
Chemotherapeutic drug–DNA hybrid nanostructures for anti-tumor therapy
Xiangang
Huang
ab,
Nicholas Thomas
Blum
a,
Jing
Lin
 a,
Jinjun
Shi
*b,
Chuan
Zhang
*c and
Peng
Huang
*a
a,
Jinjun
Shi
*b,
Chuan
Zhang
*c and
Peng
Huang
*a
aMarshall Laboratory of Biomedical Engineering, International Cancer Center, Laboratory of Evolutionary Theranostics (LET), School of Biomedical Engineering, Health Science Center, Shenzhen University, Shenzhen, 518060, China. E-mail: peng.huang@szu.edu.cn
bCenter for Nanomedicine and Department of Anesthesiology, Brigham and Women's Hospital, Harvard Medical School, Boston, MA 02115, USA. E-mail: jshi@bwh.harvard.edu
cSchool of Chemistry and Chemical Engineering, State Key Laboratory of Metal Matrix Composites, Shanghai Jiao Tong University, Shanghai, 200240, China. E-mail: chuanzhang@sjtu.edu.cn
First published on 19th August 2020
Abstract
Compared to traditional drug delivery systems, DNA nanostructure-based drug delivery systems have several advantages including programmable sequences, precise size and shape, high drug payloads, excellent biocompatibility and biodegradability. To date, a wide range of chemotherapeutic drug–DNA hybrid nanostructures have been developed for anti-tumor therapy. In this review, the constructions of various DNA nanostructures for anticancer drug delivery are firstly summarized. Next, the anticancer drug loading methods for DNA nanostructures are presented. Then, the recent applications of chemotherapeutic drug–DNA hybrid nanostructures for drug delivery are highlighted. In the end, the challenges and opportunities of the chemotherapeutic drug–DNA hybrid nanostructure-based delivery system are discussed. The designs of drug–DNA hybrid systems, including the constructions of nanostructures and the strategies for drug loading, largely influence the efficiency of drug delivery. Recent studies have focused on the development of novel drug–DNA hybrid systems to acquire more precise and efficient therapy for various diseases. A systematic review of the design strategies of chemotherapeutic drug–DNA hybrid nanostructures will benefit the innovation and development of the chemotherapeutic drug-based chemotherapy in clinics.
 Xiangang Huang | Xiangang Huang obtained his PhD in Pharmaceutical Science from ETH Zurich under the supervision of Prof. Jean-Christophe Leroux in 2016. Then he worked with Prof. Xinyuan Zhu and Prof. Chuan Zhang at Shanghai Jiao Tong University as a postdoctoral fellow in 2017, and worked with Prof. Peng Huang at Shenzhen University and Prof. Jinjun Shi at Harvard Medical School as a postdoctoral fellow in 2019. His research interest focuses on the development of multi-functional nanosystems for biomedical applications. |
 Nicholas Thomas Blum | Nicholas Thomas Blum received his Bachelor's degree in chemical engineering from The Ohio State University in 2014, before moving to the University of Colorado, Boulder, to begin his post-graduate studies. There he studied under Prof. Andrew Goodwin and developed an expertise in high intensity focused ultrasound (HIFU) responsive micro- and nano-materials as contrast agents and therapeutic adjuvants. He graduated with his PhD in 2019 before moving to a postdoctoral position in Shenzhen University, Guangdong, China, to continue working on advanced applications of HIFU micro/nanomaterials as theranostic agents. |
 Jing Lin | Jing Lin is a Distinguished Professor at the School of Biomedical Engineering, Shenzhen University Health Science Center, China. She received her PhD in Organic Chemistry from the Donghua University and Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, in 2010. Then she joined the PharmaResources (Shanghai) Co., Ltd as a group leader. After two years, she moved to the United States of America and spent 4 years as a postdoc at the University of Maryland and the National Institutes of Health (NIH). She joined the faculty of Shenzhen University (SZU) in 2016 and was promoted as a Distinguished Professor in 2018. Her research focuses on molecular imaging, nanomedicine and theranostics. |
 Jinjun Shi | Jinjun Shi holds an academic appointment as an Associate Professor at Harvard Medical School, and is a faculty member in the Center for Nanomedicine and Department of Anesthesiology at Brigham and Women's Hospital. He has extensive experience in the research fields of nanomedicine, drug delivery, and immunotherapy. He has developed many unique nanoparticle platforms for delivery of various therapeutic molecules and for biomedical applications. His current research focuses include (i) RNAi nanotechnology for gene silencing; (ii) mRNA delivery for restoration of tumor suppressors; (iii) nanoparticle-mediated immune modulation; and (iv) development of stimuli-responsive nanomaterials and bioinspired antioxidative polymers. |
 Chuan Zhang | Chuan Zhang obtained his bachelor's and master's degrees in Chemistry from Tsinghua University, Beijing. After that, he continued his PhD study at Purdue University and obtained his PhD degree in Chemistry in 2011 under the supervision of Prof. Chengde Mao. Then he received postdoctoral training in the group of Prof. Chad A. Mirkin at Northwestern University, USA. He joined the School of Chemistry and Chemical Engineering, Shanghai Jiao Tong University, as an Assistant Professor in 2014. His main research interests focus on DNA supramolecular chemistry, DNA-based nanomaterials for biomedical applications, cell–material interactions, and materials engineering for immunomodulation. |
 Peng Huang | Peng Huang is a Distinguished Professor, Chief of the Laboratory of Evolutionary Theranostics (LET), and Director of the Department of Molecular Imaging, at the School of Biomedical Engineering, Shenzhen University Health Science Center, China. He received his PhD degree in Biomedical Engineering from Shanghai Jiao Tong University in 2012. He then joined the Laboratory of Molecular Imaging and Nanomedicine (LOMIN) at the National Institutes of Health (NIH) as a postdoctoral fellow. In 2015, he moved to Shenzhen University as a Distinguished Professor. His research focuses on the design, synthesis, and biomedical applications of molecular imaging contrast agents, stimuli-responsive programmed targeting drug delivery systems, and activatable theranostics. |
1. Introduction
DNA is a biopolymer consisting of DNA bases with predictable sequences, which can form double helices via Watson–Crick base pairing driven by hydrogen bonds.1 Based on this base pairing principle, DNA nanostructures with well-controlled and precise size, shape,2,3 and surface chemistry4 can be constructed using the latest DNA nanotechnology. Compared to traditional drug delivery systems, DNA nanostructure-based drug delivery systems have several advantages.5,6 First, DNA strands with specific sequences can be easily programmed and synthesized using an automated DNA synthesis machine via phosphoramidite chemistry.7 Second, the size and shape of the DNA nanostructure can be precisely and conveniently adjusted to meet the requirements of different delivery tasks, maximizing the drug delivery efficiency.8 For example, in order to protect therapeutic proteins from degradation, a DNA nanorobot was developed that could transform its planar structures into three-dimensional (3D) tubular structures.9 Third, DNA as an inherent genetic material exhibits excellent biocompatibility and biodegradability.10,11 On the other hand, some DNA nanostructures showed preferential binding to specific proteins12 or accumulations in specific organs13,14in vivo, facilitating the delivery of therapeutics to targeted sites.DNA nanostructures were initially intensively investigated for the delivery of antisense oligonucleotides (ASO),15–18 CpG oligodeoxynucleotides19,20 and small interference RNA (siRNA),21–24 and the sensing25 and imaging26 of biomolecules, since the oligonucleotides could bind to DNA nanostructures conveniently via base pairing. The rapid development of DNA nanotechnology has endowed DNA nanostructures with the ability to deliver more types of therapeutics including proteins,27 genes,28 and chemotherapeutic drugs;29 of these, the delivery of chemotherapeutic drugs is particularly attractive.30 Firstly, chemotherapeutic drugs are relatively small, hence they can be easily loaded to DNA nanostructures31 or directly replace the DNA bases.32 Secondly, chemotherapeutic drugs are generally much more stable than proteins and genes, so they will not lose their activities when loaded to the surface of DNA nanostructures.33,34
In this review, we focus on drug delivery systems based on chemotherapeutic drug–DNA hybrid nanostructures (Fig. 1). We first introduce the main chemotherapeutic drug–DNA hybrid nanostructures for chemotherapeutic drug delivery, including the main building elements and driving forces for the construction of these nanostructures. Then, we move to the chemotherapeutic drug loading methods where we compare the advantages and disadvantages of each drug loading method. In the end, we highlight the latest developments of the drug delivery systems based on chemotherapeutic drug–DNA hybrid nanostructures, including the drug-hybridized aptamers, spherical nucleic acids (SNAs), DNA origami, DNA cages, DNA micelles, DNA nanogels, and so on. The origin of DNA nanotechnology, different techniques for construction of complicated DNA nanostructures,5 the biomedical applications of DNA nanostructures for the delivery of genes,35 and the sensing and imaging of biomolecules36 have been comprehensively reviewed in the literature and have thus not been included here.
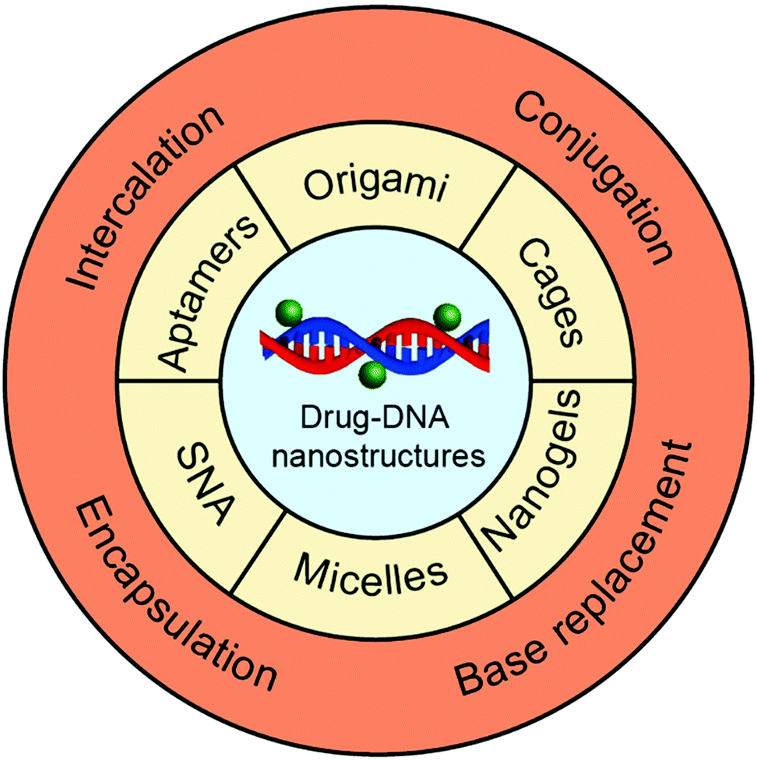 | ||
| Fig. 1 DNA nanostructures used for chemotherapeutic drug delivery and the methods for loading chemotherapeutic drugs. The representative chemotherapeutic drug–DNA hybrid nanostructures contain drug-hybridized DNA origami, cages, nanogels, micelles, spherical nucleic acid (SNA) and aptamers. The main drug loading strategies are intercalation, conjugation, base replacement and encapsulation. | ||
2. Construction of chemotherapeutic drug–DNA hybrid nanostructures
Numerous types of complicated DNA nanostructures have been developed using the latest DNA nanotechnology.37 Many of them are suitable for digital data storage,38 preparation of movable devices,39 spatial organization of enzymes40 and fabrication of plasmonic materials,41 but none quite so prolific as drug delivery. Generally, an ideal drug carrier candidate should meet several criteria. First, chemotherapeutic drugs can be easily loaded to the carrier with high efficiency and capacity. Second, the carriers should have prolonged stability during the blood circulation to ensure high accumulation of the drugs in the tumor sites. Third, the carriers should have appropriate surface properties that facilitate their cellular uptake. This section describes the construction of different drug–DNA hybrid nanostructures for chemotherapeutic drug delivery. The most representative drug–DNA nanostructures are categorized into several subcategories based on the driving force for the formation of the nanostructures: the formation of drug–SNA nanostructures is driven by chemical conjugation; the formation of drug–DNA micelle nanostructures is driven by hydrophobic interactions; the formation of drug–aptamers, drug–DNA nanogels, drug–DNA cages and drug–DNA origami nanostructures is driven by base pairing. Of course, there are other miscellaneous drug–DNA nanostructures whose construction is driven by other forces. Construction of drug–DNA nanostructures can be simple or complicated depending on the complexity of the driving force and of the components (DNA sequences) that are being utilized (Fig. 2).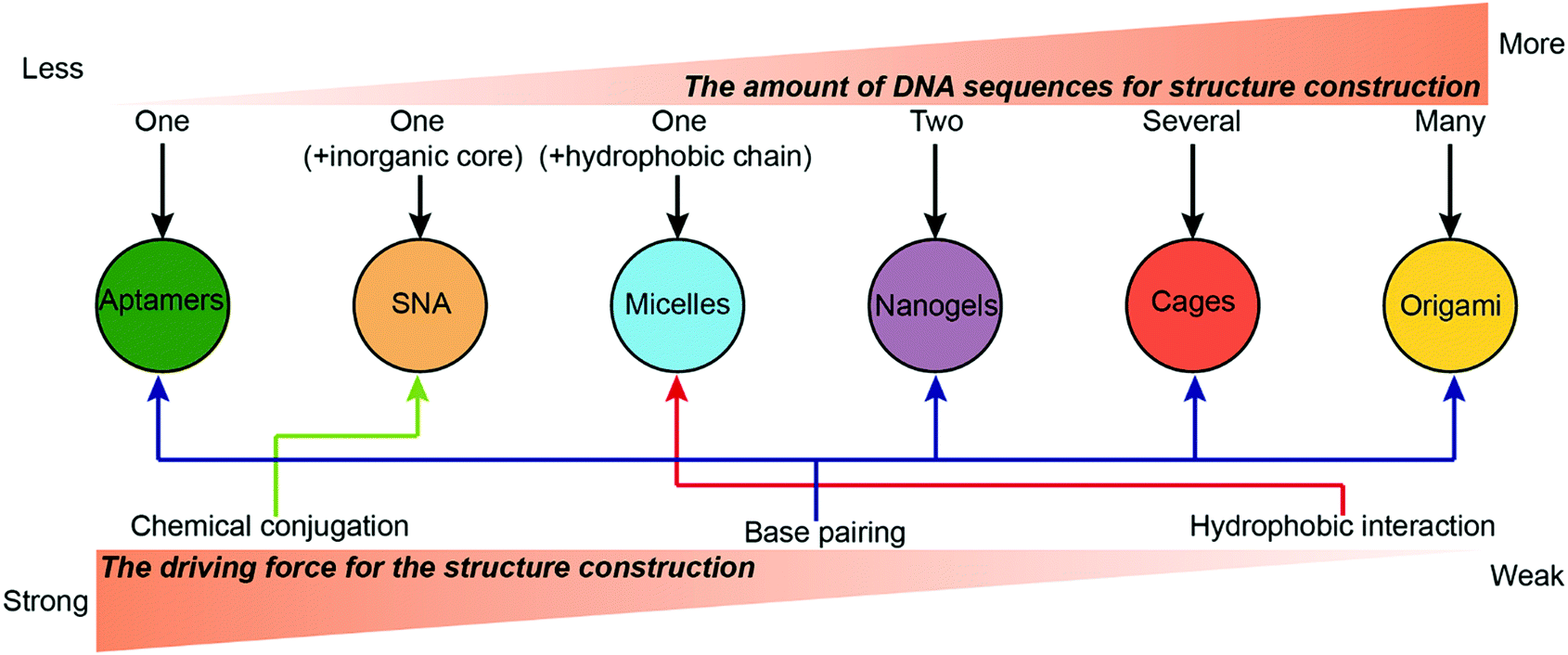 | ||
| Fig. 2 The driving forces and building components (DNA sequences) for the construction of drug–DNA hybrid nanostructures. The nanostructures are organized by the number of DNA sequences required for their construction (one to many), and the corresponding driving force is indicated based on strength of the interaction (strong to weak). | ||
2.1. Chemical conjugation
Zheng and co-workers reported high-performance drug delivery of drug–SNA nanostructures in cancer therapy (Fig. 3A).42 Gold nanoparticles (AuNPs) or super-paramagnetic magnetic nanoparticles (MNPs) were employed as the inorganic core. The SNA was obtained by attaching the DNA strands to the core via thiol–gold chemistry. More DNA strands and aptamer ligand AS1411 targeting nucleolin protein were introduced to the SNA via DNA hybridization to yield the AS1411/MNP-SNAs. Then, a widely used photodynamic therapy (PDT) reagent 5,10,15,20-tetrakis(4-N-methylpyridiniumyl)porphyrin (TMPyP4) was loaded to the G-quadruplex structure of AS1411. The confocal microscopy images confirmed that AS1411/MNP-SNAs were internalized into the human ovarian carcinoma (SKOV3) cells, while very few of the non-targeting MNP-SNAs were observed in the cells. In contrast to conventional SNA nanostructures, the length and density of the DNA strands on the surface of the present SNA were dramatically increased by the polymerization of two DNA monomers, offering more spaces for drug loading. The so-called polymerization of DNA monomers was driven by the DNA hybridization rather than the formation of covalent bonds.
