
A self-reinforcing and self-healing elastomer with high strength, unprecedented toughness and room-temperature reparability†
Yuhan
Li
a,
Wenjuan
Li
a,
Ailing
Sun
a,
Mengfan
Jing
b,
Xingjiang
Liu
 a,
Liuhe
Wei
*a,
Kai
Wu
*c and
Qiang
Fu
*c
a,
Liuhe
Wei
*a,
Kai
Wu
*c and
Qiang
Fu
*c
aCollege of Chemistry and Green Catalysis Center, Zhengzhou Key Laboratory of Elastic Sealing Materials, Zhengzhou University, Zhengzhou 450001, China. E-mail: weiliuhe@zzu.edu.cn
bKey Laboratory of Materials Processing and Mold, National Engineering Research Center for Advanced Polymer Processing Technology, Zhengzhou University, Zhengzhou 450001, China
cCollege of Polymer Science and Engineering, State Key Laboratory of Polymer Materials and Engineering, Sichuan University, Chengdu 610065, China. E-mail: kaiwu@scu.edu.cn; qiangfu@scu.edu.cn
First published on 2nd November 2020
Abstract
The development of intrinsic self-healing elastomers with simultaneous high mechanical strength, toughness and room-temperature reparability remains a formidable challenge. Herein, we report a mechano-responsive strategy, known as strain induced crystallization, to address the above issue, whereby synthesized elastomers with unprecedented high mechanical performances are bestowed with room-temperature self-healing materials, achieving tensile strength, toughness and fracture energy values of 29.0 MPa, 121.8 MJ m−3 and 104.1 kJ m−2, respectively.
New conceptsHigh molecular rigidity and bonding interactions for strength are inherently contradictory with favorable molecular arrangement or bonding dissociation for stretchability, and high mobility and exchange of reversible bonds for self-healing, resulting in room-temperature self-healing elastomers suffering from limited mechanical enhancements, i.e. tensile strength <15 MPa, toughness <80 MJ m−3 or fracture energy <50 kJ m−2. Herein, in contrast to conventional compromised strategies, we report a slow but reversible self-reinforcing effect based on strain-induced crystallization (SIC), in order to engineer a novel polyurethane material, which for the first-time behaves with unprecedented high mechanical performance compared to all of the room-temperature self-healing materials, with tensile strength, toughness and fracture energies up to 29.0 MPa, 121.8 MJ m−3 and 104.1 kJ m−2, respectively. Its superior performances can be attributed to rational optimization of the effective soft segment, and the elaborate design of loosely packed and hierarchical H-bonding interactions by the following: (i) a crystallizable soft segment, with a suitable effective length, provides a much lower crystallization energy barrier during the uniaxial stretching process, and this self-reinforces the elastomers while keeping its viscoelasticity at its original or retracting state; (ii) non-covalent hydrogen-bonding interactions with a low binding energy in the hard domains serve as sacrificial and dynamic bonds, not only for dissipating stress energy, maintaining the crystalline configuration, but also for offering effective exchange of dynamic bonds upon being damaged. |
1. Introduction
Self-healing materials, capable of spontaneously repairing physical damages and recovering mechanical performance in order to prolong their lifespan, have promising vital applications in cutting-edge modern technologies, including in wearable electronics, energy conversion devices, robotics, sensors and protective coatings, etc.1,2 Whilst extrinsic self-healing materials have been extensively investigated for decades, they are inherently imperfect at exerting a durable healing function due to the consumption of healing reagents. Currently, attention into self-healing materials has been focused on developing intrinsic pathways through reversible covalent bonds, such as with Diels–Alder (DA) reactions,3,4 disulfide bonds,5–7 diselenide bonds,8 oxime–carbamate bonds,9,10 B–O bonds,11 hindered urea bonds,12 phenol–carbamate bonds,13 acylhydrazone bonds,14 reversible C–C bonds,15 imine bonds,16 or noncovalent interactions such as hydrogen bonds (H-bonds),17–22 coordination bonds,23,24 π–π stacking interactions,25 ionic interactions,26,27 and host–guest interactions.28 However, simply utilizing these dynamic bonds to confer the ability to self-heal, regardless of mechanical performance and healing conditions, hardly provides impetus for their use in widespread applications. For instance, strong and tough self-healing materials are essential to ensure functional reliability and durability of industrial products, and extra stimuli such as heat or light are inconvenient because of their unavailability at ambient conditions in real life. Therefore, it is highly desirable to explore novel materials that can be automatically healed at room temperature and that exhibit simultaneous high strength and great toughness.However, high chain rigidity, intermolecular interactions and crystallinity to improve the strength contradict with favorable chain arrangement and the dissociation of bonds for stretchability, as well as high chain mobility and the exchange of dynamic bonds for self-healing. Therefore, simultaneous strengthening and toughening of room-temperature self-healing materials is always a formidable challenge. Previously, several academics have proposed ingenious tactics to address this issue. For example, an asymmetric alicyclic structure adjacent to an aromatic disulfide constructed hard segment was tactfully tailored, in which a loosely-packed configuration derived from the asymmetric structure provided optimal exchange efficiency for reversible disulfide bonds, yielding a simultaneous achievement of high tensile strength (6.8 MPa), toughness (26.9 MJ m−3) and quick self-healing at room-temperature.7 Another enlightening study reported a “dynamic hard domain” strategy to balance the self-healing property, mechanical strength and toughness; this included the features of a loosely-packed structure, a lower binding energy with high mobility and sequential dissociation with fast rearrangement, resulting in a high tensile strength of ∼5 MPa, a toughness of 65.49 MJ m−3 and good room-temperature repairability.18 Recently, previous high recordings have even been consistently broken by adopting a “synergistic triple dynamic bonds” method, in which the coordination of Cu2+ exerts the key effect of reducing the energy barrier for dynamic dissociation–association of oxime–carbamate, therefore a mechanical tensile strength and toughness up to 14.8 MPa and 87.0 MJ m−3, respectively, could be obtained for a room-temperature self-healing chemically-crosslinked elastomer.9 Although these satisfactory progresses have already been achieved, the mechanical performance, typically the tensile strength, still suffers from limited enhancement (<15 MPa), due to the inevitable trade-off between the high bonding energy for strength, and efficient chain diffusion and exchange of the reversible bonds for the ability to self-heal. Generally, a room-temperature self-healing elastomer with a high toughness value, large deformability and, more importantly, plastic-like strength (i.e. polyprene, ≈30 MPa) still attracts research attention and is highly pursued, i.e. in applications of puncture-resistant tire sealants of military-grade automobiles, tough and resilient sports-ware materials, durable protective coatings, and self-sealing elastic materials of pressured inflatable or hazardous liquid containers. This aim requires original innovation to provide further delicate regulation of the dynamic topological structures of artificial elastomers.
