
Synthesis and application of core–shell liquid metal particles: a perspective of surface engineering
Yong
Liu
 *ab,
Wei
Zhang
ab and
Hao
Wang
*ab
*ab,
Wei
Zhang
ab and
Hao
Wang
*ab
aCAS Key Laboratory for Biomedical Effects of Nanomaterials and Nanosafety, CAS Center for Excellence in Nanoscience, National Center for NanoScience and Technology, No. 11 Zhongguancun Beiyitiao, Beijing 100190, P. R. China. E-mail: liuyong1@nanoctr.cn; wanghao@nanoctr.cn
bUniversity of Chinese Academy of Sciences, No. 19A Yuquan Road, Beijing 100049, P. R. China
First published on 24th August 2020
Abstract
Liquid metal micro/nano particles (LMPs) from gallium and its alloys have attracted tremendous attention in the last decade due to the unique combination of their metallic and fluidic properties at relatively low temperatures. Unfortunately, there is limited success so far in realizing the highly controllable fabrication and functionalization of this emerging material, posing great obstacles to further promoting its fundamental and applied studies. This review aims to explore solutions for the on-demand design and manipulation of LMPs through physicochemically engineering their surface microenvironment, including compositions, structures, and properties, which are featured by the encapsulation of LMPs inside a variety of synthetic shell architectures. These heterophase, core–shell liquid metal composites display adjustable size and structure–property relationships, rendering improved performances in several attractive scenarios including but not limited to soft electronics, nano/biomedicine, catalysis, and energy storage/conversion. Challenges and opportunities regarding this burgeoning field are also disclosed at the end of this review.
 Yong Liu | Yong Liu obtained his BS in chemistry at Wuhan University and after that he joined the National Center for NanoScience & Technology (NCNST) of China in 2015 as a PhD candidate. He worked with Prof. Xingyu Jiang and Prof. Wei Zhang in 2015–2019, and has been working with Prof. Hao Wang since 2020. His current scientific interest is focused on developing liquid metal-based materials for electronic and biomedical devices. |
 Wei Zhang | Wei Zhang obtained his PhD at Tsinghua University in 2004, majoring in materials engineering. He then became a post-doctoral researcher at University at Buffalo, the State University of New York (2004–2006). Afterwards he worked as a project leader at Research Institute of Tsinghua University in Shenzhen (2006–2008). He joined NCNST in 2008 as an associate professor and was promoted to a full processor in 2014. His research interests include liquid metal materials and biomedical microdevices. |
 Hao Wang | Hao Wang obtained his BS and PhD from the Department of Chemistry at Nankai University in 2000 and 2005, respectively. Afterwards, he moved to Universät Würzburg in Germany (2005–2007). Then he was a post-doctoral researcher at Department of Molecular and Medical Pharmacology in UCLA (2007–2010). In 2011, he started his independent career in NCNST. His current research interests are to (i) develop polymeric biomaterials; (ii) study their bio-effect and further regulate biological behavior; (iii) explore functional nanomaterials for bioimaging and drug delivery. |
1. Introduction
“Liquid metals” broadly refer to metals and metal alloys that display a much lower melting point (m.p., typically <300 °C) compared to conventional metals, among which three elements have witnessed an explosive increase in recent studies, i.e., gallium (Ga), indium (In), and tin (Sn).1 Specifically, Ga and its eutectic alloys with In and Sn such as eutectic gallium indium (EGaIn, 75% Ga, 25% In by weight) and galinstan (68.5% Ga, 21.5% In, 10% Sn by weight) have drawn broad scientific interest because of their liquid nature near room temperature or even below 0 °C (m.p. 29.8, 15, and −19 °C for Ga, EGaIn, and galinstan respectively).2 Additionally, these liquid metals have negligible vapour pressure (∼10−35 Pa at 29.9 °C, for Ga) compared to the most familiar liquid metal mercury (1 Pa at 42 °C), and thus are much safer. Downsizing liquid metals into smaller length scales, i.e., liquid metal micro/nanoparticles (LMPs) featuring a liquid core wrapped by a solid interface, has dramatically expanded their application horizons primarily due to the size effect of micro/nanomaterials.3–5 For instance, the reduced size of LMPs leads to suppressed freezing and melting temperatures compared to bulk liquid metals, which can thus expand the working temperature of liquid metal-based devices.6 The small-sized LMPs are also more compliant to solution processing and are easier to be incorporated into bulk matrices, e.g., elastomers and hydrogels for developing functional composites. Other attractive properties of LMPs include stimuli-responsive coalescence, self-healing behaviour, broad surface plasmon resonance (SPR), catalytic potential, alloying capacity, and so forth. All these advantages have made liquid metals a hot topic in micro/nanoscience and technology, which have triggered great innovations in many fields including materials engineering,7,8 electronics,9 microfluidics,2,10 biomedicine,11 catalysis,12 and energy.13LMPs typically display a core–shell structure in which the metallic core is encapsulated in a naturally formed oxide skin. The oxide skin is thin but essential to maintaining the structural integrity of LMPs, otherwise they would coalesce back into large droplets. Oxide skin-stabilized LMPs represent the simplest and also the earliest form of liquid metal micro/nanomaterials, and have inspired many pioneering works in liquid metal nanotechnology. However, they do not generally suit all applied scenarios due to the fragility and poor modifiability of the oxide skin, which has failed to achieve long-term colloidal stability, favourable durability against solution processing and external disturbance, and designable multifunctionality. New toolkits for manipulating the surface microenvironment of LMPs are thus urgently required to accommodate the ever growing interest from various fields. A feasible, and also effective strategy is to reinforce the armour of LMPs by constructing an extra protective layer outside the oxide skin, which consequently produces core–shell composite materials with improved stability and functionality. Advances in the past decade have witnessed the evolution of this concept from a simple ligand layer to multilevel synthetic architectures, including both polymers and inorganic coatings. These hybrid LMPs exhibit strengthened processability and also tailorable responses to mechanical, electrical, optical, thermal, and even magnetic manipulations, and are thus more competent in practical applications compared to unmodified ones. Despite these benefits, the surface engineering of LMPs has not been methodologically discussed and timely summarized.
As a remedy for existing reviews that have rarely sketched the landscape near the liquid metal surface, our perspective herein is concentrated on the trend of applying established chemistries in manipulating the surface microenvironment of LMPs to bridge their incorporations with diverse shell components. To make the scope topical and succinct, only metals and alloys from Ga, In, and Sn are involved in our discussions. As shown in Fig. 1, we will start with a fundamental introduction to the liquid metal chemistry as well as the sonochemical synthesis that has been commonly used for preparing LMPs. The story will then flow to the surface encapsulation of LMPs where representative core–shell composites are highlighted and the underlying principles of selecting appropriate building blocks and synthetic methods are summarized. Subsequently, the outcome of shell encapsulation, reflected in the tunable properties and improved performances of LMPs, will be demonstrated in several applied fields. Future directions regarding the advanced manufacturing, structure–property relations, and broad uses of LMPs will also be investigated. Placing a unique view near the surface of LMPs, we expect to provide an accessible guidebook for engineering this material toward future innovations.
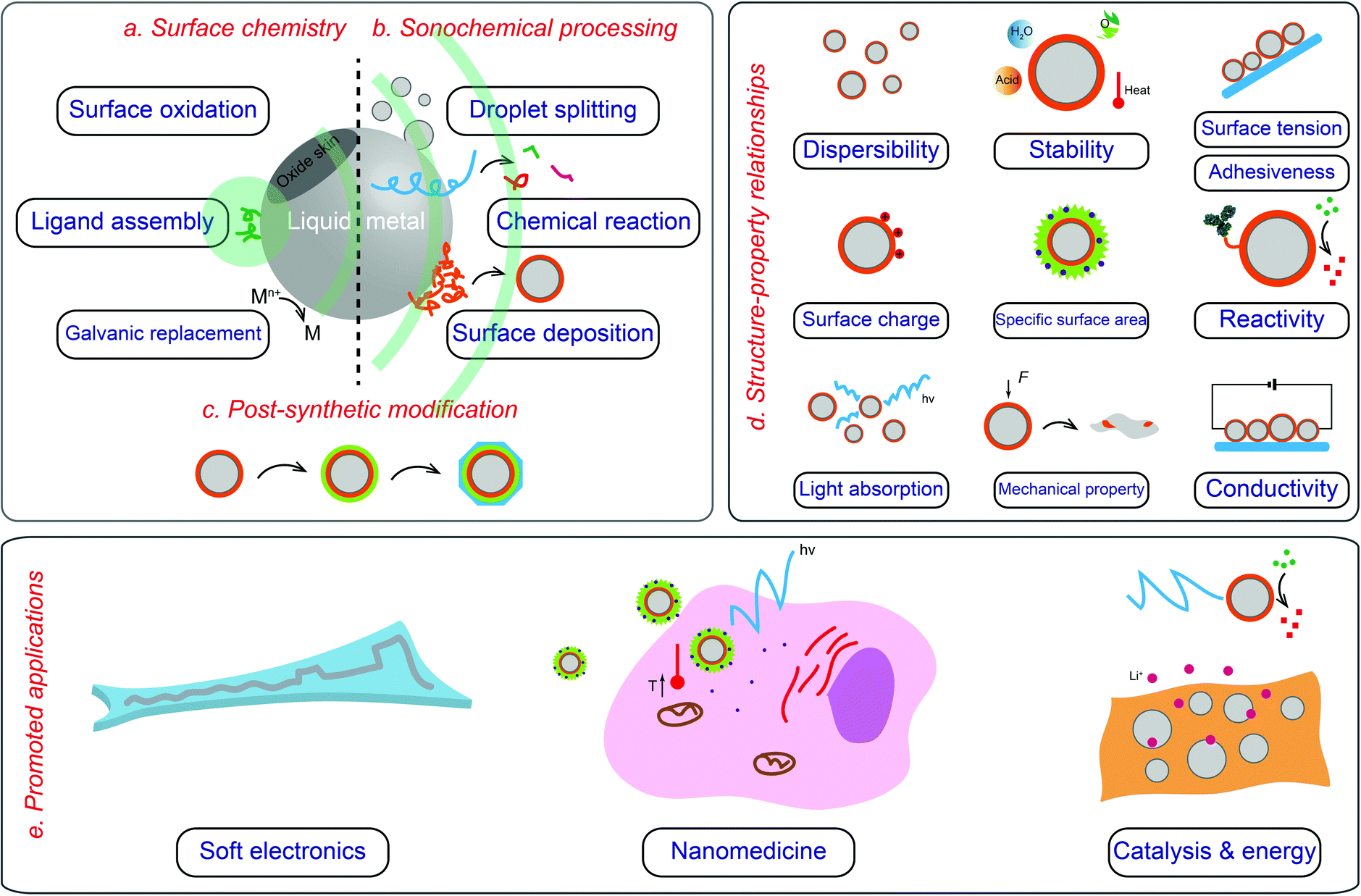 | ||
| Fig. 1 Overview of core–shell LMPs including their surface chemistry (a), fabrication (b and c), properties (d), and application (e). | ||
2. Chemistry and processing of liquid metals
Unlike conventional metallic nanomaterials that can be facilely prepared by bottom-up synthesis, top-down approaches are much more favored by liquid metals to obtain LMPs due to the difficulty in reducing Ga3+. Though thermal decomposition of Ga alkylamides yields Ga NPs, the reaction temperature exceeds 250 °C.14 LMPs are generally produced by ultrasonic secession of large liquid metal droplets in an immiscible solvent such as ethanol,15 though other top-down methods that are effective but less used also exist, such as mechanical shearing,16 flow focusing,17 and molding.18 In this review, we concentrate on the sonochemical synthesis method because of its robustness in triggering various physicochemical processes that are useful for engineering the liquid metal surface. Other methods may also be consulted if the obtained LMPs are highlighted in an important application.Engineering the interface of LMPs linking their electron-rich liquid core with the surroundings can modulate their stability and intrinsic properties as well as responses to external stimuli. This depends on several chemistries occurring at the liquid metal surface, i.e., oxidation, ligand assembly, and Galvanic replacement (Fig. 1a). These phenomena commonly exist in both bulk liquid metal droplets and LMPs, but appear more complex and changeable for the latter because of the chaotic ultrasonic synthesis.
2.1. Featured surface chemistry
| 4Ga + 3O2 = 2Ga2O3 |
This oxide skin impedes the further oxidation of liquid metals but is fragile to acid or base treatment.20,21 It also helps depress the high surface/interface tension of liquid metals (>500 mN m−1) that hampers the breakup of liquid metal droplets into LMPs.22 For Ga-based eutectic alloys, the composition of the oxide skin is determined by the Gibbs free energy of metal oxides derived from different elements that constitute the core, i.e., −998.3, −830.7, and −515.8 kJ mol−1 for Ga2O3, In2O3, and SnO2, respectively.23 Thus, all eutectic alloys from Ga, In, and Sn have a Ga2O3 skin, which can be directly visualized by a touch-and-print method.23,24 Similarly, LMPs produced by ultrasonication are wrapped by a Ga2O3 skin as well, which offers a protective interface for their colloidal stability.15 However, the pristine liquid metal can be further oxidized into GaOOH in the presence of water molecules, from either the solvent or the humid air, which is possibly through the following pathways:25
| 4Ga + 2H2O + 3O2 = 4GaOOH |
| 2Ga + 4H2O = 2GaOOH + 3H2↑ |
These reactions may cause the phase separation and shape transformation of LMPs because the product GaOOH has a rod-like morphology. Thus, it has been challenging to stabilize LMPs in aqueous solutions.
Besides sulfhydryl compounds, other types of molecules including polyphenols,30 surfactants,31,32 silanes,33 polymers,34 and proteins,35 have also been developed as potential ligands for LMPs. Such an expansion of available ligand molecules allows the stabilization of LMPs in various solvents. However, their interactions with the liquid metal surface have remained elusive. Electrostatic adsorption has been proved as a dominant factor in screening proper ligands,35 but does not always work in different cases. Other interactions, e.g., coordination, electron/free radical transfer, and even gelation, may also contribute to the ligand decoration on LMPs and will be independently discussed where the corresponding examples are referred to.
| Ga0 + AuBr4− = Ga3+ + Au0 + 4Br− |
| Ga0 + 3Ag+ = Ga3+ + 3Ag0 |
These reactions can take place on the surface of both bulk liquid metals and LMPs,38 even during sonochemical synthesis.39 Based on Galvanic replacement, metal/metal oxide structures can be constructed around LMPs, which can not only improve their stability under harsh conditions (e.g., upon heavy oxidation and acid etching), but also regulate their basic attributes, e.g., thermal/electrical conductivity, catalytic activity, and SPR absorption.
