
Significantly improving the efficiency of polymer solar cells through incorporating noncovalent conformational locks†
Lei
Lv‡
a,
Xiaofen
Wang‡
ab,
Tao
Dong
a,
Xinlong
Wang
a,
Xiaoxi
Wu
a,
Lei
Yang
a and
Hui
Huang
*a
aCollege of Materials Science and Opto-Electronic Technology & CAS Key Laboratory of Vacuum Physic, University of Chinese Academy of Sciences, Beijing 100049, P. R. China. E-mail: huihuang@ucas.ac.cn
bBeijing Key Laboratory of Function Materials for Molecular & Structure Construction, School of Materials Science and Engineering, University of Science and Technology Beijing, Beijing, 100083, P. R. China
First published on 6th February 2017
Abstract
Noncovalent conformational locks have been widely employed to construct highly planar and rigid conjugated systems for organic electronics. In this paper, two conjugated polymers (PDTffBT–TVT and PDTffBT–TVTOEt) were synthesized through the Stille coupling of 4,7-di(thien-2-yl)-5,6-difluoro-2,1,3-benzothiadiazole (DTffBT) with (E)-2-(2-(thiophen-2-yl)vinyl)thiophene (TVT) and (E)-1,2-diethoxy-1,2-di(thiophen-2-yl)ethane (TVTOEt), respectively, to investigate the effect of incorporation of the S⋯O noncovalent conformational locks on the performance of the polymer based bulk heterojunction solar cells. The physicochemical properties and photovoltaic characteristics of the conjugated polymers were fully investigated with different characterization techniques, which demonstrated that incorporation of the noncovalent conformational locks improved the rigidity of the backbone, leading to enhanced charge transport mobilities, and thus higher Jsc and FF. As a result, the efficiencies of the solar cells were significantly improved from 2.59% (PDTffBT–TVT) to 6.16% (PDTffBT–TVTOEt).
Introduction
Bulk heterojunction polymer solar cells (PSCs) have attracted much attention due to their advantages of mechanical flexibility, light weight, solution-processability, large surface areas and cost-effective fabrication.1,2 Over the past decades, great efforts have been made to achieve high performance PSCs, including material design and synthesis,3–5 interfacial layer modification6,7 and device engineering.8,9 Generally, the design and synthesis of novel conjugated materials are critical to achieve high performance solar cells. Through molecular engineering, thousands of conjugated polymers with high rigidity/planarity have been synthesized in order to achieve high charge transport properties and high power conversion efficiencies (PCE).5,10There are two general methods to improve the rigidity of the backbone of conjugated systems. One is to covalently bond the neighboring aromatic rings with atoms of carbon, nitrogen, silicon etc., which is usually synthetically challenging. The second one is to employ noncovalent “conformational locks” to restrict the free rotation of the neighboring aromatic systems. In 2012, we designed and synthesized an easily accessible, electron-neutral, and highly planar building block, (E)-1,2-diethoxy-1,2-di(thiophen-2-yl)ethane (TVTOEt), having intramolecular S⋯O noncovalent “conformational locks”. Based on this building block, several high performing conjugated polymers for organic thin film transistors (OTFTs) and organic solar cells have been reported.11,12 Since then, various “conformational locks” including S⋯O,13–15 S⋯X, (X = F, Br etc.),16 S⋯N17etc. have been employed to build highly planar and rigid conjugated systems. In 2014, Woo et al. introduced fluorine or alkoxy substituents into the benzotriazole unit to enhance the molecular ordering through intra- and intermolecular F⋯S, F⋯H–C, C–F⋯πF, or S⋯O interactions. With that, both the fluorinated- and alkoxy-substituted polymers demonstrated enhanced hole mobilities.16 In 2016, Yu and coworkers synthesized and characterized a series of EDTE-based copolymers with multiple O⋯H–C and S⋯O conformational locks and achieved a hole mobility of up to 5.37 cm2 V−1 s−1. They illustrated the significance of conformational locks in tuning the optical, electrochemical, and thermal properties, and intra- and intermolecular interactions of semiconducting copolymers.15
Recently, incorporation of fluorine atoms into the backbone of conjugated polymers has shown great potential for enhancing the efficiency of BHJ PSCs.18–22 In some cases, attaching strong electron-withdrawing fluorine atoms to electron-accepting units can simultaneously enhance the key factors of power conversion efficiency (PCE) including the open-circuit voltage (Voc), short-circuit current density (Jsc), and fill factor (FF).19 Among them, 4,7-di(thien-2-yl)-5,6-difluoro-2,1,3-benzothiadiazole (DTffBT), an easily accessible electron accepting unit, has been widely applied to construct p-type conjugated polymers for high performance photovoltaic devices.18–21,23–28 You and co-workers firstly employed DTffBT to synthesize a series of copolymers for PSCs, reaching a PCE as high as 7.2%.19 Jiang et al. developed a series of DTffBT-based copolymers with different side chains, which afforded high performing PSCs with a PCE of 8.30%.23 Furthermore, Yan et al. reported a series of polymers based on DTffBT and quaterthiophene (T4), which were employed to fabricate PSCs to afford an efficiency over 10%.21
In this contribution, we employed the fluorinated building block DTffBT to copolymerize with two building blocks TVT and TVTOEt, resulting in two conjugated polymers (PDTffBT–TVT and PDTffBT–TTVOEt). The physicochemical properties of these two conjugated polymers were systematically investigated. Significantly, through introducing S⋯O noncovalent conformational locks into the backbone of the conjugated systems, the charge carrier mobilities were obviously enhanced. As a result, the fill factor and power conversion efficiency of the PSCs were significantly increased, leading to a maximum efficiency as high as 6.2%.