Natural rubber undergoes typical strain-induced crystallization (SIC), which can be attributed to the aggregation state transformation of cis-1,4-polyisoprene chains upon unidirectional deformation, and thus can exhibit a simultaneous strengthening and toughening behavior, but also remain soft and extremely stretchable.29,30 With this in mind, herein, a slow but reversible self-reinforcing strategy based on SIC is adopted to develop a mechanically strong, highly stretchable, unprecedentedly tough and room-temperature self-repairable thermoplastic polyurethane elastomer. More precisely, its superior comprehensive performances are attributed to rational optimization of the effective soft chain length and elaborate design of the hierarchical H-bonding interactions in loosely-packed hard domains by the following: (i) polytetramethylene ether glycol (PTMEG), a crystallizable soft segment, with suitable effective length, provides a much lower crystallization energy barrier during the uniaxial stretching process to enable a lagging but reversible SIC phenomenon, which could self-reinforce the elastomers following large deformations, while maintaining the random-coils and viscoelasticity at the original or retracting states; (ii) the co-operation of hierarchical H-bonding interactions as sacrificial and dynamic bonds, with low binding energy, in hard domains not only dissipates the stress energy and maintains the crystalline configuration, but it also offers the effective dynamic exchange of H-bonds upon damage. As a result, the synergistic effect of the dynamic topological structures produces unprecedented tensile strength (29.0 MPa), toughness (121.8 MJ m−3) and fracture energy (104.1 kJ m−2) among all room-temperature self-healing materials.
2. Results and discussion
2.1 Molecular design and mechanical properties
As seen in Fig. 1a, our self-healable elastomers were synthesized by a two-step prepolymer method from commercially available raw materials. Typically, PTMEG, a crystallizable soft segment, was mixed with R (molar ratio of isocyanate groups to hydroxyl groups of PTMEG, [NCO]/[OH]) stoichiometric equivalent weight of asymmetric and bulky isophorone diisocyanate (IPDI). The reaction was catalyzed by dibutyltin dilaurate (DBTDL) at 80 °C to obtain prepolymer (PTMEG capped with NCO) mixing with marginal IPDI. Toluene was added to obtain correct viscosity and to ensure fast kinetics of the reaction. 2,6-Pyridinedimethanol (PDM), a chain extender whose nitrogen atom in the pyridine ring can generate additional hierarchical H-bonding with carbamate, was introduced to react with both the prepolymer and marginal IDPI, resulting in linear polyurethane containing two distinct types of blocks. Altering the R value changes the ratio of y and z (as defined in Fig. 1a), and this actually tunes the total content of the hard segments and thus the dimensions of the hard domains to optimize the mechanical and healing properties. As illustrated in Fig. 1b, for a low R value, the hard segments are likely to assemble into small-sized hard domains through carbamate–carbamate and pyridine–carbamate H-bonding, while large-sized hard domains may be generated for a high R value. Based on this, a series of elastomers with R values varying from 2.1 to 2.7 were successfully synthesized by referring to the 1H nuclear magnetic resonance (1H NMR) (Fig. S4, ESI†), Fourier transform infrared spectroscopy (FTIR) (Fig. S5 and Table S1, ESI†) and gel permeation chromatography (GPC) data (Fig. S6 and Table S2, ESI†). To our surprise, the as-synthesized amorphous (differential scanning calorimetry (DSC) curves in Fig. S7 of the ESI†) and transparent (Fig. S8, ESI†) elastomer with the optimized R value of 2.5 exhibited extraordinary comprehensive properties, including simultaneous enhanced strength and toughness, and high self-healing efficiency (SE). For example, the elastomer is able to lift a heavy bucket (15.21 kg) whose weight is more than 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 times that of its own weight (Fig. 1c), and it can withstand a large-displacement needle puncture with a puncture energy of 788 mJ (Fig. 1d). More interestingly, optical microscopic images (Fig. 1e) demonstrate that PDM-2.5 is also healable at room-temperature, and this could spontaneously repair the cut gap after 24 h at 25 °C. To the best of our knowledge, this room-temperature self-healing elastomer with tensile strength of 29.0 ± 0.9 MPa and toughness of 121.8 ± 8.5 MJ m−3 is unprecedented, and it even outperforms all of the other room-temperature self-healing materials recorded in the previous literature (Fig. 1f and Table S3 in the ESI†).
000 times that of its own weight (Fig. 1c), and it can withstand a large-displacement needle puncture with a puncture energy of 788 mJ (Fig. 1d). More interestingly, optical microscopic images (Fig. 1e) demonstrate that PDM-2.5 is also healable at room-temperature, and this could spontaneously repair the cut gap after 24 h at 25 °C. To the best of our knowledge, this room-temperature self-healing elastomer with tensile strength of 29.0 ± 0.9 MPa and toughness of 121.8 ± 8.5 MJ m−3 is unprecedented, and it even outperforms all of the other room-temperature self-healing materials recorded in the previous literature (Fig. 1f and Table S3 in the ESI†).
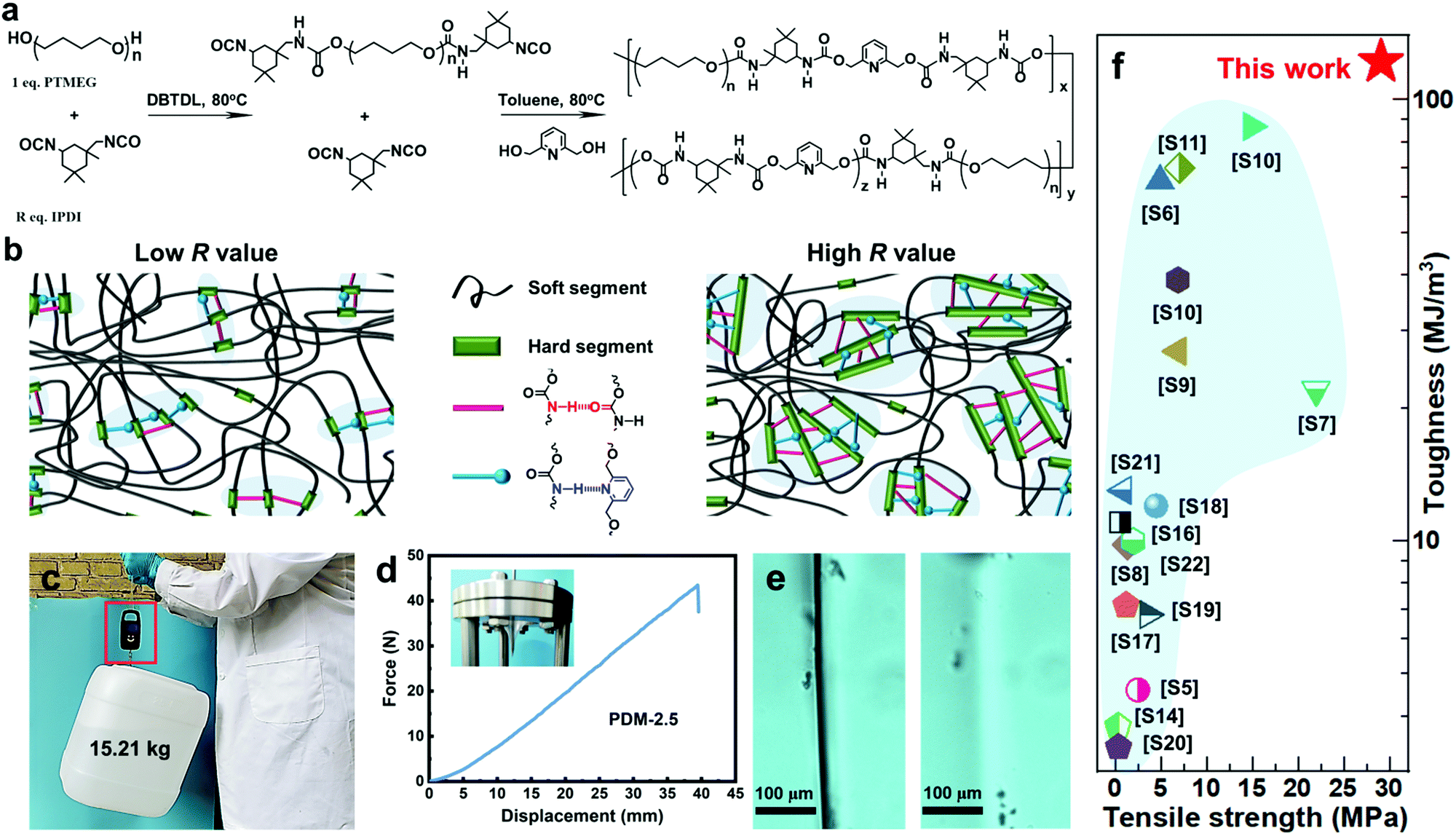 | ||
