
Understanding the influence of carboxylate substitution on the property of high-performance donor polymers in non-fullerene organic solar cells†
Guofang
Yang‡
 ab,
Jing
Liu‡
a,
Lik-Kuen
Ma
a,
Shangshang
Chen
a,
Joshua Yuk Lin
Lai
a,
Wei
Ma
*b and
He
Yan
*ac
ab,
Jing
Liu‡
a,
Lik-Kuen
Ma
a,
Shangshang
Chen
a,
Joshua Yuk Lin
Lai
a,
Wei
Ma
*b and
He
Yan
*ac
aDepartment of Chemistry and Energy Institute, The Hong Kong University of Science and Technology, Clear Water Bay, Hong Kong, P. R. China. E-mail: hyan@ust.hk
bState Key Laboratory for Mechanical Behavior of Materials, Xi’an Jiaotong University, Xi’an 710049, P. R. China. E-mail: msewma@mail.xjtu.edu.cn
cThe Hong Kong University of Science and Technology-Shenzhen Research Institute, No. 9 Yuexing 1st RD, Hi-tech Park, Nanshan, Shenzhen 518057, China
First published on 11th May 2018
Abstract
Carboxylate substitution is a common approach to tune the energy level of donor polymers for organic solar cells. However, the influence of carboxylate substitution on the morphological and electronic properties of donor polymers is not well understood. In this paper, we study two pairs of structurally similar terthiophene or quarterthiophene donor polymers with partial or complete carboxylate substitution on the alkyl side chains. It is found that the carboxylate substitution can enhance the crystallinity of the donor polymers and introduce larger and purer domains. Moreover, the polymers with the carboxylate substitution exhibit reduced bimolecular recombination due to the improved morphology. For device efficiencies, the terthiophene-based polymer, P3TEA (with 50% carboxylate substitution), exhibits the best performance. The alkyl side chains on P3TEA provide a typical temperature-dependent aggregation property, allowing for effective morphology control, while the carboxylate substitution deepens the HOMO level and enhances the crystallinity of the polymer. These benefits yield a near optimal morphology and high Voc value, and thus the best device efficiency among the polymers studied.
Introduction
The organic solar cell (OSC) is considered as a promising solar energy conversion technology due to its merits of low cost, light weight and mechanical flexibility.1–9 High-performance OSCs are based on bulk-heterojunction (BHJ) active layers containing electron donor and acceptor materials. Fullerene derivatives are traditional acceptor materials, and the power conversion efficiency (PCE) of fullerene-based single junction OSCs has exceeded 11%.10 However, fullerene derivatives have several limitations including: high-cost production, poor light absorption and low stability.11–13 Therefore, alternative acceptor materials such as small molecule acceptors (SMA) have attracted more research interest, with SMA-based devices achieving the most efficient OSC device today.14–18 SMAs offer the advantages of low production costs, broad absorption spectra, good stability, easily tunable energy levels, and most importantly, low voltage loss.To develop efficient non-fullerene OSCs, it is not only important to synthesize high-performance non-fullerene acceptors, but also significant to design matching donor polymers to maximize the PCE of OSC devices. Several factors should be considered when designing a high-performance donor polymer. One is tuning the energy level to maximize the Voc and minimize the voltage loss.19–21 There are a variety of chemical strategies to tune the energy levels of OSC materials. Besides the commonly used fluorination strategy,22–26 another approach is to attach carboxylate groups,27,28 which is also electron withdrawing and thus can modulate the energy levels effectively. For example, a state-of-art donor polymer named P3TEA has been reported by partially attaching the carboxylate substitution on the alkyl side chains.19 This modification introduces an excellent device performance including a record-high Voc value of 1.11 V and a low voltage loss of 0.55 V. However, the influence of the carboxylate substitution on the morphological and electronic properties of the polymer is not well understood.
In this paper, we study two pairs of structurally similar terthiophene or quarterthiophene donor polymers with partial or complete carboxylate substitution on the alkyl side chains. It is found that carboxylate substitution can enhance the crystallinity of the donor polymers and introduce larger and purer domains. Moreover, light intensity dependent photocurrent measurement reveals that the polymers with the carboxylate substitution exhibit reduced bimolecular recombination due to the improved morphology. For device efficiencies, the terthiophene-based polymer, P3TEA (with 50% carboxylate substitution), exhibits the best performance. The alkyl side chains on P3TEA provide a typical temperature-dependent aggregation (TDA) property, allowing for effective morphology control, while the carboxylate substitution deepens the HOMO level and enhances the crystallinity of the polymer. These benefits yield a near optimal morphology and high Voc value, and thus the best device efficiency among the polymers studied.