Results and discussion
Synthesis and characterization of polymers
Two p-type copolymers were synthesized by the Stille coupling of 5,6-difluoro-4,7-bis(5-bromo-4-(2-octyldodecyl)-2-thienyl)-2,1,3-benzothiadiazole (DTffBT-Br2)21 with (E)-1,2-bis(5-(trimethylstannyl)thiophen-2-yl)ethane29 or (E)-1,2-diethoxy-1,2-bis(5-(trimethylstannyl)thiophen)-2-ylethane,11 respectively, in high yields of about 90% (as shown in Scheme 1). The copolymers were confirmed by 1H NMR (Fig. S1 and S2, ESI†) and elemental analysis. The number-average molecular weights (Mn) and the polydispersity index (PDI) of the polymers PDTffBT–TVT and PDTffBT–TVTOEt, estimated by gel permeation chromatography (GPC), are 36.0 kDa (PDI = 1.55) and 37.9 kDa (PDI = 1.65), respectively. Thermogravimetric analysis (TGA) showed that these polymers have excellent thermal stability with high onset decomposition temperatures (Td) over 390 °C at 5% weight loss under a nitrogen flow (Fig. S3, ESI†).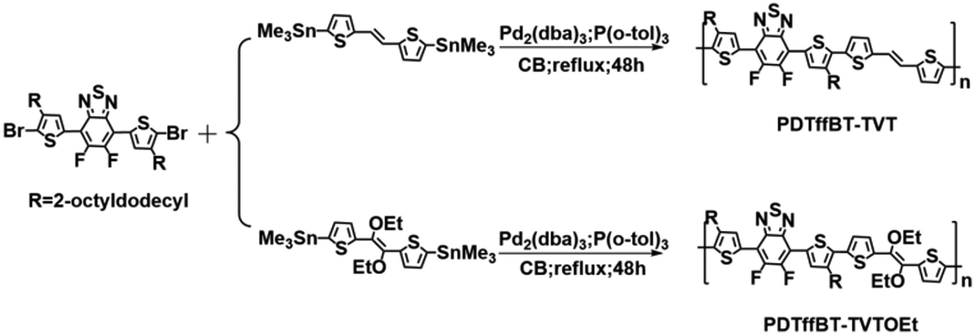 | ||
| Scheme 1 Chemical structures and synthesis routes of the polymers. | ||
Optical absorption properties and DFT calculations
To investigate the characteristics of S⋯O conformational locks, the UV-vis and photoluminescence emission spectra in solution of the monomers TVT and TVTOEt were recorded and are shown in Fig. S4 (ESI†), and the corresponding data are listed in Table S1 (ESI†). The UV-vis absorption peaks of TVT and TVTOEt are located at 345 and 343 nm, respectively. It is well recognized that 0–0 transitions are rarely observed in room temperature solution spectra, so it is acceptable to construct Δ = λem − λabs as an estimation of the magnitude of the Stokes shift,30 which could be used to evaluate the rigidity of the conjugated backbone.31 TVT and TVTOEt solutions emit violet-blue light with maxima at 416 and 402 nm, respectively, which are assigned to the 0–1 transition.30 It is obvious that the Stokes shift of TVTOEt (59 nm) is smaller than that of TVT (71 nm), suggesting that the backbone of TVTOEt is more rigid due to the presence of S⋯O conformational locks.Fig. 1 shows the optical absorption of these polymers in dilute 1,3-dichlorobenzene (DCB) solution and as thin films, investigated by UV-vis spectroscopy, and the detailed parameters are listed in Table 1. The two polymers PDTffBT–TVT and PDTffBT–TVTOEt show similar absorption bands in DCB solution with maximum absorption peaks at 599 nm and 574 nm, respectively. The typical lower-energy bands can be attributed to the intramolecular charge transfer (ICT) interaction between electron-rich and electron-deficient units.23 Compared to the solution spectra, those of PDTffBT–TVT and PDTffBT–TVTOEt films became broader and red-shifted towards longer wavelengths with maximum absorption peaks at 645 nm and 619 nm, respectively. The large red shifts (46 nm for PDTffBT–TVT and 45 nm for PDTffBT–TVTOEt) from solution to the solid state indicate their coplanar structures and stronger interchain π–π stacking in the solid state. Based on the low-edge onset of absorption, the optical band gaps (Eoptg) of PDTffBT–TVT and PDTffBT–TVTOEt were calculated to be 1.56 eV and 1.60 eV, respectively.