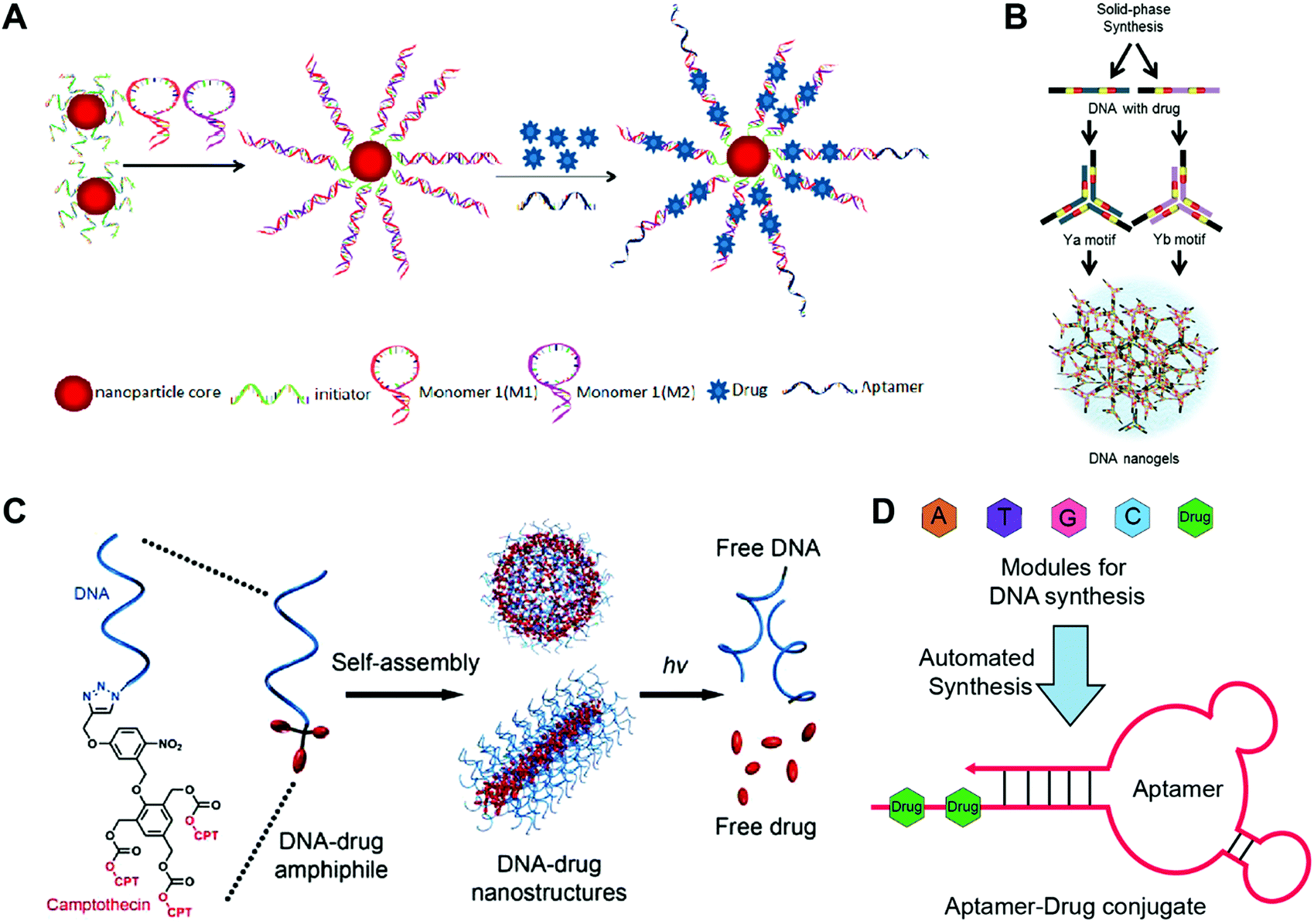 | ||
| Fig. 3 Construction of drug-hybridized DNA aptamers, micelles, SNA and nanogel hybrid nanostructures. (A) The SNA was prepared via the polymerization of DNA monomers 1 and 2 initiated by the DNA initiators, followed by the decoration of the AS1411 aptamer ligands and the accommodation of drugs. Reproduced with permission from ref. 42. Copyright 2013 American Chemical Society; (B) drug fluorouracil (5-FU)-integrated DNA motifs Ya and Yb were prepared, and the DNA nanogels were obtained by the assembly of the Y motifs. Reproduced with permission from ref. 56. Copyright 2018 The Royal Society of Chemistry; (C) the drug camptothecin (CPT) was conjugated to the DNA strands via photo-sensitive linkers, and the spherical and tubular DNA–drug nanostructures that assembled from the DNA–drug conjugates could release the free drugs under UV irradiation. Reproduced with permission from ref. 47. Copyright 2015 American Chemical Society; (D) drugs were introduced into the DNA chain directly via the DNA automated synthesis, and aptamer–drug conjugates with drugs in the tail DNA chain were obtained via self-assembly; 5-FU was used. Reproduced with permission from ref. 53. Copyright 2014 American Chemical Society. | ||
In a classical Au–SNA system, the inorganic Au cores have two functions. One is that their physical and chemical properties (e.g. quenching) can be employed for biodetection applications. The other one is that they act as scaffolds for assembling DNA strands into a dense and highly oriented arrangement, which determines the functional properties of SNA (e.g. cellular uptake).43 One concern of the SNA nanostructures is that the poor biodegradability of the inorganic core may cause toxicity in vivo. To address this potential problem, a coreless form of SNAs has been developed via crosslinking the DNA shell of SNAs, and then dissolving the Au core. The new coreless SNAs have retained most of the characteristic properties of original SNAs such as base paring and efficient cellular uptake, indicating that many properties of SNAs are core-independent.44 Besides inorganic cores, organic liposomes have also been used as cores for SNAs. One advantage of liposomal SNAs is the high cargo-loading capability of the liposome cores, broadening the applications of SNAs.45,46
2.2. Hydrophobic interaction
Tan and co-workers have developed light-sensitive, self-immolative drug–DNA micelles nanostructures for controlled drug delivery (Fig. 3C).47 In this study, the hydrophobic chain was a photolabile 2-nitrobenzyl ether moiety containing three camptothecin (CPT) molecules. After conjugating this hydrophobic chain to the DNA strands, the DNA micelles were obtained via self-assembly. Interestingly, the DNA micelles could transform into DNA tubes by increasing the length of the DNA strands. In this system, the use of the hydrophobic drugs as the hydrophobic part of the micelles not only guarantees the high drug loading capacity, but also avoids the introduction of additional hydrophobic polymers. The introduction of photo-sensitive bonds to the system allows on-demand drug release.
Driven by hydrophobic interactions, a DNA micelle is one of the most easily obtained DNA–drug nanostructures. Micelles assembled from hydrophobic drug–DNA conjugates or hydrophobic polymer/lipid–DNA conjugates represent two main types of drug–DNA micelles. For the construction of hydrophobic drug-based DNA micelles, the drugs or polydrugs are covalently conjugated to DNA strands, ensuring the stability of the loaded drugs.48 However, a degradable bond between the drugs and the carriers is needed to facilitate efficient drug release. For the construction of hydrophobic polymer/lipid-based DNA micelles, the drugs are encapsulated into the hydrophobic core of the micelles, normally with a high drug loading content.49 This type of micelle is especially useful for drugs that are not able to be chemically modified.
2.3. Base pairing
Wang and co-workers have prepared an aptamer–drug conjugate for targeted drug delivery (Fig. 3D).53 The chemotherapeutic drug fluorouracil (5-FU) was chosen as a model drug, which was firstly modified with solid-phase functionalities including different protecting groups and a phosphoramidite group. The molecules with solid-phase functionalities could be integrated into the DNA chain using a DNA synthesizer via a solid-phase synthesis method. Then the drug and DNA base modules were loaded to the DNA synthesizer to produce the DNA chain with drug in the tail. The aptamer–drug conjugates were obtained via the self-assembly of the DNA chain. In this study, the drug-integrated DNA strands can be easily synthesized by the DNA synthesizer, and directly assemble to ready-to-use drug-loaded aptamers without post-modifications. More importantly, the number and position of the drug molecules in the aptamers can be precisely controlled. Indeed, the location of drugs at the tail chains of the aptamers avoids interference on the targeting ability of the aptamers.
Li and co-workers have prepared a nucleolin aptamer–paclitaxel (PTX) conjugate for ovarian cancer therapy.54 The PTX was firstly modified with a cathepsin B sensitive dipeptide through its active hydroxyl group at the 2′ position, and a tumor-targeting nuceleolin aptamer containing an amine group was conjugated to PTX via the carboxyl group of the dipeptide subsequently. In order to monitor the release of PTX from the aptamer–PTX conjugate, 6-carboxyfluorescein (FAM) and rhodamine B (Rh) were introduced to the conjugate with a fluorescence resonance energy transfer (FRET) effect existing between the two fluorophores. This study demonstrates an efficient aptamer–drug system with multiple functions, including targeted delivery of PTX to the tumor site, responsive release of PTX in the presence of cathepsin B in lysosomes, and precise monitoring of drug release via the FRET effect. In another study, Yang and co-workers have prepared an aptamer–mitomycin C (MMC) conjugate for synergistic cancer therapy.55 The aptamer–drug conjugate was prepared by the conjugation of mitomycin C to an aptamer via the reductant-responsive disulfide linker.
Ma and co-workers reported floxuridine (Fu)-containing DNA nanogels for chemotherapeutic drug delivery (Fig. 3B).56 In this design, one Y motif was used as the DNA backbone and the other Y motif was used as the DNA crosslinker. The DNA nanogels were prepared via three steps. First, the chemotherapeutic drug floxuridine (Fu), a nucleoside analogue, was converted to a phosphoramidite monomer (Fu-phosphoramidite) before integration into the DNA strands through solid-phase synthesis. Second, the Fu-containing DNA strands were annealed to produce two Y shaped motifs with three sticky ends, denoted as Ya and Yb. Third, the Ya motifs that functioned as the DNA backbones were crosslinked by the Yb motifs that functioned as the DNA crosslinker to construct the DNA nanogels. This is a good example that demonstrates that the introduction of an appropriate amount of drug molecules to the DNA strands does not disturb the base pairing of the DNA strands. The system also has high biocompatibility since only DNA materials are used for the construction of the nanogels.
Similar to the conventional nanogels, the size and morphology of DNA nanogels can be precisely controlled by adjusting the parameters such as the self-assembly time, concentration and crosslinker ratio. Short self-assembly time and low concentration normally yield small DNA nanogels, whereas large DNA nanogels can be obtained by increasing the self-assembly time and concentration.57 Further increasing the self-assembly time, concentration or crosslinker ratio results in DNA hydrogels.58 In addition, many functional DNA sequences like pH-sensitive i-motifs, immunostimulatory CPGs, and targeting aptamers can be conveniently introduced to the nanogels during their construction without disturbing their self-assembly process.59 One advantage of DNA nanogels is their controllable stability. The stability of the DNA duplex is proportional to the length and G–C base pair content of the oligonucleotides,60 therefore, the stability of DNA nanogels can be easily manipulated through changing the length and G–C base pair content of the crosslinker.
The tetrahedron is the simplest polyhedron. The DNA tetrahedron has been widely used as a drug carrier due to its simple structure and easy preparation procedure. Kim and co-workers reported mirror (chirality) DNA tetrahedrons for tumor-specific delivery of anticancer drugs (Fig. 4A).66 According to Seeman's tile-based bottom-up method, the tetrahedrons were prepared via the self-assembly of four DNA strands with programmed sequences. The anticancer drug DOX was then loaded to the double strands of the tetrahedron by intercalation. The mirror tetrahedrons were obtained using D-DNA or L-DNA strands. The biodistribution of the tetrahedrons was determined by tail vain injection of fluorescence-labeled tetrahedrons to mice bearing xenograft SCC7 (a murine squamous cell carcinoma) tumors. Interestingly, the L-tetrahedrons accumulated in the tumor sites after 7 or 9 h, while the D-tetrahedrons were immediately removed from the body after the administration, displaying much weaker signals in the tumors. This could be ascribed to the high serum stability and the bio-orthogonal base-pairing system of the L-DNA.67,68 This is an interesting study about how the chirality of the DNA strands influences the behavior of DNA nanostructures in vivo, while similar studies have been rarely reported. More studies should focus on the mechanism of how the chirality of DNA nanostructures influences their behaviors in vivo.
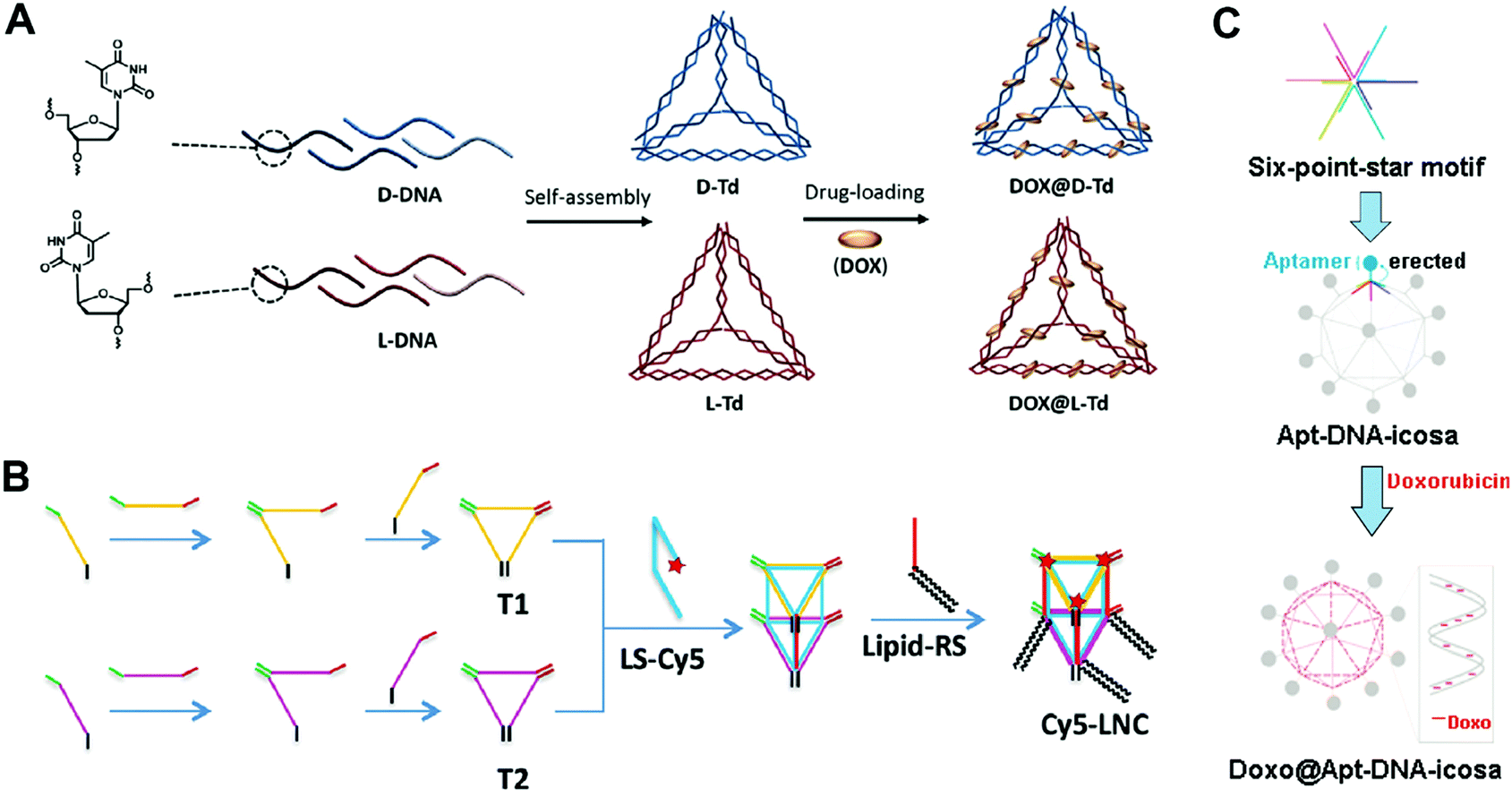 | ||
| Fig. 4 Construction of drug–DNA cage hybrid nanostructures. (A) The mirror DNA tetrahedrons were assembled from the L-DNA and D-DNA strands, followed by the introduction of drug DOX. Reproduced with permission from ref. 66. Copyright 2016 Elsevier; (B) the DNA pentahedrons were first prepared via the assembly of DNA triangles T1 and T2, and then the introduction of the lipid–DNA strands yielded the amphiphilic DNA pentahedrons. The drug DOX was loaded to the DNA double strands of the pentahedrons. Reproduced with permission from ref. 69. Copyright 2016 Wiley-VCH; (C) the DNA icosahedrons were prepared from the assembly of the six-point-star motifs and connecting DNA strands; then the aptamer ligands and DOX were introduced to the nanostructures. Reproduced with permission from ref. 71. Copyright 2011 American Chemical Society. | ||
DNA polyhedrons with more faces such as DNA pentahedrons have also been used for drug delivery. Chan and co-workers reported amphiphilic DNA pentahedrons for mitochondrial delivery of anticancer drugs (Fig. 4B).69 To prepare the amphiphilic pentahedrons, two DNA triangles T1 and T2 were first assembled from three DNA strands, respectively. Then, DNA strands that contained complementary sequences of T1 and T2 were used to connect T1 and T2 to produce the pentahedrons. The amphiphilic lipid-functionalized pentahedrons were produced by attaching the lipid to the pentahedrons via a complementary DNA strand. Finally, chemotherapeutic drug DOX was loaded to double strands of the pentahedrons through intercalation. The confocal fluorescence images showed that the pentahedrons exclusively localized in mitochondria even after 72 h of incubation, making them ideal for mitochondria-targeted drug delivery. The cell viability assay demonstrated that the free DOX was insufficient to induce cell death at low drug concentration, while the DOX loaded pentahedrons significantly enhanced cell death at the same drug concentration after 72 h of incubation. The selective accumulation of the pentahedrons in the mitochondria could be due to the insertion of lipids of the pentahedrons into phospholipid bilayers membrane of mitochondria.70 To achieve the optimal therapeutic effect, different functional moieties can be introduced to the DNA cages.