| Fig. 1 (a) Synthetic route of the PDM elastomers. (b) A sketch illustrating the aggregation of hard segments of a PDM elastomer with a low or high R value via hierarchical H-bonding assembly. (c) A 15.21 kg bucket lifted by a strip of the PDM-2.5 elastomer. (d) Force–displacement curve from a puncture test; the inset photo shows the puncture resistance of the PDM-2.5 elastomer by pressing the film against a needle. (e) Evolution of the cutting gap of the PMD-2.5 film before (left) and after (right) being healed at 25 °C for 24 h. (f) Comparison of tensile strength and toughness of our PDM-2.5 elastomer with other room-temperature self-healable materials (detailed data are listed in Table S3 in the ESI†). | ||
In order to analyze the mechanism, stress–strain curves of the polyurethane elastomers with various R values extended by PDM are compared in Fig. 2a. It is clear that the stretchability decreases with an increase in R value, while the ultimate tensile strength is augmented with increasing R value. The specific mechanical properties of these samples are listed in Table S4 (ESI†). In particular, both the PDM-2.5 and PDM-2.7 elastomers showed tremendous increases in tensile strength when the strain reached a certain range, and this is actually a typical sign of strain hardening. In contrast, the PDM-2.1 elastomer had a tensile yield point similar to the mechanical behavior of glassy polymers and its stress slightly decreased in the following strains until ∼2600%. As the R value increased to 2.3, the stress did not decrease, but it slowly increased in the following strain with 7.3 ± 0.4 MPa tensile strength, 1902 ± 38% elongation and 52.8 ± 4.0 MJ m−3 toughness. Although this is apparently strengthened compared to the PDM-2.1 elastomer, the sample seems to break right before the occurrence of strain hardening, signifying the importance of rational regulation of the R value. In order to better understand strain hardening to reveal the mechanical behavior in detail, the stress–strain curve of PDM-2.5 was taken as an example, and it was divided into three characteristic regimes, as illustrated in Fig. 2b. Regime 1 refers to the range in the strain with a slow increase of strength, and the strength changes in a quasi-linear way within a wide range of the strain. Regime 2 is the intermediate range of the strain that undergoes an incremental transition of the derivative stress (Fig. S9, ESI†), and Regime 3 is attributed to the range of strain that has a sharp quasi-linear increase of tensile strength, both of which are critically important for the ultimate mechanical properties and require in-depth analysis. According to the stress–strain curve and the derivative stress, a hump is present in Regime 1 that likely correlates to the rearrangement of the chain conformation, disentanglement and alignment of the soft segments, and evolution of the aggregation state. This conjecture is reasonable, because we found that the transparent sample gradually decreased in its transmittance with the appearance of distinguishable minute whitish spots as it was stretched to ∼500% (Fig. 2c). Upon further stretching to ∼1400% (Regime 3), the sample appeared even more whitish and the slope of the stress–strain curve was even larger. After being released for 1 h, the whitish spots disappeared and the sample was as transparent as in its original state. This phenomenon indicates that a certain phase with a distinct refractive index was formed during large deformations, and this is derived from the crystallization of soft segments between associative hard domains, like the SIC characteristic of natural rubber, and is responsible for the clearly improved strength. Wide-angle X-ray scattering (WAXS) tests were then performed to verify the SIC hypothesis. Fig. 2d shows the one-dimensional WAXS (1D WAXS) results of PDM-2.5 stretched to various strains. The accumulated data can be fit and split into two peaks for the curves of the samples stretched to 1200%, 1400% and 1600%. Peak 1 at the vector q = 13.5 nm−1 can be assigned to the (020) plane in the triclinic cell of the hard domains.31 It can be clearly seen that the shape and peak width at half height remain almost constant for 100%, 400% and 800% stretching (in Regime 1), while an additional shoulder peak appears at the vector q = 16.4 nm−1 (peak 2) as the strain increases to 1200%, 1400% and 1600% (Regime 2). Moreover, as the strain increases, the relative peak intensity and integrated area of the peak also exhibit a gradual increase. The two-dimensional WAXS (2D WAXS) images in Fig. 2e also validate the formation of a certain phase along the stretching direction, as a small spot (corresponding to vector q = 16.4 nm−1) emerges for the strains of 1200% and 1600%, which can be assigned to the (110) plane in a monoclinic cell of crystallized PTMEG chains.32 Fig. S10 (ESI†) further shows the reversibility of SIC induced PTMEG crystalline domains at room temperature. It also reveals that the stress whitening (Fig. 2c) is caused by SIC instead of by micro-voids such as glassy polymers. Polarized microscope observations further demonstrate this analysis, as blank regions of the original PDM-2.5 sample under polarized light (Fig. 2f) show numerous crystal chips (Fig. 2g) with a large deformation of 1400% stretching, and this eventually changes back to be unobservable after being released for 15 min (Fig. 2h). Therefore, this confirms the reversibility of such SIC, and explains that the whitish sample could be restored to its transparent state again, suggesting that this mechano-responsive strengthening could be slow and reversible. Therefore, stretching could induce the PDM-2.5 elastomer to be strong, while keeping its viscoelasticity at the original or retracting state. Thus, this strengthening trait, derived from the rearrangement and assembly of soft segments, does not contradict with additional toughening and room-temperature self-healing, because of the adequate regulatory space for tailoring the chemical molecular structure of hard segments.
