
Optimization of the overall water-splitting performance of N, S co-doped carbon-supported NiCoMnSx−10 at high current densities by the introduction of sulfur defects and oxygen vacancies†
Runzhi
Zhang
a,
Zebin
Yu
 *ab,
Ronghua
Jiang
c,
Jun
Huang
de,
Yanping
Hou
a,
Qiuyue
Zhou
a,
Shiyu
Zhu
a,
Xiaocan
Huang
a,
Feng
Zheng
a and
Zhao
Luo
a
*ab,
Ronghua
Jiang
c,
Jun
Huang
de,
Yanping
Hou
a,
Qiuyue
Zhou
a,
Shiyu
Zhu
a,
Xiaocan
Huang
a,
Feng
Zheng
a and
Zhao
Luo
a
aSchool of Resources, Environment and Materials, Guangxi University, Nanning 530004, PR China. E-mail: xxzx7514@hotmail.com; yuzebin@gxu.edu.cn; Tel: + 8613877108420
bGuangxi Key Laboratory of Clean Pulp & Papermaking and Pollution Control, Nanning 530004, PR China
cSchool of Chemical and Environmental Engineering, Shaoguan University, Shaoguan 512005, P. R. China
dCollege of Civil Engineering and Architecture, Guangxi University, Nanning 530004, P. R. China
eHualan Design & Consulting Group, Nanning 530004, P. R. China
First published on 20th August 2020
Abstract
Designing an electrocatalyst with excellent performance for both the hydrogen evolution reaction and oxygen evolution reaction in overall water splitting is essential to promote the development of electrolyzed hydrogen production. Defect engineering is a promising method to improve the catalytic performance of target materials. In this study, we used a metal–organic framework as a precursor to establish oxygen-vacancy-rich metal oxide groups supported by a defect-rich carbon framework via calcination. Further, thermal ion exchange was performed to introduce sulfur ions to generate NiCoMnSx−10 with both oxygen vacancies and sulfur defects. Due to the synergistic effect between oxygen vacancies and sulfur defects as well as the good morphology, NiCoMnSx−10 exhibited good catalytic performance at high current densities. The NiCoMnSx−10 cell could achieve a current density of 10 mA cm−2 at a cell voltage of 1.506 V and a current density of 100 mA cm−2 at an overpotential of only 1.94 V, which exceeds that of most materials reported to date. Due to the supporting effect of N, S co-doped carbon, the NiCoMnSx−10 cell maintained stable performance during a 100 h operation at high current. This work provides an innovative method for the preparation of highly efficient electrocatalysts for overall water splitting.
Introduction
Continuous attention on global energy and environmental issues has stimulated comprehensive research on efficient, natural, environment friendly, and sustainable alternate energy systems.1,2 It has been found that the electrocatalytic overall water splitting to H2 and O2 through the hydrogen evolution reaction (HER) and oxygen evolution reaction (OER) is a promising technology to expand the production of clean and renewable energy.3–5 This technology requires highly efficient HER and OER catalysts to overcome the high overpotential generated by the two half reactions. Pt and RuOx are used as traditional benchmark electrocatalysts for the HER and OER, respectively. However, their scarcity and high cost hinder their widespread application. Based on the current situation, the search for an efficient, stable, and noble metal-free bifunctional electrocatalyst is booming in the field of electrocatalytic overall water splitting.6–8In recent years, transition metal chalcogenide materials have been widely studied for overall water splitting due to their remarkable electroactivity, lower metal-H adsorption bond energy, and higher hydrogen dissociation properties.9,10 However, structural problems, including single active sites and excessive order, are great challenges in the practical application of single metal sulfides as they significantly limit the catalytic performance. To solve this problem, researchers have tried to construct polymetallic hybrid sulfides to modify the interface and surface defects of sulfides and adjust their electronic structure.11,12 The introduction of a disordered structure into a catalyst may be a prospective way to improve the intrinsic catalytic performance. Oxygen vacancies can cause the redistribution of electrons on the catalyst surface during the catalytic process; this further improves the catalytic performance of the catalysts. Moreover, oxygen vacancies can reduce the adsorption barrier of the OH− anions generated during the catalytic reaction and can significantly improve the reactivity of the catalytic active sites;13,14 accordingly, the OER activity of the catalyst is substantially improved rather than the HER activity. However, sulfur defects, which are common in transition metal chalcogenide materials, have a significant impact on the HER catalytic activity as they expose other active edges.15,16 Herein, we proposed a hypothesis that by simultaneously introducing oxygen vacancies and sulfur defects into a material, the hydrogen and oxygen evolution activities of the material can be excellently adjusted. Studies have shown that the oxide lattice breaks into a disordered distribution after the electronegative sulfur atom is incorporated into the material to produce more defect sites and enhance the active of low coordination oxygen.17,18 Therefore, we can choose a multi-metal hybrid oxide as a precursor for thermal ion exchange to introduce sulfur atoms instead of oxygen atoms for the preparation of multi-metal hybrid sulfides with high-density oxygen vacancies and sulfur defects.
In this study, we introduced a metal–organic framework (MOF) material with a specific structure and used its organic part to construct a well-structured carbon skeleton as a support for active materials by carbonization. Furthermore, its metal oxide groups were used to prepare oxygen-vacancy-containing metal oxide precursors. Then, along with the introduction of a sulfur source, thermal ion exchange was performed at high temperatures to prepare a polymetallic hybrid sulfide anchored by a N, S co-doped carbon skeleton. The obtained catalytic material not only retained the original structure of the MOF with N, S co-doped carbon skeleton and high-speed electron transport capability, but also had a high concentration of oxygen vacancies and sulfur defects that would synergistically promote the overall water-splitting reaction. Results shows good catalytic effect of this material, and it remained stable during operation at high current densities for a long time. Our study provides an innovative method for the preparation of highly efficient electrocatalysts.