2.2. Sonochemical processing
As a commonly used method for preparing LMPs and also a popular strategy for synthesizing and functionalizing many known materials, sonochemical synthesis refers to the use of ultrasonic power to trigger physicochemical reactions and transformations. The experimental setups of sonochemical synthesis include bath sonication,27,40 probe sonication,15 nebulization,41etc. During ultrasonication, many highly energetic phenomena, both physical and chemical, can take place, and the most notable one is acoustic cavitation, i.e., the formation, growth, and implosive collapse of bubbles. This phenomenon is accompanied by the generation of high-temperature (5000 K) and high-pressure (2000 atm) fields particularly at the centers of the bubbles and the sonolysis of solvent vapors into free radicals that can also diffuse to the liquid phase.42 These factors together have enabled various chemistries, e.g., mechanochemistry, free radical chemistry, and redox chemistry for synthesizing functional molecules and materials.Compared to post-synthetic modifications, encapsulation of LMPs in a single co-sonication step, i.e., putting all reactants in a pot and exposing them to the ultrasonic power, is more convenient and cost-effective. However, it lacks flexibility in designing the liquid metal surface because many precursors could be damaged upon the exposure to high-power ultrasonication. The synthesis is typically performed in an ice bath to counteract the abundant heat generation that may cause the over-oxidation of liquid metals and damage the molecular reactants involved for surface decoration. However, for preparing Ga NPs, a properly elevated temperature (e.g., 60 °C) is required to ensure a favorable yield because of the higher m.p. of Ga than its eutectic alloys.30 Despite some success of applying the co-sonication strategy in functionalizing LMPs, studies on the underlying mechanisms have remained scarce. We herein conceive a simplified idea to model this process by studying the responses of different components to the ultrasonic power, including liquid metals, stabilizing agents, and even solvent molecules (Fig. 1b).
| H2O = H˙ + ˙OH |
| H˙ + H˙ = H2 |
| ˙OH + ˙OH = H2O2 |
| H˙ + O2 = HO2 |
| H˙ + HO2˙ = H2O2 |
| HO2˙ + HO2˙ = H2O2 + O2 |
The generated reactive species are responsible for redox reactions such as the surface oxidation of carbon nanomaterials47 and the reduction of metal salts into metallic nanoparticles,48,49 and can also function as initiators for free radical reactions such as free radical polymerization.50
Polymers, both natural and synthetic, may degrade into smaller fragments via ultrasonic breakage of C–C bonds.51 These fragments may appear as macroradicals, macromolecular ions, and macromolecules depending on the nature of degradation, and can recombine to generate macromolecules of the same or different lengths.52 Some molecules, e.g., surfactants, can form emulsions upon sonication in an appropriate solvent. In a word, ultrasonic processing creates an extensive molecular library for engineering the liquid metal surface.
3. Encapsulation of LMPs in synthetic architectures
Facilitated by the surface chemistry of liquid metals, various shell structures have been developed to encapsulate LMPs in the form of core–shell composites, rendering improved stability and multifunctionality. Unlike conventional metallic materials, the encapsulation of LMPs faces several challenges. (1) The energetic environment created by ultrasonication may be detrimental to in situ encapsulation because of the unpredictable side reactions of the shell building blocks. (2) The as-prepared LMPs are chemically and colloidally unstable, e.g., in the presence of water, oxygen, acid, base, and heat, making it hard for post-synthetic modifications. An essential point to solve these problems is selecting appropriate building blocks, typically ligand molecules that can tolerate the chaotic reaction system, to construct a primary stabilizing layer around LMPs. This step can also create a functional interface for post-synthetic fabrication of multilevel architectures through surface-supported reactions, e.g., polymerization, deposition, hydrolysis, and self-assembly (Fig. 1c). In this section, we will introduce the design, fabrication, and structures of different core–shell LMPs categorized by their shell compositions.3.1. Oxide skin
The simplest encapsulation form of LMPs is by their native oxide skin, forming an LM@Ga2O3 structure. Such an oxide skin is universal to LMPs synthesized by different methods and in different solvents, which appears as a complete surface coating and enables ligand-free stabilization of LMPs. As a model system for preparing LMPs, sonicating a liquid metal droplet in ethanol produces LMPs with an explicit core–shell structure (Fig. 2a).15 Interestingly, the shell can be divided into two layers, i.e., an oxide skin layer (∼3 nm) and a carbon layer, the latter of which is hypothesized to originate from the side reactions of ethanol molecules. This double layered shell is essential to protect LMPs from coalescing back into large aggregates, and stable suspensions of LMPs can thus be obtained for further use, e.g., as printable inks. Note that for some other solvents like dodecane and water, the carbon layer is not observed, which explains the higher stability of LMPs in ethanol over many hours. | ||
| Fig. 2 (a) TEM images and EDS mapping of EGaIn NPs stabilized by the native oxide skin. The LMPs are synthesized by sonication in ethanol. Scale bar: 10 nm. Reproduced with permission.15 Copyright 2015, Wiley-VCH. (b) TEM images of EGaIn NPs stabilized by ligand molecules C12 and 1ATC9. Scale bar: 50 nm. Reproduced with permission.27 Copyright 2011, American Chemical Society. (c) HRTEM images of EGaInSn NPs capped with a ligand layer of ethyl-3-mercaptopropionate. Scale bar: 2 nm. Reproduced with permission.6 Copyright 2016, Wiley-VCH. | ||
The degree of surface oxidation depends on many factors including particle size,53 oxygen content, ligands,28,54 temperature,35,55,56 sonication time/power, and solvent.15 Adjusting these conditions can alter the thickness, morphology, and composition of the oxide skin, and even transform LMPs into other materials due to intense over-oxidation that occurs at not only the surface, but also the interior. For example, sonicating a molten Ga droplet in water (55 °C, 2 h, 40% power) produces GaOOH nanorods, not Ga NPs.55 In addition, annealing dry powders of Ga NPs under 700 °C enables complete oxidation and crystallization of these particles into Ga2O3 NPs.56 These results also suggest that liquid metals and LMPs can serve as starting materials for Ga-based oxide and hydroxide materials.
Despite the straightforwardness of oxide skin encapsulation, several challenges exist. (1) The stabilizing effect of the instantly formed oxide skin is highly dependent on the solvent used for the synthesis. Only a few solvents, typically alcohols, have been proved effective in making stable suspensions of LMPs possibly due to their ability to generate a carbon surfactant layer upon ultrasonication. (2) Even in alcohol solutions, LMPs protected by the oxide skin show transient stability because the shell is too thin against continuous oxidation, fragile to force damage, and chemically sensitive to both acid and base etching. (3) It is hard to control the size and surface property of LMPs using the oxide skin as the only regulatory factor. In particular, the yield of LMPs whose size is below the microscale (<1 μm) is extremely low. Scientists have thus endeavored to develop alternative tools for stabilizing and manipulating LMPs, e.g., by ligand decoration.
3.2. Ligand molecule
Molecular ligands have long been used in endowing nanomaterials, both organic and inorganic, with colloidal stability and directing their growth and assembly toward a designed morphology. Ligand-capped LMPs can be easily fabricated by the co-sonication strategy. The important role of ligands is reflected in three aspects. (1) Stabilization. Ligand molecules can produce a protective shell around LMPs during co-sonication, offering an extra stability beyond the native oxide skin. (2) Manipulation. Ligand molecules help control the yield, size, and morphology of LMPs by cooperating with other parameters such as the reaction temperature. (3) Functionalization. The intrinsic functionalities of ligand molecules, e.g., molecular polarity, flexibility, and reactivity, are inherited in LMPs that facilitate their subsequent modifications and applications.The dispersibility of LMPs in solvents largely depends on their surface property and is tunable by ligand decoration. Alcohols, e.g., methanol, ethanol, and 2-propanol, are frequently used solvents for sulfhydryl ligands. The obtained LMPs are unstable when resuspended in aqueous solutions and rapidly form aggregates. Improving this dilemma can broaden the applicability of LMPs especially in biological systems. One solution is using sulfhydryl molecules conjugated with a hydrophilic skeleton or tail, e.g., 16-mercapto-hexadecanoic acid59 and methoxypoly(ethyleneglycol)thiol (mPEG-SH).60 These reactions still proceed in alcohol solutions but the products are dispersible in water.
Using water-soluble ligands is more straightforward to make aqueous suspensions of LMPs, which is challenging because of the uncontrollable oxidation of Ga into GaOOH in the presence of water. Such ligands must have strong affinity to the liquid metal surface and importantly, be able to prevent its over-oxidation in aqueous environments. Generally, positively charged molecules are preferred because the metal/metal oxide surface is typically negatively charged. Currently, the ligand pool for aqueous dispersions of LMPs is under exploration and some representative candidates include: (1) polymers and their derivatives that are soluble in water, especially those with abundant hydroxyl, ether, carbonyl, and amine groups that can strongly interact with the metal/metal oxide surface. Examples include poly(vinyl pyrrolidone) (PVP),38 poly(vinyl alcohol) (PVA),61 poly(ethylene glycol) (PEG),29 and polysaccharides such as alginate25 and cellulose;62 (2) surfactant molecules, typically cationic surfactants such as cetyl trimethyl ammonium bromide (CTAB);31,35 (3) phenolic compounds rich in catechol and pyrogallol groups that can chelate the liquid metal surface and alleviate its oxidation. Examples include dopamine,63 gallic acid,30 and even crude extracts from green tea. Note that by using gallic acid, Ga NPs are obtained under a high temperature of 60 °C without heavy oxidation due to the intrinsic anti-oxidation property of phenolic compounds; (4) biomacromolecules such as proteins. In particular, proteins with high isoelectric points (IEP) are greatly favored because of their positive charges under normal aqueous conditions, e.g., lysozyme.35,38
Under the ultrasonic environment, ligand molecules unavoidably experience complex physicochemical transformations. Nevertheless, the functionalities of these molecules are (at least partially) preserved as indicated by the dispersibility, surface charge, and surface reactivity of LMPs. For example, LMPs stabilized by carboxyl-terminated PEG can be modified with antibodies through carbodiimide-mediated chemical crosslinking.64
Ligand decoration of LMPs is also accomplishable by post-synthetic modifications, through physical adsorption31 or chemical conjugation.33 A recent work applies silanization in functionalizing the surface of LMPs.33 The hydroxylated surface of LMPs synthesized in ethanol can directly react with trialkoxysilanes with tailorable functional groups to produce a silanized surface. This strategy creates a highly designable surface for post-modifications. For example, using 3-(glycidyloxypropyl)trimethoxysilane as the ligand makes the LMPs polymerizable in air to form interparticle crosslinking.
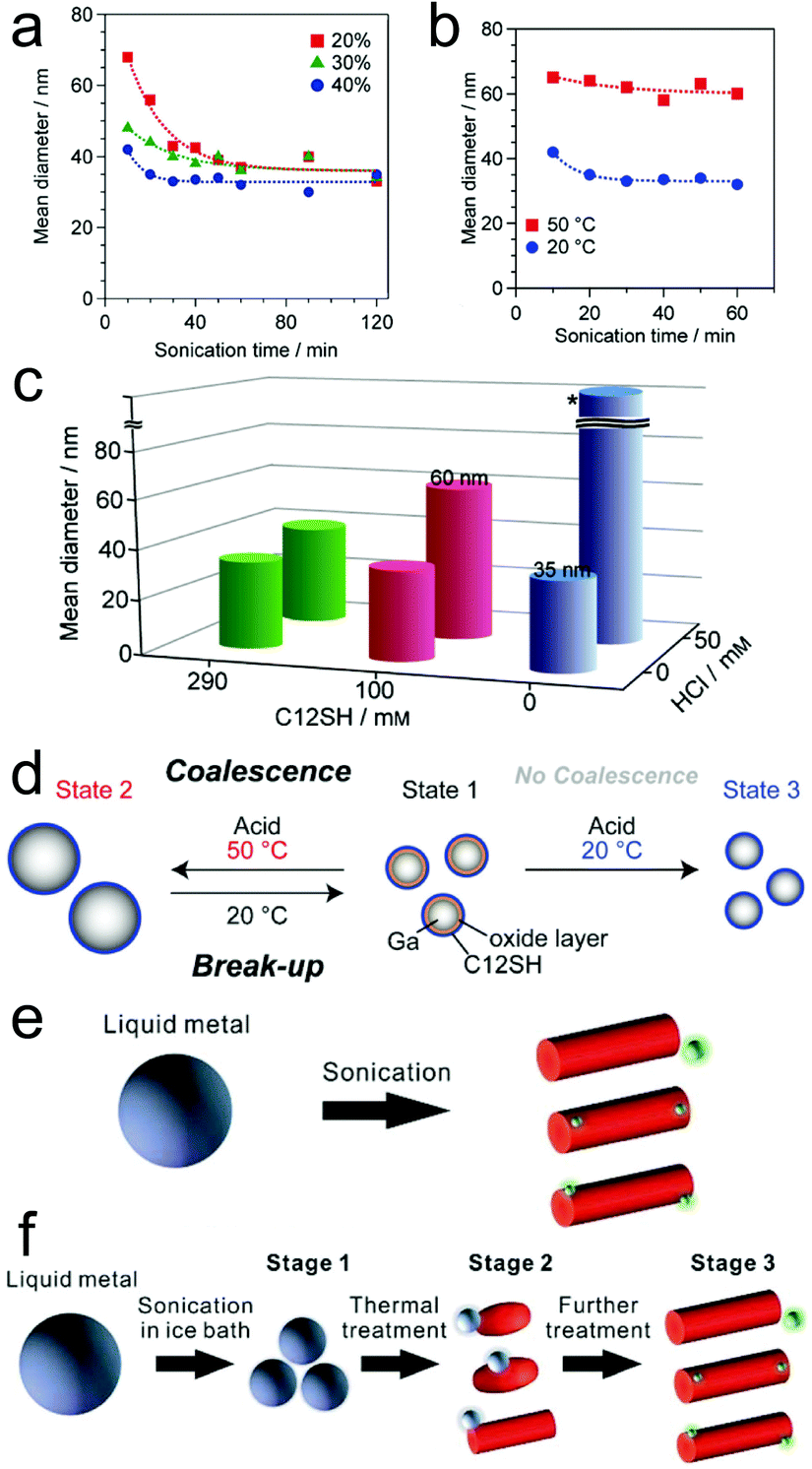 | ||
| Fig. 3 Size and morphology adjustment of LMPs. (a–c) The impact of different parameters (sonication time, power, reaction temperature, addition of acid) on the size of Ga NPs. The synthesis is performed in 2-propanol. (d) Reversible size control of Ga NPs. Reproduced with permission.57 Copyright 2015, Wiley-VCH. (e and f) Morphological transformation of LMPs in water by temperature control. Reproduced with permission.35 Copyright 2018, The Royal Society of Chemistry. | ||
The landscape of tuning the basic attributes of LMPs becomes more diverse when the solvent changes to water, harnessing the oxidation of Ga into GaOOH that causes both compositional and morphological transformations of the pristine liquid metal. For example, though ligand molecules (e.g., lysozyme and CTAB) can stabilize LMPs in a spherical core–shell configuration, this effect is conquered by heat-accelerated oxidation under a high reaction temperature (∼80 °C), producing GaOOH nanorods (Fig. 3e).35 For Ga-based alloys like EGaIn and galinstan, this process is ulteriorly accompanied by phase separation that generates progeny nanoparticles rich in In and Sn (green dots in Fig. 3e). Note that these phenomena are ligand-dependent, i.e., using gallic acid or HS-PEG-SH still produces LMPs but not GaOOH rods at high reaction temperatures.30,31 The shape morphing behavior can also be triggered by heating pre-synthesized LMPs (Fig. 3f). However, when the ligand molecules are crosslinked on the liquid metal surface, e.g., by glutaraldehyde, the morphological change is prohibited due to the improved protection from the crosslinked shell structure. These observations together suggest an efficient strategy for fabricating shape-transformable LMPs by ligand encapsulation and temperature control.