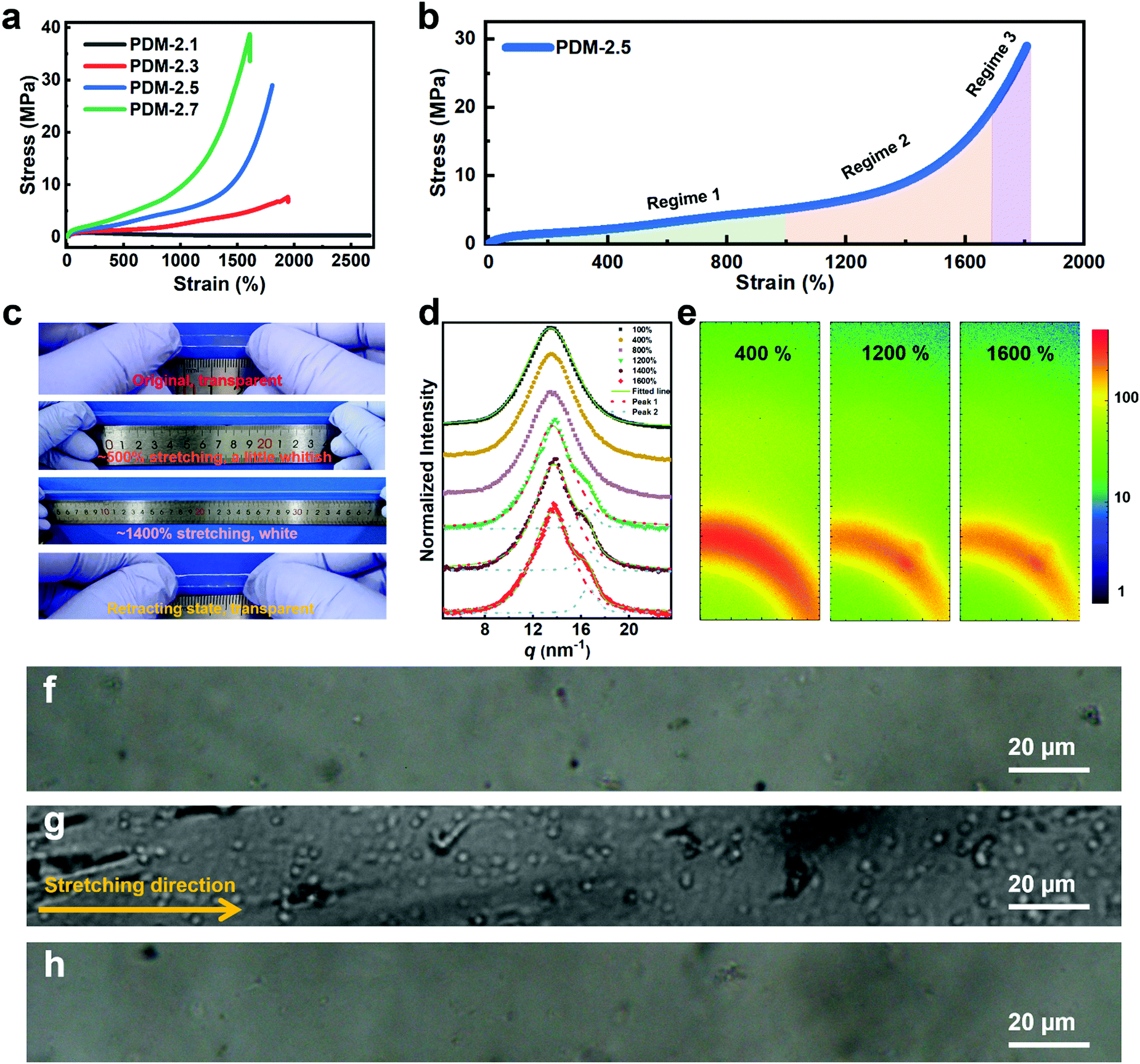 | ||
| Fig. 2 (a) Typical stress–strain curves of polyurethane elastomers with various R values extended by PDM. (b) Stress–strain curve of the PDM-2.5 elastomer divided into three regimes. (c) Optical photos showing the whitening process of the PDM-2.5 elastomer manually stretched to ∼500% and ∼1400% and its retracting state (released for 1 h). (d) 1D WAXS results of PDM-2.5 stretched to various strains. (e) 2D WAXS patterns of the PDM-2.5 elastomer stretched to ∼400%, ∼1200% and ∼1600%. (f) Original sample of the PDM-2.5 elastomer and (g) an identical sample stretched to ∼1400% and (h) released for 15 min under a polarized microscope. | ||
In the following tests, the successful SIC strengthening along with the simultaneous toughening in our polyurethane elastomer was further analyzed. The success is considered to mainly be attributed to the cooperative effect, rather than the exclusive effect, that occurs during the rational optimization of the average constraint molecular weight between the physical crosslinking junctions 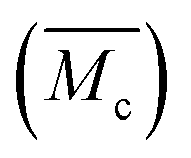 , which are, respectively, 9980, 6503, 5518 and 3372 g mol−1 for the PDM-2.1, PDM-2.3, PDM-2.5, and PDM-2.7 elastomers (see the detailed deduction in Fig. S11 in the ESI†). The average size of the hard domains gradually increases as the peak q vector in the SAXS results (Fig. S12, ESI†) undergoes a downshift, and this also confirms that
, which are, respectively, 9980, 6503, 5518 and 3372 g mol−1 for the PDM-2.1, PDM-2.3, PDM-2.5, and PDM-2.7 elastomers (see the detailed deduction in Fig. S11 in the ESI†). The average size of the hard domains gradually increases as the peak q vector in the SAXS results (Fig. S12, ESI†) undergoes a downshift, and this also confirms that 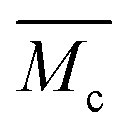 is reduced by adjusting the R value. Therefore, rational selection of PTMEG as the soft segment and simultaneous regulation of R to be 2.5 results in the suitable effective length of the crystallizable segments containing several PTMEG chain blocks, which provide a much lower crystallization energy barrier during the uniaxial stretching process, due to the favorable rearrangement of the chain conformation, and disentanglement and alignment of the molecular chains. In addition, the prominent toughening via SIC at large deformations also requires the cooperation of hierarchical non-covalent interactions, as the dynamic sacrificial bonds not only dissipate the stress energy, but also maintain the crystalline configuration, and this will subsequently be discussed.
is reduced by adjusting the R value. Therefore, rational selection of PTMEG as the soft segment and simultaneous regulation of R to be 2.5 results in the suitable effective length of the crystallizable segments containing several PTMEG chain blocks, which provide a much lower crystallization energy barrier during the uniaxial stretching process, due to the favorable rearrangement of the chain conformation, and disentanglement and alignment of the molecular chains. In addition, the prominent toughening via SIC at large deformations also requires the cooperation of hierarchical non-covalent interactions, as the dynamic sacrificial bonds not only dissipate the stress energy, but also maintain the crystalline configuration, and this will subsequently be discussed.