 | ||
| Fig. 1 Optical absorption spectra of the polymers PDTffBT–TVT and PDTffBT–TVTOEt in DCB solution (a); as-cast pristine films (b); and blend films on glass substrates (c). | ||
| Polymers |
E
optg![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a [eV] a [eV] |
E red-1/2 [eV] | E ox-1/2 [eV] | HOMOb [eV] | LUMOc [eV] | λ solutionmax [nm] | λ thin-filmmax [nm] |
|---|---|---|---|---|---|---|---|
| a Determined from the onset of UV-vis absorption spectra. b Calculated from HOMO = −Eox-1/2 − 4.44 eV. c Calculated from LUMO = HOMO + Eoptg. d Determined from the onset of reduction potential of a cyclic voltammogram. | |||||||
| PDTffBT–TVT | 1.56 | −1.21 | 1.04 | −5.48 |
−3.92c
(−3.23)d |
474, 599, 707 | 473, 645, 709 |
| PDTffBT–TVTOEt | 1.60 | −1.22 | 1.01 | −5.45 |
−3.85c
(−3.22)d |
324, 496, 574, 685 | 327, 464, 619, 678 |
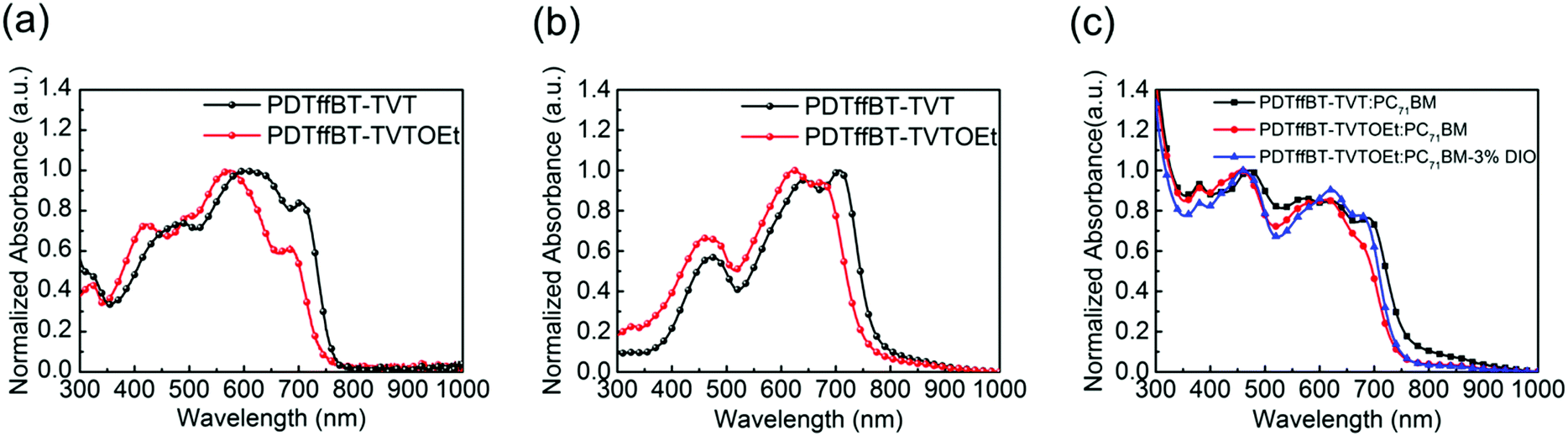
Structurally, the PDTffBT–TVT and PDTffBT–TVTOEt polymers differ from each other based on the identity of the donating building blocks (TVT and TVTOEt) conjugated with the DTffBT unit. Two parameters may decide the difference in absorption spectra: the electron donating capability of the moieties (TVT and TVTOEt) neighboring DTffBT along the backbone and the dihedral angle between the DTffBT unit and its neighboring units (TVT and TVTOEt). On the basis of these considerations, DFT (TDDFT)32 calculations at the B3LYP/6-31G** level were carried out on the polymer building blocks to provide insights into the electronic structure of the corresponding copolymers. As shown in Fig. 2a, the optimized geometries demonstrated that the dihedral angles between DTffBT and TVT units were calculated to be 0° and 0°, smaller than DTffBT–TVTOEt (2.3° and 12.5°), leading to stronger interchain π–π stacking in the solid state. The larger dihedral angles between the DTTffBT and TVTOEt units may be ascribed to the intramolecular S⋯O interactions.15,33 Although PDTffBT–TVT showed a more planar structure, the noncovalent interactions introduced into PDTffBT–TVTOEt could act as “conformational locks” to limit the free rotation of aromatic rings and improve the rigidity of the backbone.15 It is found that the rigidification of the