Experimental
Materials
Cobalt acetate tetrahydrate (C4H6CoO4·4H2O), manganese acetate tetrahydrate (C4H6MnO4·4H2O), urea, ammonium fluoride (NH4F), thioacetamide (TAA), 2-aminoterephthalic acid, and N,N-dimethylformamide (DMF) were supplied by Lantian Co., Ltd. (Nanning, China). Nickel foam (thickness: 1.7 mm, pores per linear inch: 120 ppi) was purchased from Suzhou Jialongde Foam Metals Co., Ltd. (Suzhou, China). All reagents were of analytical grade and directly used without further purification.Synthesis
Physicochemical characterizations
The synthesized materials were characterized by various techniques. The crystal structures of the materials were characterized by X-ray diffraction (XRD; Rigaku D/MAX-2500V, Rigaku Corporation) in the 2θ range of 5–90° at the scanning rate of 10° min−1. The degree of graphitization of the prepared electrodes was determined using a Raman spectrometer (Renishaw invia Reflex) at room temperature with a 532 nm laser and a source spectral range of 1000–2000 cm−1. The samples were subjected to Fourier transform infrared (FTIR) spectroscopy using a Fourier transform infrared spectrometer (Nicolet iS 50, Somerfly, USA) with KBr as a diluent. X-ray photoelectron spectroscopy (XPS) was performed using an X-ray photoelectron spectrometer (ESCALAB 250XI+, Thermo Fisher Scientific) equipped with an Al (single) X-ray source and calibrated against a variable carbon signal (binding energy = 284.8 eV). The element content of the materials was analyzed using an ICP-AES inductively coupled plasma emission spectrometer (model ICPS-7510, Shimadzu Corporation). Scanning electron microscopy (SEM) images were acquired using an SU8020 (Hitachi High-tech) instrument at an accelerating voltage of 10 kV. Transmission electron microscopy (TEM) images were obtained by a FEI TECNAI G2 F30 instrument. The hydrophilicity of the materials was tested using a contact-angle measuring instrument (model DSA100E, German KYUSS company). Using the OmniFluo-960 fluorescence spectrometer, photoluminescence (PL) spectra were obtained at the excitation wavelengths of 285 nm and 532 nm.Electrochemical characterization
An electrochemical workstation (CHI 660E, China) with a typical three-electrode system was used for all the electrochemical tests. Herein, 1 M KOH was used as an electrolyte for all the electrochemical tests. The prepared catalyst materials were used as the working electrode, a graphite rod electrode was used as the counter electrode, and saturated Hg|HgO|OH− was employed as the reference electrode to examine the performance of HER and OER. The prepared electrode materials were simultaneously used as an anode and a cathode, and the performance of overall water splitting was obtained using a two-electrode system. Before the electrochemical tests, the electrolyte was purged with N2 for 30 min to remove O2. For parallel comparison with literature values, the potentials were measured vs. Hg|HgO|OH− and converted to the reversible hydrogen electrode (RHE) scale according to the Nernst equation:| E(RHE) = E(vs. Hg|HgO|OH−) + 0.059 pH + 0.098 V |
Results and discussion
Physical properties
As shown in Scheme 1, 2-aminoterephthalic acid acted as an organic ligand, and nickel foam acted as a nickel source; cobalt and manganese salts were added to provide cobalt and manganese for the synthesis of NCM(BDC)x. Then, NCM(BDC)x was carbonized to obtain a well-structured N-doped carbon skeleton-supported NCMOx with oxygen vacancies. After this, the sulfur source was introduced for the thermal ion-exchange reaction, and N, S co-doped carbon skeleton-anchored NCMSx rich in oxygen vacancies and sulfur defects was obtained.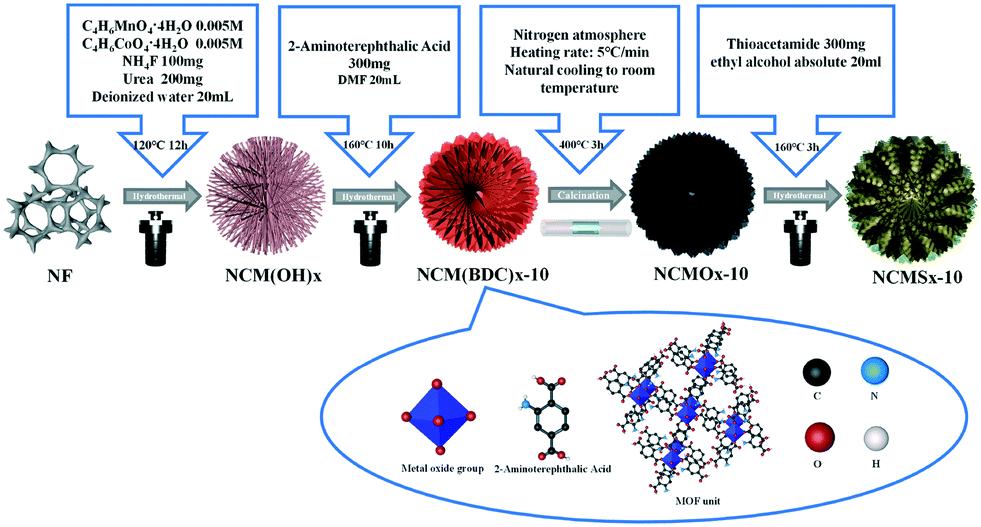 | ||
| Scheme 1 Using NiCoMn(BDC)x as a precursor, an anion exchange method was used to synthesize highly disordered NCMSx compounds. | ||
As shown in Fig. 1 and S1,† the morphologies of the synthesized materials were characterized by a scanning electron microscope (SEM). It can be observed from Fig. S1a–j† that when the reaction time was 5 h, the resulting material NCM(BDC)x−5 retained the size of NCM(OH)x and showed agglomeration. With an increase in the reaction time, dispersed flower-like NCM(BDC)x−10 was obtained. With a further increase in the reaction time, the petals continued to thicken, and NCM(BDC)x−15 and NCM(BDC)x−20 were achieved. Fig. 1a–d show the morphology of the pyrolysis products NCMOx; it can be observed that the pyrolysis products maintained the basic morphology of the original MOF; however, the petals became thinner. Among them, NCMOx−10 exhibited well-connected three-dimensional flower-like sheets, which are conducive for a full contact between the active sites and the electrolyte and the conduction of electrons inside the material.
 | ||
| Fig. 1 SEM images of (a) NCMOx−5; (b) NCMOx−10; (c) NCMOx−15; (d) NCMOx−20; (e) NCMSx−5; (f) NCMSx−10; (g) NCMSx−15; and (h) NCMSx−20. | ||
Fig. 1e–h show the morphology of the product obtained after vulcanization. Many thin, transparent, and small-sized sheets were noticed on the petals after vulcanization. NCMSx−5 still showed an aggregated form (Fig. 1e), which was not conducive for the participation of deep active sites in the reaction. For NCMSx−15 and NCMSx−20 (Fig. 1g and h), as the petals of their precursors were too thick, only a few thin slices could be generated on the surface of these petals, and more sites were wrapped in them. The difference is that under the advantages of the precursor's just dispersion and proper thickness, NCMSx−10 (Fig. 1f) shows a uniform state of small sheets intertwined after vulcanization. This morphology significantly increases the contact area between the material and the electrolyte, leading to the participation of more active sites in the reaction. In addition, under the combined effect of the high winding degree of the sheet structure and the carbon skeleton, the electron transfer within the material will be more convenient and fast.
In order to characterize the synthesis of the materials, XRD tests were performed on the materials, and the results are shown in Fig. S2a† and 2a–b. Fig. S2a† shows that the diffraction peaks in the XRD pattern of NCM(BDC)x basically correspond to Ni(BDC), Co(BDC), and Mn(BDC). However, due to the interaction of different metal ions, a small shift was observed in some peaks of the polymetallic MOF as compared to the case of the monometallic MOF, which further verifies that the MOF synthesized herein is polymetallic rather than a simple mixture of the three monometallic MOFs. As can be noticed in Fig. 2a and b, the characteristic diffraction peaks attributed to the MOF were retained in the patterns of both the oxide and sulfide; this indicates that even after pyrolysis and vulcanization, the characteristics of NCM(BDC)x were retained. In addition, the products achieved after pyrolysis, as shown in Fig. 2a, were a mixture of NiO, CoO, and Mn3O4; this proves the successful synthesis of oxide precursors. Similarly, as shown in Fig. 2b, after the thermal ion-exchange reaction, the sulfur atoms successfully replaced the oxygen atoms. The products obtained were a mixture of Ni3S2, Co3S4, and MnS, which further proved the successful synthesis of NCMSx.
