A. Mazumdar, R. Naftalin, M. Pickholz, S. Piotto, P. Pohl, P. Quinn, M. J. Ramos, B. Schiøtt, D. Sengupta, L. Sessa, S. Vanni, T. Zeppelin, V. Zoni, A.-N. Bondar and C. Domene, J. Membr. Biol., 2018, 251, 609–631 CrossRef CAS.
- A. E. Nel, L. Mädler, D. Velegol, T. Xia, E. M. V. Hoek, P. Somasundaran, F. Klaessig, V. Castranova and M. Thompson, Nat. Mater., 2009, 8, 543–557 CrossRef CAS.
- V. P. Torchilin, Nat. Rev. Drug Discovery, 2005, 4, 145–160 CrossRef CAS.
- D. C. Drummond, O. Meyer, K. Hong, D. B. Kirpotin and D. Papahadjopoulos, Pharmacol. Rev., 1999, 51, 692–743 Search PubMed.
- T. M. Allen and P. R. Cullis, Adv. Drug Delivery Rev., 2013, 65, 36–48 CrossRef CAS.
- G. T. Noble, J. F. Stefanick, J. D. Ashley, T. Kiziltepe and B. Bilgicer, Trends Biotechnol., 2014, 32, 32–45 CrossRef CAS.
- B. S. Pattni, V. V. Chupin and V. P. Torchilin, Chem. Rev., 2015, 115, 10938–10966 CrossRef CAS.
- G. v. Meer, D. R. Voelker and G. W. Feigenson, Nat. Rev. Mol. Cell Biol., 2008, 9, 112–124 CrossRef.
- E. T. Castellana and P. S. Cremer, Surf. Sci. Rep., 2006, 61, 429–444 CrossRef CAS.
- D. L. Gettel, J. Sanborn, M. A. Patel, H.-P. d. Hoog, B. Liedberg, M. Nallani and A. N. Parikh, J. Am. Chem. Soc., 2014, 136, 10186–10189 CrossRef CAS.
- R. Koynova and M. Caffrey, Biochim. Biophys. Acta, 1998, 1376, 91–145 CrossRef CAS.
- G. Cevc and D. Marsh, Phospholipid Bilayers, Wiley-Interscience, New York, 1987 Search PubMed.
- S. Chen, J. Zheng, L. Li and S. Jiang, J. Am. Chem. Soc., 2005, 127, 14473–14478 CrossRef CAS.
- J. B. Schlenoff, Langmuir, 2014, 30, 9625–9636 CrossRef CAS.
- H. M. Seeger, G. Marino, A. Alessandrini and P. Facci, Biophys. J., 2009, 97, 1067–1076 CrossRef CAS.
- G. Lindblom and G. Orädd, Biochim. Biophys. Acta, Biomembr., 2009, 1788, 234–244 CrossRef CAS.
- J. E. Vance and G. Tasseva, Biochim. Biophys. Acta, 2013, 1831, 543–554 CrossRef CAS.
- M. Raja, J. Membr. Biol., 2011, 242, 137–143 CrossRef CAS.
- N. Kučerka, M.-P. Nieh and J. Katsaras, Biochim. Biophys. Acta, Biomembr., 2011, 1808, 2761–2771 CrossRef.
- A. M. Sendecki, M. F. Poyton, A. J. Baxter, T. Yang and P. S. Cremer, Langmuir, 2017, 33, 13423–13429 CrossRef CAS.
- E. v. d. B.-v. d. Laan, J. A. Killian and B. d. Kruijff, Biochim. Biophys. Acta, 2004, 1666, 275–288 Search PubMed.
- J. G. Kay and G. D. Fairn, Cell Commun. Signaling, 2019, 17, 126 CrossRef.
- T. Skotland and K. Sandvig, Nat. Commun., 2019, 10, 2752 CrossRef.
- H. Antila, P. Buslaev, F. Favela-Rosales, T. M. Ferreira, I. Gushchin, M. Javanainen, B. Kav, J. J. Madsen, J. Melcr, M. S. Miettinen, J. Määttä, R. Nencini, O. H. S. Ollila and T. J. Piggot, J. Phys. Chem. B, 2019, 123, 9066–9079 CrossRef CAS.
- S. Nagata, J. Suzuki, K. Segawa and T. Fujii, Cell Death Differ., 2016, 23, 952–961 CrossRef CAS.
- I. Vermes, C. Haanen, H. Steffens-Nakken and C. Reutelingsperger, J. Immunol. Methods, 1995, 184, 39–51 CrossRef CAS.