molecule is beneficial for reducing the reorganisation energy,34 which is inversely proportional to the mobility of charge carriers.35,36 The molecular orbital distributions (Fig. 2b) showed that the HOMO/LUMO energy levels of PDTffBT–TVT and PDTffBT–TVTOEt are −4.89/−2.77 and −4.83/−2.72 eV, respectively. As a result, the calculated HOMO–LUMO gap (2.11 eV) of PDTffBT–TVTOEt is intrinsically similar to that of PDTffBT–TVT (2.12 eV), which indicates that TVTOEt may have a similar electron donating strength to TVT, similar to the reported results.11 However, the experimental Eoptg of PDTffBT–TVT is slightly smaller than that of PDTffBT–TVTOEt, reasonably attributed to its stronger π–π stacking. As shown in Fig. 1c, UV-vis absorption of both the PDTffBT–TVT/PDTffBT–TVTOEt:PC71BM blend films have a broad coverage from 300 to 780 nm, showing that the blend films may be excellent light absorbers.
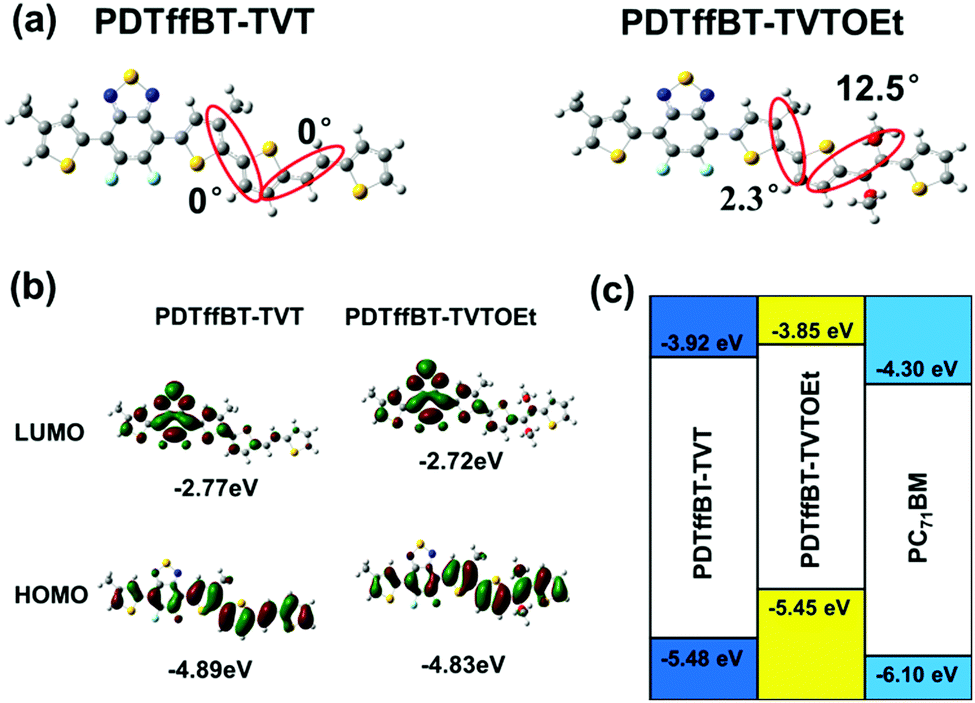 | ||
| Fig. 2 The optimized geometries of PDTffBT–TVT and PDTffBT–TVTOEt based on computation at the DFT/B3LYP/6-31G** level (a), the corresponding frontier orbital density distributions (b), and experimentally determined energy levels (c). | ||
Electrochemical properties
The ionization potential and electron affinity of the polymers were investigated by cyclic voltammetry (CV) using ferrocene as the internal standard, and the results are summarized in Table 1; their CV curves are shown in Fig. S5 (ESI†). Based on the onset of oxidation potential (Eox-1/2) of the polymers observed from the CV curve, the HOMO energy levels were estimated to be −5.48 eV for PDTffBT–TVT and −5.45 eV for PDTffBT–TVTOEt. By subtracting Eoptg from the HOMO, the LUMO energy levels of the corresponding polymers are −3.92 eV and −3.85 eV, respectively.Photovoltaic properties
To investigate the photovoltaic performance of PDTffBT–TVT and PDTffBT–TVTOEt, PSCs with a device architecture of ITO/ZnO/polymer:PC71BM/MoO3/Ag were fabricated. Thermal spin coating was employed to completely dissolve the polymers and control the aggregation.21 The J–V curves of the optimized devices under simulated solar illumination (AM 1.5G, 100 mW cm−2) are shown in Fig. 3a, and the device parameters