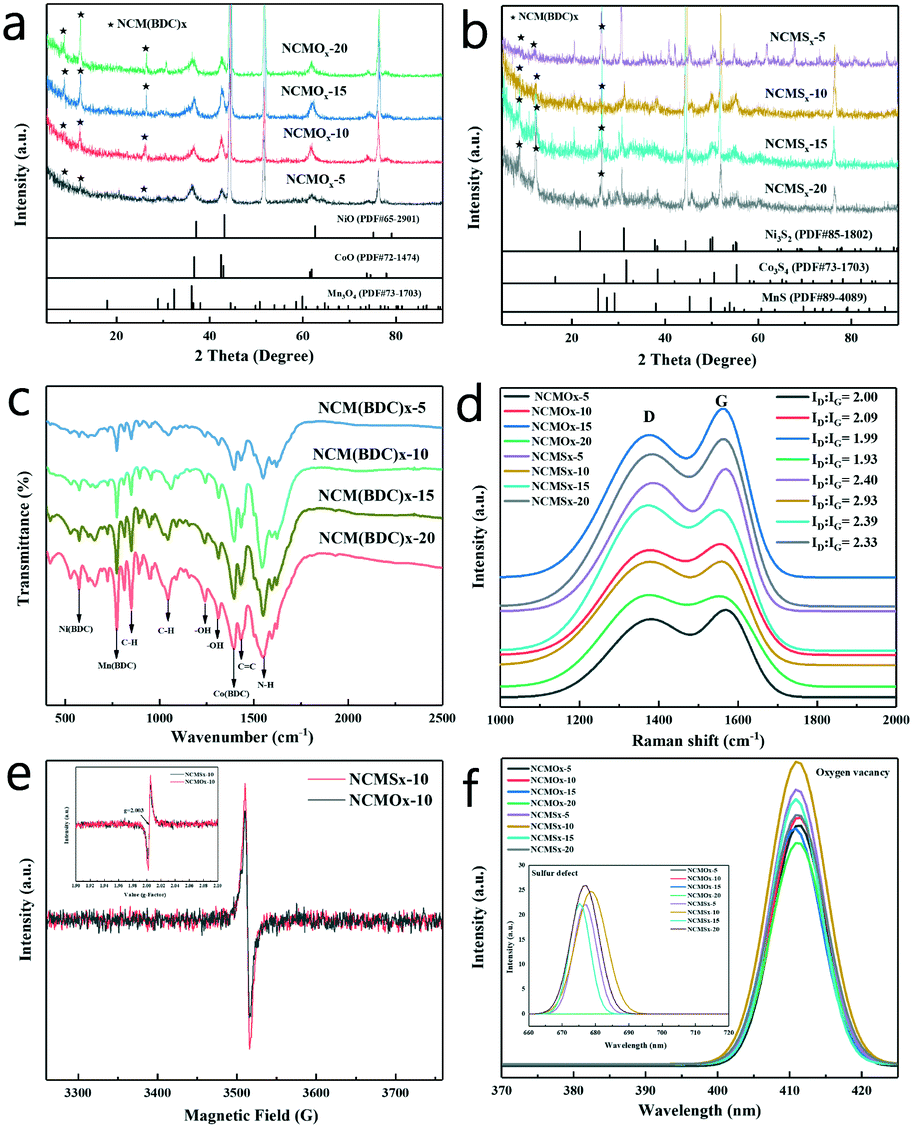 | ||
| Fig. 2 (a) XRD patterns of NCMOx; (b) XRD patterns of NCMSx; (c) FTIR spectra of NCM(BDC)x; (d) Raman spectra of NCMOx and NCMSx; (e) EPR spectra of NCMOx−10 and NCMSx−10; and (f) PL spectra of NCMOx and NCMSx. | ||
In order to confirm the synthesis of MOFs and to characterize the functional groups in the precursor materials, FTIR was performed on NCM(BDC)x, and the FTIR spectra are shown in Fig. 2c. The peak observed at 1380 cm−1 corresponds to the symmetric tensile vibration of the coordinated carboxylate group, indicating the successful coordination of Co2+ ions with BDC ligands, proving the successful synthesis of Co(BDC).19,20 Furthermore, a peak corresponding to Ni(BDC) was observed at 508 cm−1.21 In addition, the spectrum displayed a peak at 748 cm−1, which is attributed to Mn(BDC).22 The peaks related to the functional groups of organic ligands were also obtained in the spectra, in which the two bands at 1032 and 808 cm−1 are attributed to the C–H tensile band.23 Moreover, two obvious peaks were found in the range of 1410–1260 cm−1, indicating the presence of –OH. The band at 1450 ± 20 cm−1 is a sign of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C tensile vibration in the presence of an aromatic carbon network. The peak of the combination of N–H in-plane bending and C–N stretching was found at 1548 cm−1, demonstrating the retention of amino groups in the ligand. The analysis of the FTIR spectra further proves the successful synthesis of the MOF precursors.
C tensile vibration in the presence of an aromatic carbon network. The peak of the combination of N–H in-plane bending and C–N stretching was found at 1548 cm−1, demonstrating the retention of amino groups in the ligand. The analysis of the FTIR spectra further proves the successful synthesis of the MOF precursors.
The concentration of surface defects has a non-negligible effect on the performance of the material. In order to characterize the defects of the materials and prove the high disorder of the materials, Raman, PL, and EPR spectroscopy tests were performed on the synthesized NCMOx and NCMSx. It can be noticed from the Raman spectrum shown in Fig. 2d that a defect-rich carbon skeleton with ID![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :IG > 1 was successfully prepared after calcination. Among them, the carbon defect degree of NCMOx−10 was greater than that of the other three oxides. After vulcanization, due to the doping effect of sulfur atoms, the defect degree of the carbon skeleton was further increased; this implies the successful synthesis of N, S co-doped carbon. It can also be observed that the defect degree of NCMSx−10 is slightly larger than those of the other materials.
:IG > 1 was successfully prepared after calcination. Among them, the carbon defect degree of NCMOx−10 was greater than that of the other three oxides. After vulcanization, due to the doping effect of sulfur atoms, the defect degree of the carbon skeleton was further increased; this implies the successful synthesis of N, S co-doped carbon. It can also be observed that the defect degree of NCMSx−10 is slightly larger than those of the other materials.
In addition, as shown in Fig. 2e, the EPR curves prove that there are oxygen vacancies in NCMOx−10, whereas there may be both oxygen vacancies and sulfur defects in NCMSx−10.24–27 It can be verified that the synthesized NCMOx−10 and NCMSx−10 are rich in defects and show a high disorder. After this, the specific defects of the materials were characterized by PL spectroscopy. As can be observed from the small graph shown in Fig. 2f, at an excitation wavelength of 532 nm, the spectrum of NCMSx showed a peak at 680 nm that was attributed to the S defects.28–30 However, the spectrum of NCMOx does not have this peak, indicating that the NCMSx compounds containing sulfur defects were successfully prepared. However, at the excitation wavelength of 285 nm, the spectra of all the NCMOx and NCMSx compounds showed a peak at 410 nm corresponding to oxygen vacancies.31,32 Interestingly, the peak intensity of the vulcanized product was higher than that of the oxide precursor; this shows that the concentration of the oxygen vacancies increases after vulcanization. This further verifies that after the introduction of negatively charged sulfur, the oxide lattice is decomposed into a disordered arrangement, thereby generating more defect sites and enhancing the active of low coordination oxygen. These characteristics of the materials rich in oxygen vacancies and sulfur defects are very beneficial for achieving efficient overall water splitting.
XPS was performed to explore the elemental characteristics of the material, and the results are shown in Fig. 3. As shown in Fig. 3a, the peaks at 284.5, 285.1, and 288.3 eV correspond to the C–C, CC, and CO bonds.33,34 Furthermore, the binding energy peaks at 284.1 and 285.5 eV can be assigned to C–S and C–N, respectively.33,35 This proves the successful synthesis of the N-doped carbon skeleton in NCMOx−10 and the successful synthesis of N, S co-doped carbon in NCMSx−10. The peaks corresponding to pyridinic N (398.3 eV), pyrrolic N (399.4 eV), and graphitic N (400.7 eV) were found in the high-resolution N 1s spectrum (Fig. 3b),35–37 and the presence of the graphitic N peak further proves that N was doped into the carbon skeleton. We found that pyridinic N and pyrrolic N coexist in NCMSx−10, whereas only pyridinic N exists in NCMOx−10. The presence of pyrrolic N and pyridinic N proves that the material can exhibit improved electrocatalytic activity by interacting with H+ for the HER or bind to OER intermediates (O*, OOH*, or OH*) under appropriate force.38,39 Therefore, the performance of the material having both pyrrolic N and pyridinic N will be more excellent. Thus, it is inferred that the electrocatalytic performance of NCMSx−10 will be better than that of NCMOx−10. In addition, there are peaks at 531.4 eV, 530.2 eV, and 532.2 eV, attributed to lattice oxygen, oxygen vacancies, and surface oxygen adsorption, in the O 1s spectrum (Fig. 3c) of the materials.40,41 The analysis of the O 1s spectrum shows that there are oxygen vacancies in both NCMOx−10 and NCMSx−10. From the peak areas, it can be analyzed that the concentration of oxygen vacancies in NCMSx−10 is even higher than that in NCMOx−10, and this conclusion agrees well with the PL results.