Result
Shown in Fig. 1 are the chemical structures of the four polymers of interest. For the quarterthiophene system, a state-of-the-art donor polymer PffBT4T-2DT (renamed as P4TA for the ease of comparison in this paper) is studied,29 which exhibits the typical TDA property and high device performance with fullerene acceptors. For comparison, we synthesized a new analogous polymer P4TE, by attaching carboxylate groups on 100% alkyl side chains of P4TA. Next, for the terthiophene based polymers, we choose to study two high performance polymers, PffBT-T3(1,3)-2(100% alkyl side chain, renamed as P3TA in this paper) and P3TEA (by attaching carboxylate groups on 50% alkyl side chains).19,30 While the quarterthiophene-based polymers were shown to match well with the fullerene acceptors, the terthiophene-based polymers were found to better match with the emerging non-fullerene acceptors according to our experience.10,19,29 Therefore, the choice of these quarterthiophene and terthiophene polymers allows us to understand the performance of materials under different scenarios.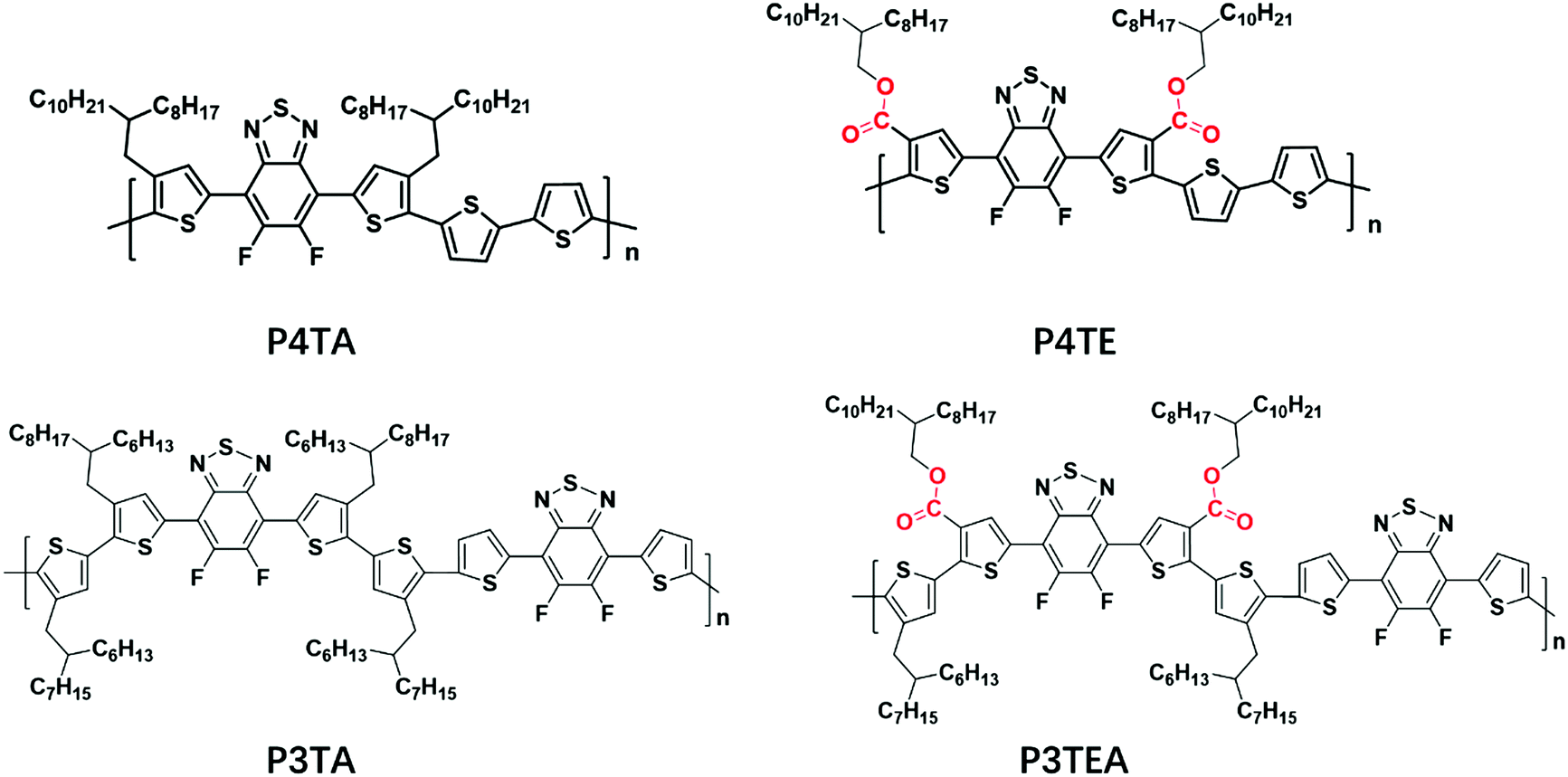 | ||
| Fig. 1 Chemical structures of the four polymers studied. | ||
Grazing incident wide angle X-ray scattering (GIWAXS) is utilized to characterize the molecular ordering of the polymers, with the profile shown in Fig. 2 and the 2D patterns displayed in the (ESI†). For the quarterthiophene system, it can be clearly seen that, in both pure and blend films, the q value of the (010) peak of P4TE is larger than that of P4TA, indicating a smaller d spacing value and closer π–π stacking. In the pure film, the π–π stacking d spacing value is 3.62 Å for P4TA, but 3.55 Å for P4TE. In the blend film, the π–π stacking d spacing of the P4TA polymer is estimated to be 3.59 Å, while that of the P4TE polymer is about 3.53 Å. In addition, the (010) peak of P4TE is sharper than that of P4TA, in both pure and blend film. As a result, P4TE has a larger coherence length (CL) than its analogues polymer P4TA. In the pure