More complicated DNA polyhedrons such as DNA icosahedrons have also been used for drug delivery. Chang and co-workers reported targeted DNA icosahedrons as a carrier of DOX for cancer therapy (Fig. 4C).71 To prepare the targeted DNA icosahedrons, six individual strands were firstly assembled to form the sticky-ended six-point-star motifs, which were then used as building blocks for the construction of aptamer-modified icosahedrons. The DOX was loaded into the double strands of the icosahedrons. Flow cytometry data showed the higher cellular uptake efficiency of aptamer–icosahedrons than icosahedrons in target human breast cancer cells (MCF-7) but not in non-target Chinese hamster ovary cells (CHO-K1), suggesting an aptamer ligand-mediated internalization of DNA icosahedrons. In the cytotoxicity study, the free DOX and DOX-aptamer-icosahedron treated CHO-K1 exhibited similar cell viabilities; however, in MCF-7 cells, the DOX-aptamer-icosahedrons displayed greater cytotoxicity than free DOX. Considering that the uptake efficiency of DNA cages is suboptimal, modification with a target aptamer ligand is necessary for the efficient delivery of DOX.
A triangular DNA origami is one of the most widely used DNA nanostructures for drug delivery. Jiang and co-workers reported triangular and tubular DNA origamis as drug carriers to alleviate drug resistance (Fig. 5A).76 Drug resistance is the phenomenon where cells continually pump drugs out of the cells.77 Upon internalization by the cells, the DNA origami can improve the drug retention in the cells by slowly releasing the intercalated drug daunorubicin to cells, reversing drug resistance. The scaffold DNA M13mp18 and different short staple DNA strands were annealed together to generate the triangular and tubular DNA origamis according to Rothemund's method that are described above. Then, chemotherapeutic drug DOX was intercalated into the double helix of the origamis. The cell viability assay showed that the free DOX and DOX-triangular/tubular origami induced similar amounts of cell death in regular MCF-7 cells. In contrast, the DOX-triangular/tubular origami resulted in much higher levels of cell death than the free DOX in DOX-resistant MCF-7 cells, indicating that the DNA origami-based drug carriers could alleviate drug resistance. This is one of the earliest DNA origami-based system for the delivery of chemotherapeutic drugs. The triangular DNA origami is relatively simple to prepare compared to other complicated DNA origamis, yet it is robust. Therefore, it has been widely used for the delivery of therapeutics like DOX.
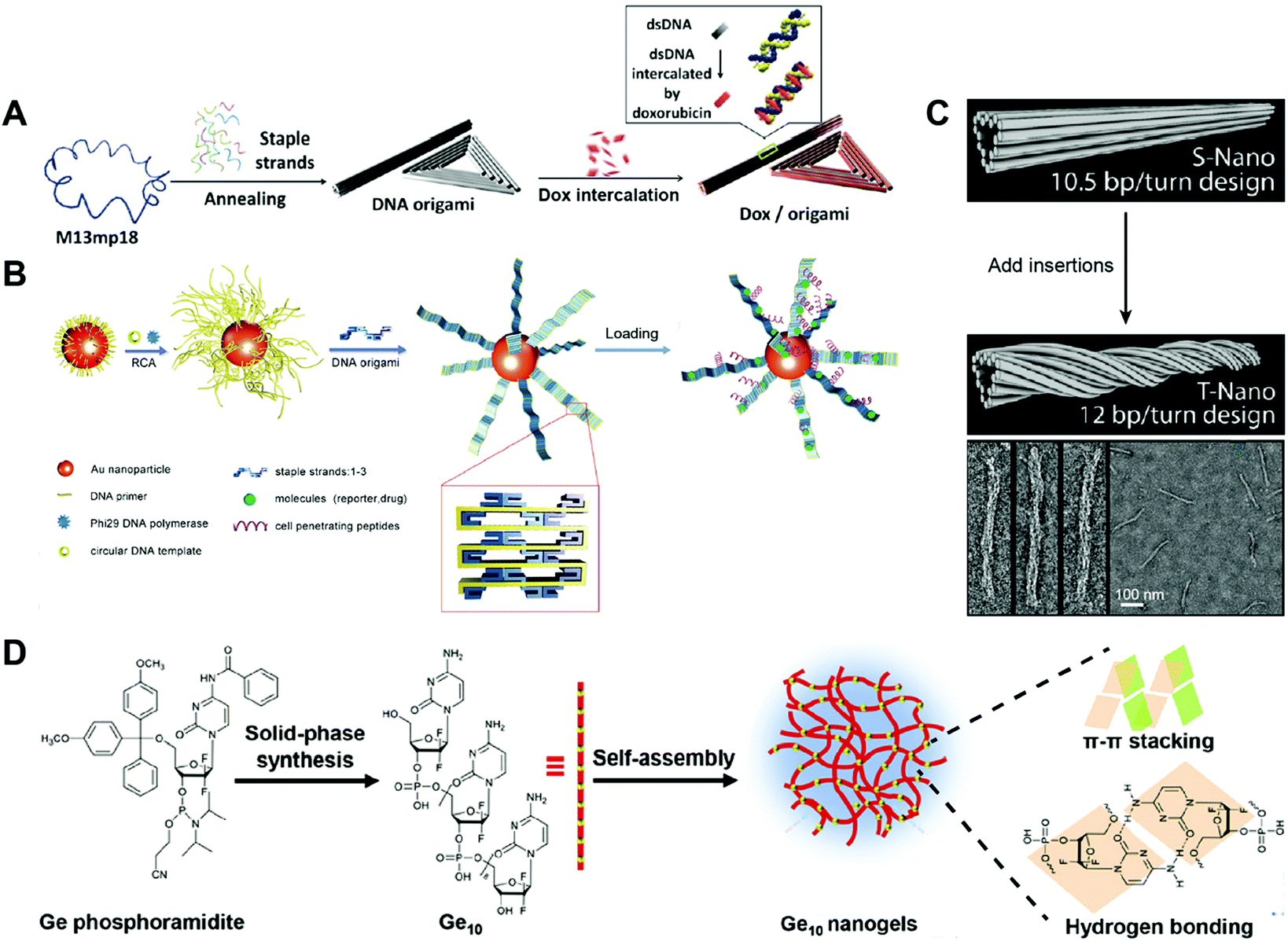 | ||
| Fig. 5 Construction of drug–DNA origami and other hybrid nanostructures. (A) The annealing of M13mp18 and staple DNA strands generated a triangular DNA origami and a tubular DNA origami, followed by the loading of the drug DOX into the DNA double strands. Reproduced with permission from ref. 76. Copyright 2012 American Chemical Society; (B) long DNA strand coated gold (Au) nanoparticles were first prepared by rolling circle amplification (RCA), then, the DNA origami belts were obtained using the staple DNA strands, and the peptides and drugs were loaded subsequently. Reproduced with permission from ref. 78. Copyright 2014 Wiley-VCH; (C) the tubular DNA origami was twisted by the addition of DNA insertions. Reproduced with permission from ref. 79. Copyright 2012 American Chemical Society; (D) Ge conjugates containing ten Ge (Ge10) were prepared by solid-phase synthesis, then the Ge-integrated DNA-like nanogels were obtained by the self-assembly of Ge10 driven by the π–π stacking interaction and hydrogen bond. Reproduced with permission from ref. 89. Copyright 2019 The Royal Society of Chemistry. | ||
Another DNA origami that has been used for drug delivery is the DNA origami belt. Yan and co-workers reported DNA origami belts as drug carriers for high-efficiency cellular imaging and drug delivery (Fig. 5B).78 The DNA origami belts were prepared using Rothemund's strategy with some modifications. To be specific, the DNA primer was firstly attached to gold (Au) nanoparticles followed by rolling circle amplification (RCA) to produce long DNA strands on the surface of the gold particles. Then, these long DNA strands acted as DNA scaffolds and were folded into DNA origami belts by hybridization with staple DNA strands. The chemotherapeutic drug DOX was loaded into the DNA double helix by intercalation. Upon incubating the free DOX and the DOX-Au-origami belts with the human glioblastoma multiforme (U87MG) cells, the DOX-Au-origami belts induced 2.5 times higher cell killing efficiency than that of the free DOX. The Au-origami belts could also be used for cellular imaging purposes by loading quantum dots (QDs) to the system. The combination of inorganic particles and DNA origami for drug delivery is not so common; however, this SNA-like structure of the Au-origami belts may enhance the cellular uptake of the origami belts. A twisted tubular DNA origami has also been used for drug delivery. Yan and co-workers reported a twisted tubular DNA origami as drug carriers for cancer therapy (Fig. 5C).79 The twisted tubular DNA origami was prepared using Shih's strategy. First, an 8634-nucleotide DNA scaffold was folded into a tubular DNA origami with designed staple DNA strands. Then, at every seventh base pair, an insertion was introduced in order to fabricate the twisted tubular DNA origami. The chemotherapeutic drug DOX was loaded into the DNA double helix by intercalation. The cytotoxicity of the free DOX and DOX-twisted tubular DNA origami was evaluated in three different breast cancer cell lines including MDA-MB-231, MDA-MB-468, and MCF-7. The half-maximal inhibitory concentration, IC50, of the DOX–twisted tubular DNA origami was much lower than the free DOX. Compared to the tubular DNA origami, the twisted DNA origami may have better stability which promotes the delivery efficiency of the drugs. However, how these twisted structure influences the cellular uptake and endosome escape of the DNA origami needs to be studied.
2.4. Other driving forces
The construction of drug–DNA hybrid nanostructures could be driven by other driving forces. For example, Ma and co-workers reported Gemcitabine (Ge)-integrated DNA-like nanogels with accelerated drug activation for cancer therapy (Fig. 5D),89 in which the formation of the Ge-based DNA-like nanogels was driven by the π–π stacking interaction and hydrogen bonds. Here, conjugates containing 10 Ge are selected for the constructions of the Ge-nanogels. The stability of these nanogels should be compared with that of the crosslinked DNA nanogels. Also, how the number of the Ge in the conjugates influences the formation of nanogels is also worthy to be systematically investigated.Overall, the strength hierarchy of driving force for the construction of DNA nanostructures is chemical conjugation > base paring > hydrophobic interaction (Fig. 2). Compared to DNA nanostructures driven by base paring or hydrophobic interactions, nanostructures (e.g. SNA) driven by chemical conjugation exhibit higher stability, making them promising drug carrier candidates with long blood circulation in vivo. Nanostructures driven by base paring (e.g. DNA cages) have many unique advantages, including specific shape, precise size, and controllable drug loading content. The precise control on the size and drug loading content of DNA nanostructures can eliminate the variations of drug administration from batch to batch in clinics, a benefit that cannot be achieved by nanostructures driven by other driving forces. Nanostructures driven by hydrophobic interactions (e.g. DNA micelles) have a higher drug loading content than nanostructures driven by the other two driving forces. However, they may suffer from instability problems in vivo when the concentrations of micelles are lower than the critical micelle concentration (CMC).
3. Chemotherapeutic drug loading strategies
Chemotherapeutic drugs can be introduced to DNA nanostructures by various strategies with different driving forces (Table 1). This section highlights various types of chemotherapeutic drug loading methods for DNA nanostructures. We categorize the chemotherapeutic drug–DNA hybrid nanostructure-based drug delivery systems into four subtypes according to their drug loading methods, which are intercalation, conjugation, encapsulation, and base replacement. The advantages and disadvantages of each drug loading method are compared.| Loading method | Chemotherapeutic drug | DNA nanostructure | Driving force | Precision | Ref. |
|---|---|---|---|---|---|
| Abbreviations: Ref., references. | |||||
| Intercalation | Doxorubicin (DOX) | Cage | π–π stacking interaction | Low | 80 |
| Origami | 31 | ||||
| Aptamer | 81 | ||||
| Daunorubicin | Origami | 82 | |||
| Platinum drug | Cage | 83 | |||
| Conjugation | Paclitaxel (PTX) | Micelle | Covalent bond | Medium | 84 |
| Camptothecin (CPT) | Cage | High | 33 | ||
| DOX | SNA | Medium | 85 | ||
| Encapsulation | Platinum drug | Cage | Contained in cages | Low | 86 |
| BKM120 | Micelle | Hydrophobic interaction | 49 | ||
| Base replacement | Gemcitabine (Ge) | Nanogel | Covalent bond | Medium | 87 |
| Floxuridine (Fu) | Cage | High | 88 | ||
| Cage | High | 32 | |||
3.1. Drug loading by intercalation
Some of the chemotherapeutic drugs can be loaded to the DNA duplex by intercalation, where the planar moiety of the drug molecule inserts between the DNA base pairs. The main driving force for chemotherapeutic drug intercalation is thought to be π–π stacking interactions, with contributions from van der Waals and hydrophobic interactions. Drugs that can intercalate into the DNA duplex normally contain a planar moiety (Fig. 6). So far, the intercalation method has been the dominant drug loading method for drug–DNA hybrid nanostructure-based drug delivery systems due to its simplicity. The biggest advantage of the intercalation method is that the loading of drug does not interfere with the construction of the DNA nanostructure because drugs can be loaded to the carrier after the construction of the DNA nanostructure. Therefore, theoretically, any DNA nanostructures containing DNA double strands can be used as drug carriers using the intercalation drug loading method. One problem of the intercalation method is the loading of drugs cannot be precisely controlled. The total amount of the loaded drugs may be able to be calculated, but the exact locations of drugs in DNA nanostructures cannot be known. Furthermore, only a few drugs with planar moieties are able to be loaded to DNA nanostructures by intercalation.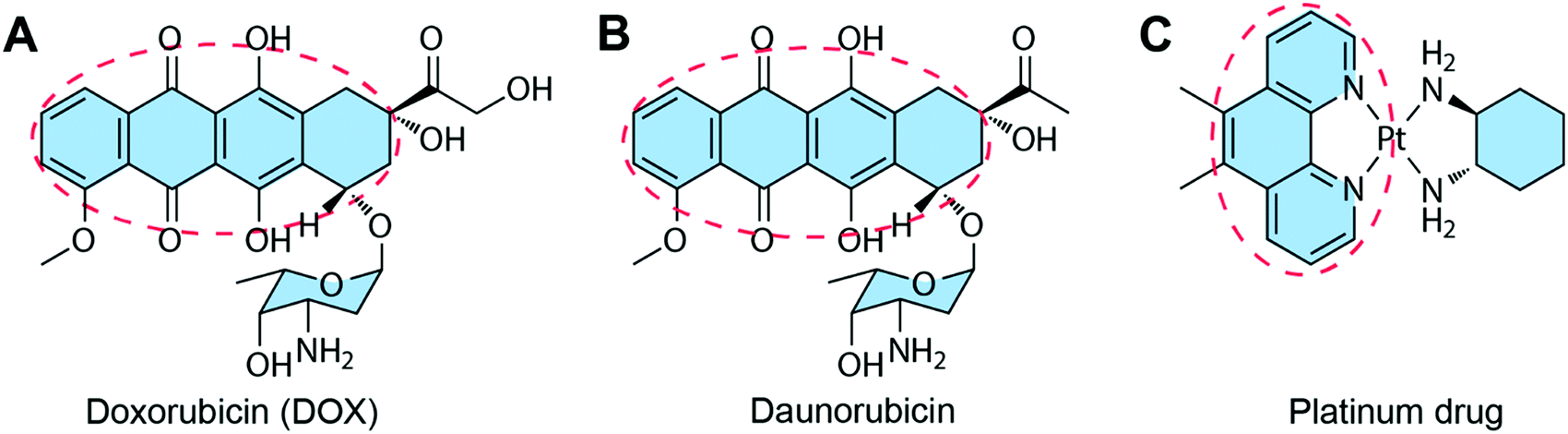 | ||
| Fig. 6 Chemotherapeutic drugs that can intercalate to DNA nanostructures. The red circles show the planar moieties for drug intercalation. (A) DOX. Reproduced with permission from ref. 81. Copyright 2013 National Academy of Sciences; (B) Daunorubicin. Reproduced with permission from ref. 82. Copyright 2016 Wiley-VCH; (C) Platinum drug. Reproduced with permission from ref. 83. Copyright 2019 Wiley-VCH. | ||
Besides the three drugs mentioned here, many other anticancer drugs containing planar moieties can also be loaded to the DNA nanostructures via intercalation, including epirubicin, idarubicin, aclarubicin, actinomycin-D, and mitoxantrone.90 In order to accommodate drugs, a DNA duplex needs to increase the vertical distance between the base pairs to generate a space for the incoming drugs. The double helix is thereby partially unwound, resulting in distortions of the sugar-phosphate backbone and alterations in the twist angle of base pairs.91 For example, the intercalation of DOX to DNA leads to alterations of the DNA structure with a partial B-form to A-form DNA transition.92 Therefore, the amounts of drugs intercalated to DNA nanostructures needs to be optimized to avoid the instability problems of the resulted drug–DNA nanostructures. The drug loading capacity can be enhanced by introducing more GC-pair sequences to DNA nanostructures due to the intercalation preference of drugs to the GC regions.93–95 Additionally, DNA nanostructures with multiple layers of double helices (e.g. DNA origami) are able to accommodate therapeutic or high levels of drugs, making them better candidates for drug intercalation than DNA nanostructures with fewer double helices (e.g. DNA cages).