are summarized in Table 2. The devices fabricated from the blend of PDTffBT–TVT:PC71BM afforded an average Voc of 0.68 eV, a Jsc of 6.77 mA cm−2, a FF of 48.61%, and a correspondingly average PCE of 2.39%. For the PDTffBT–TVTOEt:PC71BM based device, although Voc slightly decreased to 0.66 eV, the Jsc, FF and PCE improved to 8.69 mA cm−2, 60.00%, and 3.56%, respectively. Furthermore, DIO (3.0%, by volume) was used as the processing additive to tune the morphology and device performances. As a result, the Jsc and FF were significantly improved to 13.00 mA cm−2 and 68.74%, respectively, while Voc stayed the same, leading to an average PCE of 5.98% with a maximum efficiency of 6.16%. External quantum efficiency (EQE) curves of the devices are shown in Fig. 3b. The EQE plots of PDTffBT–TVT and PDTffBT–TVTOEt (w/o 3% DIO) based solar cells cover from 300 to 800 nm, consistent with the UV-vis absorption spectra of the blend films. The calculated Jsc values based on the EQE results are in accordance with those of the corresponding PSCs. For example, the integral current density of solar cells based on PDTffBT–TVTOEt:PC71BM (with 3% DIO) is 12.36 mA cm−2, the highest among different systems, in accordance with the value of J–V measurements (within 10% error).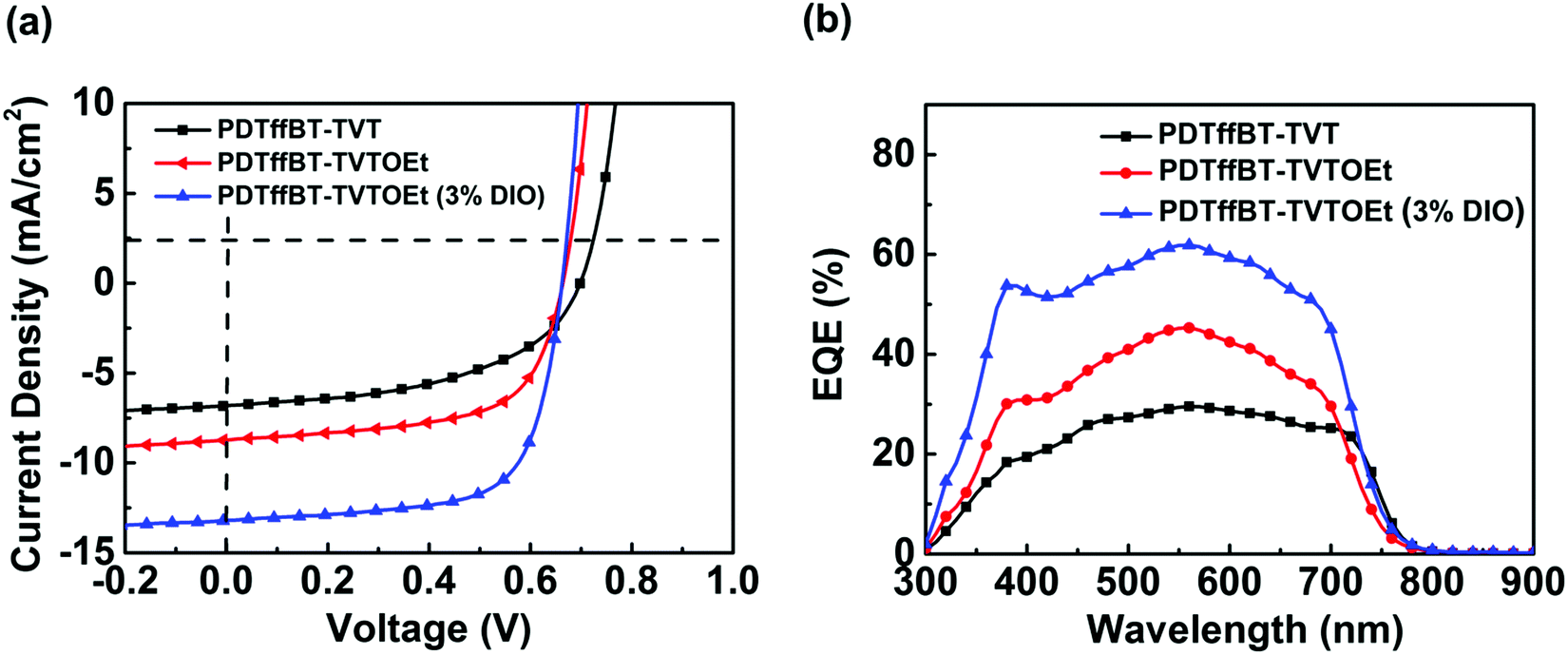 | ||
| Fig. 3 The typical J–V curves (a) and EQE plots (b) of organic solar cells with different active layers. | ||
| Polymer:PC71BM | V oc [V] | J sc [mA cm−2] | FF [%] | PCEmax (ave)a [%] | μ h [cm2 V−1 s−1] |
|---|---|---|---|---|---|
| a The values of Voc, Jsc, and FF are the average values of about 10 devices. b Integrated from EQE data. | |||||
| PDTffBT–TVT | 0.68 ± 0.02 | 6.77 ± 0.04 (6.20)b | 48.61 ± 2.71 | 2.59 (2.39)a | 3.57 × 10−3 |
| PDTffBT–TVTOEt | 0.66 ± 0.02 | 8.69 ± 0.03 (8.47)b | 60.00 ± 4.00 | 3.62 (3.56)a | 6.72 × 10−3 |
| PDTffBT–TVTOEt (3% DIO) | 0.66 ± 0.01 | 13.00 ± 0.24 (12.36)b | 68.74 ± 0.16 | 6.16 (5.98)a | 8.99 × 10−3 |
Photoluminescence of BHJ solar cells