film, the CL is ∼47 Å for the P4TA polymer and ∼55 Å for the P4TE polymer. In the blend film, the CL of the P4TA polymer is estimated to be 37 Å, while that of the P4TE polymer is about 42 Å. These data all indicate that attaching the carboxylate substitution on the alkyl side chains can enhance the crystallinity of the polymer. For the terthiophene system, similar phenomenon was observed. In both pure and blend film, the q location of the (010) peak of P3TEA is larger than that of P3TA, showing that its d spacing is shorter and π–π stacking is closer. What's more, the (010) peak of P3TEA is sharper than that of P3TA, indicating that P3TEA has a longer CL. All the above results reveal that the carboxylated polymers have higher crystallinity in the terthiophene system, which is consistent with the conclusion in the quarterthiophene system.
 | ||
| Fig. 2 GIWAXS profiles of the investigated films. | ||
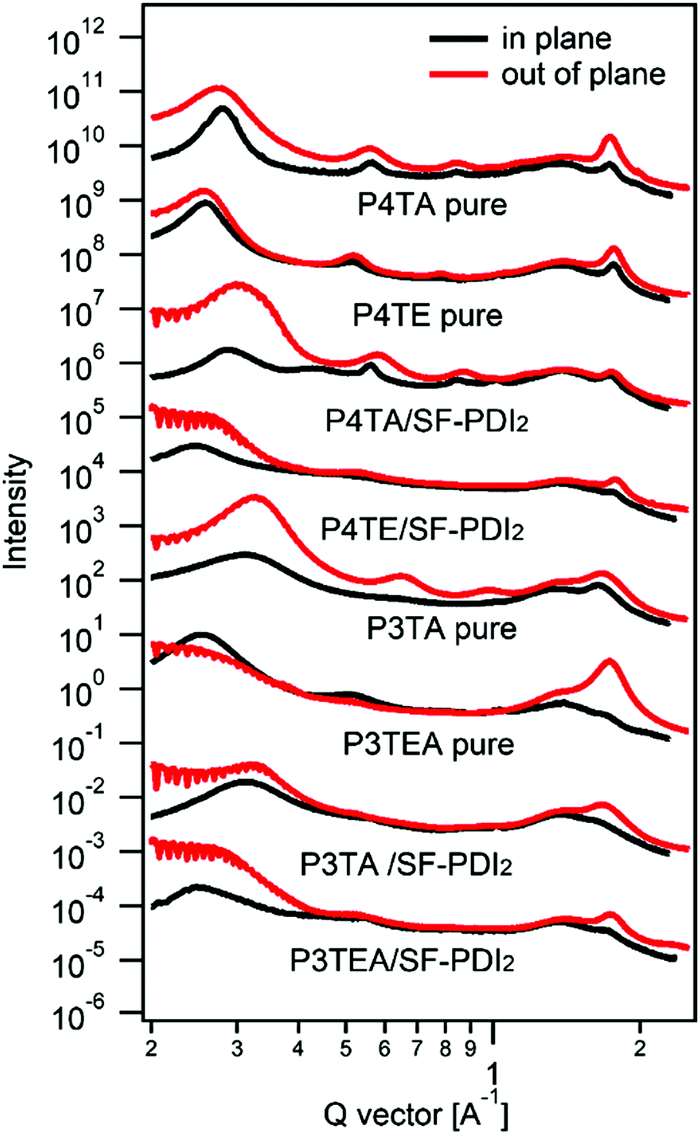
Resonant soft X-ray scattering (R-SoXS) is a powerful tool for probing the in-plane composition of the blend films.31 A photon energy of 284.8 eV is selected for the R-SoXS measurement to maximize the contrast between the donor/accepter materials. The values of domain size and relative domain purity of the four blend films (listed in Table 1) are extracted from the R-SoXS profiles, as shown in Fig. 3. For the quarterthiophene system, the average domain size of the P4TA/SF-PDI2 blend film is about 15 nm, and the relative domain purity is a little low at ∼0.34. When it turns to the P4TE/SF-PDI2 blend film, the domain size is much larger at ∼44 nm, and the relative domain purity is also higher at ∼0.57. For the terthiophene system, similar trend was observed. Domain size of the P3TA/SF-PDI2 blend film is about 7.7 nm and its domain purity is about 0.64. Yet for its carboxylated counterpart P3TEA, domain size of the blend film is ∼13 nm and the relative domain purity is 1.00. What's more, AFM data (Fig. S5, ESI†) shows that the carboxylated polymers could have larger domains in the blend films. These results indicate that after attaching the carboxylate substitution on the alkyl side chains, the domains of the blend films are larger and purer.