- N. S. Templeton, D. D. Lasic, P. M. Frederik and G. N. Pavlakis, Nat. Biotechnol., 1997, 15, 647–652 CrossRef CAS.
- A. D. Miller, Angew. Chem., Int. Ed., 1998, 37, 1768–1785 CrossRef.
- C. Lonez, M. Vandenbranden and J.-M. Ruysschaert, Prog. Lipid Res., 2008, 47, 340–347 CrossRef CAS.
- E. J. Fernández-Pérez, F. J. Sepúlveda, C. Peters, D. Bascuñán, N. O. Riffo-Lepe, J. González-Sanmiguel, S. A. Sánchez, R. W. Peoples, B. Vicente and L. G. Aguayo, Front. Aging Neurosci., 2018, 10, 226 CrossRef.
- R. Dawaliby, C. Trubbia, C. Delporte, C. Noyon, J.-M. Ruysschaert, P. V. Antwerpen and C. Govaerts, J. Biol. Chem., 2016, 291, 3658–3667 CrossRef CAS.
- F. d. Meyer and B. Smit, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 3654–3658 CrossRef.
- E. K. Perttu, A. G. Kohli and J. F. C. Szoka, J. Am. Chem. Soc., 2012, 134, 4485–4488 CrossRef CAS.
- Y. Liu, F. Wang and J. Liu, Langmuir, 2018, 34, 9337–9348 CrossRef CAS.
- S. Li, F. Wang, X. Li, J. Chen, X. Zhang, Y. Wang and J. Liu, ACS Appl. Mater. Interfaces, 2017, 9, 17736–17744 CrossRef CAS.
- W. Wang, B. Wang, S. Liu, X. Shang, X. Yan, Z. Liu, X. Ma and X. Yu, ACS Appl. Mater. Interfaces, 2017, 9, 15986–15994 CrossRef CAS.
- X. Yu, Z. Liu, J. Janzen, I. Chafeeva, S. Horte, W. Chen, R. K. Kainthan, J. N. Kizhakkedathu and D. E. Brooks, Nat. Mater., 2012, 11, 468–476 CrossRef CAS.
- T. Aikawa, K. Yokota, T. Kondo and M. Yuasa, Langmuir, 2016, 32, 10483–10490 CrossRef CAS.
- T. Aikawa, H. Okura, T. Kondo and M. Yuasa, ACS Omega, 2017, 2, 5803–5812 CrossRef CAS.
- Y. Du, W. He, Q. Xia, W. Zhou, C. Yao and X. Li, ACS Appl. Mater. Interfaces, 2019, 11, 37411–37420 CrossRef CAS.
- L. A. Meure, N. R. Foster and F. Dehghani, AAPS PharmSciTech, 2008, 9, 798–809 CrossRef CAS.
- D. Zucker, D. Marcus, Y. Barenholz and A. Goldblum, J. Controlled Release, 2009, 139, 73–80 CrossRef CAS.
- W. Zhang, G. Wang, J. R. Falconer, B. C. Baguley, J. P. Shaw, J. Liu, H. Xu, E. See, J. Sun, J. Aa and Z. Wu, Pharm. Res., 2015, 32, 1451–1461 CrossRef CAS.
- N. Dimov, E. Kastner, M. Hussain, Y. Perrie and N. Szita, Sci. Rep., 2017, 7, 12045 CrossRef.
- K. Otake, T. Shimomura, T. Goto, T. Imura, T. Furuya, S. Yoda, Y. Takebayashi, H. Sakai and M. Abe, Langmuir, 2006, 22, 2543–2550 CrossRef CAS.
- F. Szoka, Jr. and D. Papahadjopoulos, Proc. Natl. Acad. Sci. U. S. A., 1978, 75, 4194–4198 CrossRef.
- B. Zadi and G. Gregoriadis, J. Liposome Res., 2000, 10, 73–80 CrossRef CAS.
- J. O. Eloy, M. C. d. Souza, R. Petrilli, J. P. A. Barcellos, R. J. Lee and J. M. Marchetti, Colloids Surf., B, 2014, 123, 345–363 CrossRef CAS.
- D. C. Drummond, C. O. Noble, Z. Guo, K. Hong, J. W. Park and D. B. Kirpotin, Cancer Res., 2006, 66, 3271–3277 CrossRef CAS.
- H. Tamam, J. Park, H. H. Gadalla, A. R. Masters, J. A. Abdel-Aleem, S. I. Abdelrahman, A. A. Abdelrahman, L. T. Lyle and Y. Yeo, Mol. Pharmaceutics, 2019, 16, 2858–2871 CrossRef CAS.