Photoluminescence (PL) spectroscopy is employed to explore the exciton quenching of the blend films (as shown in Fig. S6, ESI†). All the blend films possessed excellent quenching efficiency higher than 80%. The blend film PDTffBT–TVT:PC71BM showed a quenching efficiency of 83%. In comparison, the blend films PDTffBT–TVTOEt:PC71BM with or without DIO demonstrated a relatively higher quenching efficiency, 84% or 93%, indicative of higher Jsc values, in accordance with the photovoltaic performances.Morphological characterization
Atomic force microscopy (AFM) was employed to gain insight into the morphology of the blend films coated by the polymers and the fullerene acceptor. As shown in Fig. 4a and d, PDTffBT–TVT:PC71BM showed a relatively smooth morphology with a mean-square surface roughness (Rq) of 3.51 nm. In comparison, the morphologies of PDTffBT–TVTOEt:PC71BM were relatively rough and the Rq of the blend film with 3% DIO reached as high as 5.99 nm. It is of great interest to observe that the rougher surface of the film resulted in higher performance of the polymer solar cells. The reason for this phenomenon may be that the rougher surface can ensure more contact areas and a stronger interaction between the active layer and the top electrodes.37 To achieve straightforward observation of the phase separation, the blend films were also studied by transmission electron microscopy (TEM). As shown in Fig. 5a, the PDTffBT–TVT:PC71BM blend film displayed homogeneous features with less miscibility of PC71BM. In comparison, the PDTffBT–TVTOEt:PC71BM blend films demonstrated clear phase separation. Moreover, with addition of DIO, the blend film exhibited entangled nanofibrils, beneficial to charge generation and transport. These results are consistent with the photovoltaic performances in that PDTffBT–TVTOEt:PC71BM based solar cells exhibit relatively high Jsc and FF. | ||
| Fig. 4 AFM topography and phase images (5 μm × 5 μm) of blend films of PDTffBT–TVT:PC71BM (a and d), PDTffBT–TVTOEt:PC71BM (b and e) and PDTffBT–TVTOEt:PC71BM (with 3% DIO) (c and f). | ||
 | ||
| Fig. 5 TEM images of the blend films of PDTffBT–TVT:PC71BM (a), PDTffBT–TVTOEt:PC71BM (b) and PDTffBT–TVTOEt:PC71BM (with 3% DIO) (c). | ||
Bulk charge transport in BHJ solar cells