| q location (Å−1) | d spacing (Å) | Coherence length (Å) | Domain size (nm) | Domain purity (a.u.) | |
|---|---|---|---|---|---|
| P4TA pure | 1.7344 | 3.62 | 47.32 | N.A. | N.A. |
| P4TE pure | 1.7668 | 3.55 | 55.16 | N.A. | N.A. |
| P4TA/SF-PDI2 | 1.7483 | 3.59 | 36.99 | 15.15 | 0.3368 |
| P4TE/SF-PDI2 | 1.7803 | 3.53 | 42.06 | 44.48 | 0.5699 |
| P3TA pure | 1.6686 | 3.76 | 21.49 | N.A. | N.A. |
| P3TEA pure | 1.7300 | 3.63 | 33.05 | N.A. | N.A. |
| P3TA/SF-PDI2 | 1.6671 | 3.77 | 20.42 | 7.68 | 0.6369 |
| P3TEA/SF-PDI2 | 1.7348 | 3.62 | 28.95 | 12.92 | 1.0000 |
 | ||
| Fig. 3 R-SoXS profiles of the four blend films. | ||
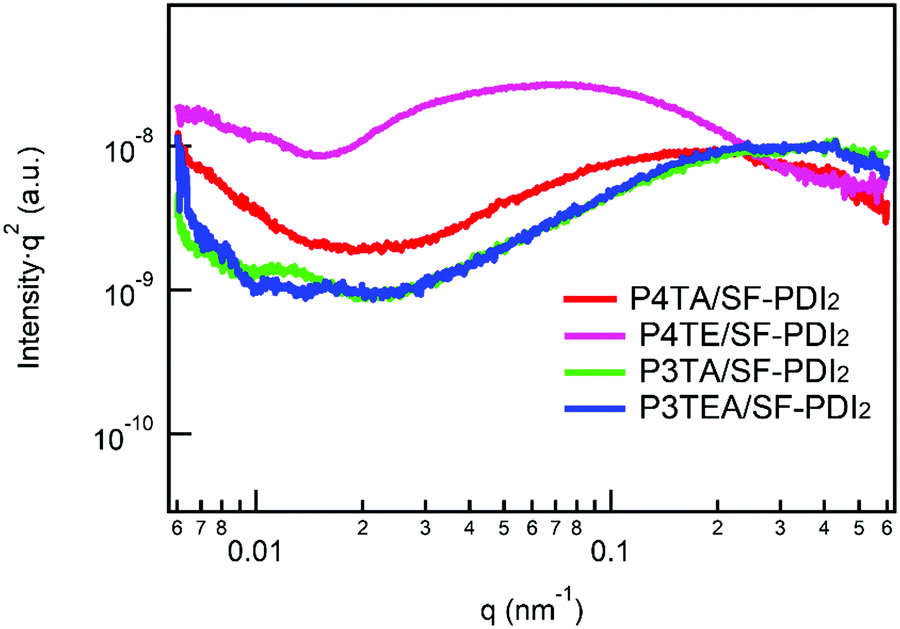
Light intensity dependent photocurrent measurement was carried out for the four devices. The photocurrent follows a power-law dependence on light intensity, which can be described as Jph ∝ Pα (Jph = JL − JD, where JL and JD are the current density under illumination and in the dark, respectively). When the space charge buildup reaches fundamental limit, the power exponent α will equal to 0.75; while there is no space charge formed, α will equal to 1.32 Therefore, corresponding to the higher α value should be the less bimolecular recombination. The experimental power-law dependence of photocurrent on the incident light intensity was shown in Fig. 4. For the quarterthiophene system, α is 0.9611 in P4TA/SF-PDI2 based device, yet 0.9977 in the counterpart P4TE/SF-PDI2 based device. For the terthiophene system, the tendency was similar. The α value is 0.9831 in the P3TA/SF-PDI2 based device, while it turns to 0.9971 in the P3TEA/SF-PDI2 based device. These results indicate that attaching the carboxylate substitution on the alkyl side chains can reduce the bimolecular recombination within the PSC devices, which could be attributed to the improved morphology mentioned above.