3.2. Drug loading by conjugation
Some chemotherapeutic drugs can be loaded into DNA nanostructures by covalent conjugation. Normally, drugs conjugate to the DNA chain via the formation of a degradable chemical bond, which can be degraded in biological medium and release the functional free drugs for anticancer therapy (Fig. 7). The biggest advantage of the conjugation method is the high retention of the loaded drugs. Drugs can stably attach to DNA nanostructures, thus they are entirely transferred to the target tumor sites without drug leaking during in vivo circulation. However, only the drugs with appropriate reactive groups can be conjugated to DNA nanostructures via covalent bonds. Some of the conjugation reactions have quite low conjugation efficiency, therefore need harsh reaction conditions, which may partially destroy the DNA materials.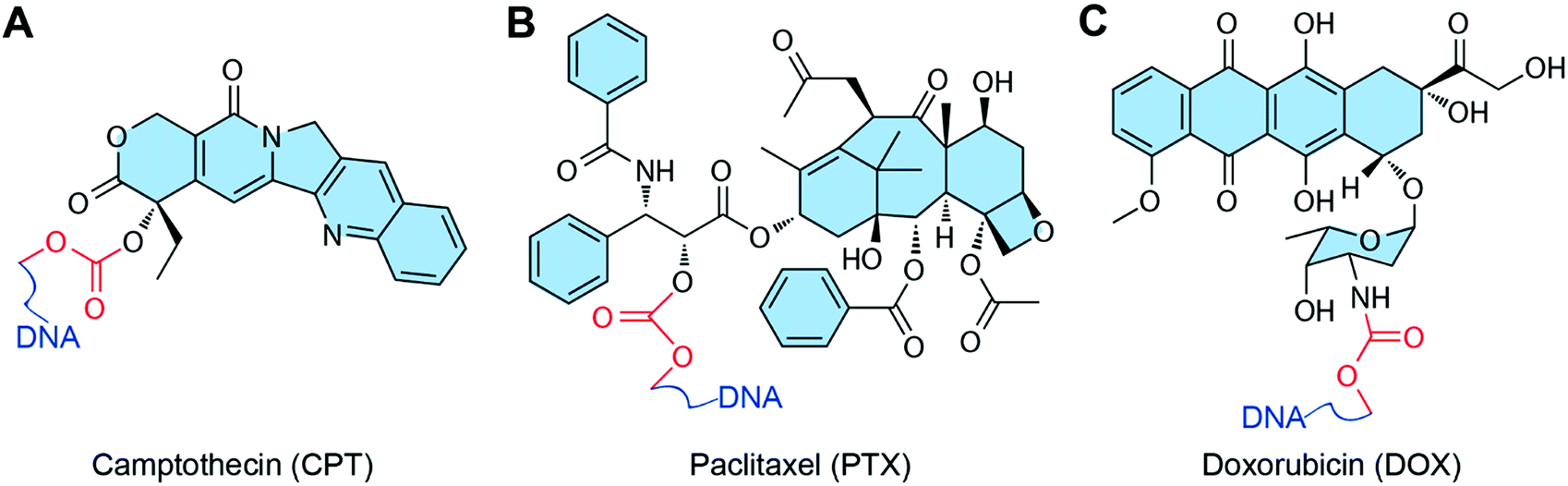 | ||
| Fig. 7 Chemotherapeutic drugs that can conjugate to DNA nanostructures. The red parts are chemical bonds for drug conjugation. (A) Camptothecin (CPT). Reproduced with permission from ref. 33. Copyright 2019 Wiley-VCH; (B) paclitaxel (PTX). Reproduced with permission from ref. 84. Copyright 2016 American Chemical Society; (C) DOX. Reproduced with permission from ref. 85. Copyright 2017 American Chemical Society. | ||
In general, several conjugation strategies have been developed to load drugs to DNA nanostructures. The modification of DNA strands with reactive groups can be easily achieved during the synthesis of DNA strands. Hence, the conjugation of drugs to DNA nanostructures through the reactive end groups of DNA is the most widely used strategy. For instance, polydrugs of DOX and CPT are efficiently conjugated to aptamers via ‘click’ chemistries (thiol maleimide and strain promoted azide alkyne click).96 The other strategy is that the drugs (PTX) are conjugated to a phosphorothiolated DNA backbone via the reaction between the phosphorothioate group (PS) of the DNA backbone and a benzyl bromide group of modified drugs.97 One advantage of this strategy is that the PS modification sites can be introduced to any desired locations of the DNA strands during their solid-phase synthesis. Another strategy is that the drugs (DOX) are conjugated directly to the reactive 2-NH2 on deoxyguanosine using formaldehyde, and the thermo-liable methylene linkage between drugs and DNA strands triggers drug release at a physiological temperature.98
3.3. Drug loading by encapsulation
For drugs that can form small nanoparticles, they can be encapsulated into the cavity of the DNA cages.86 For drugs with high hydrophobicity, they can be encapsulated into the hydrophobic core of DNA micelles via hydrophobic interactions. The biggest advantage of the encapsulation method is the high drug loading capacity. Chemotherapeutic drugs can fill the whole cavity of DNA cages or the hydrophobic core of DNA nanostructures. In addition, most of the anticancer drugs are hydrophobic, but only a few of them have planar moieties or reactive groups. Therefore, the encapsulation method has broader applications than the intercalation or conjugation methods. The encapsulation method also has the problem of low precision in the drug loading content. It is unable to adjust the locations of drugs in DNA nanostructures. In addition, the encapsulation of drugs is not stable, therefore drug leakage can easily happen under diluted conditions owing to the destabilization of DNA micelles. For these reasons, the encapsulation method has not been widely used for loading drugs to DNA nanostructures.3.4. Drug loading by base replacement
Some chemotherapeutic drugs or prodrugs have high structural similarity to natural nucleosides; therefore, these nucleoside analogue drugs can replace some DNA bases of DNA strands without disrupting the base pairing. Thus, these chemotherapeutic drugs can be loaded into DNA nanostructures by base replacement (Fig. 8). There are several advantages for the base replacement method. First, chemotherapeutic drugs can be easily and conveniently introduced to DNA strands using an automated DNA synthesizer. Second, partial replacement of the DNA bases in DNA strands with chemotherapeutic drugs does not disrupt the base pairing or function of DNA strands.99 Therefore, the drug-containing DNA strands can be used like normal DNA strands for the assembly of any DNA nanostructures. Third, drugs can be loaded to any positions of any DNA nanostructures with high precision. The drug loading contents of DNA nanostructures can be precisely controlled by sequence programming, making them ideal candidates for precision medicine. One disadvantage of the base replacement method is that only a few drugs that have similar structures to the DNA bases can be loaded to DNA nanostructures by this method. Additionally, drugs need to be transformed into the corresponding phosphoramidites before the solid-phase synthesis.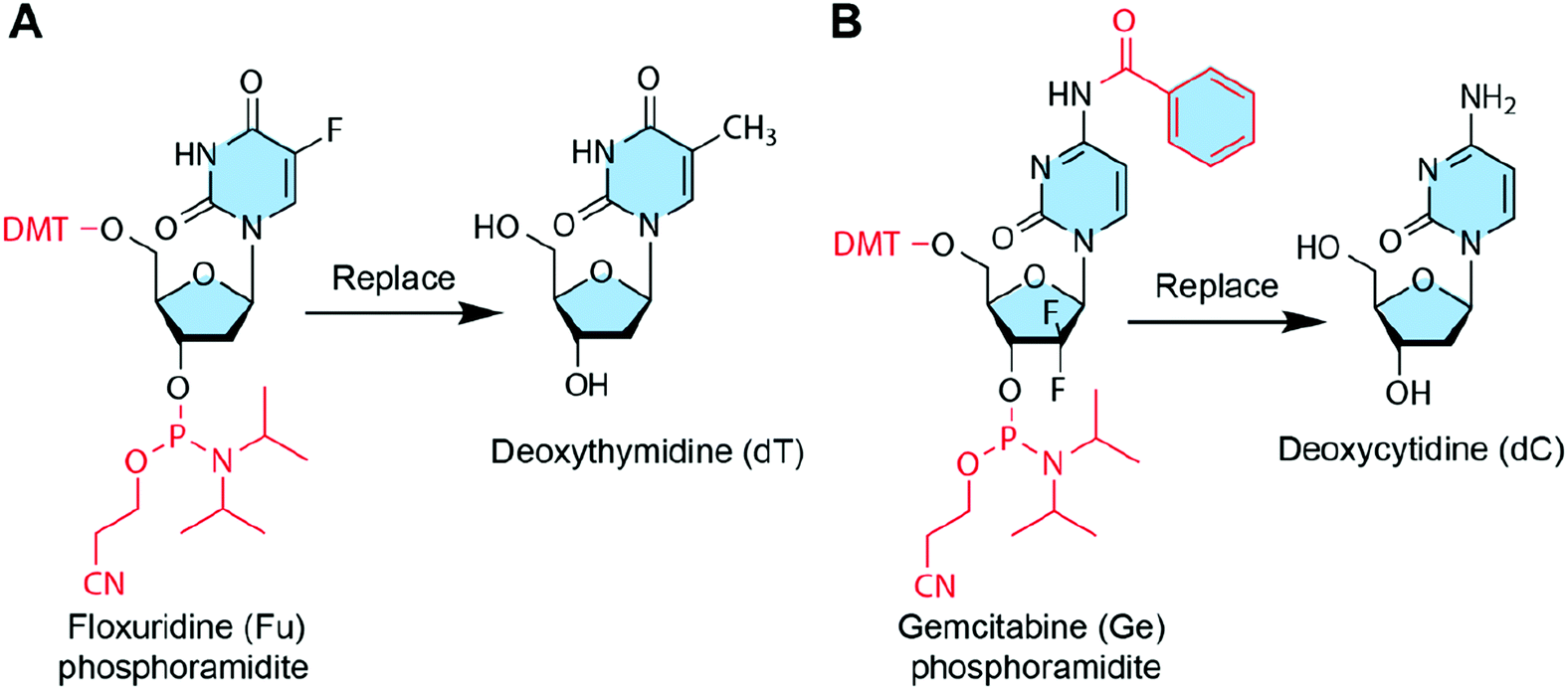 | ||
| Fig. 8 Chemotherapeutic drugs that can be introduced to DNA nanostructures by base replacement. The red parts show the protection groups and modifications to drugs so that they can replace DNA bases during the DNA synthesis. (A) Floxuridine (Fu) can replace deoxythymidine (dT). Reproduced with permission from ref. 32. Copyright 2017 Wiley-VCH; (B) Gemcitabine (Ge) can replace deoxycytidine (dC). Reproduced with permission from ref. 89. Copyright 2019. The Royal Society of Chemistry. | ||
In addition to deoxycytidine analogue Ge and deoxythymidine analogue Fu, some other anticancer nucleoside analogues could also be introduced to DNA nanostructures via base replacement, including cytarabine, 6-mercaptopurine, 8-chloro-adenosine, 8-amino-adenosine, fludarabine, capecitabine, clofarabine, and trifluorothymidine, etc.124,125
3.5. Drug loading by other methods
Chemotherapeutic drugs can also be loaded to DNA nanostructures by other methods. For example, the anticancer drug clofarabine can be loaded to DNA micelles by a molecular recognition approach, which is based on the interaction of two molecules via hydrogen bonding. Wang and co-workers reported the preparation of multifunctional nanoparticles by a molecular recognition method for targeted cancer therapy.1264. Chemotherapeutic drug–DNA hybrid nanostructures for anti-tumor therapy
This section highlights the latest development of drug delivery systems based on chemotherapeutic drug–DNA hybrid nanostructures (Table 2). We categorize drug–DNA nanostructure-based delivery systems into several subcategories according to their structure, such as DNA origami, cages, micelles, nanogels, aptamers, spherical nucleic acids (SNAs) and others. The advantages and disadvantages of each type are elaborated.| DNA structure | Drug | Tumor or cell model | Result | Ref. |
|---|---|---|---|---|
| Abbreviations: MTX, mitoxantrone; P-gp, P-glycoprotein; shRNA, small hairpin RNA; AuNRs, gold nanorods; EGFR-nb, epidermal growth factor receptor-nanobody; Ref., references. | ||||
| Aptamers | DOX | HepG2, Hu1545 cells | Selectively kill liver cancer cells HepG2 rather than Hu1545 cells | 100 |
| PTX and CPT | K562, DU145, HEK293 cells | IC50: 31.17nM, 76.63 nM (cancer cells K562 and DU145), >500 nM (normal cells HEK293) | 34 | |
| DOX | MCF-7R drug-resistant breast tumor mice | 88.2% of tumor growth inhibition, 21.6% for free DOX | 52 | |
| DOX | 4T1 breast tumor mice | Induce significantly slower tumor growth than free Dox | 101 | |
| Micelles | PTX (+ ASO) | MCF-7 breast tumor mice, drug-resistant HeLa cervical tumor mice | Accumulate in the tumor, and significantly slow the growth of the breast tumor and PTX-resistant cervical tumor | 97 |
| Fu | Drug-resistant BEL-7402 liver tumor mice | Remarkably inhibit both the subcutaneous and orthotopic tumor growth | 99 | |
| Fu | HeLa cervical tumor mice | Induce efficient cell apoptosis in tumor and efficiently inhibit the tumor growth | 102 | |
| SNA | DOX and MTX | 16 HBE, MRC 5, A549 cells | Decrease the viability of 80%, 92% and 70%, respectively, for 16 HBE, MRC 5 and A549 cells | 103 |
| DOX | HeLa cervical tumor mice | Gradually decrease the size of the tumor under the NIR laser | 104 | |
| DOX | HeLa cells | Induce significantly lower cell viability than the free DOX | 105 | |
| CPT | A549 cells | Result in 80% of cell death after 7 days | 106 | |
| DOX | KC, MCF-7, OMM1.3, Mel202 cells | Reduce the viability of the malignant cells MCF-7, OMM1.3 and Mel202 to a greater extent than normal cells KC | 107 | |
| Nanogels | Ge | A549 cells | Show much better proliferation inhibition efficacy (IC50 8 μM) than free Ge | 87 |
| DOX | CCRF-CEM, Ramos cells | Induce dose-dependent cytotoxicity only in target CEM cells, but not in non-target Ramos cells | 108 | |
| Cages | Pt NPs | BGC823/DDP Cisplatin-resistant gastric tumor mice | Overcome the drug resistance of the tumor and show potent inhibition of tumor growth | 86 |
| 56MESS (+EGFR-nb) | A431 epidermoid tumor mice | Result in marked apoptosis and down-regulation of EGFR in the tumors, exhibit a great tumor-inhibiting effect | 83 | |
| DOX | MCF-7 breast tumor mice | Induce significant inhibition of tumor growth | 109 | |
| PTX | A549 and PTX-resistant A549 cells | Downregulate MDR 1 gene and P-gp, kill cancer cells via apoptosis | 110 | |
| DOX | MCF-7 and drug-resistant MCF-7/ADR cells | Overcome drug-resistant cells with enhanced uptake and bypass the efflux process, induce significant cytotoxicity | 111 | |
| Origami | DOX (+p53 gene) | MCF-7R drug-resistant breast tumor mice | Reverse the drug-resistance of tumor and exhibit a marked and consistent inhibition of tumor growth | 112 |
| DOX (+shRNA) | MCF-7R drug-resistant breast tumor mice | Result in potent silencing effect of survivin and P-gp, show complete suppression of tumor growth | 113 | |
| DOX (+AuNRs) | Drug-resistant MCF-7/ADR cells | Inhibit P-gp with low power laser, induce obvious cell death | 114 | |
| Others | DOX | CCRF-CEM, Ramos cells | Show significantly higher cytotoxicity under UV irradiation than without UV | 115 |
| DOX | HCT 116 colon tumor mice | Display excellent antitumor capability compared to free DOX | 116 | |
| DOX | CCRF-CEM lymphoblast tumor mice | Achieve remarkable tumor regression and no obvious change in body weight | 117 | |
| DOX | CCRF-CEM lymphoblast tumor mice | Show significant accumulation of DOX in the tumor site | 118 | |
| DOX (+AuNRs) | HeLa cells | Induce much higher cytotoxicity than a non-targeted nanosystem | 119 | |
| DOX | MDA-MB-468 3D breast tumor spheroid models | Exhibit high effectiveness in killing tumor cells | 120 | |
| DOX (+AuNRs) | 4T1 breast tumor mice | Remarkably inhibit the primary orthotopic tumor growth and suppress lung metastasis | 121 | |
| DOX | CEM, HeLa, Ramos cells | Induce dose-dependent cytotoxicity in the targeted CEM and HeLa cells | 122 | |
| DOX (+AuNRs) | KB and HeLa cervical tumor mice | Induce potent inhibition of both KB and HeLa tumor growth under NIR irradiation | 123 | |
4.1. Drug–aptamer nanostructures
Aptamers have been used as drug carriers for the delivery of DOX, PTX, CPT, etc. Without the introduction of any additional targeting ligand, the aptamers themselves have strong targeting ability and can precisely deliver the chemotherapeutic drugs into the target cells or tissues, making the drug–aptamer nanostructure-based carrier a highly efficient drug delivery system. One problem of the aptamers is the low drug loading capacity. The overloading of drugs or loading drugs at inappropriate positions may reduce the targeting ability of the aptamers.Zhang and co-workers developed an aptamer-modified nanotrain assembled from six-letter DNA for the delivery of DOX (Fig. 9A).100 In addition to the 4 basic DNA bases, an additional base pair denoted as Z and P was also used to construct the aptamer-nanotrain. In vitro cytotoxicity assay showed that the DOX-loaded aptamer-nanotrain selectively killed the target liver cancer cells and did not exhibit significant toxicity to cells to which the aptamer did not bind. This work expands the toolbox of DNA bases. The introduction of the new Z and P base pairs allows a more diverse sequence for the construction of DNA nanostructures. However, how the introduction of the Z and P pairs influences the intercalation of the DOX to nanostructures needs to be investigated.