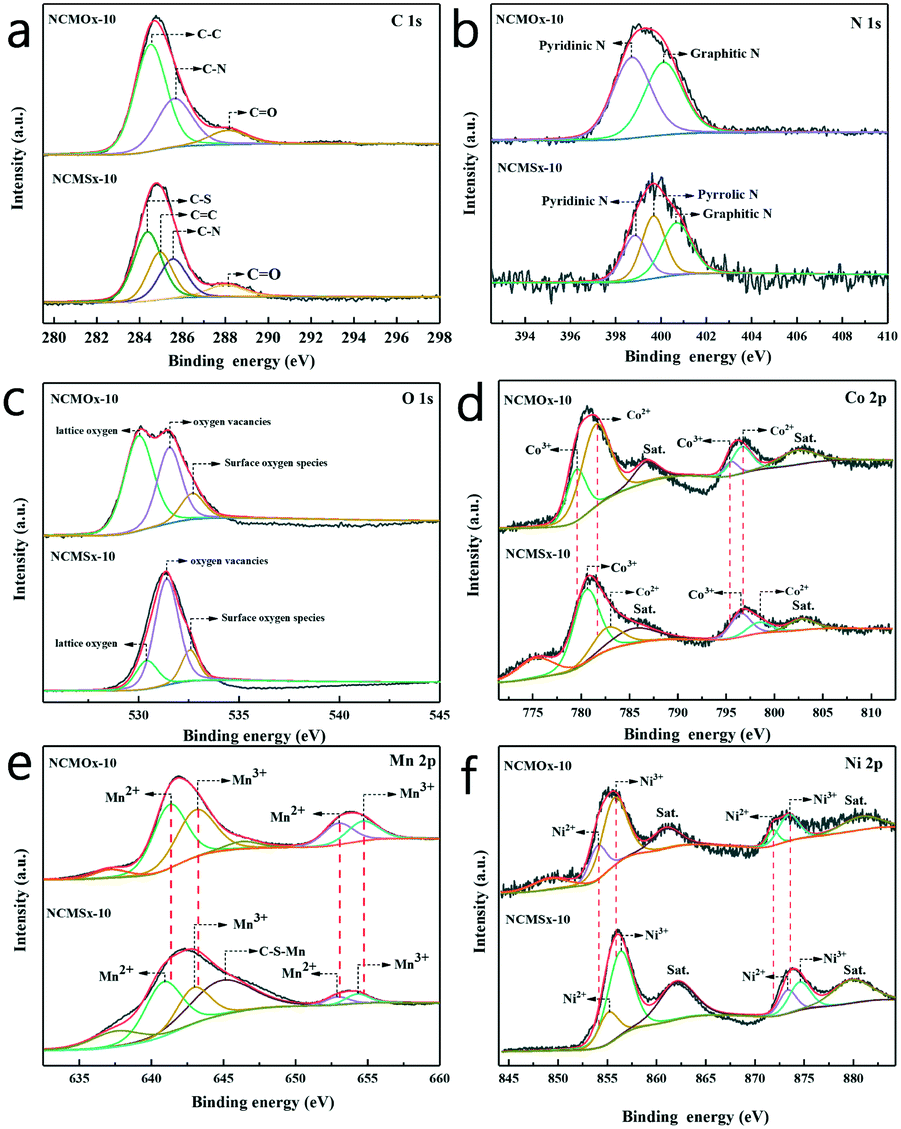 | ||
| Fig. 3 (a–f) C 1s, N 1s, O 1s, Co 2p, Mn 2p, and Ni 2p spectra of NCMOx−10 and NCMSx−10. | ||
In the Co 2p spectrum (Fig. 3d), the peaks at 779.5 and 795.1 eV are attributed to Co3+ (2p3/2) and Co3+ (2p1/2), respectively, and the two peaks at 781.3 and 795.9 eV correspond to Co2+ (2p3/2) and Co2+ (2p1/2). The two peaks obtained at 787.1 and 802.6 eV are satellite peaks.31,32,42 As shown in Fig. 3e, the two peaks at 641.0 and 652.5 eV can be assigned to Mn2+, and the other two peaks at 642.2 and 653.7 eV can be attributed to Mn3+.41,43 In addition, the peak at 644.3 eV can be assigned to C–S–Mn.44,45 The Ni 2p spectrum shown in Fig. 3f was analyzed, and the fitted peaks at 854.0 and 871.80 eV were attributed to the Ni2+ ions, whereas the fitted peaks at 855.70 and 873.40 eV were assigned to the Ni3+ ions.46,47Via a comparative analysis of the XPS spectra of NCMOx−10 and NCMSx−10, we found a regular phenomenon. When the sulfur atoms replace the oxygen atoms, the XPS peaks of different metals have different degrees of displacement; this shows that with the addition of sulfur atoms, a strong charge transfer occurs inside the material.
The morphology and lattice conditions of the materials were observed using a projection electron microscope, as shown in Fig. 4. As shown in Fig. 4a, NCMSx exhibits a cross-linked film shape, which can provide more channels for electron transfer. In the HRTEM image (Fig. 4b), lattice fringes belonging to the MnS (201) crystal plane and Ni3S2 (210) crystal plane can be observed, and there are some parts without lattice spacing, which is due to the presence of N, S co-doped carbon. Moreover, the lattice fringes in Fig. 4c can be assigned to Co3S4(311), Ni3S2(200), and MnS(110). It can be observed from the HRTEM images (Fig. 4b and c) that the lattice fringes of NCMOx−10 are highly disordered, and this high disorder is also closely related to the defect-rich features of NCMOx−10. In addition, the morphology of the other NCMSx compounds was examined. It can be noticed that the NCMSx−5 (Fig. 4d) film is relatively dispersed and flat, and the electron transfer is limited by the number of channels. Moreover, NCMSx−15 and NCMSx−20 (Fig. 4e and f) exhibit aggregation phenomena, which is not conducive for the contact between the reaction site and the electrolyte, respectively. On the other hand, the morphology of NCMSx−10 is helpful for the electrocatalytic reaction.