 | ||
| Fig. 4 Experimental power-law dependence of photocurrent on the incident light intensity for the four devices. | ||
Discussion
Device performance
Solar cell devices for the polymers were fabricated with an inverted device structure (ITO/ZnO/Polymer: SMA/V2O5/Al). The J–V characteristics of the devices are shown in the ESI† and the relevant photovoltaic device parameters are summarized in Table 2. The best performance was achieved by the terthiophene polymer, P3TEA. Compared to its counterpart polymer P3TA, all the Voc, Jsc, FF values of the P3TEA polymer are increased. The increased Voc value resulted from the deepened HOMO level due to the electron withdrawing effect of the carboxylate substitution. The higher Jsc value was caused by the optimum morphology with proper domain size and high domain purity, and the increased FF value could be explained by the enhanced crystallinity and mobility.| Blends | Hole mobility (cm2 V−1 s−1) | V oc (V) | J sc (mA cm−2) | FF | PCE (%) |
|---|---|---|---|---|---|
| a The values in parentheses stand for the average PCEs from over 20 devices. | |||||
| P4TA/SF-PDI2 | 5.7 × 10−4 | 0.96 | 8.60 | 0.398 | 3.29 (3.00)a |
| P4TE/SF-PDI2 | 7.0 × 10−4 | 1.06 | 9.27 | 0.541 | 5.34 (4.70)a |
| P3TA/SF-PDI2 | 2.7 × 10−4 | 1.02 | 10.90 | 0.488 | 5.44 (4.45)a |
| P3TEA/SF-PDI2 | 4.4 × 10−4 | 1.11 | 13.27 | 0.643 | 9.50 (8.63)a |
To better understand their morphology and device performance, TDA properties of the four polymers were tested, as shown in Fig. 5. For the quarterthiophene system, P4TA has an obvious TDA behavior as expected. The UV-Vis absorption spectra show a distinct red-shift when the temperature is lowered. Solution at 100 °C has an absorption maximum at ∼650 nm, while the solution at 20 °C is greatly red-shifted with a maximum at ∼770 nm. Moreover, at high temperatures, P4TA is well dissolved and disaggregated in solution. When the temperature is slowly reduced, a strong 0–0 transition peak at ∼700 nm gradually emerges. These phenomena reveal that P4TA has a typical TDA property. However, its analogues polymer P4TE does not own a notable TDA property. Obvious red-shift is not found when the solution temperature declines and even in low temperature as 20 °C, 0–0 transition peak is quite weak. Therefore, the ability of P4TE to control the morphology is poor, leading to excessively strong crystallinity, large domains (∼44 nm) and thus, low device performance. In the terthiophene system, obvious TDA behavior is found for the P3TA polymer. When the temperature falls, the red-shift of the UV-Vis absorption spectra is distinct and the absorption onset is markedly shifted from ∼650 nm at 100 °C to ∼770 nm at 20 °C. Interestingly, the counterpart polymer, P3TEA, keeps the TDA property. When the temperature decreases slowly, the red-shift of the absorption spectra is remarkable and the absorption maximum is greatly shifted from ∼650 nm at 100 °C to ∼770 nm at 20 °C. These phenomena reveal that the P3TEA polymer shows an obvious TDA behavior, and thus, an excellent ability of morphology control. What's more, the carboxylate substitution endows the P3TEA polymer an enhanced crystallinity compared with the counterpart polymer P3TA. Consequently, the P3TEA polymer owns a near-perfect morphology containing strong crystallinity, proper domain size and high domain purity. Combining its ultrahigh Voc value, it achieves the excellent device performance.