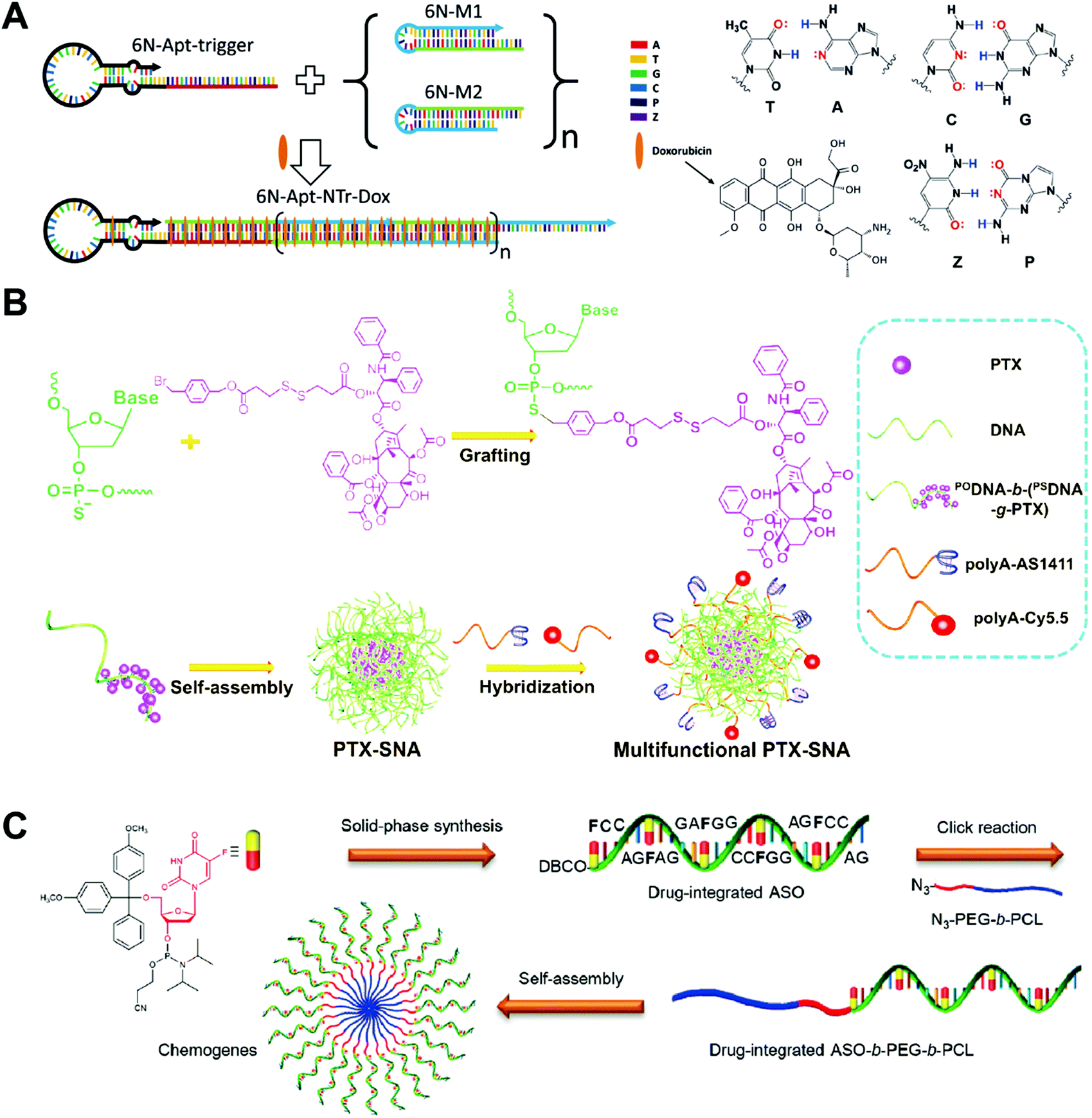 | ||
| Fig. 9 Biomedical application of drug–DNA aptamer and micelle hybrid nanostructures. (A) (left) The aptamer-nanotrain was prepared by polymerization of six-letter DNA monomers M1 and M2 initiated by the aptamer-trigger. The DOX was then loaded to the nanotrain by intercalation; (right) the structures of the six DNA bases including the two new bases Z and P. Reproduced with permission from ref. 100. Copyright 2014 Wiley-VCH; (B) PTX was first grafted to the DNA chain, which assembled to the SNA-like PTX micelles, and then the PTX micelles were further functioned with ligands or dyes. Reproduced with permission from ref. 97. Copyright 2019 Wiley-VCH; (C) the drug-integrated ASO was prepared directly by solid-phase synthesis, followed by conjugation to the hydrophobic polymer poly(ε-caprolactone) (PCL). The chemogene-micelles were obtained via the self-assembly of ASO-PCL. Reproduced with permission from ref. 99. Copyright 2019 American Chemical Society. | ||
Zhou and co-workers described a bivalent aptamer–drug conjugate with tunable drug ratios for drug combination targeted cancer therapy.34 Two aptamers targeting transferrin receptor 1 were modified with different amounts of CPT and PTX molecules separately, and the conjugation of these two drug-modified aptamers by DNA ligation produced circular bivalent aptamer–drug conjugates. The bivalent aptamer–drug conjugates presented high cytotoxicity on transferrin receptor 1-positive (aptamer-targeted) DU145 cells and weak cytotoxicity on transferrin receptor 1-negative HEK293 cells. The present system has several advantages including modular preparation, accurately tunable drug ratios, coordinated cargo delivery, cancer-targeted binding and uptake, esterase-triggered cargo release, and capability of releasing multiple drug combinations. Other drug–aptamer hybrid nanostructures such as aptamer-based nanoparticles52 and aptamer-based three-way junction pocket nanostructures101 have also been developed for DOX delivery in vitro and in vivo.
4.2. Drug–DNA micelle nanostructures
DNA micelles have been used as drug carriers for the delivery of PTX, 5-Fu, etc. DNA micelles normally have a simple preparation procedure and high drug loading capacity, making them appealing for chemotherapeutic drug delivery. The major issue of DNA micelles is their instability; drugs are loaded in the hydrophobic core of the micelles, and so the destabilization of the micelles triggers the burst release of the drugs during the circulation in off-target areas. For those drugs which are conjugated to DNA strands, the destabilization of the micelles does not trigger the immediate release of drugs, but does significantly reduce the accumulation of drugs in the tumor sites.Guo and co-workers prepared DNA–drug conjugates for the delivery of anticancer drug PTX (Fig. 9B).97 As a proof of concept, benzyl-bromide-modified paclitaxel (PTX) was grafted onto the phosphorothiolated DNA backbone at the phosphorothioate group modified sites. Then, the PTX-DNA micelles with high drug loading capacity were prepared via the self-assembly of the resulting amphiphilic PTX-DNA conjugates. The in vivo antitumor effect of PTX-DNA micelles was evaluated in MCF-7-cell-xenografted nude mice. PTX-DNA micelles with anti-Bcl-2 sequences (Bcl2-PTX-DNA) significantly downregulated Bcl-2 gene expression and showed greater inhibitions of the growth of PTX-resistant tumor than the free PTX and PTX-DNA micelles. In most of the drug–DNA micelles, drugs are first conjugated to a polymer backbone, followed by conjugation to DNA strands. Here the drugs PTX are directly conjugated to the block DNA strands via the phosphorothioate groups without introducing additional polymers. The present system enhances the antitumor therapeutic effect of the anticancer drug by the reversal of drug resistance.
Mou and co-workers constructed micelles assembled from drug-integrated ASO to reverse drug resistance (Fig. 9C).99 A 5-Fu drug-integrated ASO was first prepared by the solid-phase synthesis using a DNA synthesizer. After conjugating the 5-Fu ASO to a hydrophobic polymer chain poly(ε-caprolactone) (PCL), the resulting amphiphilic 5-Fu ASO-PCL assembled into DNA micelles. The western blot assay showed that the drug-resistant human liver carcinoma (BEL-7402) cells treated with Bcl-2 micelles displayed downregulated Bcl-2 gene expression. The in vivo antitumor effect of the Bcl-2 micelles was evaluated in mice bearing orthotopic drug-resistant BEL-7402 tumors. The T2-weighted magnetic resonance imaging (MRI) data showed that the tumor growth of the mice was significantly inhibited by the treatment of Bcl-2 5-Fu micelles. This study demonstrates that appropriate integration of 5-Fu drug to ASOs does not influence the biological silencing function of the ASO. Based on this, it should be much easier to develop combined chemo- and gene therapies for an enhanced anti-tumor effect with ASOs.
Jin and co-workers reported that DNA micelles bound with albumin in situ for the delivery of the 5-Fu drug.102 DNA micelles were assembled from the lipid modified 5-Fu oligonucleotide (LFU20). Upon intravenous injection, the LFU20 detaches from DNA micelles and then forms an LFU20/albumin complex via the hydrophobic interactions between the lipid and the hydrophobic cave of albumin. DNA micelles showed a much stronger tumor inhibition effect in the mice than the free drugs. This is an interesting study which takes advantage of the instability of DNA micelles. The drug-integrated DNA strands detach from the micelles and rapidly bind to the albumin, and the resulted LFU20/albumin complex accumulate in the tumor via the EPR effect.
4.3. Drug–SNA nanostructures
The SNA has been used as drug carriers for the delivery of DOX, mitoxantrone (MTX), etc. One of the advantages of the SNA-based drug delivery system is its efficient cellular internalization. The SNA structure can be endocytosized by the cells via the scavenger receptor-mediated pathway. The problem of the SNA nanostructure is the limited drug loading capacity. Normally, drugs can only be loaded to outer layer DNA strands. Since the density and orientation of the surface DNA strands of the SNA are crucial for their efficient cellular uptake, the high amounts of drugs might change the surface properties of the SNA, therefore influencing their cellular uptake.Kyriazi and co-workers developed gold-based SNA dimers for mRNA sensing and targeted drug delivery (Fig. 10A).103 Two gold (Au)-based SNAs were first prepared, and then, two different mRNA probes and anticancer drugs DOX and MTX were loaded into each SNA. The conjugation of the two SNAs via DNA hybridization yielded the SNA dimers. The mRNA sensing and drug delivery ability of the SNA dimers were evaluated by confocal microscopy. In the human lung adenocarcinoma (A549) cells that contained both keratin 8 and vimentin mRNAs, both SNAs of the SNA dimers were activated, showing yellow fluorescence that merged from green and red fluorescence, and released the drugs DOX and MTX. The study develops a SNA-based multifunctional platform for the sensing of multiple mRNAs and delivery of multiple drugs.
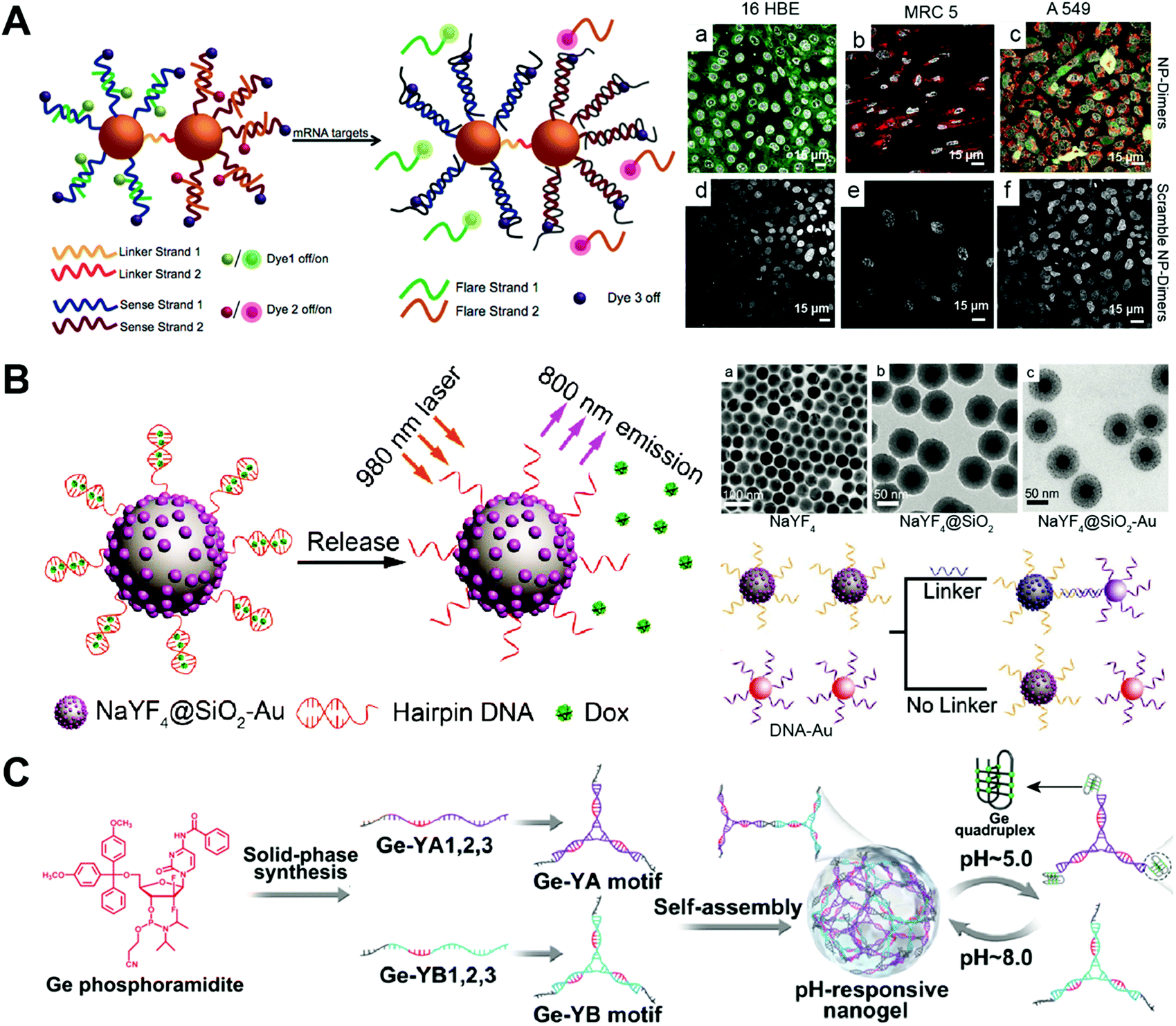 | ||
| Fig. 10 Biomedical application of drug–SNA and DNA nanogel hybrid nanostructures. (A) (left) The gold-based SNA-dimers could sense the mRNA target and activate the dyes. (right) Microscopy images of SNA (NP)-dimers incubated with 16 HBE, MRC 5 and A549 cells. Reproduced with permission from ref. 103. Copyright 2018 American Chemical Society; (B) (left) the SNA containing an upconversion nanoparticle (UCNP) core released the DOX from the hairpin DNA structure when exposed to 980 nm light due to the photothermal effect; (right) TEM images of the UCNP with different coatings and the UCNP-SNA or Au-SNA could form conjugates with a DNA linker. Reproduced with permission from ref. 104. Copyright 2017 Wiley-VCH; (C) the Ge-integrated YA and YB motifs containing pH-sensitive i-motif sequences were prepared from different Ge–DNA strands, and then the self-assembly of YA and YB motifs resulted in the pH-responsive nanogels, which could disassemble at pH 5.0 due to the formation of the G-quadruplex of the i-motifs. Reproduced with permission from ref. 87. Copyright 2019 American Chemical Society. | ||
Han and co-workers described the upconversion nanocrystal-based SNA for imaging and in vivo drug delivery (Fig. 10B).104 The upconversion nanoparticles (UCNP) were first coated with Au nanoparticles, followed by the immobilization of the highly thermally sensitive hairpin DNA strands to the Au nanoparticles via Au–thiol chemistry. The drug-loaded UCNP-SNA was obtained after loading the chemotherapeutic drug DOX into the double stranded structure of the hairpin DNA by intercalation. The in vivo therapy ability of the UCNP-SNA was evaluated in a mouse model containing a subcutaneous tumor. Upon local injection of UCNP-SNA to the tumor, the upconverted NIR emission was observed at the injection site under 980 nm laser irradiation. The laser irradiation resulted in a photothermal effect which subsequently triggered the DOX release from the hairpin DNA, dramatically inhibiting the tumor growth of the mouse. This is a good example of DNA nanostructures with on-demand drug release properties. The duplex structure of the hairpin DNA can be destroyed at a certain temperature. The laser irradiation of the SNA rises the temperature of the hairpin DNA via the photothermal effect, triggering the release of the intercalated DOX. Other drug–SNA hybrid nanostructures such as pH-responsive SNA,105 SNA conjugates,106 and targeted SNA107 have also been reported for cancer therapy.
4.4. Drug–DNA nanogel nanostructures
DNA nanogels have been used as drug carriers for the delivery of Ge, DOX, etc. One advantage of the DNA nanogel-based drug delivery system is that the size of the nanogels can be easily controlled by adjusting the linker ratio. Another advantage is that the drug cargo is loaded inside the nanogels, avoiding the interaction of the cargo with other components in the biological medium. One major problem of DNA nanogels is their propensity for aggregation. The size of DNA nanogels not only depends on the ratio of the backbone and the crosslinker, but also the concentration, crosslinking time, etc. Further crosslinking of the nanogels may happen when the nanogels are concentrated or incubated for too long, resulting in the aggregation of the nanogels.Pan and co-workers prepared chemotherapeutic drug Ge-integrated DNA nanogels for pH-responsive drug delivery (Fig. 10C).87 The Ge-integrated DNA strands containing pH-sensitive i-motif sequences were first prepared by solid-phase synthesis using the DNA synthesizer. The self-assembly of these DNA strands generated two Y motifs denoted YA and YB, which further assembled into the Ge-integrated DNA nanogels. The pH-responsive dissociation of DNA nanogels was confirmed by confocal microscopy, in which, a fluorescence resonance energy transfer (FRET) effect of DNA nanogels was observed in the acidic environment of the cells. Both cell viability and apoptosis experiments demonstrated that the i-motif DNA nanogels induced more cell death compared to free drug and the non-pH-responsive DNA nanogels. This study shows that appropriate integration of Ge to DNA strands does not significantly disturb the base pairing function of DNA strands. Furthermore, the on-demand destabilization of the nanogels in cells can enhance the anticancer effect of drugs.
Wu and co-workers developed the DOX-loaded DNA nanogels for targeted cancer therapy.108 The Y-shaped functional DNA domains and the X-shaped DNA core connectors were first prepared. These two DNA structures assembled to building units which further assembled to DNA nanogels by polymerization under UV irradiation. The confocal microscopy images showed that DNA nanogels specifically internalized into the target CEM cells rather than the non-target Ramos cells. This study demonstrates the use of the polymerization method to prepare DNA nanogels. The building blocks consisting of DNA strands are crosslinked by the UV irradiation-initiated polymerization.