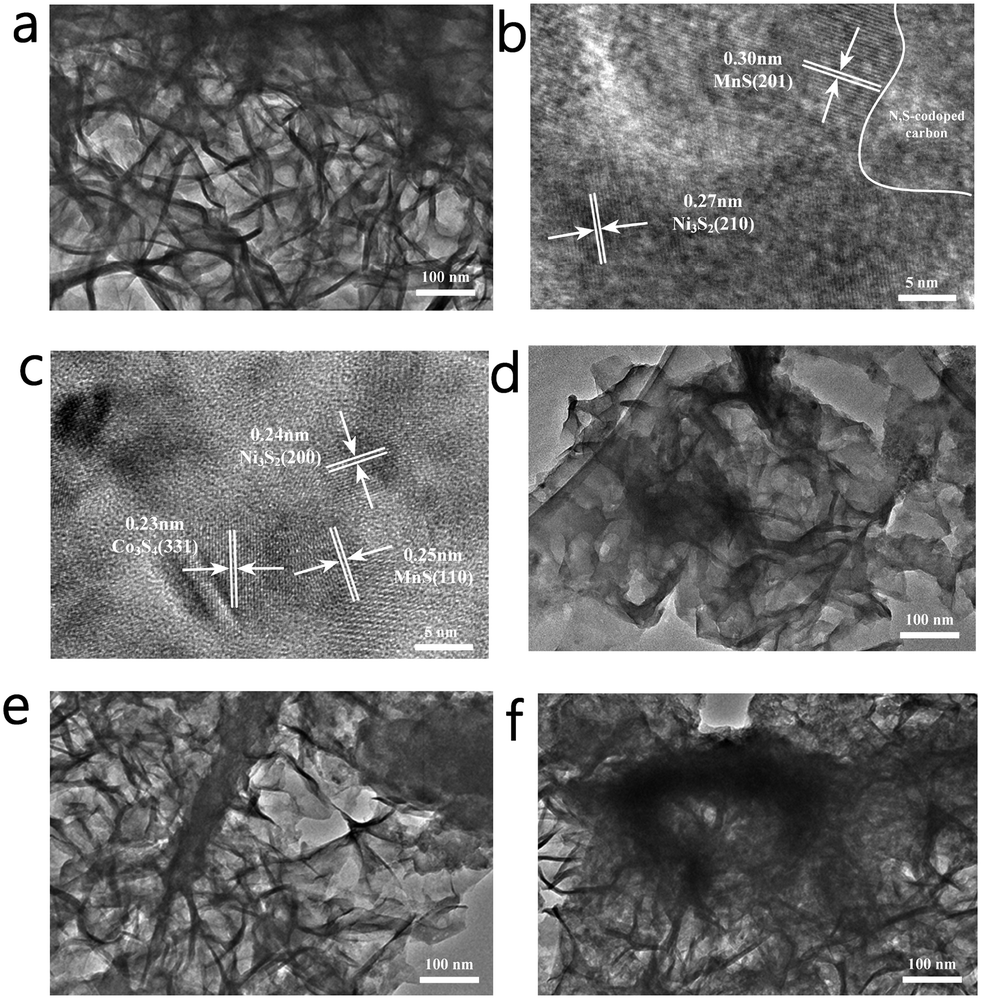 | ||
| Fig. 4 (a) TEM image of NCMSx−10; (b and c) HRTEM images of NCMSx−10; (d) TEM image of NCMSx−5; (e) TEM image of NCMSx−15; and (f) TEM image of NCMSx−20. | ||
In addition, the atomic ratios of Co and Mn in NCM(BDC)x prepared at different reaction times were analyzed by the ICP-OES analysis (the proportion of the Ni element could not be accurately estimated due to the influence of the nickel substrate), and the results are shown in Table S1.† The atomic ratios of Co and Mn in NCM (BDC)x−5, NCM (BDC)x−10, NCM (BDC)x−15, and NCM (BDC)x−20 are 1:0.88, 1:0.99, 1:1.01, and 1:0.82; it can be observed that the atomic ratios of Co and Mn in different materials are close to 1. In addition, the hydrophilicity of different NCMSx was investigated. As shown in Fig. S3,† the contact angle of NCMSx−10 is the smallest and its hydrophilicity is the best, which is most conducive for the full contact of the material with the electrolyte. This feature is also one of the reasons for the best catalytic performance of NCMSx−10.
HER performance
Using a typical three-electrode system in 1.0 M KOH with the obtained materials as the working electrodes, the electrocatalytic performance of these materials for the HER was evaluated at a scan rate of 2 mV s−1. In order to verify the applicability of these materials in the industry, we compared the performances of the materials at high current densities. It can be observed from Fig. 5a and Table S4† that when the current density was 10 mA cm−2, the overpotential of NCMSx−10 was only −105 mV, which was far lower than those of other comparative materials. Similarly, when the current density reached the industrial application standard of 100 mA cm−2, the required overpotential of NCMSx−10 (−330 mV) was substantially smaller than those of NCMOx−5 (−429 mV), NCMOx−10 (−401 mV), NCMOx−15 (−423 mV), NCMOx−20 (−448 mV), NCMSx−5 (−352 mV), NCMSx−15 (−365 mV), and NCMSx−20 (−372 mV). This performance gap is mainly because the well-crosslinked film morphology of the material provides a fast electron channel, and the rich sulfur defects are also beneficial for the HER.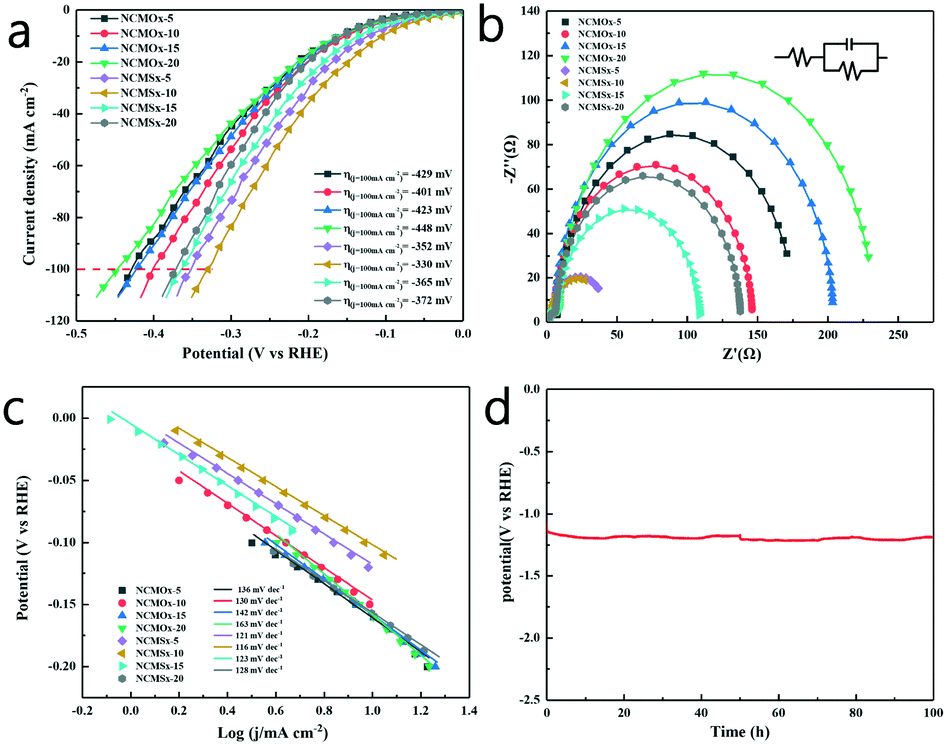 | ||
| Fig. 5 (a) HER polarization curves (iR-corrected) obtained at a scan rate of 2 mV s−1; (b) HER electrical impedance maps; (c) the Tafel slopes of the materials; and (d) results of the 100 h chronopotentiometry test of NCMSx under HER. | ||
Fig. 5b shows the EIS curves of the materials and the corresponding fitted circuit diagrams, and the EIS-related data of the materials is provided in Table S2.† From these data, we can observe that the Rct (charge transfer resistance) of NCMSx−10 (37.37 Ω) is significantly smaller than those of NCMOx−5 (168.10 Ω), NCMOx−10 (129.80 Ω), NCMOx−15 (195.80 Ω), NCMOx−20 (222.1 Ω), NCMSx−5 (38.07 Ω), NCMSx−15 (99.24 Ω), and NCMSx−20 (129.20 Ω). This is due to the presence of the N, S co-doped carbon framework and the well-crosslinked morphology of the material. Fig. 5c presents the Tafel slopes of these electrodes. Compared with NCMOx−5 (136 mV dec−1), NCMOx−10 (130 mV dec−1), NCMOx−15 (142 mV dec−1), NCMOx−20 (163 mV dec−1), NCMSx−5 (121 mV dec−1), NCMSx−15 (123 mV dec−1), and NCMSx−20 (128 mV dec−1), NCMSx−10 has the smallest Tafel slope (116 mV dec−1). This indicates that the rate of apparent hydrogen formation rapidly increases with an increase in the applied potential. In order to explore the stability of the material at high current densities, NCMSx−10 was subjected to chronopotentiometry for 100 hours under the HER reaction. Fig. 5d shows that the change in the overpotential was negligible after 100 h, indicating that NCMSx−10 has good stability.