- A. Nagayasu, K. Uchiyama and H. Kiwada, Adv. Drug Delivery Rev., 1999, 40, 75–87 CrossRef CAS.
- M. E. Davis, Z. G. Chen and D. M. Shin, Nat. Rev. Drug Discovery, 2008, 7, 771–782 CrossRef CAS.
- J. W. Nichols and Y. H. Bae, J. Controlled Release, 2014, 190, 451–464 CrossRef CAS.
- F. Danhier, J. Controlled Release, 2016, 244, 108–121 CrossRef CAS.
- F. Yuan, M. Dellian, D. Fukumura, M. Leunig, D. A. Berk, V. P. Torchilin and R. K. Jain, Cancer Res., 1995, 55, 3752–3756 CAS.
- M. Longmire, P. L. Choyke and H. Kobayashi, Nanomedicine, 2008, 3, 703–717 CrossRef CAS.
- T. M. Allen and A. Chonn, FEBS Lett., 1987, 223, 42–46 CrossRef CAS.
- A. Gabizon and D. Papahadjopoulos, Proc. Natl. Acad. Sci. U. S. A., 1988, 85, 6949–6953 CrossRef CAS.
- N. D. Santos, C. Allen, A.-M. Doppen, M. Anantha, K. A. K. Cox, R. C. Gallagher, G. Karlsson, K. Edwards, G. Kenner, L. Samuels, M. S. Webb and M. B. Bally, Biochim. Biophys. Acta, 2007, 1768, 1367–1377 CrossRef.
- Q. Shao and S. Jiang, Adv. Mater., 2015, 27, 15–26 CrossRef CAS.
- T. S. Levchenko, R. Rammohan, A. N. Lukyanov, K. R. Whiteman and V. P. Torchilin, Int. J. Pharm., 2002, 240, 95–102 CrossRef CAS.
- H. S. S. Qhattal, T. Hye, A. Alali and X. Liu, ACS Nano, 2014, 8, 5423–5440 CrossRef CAS.
- J. A. Kulkarni, P. R. Cullis and R. v. d. Meel, Nucleic Acid Ther., 2018, 28, 146–157 CrossRef CAS.
- E. Blanco, H. Shen and M. Ferrari, Nat. Biotechnol., 2015, 33, 941–951 CrossRef CAS.
- F. Wang and J. Liu, Nanoscale, 2015, 7, 15599–15604 RSC.
- A. Blicher, K. Wodzinska, M. Fidorra, M. Winterhalter and T. Heimburg, Biophys. J., 2009, 96, 4581–4591 CrossRef CAS.
- Z. Al-Ahmady and K. Kostarelos, Chem. Rev., 2016, 116, 3883–3918 CrossRef CAS.
- M. Loew, R. Springer, S. Scolari, F. Altenbrunn, O. Seitz, J. Liebscher, D. Huster, A. Herrmann and A. Arbuzova, J. Am. Chem. Soc., 2010, 132, 16066–16072 CrossRef CAS.
- N. Dave and J. Liu, Chem. Commun., 2012, 48, 3718–3720 RSC.
- Q. Luo, Z. Shi, Y. Zhang, X.-J. Chen, S.-Y. Han, T. Baumgart, D. M. Chenoweth and S.-J. Park, J. Am. Chem. Soc., 2016, 138, 10157–10162 CrossRef CAS.
- M. Ma, Y. Gong and D. Bong, J. Am. Chem. Soc., 2009, 131, 16919–16926 CrossRef CAS.
- L. Wu, H. Ding, X. Qu, X. Shi, J. Yang, M. Huang, J. Zhang, H. Zhang, J. Song, L. Zhu, Y. Song, Y. Ma and C. Yang, J. Am. Chem. Soc., 2020, 142, 4800–4806 CrossRef CAS.
- J. Bompard, A. Rosso, L. Brizuela, S. Mebarek, L. J. Blum, A.-M. Trunfio-Sfarghiu, G. Lollo, T. Granjon, A. s. Girard-Egrot and O. Maniti, Langmuir, 2020, 36, 5134–5144 CrossRef CAS.
- I. Bacia, P. Schwille and T. Kurzchalia, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 3272–3277 CrossRef.
- T. Baumgart, S. T. Hess and W. W. Webb, Nature, 2003, 425, 821–824 CrossRef CAS.