For further comprehension, two control polyurethanes with identical R values, extended by 1,3-benzenedimethanol and 2,6-diaminopyridine (denoted as BDM-2.5 and DAP-2.5, respectively) were also synthesized and used for comparison (Fig. 3a). Their 1H NMR, FTIR and molecular weight information are provided in Fig. S13 and in Tables S5–S7 (ESI†) to demonstrate their successful synthesis. As shown in Fig. 3a, PDM-2.5 is endowed with the characteristic hierarchical H-bonds created by carbamate–carbamate and pyridine–carbamate H-bonding interactions, and this can be validated by two-dimensional correlated FTIR spectroscopy (2D-FTIR), in which two correlation cross peaks at Φ (1701 and 3327) and Φ (1598 and 3327) can be seen in the synchronous spectrum (Fig. 3b) and then vanish in the corresponding asynchronous spectrum (Fig. 3c). However, it can be assumed that in the structure of BDM-2.5 the pyridine ring of PDM-2.5 is replaced with an aromatic ring, hence it contains only carbamate–carbamate H-bonds within its loosely-packed hard domains. Meanwhile, it is thought that for DAP-2.5 partial carbamate groups are replaced by urea, and so it also possesses hierarchical H-bonds among the carbamate, urea and pyridine groups, but especially in the tightly-packed hard domains, for comparison. In Fig. S14 (ESI†), the above dynamic non-covalent H-bonding interactions are first verified by the variable-temperature 1H NMR data. In addition to the differences in quantity and type of H-bonds, results of the quantum chemistry calculations (as shown in the ESI†) reveal (Fig. 3d) that the binding energy of the three types of H-bond ranks are in the sequence of: carbamate–carbamate (3.663 kcal mol−1) < pyridine–carbamate (7.686 kcal mol−1) < urea–urea (13.447 kcal mol−1). Therefore, these discrepancies of dynamic H-bonding interactions in the quantities, types and binding energies are expected to result in different aggregation states among the elastomers. Atomic force microscopy (AFM) images (Fig. 3e and Fig. S15, S16 in the ESI†) reflect that both PDM-2.5 and BDM-2.5 elastomers have nano-scale sized and loosely-packed hard domains due to the selection of IPDI as their partial hard segments and the relatively low binding energy of the H-bonds, while the aggregation of the hard segments is seemingly more compact for DAP-2.5, due to the much higher urea–urea interactions (Fig. S17 in the ESI†). The binding energy of the hierarchical H-bonds together with the aggregation state of the hard segments significantly influences the dissociation and association of non-covalent H-bonding interactions within the hard domains. As shown in Fig. 3f and g, a molecular simulation method (as shown in the ESI†) was then carried out to calculate the binding energy of the hard domains within the three different polyurethanes, and the results indicated that the hard domains in PDM-2.5 and BDM-2.5 possess similar binding energies that behave with a similar ability for the dissociation and association of H-bonds upon large deformations, while the binding energy for DAP-2.5 is much higher and so its dissociation of H-bonds is more difficult. Therefore, this obvious discrepancy may serve as an insight into the occurrence of SIC, and the effect of mechanical toughening.
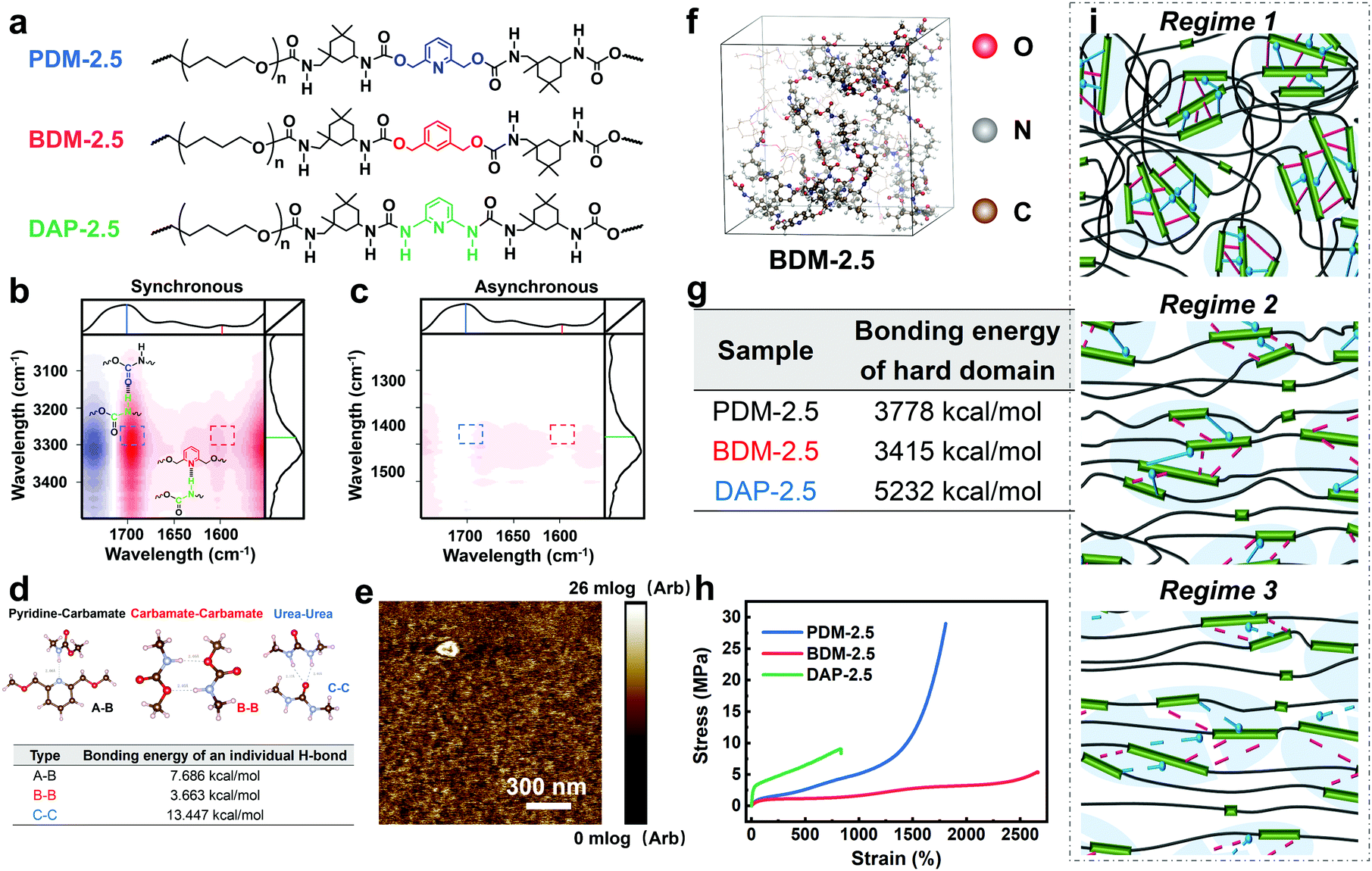 | ||
| Fig. 3 (a) Structural comparison of PDM-2.5, BDM-2.5 and DAP-2.5. (b) Synchronous and (c) asynchronous 2D-FTIR images of PDM-2.5. (d) Bonding energy of three types of individual H-bond, including pyridine–carbamate interaction, carbamate–carbamate interaction and urea–urea interaction. (e) AFM modulus image of PDM-2.5 reflecting the phase morphology. (f) Visualized representation of modelling the hard segments of polyurethane in an equilibrium state, taking PDM-2.5 as an example. (g) Binding energies of the hard domains of PDM-2.5, BDM-2.5 and DAP-2.5. (h) Typical stress–strain curves of PDM-2.5, BDM-2.5 and DAP-2.5. (i) Schematic modeling showing the evolution of the dynamic topological structures of PDM-2.5 during deformations. | ||