OER performance
Similarly, the OER performance of the catalyst was evaluated at a scan rate of 2 mV s−1 in a 1.0 M KOH solution. Consistent with the results of HER, NCMSx−10 exhibited better performance than other materials due to the synergetic effect of good morphology, more abundant defects, and N, S co-doped carbon. As shown in Fig. 6a and Table S4,† NCMSx−10 requires an overpotential of only 226 mV to achieve a current density of 10 mA cm−2. Moreover, the current density could reach 100 mA cm−2 at a voltage of 417 mV, which was better than those of NCMOx−5 (564 mV), NCMOx−10 (557 mV), NCMOx−15 (568 mV), NCMOx−20 (579 mV), NCMSx−5 (440 mV), NCMSx−15 (453 mV), and NCMSx−20 (472 mV).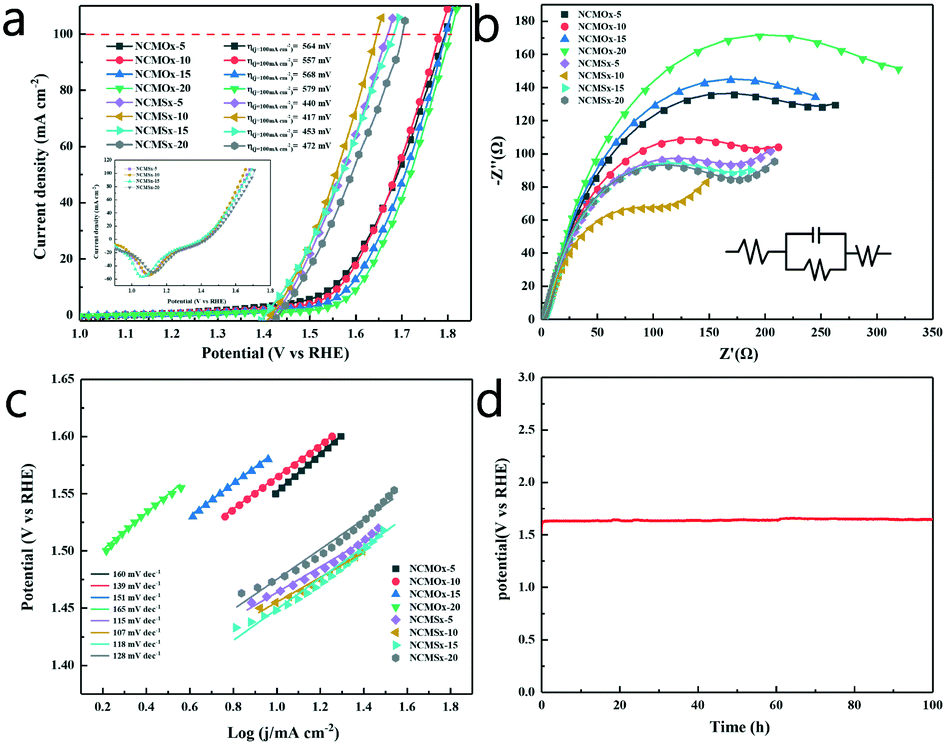 | ||
| Fig. 6 (a) OER polarization curves (iR-corrected) at a scan rate of 2 mV s−1; (b) OER electrical impedance maps; (c) the Tafel slopes of the materials; and (d) results of the 100 h chronopotentiometry test of NCMSx−10 under the OER. | ||
Fig. 6b shows the EIS curves of the materials and the corresponding fitted circuit diagrams, and the EIS-related data of the materials is provided in Table S3.† It can be noticed from the comparison that the Rct (74.73 Ω) of NCMSx−10 is smaller than those of NCMOx−5 (179.11 Ω), NCMOx−10 (142.70 Ω), NCMOx−15 (199.92 Ω), NCMOx−20 (243.84 Ω), NCMSx−5 (123.21 Ω), NCMSx−15 (124.22 Ω), and NCMSx−20 (127.80 Ω); this shows that NCMSx−10 has ultra-fast charge transfer capability and can lead to a higher reaction rate. It can be noticed in Fig. 6c that NCMSx−10 has a Tafel slope of 107 mV dec−1, which is lower than those of NCMOx−5 (160 mV dec−1), NCMOx−10 (139 mV dec−1), NCMOx−15 (151 mV dec−1), NCMOx−20 (165 mV dec−1), NCMSx−5 (115 mV dec−1), NCMSx−15 (118 mV dec−1), and NCMSx−20 (128 mV dec−1). This indicates that NCMSx−10 is kinetically advantageous for the OER. In order to explore the stability of NCMSx−10 at high current densities in the OER, a 100 h chronopotentiometry test was performed at a current density of 100 mA cm−2, and the overpotential curve showed a slight change (Fig. 6d), confirming the stability of NCMSx−10 in the OER.
Overall water-splitting performance
By conducting the HER and OER using the proposed materials, we determined the feasibility of using NCMSx−10 as a bifunctional catalyst. In order to explore its overall water-splitting performance, we simultaneously used NCMSx−10 as a positive electrode and negative electrode in an alkaline electrolysis cell at a scan rate of 2 mV s−1 and conducted the overall water-splitting reaction. Fig. 7a and Table S4† show that the NCMSx−10 cell can achieve the current density of 10 mA cm−2 at the cell voltage of 1.506 V, obtaining a current density of 100 mA cm−2 at an overpotential of only 1.94 V. It can also be observed in Fig. 7a that when the current density is 100 mA cm−2, the NCMSx−10 cell exhibits the smallest voltage value when compared with the NCMOx−5 cell (2.24 V), NCMOx−10 cell (2.23 V), NCMOx−15 cell (2.28 V), NCMOx−20 cell (2.32 V), NCMSx−5 cell (2.10 V), NCMSx−15 cell (2.05 V), and NCMSx−20 cell (2.14 V). Upon further comparing NCMSx−10 with the current noble-metal-free catalysts (Table S5†), it was found that NCMSx−10 has the best overall water-splitting catalytic performance. Thus, NCMSx−10 anchored by N, S co-doped carbon and rich in oxygen vacancies and sulfur defects exhibits excellent performance in catalyzing the overall water-splitting reaction. As shown in Fig. 7b, a 100 h chronopotentiometry test was performed at a current density of 100 mA cm−2, and the overpotential curve showed a slight change, confirming the stability of NCMSx−10 in the overall water-splitting reaction.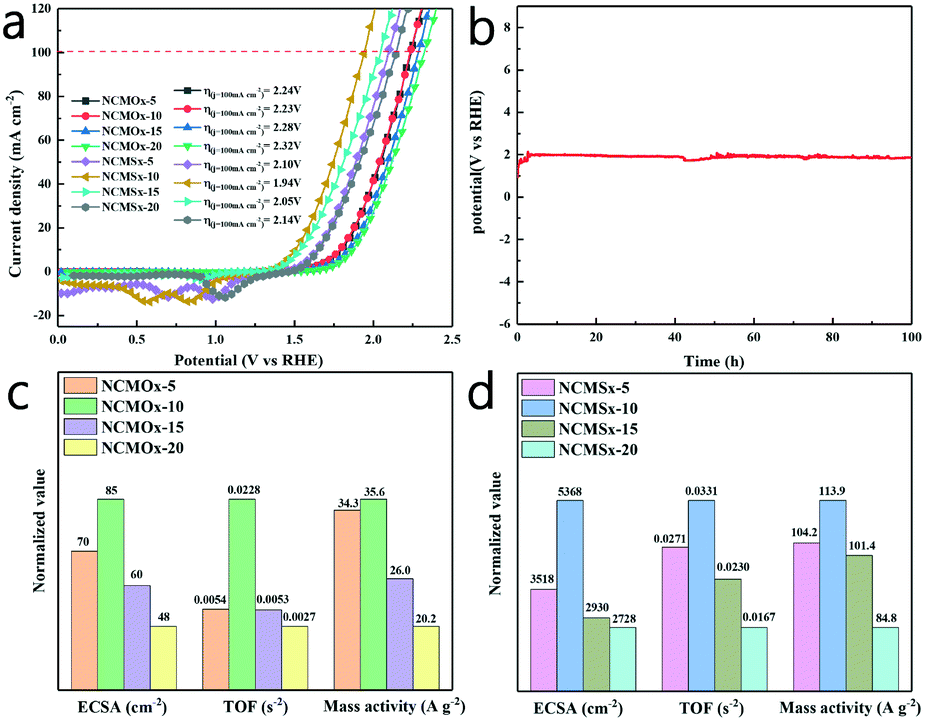 | ||
| Fig. 7 (a) Polarization curves (iR-corrected) in the two-electrode configuration; (b) chronopotentiometry test of NCMSx−10 under overall water splitting; (c) comparison between the activities of different NCMOx materials in 1.0 M KOH; and (d) comparison between the activities of different NCMSx materials in 1.0 M KOH. | ||
In order to more comprehensively evaluate and compare the performances of these materials in overall water splitting, different electrochemical parameters were evaluated using different electrochemical parameter calculation methods (supporting materials). As shown in Fig. 7c and d, the mass activity, ECSA (electrochemically active surface area), and TOF (turn over frequency) of NCMOx and NCMSx were investigated to evaluate the properties of these materials. We calculated the double-layer capacitance (Cdl) of these materials based on their CV curves (Fig. S4†) and then evaluated the ECSA of these materials to determine their intrinsic activity (Fig. S5†). As shown in Fig. 7c and d, the ECSA of NCMSx−10 is the largest (5368 cm−2). The ECSAs of other materials are as follows: NCMOx−5 (70 cm−2), NCMOx−10 (85 cm−2), NCMOx−15 (60 cm−2), NCMOx−20 (48 cm−2), NCMSx−5 (3518 cm−2), NCMSx−15 (2930 cm−2), and NCMSx−20 (2728 cm−2). Moreover, via the values of mass activity, the performances of the electrocatalysts can be determined. The values of mass activity are as follows: NCMSx−10 (113.9 A g−1), NCMOx−5 (34.3 A g−1), NCMOx−10 (35.6 A g−1), NCMOx−15 (26.0 A g−1), NCMOx−20 (20.2 A g−1), NCMSx−5 (104.2 A g−1), NCMSx−15 (101.4 A g−1), and NCMSx−20 (84.8 A g−1). The ECSA and mass activity results are consistent with those obtained via polarization curves. In addition, the TOF of NCMSx−10 is also the largest, which reached 0.0331 s−1, whereas the TOFs of NCMOx−5, NCMOx−10, NCMOx−15, NCMOx−20, NCMSx−5, NCMSx−15, and NCMSx−20 are 0.0054 s−1, 0.0228 s−1, 0.0053 s−1, 0.0027 s−1, 0.0271 s−1, 0.0230 s−1, and 0.0167 s−1, respectively. Based on these parameters, we can conclude that NCMSx−10 has better performance than other materials in all aspects due to the synergistic effect of good morphology, abundant oxygen vacancies, and sulfur defects.