3.3. Polymer
Polymers represent a distinct type of ligand due to their large molecular weights and strong intra/intermolecular interactions. Their designable chain structures and abundant chemical groups can enrich the surface diversity of LMPs, creating a tailorable interface for further modification. Using polymers for liquid metal encapsulation has just started in the past few years. Methods to accomplish this include direct sonochemical assembly using pre-synthesized or commercially available polymers, and surface-supported polymerization following the pre-adsorption of monomers or initiators on LMPs (Fig. 4a). The former is highly straightforward and can be completed in one step, in which the polymers behave as a ligand and directly interact with the liquid metal surface.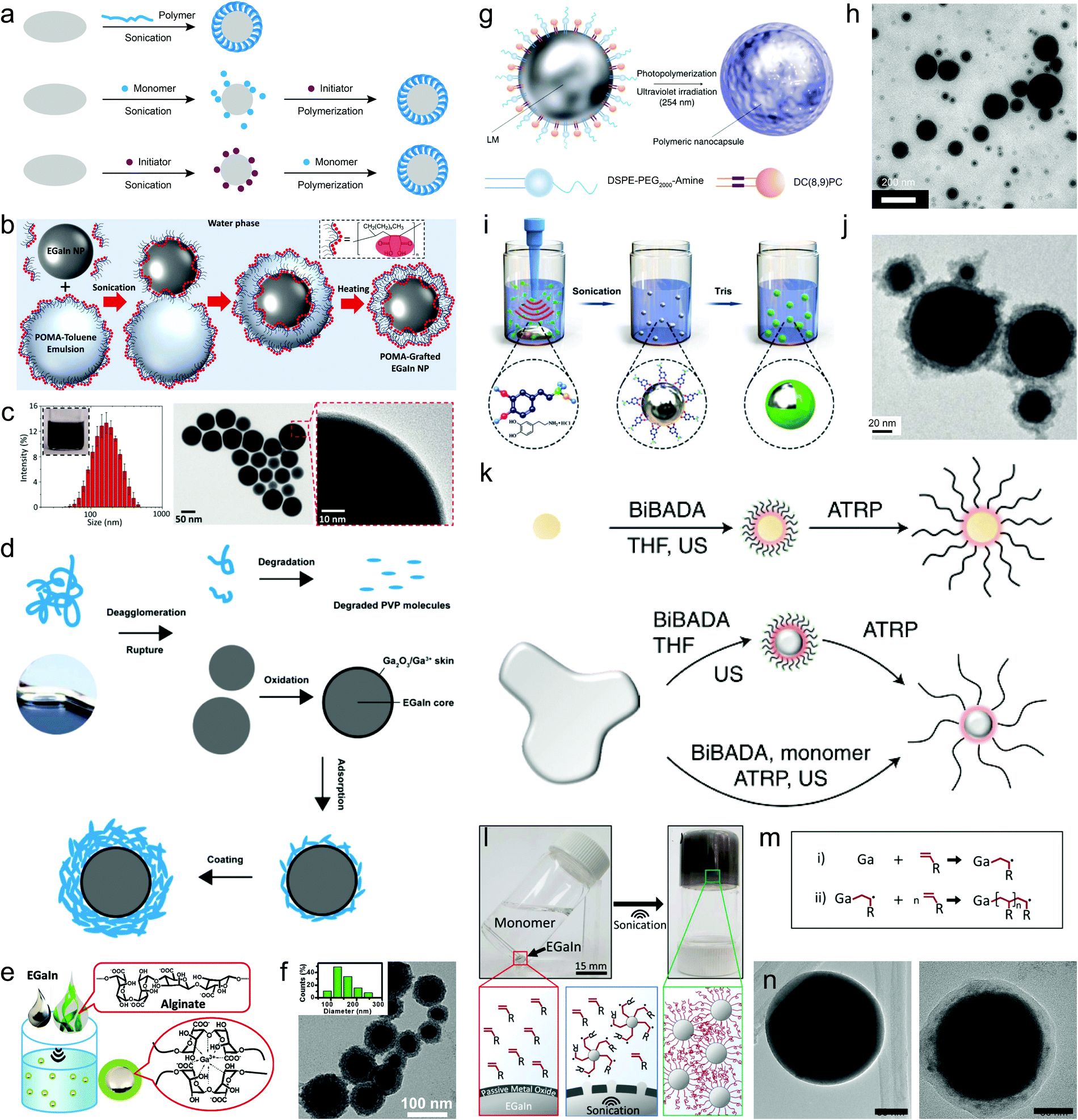 | ||
| Fig. 4 Polymer encapsulation of LMPs. (a) Three routes for the synthesis of polymer-wrapped LMPs. (b) Experimental setup of a sonication–emulsification method for preparing hydrophobic polymer-encapsulated EGaIn NPs (EGaIn@POMA NPs) in a biphasic solvent system. (c) Size distribution and TEM images of EGaIn@POMA NPs. Scale bar: 50 nm (middle) and 10 nm (right). Reproduced with permission.34 Copyright 2018, The Royal Society of Chemistry. (d) A schematic model showing sonication-induced dispersion and degradation of PVP macromolecules and their interactions with LMPs. Reproduced with permission.29 Copyright 2020, The Royal Society of Chemistry. (e) Schematic picture of LMPs produced by polysaccharide (alginate) encapsulation. (f) Size distribution and TEM image of EGaIn@alginate NPs. Scale bar: 100 nm. Reproduced with permission.25 Copyright 2018, Wiley-VCH. (g) Schematic illustration of encapsulating LMPs by light-triggered polymerization around the liquid metal surface. The ligand DC(8,9)PC contains light-responsive polymerizable units. (h) TEM image of LM NPs in (f). Scale bar: 200 nm. Reproduced with permission.65 Copyright 2017, Springer Nature. (i) Experimental setup of synthesizing polydopamine-coated LM NPs by a two-stage sonication-polymerization method. (j) TEM image of polydopamine-coated EGaIn NPs. Scale bar: 20 nm. Reproduced with permission.63 Copyright 2019, Wiley-VCH. (k) Schematic illustration of the surface atom transfer radical polymerization (ATRP) strategy for polymer encapsulation of LMPs. The initiator BiBADA is first modified onto the liquid metal surface by sonication that can trigger the following ATRP reaction to generate the polymer shell. Reproduced with permission.69 (l) Liquid metal-initiated polymerization of a vinyl monomer, acrylamide. (m) The proposed mechanism of Ga-triggered free radical polymerization. (n) TEM images of bare LMPs (left) and LMPs encapsulated in polymers (right). Scale bar: 15 mm in (l), 50 nm in (n). Reproduced with permission.71 Copyright 2019, American Chemical Society. | ||
Using hydrophilic polymers as coating ligands avoids the generation of metastable emulsions and the use and removal of toxic solvents. PVP, a frequently used stabilizer and dispersant for traditional metallic nanomaterials, can be directly deposited onto LMPs by co-sonication, forming a shell up to ∼20 nm that helps improve their stability in water to 30 days without extra protection.29 During sonication, the polymer chains are proved to undergo dispersion, secession, and recombination, and eventually assemble into a thick coating (Fig. 4d). Polysaccharides, e.g., alginate, can also form a densely packed coating around LMPs (Fig. 4e).25 Alginate can chelate the surface Ga3+ and form a microgel network to stabilize LMPs. The particle size and shell thickness can be controlled by varying the concentration of alginate and the shell can reach up to 20 nm when 1 wt% alginate is used (Fig. 4f). Compared to small molecules, polymer ligands produce much thicker shell structures (tens of nanometers) that can be clearly distinguished even under low magnification TEM.
![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C–CC–) modules that can polymerize upon ultraviolet light irradiation.65 The monomer, namely 1,2-bis(10,12-tricosadiynoyl)-sn-glycero-3-phosphocholine (DC(8,9)PC), is used together with 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[amino(polyethylene glycol)-2000] (DSPE-PEG2000-amine) to pre-stabilize EGaIn NPs by co-sonication (Fig. 4g). Afterwards, the mixture is exposed to a 254 nm lamp under room temperature to initiate the polymerization of DC(8,9)PC and its assembly with DSPE-PEG2000-amine into a compact polymer coating (Fig. 4h). The PEG moieties and cationic trimethyl amino groups endow the LMPs with good dispersibility in aqueous solvents, which can benefit their biomedical applications.
C–CC–) modules that can polymerize upon ultraviolet light irradiation.65 The monomer, namely 1,2-bis(10,12-tricosadiynoyl)-sn-glycero-3-phosphocholine (DC(8,9)PC), is used together with 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[amino(polyethylene glycol)-2000] (DSPE-PEG2000-amine) to pre-stabilize EGaIn NPs by co-sonication (Fig. 4g). Afterwards, the mixture is exposed to a 254 nm lamp under room temperature to initiate the polymerization of DC(8,9)PC and its assembly with DSPE-PEG2000-amine into a compact polymer coating (Fig. 4h). The PEG moieties and cationic trimethyl amino groups endow the LMPs with good dispersibility in aqueous solvents, which can benefit their biomedical applications.
The pre-adsorbed monomers on LMPs can be crosslinked by chemical initiators as well. A simple demonstration is polydopamine-coated EGaIn NPs,63 in which EGaIn is first sonicated in a dopamine chloride solution and then a base is introduced to increase the pH and initiate dopamine polymerization (Fig. 4i). A polydopamine shell is found under TEM whose thickness can reach ∼20 nm after 6 h polymerization (Fig. 4j). Despite the robustness of dopamine chemistry, the need of alkaline pH for initiating the polymerization may damage the encapsulation efficiency because it can destroy the oxide skin of LMPs. In another work, a fibrous polyaniline (PANI) shell is fabricated around LMPs by ammonium persulfate-initiated polymerization of aniline.68 The key to this design lies in the first step of immobilizing a comonomer p-phenylenediamine (PPD) on LMPs by co-sonication, which functions as a polymerization enhancer that significantly reduced the acid concentration needed for synthesizing PANI from ≥0.1 M to <0.01 M. This is extremely important to protecting the liquid metal core that is sensitive to acid etching.
To develop a tailorable surface for customized polymer grafting, liquid metal EGaIn is co-sonicated with an initiator 12-(2-bromoisobutyramido)dodecanoic acid (BiBADA) that can trigger atom transfer radical polymerization (ATRP) of added monomers (Fig. 4k).69 The produced LMPs can initiate the polymerization of various monomers on their surfaces including poly(methyl methacrylate) (PMMA), poly(n-butyl acrylate) (PBMA), poly(2-(dimethylamino)ethyl methacrylate) (PDMAEMA), and poly(n-butyl acrylate-block-methyl methacrylate) (PBA-b-PMMA), forming a well-defined core–shell structure. Given the broad applicability of ATRP, a wide range of polymeric structures are expected to be incorporated with LMPs for desired functionalization. However, the entire workflow of this approach proceeds in hazardous organic solvents and requires ultrafine control of reaction parameters such as complete degassing of oxygen.
3.4. Inorganic coating
Surface-initiated Galvanic replacement can also produce metal/metal oxide shells, which can occur both during the ultrasonication of bulk droplets and on the surface of pre-stabilized LMPs. The Galvanic replacement of liquid metals (typically Ga) was first disclosed in 2017 exploring their interactions with AuBr4− and Ag+,37 and has extended to other metals including Al,74 Mn,39 Cu,75 Pt,76 and Pd.77 Sonicating liquid metals in a solution of KAuBr4 or AgNO3 produces rice-shaped alloys, not core–shell particles.37 However, these reactions proceed differently in a dip coating scenario, i.e., immersing liquid metal droplets or LMPs in a metal salt solution, producing a rough metallic coating. For example, LMPs pre-stabilized with lysozyme or PVP can react with KAuBr4 to form the Au shell (Fig. 5a and b).38 The shell composition is tunable by the reaction kinetics,78i.e., Au for a fast reaction rate and AuGa2 for a slow reaction rate. Similarly, dispersing LMPs in a CuSO4 solution generates a CuxO shell whose composition and morphology can be tuned by pH.75 Though the acquisition of the well-defined Au/Ag shells through co-sonication has been a failure, a recent example successfully fabricated an EGaIn@MnO2 composite by sonicating a liquid metal droplet in the KMnO4 solution. The LMPs possess a double shell comprising an inner GaOOH layer encapsulated by a laminated MnO2 layer (Fig. 5c and d).39 Note that powerful sonication can also reduce metal salts into nanoparticles,48 which may compete with the Galvanic replacement on LMPs and affect the shell structure.