- Z. Dai, M. Yu, X. Yi, Z. Wu, F. Tian, Y. Miao, W. Song, S. He, E. Ahmad, S. Guo, C. Zhu, X. Zhang, Y. Li, X. Shi, R. Wang and Y. Gan, ACS Nano, 2019, 13, 7676–7689 CrossRef CAS.
- M. Doktorova, D. Harries and G. Khelashvili, Phys. Chem. Chem. Phys., 2017, 19, 16806–16818 RSC.
- H. A.-M. D. d. Silva, M. Poley, A. Zinger, E. Goldman, N. Krinsky, R. Kleiner, G. Shenbach, J. E. Schroeder, J. Shklover, J. Shainsky-Roitman and A. Schroeder, J. Controlled Release, 2019, 307, 331–341 CrossRef.
- Y. Takechi-Haraya, Y. Goda and K. Sakai-Kato, Mol. Pharmaceutics, 2017, 14, 2158–2165 CrossRef CAS.
- L. V. Chernomordik and M. M. Kozlov, Nat. Struct. Mol. Biol., 2008, 15, 675–683 CrossRef CAS.
- G. Stengel, R. Zahn and F. Höök, J. Am. Chem. Soc., 2007, 129, 9584–9585 CrossRef CAS.
- Y.-H. M. Chan, B. v. Lengerich and S. G. Boxer, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 979–984 CrossRef CAS.
- Y.-H. M. Chan, B. v. Lengerich and S. G. Boxer, Biointerphases, 2008, 3, FA17–FA21 CrossRef CAS.
- L. Kong, S. H. C. Askes, S. Bonnet, A. Kros and F. Campbell, Angew. Chem., Int. Ed., 2016, 55, 1396–1400 CrossRef CAS.
- F. Versluis, J. Voskuhl, B. v. Kolck, H. Zope, M. Bremmer, T. Albregtse and A. Kros, J. Am. Chem. Soc., 2013, 135, 8057–8062 CrossRef CAS.
- H. R. Marsden, N. A. Elbers, P. H. H. Bomans, N. A. J. M. Sommerdijk and A. Kros, Angew. Chem., Int. Ed., 2009, 48, 2330–2333 CrossRef.
- D. Dutta, A. Pulsipher, W. Luo, H. Mak and M. N. Yousaf, Bioconjugate Chem., 2011, 22, 2423–2433 CrossRef CAS.
- A. Pulsipher, D. Dutta, W. Luo and M. N. Yousaf, Angew. Chem., Int. Ed., 2014, 53, 9487–9492 CrossRef CAS.
- D. Dutta, A. Pulsipher, W. Luo and M. N. Yousaf, J. Am. Chem. Soc., 2011, 133, 8704–8713 CrossRef CAS.
- T. C. Südhof and J. E. Rothman, Science, 2009, 323, 474–477 CrossRef.
- J. Yang, A. Bahreman, G. Daudey, J. Bussmann, R. C. Olsthoorn and A. Kros, ACS Cent. Sci., 2016, 2, 621–630 CrossRef CAS.
- J. Yang, J. Tu, G. E. M. Lamers, R. C. L. Olsthoorn and A. Kros, Adv. Healthcare Mater., 2017, 6, 1700759 CrossRef.
- M. F. Attia, N. Anton, J. Wallyn, Z. Omran and T. F. Vandamme, J. Pharm. Pharmacol., 2019, 71, 1185–1198 CrossRef CAS.
- V. Torchilin, Adv. Drug Delivery Rev., 2011, 63, 131–135 CrossRef CAS.
- R. Ngoune, A. Peters, D. v. Elverfeldt, K. Winkler and G. Pütz, J. Controlled Release, 2016, 238, 58–70 CrossRef CAS.
- P. P. Deshpande, S. Biswas and V. P. Torchilin, Nanomedicine, 2013, 8, 1509–1528 CrossRef CAS.
- G. Kibria, H. Hatakeyama, N. Ohga, K. Hida and H. Harashima, Biomaterials, 2013, 34, 5617–5627 CrossRef CAS.
- H. Ren, Y. He, J. Liang, Z. Cheng, M. Zhang, Y. Zhu, C. Hong, J. Qin, X. Xu and J. Wang, ACS Appl. Mater. Interfaces, 2019, 11, 20304–20315 CrossRef CAS.
- X. Li, Y. Zhao, W. Jiang, S. Li, M. Zhan, H. Liu, C. Zhang, H. Liang, H. Liu, L. Lu and Y. Wang, Biomater. Sci., 2019, 7, 1335–1344 RSC.