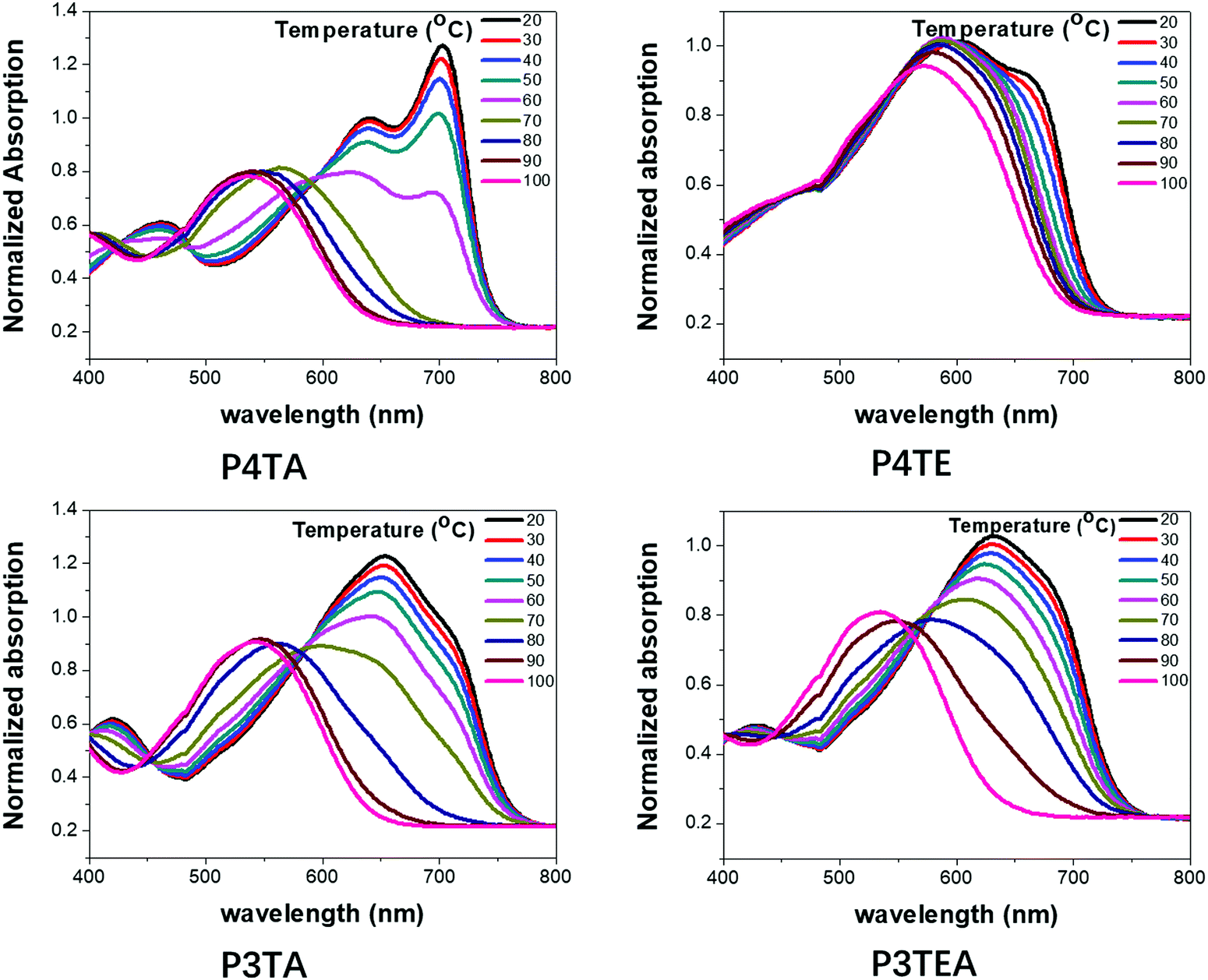 | ||
| Fig. 5 Ultraviolet-Visible (UV-Vis) absorption spectra of the four polymer solutions (0.02 mg mL−1 in CB) at temperatures as indicated: (a) P4TA, (b) P4TE, (c) P3TA and (d) P3TEA. | ||
Conclusions
In summary, we studied the effect of the carboxylate substitution on the property of donor polymers in non-fullerene organic solar cells. GIWAXS and R-SoXS analysis reveal that the carboxylate substitution can introduce enhanced crystallinity, larger and purer domains. Moreover, light intensity dependent photocurrent measurement shows that polymers with the carboxylate substitution have reduced bimolecular recombination due to the improved morphology. For the device efficiency, only the polymer with half carboxylate substitution can achieve high efficiency when combining with non-fullerene acceptors. TDA property is tested to explain the relationship between their morphology and device performance. For the polymer with 100% carboxylate substitution, it does not show a typical TDA behaviour. Thus, its ability to control the morphology is poor, leading to excessively strong crystallinity and large domains. For the polymer with half carboxylate substitution, the alkyl chain endows the polymer an obvious TDA property (and thus, a good morphology control ability), and the carboxylate substitution affords the polymer with an enhanced crystallinity. Therefore, it possesses a near-perfect morphology with strong crystallinity, proper domain size and high domain purity. Combining the near optimal morphology and an ultrahigh Voc value, P3TEA/SF-PDI2 blend finally achieves an excellent device performance. Our study shows that attaching the carboxylate substitution on the alkyl side chains can not only enlarge the Voc value, but also improve the blend morphology to afford higher Jsc and FF values. Proper extent of carboxylate substitution could be an effective way to design new donor polymers to achieve higher efficiency in the field of polymer solar cells.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
