
Liquid–liquid interfaces: a unique and advantageous environment to prepare and process thin films of complex materials

Abstract
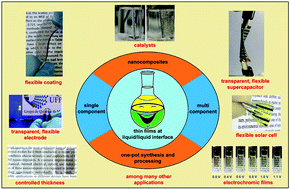
Thin film technology is pervasive for many fields with high impact in our daily lives, which makes processing materials such as thin films a very important subject in materials science and technology. However, several paramount materials cannot be prepared as thin films through the well-known and consolidated deposition routes, which strongly limits their applicability. This is particularly noticeable for multi-component and complex nanocomposites, which present unique properties due to the synergic effect between the components, but have several limitations to be obtained as thin films, mainly if homogeneity and transparence are required. This review highlights the main advances of a novel approach to both process and synthesize different classes of materials as thin films, based on liquid/liquid interfaces. The so-called liquid/liquid interfacial route (LLIR) allows the deposition of thin films of single- or multi-component materials, easily transferable over any kind of substrate (plastics and flexible substrates included) with precise control of the thickness, homogeneity and transparence. More interesting, it allows the in situ synthesis of multi-component materials directly as thin films stabilized at the liquid/liquid interface, in which problems related to both the synthesis and processing are solved together in a single step. This review presents the basis of the LLIR and several examples of thin films obtained from different classes of materials, such as carbon nanostructures, metal and oxide nanoparticles, two-dimensional materials, organic and organometallic frameworks, and polymer-based nanocomposites, among others. Moreover, specific applications of those films in different technological fields are shown, taking advantage of the specific properties emerging from the unique preparation route.
- This article is part of the themed collections: Materials Horizons 10th anniversary regional spotlight collection: The Americas and 2021 Materials Horizons Advisory Board collection
 Please wait while we load your content...
Please wait while we load your content...