It is well known that the charge transport characteristic is critical for the FF of PSCs. The space charge limit current (SCLC) method was used to estimate the bulk charge transport characteristics of blend films. Herein, hole mobilities were measured with the device structure of ITO/PEDOT:PSS/active layer/MoO3/Ag. The J1/2vs. V curves are shown in Fig. S7 (ESI†), and the calculated hole (μh) mobilities of the blend films are summarized in Table 2. The PDTffBT–TVT:PC71BM blend film demonstrated a μh of 2.39 × 10−3 cm2 V−1 s−1, while the PDTffBT–TVTOEt:PC71BM blend film exhibited an enhanced μh of 6.72 × 10−3 cm2 V−1 s−1. Furthermore, with addition of 3% DIO, the μh was further increased to 8.99 × 10−3 cm2 V−1 s−1. These results are consistent with the photovoltaic performances in that both the Jsc and FF of PDTffBT–TVTOEt:PC71BM based PSCs are higher than those of PDTffBT–TVT:PC71BM based ones.Exciton dissociation and bimolecular recombination kinetics
To gain more insight into light absorption and the exciton dissociation process, the charge generation and extraction properties were studied. Fig. 6a shows the photocurrent density (Jph) versus effective voltage (Veff) curves of the solar cells. In general, Jph is determined as Jph = JL − JD, where JL and JD are the photocurrent densities under illumination and in the dark, respectively. Veff is defined as Veff = V0 − Va, where V0 is the voltage when Jph is zero and Va is the applied bias voltage. At a large reverse voltage (i.e., Veff ≥ 2 V), Jph reaches saturation (Jsat), indicating that all of the photogenerated excitons split into free charges and reached the electrodes. Thus, the exciton dissociation and charge collection efficiency can be estimated using the Jph/Jsat ratio.38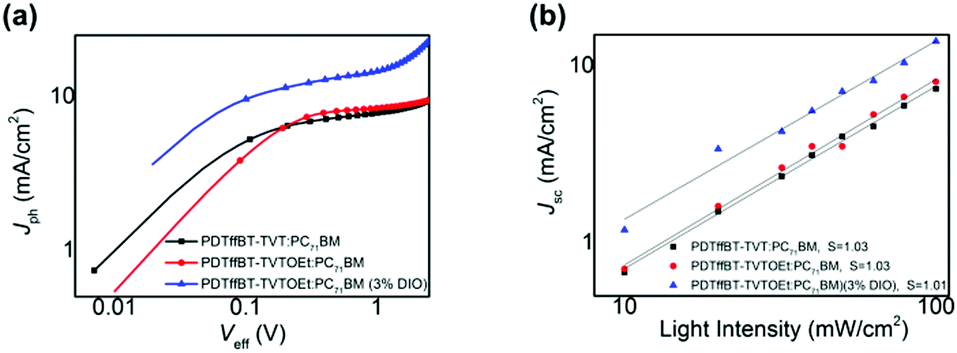 | ||
| Fig. 6 (a) Photocurrent density (Jph) versus effective voltage (Veff) characteristics. (b) Short current density (Jsc) versus light intensity for the PDTffBT–TVT–TVTOEt (3% DIO):PC71BM based solar cells. | ||
PDTffBT–TVTOEt:PC71BM blend films showed a higher ratio (85.75%) than those of PDTffBT–TVT:PC71BM (84.5%), indicative of efficient exciton dissociation at the interface of the PDTffBT–TVTOEt:PC71BM active layer. These results supported that the PDTffBT–TVTOEt-based solar cells possessed higher Jsc in comparison to PDTffBT–TVT-based ones.