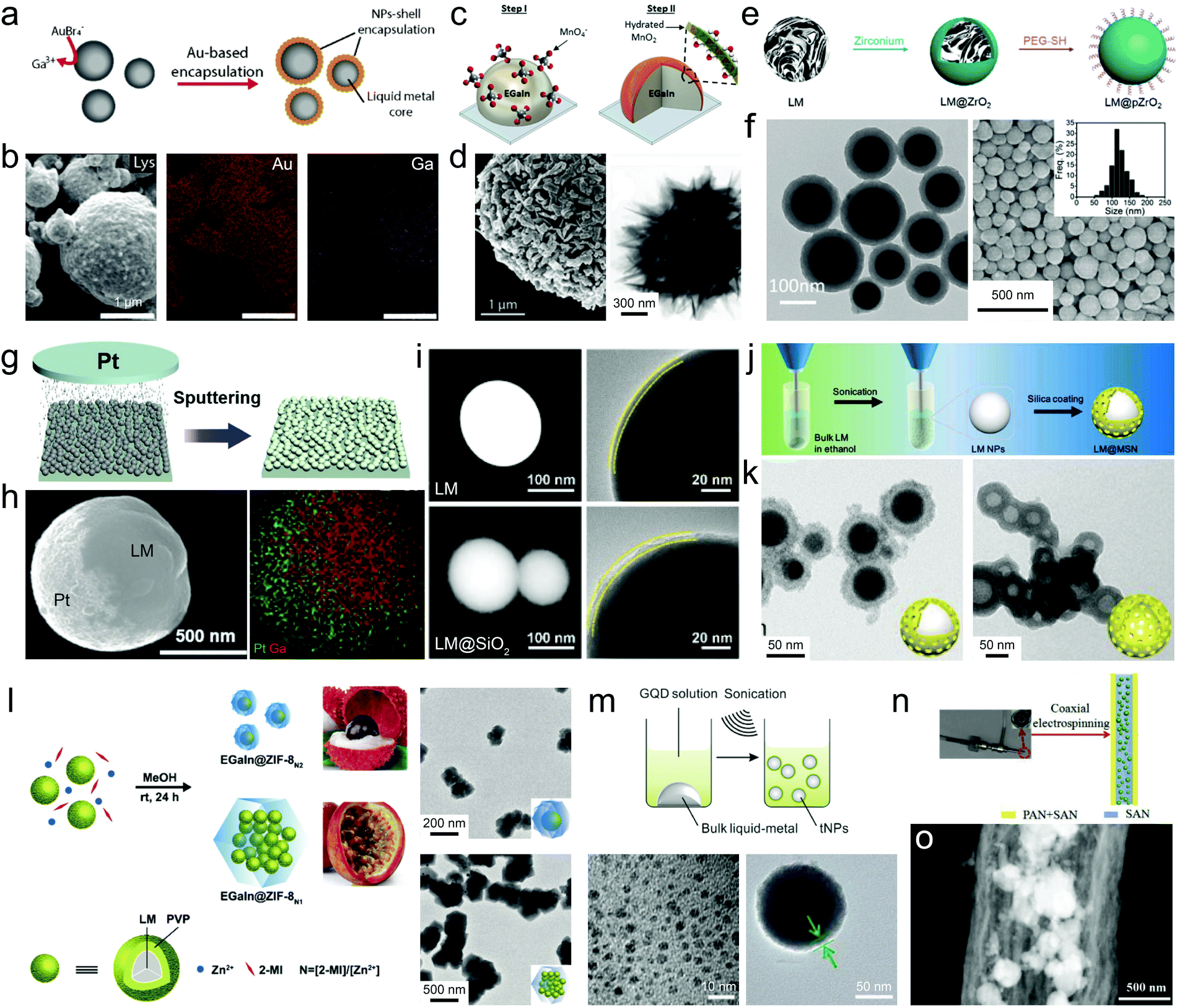 | ||
| Fig. 5 LMPs encapsulated in inorganic coatings. (a) Schematic illustration of the synthesis of Au-coated EGaInSn NPs by Galvanic replacement. (b) SEM images and element mapping of LM@Au NPs. Scale bar: 1 μm. Reproduced with permission.38 Copyright 2019, The Royal Society of Chemistry. (c) Schematic illustration of the synthesis of a MnO2 shell around EGaIn NPs by Galvanic replacement. (d) SEM and TEM images of EGaIn@MnO2 NPs. Scale bar: 1 μm (left) and 300 nm (right). Reproduced with permission.39 Copyright 2019, Wiley-VCH. (e) Schematic illustration of the synthesis of a ZrO2 shell around EGaIn NPs by a hydrolysis method. (f) TEM and SEM images of EGaIn@ZrO2 NPs. Scale bar: 100 nm (left) and 500 nm (right). Reproduced with permission.80 Copyright 2019, The Royal Society of Chemistry. (g) Deposition of a Pt layer on LMPs by sputtering. (h) SEM image and EDS mapping of LMPs half-coated with Pt. Scale bar: 500 nm. Reproduced with permission.81 Copyright 2019, Wiley-VCH. (i) SEM and TEM images of bare LMPs and LM@SiO2 NPs. The silica coating is fabricated by hydrolysis of TEOS. Scale bar: 100 nm in the left three columns, 20 nm in the right column. Reproduced with permission.43 Copyright 2019, American Chemical Society. (j) Experimental setup of the synthesis of a mesoporous silica shell around EGaIn NPs by hydrolysis method. (k) TEM images of EGaIn@mSiO2 NPs (left) and the mSiO2 shell after etching the core (right). Scale bar: 50 nm. Reproduced with permission.83 Copyright 2019, Elsevier. (l) Schematic illustration of the synthesis of a MOF shell around EGaIn NPs by polymer-bridged growth. TEM images show different configurations of EGaIn@ZIF-8 NPs by tuning the ligand to metal ion ratio. Scale bar: 200 nm (top) and 500 nm (bottom). Reproduced with permission.85 Copyright 2020, The Royal Society of Chemistry. (m) Schematic illustration of the synthesis of a shell composed of assembled GQDs by co-sonication. TEM images show GQDs (left) and EGaIn@GQDs NPs (right). Scale bar: 10 nm (left) and 50 nm (right). Reproduced with permission.86 Copyright 2017, American Chemical Society. (n) Experimental setup for preparing EGaSn NPs encapsulated in hollow carbon nanofibers by electrospinning. (o) TEM image of LMPs in carbon nanofibers. Scale bar: 500 nm. Reproduced with permission.88 Copyright 2019, Elsevier. | ||
Despite the robustness of Galvanic replacement, its heterogeneous feature causes anisotropic growth of the shell, producing porous lamination structures with improved surface area but impaired stability against a harsh environment. Besides, unpredictable side reactions accompanying this process, e.g., alloying and dealloying, have not been fully understood, which can introduce undesired modifications to the product.
Post-synthetic strategies such as wet-chemical deposition using pre-synthesized LMPs as seeds enable continuous, densely packed metal/metal oxide shells. This can be achieved by adding precipitants together with metal salts into a dispersion of LMPs (e.g., glucose and Ag(NH3)2OH, the well-known silver mirror reaction).79 Hydrolysis of metal alkoxides can generate an isotropic metal oxide coating around LMPs. For example, zirconium n-propoxide that can hydrolyze into ZrO2 in the presence of ammonia has been used to fabricate EGaIn@ZrO2 NPs (Fig. 5e).80 The ZrO2 shell whose thickness can exceed 20 nm displays a dense smooth morphology, which allows long-term stabilization of the inner liquid metal (Fig. 5f).
Physical methods are also applicable to fabricating metal/metal oxide coatings. In a recent work, Au, Ag, and Pt are coated onto the hemisphere of the pre-synthesized LMPs by a sputtering method (Fig. 5g), forming a Janus-type structure (Fig. 5h).81 However, this method requires good arrangement of LMPs into a monolayer, which is hard to achieve for large scale manipulations.
4. Promoted applications of core–shell LMPs
Liquid metals have become a hot topic in nanoscience and nanotechnology. To name a few, their electrical/thermal conductivities combined with liquid-like fluidity make it promising for soft electrical/thermal conductors and devices. Their acid-sensitive skin and photothermal conversion capacity are useful for controlled drug release in tumor microenvironments and photothermal therapy. They are also potential catalysts and energy storage materials. The encapsulation of LMPs in designed shell structures endows them with enhanced stability and tunable even unexpected surface, optical, mechanical, and electrical properties, creating complex and innovative functionalities for various applications (Fig. 1d and e). These include but not limited to robust processability and stability even under extreme environments, good printability, diverse sintering behaviors, enhanced targeting ability, photothermal efficacy, catalytic activity, and energy conversion capability. In this section, we will discuss how the fundamental properties of LMPs are tuned by surface encapsulation and how this can benefit their applications.4.1. Soft electronics
Liquid metals with high conductivity, liquid-like fluidity, and self-healing properties are dream materials for soft, flexible, and stretchable electronic devices, including conductors, energy harvesters, solders, and even insulators.9 Due to the robust processability, adjustable size and surface structure, and most importantly, the capability to merge into continuous liquid metal paths, LMPs have become a promising alternative of bulk liquid metals in these applications. LMPs are typically used in the form of solution processable, printable inks or fillers in polymer matrices.LMPs typically prepared in solutions are compatible with industrialized printing techniques, e.g., inkjet printing94 and screen printing.95 By rationally designing their surface, the dispersibility and stability of LMPs in various solvents can be tuned and thus both oily and aqueous electronic inks can be obtained, in which the latter is greener and more sustainable. For example, LMPs encapsulated in an alginate-derived nanogel show improved stability in aqueous solutions and can be used as printable inks for soft circuits.25 Also in this work, the adhesive alginate shell endows LMPs with strong interactions with various substrate materials and good film-forming properties upon solvent evaporation, which facilitates the formation of circuit patterns on different surfaces including polyethylene terephthalate, cellulose, polyvinylidene fluoride, nylon, leaf, and skin.
The operation of LMP-based conductors is generally based on the stimuli-responsive, and sometimes reversible transformation of LMPs from particulate insulators to fluidic conductors. After printing, LMPs have to be sintered into a continuous liquid metal phase to acquire conductivity due to their insulating oxide skin, which can be achieved by applying force (e.g., compressional and frictional force),15,94 heat,96 and laser irradiation.97–100 Some pioneering works done in 2015 employed oxide skin-stabilized LMPs, either dispersed in ethanol94 or as liquid inclusions in PDMS matrices,101 in developing mechanically sintered conductors. Later, the size-dependent response of LMPs to mechanical sintering is also revealed.102 These early attempts formulate a successful paradigm for sintering LMPs by breaking up the insulating oxide skin, and since then LMPs-incorporated conductors have entered an age of rapid development.
Methods for sintering oxide skin-stabilized LMPs are readily extendable to LMPs encapsulated in multilevel shell structures using modified parameters. However, what attracts more attention is that the unique responsiveness of a shell structure can expand the sintering techniques for LMPs and simplify or even eliminate this procedure. This is illustrated in several representative examples. In the first one, EGaIn NPs are stabilized by a bifunctional ligand 11-phosphono-undecylacrylate whose phosphonic acid head can bind the oxide skin and the acrylate tail is polymerizable upon photoinitiation (Fig. 6a).66 The polymer shell allows efficient interparticle crosslinking into polymerized liquid metal networks (Poly-LMNs) that can be cast as a densely packed layer on a stretchable substrate. The insulating Poly-LMNs are responsive to uniaxial elongation and become conductive after stretching-induced rupture (108-fold increase in conductivity upon 300% strain), which does not occur for unpolymerized particles. Importantly, the acquired conductivity is well retained during multiple stretch–release cycles (up to 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles under 100% strain) due to the autonomously formed hierarchical structures after the first cycle. This design inspires a new activation mode of liquid metal particles, i.e., by stretching, which is promising for next-generation stretchable conductors and devices.
000 cycles under 100% strain) due to the autonomously formed hierarchical structures after the first cycle. This design inspires a new activation mode of liquid metal particles, i.e., by stretching, which is promising for next-generation stretchable conductors and devices.
 | ||
| Fig. 6 LMPs for soft electronics. (a) Schematic illustration of the synthesis of polymerized liquid metal networks (Poly-LMNs) for stretching-responsive conductors. Reproduced with permission.66 Copyright 2019, Wiley-VCH. (b) Proposed mechanism of CNFs-assisted sintering by increasing the capillary force during solvent evaporation. SEM images show the cross sections of the paths composed of LMPs with (top) and without (bottom) the CNFs shell. Scale bar: 1 μm (top) and 2 μm (bottom). Reproduced with permission.62 Copyright 2019, Springer Nature. (c) Humidity-responsive sintering of PVP-coated LMPs. Reproduced with permission.103 Copyright 2020, The Royal Society of Chemistry. (d) SEM image of LMPs encapsulated in a Ag shell to provide initial conductivity. Scale bar: 1 μm. Reproduced with permission.79 Copyright 2020, Wiley-VCH. (e) The influence of particle size on the break down strength and stretchability of LMP-included dielectric elastomers. Reproduced with permission.107 Copyright 2019, Wiley-VCH. (f) The influence of particle size on the freezing and melting temperatures of liquid metals and liquid metal-polymer composites. Reproduced with permission.108 Copyright 2019, Wiley-VCH. (g) SEM images of the secreted liquid metal solders from PVP-coated LMPs. Scale bar: 0.5 mm. (h) Liquid metal motors for welding Ag nanowires. Scale bar: 500 nm. Reproduced with permission.81 Copyright 2020, Wiley-VCH. (i) The corrosion of Al plates in contact with LMPs and bulk liquid metal droplets respectively. Reproduced with permission.115 Copyright 2018, The Royal Society of Chemistry. | ||
Encapsulation of LMPs in a nanofibril shell enables an evaporation-induced sintering technique.62 The liquid metal ink is produced by sonicating EGaIn along with cellulose nanofibrils (CNFs) in water. The nanofibril shell, as well as free CNFs in the solution, are essential to the increased capillary force generated by solvent evaporation that can rupture the encapsulated LMPs to acquire conductivity. This is possibly mediated by decreasing the size and contact angle of LMPs, and splitting the liquid bridges between the droplets (Fig. 6b). Consequently, the LM/CNFs composites are sintered into a bilayer structure in which a CNF layer floats on a continuous liquid metal film, but the control group using unmodified LMPs remains undisturbed. This phenomenon, namely evaporative sintering is extendable to other fibrils such as silk NFs and amyloid NFs, but the underlying mechanism especially the crucial role of free CNFs remains to be further resolved. Besides, the time required to complete the sintering process is much longer than other methods (up to 12 h) to ensure a reasonable evaporation rate. Recently, a humidity-responsive liquid metal circuit is developed based on LMPs encapsulated in hydroscopic polymers such as PVP.103 The printed LMPs become conductive after a wet–dry cycle due to the shrinkage of PVP molecules that generates a compressive force (Fig. 6c). In contrast, bare LMPs show no response. This work offers a new route for sintering LMPs that is also potentially useful in humidity sensing.104
It is possible to eliminate the sintering step by encapsulating inactivated LMPs in an intrinsically conductive shell composed of metals79 or conductive polymers.68 For example, immersing a printed path of LMPs in KAuBr4 triggers Galvanic replacement and produces a conductive coating of Au, which significantly reduces its resistance from 11.09 MΩ to 4.3 Ω after the reaction for 60 min due to the generation and electrical contact among the surface Au NPs.38 A recent work further advances this concept by depositing a conductive Ag layer onto pre-stabilized LMPs to make an intrinsically conductive liquid metal ink (Fig. 6d).79 The Ag coating endows LM NPs with an initial conductivity after printing that depends on the shell thickness, e.g., 0.2 Ω sq−1 for 259 nm. When a force is applied, the Ag shell can be ruptured and the encapsulated liquid metals are released, providing a self-repairing property for the conductive filler. A bending test shows that the circuit made of LM@Ag particles is much more stable than that made of commercial Ag particles during 5 bending cycles, showing a resistance change (R/R0) of only 1.2. This design strategy provides an efficient solution to flexible electronics by the synergy of inflexible conductive fillers with flexible liquid metals to achieve both high initial conductivity and flexibility, akin to the co-printing of LMPs and silver microparticles.105 Besides metallic coatings, conductive polymers such as PANI have also been exploited to encapsulate LMPs to achieve sintering-free conductivity.68 These attempts together suggest the power of surface engineering in tuning the sintering behaviors of LMP-based electronics.
In an early work of dielectric elastomers, doping LMPs (∼15 μm in diameter) in PDMS at a high volume percent of 50% results in an over 400% increase in the dielectric constant and a lower dielectric dissipation factor.106 Importantly, the hyperelastic dielectric composite can undergo significant deformations without the degradation of electrical and mechanical properties. However, using microparticles results in a low electrical breakdown strength. Recently, the same research group has systematically investigated the size effect in tuning the performances of dielectric elastomers with liquid metal inclusions.107 It is found that smaller LMPs (1 and 0.1 μm) yield higher breakdown strength and also higher mechanical stretchability than larger particles (10 μm) (Fig. 6e). This is because smaller particles can reduce the intensity of internal charge and field concentrations that lead to electrical breakdown. The possible explanations for these phenomena are also given by calculation and simulation.
Adjusting the size of LMPs can modulate their phase transformation behaviors by changing the freezing and melting points of liquid metals. These two values (f.p. and m.p.) do not match because of the supercooling effect of liquid metals. The f.p. of EGaInSn nanoparticles (∼100 nm in diameter) is depressed by 150 °C (from 10 to −140 °C) compared to bulk EGaInSn.6 This phenomenon is valuable for liquid metals to preserve their fluidity, softness, and compliance to mechanical deformations under ultralow working temperatures. One of the most promising applications is to develop highly durable liquid metal-embedded polymer composites. By reducing the size of EGaIn to ∼2 μm, its f.p. decreases from −5.9 to −84.1 °C and its m.p. decreases from 17.8 to −25.6 °C.108 Embedding these LMPs in elastomer matrices generates a thermoelectric energy harvester that can operate and still remains stretchable under low temperatures (Fig. 6f). This work demonstrates a new benefit of LMPs in developing durable devices for extreme cold conditions such as in the deep sea and even in space. However, the accurate and even continuous modulation of the f.p. and m.p. of liquid metals by size adjustment has remained elusive, leaving much room for future exploitations.