The work described in this paper was partially supported by the National Basic Research Program of China (973 Program project numbers 2013CB834701 and 2014CB643501), the ShenZhen Technology and Innovation Commission (project number JCYJ20170413173814007), the Hong Kong Research Grants Council (project numbers T23-407/13 N, N_HKUST623/13, 16305915, 16322416, 606012, and 16303917), HK JEBN Limited, HKUST president's office (Project FP201) and the National Science Foundation of China (#21374090). We especially thank Hong Kong Innovation and Technology Commission for the support through projects ITC-CNERC14SC01 and ITS/083/15.References
- S. H. Park, A. Roy, S. Beaupré, S. Cho, N. Coates, J. S. Moon, D. Moses, M. Leclerc, K. Lee and A. J. Heeger, Nat. Photonics, 2009, 3, 297–302 CrossRef.
- Z. He, C. Zhong, S. Su, M. Xu, H. Wu and Y. Cao, Nat. Photonics, 2012, 6, 593–597 Search PubMed.
- G. Li, V. Shrotriya, J. Huang, Y. Yao, T. Moriarty, K. Emery and Y. Yang, Nat. Mater., 2005, 4, 864–868 CrossRef.
- C. J. Brabec, S. Gowrisanker, J. J. Halls, D. Laird, S. Jia and S. P. Williams, Adv. Mater., 2010, 22, 3839–3856 CrossRef PubMed.
- A. J. Heeger, Adv. Mater., 2014, 26, 10–28 CrossRef PubMed.
- G. Li, V. Shrotriya, J. Huang, Y. Yao, T. Moriarty, K. Emery and Y. Yang, Nat. Mater., 2005, 4, 864 CrossRef.
- J. Y. Kim, K. Lee, N. E. Coates, D. Moses, T.-Q. Nguyen, M. Dante and A. J. Heeger, Science, 2007, 317, 222–225 CrossRef PubMed.
- J. Peet, J. Y. Kim, N. E. Coates, W. L. Ma, D. Moses, A. J. Heeger and G. C. Bazan, Nat. Mater., 2007, 6, 497 CrossRef PubMed.
- H.-Y. Chen, J. Hou, S. Zhang, Y. Liang, G. Yang, Y. Yang, L. Yu, Y. Wu and G. Li, Nat. Photonics, 2009, 3, 649 CrossRef.
- J. Zhao, Y. Li, G. Yang, K. Jiang, H. Lin, H. Ade, W. Ma and H. Yan, Nat. Energy, 2016, 1, 15027 CrossRef.
- G. Sauvé and R. Fernando, J. Phys. Lett., 2015, 6, 3770–3780 Search PubMed.
- Y. Lin and X. Zhan, Acc. Chem. Res., 2016, 49, 175–183 CrossRef PubMed.
- Y. Lin, Y. Li and X. Zhan, Chem. Soc. Rev., 2012, 41, 4245–4272 RSC.
- Y. Zhong, M. T. Trinh, R. Chen, G. E. Purdum, P. P. Khlyabich, M. Sezen, S. Oh, H. Zhu, B. Fowler, B. Zhang, W. Wang, C.-Y. Nam, M. Y. Sfeir, C. T. Black, M. L. Steigerwald, Y.-L. Loo, F. Ng, X. Y. Zhu and C. Nuckolls, Nat. Commun., 2015, 6, 8242 CrossRef PubMed.