- K. Simons and W. L. C. Vaz, Annu. Rev. Biophys. Biomol. Struct., 2004, 33, 269–295 CrossRef CAS.
- M. R. Krause and S. L. Regen, Acc. Chem. Res., 2014, 47, 3512–3521 CrossRef CAS.
- D. E. Vance and J. E. Vance, Biochemistry of Lipids, Lipoproteins and Membranes, Elservier, Amsterdam, 1996 Search PubMed.
- A. L. Klibanov, K. Maruyama, V. P. Torchilin and L. Huang, FEBS Lett., 1990, 268, 235–237 CrossRef CAS.
- D. Papahadjopoulos, T. M. Allen, A. Gabizon, E. Mayhew, K. Matthay, S. K. Huang, K.-D. Lee, M. C. Woodle, D. D. Lasic, C. Redemann and F. J. Martin, Proc. Natl. Acad. Sci. U. S. A., 1991, 88, 11460–11464 CrossRef CAS.
- J. F. Stefanick, J. D. Ashley, T. Kiziltepe and B. Bilgicer, ACS Nano, 2013, 7, 2935–2947 CrossRef CAS.
- T. M. Allen, C. Hansen, F. Martin, C. Redemann and A. Yau-young, Biochim. Biophys. Acta, Biomembr., 1991, 1066, 29–36 CrossRef CAS.
- D. Pozzi, V. Colapicchioni, G. Caracciolo, S. Piovesana, A. L. Capriotti, S. Palchetti, S. D. Grossi, A. Riccioli, H. Amenitschf and A. Laganà, Nanoscale, 2014, 6, 2782–2792 RSC.
- T. Ishida, M. Ichihara, X. Wang and H. Kiwada, J. Controlled Release, 2006, 115, 243–250 CrossRef CAS.
- X. Wang, T. Ishida and H. Kiwada, J. Controlled Release, 2007, 119, 236–244 CrossRef CAS.
- Z. I. Imam, L. E. Kenyon, G. Ashby, F. Nagib, M. Mendicino, C. Zhao, A. K. Gadok and J. C. Stachowiak, Cell. Mol. Bioeng., 2017, 10, 387–403 CrossRef CAS.
- J. F. Stefanick, J. D. Ashley and B. Bilgicer, ACS Nano, 2013, 7, 8115–8127 CrossRef CAS.
- M. Hadjidemetriou, Z. Al-Ahmady, M. Mazza, R. F. Collins, K. Dawson and K. Kostarelos, ACS Nano, 2015, 9, 8142–8156 CrossRef CAS.
- G. Caracciolo, D. Pozzi, A. L. Capriotti, C. Cavaliere, S. Piovesana, G. La Barbera, A. Amici and A. Laganà, J. Mater. Chem. B, 2014, 2, 7419–7428 RSC.
- A. Bigdeli, S. Palchetti, D. Pozzi, M. R. Hormozi-Nezhad, F. B. Bombelli, G. Caracciolo and M. Mahmoudi, ACS Nano, 2016, 10, 3723–3737 CrossRef CAS.
- G. Caracciolo, S. Palchetti, L. Digiacomo, R. Z. Chiozzi, A. L. Capriotti, H. Amenitsch, P. M. Tentori, V. Palmieri, M. Papi, F. Cardarelli, D. Pozzi and A. Laganà, ACS Appl. Mater. Interfaces, 2018, 10, 22951–22962 Search PubMed.
- M. Hadjidemetriou, Z. Al-Ahmady and K. Kostarelos, Nanoscale, 2016, 8, 6948–6957 RSC.
- J. Guan, Q. Shen, Z. Zhang, Z. Jiang, Y. Yang, M. Lou, J. Qian, W. Lu and C. Zhan, Nat. Commun., 2018, 9, 2982 CrossRef.
- J. Guan, Z. Jiang, M. Wang, Y. Liu, J. Liu, Y. Yang, T. Ding, W. Lu, C. Gao, J. Qian and C. Zhan, Mol. Pharmaceutics, 2019, 16, 907–913 CrossRef CAS.
- Z. Zhang, J. Guan, Z. Jiang, Y. Yang, J. Liu, W. Hua, Y. Mao, C. Li, W. Lu, J. Qian and C. Zhan, Nat. Commun., 2019, 10, 3561 CrossRef.
- J. Yang, Y. Shimada, R. C. L. Olsthoorn, E. Snaar-Jagalska, H. P. Spaink and A. Kros, ACS Nano, 2016, 10, 7428–7435 CrossRef CAS.