4.5. Drug–DNA cage nanostructures
DNA cages have been used as drug carriers for the delivery of platinum drug, CPT, etc. One advantage of the DNA cage-based drug delivery system is the high precision of drug loading. Both the amount of loaded drug and the position of drug in DNA cages can be precisely controlled. Another advantage is the high uniformity of the size of DNA cages. However, the drug loading capacity of DNA cages may be limited by their special structure. The drugs can be only introduced to the surface DNA strands of the cages and the interior remaining empty. Therefore, DNA cages have much lower drug loading capacity than DNA nanogels with the same size.Ma and co-workers constructed the enzyme-responsive DNA icosahedron-based platinum drug delivery system for cisplatin-resistant cancer therapy (Fig. 11A).86 The construction of DNA icosahedron required two building blocks, the pyramidal DNA cages and the telomerase-responsive connector. The drug-loaded DNA icosahedrons were obtained by connecting two pyramidal cages together using the telomerase-responsive connector and loading the platinum drug to the cages. Pt NPs with different sizes (2, 4 and 10 nm) were prepared, and the small Pt NPs with size of 2 nm were chosen as appropriate nanodrugs for encapsulation into the DNA icosahedra due to their higher loading amounts and more efficient Pt ion release. After the incubation with DOX intercalated DNA icosahedrons, the cancer cells U87MG and HepG2 that contained telomerase exhibited red fluorescence of the released DOX as demonstrated by confocal microscopy, while the normal human normal hepatocyte (L02) cells without telomerase did not, indicating that the telomerase triggered disassembly of DNA icosahedrons. The drug-loaded DNA icosahedrons displayed significantly higher tumor growth inhibition in BCG823/DDP tumor-bearing mice than the free platinum drug nanoparticles and the free cisplatin controls. This is an interesting study that the drug nanoparticles are encapsulated in DNA cages directly without hydrophobic interactions or covalent conjugation. However, encapsulation of the free drugs by the cages is not possible since small drug molecules can easily escape from the cages through the large holes. This system may be an ideal platform for the delivery of large therapeutics like proteins.
 | ||
| Fig. 11 Biomedical application of drug–DNA cage hybrid nanostructures. (A) (left) The platinum drugs Pt NPs were encapsulated into DNA icosahedrons during the formation of the cages, and the release of drugs was triggered by telomerase which could cut specific DNA sequences in the cages; (right) TEM images of platinum drugs and drug-loaded cages with or without telomerase. Reproduced with permission from ref. 86. Copyright 2018 Wiley-VCH; (B) (left) the tetrahedrons were first modified with anti-epidermal growth factor receptor (EGFR) nanobody, followed by the intercalation of the drugs 56MESS. The resulted tetrahedrons could target the cells via the EGFR; (right-up) gel electrophoresis of the tetrahedrons; (right-down) AFM images of the tetrahedrons. Reproduced with permission from ref. 83. Copyright 2019 Wiley-VCH. | ||
Wu and co-workers described the nanobody-modified DNA tetrahedrons for targeted delivery of platinum drug (Fig. 11B).83 The targeted DNA tetrahedrons were prepared by the conjugation of an anti-epidermal growth factor receptor (EGFR) nanobody to the tetrahedrons. The nanobody was used not only as a target ligand to facilitate the cellular uptake of the tetrahedrons, but also as an inhibitor to block the signal transduction of EGFR in the cells with EGFR overexpression. The cytotoxicity of DNA tetrahedrons was evaluated in human epidermoid carcinoma (A431) cells with high EGFR expression. The drug-loaded nanobody-tetrahedrons exhibited the greatest tumor growth inhibitory effect. In vivo data showed that the drug-loaded nanobody-tetrahedrons almost completely inhibit the tumor growth of the A431-tumor-bearing mice for 20 days. This study demonstrated a DNA tetrahedron-based system for combined chemo and protein therapies. The drugs are easily loaded to the tetrahedrons by intercalation and the proteins are conveniently attached to the tetrahedrons by DNA hybridization. Other drug–DNA cage hybrid nanostructures such as targeted tetrahedrons,109 PTX-tetrahedrons110 and self-assembled DOX-tetrahedrons111 have also been produced for anti-tumor therapy and overcoming drug-resistance in different cancers.
4.6. Drug–DNA origami nanostructures
Compared to DNA cages, DNA origami has more complicated and varied shapes and structures. DNA origami has been used as drug carriers for the delivery of DOX. Like DNA cages, one advantage of the DNA origami-based drug delivery system is the high uniformity in the size of DNA origami. Another advantage is that DNA origami can have various structures with different shapes and sizes which are adjustable for the purpose of efficient drug delivery. In addition, the excellent mechanical rigidity of DNA origami may facilitate its cellular uptake. Compared to DNA nanostructures that are described above, DNA origami is the costliest nanostructure since it requires a large amount of well-designed DNA strands for its construction. In addition, not all drug loading methods are suitable for DNA origami, and intercalation is the only method that has been widely reported for the construction of hybrid drug–DNA origami nanostructures.Liu and co-workers reported a triangular DNA origami-based system for combined gene and anticancer drug delivery (Fig. 12A).112 The triangular DNA origami (TO) was first modified with an aptamer targeting ligand, and then DOX was loaded to the TO followed by the conjugation of the p53 gene to the TO by DNA hybridization. The kite-like structure of the DOX-p53-TO was determined by atomic force microscopy (AFM). A DOX-resistant MCF-7 tumor-bearing mouse model was used to evaluate the antitumor efficacy of the DOX-p53-TO. During a treatment period of 24 days, tumor growth was largely inhibited by the combined therapy of DOX-p53-TO, while only partial tumor growth inhibitions were observed for the free DOX and treatments with DOX-p53. This is a good example of the DNA origami-based system for combined chemo and gene therapies. Normally, the large functional genes (p53) have difficulty entering cells unless they are condensed by cationic materials. However, the large p53 gene can be efficiently internalized by cells with the help of DNA origami (TO). Therefore, this system also represents a non-cationic system for the delivery of large functional genes.
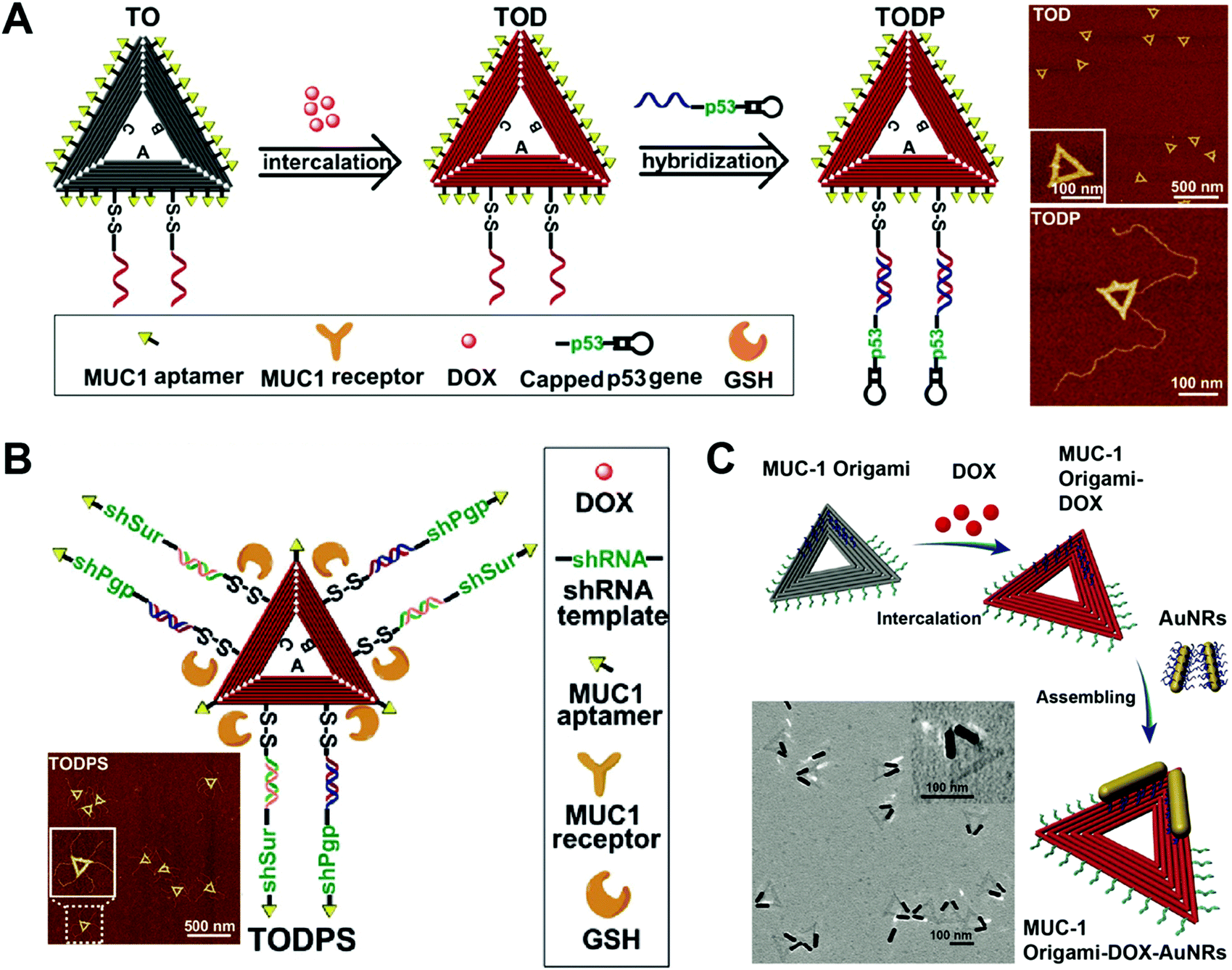 | ||
| Fig. 12 Biomedical application of DNA origami. (A) (left) Intercalation of DOX with the aptamer-modified triangular origami (TO) complex yielded the DOX-loaded TO (TOD), which was further modified with the anti-tumor gene p53; (right) AFM images of the TO. Reproduced with permission from ref. 112. Copyright 2018 American Chemical Society; (B) the triangular DNA origami-based nanoplatform containing the target aptamer ligands, the genes encoding siRNAs targeting P-gp and survivin, and the anticancer drug DOX is presented; (inset) the AFM image of the TO. Reproduced with permission from ref. 113. Copyright 2018 Wiley-VCH; (C) DOX was intercalated to the aptamer-decorated TO, followed by the introduction of gold nanorods (AuNRs) via assembly; (inset) TEM image of the TO. Reproduced with permission from ref. 114. Copyright 2017 The Royal Society of Chemistry. | ||
Liu and co-workers prepared triangular DNA origami-based nanoplatform for multidrug-resistant cancer therapy (Fig. 12B).113 A triangular origami-based drug and gene combined system DOX-shRNA-TO was prepared. After uptake, the DOX-shRNA-TO was designed to release gene which codes for the production of siRNAs which target P-gp and survivin. In vivo data demonstrated that the combined therapy of chemotherapeutic drug DOX and double siRNAs significantly inhibited tumor growth of DOX resistant MCF-7 tumor-bearing mice, while other treatments showed lower antitumor efficacy. Instead of a combination of chemo- and gene therapy, this study demonstrates a DNA origami-based system for synergistic chemo-/gene therapy. The siRNAs targeting the P-gp knocked down the P-gp expression on the cells, dramatically enhancing the anti-tumor effect of the DOX by the reversal of the drug-resistance of the cancer cells.
Song and co-workers circumvented drug resistance by constructing hybrid nanostructures consisting of triangular DNA origami and gold nanorods (Fig. 12C).114 In this case, the DOX-TO and DNA gold nanorods (AuNRs) were first prepared, and then the DOX-TO–AuNR hybrid nanostructures were obtained by conjugating the AuNR to the DOX-TO via DNA hybridization. The hybrid structure of the DOX-TO–AuNR was determined by transmission electron microscopy (TEM). In cell viability assay, the combined treatment of chemotherapeutic drug DOX and photothermal therapy led to about 80% cell death. This study shows a DNA origami-based system for combined chemo and photothermal therapy. The high programmability of the DNA origami allows the attachment of the photothermal AuNRs to any precise position within the DNA origami, achieving the maximal therapeutic effect.
4.7. Other drug–DNA nanostructures
Besides the DNA structures described above, there are also some other DNA structures used as nanocarriers for the delivery of chemotherapeutic drugs.Liu and co-workers prepared an aptamer-conjugated hyperbranched polymer assembly for targeted delivery of DOX (Fig. 13A).115 The aptamer DNA was first conjugated to the hyperbranched polymers (HBP), and then the self-assembly of the aptamer-HBP and DOX formed the aptamer DOX-HBP nanoparticles. Photosensitive nitrobenzyl groups were introduced to the aptamer DOX-HBP nanoparticles to trigger drug release from the nanoparticles under UV irradiation. In cell viability experiments, the aptamer nanoparticles showed the strongest cell killing capability under UV irradiation. In this study, the aptamers function as the hydrophilic outer layers and the targeting ligands of the nanoparticles. Nitrobenzyl group-based photo-sensitive bonds allows for release of DOX from the nanoparticles under external stimuli.
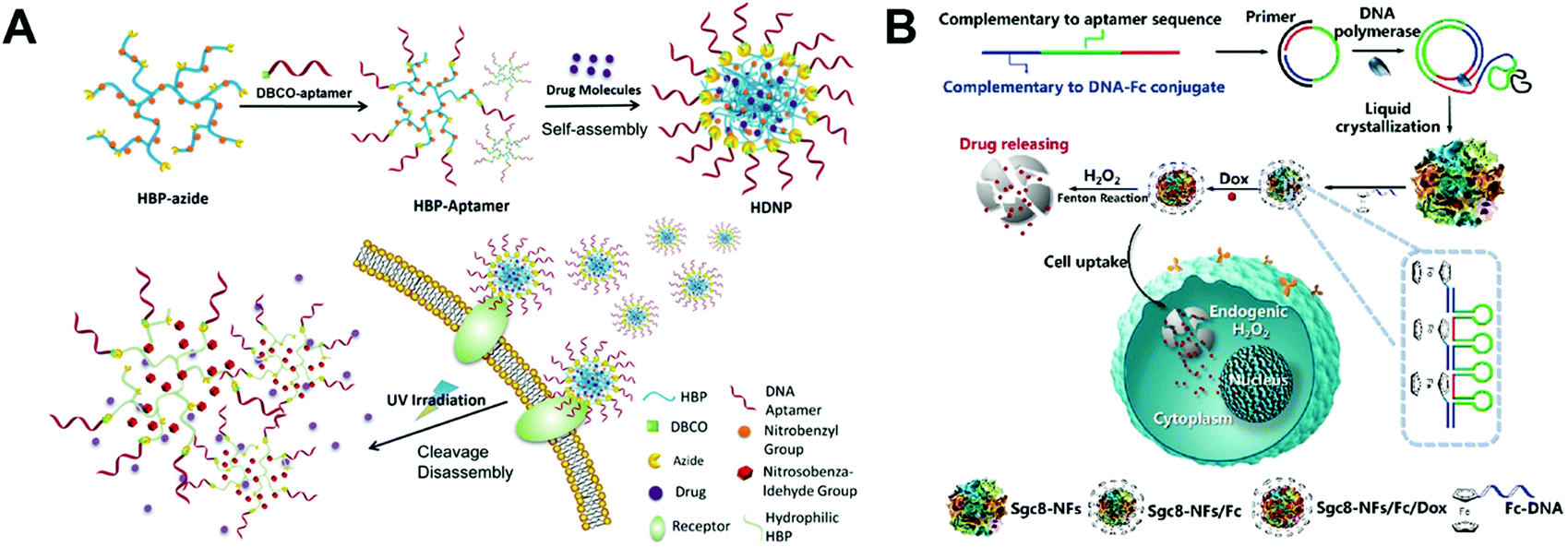 | ||
| Fig. 13 Biomedical application of other drug–DNA hybrid nanostructures. (A) The hyperbranched polymers (HBP) containing photosensitive moieties were first modified with aptamers and drugs were loaded to DNA nanostructures via self-assembly. The resulted nanostructures can target the cells via the receptors and release drugs under UV irradiation. Reproduced with permission from ref. 115. Copyright 2018 Wiley-VCH; (B) long DNA strands were obtained by the DNA polymerase, and the liquid crystallization of these strands resulted in DNA nanoflowers, which were further modified with ferrocene (Fc) and loaded with DOX. The release of drugs were triggered by the endogenic H2O2via the Fenton reactions. Reproduced with permission from ref. 116. Copyright 2019 American Chemical Society. | ||
Liu and co-workers reported a bioinspired, self-degradable DNA nanoflower-based drug delivery system for targeted cancer therapy (Fig. 13B).116 Long DNA building blocks were first generated by rolling circle replication. Then, the crystallization, nucleation, and growth of the long DNA building blocks yielded DNA nanoflowers which were subsequently modified with ferrocene (Fc). The structure of the obtained DNA nanoflowers was determined by TEM. The in vivo antitumor efficacy of the DOX loaded Fc-nanoflowers was evaluated in a mouse model bearing HCT-116 tumors. Among all the formulations, the DOX-Fc-nanoflowers displayed the greatest antitumor efficacy in mice. This study introduces a new method for controlled drug release from DNA nanostructures. With modification of the Fc molecules, DNA nanoflowers are able to release their DOX in the presence of the endogenic H2O2via the Fenton reaction. Other drug–DNA hybrid nanostructures such as missile-like nanostructures,117 magnesium-stabilized nanostructures,118 photocontrollable nanosystems,119 nanosheet-DNA nanostructures,120 DNA–gold nanorods,121 DNA nanoflowers,122 and DNA–gold nanoparticles123 have also been prepared for anticancer drug delivery.