Discussion of stability
In order to verify the stability and explore the change in the valence band of the material after different reaction processes to infer the possible reaction active sites, we performed XRD, PL, SEM, and XPS characterizations of the material after 100 hours of OER and HER chronopotentiometry test. It can be observed that there is no obvious change in the XRD pattern of the material (Fig. S6a†) after the reactions; this shows that neither the OER nor the HER affected the crystal structure of the material; this further illustrates the stability of the material. In addition, it can be noticed in Fig. S6b and c† that the intensity of the PL peaks of sulfur defects and oxygen vacancies slightly increased after the reaction; this may be caused by the leaching of a small amount of sulfide ions and oxidation reactions during the test. However, this change is so tiny that it can even be ignored. This indicates that the content of oxygen vacancies and sulfur defects in the material is basically maintained before and after the reaction, which further illustrates the stability of the material.The XPS spectrum (Fig. S7†) shows the peaks of the elements with relatively obvious changes. It can be observed that compared with those before the reaction, the valence state of the metal elements and the ratio of different valence states had no obvious changes after the HER, and no new substances were produced; however, there are different degrees of deviation. This shows that the material could still maintain a relatively stable state after 100 hours of high-current hydrogen evolution reaction. After the OER, although the valence state of the metal element did not significantly change, the valence state ratio of the ion changed when compared with that before the reaction. Furthermore, the peak of CoO was observed in the Co 2p spectrum, which indicates that during the OER, the surface of the material was partially oxidized, and metal oxides with strong oxygen evolution properties were generated. This change could promote the OER.
Via the analysis of the S 2p spectrum, it can be found that the noise level of the S 2p spectrum increased after the HER and OER; this indicates that the leaching of the S element occurred during these two half reactions, and the leaching of S during the OER was most obvious. The stability test results show that the OER performance (Fig. 6d) is very stable, indicating that the leaching of sulfur ions does not affect the progress of the OER. Although there is leaching of only a small amount of S ions during the HER (Fig. 5d), the stability of the HER performance still significantly fluctuated. This shows that the S ion has a significant effect on the HER performance, whereas the effect on the OER performance is negligible.
As shown in Fig. S8a,† NCMSx−10 slightly aggregated after the HER; however, the basic morphology of NCMSx−10 was retained. Moreover, the SEM image (Fig. S8b†) obtained after the OER showed that the flower-like structure of NCMSx−10 was destroyed, the pore size of the material became larger, and the interlacing degree of the sheet structure reduced. By combining the analysis results of PL and XPS acquired after the reaction, it can be inferred that this phenomenon may be due to the leaching of the S element and the formation of an oxide film during the OER.
Conclusions
In summary, herein, we used an MOF material as a precursor and performed calcination and thermal ion exchange to successfully prepare N, S co-doped carbon framework-anchored NCMSx−10 with abundant oxygen vacancies and sulfur defects. The oxygen vacancies and sulfur defects contributed to the improvement of the OER and HER performances, respectively. Due to the synergistic effect of oxygen vacancies and sulfur defects and morphology control, the material showed excellent performance. The NCMSx−10 cell could achieve a current density of 100 mA cm−2 at an overpotential of only 1.94 V. Moreover, at the high current density of 100 mA cm−2, this cell could operate stably for 100 h, indicating that NCMSx−10 is an excellent electrocatalytic material. This study provides an available solution for further promoting the development of non-noble metal catalysts.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge the support provided by the Guangxi Natural Science Foundation (No. 2017GXNSFBA198186, 2018GXNSFAA281290, and 2018GXNSFAA294062), National innovation program for College Students (202010593047), China Post-Doctoral Science Foundation Grant (No. 2018M633295), Young Teachers Innovation Cultivation Program (BRP180261) from the Guangxi Bossco Environmental Protection Technology Co., Ltd., and Open Fund of Guangxi Key Laboratory of Clean Pulp & Papermaking and Pollution Control (No. 2019KF19).References
- J. Yang, X. Wang, B. Li, L. Ma, L. Shi, Y. Xiong and H. Xu, Adv. Funct. Mater., 2017, 27, 1606497 CrossRef.
- F. Si, C. Tang, Q. Gao, F. Peng, S. Zhang, Y. Fang and S. Yang, J. Mater. Chem. A, 2020, 8, 3083–3096 RSC.
- B. Ye, L. Huang, Y. Hou, R. Jiang, L. Sun, Z. Yu, B. Zhang, Y. Huang and Y. Zhang, J. Mater. Chem. A, 2019, 7, 11379–11386 RSC.
- B. Ye, R. Jiang, Z. Yu, Y. Hou, J. Huang, B. Zhang, Y. Huang, Y. Zhang and R. Zhang, J. Catal., 2019, 380, 307–317 CrossRef CAS.
- J.-G. Wang, W. Hua, M. Li, H. Liu, M. Shao and B. Wei, ACS Appl. Mater. Interfaces, 2018, 10, 41237–41245 CrossRef CAS.
- R. Zhang, L. Huang, Z. Yu, R. Jiang, Y. Hou, L. Sun, B. Zhang, Y. Huang, B. Ye and Y. Zhang, Electrochim. Acta, 2019, 323, 134845 CrossRef CAS.
- Y. Huang, L. Sun, Z. Yu, R. Jiang, J. Huang, Y. Hou, F. Yang, B. Zhang, R. Zhang and Y. Zhang, Catal. Sci. Technol., 2020, 10, 2627–2643 RSC.