Fig. 3h and Table S9 (ESI†) show the distinct mechanical behaviors of these three elastomers. For BDM-2.5, a slow increase in tensile strength is shown for a wide range of strain and no sign of obvious strain hardening is observed. The derivative stress of BDM-2.5 (Fig. S14c in the ESI†) shows a hump within a certain range of strain, and this reveals the oriented alignment of the soft segments and provides the potential possibility of SIC. Meanwhile, unfortunately, its carbamate–carbamate interactions within the hard domains are not strong enough to maintain the oriented soft segments (due to the decreased tendency of derivative stress within some deformation ranges, as shown in Fig. S14c in the ESI†). Therefore, both the hard and soft segments continuously perform mutual displacement and, as a result, the PTMEG chains fail to align into SIC domains in sufficient amounts and in large sizes. For DAP-2.5, the hard domains are too strong to efficiently dissipate the stress energy during stretching. The soft phase fails to withstand the external stress and breaks at a short elongation before the PTMEG chains are thoroughly disentangled or uniaxially rearranged. The derivative stress does not display a hump or a surge (Fig. S14d in the ESI†), indicating that the soft phase dissipates most of the energy until it breaks. Thus, DAP-2.5 also fails to induce SIC domains resulting from the sufficient dissociation of their hierarchical H-bonds. In contrast, as PDM-2.5 benefits from loosely packed but hierarchically associated H-bonds, the stress–strain behavior exhibits the typical phenomenon of strain hardening. As illustrated in Fig. 3i, the relatively low binding energy of the carbamate–carbamate H-bonds (Regime 2) is beneficial for dissipating stress energy during stretching. In the meantime, as the strain increases, the PTMEG chains gradually undergo alignment along the stretching direction, and this results in crystalline domains as another hard phase. The newly-formed hard domains of the PTMEG crystalline act as crosslinking junctions and a self-reinforcing phase, so the PDM-2.5 elastomer exhibits an abrupt increase in tensile strength; this is mainly attributed to the strong pyridine–carbamate H-bonding energy that delays its own mutual dissociations and maintains the configuration of the PTMEG crystalline. Once the pyridine–carbamate H-bonds are not able to withstand the surging external force during tremendous deformations (Regime 3), they dissociate and fail to dissipate more energy, creating enough mechanical toughness, and followed by material breaking.
2.2 Self-healing properties
As discussed above, SIC strengthening and toughening is a slow and reversible strategy. Its key point is to adjust the topological structure of the effective soft segment length, whilst providing enough regulatory space for achieving the result of high chain mobility of the hard segments and effective dissociation and association of dynamic bonds. Therefore, as it benefits from the loosely packed configuration of hard domains and their low binding energy, in addition to being simultaneously strong and tough, it is noteworthy that our PDM-2.5 elastomer also exhibits excellent room-temperature self-healing performance. As shown in Fig. 4a, as a result of 24 h automatic repair without any external stimulus, the typical stress–strain curve of the healed samples overlaps well with that of the original. Herein, considering the strain hardening of the SIC effect, it can be noted that SEs were synthetically evaluated by the repairing of their tensile strength, elongation and toughness, instead of an individual property, and the results are also compared with PDM-based polyurethanes with various R values (Fig. 4b and Tables S10–S12 in the ESI†). It is reasonable that a higher R value give rises to inferior SEs, due to the restricted chain mobility and higher binding energy of the hard domains. However, to our delight, the PDM-2.5 elastomer exhibited the ability to synchronously self-heal its strength, elongation and toughness, with a high level of SEs that were all more than 80%, while the PDM-2.7 elastomer did not have this ability, signifying rational regulation of the molecular topological structure.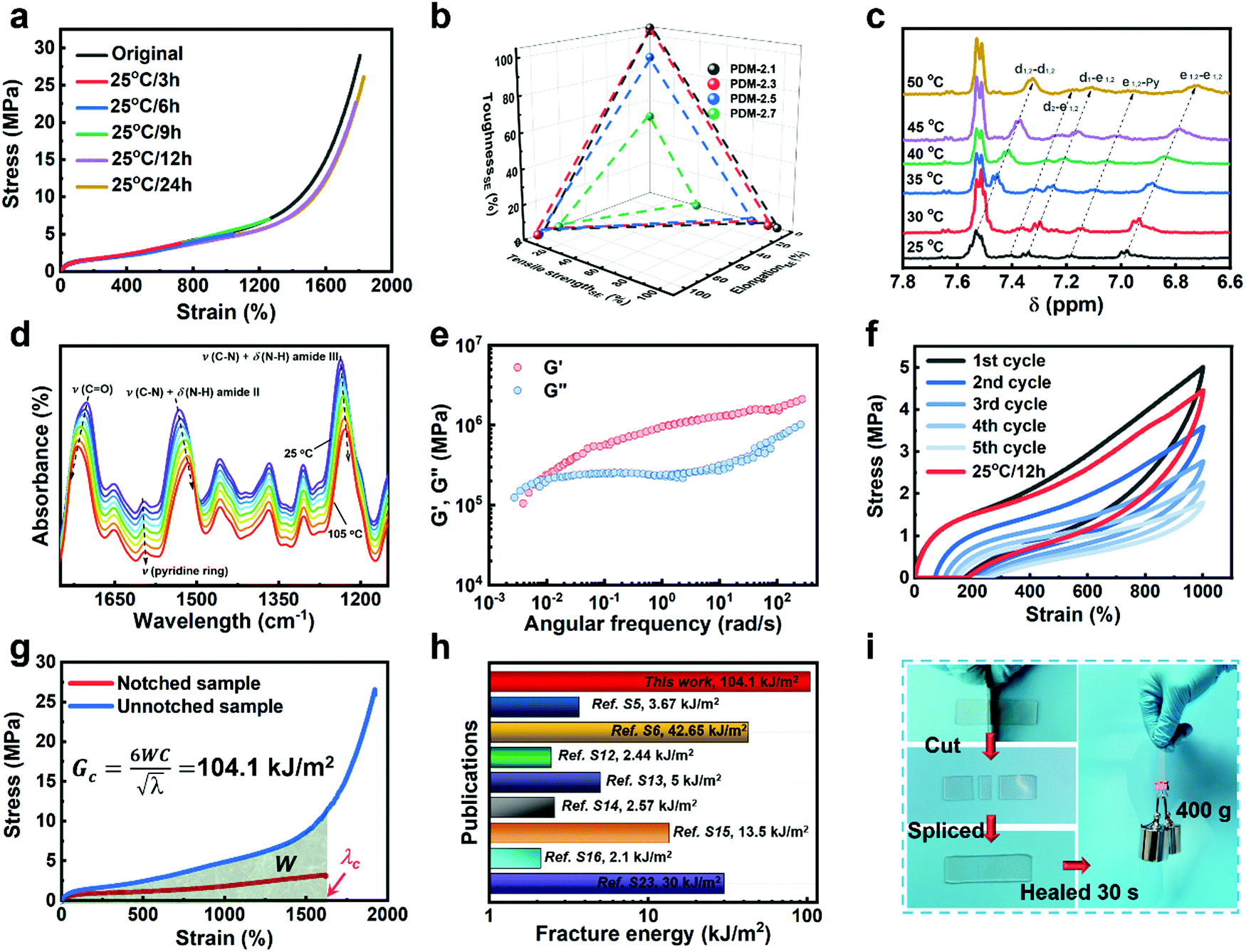 | ||
| Fig. 4 (a) Stress–strain curves of PDM-2.5 original samples and healed samples at 25 °C after various times. (b) SEs calculated by tensile strength, elongation after break and toughness for the PDM-based polyurethane elastomers, healed at 25 °C for 24 h. (c) Evolution of characteristic peaks in the 1H NMR spectra, related to H-bonding at various temperatures. (d) Evolution of characteristic peaks in variable-temperature FTIR data, related to H-bonding with temperatures varying from 25 °C to 105 °C. (e) Master curves of G′ and G′′ obtained from rheology tests for PDM-2.5. (f) Cyclic tensile tests of PDM-2.5 with consecutive five cycles and an additional cycle after waiting for 12 h at 25 °C. (g) Stress–strain curves of notched and unnotched PDM-2.5 samples for calculating the fracture energy. (h) Comparison of the fracture energy of PDM-2.5 to previously reported room-temperature self-healable materials (see details in Table S3 in the ESI†). (i) Photos showing the instant self-healing feature of PDM-2.5 by cutting a rectangular sample into three parts and immediately being spliced together and then lifting a 400 g load. | ||