Liquid metals have also been exploited as thermal conductors for cooling electronic devices such as chips. In these applications, the liquid metal material should be in close contact with the chip packaging material, usually aluminum silicon alloy. However, the highly reactive Ga-based alloys can etch the oxide skin of Al and thus Al will react with the oxygen molecules in the air, which generates much alumina and causes Al corrosion. Using LMPs (embedded in polymer matrices) that are thermally conductive but electrically insulating significantly alleviates this problem (Fig. 6i).115 Besides, nanoscale LMPs are superior than microscale LMPs because if larger particles are used, liquid metal droplets may extrude from the matrix upon strong compression. This design is also applicable in other scenarios where thermal conductivity is required but electrical conductivity is strictly forbidden.
4.2. Nanomedicine
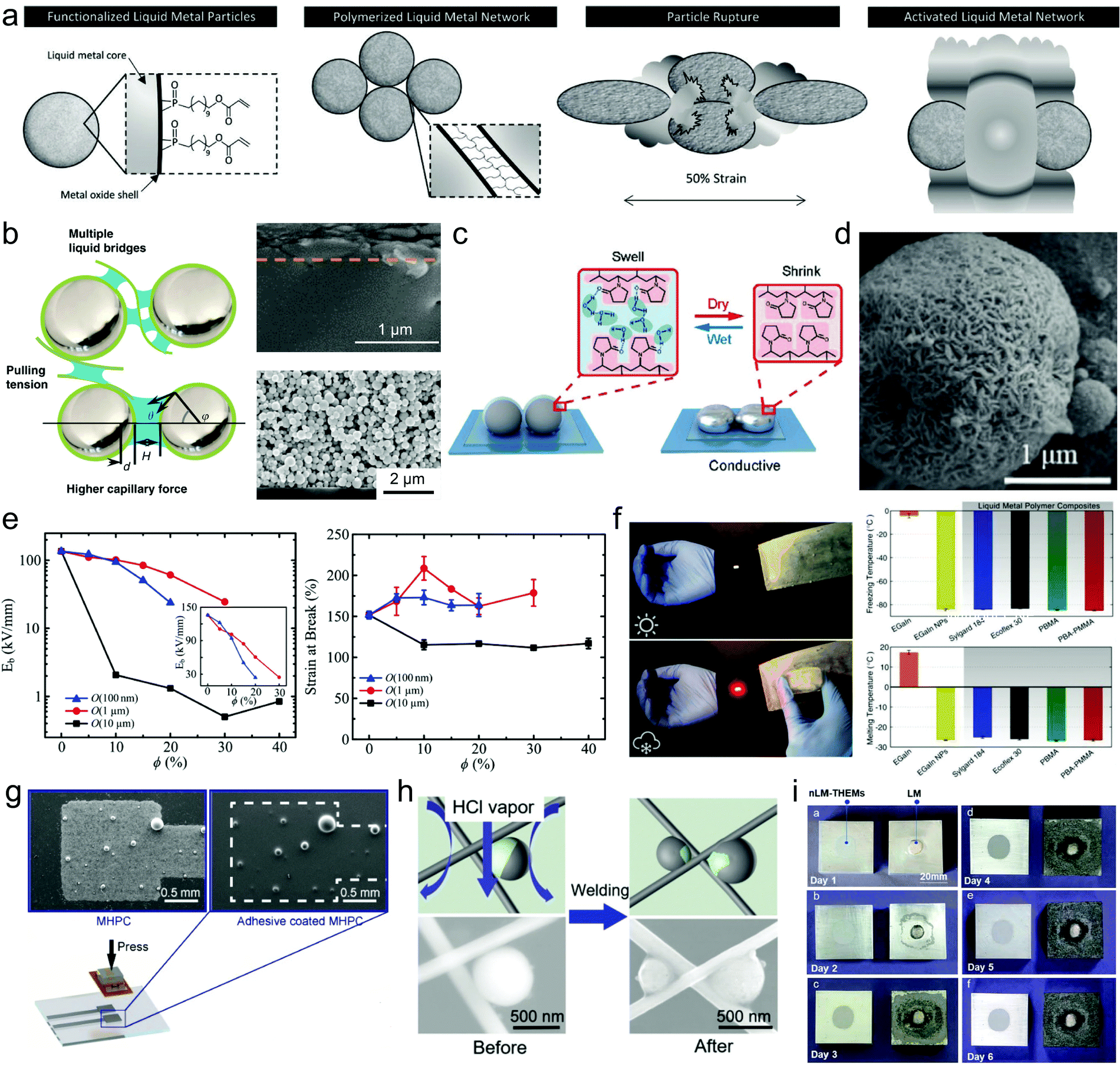
LMPs are promising for nano–bio technology due to some featured properties such as pH-responsive particle collapse, oxidation-triggered morphing, and photothermal conversion capacity. They have emerged as an innovative nanomedicine for cancer treatment. LMPs tend to form an unstable nano–bio interface in biological media because of their poor colloidal stability in an aqueous environment. Hydrophilic ligands, e.g., mPEG-SH, are thus commonly used to stabilize LMPs in aqueous formulations, which also help reduce their size to the nanoscale, a reasonable length scale for biomedicine.60 LMPs equipped with abundant surface groups from their shell structures are modifiable with functional biomolecules for targeting, delivery, and therapeutics, which can be realized by various types of interactions including covalent conjugation, electrostatic/hydrophobic adsorption, and host–guest recognition. For example, LMPs encapsulated in a polymeric skeleton with an amine tail are compatible with carbodiimide-mediated conjugation chemistry and can be modified with avidin and then biotin-labeled antibodies (Fig. 7a).65 LMPs coated with mPEG-SH possess a positive surface charge of 14.3 eV. Thus, negatively charged biomolecules, e.g., glucose oxidase (GOX) with an isoelectric point of ∼4.9, can adsorb onto their surface by electrostatic interaction (Fig. 7b).60 Encapsulation of LMPs in a mesoporous SiO2 shell allows the adsorption of doxorubicin (Dox) in the pore structures.83 A thiolated (2-hydroxypropyl)-β-cyclodextrin (MUA-CD) shell can serve as the host for the guest molecule triethanolamine-treated Dox, realizing an optimized loading capacity of 24% (Fig. 7c).58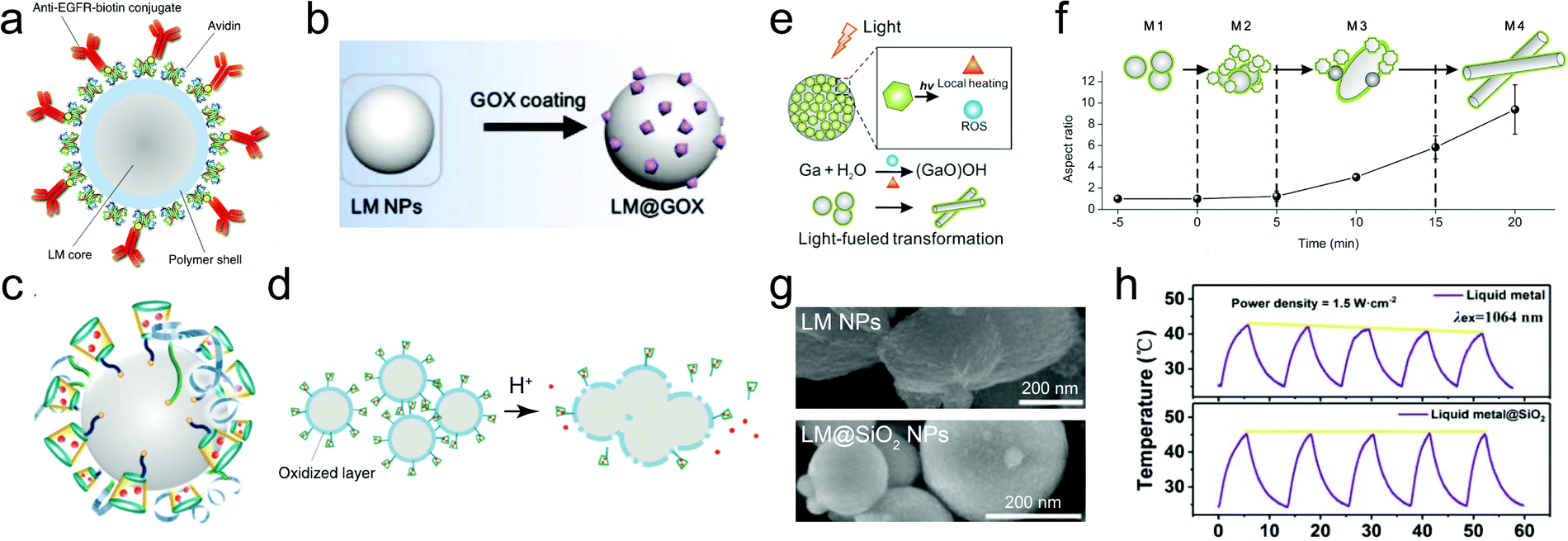 | ||
| Fig. 7 LMPs for nanomedicine. (a) Polymer-encapsulated LMPs for bioconjugation with antibodies. Reproduced with permission.65 Copyright 2017, Springer Nature. (b) LMPs encapsulated in mPEG for enzyme loading by electrostatic adsorption. Reproduced with permission.60 Copyright 2019, Elsevier. (c) LMPs encapsulated in MUA-CD for drug loading by host–guest interaction. (d) LMPs for controlled drug release by acid-triggered particle fusion. Reproduced with permission.58 Copyright 2015, Springer Nature. (e) Schematic illustration of GQD-induced generation of heat and reactive oxygen species upon light irradiation, which cause the morphological change of LMPs for controlled drug release and enhanced endosomal escape. (f) Variation of the aspect ratio of LMPs upon light irradiation, indicating the phase separation and shape morphing process. Reproduced with permission.86 Copyright 2017, American Chemical Society. (g) SEM images of bare LMPs and LM@SiO2 NPs after NIR light irradiation. The silica coating can protect the liquid metal core against oxidation and dealloying during photothermal conversion. Scale bar: 200 nm. (h) Cycling performance of LMPs and LM@SiO2 NPs during photothermal conversion. LM@SiO2 NPs exhibit improved photothermal stability than unprotected particles. Reproduced with permission.43 Copyright 2019, American Chemical Society. | ||
Upon arrival at the target sites such as tumors, LMPs allow controlled release of loaded drugs by external stimuli such as pH gradient and light irradiation. The oxide skin of LMPs can be etched by acid and alkali and after that particle fusion appears. This is detrimental for liquid metal-based circuits because it may destroy the contact between the printed patterns and the substrate. However, the instantly formed, pH-responsive shell is valuable for controlled drug release in cancer therapeutics due to the acidic tumor microenvironment (Fig. 7d).58 The fusion of LMPs can be stimulated by light irradiation as well, which causes thermal expansion and collapse of the particles, enabling light-controlled drug release.43,60,65,83,86 LMPs exhibit a broad light absorption band throughout the ultraviolet/visible/near-infrared (UV/vis/NIR) region due to their large polydispersity in size. Thus, NIR light is typically used because of its long penetration depth in tissues.
Note that light-triggered collapse of LMPs is commonly accompanied by two other processes. One is heat generation and the other is oxidation of Ga. The generated heat is applicable in photothermal therapy and also accelerates the oxidation kinetics of Ga. Consequently, LMPs transform into rod-like GaOOH particles with an enhanced aspect ratio. This shape morphing behavior is valuable in modulating the bio–nano interactions of liquid metal nanomaterials. Recently, researchers have attempted to manipulate this process by fabricating a light-responsive shell around LMPs for enhanced energy conversion and on-demand particle oxidation.86 GQDs are used as building blocks for a carbon shell, which serve as energy collectors to absorb photoenergy and convert it to heat and reactive oxygen species (ROS) (Fig. 7e), driving the phase separation and morphological change of the EGaIn core (Fig. 7f). Upon light irradiation, In separates from the core into spherical nanoparticles and Ga is oxidized into GaOOH rods. This transformation not only allows light-controlled drug release, but also facilitates endosomal escape after cellular uptake by physically disrupting the endosome membrane, offering new possibilities for the dynamic morphological control of LMPs in living organisms.