5. Conclusions and perspectives
Chemotherapeutic drug–DNA hybrid nanostructure-based anti-tumor therapy represents one of the most successful applications of DNA nanostructures, drawing increasing attention due to their high programmability.127 Compared to other traditional nanosized systems for biomedical applications, the DNA nanostructure-based systems have several advantages.5 One is the intrinsic biocompatibility and biodegradability,10,11 making them ideal platforms for the manipulation of different biological signals128,129 and the delivery of various therapeutics.130 The toxicity of nanomaterials is a major issue that needs to be addressed before translation to clinics.131 The other advantage is the modularity;132 not only can the shape, size and rigidity be well controlled, but the amounts of loading therapeutics and the distributions and positions of the cargos can also be precisely adjusted as needed.133 These unique and controllable properties make DNA nanostructures a powerful biomedical platform; they can precisely sense or image specific biomolecules and are capable of targeted delivery of various therapeutics including genes, proteins and chemotherapeutic drugs to the lesion sites as well as releasing the therapeutics in a controlled mode.134 Therefore, diverse and well-designed DNA nanostructures not only help to ensure safe and effective delivery of chemotherapeutic drugs, but also have an impact on the translation of various novel therapeutic platforms into clinical therapy.Despite the progress that has been made in this field, there are still some challenges that need to be tackled before the clinical use of DNA nanostructure-based drug delivery systems.135 Firstly, DNA nanostructures have low in vivo stability.136 Their biomedical performance depends on the stability and integrity in a biological environment. A long blood circulation time is crucial for the accumulation of therapeutics like chemotherapeutic drugs in the tumor. However, existing DNA nanostructure-based delivery systems normally have short circulation half-lives due to their vulnerabilities to enzymatic degradation. DNA nanogels has a blood half-life of about 1 h.28 New structure designs or protection methods are needed to enhance the stability. For example, the oligolysine-PEG coating has significantly protected DNA origami from denaturation and degradation,137 and the dendritic oligonucleotide-modified DNA brick nanostructures have exhibited enhanced stability.138
Secondly, most DNA nanostructures exhibit relatively poor cellular uptake and endosome escape efficiency owing to their negative charges.139 Aptamers have good cellular uptake owing to their targeting abilities.140 SNA nanocarriers have been reported that can efficiently enter the cells via scavenger receptors.141 Except for these two structures, other structures have poor cellular uptake due to their negative surface charges, which is not favorable for the negatively charged cell membranes.142 The inefficient endosome escape of the present DNA nanostructures is also a big issue. The efficient cellular delivery of the therapeutics by the nanoparticles depends on their capability of rapid endosome escape. Membrane fusion, osmotic rupture, particle swelling and membrane destabilization are the most representative mechanisms of endosome escape.143 However, none of these mechanisms is applicable for DNA nanostructures. Therefore, novel targeting and endosome disrupting strategies are required to improve cellular uptake and endosome escape. For instance, the cellular uptake of DNA origamis can be enhanced by increasing their size and compactness.144
Thirdly, DNA nanostructures have potential to generate immune response in vivo.145 In most of the studies, only the antitumor effect of drugs is studied. The immune response aroused by the DNA materials may be unobvious, therefore it has been neglected. It is noteworthy that the immune response may be quite different for different DNA sequences and nanostructures. The relationship of the DNA sequences/structures and the immune response needs to be comprehensively studied and illustrated. Thereafter, unwanted immune responses can be avoided, and desired immune responses can be employed for the purpose of immunotherapy. For example, the CPG sequences integrated DNA nanostructures have been exploited for immunotherapy via the activation of spleen dendritic cells (DCs).146
Finally, the preparation of DNA nanostructures is costly. The synthesis of the DNA building blocks is expensive and limited by synthetic capacity. In 2020, the price for 1 mg of commercial DNA oligos is 100–200 $. The high cost and low synthesis capacity significantly hinder the development of DNA nanostructure-based delivery systems from bench to clinics. New technologies and strategies are needed to reduce the cost and improve the synthetic capacity of the DNA materials. For instance, recently a biotechnological method has been developed for the mass production of DNA origamis,147 expanding the use of DNA nanotechnology in many areas.
Future studies need to focus on the unique properties of DNA nanostructures to maximize their efficacy in biomedical application. First, precision medicine can be achieved by DNA nanostructures, and is a preferred route of treatment due to the expected better outcome of the treatments.148,149 With precise control over the therapeutic-loading ratio and structure of the carriers, DNA nanostructures offer more reproducible formulations for precision medicine.32 Second, targeted delivery can be achieved by precise control over the shape of DNA nanostructures. DNA origami with rectangular, triangular and tubular shapes have been reported to accumulate preferentially in the kidneys.13 Therefore, it might be possible to realize targeted delivery of therapeutics to different organs in vivo by precise control over the shape of DNA nanostructures. Since the federal approval of the first aptamer drug pegaptanib (Macugen; Pfizer/Eyetech), an anti-vascular endothelial growth factor therapy for neovascular age-related macular degeneration (AMD),150 an increasing number of DNA aptamer drugs have been investigated for the treatment of different diseases in clinical trials.151 Encouraged by these clinical studies, it is highly expected that more and more clinically effective DNA nanostructure-based therapies will be developed in the near future.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by National Key R&D Program of China (2018YFA0704000), Basic Research Program of Shenzhen (JCYJ20170412111100742, JCYJ20180507182413022), Shenzen Science and Technology Program (KQTD20190929172538530), Fok Ying-Tong Education Foundation for Young Teachers in the Higher Education Institutions of China (161032), and Guangdong Province Natural Science Foundation of Major Basic Research and Cultivation Project (2018B030308003).References
- J. D. Watson and F. H. C. Crick, Nature, 1953, 171, 737–738 CrossRef CAS.
- G. Tikhomirov, P. Petersen and L. Qian, Nature, 2017, 552, 67–71 CrossRef CAS.
- P. W. K. Rothemund, Nature, 2006, 440, 297–302 CrossRef CAS.
- N. V. Voigt, T. Tørring, A. Rotaru, M. F. Jacobsen, J. B. Ravnsbæk, R. Subramani, W. Mamdouh, J. Kjems, A. Mokhir, F. Besenbacher and K. V. Gothelf, Nat. Nanotechnol., 2010, 5, 200–203 CrossRef CAS.
- Q. Hu, H. Li, L. Wang, H. Gu and C. Fan, Chem. Rev., 2019, 119, 6459–6506 CAS.
- Y.-J. Chen, B. Groves, R. A. Muscat and G. Seelig, Nat. Nanotechnol., 2015, 10, 748–760 CrossRef CAS.
- J. Y. Kishi, T. E. Schaus, N. Gopalkrishnan, F. Xuan and P. Yin, Nat. Chem., 2018, 10, 155–164 CrossRef CAS.
- N. C. Seeman and H. F. Sleiman, Nat. Rev. Mater., 2017, 3, 17068–17090 CrossRef.
- S. Li, Q. Jiang, S. Liu, Y. Zhang, Y. Tian, C. Song, J. Wang, Y. Zou, G. J. Anderson, J. Y. Han, Y. Chang, Y. Liu, C. Zhang, L. Chen, G. Zhou, G. Nie, H. Yan, B. Ding and Y. Zhao, Nat. Biotechnol., 2018, 36, 258–264 CrossRef CAS.
- K. Xia, H. Kong, Y. Cui, N. Ren, Q. Li, J. Ma, R. Cui, Y. Zhang, J. Shi, Q. Li, M. Lv, Y. Sun, L. Wang, J. Li and Y. Zhu, ACS Appl. Mater. Interfaces, 2018, 10, 15442–15448 CrossRef CAS.
- S. Ko, H. Liu, Y. Chen and C. Mao, Biomacromolecules, 2008, 9, 3039–3043 CrossRef CAS.
- A. Lacroix, T. G. W. Edwardson, M. A. Hancock, M. D. Dore and H. F. Sleiman, J. Am. Chem. Soc., 2017, 139, 7355–7362 CrossRef CAS.
- D. Jiang, Z. Ge, H. J. Im, C. G. England, D. Ni, J. Hou, L. Zhang, C. J. Kutyreff, Y. Yan, Y. Liu, S. Y. Cho, J. W. Engle, J. Shi, P. Huang, C. Fan, H. Yan and W. Cai, Nat. Biomed. Eng., 2018, 2, 865–877 CrossRef CAS.
- D. Jiang, N. Dalong, B. Yu, L. Kang, E. Ehlerding, J. Engle and W. Cai, J. Nucl. Med., 2018, 59, 261 Search PubMed.
- J. Liu, T. Wu, X. Lu, X. Wu, S. Liu, S. Zhao, X. Xu and B. Ding, J. Am. Chem. Soc., 2019, 141, 19032–19037 CrossRef CAS.
- Y. Zhang, W. Ma, Y. Zhu, S. Shi, Q. Li, C. Mao, D. Zhao, Y. Zhan, J. Shi, W. Li, L. Wang, C. Fan and Y. Lin, Nano Lett., 2018, 18, 5652–5659 CrossRef CAS.
- A. E. Prigodich, A. H. Alhasan and C. A. Mirkin, J. Am. Chem. Soc., 2011, 133, 2120–2123 CrossRef CAS.
- A. H. Alhasan, P. C. Patel, C. H. J. Choi and C. A. Mirkin, Small, 2014, 10, 186–192 CrossRef CAS.
- J. Li, H. Pei, B. Zhu, L. Liang, M. Wei, Y. He, N. Chen, D. Li, Q. Huang and C. Fan, ACS Nano, 2011, 5, 8783–8789 CrossRef CAS.
- K. Mohri, M. Nishikawa, N. Takahashi, T. Shiomi, N. Matsuoka, K. Ogawa, M. Endo, K. Hidaka, H. Sugiyama, Y. Takahashi and Y. Takakura, ACS Nano, 2012, 6, 5931–5940 CrossRef CAS.
- H. Lee, A. K. Lytton-Jean, Y. Chen, K. T. Love, A. I. Park, E. D. Karagiannis, A. Sehgal, W. Querbes, C. S. Zurenko and M. Jayaraman, Nat. Nanotechnol., 2012, 7, 389–393 CrossRef CAS.
- K. E. Bujold, J. C. C. Hsu and H. F. Sleiman, J. Am. Chem. Soc., 2016, 138, 14030–14038 CrossRef CAS.
- D. Zheng, D. A. Giljohann, D. L. Chen, M. D. Massich, X.-Q. Wang, H. Iordanov, C. A. Mirkin and A. S. Paller, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 11975–11980 CrossRef CAS.
- S. A. Jensen, E. S. Day, C. H. Ko, L. A. Hurley, J. P. Luciano, F. M. Kouri, T. J. Merkel, A. J. Luthi, P. C. Patel, J. I. Cutler, W. L. Daniel, A. W. Scott, M. W. Rotz, T. J. Meade, D. A. Giljohann, C. A. Mirkin and A. H. Stegh, Sci. Transl. Med., 2013, 5, 209ra152 Search PubMed.
- J. Lu, J. Wang, X. Hu, E. Gyimah, S. Yakubu, K. Wang, X. Wu and Z. Zhang, Anal. Chem., 2019, 91, 7353–7359 CrossRef CAS.
- X. He, T. Zeng, Z. Li, G. Wang and N. Ma, Angew. Chem., Int. Ed., 2016, 55, 3073–3076 CrossRef CAS.
- K. Lee, M. Rafi, X. Wang, K. Aran, X. Feng, C. Lo Sterzo, R. Tang, N. Lingampalli, H. J. Kim and N. Murthy, Nat. Mater., 2015, 14, 701–706 CrossRef CAS.
- F. Ding, Q. Mou, Y. Ma, G. Pan, Y. Guo, G. Tong, C. H. J. Choi, X. Zhu and C. Zhang, Angew. Chem., Int. Ed., 2018, 130, 3118–3122 CrossRef.
- W. Sun, T. Jiang, Y. Lu, M. Reiff, R. Mo and Z. Gu, J. Am. Chem. Soc., 2014, 136, 14722–14725 CrossRef CAS.
- Q. Jiang, S. Liu, J. Liu, Z.-G. Wang and B. Ding, Adv. Mater., 2019, 31, 1804785 CrossRef CAS.
- Q. Zhang, Q. Jiang, N. Li, L. Dai, Q. Liu, L. Song, J. Wang, Y. Li, J. Tian, B. Ding and Y. Du, ACS Nano, 2014, 8, 6633–6643 CrossRef CAS.
- Q. Mou, Y. Ma, G. Pan, B. Xue, D. Yan, C. Zhang and X. Zhu, Angew. Chem., Int. Ed., 2017, 129, 12702–12706 CrossRef.
- J. Zhang, Y. Guo, F. Ding, G. Pan, X. Zhu and C. Zhang, Angew. Chem., Int. Ed., 2019, 58, 13794–13798 CrossRef CAS.
- F. Zhou, P. Wang, Y. Peng, P. Zhang, Q. Huang, W. Sun, N. He, T. Fu, Z. Zhao, X. Fang and W. Tan, Angew. Chem., Int. Ed., 2019, 131, 11787–11791 CrossRef.
- Q. Hu, S. Wang, L. Wang, H. Gu and C. Fan, Adv. Healthcare Mater., 2018, 7, 1701153 CrossRef.
- H.-M. Meng, H. Liu, H. Kuai, R. Peng, L. Mo and X.-B. Zhang, Chem. Soc. Rev., 2016, 45, 2583–2602 RSC.
- L. L. Ong, N. Hanikel, O. K. Yaghi, C. Grun, M. T. Strauss, P. Bron, J. Lai-Kee-Him, F. Schueder, B. Wang, P. Wang, J. Y. Kishi, C. Myhrvold, A. Zhu, R. Jungmann, G. Bellot, Y. Ke and P. Yin, Nature, 2017, 552, 72–77 CrossRef CAS.
- K. Chen, J. Kong, J. Zhu, N. Ermann, P. Predki and U. F. Keyser, Nano Lett., 2019, 19, 1210–1215 CrossRef.
- J. Valero, N. Pal, S. Dhakal, N. G. Walter and M. Famulok, Nat. Nanotechnol., 2018, 13, 496–503 CrossRef CAS.
- J. Fu, M. Liu, Y. Liu, N. W. Woodbury and H. Yan, J. Am. Chem. Soc., 2012, 134, 5516–5519 CrossRef CAS.
- R. Schreiber, N. Luong, Z. Fan, A. Kuzyk, P. C. Nickels, T. Zhang, D. M. Smith, B. Yurke, W. Kuang, A. O. Govorov and T. Liedl, Nat. Commun., 2013, 4, 2948–2953 CrossRef.
- J. Zheng, G. Zhu, Y. Li, C. Li, M. You, T. Chen, E. Song, R. Yang and W. Tan, ACS Nano, 2013, 7, 6545–6554 CrossRef CAS.
- J. I. Cutler, E. Auyeung and C. A. Mirkin, J. Am. Chem. Soc., 2012, 134, 1376–1391 CrossRef CAS.
- J. I. Cutler, K. Zhang, D. Zheng, E. Auyeung, A. E. Prigodich and C. A. Mirkin, J. Am. Chem. Soc., 2011, 133, 9254–9257 CrossRef CAS.
- R. J. Banga, N. Chernyak, S. P. Narayan, S. T. Nguyen and C. A. Mirkin, J. Am. Chem. Soc., 2014, 136, 9866–9869 CrossRef CAS.
- J. R. Ferrer, A. J. Sinegra, D. Ivancic, X. Y. Yeap, L. Qiu, J.-J. Wang, Z. J. Zhang, J. A. Wertheim and C. A. Mirkin, ACS Nano, 2020, 14, 1682–1693 CrossRef CAS.
- X. Tan, B. B. Li, X. Lu, F. Jia, C. Santori, P. Menon, H. Li, B. Zhang, J. J. Zhao and K. Zhang, J. Am. Chem. Soc., 2015, 137, 6112–6115 CrossRef CAS.
- J. Tan, H. Li, X. Hu, R. Abdullah, S. Xie, L. Zhang, M. Zhao, Q. Luo, Y. Li, Z. Sun, Q. Yuan and W. Tan, Chem, 2019, 5, 1775–1792 CAS.
- D. Bousmail, L. Amrein, J. J. Fakhoury, H. H. Fakih, J. C. C. Hsu, L. Panasci and H. F. Sleiman, Chem. Sci., 2017, 8, 6218–6229 RSC.
- G. Zhu and X. Chen, Adv. Drug Delivery Rev., 2018, 134, 65–78 CrossRef CAS.
- W. Xuan, Y. Peng, Z. Deng, T. Peng, H. Kuai, Y. Li, J. He, C. Jin, Y. Liu, R. Wang and W. Tan, Biomaterials, 2018, 182, 216–226 CrossRef CAS.
- J. Liu, T. Wei, J. Zhao, Y. Huang, H. Deng, A. Kumar, C. Wang, Z. Liang, X. Ma and X. Liang, Biomaterials, 2016, 91, 44–56 CrossRef CAS.
- R. Wang, G. Zhu, L. Mei, Y. Xie, H. Ma, M. Ye, F. Qing and W. Tan, J. Am. Chem. Soc., 2014, 136, 2731–2734 CrossRef CAS.
- F. Li, J. Lu, J. Liu, C. Liang, M. Wang, L. Wang, D. Li, H. Yao, Q. Zhang and J. Wen, Nat. Commun., 2017, 8, 1–14 CrossRef.
- Q. Yang, Z. Deng, D. Wang, J. He, D. Zhang, Y. Tan, T. Peng, X.-Q. Wang and W. Tan, J. Am. Chem. Soc., 2020, 142, 2532–2540 CrossRef CAS.
- Y. Ma, H. Liu, Q. Mou, D. Yan, X. Zhu and C. Zhang, Nanoscale, 2018, 10, 8367–8371 RSC.
- B. J. Hong, I. Eryazici, R. Bleher, R. V. Thaner, C. A. Mirkin and S. T. Nguyen, J. Am. Chem. Soc., 2015, 137, 8184–8191 CrossRef CAS.
- Y. Shao, H. Jia, T. Cao and D. Liu, Acc. Chem. Res., 2017, 50, 659–668 CrossRef CAS.
- H. Wei, Z. Zhao, Y. Wang, J. Zou, Q. Lin and Y. Duan, ACS Appl. Mater. Interfaces, 2019, 11, 46479–46489 CrossRef CAS.
- R. Owczarzy, B. G. Moreira, Y. You, M. A. Behlke and J. A. Walder, Biochemistry, 2008, 47, 5336–5353 CrossRef CAS.
- N. C. Seeman, J. Theor. Biol., 1982, 99, 237–247 CrossRef CAS.
- T. H. LaBean, H. Yan, J. Kopatsch, F. Liu, E. Winfree, J. H. Reif and N. C. Seeman, J. Am. Chem. Soc., 2000, 122, 1848–1860 CrossRef CAS.