In order to validate the room-temperature self-healing origin, we must confirm the ability of the reversible bonds to dissociate and associate in ambient conditions, and the chain mobility for dynamic exchanges. Therefore, first, variable-temperature 1H NMR experiments, along with variable-temperature FTIR characterizations, were respectively performed. According to Fig. 4c, the 1H NMR spectra show a clear chemical shift of the H-bonding related groups of PDM-2.5 (Fig. S18 and Table S8 in the ESI†) as the temperature increases from 25 °C to 50 °C, and this is consistent with the variable-temperature FTIR result (Fig. 4d and Fig. S20 in the ESI†), profoundly proving the room-temperature reversibility of H-bonds in PDM-2.5. Additionally, the chain mobility, another prerequisite of self-healing, was also investigated and is subsequently discussed. Rheological tests (Fig. 4e and Fig. S21 in the ESI†) were carried out to quantify the characteristic relaxation time (τc), which is a parameter that is highly correlated to the mobility of chains and the reversibility of the network, and this was calculated to be 924.9 s and 810.3 s for PDM-2.5 and BDM-2.5, respectively, signifying the fast rearrangement of hard segments at ambient conditions without any external motivations. In contrast, DAP-2.5 has no crossover point, due to intimately compacted hard segments. Therefore, due to the loosely-packed hard domains with relatively low binding energy, it is clear that the hard segments within PDM-2.5 can readily diffuse in a certain vicinity, facilitating the exchange of dynamic bonds and then promoting their dissociation and association. The above analysis can also be verified by another powerful evidence, as the stress relaxation results of PDM-based polyurethanes with different R values also describe a similar tendency (Fig. S22 in the ESI†).
Due to the effective chain mobility and dynamic dissociation and association of hierarchical H-bonding interactions, the PDM-2.5 elastomer also possesses unique mechanical recovery, unparalleled fracture energy and an instant room-temperature self-healing feature. As shown in Fig. 4f, the elastomer could be consecutively stretched to a strain as high as 1000% and then almost recovered to its original state after waiting for 12 h at 25 °C. Moreover, fracture energy tests were performed, and the results are shown in Fig. 4g and Fig. S23 (ESI†). The photos demonstrate its extraordinary notch-insensitive feature, as the notch does not propagate even at ∼800%, and the fracture energy achieved is as high as 104.1 kJ m−2, which outperforms other room-temperature self-healable materials recorded in previous literature (Fig. 4h). Despite the fact that it takes 24 h to fully recover its mechanical properties, it is interesting that a spliced sample of PDM-2.5 is able to lift a 400 g load upon contact for only 30 s (Fig. 4i). This indicates that the well-designed hard segments and hard phase of the PDM-2.5 elastomer have sufficient quantities of hydrogen bonds with a high chain mobility for chain diffusion across the splicing interfaces for fast inter-formation of new hydrogen bonds.
3. Conclusions
In summary, we have tailored a novel polyurethane material with extraordinary room-temperature self-healing and mechanical properties. Due to the hierarchical H-bonds in the loosely packed hard domains for self-healing and the SIC of soft segments for strengthening and toughening, this self-reinforcing elastomer performs with giant tensile strength (29.0 ± 0.9 MPa), unprecedented toughness (121.8 ± 8.5 MJ m−3) and unparalleled fracture energy (104.1 kJ m−2) among the state-of-the-art room-temperature self-healable polymeric materials. This may pave the way for leading-edge applications of self-healing macromolecules in broader fields, i.e. for puncture-resistant tire sealants, tough and resilient sportswear materials, and in durable protective coatings.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by China's Post-Doctoral Science Foundation (Grant No. 2019M652572), the Zhengzhou Major Collaborative Innovation Project (Zhengzhou University, Grant No. 18XTZX12001), the Natural Science Foundation of Jiangsu Province (2020, Kai Wu), and the National Natural Science Foundation of China (Grant No. 51573102 and 51421061). Many thanks to Prof. Bin Zhang (School of Materials Science and Engineering, Zhengzhou University, Zhengzhou, 450001, P. R. China) for his kind assistance in WAXS, AFM and rheological tests.Notes and references
- T. Chang, F. Panhwar and G. Zhao, Adv. Mater. Interfaces, 2020, 1901959 CrossRef.