A severe problem accompanying the shape transformation of LMPs is that it leads to irreversible loss of photothermal conversion efficiency due to phase separation and change in particle composition. This situation is relieved in two recent works by silica coating.43,83 The dense, rigid silica shell around LMPs that not only prevents the penetration of oxygen and water but also restricts the thermal expansion of the core (Fig. 7g). Improved photothermal stability is thus achieved for EGaIn@SiO2 NPs during five on/off cycles in which no significant deterioration of the temperature is observed (Fig. 7h). This work repairs a fetal defect of liquid metal-based photothermal nanoagents, i.e., poor recyclability. Importantly, the silica coating enhances the light absorption of LMPs in the NIR-I to NIR-II region and an enhanced photothermal conversion efficiency is observed for EGaIn@SiO2 NPs (22.43%) compared to uncoated LMPs (14.12%). Similar results are also obtained by using a light-absorbing shell, polydopamine.63
4.3. Catalysis and energy
Early reports have employed bulk liquid metal droplets and also LMPs (typically from Ga only) as immobilized catalysts for synthesizing graphene,116 carbon nanotubes,117 and GaN118 and SiO2 nanowires119 by chemical vapor deposition. This field has recently been revisited in preparing large-area two-dimensional materials.24,120,121 Recently, there is a trend to incorporate metal/metal oxide nanostructures onto the surface of LMPs to make them catalytically reactive, whose performances benefit from not only the introduced shell structures, but also the synergistic interaction between the core and the shell. LMPs containing alloyed Au and Ag are reactive for reducing methylene blue in the presence of NaBH4.37 Also by Galvanic replacement, a surface layer of nanostructured Pd can be anchored on LMPs immobilized on a macroporous glass substrate, forming a supported liquid catalytic system (Fig. 8a and b).77 This catalyst is applied to butane dehydrogenation and shows improved conversion, selectivity, and also less sensitive deactivation compared to traditional solid phase catalysts. The highly dynamic liquid core is essential to homogenizing the active sites and preserving the activity against coking. Despite rigorous studies on the phase state of this catalyst, further classifications on its structure perhaps by microscopic observations are required.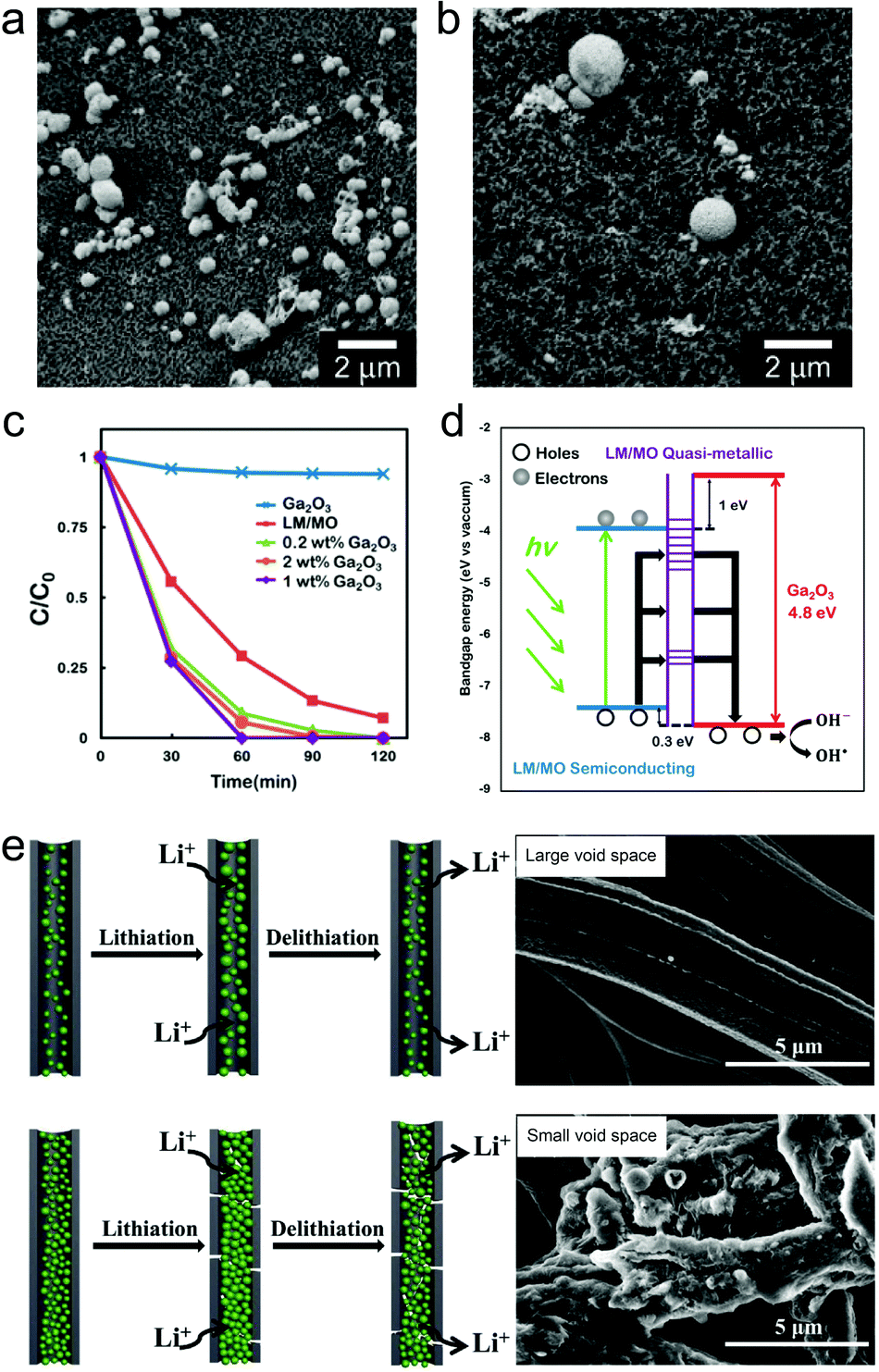 | ||
| Fig. 8 LMPs for catalysis and energy. (a and b) SEM images of LMPs before (a) and after (b) loading the Pd nanocatalysts by Galvanic replacement at the surface. Scale bar: 2 μm. Reproduced with permission.77 Copyright 2017, Springer Nature. (c) Catalytic degradation of Congo red by LMPs with a shell composed of pre-synthesized Ga2O3 NPs. (d) Proposed mechanism of the enhanced catalytic activity of Ga2O3 by electron–hole coupling with the liquid metal core. Reproduced with permission.73 Copyright 2015, American Chemical Society. (e) LMPs encapsulated in hollow carbon nanofibers for lithium-ion batteries. The void space is critical for tolerating the volume expansion of LMPs during cycling. SEM images show fibers with different void space. Scale bar: 5 μm. Reproduced with permission.88 Copyright 2019, Elsevier. | ||
LM/MO frameworks, i.e., LMPs coated with nanostructured metal oxides such as Ga2O3,40,73 MnO2,39 and CuO,75 have emerged as potential photocatalysts. LMPs synthesized in water are covered by an incomplete shell of scattered Ga2O3 NPs, which demonstrates an activity in the photocatalytic degradation of Congo red (CR).40 The catalytic efficiency can be further enhanced by assembling an additional shell composed of the pre-synthesized Ga2O3 NPs (Fig. 8c).73 This is attributed to several synergistic effects from core–shell interactions because free Ga2O3 NPs alone show limited catalytic activity. (1) The broad light absorption of LMPs across the UV/vis/NIR region allows efficient utilization of the photoenergy from solar light especially in the visible region. (2) LMPs contains a quasi-metallic part with dominant sub-bandgap states, which can form pseudo-Ohmic contacts between the semiconducting part and the surface Ga2O3 NPs (Fig. 8d). This structure facilitates nearly free transfer of holes with higher oxidizing potential from LMPs to the valence band of Ga2O3, contributing to the promoted decomposition of organic species. Coupling of a metal oxide shell such as MnO2 can also reduce the bandgap of LMPs that expands the photocatalytic reaction toward the visible light range with higher rate of exciton photogeneration.39
Liquid metals are functional materials for energy storage and conversion devices such as batteries and supercapacitors.87,122–124 Their unique properties such as inherent deformability and self-repairing potential provide new solutions to problems faced by conventional energy materials.125 Researchers have employed nanoparticles of Ga-based alloys as self-healing electrode materials for lithium-ion batteries (LIBs) due to the high lithiation capacity of Ga (769 mA h g−1), In (1011 mA h g−1), and Sn (993 mA h g−1).123,124 However, these applications have been challenged by the insufficient surface protection of LMPs, which may cause irreversible collapse and aggregation of particles during electrode fabrication and lithiation-induced volume expansion. A PVP shell allows good dispersion of LMPs in the complex electrode matrix facilitated by their strong interactions with binders and conductive additives.29 The conventionally used current collectors increase not only the workload of electrode preparation, but also the risk of particle detachment during cycling. To address these problems, a free-standing liquid metal electrode is designed by positioning LMPs (alloy of Ga and Sn) inside carbon nanofibers.88 The void space is tunable by varying the content of LM NPs and it is confirmed that an adequate void space is essential for tolerating the volume expansion and promoting the cycling performance (Fig. 8e). This concept may be extended to individual LMPs for energy storage, i.e., core@void@shell LMPs, which remains to be studied in the future.
5. Summary and outlook
We have introduced a batch of representative studies of advanced liquid metal nanomaterials featuring a designable core–shell structure. The versatile shell structure together with its synergy with the core can impart multifunctionality to LMPs that is essential for promoting their applications (Table 1). However, it should be noted that the performances of LMPs do not always benefit from a smaller size or an extra shell protection. An obvious phenomenon is that larger, bare LMPs are easier to be sintered by compression than smaller, protected ones.102| Core | Shell | Synthesis | Featured property | Application | Ref. |
|---|---|---|---|---|---|
| a Photo-initiated polymerization. b Atom transfer radical polymerization. c Hydrolysis of metal precursors. d Reduction of metal salts. e Hydrolysis of silica precursors. f Surface assembly of MOF precursors. g Graphene quantum dots. h Carbon nanofibers. | |||||
| EGaIn | Oxide skin | Sonication in ethanol | Compression-induced sintering | Soft antenna | 15 |
| EGaIn | Thiolated ligand (1ATC9, C12) | Sonication in ethanol | Tunable SPR property | — | 27 |
| Ga | Thiolated ligand (C12) | Sonication in 2-propanol | Reversible size control | — | 57 |
| EGaIn | Thiolated ligand (MUA-CD, m-HA) | Sonication in ethanol | Host–guest recognition, acid-triggered particle fusion | Anticancer drug delivery | 58 |
| Ga, EGaIn | Molecular ligand (e.g., CTAB) | Sonication in water | Heat-induced phase separation and morphing | — | 35 |
| EGaIn | Polymer (DSPE-PEG 2000-amine, DC(8,9)PC) | Post-modificationa | Bioconjugation, photothermal activity, enhanced X-ray contrast | Photothermal therapy, CT imaging | 65 |
| EGaIn | Polymer (acrylate) | Post-modificationa | Stretching-induced sintering | Stretchable heater | 66 |
| EGaIn | Polymer (e.g., POMA) | Sonication in water/toluene | Stable in aqueous solutions | — | 34 |
| EGaIn | Polymer (cellulose) | Sonication in water | Solvent evaporation-induced sintering | Soft actuator | 62 |
| EGaIn | Polymer (e.g., PMMA) | Post-modificationb | Processability in polymer matrices | Soft composites | 69 |
| EGaIn | Metal oxide (MnO2) | Galvanic replacement | Tunable bandgap and catalytic activity | Photocatalysis | 39 |
| EGaIn | Metal oxide (ZrO2) | Post-modificationc | Photothermal activity and stability, enhanced X-ray contrast | Photothermal therapy, CT imaging | 80 |
| EGaIn | Metal (Ag) | Post-modificationd | Intrinsic conductivity | Soft circuit | 79 |
| EGaIn | Silica | Post-modificatione | Photothermal activity and stability | Photothermal therapy | 43 and 83 |
| EGaIn | MOFs (ZIF-8) | Post-modificationf | Tunable SPR property | — | 85 |
| EGaIn | Carbon (GQDs)g | Sonication in water | Photothermal activity, light-induced phase separation and morphing | Anticancer drug delivery | 86 |
| EGaSn | Carbon (CNFs)h | Electrospinning | Void space, self-healing property | Lithium ion battery | 88 |
The surface engineering of LMPs is still at an early stage of development and there is much room for further exploring this field. Some important topics are discussed in the following paragraphs.
Manufacturing
A handbook is required to guide the reproducible, reliable, and predictable engineering of the liquid metal surface. The first priority is to screen potential ligand molecules and reaction systems that can yield stable LMPs in a desired solvent, which will provide a rich source of information about the fundamentals of their ligand chemistry and size/structure control. This is also important to transplanting established synthetic chemistries to the surface of LMPs and will ultimately benefit the multilevel encapsulation of LMPs, e.g., multishell structure, yolk–shell structure, and Janus structure. Nanomaterials, e.g., quantum dots and 2D materials such as graphene,126 can also function as surface stabilizers for LMPs. Exploring the core–shell interactions in such LMPs may help develop novel structures and functionalities.Scientists are encouraged to develop new concepts, methods, and devices for synthesizing LMPs and functionalizing their surface. A promising direction is solvent-free mechanochemistry,127e.g., grinding128 and templated filtration,18 which is green and easily scalable. Ligand exchange reactions are valuable for manipulating the surface of LMPs on demand, which has not been achieved yet due to the complexity in their surface interactions. Flow synthesis in microfluidic devices is also applicable to LMPs.17,64 Its unique advantages such as precise adjustment of reaction parameters, continuous processing, and high throughput can benefit the screening of parallel reactions.129
Compared to bottom-up synthesis, top-down synthesis of LMPs capable of gram-scale processing is more feasible for industrialization. However, the obtained LMPs show unevenly distributed size, quality, and properties, which cannot satisfy real applications. Finding appropriate stabilizers that can yield a narrow size distribution is important. Size selection technologies are also helpful to solve this dilemma, e.g., by centrifugal130 and chromatographic methods.131 We emphasize this as a very important topic. Unfortunately, to our knowledge, little has been carried out in this area.
Structure–property relationship
Disclosing the regulating effect of shell encapsulation on the basic attributes of LMPs can in return promote their design and applications. Opportunities exist in tuning the responses of LMPs to optical, mechanical, electrical, and thermal stimuli, and exploring novel properties and applications based on these interplays.The first topic focuses on the phase state of LMPs. Unlike conventional materials, Ga is featured by supercooling and superheating behaviors.132 The phase states of LMPs are determined by their composition, size, and the temperature of their surroundings. However, the underlying correlations still remain disputable.133 Adjusting the particle size of LMPs through ligand manipulation may help resolve the size-dependent phase diagrams of liquid metals. In addition, the phase behaviors of LMPs subjected to nanoshell confinement may differ from unrestrained particles, particularly in the cases of metal/metal oxide shells that may alter the lattice structures at the core–shell interface.
The phase separation of LMPs represents another fertile area. Previous studies are focused on over oxidation-induced phase separation of LMPs, together with the shape morphing from particles into rods. It seems attractive to obtain core–shell particles merely by phase engineering. A very recent study employs an in situ cooling method that helps uncover the co-existence of a solid In core wrapped by a liquid Ga shell after phase separation.134 New core–shell structures with unexplored properties are highly anticipated if this phenomenon could be expanded to batch synthesis.
Similar to Al, Ga-based LMPs are UV plasmonic materials whose SPR band is located in the UV region.135 There are few reports describing the SPR property of LMPs dispersed in solutions due to the difficulty in preparing monodispersed LMPs.136 The SPR band of LMPs can be tuned by surface encapsulation, e.g., a nanocoating of MOFs,85 which can guide their applications in plasmonic sensing and photothermal conversion.137 Opportunities also exist in tuning the response of LMPs to mechanical, electrical, and thermal stimuli by engineering their composition, size, and surface.
The behaviors of liquid metals in magnetic fields have not been fully understood. Liquid metal droplets are found to be magnetocaloric materials.138,139 By incorporating with magnetic particles such as Fe3O4 and Ni, liquid metal-based magnetofluids have been engineered for magnetically controlled motion and printing.140 It is appealing to duplicate these concepts to LMPs by monitoring their responses to magnetic stimulation and by surface decoration with magnetic components, which may inspire their use in magnetically responsive imaging, drug delivery, and therapy.
Another topic is the safety of LMPs. The systematic toxicity of LMPs to cells, tissues, organs, and animal bodies has remained ambiguous, though liquid metals especially Ga are generally accepted as nontoxic materials.141 Exploring the factors determining the bioeffect and biosafety of LMPs is valuable for their safe use in biomedicine. This may start with their surface engineering that is responsible for adjusting their size and surface property as well as formatting the material–bio interface upon their first contact with biosystems. The safety of LMPs used in other scenarios also deserves attention. For example, electronic inks composed of LMPs should be nontoxic to both the human body and the environment.
An important, and also useful paradigm to study the structure-determined properties and functions of LMPs is by combining theoretical simulations with experimental measurements. Some groups have applied multiphysics methods in predicting the mechanical and electrical performances of liquid metal-embedded elastomers. However, before applying these models in core–shell LMPs, elaborate descriptions on the surface/interface parameters of LMPs should be provided. In addition, the unevenly distributed size of LMPs could further complicate the simulation process. Molecular dynamics simulation, which is frequently used in chemistry and biology, may help uncover the submicroscopic details of the formation as well as interfacial behaviors of core–shell LMPs. However, a few studies have appeared in this field, i.e., simulating the chemical processes at the surface/interface of LMPs, which may direct the research of predictable LMPs in the near future.
Application
Offering improved stability and tailorablity, surface encapsulation can dramatically expand the application horizons of LMPs in many hot areas. Liquid metal-involved polymer composites are emerging materials for thermal management, electronics, robotics, and injectable biomedicine.142–147 Conventional fabrication of such composites relies on the mechanical shearing of liquid metal droplets in viscous polymer matrices, which suffers from large, uneven particle size and poor colloidal stability. Surface-engineered LMPs provide an excellent alternative for liquid metal/polymer composites because their size and surface chemistry are highly controllable.69 Importantly, LMPs decorated with polymerizable or crosslinkable surfaces can participate in the in situ formation of polymer matrices.67,144 In a recent work, LMPs stabilized by an alginate nanogel could form hydrogels in contact with Ca2+, and are used as implantable radiopaque embolic materials for tumor embolotherapy.144Surface decoration of stimuli-responsive components that help produce a propelling force have enabled micro/nanomotors based on LMPs.81 Compared to larger-sized liquid metal marbles,113,148 these materials are superior for microscopic scenarios, e.g., microwelding and nanomedicine. However, the design and function of such materials are limited. Future efforts are directed to coupling various components to the surface of LMPs to enable multiple modes of manipulation, e.g., by chemical gradient, light, and electrical/magnetic field.