- Z. Chen, P. Cai, J. Chen, X. Liu, L. Zhang, L. Lan, J. Peng, Y. Ma and Y. Cao, Adv. Mater., 2014, 26, 2586–2591 CrossRef PubMed.
- Z. Mao, W. Senevirathna, J.-Y. Liao, J. Gu, S. V. Kesava, C. Guo, E. D. Gomez and G. Sauvé, Adv. Mater., 2014, 26, 6290–6294 CrossRef PubMed.
- J. Zhao, Y. Li, H. Lin, Y. Liu, K. Jiang, C. Mu, T. Ma, J. Y. Lin Lai, H. Hu, D. Yu and H. Yan, Energy Environ. Sci., 2015, 8, 520–525 Search PubMed.
- H. Lin, S. Chen, Z. Li, J. Y. L. Lai, G. Yang, T. McAfee, K. Jiang, Y. Li, Y. Liu, H. Hu, J. Zhao, W. Ma, H. Ade and H. Yan, Adv. Mater., 2015, 27, 7299–7304 CrossRef PubMed.
- J. Liu, S. Chen, D. Qian, B. Gautam, G. Yang, J. Zhao, J. Bergqvist, F. Zhang, W. Ma, H. Ade, O. Inganäs, K. Gundogdu, F. Gao and H. Yan, Nat. Energy, 2016, 1, 16089 CrossRef.
- S. Chen, Y. Liu, L. Zhang, P. C. Y. Chow, Z. Wang, G. Zhang, W. Ma and H. Yan, J. Am. Chem. Soc., 2017, 139, 6298–6301 CrossRef PubMed.
- J. Zhang, Y. Li, J. Huang, H. Hu, G. Zhang, T. Ma, P. C. Y. Chow, H. Ade, D. Pan and H. Yan, J. Am. Chem. Soc., 2017, 139, 16092–16095 CrossRef PubMed.
- J. W. Jung, J. W. Jo, C. C. Chueh, F. Liu, W. H. Jo, T. P. Russell and A. K. Jen, Adv. Mater., 2015, 27, 3310–3317 CrossRef PubMed.
- Z. Li, H. Lin, K. Jiang, J. Carpenter, Y. Li, Y. Liu, H. Hu, J. Zhao, W. Ma, H. Ade and H. Yan, Nano Energy, 2015, 15, 607–615 CrossRef.
- A. C. Stuart, J. R. Tumbleston, H. Zhou, W. Li, S. Liu, H. Ade and W. You, J. Am. Chem. Soc., 2013, 135, 1806–1815 CrossRef PubMed.
- J. W. Jo, S. Bae, F. Liu, T. P. Russell and W. H. Jo, Adv. Funct. Mater., 2015, 25, 120–125 CrossRef.
- H. Bronstein, J. M. Frost, A. Hadipour, Y. Kim, C. B. Nielsen, R. S. Ashraf, B. P. Rand, S. Watkins and I. McCulloch, Chem. Mater., 2013, 25, 277–285 CrossRef.
- M. Zhang, X. Guo, Y. Yang, J. Zhang, Z.-G. Zhang and Y. Li, Polym. Chem., 2011, 2, 2900–2906 RSC.
- L. Huo, T. L. Chen, Y. Zhou, J. Hou, H.-Y. Chen, Y. Yang and Y. Li, Macromolecules, 2009, 42, 4377–4380 CrossRef.
- Y. Liu, J. Zhao, Z. Li, C. Mu, W. Ma, H. Hu, K. Jiang, H. Lin, H. Ade and H. Yan, Nat. Commun., 2014, 5, 5293 CrossRef PubMed.
- H. Hu, K. Jiang, G. Yang, J. Liu, Z. Li, H. Lin, Y. Liu, J. Zhao, J. Zhang, F. Huang, Y. Qu, W. Ma and H. Yan, J. Am. Chem. Soc., 2015, 137, 14149–14157 CrossRef PubMed.
- S. Swaraj, C. Wang, H. Yan, B. Watts, J. Luning, C. R. McNeill and H. Ade, Nano Lett., 2010, 10, 2863–2869 CrossRef PubMed.
- M. Lenes, M. Morana, C. J. Brabec and P. W. M. Blom, Adv. Funct. Mater., 2009, 19, 1106–1111 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8qm00101d |
| ‡ These authors contributed equally to this work. |
| This journal is © the Partner Organisations 2018 |