- E. Acome, S. K. Mitchell, T. G. Morrissey, M. B. Emmett, C. Benjamin, M. King, M. Radakovitz and C. Keplinger, Science, 2018, 359, 61–65 CrossRef CAS.
- C. e. Yuan, M. Z. Rong and M. Q. Zhang, Polymer, 2014, 55, 1782–1791 CrossRef CAS.
- Y. Peng, Y. Yang, Q. Wu, S. Wang, G. Huang and J. Wu, Polymer, 2018, 157, 172–179 CrossRef CAS.
- A. Rekondo, R. Martin, A. Ruiz de Luzuriaga, G. Cabañero, H. J. Grande and I. Odriozola, Mater. Horiz., 2014, 1, 237–240 RSC.
- Y. Lai, X. Kuang, P. Zhu, M. Huang, X. Dong and D. Wang, Adv. Mater., 2018, 30, 1802556 CrossRef.
- S.-M. Kim, H. Jeon, S.-H. Shin, S.-A. Park, J. Jegal, S. Y. Hwang, D. X. Oh and J. Park, Adv. Mater., 2018, 30, 1705145 CrossRef.
- S. Ji, W. Cao, Y. Yu and H. Xu, Angew. Chem., Int. Ed., 2014, 53, 6781–6785 CrossRef CAS.
- L. Zhang, Z. Liu, X. Wu, Q. Guan, S. Chen, L. Sun, Y. Guo, S. Wang, J. Song, E. M. Jeffries, C. He, F.-L. Qing, X. Bao and Z. You, Adv. Mater., 2019, 1901402 CrossRef.
- D. Fu, W. Pu, Z. Wang, X. Lu, S. Sun, C. Yu and H. Xia, J. Mater. Chem. A, 2018, 6, 18154–18164 RSC.
- A. P. Bapat, B. S. Sumerlin and A. Sutti, Mater. Horiz., 2020, 7, 694–714 RSC.
- H. Ying, Y. Zhang and J. Cheng, Nat. Commun., 2014, 5, 3218–3226 CrossRef.
- S. Sun, X. Gan, Z. Wang, D. Fu, W. Pu and H. Xia, Addit. Manuf., 2020, 33, 101176 CAS.
- N. Kuhl, S. Bode, R. K. Bose, J. Vitz, A. Seifert, S. Hoeppener, S. J. Garcia, S. Spange, S. van der Zwaag, M. D. Hager and U. S. Schubert, Adv. Funct. Mater., 2015, 25, 3295–3301 CrossRef CAS.
- Z. P. Zhang, M. Z. Rong and M. Q. Zhang, Adv. Funct. Mater., 2018, 28, 1706050 CrossRef.
- Z. Feng, B. Yu, J. Hu, H. Zuo, J. Li, H. Sun, N. Ning, M. Tian and L. Zhang, Ind. Eng. Chem. Res., 2019, 58, 1212–1221 CrossRef CAS.
- M. Li, W. Li, W. Cai, X. Zhang, Z. Wang, J. Street, W.-J. Ong, Z. Xia and Q. Xu, Mater. Horiz., 2019, 6, 703–710 RSC.
- D. Wang, J. Xu, J. Chen, P. Hu, Y. Wang, W. Jiang and J. Fu, Adv. Funct. Mater., 2019, 1907109 Search PubMed.
- Y. Song, Y. Liu, T. Qi and G. L. Li, Angew. Chem., Int. Ed., 2018, 57, 13838–13842 CrossRef CAS.
- C.-J. Fan, Z.-C. Huang, B. Li, W.-X. Xiao, E. Zheng, K.-K. Yang and Y.-Z. Wang, Sci. China Mater., 2019, 62, 1188–1198 CrossRef CAS.
- J. Kang, D. Son, G.-J. N. Wang, Y. Liu, J. Lopez, Y. Kim, J. Y. Oh, T. Katsumata, J. Mun, Y. Lee, L. Jin, J. B. H. Tok and Z. Bao, Adv. Mater., 2018, 30, 1706846 CrossRef.
- H. Wang, H. Liu, Z. Cao, W. Li, X. Huang, Y. Zhu, F. Ling, H. Xu, Q. Wu, Y. Peng, B. Yang, R. Zhang, O. Kessler, G. Huang and J. Wu, Proc. Natl. Acad. Sci. U. S. A., 2020, 202000001 Search PubMed.
- Y. Li, W. Guo, W. Li, X. Liu, H. Zhu, J. Zhang, X. Liu, L. Wei and A. Sun, Chem. Eng. J., 2020, 393, 124583 CrossRef CAS.
- Q. Zhang, S. Niu, L. Wang, J. Lopez, S. Chen, Y. Cai, R. Du, Y. Liu, J.-C. Lai, L. Liu, C.-H. Li, X. Yan, C. Liu, J. B.-H. Tok, X. Jia and Z. Bao, Adv. Mater., 2018, 30, 1801435 CrossRef.
- S. Burattini, H. M. Colquhoun, J. D. Fox, D. Friedmann, B. W. Greenland, P. J. F. Harris, W. Hayes, M. E. Mackay and S. J. Rowan, Chem. Commun., 2009, 6717–6719 RSC.
- X. Pei, H. Zhang, Y. Zhou, L. Zhou and J. Fu, Mater. Horiz., 2020, 7, 1872–1882 RSC.
- H. Guo, X. Fang, L. Zhang and J. Sun, ACS Appl. Mater. Interfaces, 2019, 11, 33356–33363 CrossRef CAS.
- Z. Wang, G. An, Y. Zhu, X. Liu, Y. Chen, H. Wu, Y. Wang, X. Shi and C. Mao, Mater. Horiz., 2019, 6, 733–742 RSC.
- M. Tang, R. Zhang, S. Li, J. Zeng, M. Luo, Y.-X. Xu and G. Huang, Angew. Chem., Int. Ed., 2018, 57, 15836–15840 CrossRef CAS.
- A. N. Gent, S. Kawahara and J. Zhao, Rubber Chem. Technol., 1998, 71, 668–678 CrossRef CAS.
- R. Rahmawati, S. Masuda, C.-H. Cheng, C. Nagano, S. Nozaki, K. Kamitani, K. Kojio, A. Takahara, N. Shinohara, K. Mita, K. Uchida and S. Yamasaki, Macromolecules, 2019, 52, 6825–6833 CrossRef CAS.
- K. Imada, T. Miyakawa, Y. Chatani, H. Tadokoro and S. Murahashi, Makromol. Chem., 1965, 83, 113–128 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Materials and experimental procedures; computational calculations, FTIR, 1H NMR, GPC, DSC, UV-vis, AFM, rheology, DMA, tensile testing data and graphs. See DOI: 10.1039/d0mh01447h |
| This journal is © The Royal Society of Chemistry 2021 |