The liquid metal surface also promises a powerful platform for running synthetic chemistries to prepare molecules and materials with potentially unexpected structures and properties. These species can be regarded as the “byproducts” of surface coating formation. Some results have been obtained in synthesizing 2D metal/metal oxide nanomaterials by surface-initiated Galvanic replacement39,75 and polymers by Ga-enhanced free radical polymerization.70,71 Further investigations and expansion of the chemical reactivity of liquid metals may create a win–win game play for surface engineering and material synthesis.
In summary, LMPs represent a unique material having different new knowledge from conventional solid counterparts. This poses challenges for their surface engineering but also implies great opportunities in developing innovative structures and functionalities. Enabled by a deep insight into liquid metal surface chemistry, there are tremendous prospects in employing customized LMPs for real applications.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the National Natural Science Foundation of China (51973045) for financial support.Notes and references
- T. Daeneke, K. Khoshmanesh, N. Mahmood, I. A. de Castro, D. Esrafilzadeh, S. J. Barrow, M. D. Dickey and K. Kalantar-zadeh, Chem. Soc. Rev., 2018, 47, 4073–4111 RSC.
- K. Khoshmanesh, S.-Y. Tang, J. Y. Zhu, S. Schaefer, A. Mitchell, K. Kalantar-Zadeh and M. D. Dickey, Lab Chip, 2017, 17, 974–993 RSC.
- K. Kalantar-Zadeh, J. Tang, T. Daeneke, A. P. O'Mullane, L. A. Stewart, J. Liu, C. Majidi, R. S. Ruoff, P. S. Weiss and M. D. Dickey, ACS Nano, 2019, 13, 7388–7395 CrossRef CAS.
- H. Song, T. Kim, S. Kang, H. Jin, K. Lee and H. J. Yoon, Small, 2020, 16, e1903391 CrossRef.
- Y. Lin, J. Genzer and M. D. Dickey, Adv. Sci., 2020, 2000192 CrossRef CAS.
- L. Ren, J. Zhuang, G. Casillas, H. Feng, Y. Liu, X. Xu, Y. Liu, J. Chen, Y. Du, L. Jiang and S. X. Dou, Adv. Funct. Mater., 2016, 26, 8111–8118 CrossRef CAS.
- M. H. Malakooti, M. R. Bockstaller, K. Matyjaszewski and C. Majidi, Nanoscale Adv., 2020, 2, 2668–2677 RSC.
- S. Chen, H. Z. Wang, R. Q. Zhao, W. Rao and J. Liu, Matter, 2020, 2, 1446–1480 CrossRef.
- M. D. Dickey, Adv. Mater., 2017, 29, 1606425 CrossRef.
- L. Zhu, B. Wang, S. Handschuh-Wang and X. Zhou, Small, 2020, 16, e1903841 CrossRef.
- J. Yan, Y. Lu, G. Chen, M. Yang and Z. Gu, Chem. Soc. Rev., 2018, 47, 2518–2533 RSC.
- S.-T. Liang, H.-Z. Wang and J. Liu, Chem. – Eur. J., 2018, 24, 17616–17626 CrossRef CAS.
- X. Guo, L. Zhang, Y. Ding, J. B. Goodenough and G. Yu, Energy Environ. Sci., 2019, 12, 2605–2619 RSC.
- M. Yarema, M. Woerle, M. D. Rossell, R. Erni, R. Caputo, L. Protesescu, K. V. Kravchyk, D. N. Dirin, K. Lienau, F. von Rohr, A. Schilling, M. Nachtegaal and M. V. Kovalenko, J. Am. Chem. Soc., 2014, 136, 12422–12430 CrossRef CAS.
- Y. Lin, C. Cooper, M. Wang, J. J. Adams, J. Genzer and M. D. Dickey, Small, 2015, 11, 6397–6403 CrossRef CAS.
- I. D. Tevis, L. B. Newcomb and M. Thuo, Langmuir, 2014, 30, 14308–14313 CrossRef CAS.
- S. Y. Tang, I. D. Joshipura, Y. Lin, K. Kalantar-Zadeh, A. Mitchell, K. Khoshmanesh and M. D. Dickey, Adv. Mater., 2016, 28, 604–609 CrossRef CAS.
- D. Wang, C. Gao, W. Wang, M. Sun, B. Guo, H. Xie and Q. He, ACS Nano, 2018, 12, 10212–10220 CrossRef CAS.
- M. Yunusa, G. J. Amador, D. M. Drotlef and M. Sitti, Nano Lett., 2018, 18, 2498–2504 CrossRef CAS.
- A. Zavabeti, T. Daeneke, A. F. Chrimes, A. P. O'Mullane, J. Zhen Ou, A. Mitchell, K. Khoshmanesh and K. Kalantar-Zadeh, Nat. Commun., 2016, 7, 12402 CrossRef CAS.
- Y. Liu, J. Li and W. Zhang, Chem. Commun., 2020, 56, 6229–6232 RSC.
- T. Y. Liu, P. Sen and C. J. C. J. Kim, J. Microelectromech. Syst., 2012, 21, 443–450 CAS.
- A. Zavabeti, J. Z. Ou, B. J. Carey, N. Syed, R. Orrell-Trigg, E. L. H. Mayes, C. Xu, O. Kavehei, A. P. O'Mullane, R. B. Kaner, K. Kalantar-zadeh and T. Daeneke, Science, 2017, 358, 332–335 CrossRef CAS.
- B. J. Carey, J. Z. Ou, R. M. Clark, K. J. Berean, A. Zavabeti, A. S. R. Chesman, S. P. Russo, D. W. M. Lau, Z.-Q. Xu, Q. Bao, O. Kevehei, B. C. Gibson, M. D. Dickey, R. B. Kaner, T. Daeneke and K. Kalantar-Zadeh, Nat. Commun., 2017, 8, 14482 CrossRef CAS.
- X. Li, M. Li, L. Zong, X. Wu, J. You, P. Du and C. Li, Adv. Funct. Mater., 2018, 28, 1804197 CrossRef.
- P. D. Jadzinsky, G. Calero, C. J. Ackerson, D. A. Bushnell and R. D. Kornberg, Science, 2007, 318, 430–433 CrossRef CAS.
- J. N. Hohman, M. Kim, G. A. Wadsworth, H. R. Bednar, J. Jiang, M. A. LeThai and P. S. Weiss, Nano Lett., 2011, 11, 5104–5110 CrossRef CAS.
- Z. J. Farrell and C. Tabor, Langmuir, 2018, 34, 234–240 CrossRef CAS.
- Y. Liu, Q. Wang, S. Bi, W. Zhang, H. Zhou and X. Jiang, Nanoscale, 2020, 12, 13731–13741 RSC.
- F. Centurion, M. G. Saborio, F.-M. Allioux, S. Cai, M. B. Ghasemian, K. Kalantar-Zadeh and M. A. Rahim, Chem. Commun., 2019, 55, 11291–11294 RSC.
- X. Sun, M. Sun, M. Liu, B. Yuan, W. Gao, W. Rao and J. Liu, Nanoscale, 2019, 11, 2655–2667 RSC.
- Y. Hou, C. Lu, M. Dou, C. Zhang, H. Chang, J. Liu and W. Rao, Acta Biomater., 2020, 102, 403–415 CrossRef CAS.
- Z. J. Farrell, C. Thrasher, A. Flynn and C. Tabor, ACS Appl. Nano Mater., 2020, 3, 6297–6303 CrossRef CAS.
- Y. Lin, J. Genzer, W. Li, R. Qiao, M. D. Dickey and S.-Y. Tang, Nanoscale, 2018, 10, 19871–19878 RSC.
- Y. Lin, Y. Liu, J. Genzer and M. D. Dickey, Chem. Sci., 2017, 8, 3832–3837 RSC.
- X. Xia, Y. Wang, A. Ruditskiy and Y. Xia, Adv. Mater., 2013, 25, 6313–6333 CrossRef CAS.
- F. Hoshyargar, J. Crawford and A. P. O'Mullane, J. Am. Chem. Soc., 2017, 139, 1464–1471 CrossRef CAS.
- R. David and N. Miki, Nanoscale, 2019, 11, 21419–21432 RSC.
- M. B. Ghasemian, M. Mayyas, S. A. Idrus-Saidi, M. A. Jamal, J. Yang, S. S. Mofarah, E. Adabifiroozjaei, J. Tang, N. Syed, A. P. O'Mullane, T. Daeneke and K. Kalantar-Zadeh, Adv. Funct. Mater., 2019, 29, 1901649 CrossRef.
- W. Zhang, J. Z. Ou, S.-Y. Tang, V. Sivan, D. D. Yao, K. Latham, K. Khoshmanesh, A. Mitchell, A. P. O'Mullane and K. Kalantar-zadeh, Adv. Funct. Mater., 2014, 24, 3799–3807 CrossRef CAS.
- S.-Y. Tang, R. Qiao, Y. Lin, Y. Li, Q. Zhao, D. Yuan, G. Yun, J. Guo, M. D. Dickey, T. J. Huang, T. P. Davis, K. Kalantar-Zadeh and W. Li, Adv. Mater. Technol., 2019, 4, 1800420 CrossRef.
- H. Xu, B. W. Zeiger and K. S. Suslick, Chem. Soc. Rev., 2013, 42, 2555–2567 RSC.
- P. Zhu, S. Gao, H. Lin, X. Lu, B. Yang, L. Zhang, Y. Chen and J. Shi, Nano Lett., 2019, 19, 2128–2137 CrossRef CAS.
- H. Robatjazi, D. Weinberg, D. F. Swearer, C. Jacobson, M. Zhang, S. Tian, L. Zhou, P. Nordlander and N. J. Halas, Sci. Adv., 2019, 5, eaav5340 CrossRef CAS.
- S. Handschuh-Wang, Y. Z. Chen, L. F. Zhu and X. C. Zhou, ChemPhysChem, 2018, 19, 1584–1592 CrossRef CAS.
- P. R. Gogate and A. L. Prajapat, Ultrason. Sonochem., 2015, 27, 480–494 CrossRef CAS.
- Y. C. Xing, L. Li, C. C. Chusuei and R. V. Hull, Langmuir, 2005, 21, 4185–4190 CrossRef CAS.
- K. Okitsu, A. Yue, S. Tanabe, H. Matsumoto and Y. Yobiko, Langmuir, 2001, 17, 7717–7720 CrossRef CAS.
- H. Xu and K. S. Suslick, ACS Nano, 2010, 4, 3209–3214 CrossRef CAS.
- H. Mohapatra, M. Kleiman and A. P. Esser-Kahn, Nat. Chem., 2017, 9, 135–139 CrossRef CAS.
- A. Mehrdad, J. Appl. Polym. Sci., 2011, 120, 3701–3708 CrossRef CAS.
- A. H. Lebovitz, M. K. Gray, A. C. Chen and J. M. Torkelson, Polymer, 2003, 44, 2823–2828 CrossRef CAS.
- E. Sutter and P. Sutter, J. Phys. Chem. C, 2012, 116, 20574–20578 CrossRef CAS.
- N. J. Morris, Z. J. Farrell and C. E. Tabor, Nanoscale, 2019, 11, 17308–17318 RSC.
- V. B. Kumar, A. Gedanken and Z. Porat, Ultrason. Sonochem., 2015, 26, 340–344 CrossRef CAS.
- S. Sudo, K. Kokado and K. Sada, RSC Adv., 2017, 7, 678–683 RSC.
- A. Yamaguchi, Y. Mashima and T. Iyoda, Angew. Chem., Int. Ed., 2015, 54, 12809–12813 CrossRef CAS.
- Y. Lu, Q. Hu, Y. Lin, D. B. Pacardo, C. Wang, W. Sun, F. S. Ligler, M. D. Dickey and Z. Gu, Nat. Commun., 2015, 6, 10066 CrossRef CAS.
- Y. Yu and E. Miyako, Angew. Chem., Int. Ed., 2017, 56, 13606–13611 CrossRef CAS.
- J.-J. Hu, M.-D. Liu, F. Gao, Y. Chen, S.-Y. Peng, Z.-H. Li, H. Cheng and X.-Z. Zhang, Biomaterials, 2019, 217, 119303 CrossRef CAS.
- M. Liao, H. Liao, J. Ye, P. Wan and L. Zhang, ACS Appl. Mater. Interfaces, 2019, 11, 47358–47364 CrossRef CAS.
- X. Li, M. Li, J. Xu, J. You, Z. Yang and C. Li, Nat. Commun., 2019, 10, 3514 CrossRef.
- T. Gan, W. Shang, S. Handschuh-Wang and X. Zhou, Small, 2019, 15, 1804838 CrossRef.
- S. Y. Tang, R. Qiao, S. Yan, D. Yuan, Q. Zhao, G. Yun, T. P. Davis and W. Li, Small, 2018, 14, e1800118 CrossRef.
- S. A. Chechetka, Y. Yu, X. Zhen, M. Pramanik, K. Pu and E. Miyako, Nat. Commun., 2017, 8, 15432 CrossRef CAS.
- C. J. Thrasher, Z. J. Farrell, N. J. Morris, C. L. Willey and C. E. Tabor, Adv. Mater., 2019, 31, e1903864 CrossRef.
- J. E. Park, H. S. Kang, J. Baek, T. H. Park, S. Oh, H. Lee, M. Koo and C. Park, ACS Nano, 2019, 13, 9122–9130 CrossRef CAS.
- C. Zhang, F.-M. Allioux, M. A. Rahim, J. Han, J. Tang, M. B. Ghasemian, S.-Y. Tang, M. Mayyas, T. Daeneke, P. Le-Clech, R. B. Kaner, D. Esrafilzadeh and K. Kalantar-Zadeh, Chem. Mater., 2020, 32, 4808–4819 CrossRef CAS.
- J. Yan, M. H. Malakooti, Z. Lu, Z. Wang, N. Kazem, C. Pan, M. R. Bockstaller, C. Majidi and K. Matyjaszewski, Nat. Nanotechnol., 2019, 14, 684–690 CrossRef CAS.
- T. Gan, S. Handschuh-Wang, W. Shang, J. Shen, L. Zhu, Q. Xiao, S. Hu and X. Zhou, Macromol. Rapid Commun., 2019, 40, e1900537 CrossRef.
- J. Ma, Y. Lin, Y.-W. Kim, Y. Ko, J. Kim, K. H. Oh, J.-Y. Sun, C. B. Gorman, M. A. Voinov, A. I. Smirnov, J. Genzer and M. D. Dickey, ACS Macro Lett., 2019, 8, 1522–1527 CrossRef CAS.
- J. Yeow, R. Chapman, A. J. Gormley and C. Boyer, Chem. Soc. Rev., 2018, 47, 4357–4387 RSC.
- W. Zhang, B. S. Naidu, J. Z. Ou, A. P. O'Mullane, A. F. Chrimes, B. J. Carey, Y. Wang, S. Y. Tang, V. Sivan, A. Mitchell, S. K. Bhargava and K. Kalantar-Zadeh, ACS Appl. Mater. Interfaces, 2015, 7, 1943–1948 CrossRef CAS.
- A. Zavabeti, B. Y. Zhang, I. A. de Castro, J. Z. Ou, B. J. Carey, M. Mohiuddin, R. S. Datta, C. Xu, A. P. Mouritz, C. F. McConville, A. P. O'Mullane, T. Daeneke and K. Kalantar-Zadeh, Adv. Funct. Mater., 2018, 28, 1804057 CrossRef.