- F. Mathieu, S. Liao, J. Kopatsch, T. Wang, C. Mao and N. C. Seeman, Nano Lett., 2005, 5, 661–665 CrossRef CAS.
- R. P. Goodman, I. A. Schaap, C. F. Tardin, C. M. Erben, R. M. Berry, C. F. Schmidt and A. J. Turberfield, Science, 2005, 310, 1661–1665 CrossRef CAS.
- Y. Zhang and N. C. Seeman, J. Am. Chem. Soc., 1994, 116, 1661–1669 CrossRef CAS.
- K.-R. Kim, H. Y. Kim, Y.-D. Lee, J. S. Ha, J. H. Kang, H. Jeong, D. Bang, Y. T. Ko, S. Kim, H. Lee and D.-R. Ahn, J. Controlled Release, 2016, 243, 121–131 CrossRef CAS.
- S. Fujimori, K. Shudo and Y. Hashimoto, J. Am. Chem. Soc., 1990, 112, 7436–7438 CrossRef CAS.
- D. J. Anderson, R. J. Reischer, A. J. Taylor and W. J. Wechter, Nucleosides Nucleotides, 1984, 3, 499–512 CrossRef CAS.
- M. S. Chan, D. Y. Tam, Z. Dai, L. S. Liu, J. W.-T. Ho, M. L. Chan, D. Xu, M. S. Wong, C. Tin and P. K. Lo, Small, 2016, 12, 770–781 CrossRef CAS.
- J. W. Conway, C. Madwar, T. G. Edwardson, C. K. McLaughlin, J. Fahkoury, R. B. Lennox and H. F. Sleiman, J. Am. Chem. Soc., 2014, 136, 12987–12997 CrossRef CAS.
- M. Chang, C. Yang and D. Huang, ACS Nano, 2011, 5, 6156–6163 CrossRef CAS.
- W. M. Shih, J. D. Quispe and G. F. Joyce, Nature, 2004, 427, 618–621 CrossRef CAS.
- P. W. Rothemund, Nature, 2006, 440, 297–302 CrossRef CAS.
- S. M. Douglas, H. Dietz, T. Liedl, B. Högberg, F. Graf and W. M. Shih, Nature, 2009, 459, 414–418 CrossRef CAS.
- D. Han, S. Pal, Y. Liu and H. Yan, Nat. Nanotechnol., 2010, 5, 712–717 CrossRef CAS.
- Q. Jiang, C. Song, J. Nangreave, X. Liu, L. Lin, D. Qiu, Z.-G. Wang, G. Zou, X. Liang, H. Yan and B. Ding, J. Am. Chem. Soc., 2012, 134, 13396–13403 CrossRef CAS.
- L. A. Garraway and P. A. Jänne, Cancer Discovery, 2012, 2, 214–226 CrossRef CAS.
- J. Yan, C. Hu, P. Wang, B. Zhao, X. Ouyang, J. Zhou, R. Liu, D. He, C. Fan and S. Song, Angew. Chem., Int. Ed., 2015, 127, 2461–2465 CrossRef.
- Y.-X. Zhao, A. Shaw, X. Zeng, E. Benson, A. M. Nyström and B. Högberg, ACS Nano, 2012, 6, 8684–8691 CrossRef CAS.
- K. R. Kim, S. J. Kang, A. Y. Lee, D. Hwang, M. Park, H. Park, S. Kim, K. Hur, H. S. Chung, C. Mao and D. R. Ahn, Biomaterials, 2019, 195, 1–12 CrossRef CAS.
- G. Zhu, J. Zheng, E. Song, M. Donovan, K. Zhang, C. Liu and W. Tan, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 7998–8003 CrossRef CAS.
- P. D. Halley, C. R. Lucas, E. M. McWilliams, M. J. Webber, R. A. Patton, C. Kural, D. M. Lucas, J. C. Byrd and C. E. Castro, Small, 2016, 12, 308–320 CrossRef CAS.
- T. Wu, J. Liu, M. Liu, S. Liu, S. Zhao, R. Tian, D. Wei, Y. Liu, Y. Zhao, H. Xiao and B. Ding, Angew. Chem., Int. Ed., 2019, 58, 14224–14228 CrossRef CAS.
- X. Tan, X. Lu, F. Jia, X. Liu, Y. Sun, J. K. Logan and K. Zhang, J. Am. Chem. Soc., 2016, 138, 10834–10837 CrossRef CAS.
- R. J. Banga, S. A. Krovi, S. P. Narayan, A. J. Sprangers, G. Liu, C. A. Mirkin and S. T. Nguyen, Biomacromolecules, 2017, 18, 483–489 CrossRef CAS.
- Y. Ma, Z. Wang, Y. Ma, Z. Han, M. Zhang, H. Chen and Y. Gu, Angew. Chem., Int. Ed., 2018, 57, 5389–5393 CrossRef CAS.
- G. Pan, Q. Mou, Y. Ma, F. Ding, J. Zhang, Y. Guo, X. Huang, Q. Li, X. Zhu and C. Zhang, ACS Appl. Mater. Interfaces, 2019, 11, 41082–41090 CrossRef CAS.
- A. F. Jorge, A. Aviñó, A. A. C. C. Pais, R. Eritja and C. Fàbrega, Nanoscale, 2018, 10, 7238–7249 RSC.
- Y. Ma, Q. Mou, L. Zhu, Y. Su, X. Jin, J. Feng, D. Yan, X. Zhu and C. Zhang, Chem. Commun., 2019, 55, 6603–6606 RSC.
- S. Nussbaumer, P. Bonnabry, J.-L. Veuthey and S. Fleury-Souverain, Talanta, 2011, 85, 2265–2289 CrossRef CAS.
- M. K. Goftar, N. M. Kor and Z. M. Kor, Int. J. Adv. Biol. Biomed. Res., 2014, 2, 811–822 Search PubMed.
- D. Agudelo, P. Bourassa, G. Bérubé and H.-A. Tajmir-Riahi, Int. J. Biol. Macromol., 2014, 66, 144–150 CrossRef CAS.
- C. A. Frederick, L. D. Williams, G. Ughetto, G. A. Van der Marel, J. H. Van Boom, A. Rich and A. H. J. Wang, Biochemistry, 1990, 29, 2538–2549 CrossRef CAS.
- J. B. Chaires, J. E. Herrera and M. J. Waring, Biochemistry, 1990, 29, 6145–6153 CrossRef CAS.
- J. Wen, W. Tao, S. Hao, S. P. Iyer and Y. Zu, Leukemia, 2016, 30, 987–991 CrossRef CAS.
- A. Pusuluri, V. Krishnan, V. Lensch, A. Sarode, E. Bunyan, D. R. Vogus, S. Menegatti, H. T. Soh and S. Mitragotri, Angew. Chem., Int. Ed., 2019, 58, 1437–1441 CrossRef CAS.
- Y. Guo, J. Zhang, F. Ding, G. Pan, J. Li, J. Feng, X. Zhu and C. Zhang, Adv. Mater., 2019, 31, 1807533 CrossRef.
- G. Zhu, S. Cansiz, M. You, L. Qiu, D. Han, L. Zhang, L. Mei, T. Fu, Z. Chen and W. Tan, NPG Asia Mater., 2015, 7, e169 CrossRef CAS.
- Q. Mou, Y. Ma, F. Ding, X. Gao, D. Yan, X. Zhu and C. Zhang, J. Am. Chem. Soc., 2019, 141, 6955–6966 CrossRef CAS.
- L. Zhang, S. Wang, Z. Yang, S. Hoshika, S. Xie, J. Li, X. Chen, S. Wan, L. Li, S. A. Benner and W. Tan, Angew. Chem., Int. Ed., 2020, 59, 663–668 CrossRef CAS.
- S. M. Taghdisi, N. M. Danesh, M. Ramezani, R. Yazdian-Robati and K. Abnous, Mol. Pharmaceutics, 2018, 15, 1972–1978 CrossRef CAS.
- C. Jin, H. Zhang, J. Zou, Y. Liu, L. Zhang, F. Li, R. Wang, W. Xuan, M. Ye and W. Tan, Angew. Chem., Int. Ed., 2018, 130, 9132–9135 CrossRef.
- M.-E. Kyriazi, D. Giust, A. H. El-Sagheer, P. M. Lackie, O. L. Muskens, T. Brown and A. G. Kanaras, ACS Nano, 2018, 12, 3333–3340 CrossRef CAS.
- S. Han, A. Samanta, X. Xie, L. Huang, J. Peng, S. J. Park, D. B. L. Teh, Y. Choi, Y.-T. Chang, A. H. All, Y. Yang, B. Xing and X. Liu, Adv. Mater., 2017, 29, 1700244 CrossRef.
- H. Li, X. Zhou, D. Yao and H. Liang, Chem. Commun., 2018, 54, 3520–3523 RSC.
- S. P. Narayan, C. H. J. Choi, L. Hao, C. M. Calabrese, E. Auyeung, C. Zhang, O. J. G. M. Goor and C. A. Mirkin, Small, 2015, 11, 4173–4182 CrossRef CAS.
- A. Latorre, C. Posch, Y. Garcimartín, A. Celli, M. Sanlorenzo, I. Vujic, J. Ma, M. Zekhtser, K. Rappersberger, S. Ortiz-Urda and Á. Somoza, Nanoscale, 2014, 6, 7436–7442 RSC.
- C. Wu, D. Han, T. Chen, L. Peng, G. Zhu, M. You, L. Qiu, K. Sefah, X. Zhang and W. Tan, J. Am. Chem. Soc., 2013, 135, 18644–18650 CrossRef CAS.
- X. Han, Y. Jiang, S. Li, Y. Zhang, X. Ma, Z. Wu, Z. Wu and X. Qi, Nanoscale, 2019, 11, 339–347 RSC.
- X. Xie, X. Shao, W. Ma, D. Zhao, S. Shi, Q. Li and Y. Lin, Nanoscale, 2018, 10, 5457–5465 RSC.
- K.-R. Kim, D.-R. Kim, T. Lee, J. Y. Yhee, B.-S. Kim, I. C. Kwon and D.-R. Ahn, Chem. Commun., 2013, 49, 2010–2012 RSC.
- J. Liu, L. Song, S. Liu, Q. Jiang, Q. Liu, N. Li, Z.-G. Wang and B. Ding, Nano Lett., 2018, 18, 3328–3334 CrossRef CAS.
- J. Liu, L. Song, S. Liu, S. Zhao, Q. Jiang and B. Ding, Angew. Chem., Int. Ed., 2018, 57, 15486–15490 CrossRef CAS.
- L. Song, Q. Jiang, J. Liu, N. Li, Q. Liu, L. Dai, Y. Gao, W. Liu, D. Liu and B. Ding, Nanoscale, 2017, 9, 7750–7754 RSC.
- L. Yang, H. Sun, Y. Liu, W. Hou, Y. Yang, R. Cai, C. Cui, P. Zhang, X. Pan, X. Li, L. Li, B. S. Sumerlin and W. Tan, Angew. Chem., Int. Ed., 2018, 57, 17048–17052 CrossRef CAS.
- L. Zhang, R. Abdullah, X. Hu, H. Bai, H. Fan, L. He, H. Liang, J. Zou, Y. Liu, Y. Sun, X. Zhang and W. Tan, J. Am. Chem. Soc., 2019, 141, 4282–4290 CrossRef CAS.
- C. Ouyang, S. Zhang, C. Xue, X. Yu, H. Xu, Z. Wang, Y. Lu and Z. Wu, J. Am. Chem. Soc., 2020, 142, 1265–1277 CrossRef CAS.
- H. Zhao, X. Yuan, J. Yu, Y. Huang, C. Shao, F. Xiao, L. Lin, Y. Li and L. Tian, ACS Appl. Mater. Interfaces, 2018, 10, 15418–15427 CrossRef CAS.
- Y. Yi, H. Wang, X. Wang, Q. Liu, M. Ye and W. Tan, ACS Appl. Mater. Interfaces, 2017, 9, 5847–5854 CrossRef CAS.
- B. Li, M. I. Setyawati, L. Chen, J. Xie, K. Ariga, C.-T. Lim, S. Garaj and D. T. Leong, ACS Appl. Mater. Interfaces, 2017, 9, 15286–15296 CrossRef CAS.
- D. Wang, Z. Xu, H. Yu, X. Chen, B. Feng, Z. Cui, B. Lin, Q. Yin, Z. Zhang, C. Chen, J. Wang, W. Zhang and Y. Li, Biomaterials, 2014, 35, 8374–8384 CrossRef CAS.
- R. Hu, X. Zhang, Z. Zhao, G. Zhu, T. Chen, T. Fu and W. Tan, Angew. Chem., Int. Ed., 2014, 53, 5821–5826 CrossRef CAS.
- Z. Xiao, C. Ji, J. Shi, E. M. Pridgen, J. Frieder, J. Wu and O. C. Farokhzad, Angew. Chem., Int. Ed., 2012, 51, 11853–11857 CrossRef CAS.
- A. Cavaliere, K. C. Probst, A. D. Westwell and M. Vlusarczyk, Future Med. Chem., 2017, 9(15), 1809–1833 CrossRef CAS.
- L. P. Jordheim, D. Durantel, F. Zoulim and C. Dumontet, Nat. Rev. Drug Discovery, 2013, 12, 447–464 CrossRef CAS.
- D. Wang, B. Liu, Y. Ma, C. Wu, Q. Mou, H. Deng, R. Wang, D. Yan, C. Zhang and X. Zhu, J. Am. Chem. Soc., 2017, 139, 14021–14024 CrossRef CAS.
- B. R. Madhanagopal, S. Zhang, E. Demirel, H. Wady and A. R. Chandrasekaran, Trends Biochem. Sci., 2018, 43, 997–1013 CrossRef CAS.
- R. O. Pedersen, E. G. Loboa and T. H. LaBean, Biomacromolecules, 2013, 14, 4157–4160 CrossRef CAS.
- R. Peng, L. Xu, H. Wang, Y. Lyu, D. Wang, C. Bi, C. Cui, C. Fan, Q. Liu, X. Zhang and W. Tan, Nat. Commun., 2020, 11, 978–987 CrossRef CAS.
- A. T. Veetil, K. Chakraborty, K. Xiao, M. R. Minter, S. S. Sisodia and Y. Krishnan, Nat. Nanotechnol., 2017, 12, 1183–1189 CrossRef CAS.
- Y. Min, J. M. Caster, M. J. Eblan and A. Z. Wang, Chem. Rev., 2015, 115, 11147–11190 CrossRef CAS.
- K. V. Gothelf, A. Thomsen, M. Nielsen, E. Cló and R. S. Brown, J. Am. Chem. Soc., 2004, 126, 1044–1046 CrossRef CAS.
- V. Linko, A. Ora and M. A. Kostiainen, Trends Biotechnol., 2015, 33, 586–594 CrossRef CAS.
- Y. Zhang, J. Tu, D. Wang, H. Zhu, S. K. Maity, X. Qu, B. Bogaert, H. Pei and H. Zhang, Adv. Mater., 2018, 30, 1703658 CrossRef.
- A. V. Pinheiro, D. Han, W. M. Shih and H. Yan, Nat. Nanotechnol., 2011, 6, 763–772 CrossRef CAS.
- J. Hahn, S. F. J. Wickham, W. M. Shih and S. D. Perrault, ACS Nano, 2014, 8, 8765–8775 CrossRef CAS.
- N. Ponnuswamy, M. M. C. Bastings, B. Nathwani, J. H. Ryu, L. Y. T. Chou, M. Vinther, W. A. Li, F. M. Anastassacos, D. J. Mooney and W. M. Shih, Nat. Commun., 2017, 8, 15654–15662 CrossRef CAS.
- Y. Kim and P. Yin, Angew. Chem., Int. Ed., 2020, 132, 710–713 CrossRef.
- A. H. Okholm and J. Kjems, Adv. Drug Delivery Rev., 2016, 106, 183–191 CrossRef CAS.
- X. Xiong, H. Liu, Z. Zhao, M. B. Altman, D. Lopez Colon, C. J. Yang, L. J. Chang, C. Liu and W. Tan, Angew. Chem., Int. Ed., 2013, 52, 1472–1476 CrossRef CAS.
- C. H. J. Choi, L. Hao, S. P. Narayan, E. Auyeung and C. A. Mirkin, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 7625–7630 CrossRef CAS.
- D. S. Lee, H. Qian, C. Y. Tay and D. T. Leong, Chem. Soc. Rev., 2016, 45, 4199–4225 RSC.
- S. A. Smith, L. I. Selby, A. P. R. Johnston and G. K. Such, Bioconjugate Chem., 2019, 30, 263–272 CrossRef CAS.
- M. M. C. Bastings, F. M. Anastassacos, N. Ponnuswamy, F. G. Leifer, G. Cuneo, C. Lin, D. E. Ingber, J. H. Ryu and W. M. Shih, Nano Lett., 2018, 18, 3557–3564 CrossRef CAS.
- S. D. Perrault and W. M. Shih, ACS Nano, 2014, 8, 5132–5140 CrossRef CAS.
- J. O. Jin, H. Park, W. Zhang, J. W. de Vries, A. Gruszka, M. W. Lee, D. R. Ahn, A. Herrmann and M. Kwak, Biomaterials, 2017, 115, 81–89 CrossRef CAS.
- F. Praetorius, B. Kick, K. L. Behler, M. N. Honemann, D. Weuster-Botz and H. Dietz, Nature, 2017, 552, 84–87 CrossRef CAS.
- E. A. Ashley, Nat. Rev. Genet., 2016, 17, 507–522 CrossRef CAS.
- A. A. Friedman, A. Letai, D. E. Fisher and K. T. Flaherty, Nat. Rev. Cancer, 2015, 15, 747–756 CrossRef CAS.
- E. W. Ng, D. T. Shima, P. Calias, E. T. Cunningham, D. R. Guyer and A. P. Adamis, Nat. Rev. Drug Discovery, 2006, 5, 123–132 CrossRef CAS.
- J. Zhou and J. Rossi, Nat. Rev. Drug Discovery, 2017, 16, 181–202 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2021 |