- H. Deng, K. Song, X. Zhao, Y. Li, F. Wang, J. Zhang, A. Dong and Z. Qin, ACS Appl. Mater. Interfaces, 2017, 9, 9315–9326 CrossRef CAS.
- J. P. M. Motion, J. Nguyen and F. C. Szoka, Angew. Chem., Int. Ed., 2012, 51, 9047–9051 CrossRef CAS.
- L. Zhang, Q. Feng, J. Wang, S. Zhang, B. Ding, Y. Wei, M. Dong, J.-Y. Ryu, T.-Y. Yoon, X. Shi, J. Sun and I. Jiang, ACS Nano, 2015, 9, 9912–9921 CrossRef CAS.
- J. Sun, L. Zhang, J. Wang, Q. Feng, D. Liu, Q. Yin, D. Xu, Y. Wei, B. Ding, X. Shi and X. Jiang, Adv. Mater., 2015, 27, 1402–1407 CrossRef CAS.
- P. Guo, D. Liu, K. Subramanyam, B. Wang, J. Yang, J. Huang, D. T. Auguste and M. A. Moses, Nat. Commun., 2018, 9, 130 CrossRef.
- K. Epler, D. Padilla, G. Phillips, P. Crowder, R. Castillo, D. Wilkinson, B. Wilkinson, C. Burgard, R. Kalinich, J. Townson, B. Chackerian, C. Willman, D. Peabody, W. Wharton, C. J. Brinker, C. Ashley and E. Carnes, Adv. Healthcare Mater., 2012, 1, 348–353 CrossRef CAS.
- C. E. Ashley, E. C. Carnes, K. E. Epler, D. P. Padilla, G. K. Phillips, R. E. Castillo, D. C. Wilkinson, B. S. Wilkinson, C. A. Burgard, R. M. Kalinich, J. L. Townson, B. Chackerian, C. L. Willman, D. S. Peabody, W. Wharton and C. J. Brinker, ACS Nano, 2012, 6, 2174–2188 CrossRef CAS.
- P. N. Durfee, Y.-S. Lin, D. R. Dunphy, A. e. J. Muñiz, K. S. Butler, K. R. Humphrey, A. J. Lokke, J. O. Agola, S. S. Chou, I.-M. Chen, W. Wharton, J. L. Townson, C. L. Willman and C. J. Brinker, ACS Nano, 2016, 10, 8325–8345 CrossRef CAS.
- Y. Min, J. M. Caster, M. J. Eblan and A. Z. Wang, Chem. Rev., 2015, 115, 11147–11190 CrossRef CAS.
- Y. C. Barenholz, J. Controlled Release, 2012, 160, 117–134 CrossRef CAS.
- H.-I. Chang and M.-K. Yeh, Int. J. Nanomed., 2012, 7, 49–60 CAS.
- P. Guaglianone, K. Chan, E. DelaFlor-Weiss, R. Hanisch, S. Jeffers, D. Sharma and F. Muggia, Invest. New Drugs, 1994, 12, 103–110 CrossRef CAS.
- M. S. Angst and D. R. Drover, Clin. Pharmacokinet., 2006, 45, 1153–1176 CrossRef CAS.
- Y. Jiang, N. Krishnan, J. Zhou, S. Chekuri, X. Wei, A. V. Kroll, C. L. Yu, Y. Duan, W. Gao, R. H. Fang and L. Zhang, Adv. Mater., 2020, 32, 2001808 CrossRef CAS.
- Q. Zhang, R. H. Fang, W. Gao and L. Zhang, Angew. Chem., Int. Ed., 2020, 59, 10461–10465 CrossRef CAS.
- J. Zhuang, Y. Duan, Q. Zhang, W. Gao, S. Li, R. H. Fang and L. Zhang, Nano Lett., 2020, 20, 4051–4058 CrossRef CAS.
- S. Liu, Y. Zhang, M. Li, L. Xiong, Z. Zhang, X. Yang, X. He, K. Wang, J. Liu and S. Mann, Nat. Chem., 2020, 12, 1165–1173 CrossRef.
- S. Wilhelm, A. J. Tavares, Q. Dai, S. Ohta, J. Audet, H. F. Dvorak and W. C. W. Chan, Nat. Rev. Mater., 2016, 1, 16014 CrossRef CAS.
- Y.-H. Cheng, C. He, J. E. Riviere, N. A. Monteiro-Riviere and Z. Lin, ACS Nano, 2020, 14, 3075–3095 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2021 |