- H. Li, R. Abbasi, Y. Wang, F. M. Allioux, P. Koshy, S. A. Idrus-Saidi, M. A. Rahim, J. Yang, M. Mousavi, J. Tang, M. B. Ghasemian, R. Jalili, K. Kalantar-Zadeh and M. Mayyas, J. Mater. Chem. C, 2020, 8, 1656–1665 RSC.
- O. Oloye, C. Tang, A. Du, G. Will and A. P. O. O’Mullane, Nanoscale, 2019, 11, 9705–9715 RSC.
- N. Taccardi, M. Grabau, J. Debuschewitz, M. Distaso, M. Brandl, R. Hock, F. Maier, C. Papp, J. Erhard, C. Neiss, W. Peukert, A. Goerling, H. P. Steinrueck and P. Wasserscheid, Nat. Chem., 2017, 9, 862–867 CrossRef CAS.
- R. David and N. Miki, Langmuir, 2018, 34, 10550–10559 CrossRef CAS.
- R. Zheng, Z. Peng, Y. Fu, Z. Deng, S. Liu, S. Xing, Y. Wu, J. Li and L. Liu, Adv. Funct. Mater., 2020, 30, 1910524 CrossRef CAS.
- N. Xia, N. Li, W. Rao, J. Yu, Q. Wu, L. Tan, H. Li, L. Gou, P. Liang, L. Li and X. Meng, Nanoscale, 2019, 11, 10183–10189 RSC.
- Y. Wang, W. Duan, C. Zhou, Q. Liu, J. Gu, H. Ye, M. Li, W. Wang and X. Ma, Adv. Mater., 2019, 31, 1905067 CrossRef CAS.
- A. Guerrero-Martinez, J. Perez-Juste and L. M. Liz-Marzan, Adv. Mater., 2010, 22, 1182–1195 CrossRef CAS.
- J.-J. Hu, M.-D. Liu, Y. Chen, F. Gao, S.-Y. Peng, B.-R. Xie, C.-X. Li, X. Zeng and X.-Z. Zhang, Biomaterials, 2019, 207, 76–88 CrossRef CAS.
- L. Chen, R. Luque and Y. Li, Chem. Soc. Rev., 2017, 46, 4614–4630 RSC.
- Y. Liu, Q. Wang, J. Deng and W. Zhang, Chem. Commun., 2020, 56, 1851–1854 RSC.
- Y. Lu, Y. Lin, Z. Chen, Q. Hu, Y. Liu, S. Yu, W. Gao, M. D. Dickey and Z. Gu, Nano Lett., 2017, 17, 2138–2145 CrossRef CAS.
- Q. Ye, Y. Wu, Y. Qi, L. Shi, S. Huang, L. Zhang, M. Li, W. Li, X. Zeng, H. Wo, X. Wang, S. Dong, S. Ramakrishna and J. Luo, Nano Energy, 2019, 61, 381–388 CrossRef CAS.
- J. Zhu, Y. Wu, X. Huang, L. Huang, M. Cao, G. Song, X. Guo, X. Sui, R. Ren and J. Chen, Nano Energy, 2019, 62, 883–889 CrossRef CAS.
- Q. Wang, Y. Yu, J. Yang and J. Liu, Adv. Mater., 2015, 27, 7109–7116 CrossRef CAS.
- Y. Zheng, Z. Z. He, J. Yang and J. Liu, Sci. Rep., 2014, 4, 4588 CrossRef.
- C. Ladd, J. H. So, J. Muth and M. D. Dickey, Adv. Mater., 2013, 25, 5081–5085 CrossRef CAS.
- X. Zhao, S. Xu and J. Liu, Front. Energy, 2017, 11, 535–567 CrossRef.
- M. R. Khan, C. B. Eaker, E. F. Bowden and M. D. Dickey, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 14047–14051 CrossRef CAS.
- J. W. Boley, E. L. White and R. K. Kramer, Adv. Mater., 2015, 27, 2355–2360 CrossRef CAS.
- L. Tang, S. Cheng, L. Zhang, H. Mi, L. Mou, S. Yang, Z. Huang, X. Shi and X. Jiang, iScience, 2018, 4, 302–311 CrossRef CAS.
- S. Liu, S. N. Reed, M. J. Higgins, M. S. Titus and R. Kramer-Bottiglio, Nanoscale, 2019, 11, 17615–17629 RSC.
- S. Liu, M. C. Yuen, E. L. White, J. W. Boley, B. Deng, G. J. Cheng and R. Kramer-Bottiglio, ACS Appl. Mater. Interfaces, 2018, 10, 28232–28241 CrossRef CAS.
- B. Deng and G. J. Cheng, Adv. Mater., 2019, 31, e1807811 CrossRef.
- C. Pan, K. Kumar, J. Li, E. J. Markvicka, P. R. Herman and C. Majidi, Adv. Mater., 2018, 30, 1706937 CrossRef.
- S. Liu, M. C. Yuen and R. Kramer-Bottiglio, Flexible Printed Electron., 2019, 4, 015004 CrossRef CAS.
- A. Fassler and C. Majidi, Adv. Mater., 2015, 27, 1928–1932 CrossRef CAS.
- T. R. Lear, S.-H. Hyun, J. W. Boley, E. L. White, D. H. Thompson and R. K. Kramer, Extreme Mech. Lett., 2017, 13, 126–134 CrossRef.
- L. Tang, L. Mou, J. Shang, J. Dou, W. Zhang and X. Jiang, Mater. Horiz., 2020, 7, 1186–1194 RSC.
- H. Ota, K. Chen, Y. Lin, D. Kiriya, H. Shiraki, Z. Yu, T.-J. Ha and A. Javey, Nat. Commun., 2014, 5, 5032 CrossRef CAS.
- J. Wang, G. Cai, S. Li, D. Gao, J. Xiong and P. S. Lee, Adv. Mater., 2018, 30, 1706157 CrossRef.
- M. D. Bartlett, A. Fassler, N. Kazem, E. J. Markvicka, P. Mandal and C. Majidi, Adv. Mater., 2016, 28, 3726–3731 CrossRef CAS.
- C. Pan, E. J. Markvicka, M. H. Malakooti, J. Yan, L. Hu, K. Matyjaszewski and C. Majidi, Adv. Mater., 2019, 31, e1900663 CrossRef.
- J. Y. Yang, D. Tang, J. P. Ao, T. Ghosh, T. V. Neumann, D. G. Zhang, E. Piskarev, T. T. Yu, V. K. Truong, K. Xie, Y. C. Lai, Y. Li and M. D. Dickey, Adv. Funct. Mater., 2020, 2002611 CrossRef CAS.
- M. H. Malakooti, N. Kazem, J. Yan, C. Pan, E. J. Markvicka, K. Matyjaszewski and C. Majidi, Adv. Funct. Mater., 2019, 29, 1906098 CrossRef CAS.
- S. Nayak, Y. D. Li, W. Tay, E. Zamburg, D. Singh, C. Lee, S. J. A. Koh, P. Chia and A. V. Y. Thean, Nano Energy, 2019, 64, 103912 CrossRef CAS.
- S. Cinar, I. D. Tevis, J. Chen and M. Thuo, Sci. Rep., 2016, 6, 21864 CrossRef CAS.
- R. Chen, Q. Xiong, R.-Z. Song, K.-L. Li, Y.-X. Zhang, C. Fang and J.-L. Guo, Adv. Mater. Interfaces, 2019, 6, 1901057 CrossRef CAS.
- V. Sivan, S.-Y. Tang, A. P. O'Mullane, P. Petersen, N. Eshtiaghi, K. Kalantar-zadeh and A. Mitchell, Adv. Funct. Mater., 2013, 23, 144–152 CrossRef CAS.
- X. Tang, S.-Y. Tang, V. Sivan, W. Zhang, A. Mitchell, K. Kalantar-zadeh and K. Khoshmanesh, Appl. Phys. Lett., 2013, 103, 174104 CrossRef.
- P. Fan, Z. Sun, Y. Wang, H. Chang, P. Zhang, S. Yao, C. Lu, W. Rao and J. Liu, RSC Adv., 2018, 8, 16232–16242 RSC.
- G. Ding, Y. Zhu, S. Wang, Q. Gong, L. Sun, T. Wu, X. Xie and M. Jiang, Carbon, 2013, 53, 321–326 CrossRef CAS.
- Z. W. Pan, S. Dai, D. B. Beach, N. D. Evans and D. H. Lowndes, Appl. Phys. Lett., 2003, 82, 1947–1949 CrossRef CAS.
- B. S. Simpkins, L. M. Ericson, R. M. Stroud, K. A. Pettigrew and P. E. Pehrsson, J. Cryst. Growth, 2006, 290, 115–120 CrossRef CAS.
- Z. W. Pan, Z. R. Dai, C. Ma and Z. L. Wang, J. Am. Chem. Soc., 2002, 124, 1817–1822 CrossRef CAS.
- Y. Chen, K. Liu, J. Liu, T. Lv, B. Wei, T. Zhang, M. Zeng, Z. Wang and L. Fu, J. Am. Chem. Soc., 2018, 140, 16392–16395 CrossRef CAS.
- R. S. Datta, N. Syed, A. Zavabeti, A. Jannat, M. Mohiuddin, M. Rokunuzzaman, B. Y. Zhang, M. A. Rahman, P. Atkin, K. A. Messalea, M. B. Ghasemian, E. Della Gaspera, S. Bhattacharyya, M. S. Fuhrer, S. P. Russo, C. F. McConville, D. Esrafilzadeh, K. Kalantar-Zadeh and T. Daeneke, Nat. Electron., 2020, 3, 51–58 CrossRef CAS.
- W. Liang, L. Hong, H. Yang, F. Fan, Y. Liu, H. Li, J. Li, J. Y. Huang, L.-Q. Chen, T. Zhu and S. Zhang, Nano Lett., 2013, 13, 5212–5217 CrossRef CAS.
- Y. Wu, L. Huang, X. Huang, X. Guo, D. Liu, D. Zheng, X. Zhang, R. Ren, D. Qu and J. Chen, Energy Environ. Sci., 2017, 10, 1854–1861 RSC.
- X. Guo, Y. Ding, L. Xue, L. Zhang, C. Zhang, J. B. Goodenough and G. Yu, Adv. Funct. Mater., 2018, 28, 1804649 CrossRef.
- B. Han, Y. Yang, X. Shi, G. Zhang, L. Gong, D. Xu, H. Zeng, C. Wang, M. Gu and Y. Deng, Nano Energy, 2018, 50, 359–366 CrossRef CAS.
- Y. Chen, T. Zhou, Y. Li, L. Zhu, S. Handschuh-Wang, D. Zhu, X. Zhou, Z. Liu, T. Gan and X. Zhou, Adv. Funct. Mater., 2018, 28, 1706277 CrossRef.
- P. Balaz, M. Achimovicova, M. Balaz, P. Billik, Z. Cherkezova-Zheleva, J. M. Criado, F. Delogu, E. Dutkova, E. Gaffet, F. J. Gotor, R. Kumar, I. Mitov, T. Rojac, M. Senna, A. Streletskii and K. Wieczorek-Ciurowa, Chem. Soc. Rev., 2013, 42, 7571–7637 RSC.
- I. A. de Castro, A. F. Chrirnes, A. Zavabeti, K. J. Berean, B. J. Carey, J. Zhuang, Y. Du, S. X. Dou, K. Suzuki, R. A. Shanks, R. Nixon-Luke, G. Bryant, K. Khoshmanesh, K. Kalantar-zadeh and T. Daeneke, Nano Lett., 2017, 17, 7831–7838 CrossRef.
- Y. Liu and X. Jiang, Lab Chip, 2017, 17, 3960–3978 RSC.
- P. Li, A. Kumar, J. Ma, Y. Kuang, L. Luo and X. Sun, Sci. Bull., 2018, 63, 645–662 CrossRef CAS.
- L. Pitkanen and A. M. Striegel, TrAC, Trends Anal. Chem., 2016, 80, 311–320 CrossRef CAS.
- A. Di Cicco and A. Filipponi, EPL, 1994, 27, 407–412 CrossRef CAS.
- G. B. Parravicini, A. Stella, P. Ghigna, G. Spinolo, A. Migliori, F. d’Acapito and R. Kofman, Appl. Phys. Lett., 2006, 89, 033123 CrossRef.
- S.-Y. Tang, D. R. G. Mitchell, Q. Zhao, D. Yuan, G. Yun, Y. Zhang, R. Qiao, Y. Lin, M. D. Dickey and W. Li, Matter, 2019, 1, 192–204 CrossRef.
- M. W. Knight, N. S. King, L. Liu, H. O. Everitt, P. Nordlander and N. J. Halas, ACS Nano, 2014, 8, 834–840 CrossRef CAS.
- P. Reineck, Y. Lin, B. C. Gibson, M. D. Dickey, A. D. Greentree and I. S. Maksymov, Sci. Rep., 2019, 9, 1–7 CrossRef CAS.
- A. Garcia Marin, T. Garcia-Mendiola, C. Navio Bernabeu, M. Jesus Hernandez, J. Piqueras, J. Luis Pau, F. Pariente and E. Lorenzo, Nanoscale, 2016, 8, 9842–9851 RSC.
- Y. Yu and E. Miyako, iScience, 2018, 3, 134–148 CrossRef CAS.
- D. Wang, W. Xie, Q. Gao, H. Yan, J. Zhang, J. Lu, B. Liaw, Z. Guo, F. Gao, L. Yin, G. Zhang and L. Zhao, Small, 2019, 15, 1900511 CrossRef.
- B. Ma, C. Xu, J. Chi, J. Chen, C. Zhao and H. Liu, Adv. Funct. Mater., 2019, 29, 1901370 CrossRef.
- J.-H. Kim, S. Kim, J.-H. So, K. Kim and H.-J. Koo, ACS Appl. Mater. Interfaces, 2018, 10, 17448–17454 CrossRef CAS.
- M. D. Bartlett, N. Kazem, M. J. Powell-Palm, X. Huang, W. Sun, J. A. Malen and C. Majidi, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 2143–2148 CrossRef CAS.
- N. Kazem, T. Hellebrekers and C. Majidi, Adv. Mater., 2017, 29, 1605985 CrossRef.
- L. L. Fan, M. H. Duan, Z. C. Xie, K. Q. Pan, X. L. Wang, X. Y. Sun, Q. Wang, W. Rao and J. Liu, Small, 2020, 16, 1903421 CrossRef CAS.
- J. Wu, S. Y. Tang, T. Fang, W. Li, X. Li and S. Zhang, Adv. Mater., 2018, 30, e1805039 CrossRef.
- J. Zhang, Y. Yao, L. Sheng and J. Liu, Adv. Mater., 2015, 27, 2648–2655 CrossRef CAS.
- E. J. Markvicka, M. D. Bartlett, X. Huang and C. Majidi, Nat. Mater., 2018, 17, 618–624 CrossRef CAS.
- S.-Y. Tang, V. Sivan, K. Khoshmanesh, A. P. O'Mullane, X. Tang, B. Gol, N. Eshtiaghi, F. Lieder, P. Petersen, A. Mitchell and K. Kalantar-Zadeh, Nanoscale, 2013, 5, 5949–5957 RSC.
| This journal is © The Royal Society of Chemistry 2021 |