- F. Li, D. Zhang, R.-C. Xu, W.-F. Fu and X.-J. Lv, ACS Appl. Energy Mater., 2018, 1, 3929–3936 CrossRef CAS.
- X. Wang, L. Li, Z. Wang, L. Tan, Z. Wu, Z. Liu, S. Gai and P. Yang, Electrochim. Acta, 2019, 326, 134983 CrossRef CAS.
- L. Peng, J. Shen, X. Zheng, R. Xiang, M. Deng, Z. Mao, Z. Feng, L. Zhang, L. Li and Z. Wei, J. Catal., 2019, 369, 345–351 CrossRef CAS.
- S. Shit, S. Chhetri, S. Bolar, N. C. Murmu, W. Jang, H. Koo and T. Kuila, ChemElectroChem, 2019, 6, 430–438 CrossRef CAS.
- S. Guan, X. Fu, Z. Lao, C. Jin and Z. Peng, Sustainable Energy Fuels, 2019, 3, 2056–2066 RSC.
- G. Huang, Z. Xiao, R. Chen and S. Wang, ACS Sustainable Chem. Eng., 2018, 6, 15954–15969 CrossRef CAS.
- F. Cheng, T. Zhang, Y. Zhang, J. Du, X. Han and J. Chen, Angew. Chem., Int. Ed., 2013, 52, 2474–2477 CrossRef CAS.
- Y. Li, J. Yin, L. An, M. Lu, K. Sun, Y.-Q. Zhao, D. Gao, F. Cheng and P. Xi, Small, 2018, 14, 1801070 CrossRef.
- Z. Xiao, Y. Wang, Y.-C. Huang, Z. Wei, C.-L. Dong, J. Ma, S. Shen, Y. Li and S. Wang, Energy Environ. Sci., 2017, 10, 2563–2569 RSC.
- X. Yu, Z.-Y. Yu, X.-L. Zhang, P. Li, B. Sun, X. Gao, K. Yan, H. Liu, Y. Duan, M.-R. Gao, G. Wang and S.-H. Yu, Nano Energy, 2020, 71, 104652 CrossRef CAS.
- J. Fu, F. M. Hassan, C. Zhong, J. Lu, H. Liu, A. Yu and Z. Chen, Adv. Mater., 2017, 29, 1702526 CrossRef.
- X. Liu, S. Min, Y. Xue, L. Tian, Y. Lei and F. Wang, New J. Chem., 2019, 43, 4152–4159 RSC.
- S. Zhao, Y. Wang, J. Dong, C.-T. He, H. Yin, P. An, K. Zhao, X. Zhang, C. Gao, L. Zhang, J. Lv, J. Wang, J. Zhang, A. M. Khattak, N. A. Khan, Z. Wei, J. Zhang, S. Liu, H. Zhao and Z. Tang, Nat. Energy, 2016, 1, 16184 CrossRef CAS.
- Q. Sun, M. Liu, K. Li, Y. Han, Y. Zuo, F. Chai, C. Song, G. Zhang and X. Guo, Inorg. Chem. Front., 2017, 4, 144–153 RSC.
- S. Yadav, S. Sharma, S. Dutta, A. Sharma, A. Adholeya and R. K. Sharma, Inorg. Chem., 2020, 59, 8334–8344 CrossRef CAS.
- C. Yang, X. Li, L. Yu, X. Liu, J. Yang and M. Wei, Chem. Commun., 2020, 56, 1803–1806 RSC.
- C. Du, Q. Zhang, Z. Lin, B. Yan, C. Xia and G. Yang, Appl. Catal., A, 2019, 248, 193–201 CrossRef CAS.
- S. Hu, X. Chen, Q. Li, Y. Zhao and W. Mao, Catal. Sci. Technol., 2016, 6, 5884–5890 RSC.
- K. Zhu, F. Shi, X. Zhu and W. Yang, Nano Energy, 2020, 73, 104761 CrossRef CAS.
- Y. Zhu, X. Zhong, S. Jin, H. Chen, Z. He, Q. Liu and Y. Chen, J. Mater. Chem. A, 2020, 8, 10957–10965 RSC.
- Q. Pan, K. Huang, S. Ni, Q. Wang, F. Yang and D. He, Mater. Lett., 2007, 61, 4773–4776 CrossRef CAS.
- G. Q. Xu, B. Liu, S. J. Xu, C. H. Chew, S. J. Chua and L. M. Gana, J. Phys. Chem. Solids, 2000, 61, 829–836 CrossRef CAS.
- Z. Qin, K. Xu, H. Yue, H. Wang, J. Zhang, C. Ouyang, C. Xie and D. Zeng, Sens. Actuators, B, 2018, 262, 771–779 CrossRef CAS.
- L. Zhuang, L. Ge, Y. Yang, M. Li, Y. Jia, X. Yao and Z. Zhu, Adv. Mater., 2017, 29, 1606793 CrossRef.
- J. Bao, X. Zhang, B. Fan, J. Zhang, M. Zhou, W. Yang, X. Hu, H. Wang, B. Pan and Y. Xie, Angew. Chem., Int. Ed., 2015, 54, 7399–7404 CrossRef CAS.
- Y. Liu, Y. Qiao, G. Wei, S. Li, Z. Lu, X. Wang and X. Lou, Energy Storage Mater., 2018, 11, 274–281 CrossRef.
- L. Ji, B. Wang, Y. Yu, N. Wang and J. Zhao, Electrochim. Acta, 2020, 331, 135348 CrossRef CAS.
- Y. Tang, F. Jing, Z. Xu, F. Zhang, Y. Mai and D. Wu, ACS Appl. Mater. Interfaces, 2017, 9, 12340–12347 CrossRef CAS.
- X. Hu, X. Sun, Q. Song, Y. Zhu, Y. Long and Z. Dong, Green Chem., 2020, 22, 742–752 RSC.
- C. Yu, Z. Liu, X. Meng, B. Lu, D. Cui and J. Qiu, Nanoscale, 2016, 8, 17458–17464 RSC.
- Y. Pan, K. Sun, S. Liu, X. Cao, K. Wu, W.-C. Cheong, Z. Chen, Y. Wang, Y. Li, Y. Liu, D. Wang, Q. Peng, C. Chen and Y. Li, J. Am. Chem. Soc., 2018, 140, 2610–2618 CrossRef CAS.
- L. Yang and L. Zhang, Appl. Catal., A, 2019, 259, 118053 CrossRef CAS.
- L. Liu, J. Sun, J. Ding, Y. Zhang, T. Sun and J. Jia, Inorg. Chem., 2019, 58, 13241–13249 CrossRef CAS.
- S. Jaiswar and K. D. Mandal, J. Phys. Chem. C, 2017, 121, 19586–19601 CrossRef CAS.
- Y. P. Zhu, T. Y. Ma, M. Jaroniec and S. Z. Qiao, Angew. Chem., Int. Ed., 2017, 56, 1324–1328 CrossRef CAS.
- C. Wang, X.-D. Zhu and P.-J. Zuo, Chem. Eng. J., 2020, 396, 125163 CrossRef CAS.
- J. Chen, J. Cong, Y. Chen, Q. Wang, M. Shi, X. Liu and H. Yang, Appl. Surf. Sci., 2020, 508, 145239 CrossRef.
- X. Gao, B. Wang, Y. Zhang, H. Liu, H. Liu, H. Wu and S. Dou, Energy Storage Mater., 2019, 16, 46–55 CrossRef.
- Y. Wang, Z. Zhang, X. Liu, F. Ding, P. Zou, X. Wang, Q. Zhao and H. Rao, ACS Sustainable Chem. Eng., 2018, 6, 12511–12521 CrossRef CAS.
- Y. Lu, L. Su, J. Qi, S. Lei, B. Liu, Q. Zang, S. Shi and X. Yan, J. Mater. Chem. A, 2018, 6, 13717–13724 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ce01075h |
| This journal is © The Royal Society of Chemistry 2020 |