We also measured photocurrent (Jsc) under different light intensities to probe the recombination kinetics. In principle, Jsc has a power-law relationship with light intensity (Jsc ∝ PSlight).39,40 In general, when the exponential factor (S) is close to 1, it would suggest weak bimolecular recombination. As shown in Fig. 6b, the S values of the PDTffBT–TVT and PDTffBT–TVTOEt based solar cells are 1.03 and 1.01, indicative of efficient sweep-out of carriers and weak bimolecular recombination in both systems.41 The slightly smaller S value of PDTffBT–TVTOEt based solar cells may indicate that they have a weaker bimolecular recombination.
Conclusions
In conclusion, we have synthesized two conjugated polymers based on TVT and TVTOEt to investigate the influence of the incorporation of S⋯O conformational locks on the charge transport properties and photovoltaic performances. The physicochemical properties of conjugated polymers were systematically characterized to understand the structure–property relationship. The photovoltaic properties of two conjugated polymers were investigated, which showed that introducing S⋯O conformational locks into the backbone can significantly improve the charge transport mobilities, leading to enhanced FF and PCE. As a result, the PDTffBT–TVTOEt:PC71BM based polymer solar cells afforded a maximum PCE of 6.16%. This contribution highlights the importance of noncovalent conformational locks to tuning the physicochemical properties, charge transport characteristics, and photovoltaic performances of conjugated polymers, which is meaningful for designing novel high performing conjugated polymers.Experiments
Materials
All chemicals were purchased from Energy Chemical and used as received. Solvents for chemical synthesis were freshly distilled prior to use. All synthetic procedures were performed under a nitrogen atmosphere. The following compounds: 5,6-difluoro-4,7-bis(5-bromo-4-(2-octyldodecyl)-2-thienyl)-2,1,3-benzothiadiazole,21 (E)-1,2-bis(5-(trimethylstannyl)thiophen-2-yl)ethane,29 and (E)-1,2-diethoxy-1,2-bis(5-(trimethylstannyl)thiophen-2-yl)ethane11 were synthesized according to the literature reported.Synthesis of polymers
Measurements and characterization
The 1H nuclear magnetic resonance (NMR) spectra were measured using a Bruker AVANCE 400 MHz NMR spectrometer with deuterated chloroform as the solvent. Tetramethylsilane (TMS) was used as an internal reference for the NMR analysis. Elemental analysis (EA) was performed on a FLASH EA 1112 Elemental Analyzer. Electrochemical cyclic voltammetry (CV) was carried out on a CHI600E electrochemical workstation with a conventional three-electrode configuration in dry acetonitrile containing 0.1 M tetrabutylammonium hexafluorophosphate (n-Bu4NPF6) as the supporting electrolyte with a scan rate of 50 mV s−1. Pt wire, glassy carbon disks, and a Ag/AgCl electrode were used as the counter, working, and reference electrodes, respectively. A ferrocene/ferrocenium redox couple was used as an external standard. The potential is located at 0.5 eV, which is assumed to have an absolute energy level of −4.44 eV under vacuum (HOMO = −Eox-1/2 − 4.44 eV. LUMO = HOMO + Eoptg). UV-vis absorption spectra were recorded using a Cary 60 UV-vis spectrophotometer at room temperature. All the thin-film samples were spin-coated on glass substrates. Gel permeation chromatography (GPC) analysis was conducted on a PL-GPC 220 system with polystyrene as a standard and 1,2,4-trichlorobenzene as an eluent at 150 °C. Thermogravimetric analysis (TGA) measurements were carried out using a Shimadzu thermogravimetric analyzer (model DTG-60) under a continuous nitrogen flow at a heating rate of 10 °C min−1. AFM measurements were performed by using a Scanning Probe Microscope-Dimension 3100 in tapping mode with all film samples spin-coated on ITO/ZnO substrates. The transmission electron microscopy (TEM) characterization was performed on a FEI Tecnai G2 F20.Fabrication of polymer solar cells
An ITO-coated glass substrate was cleaned by sequential sonication in soap DI water, DI water, acetone and isopropanol for 30 min for each step. Then it was dried at 80 °C in an oven for 1 h and treated in an ultraviolet-ozone chamber for 30 min. The ZnO precursor solution prepared from 0.5 M zinc acetate dehydrate in 0.5 M monoethanolamine and 2-methoxyethanol was spin-coated onto the ITO glass at 4500 rpm for 40 s, and annealed at 200 °C on a hot plate in air for 30 min. Subsequently, the substrate was moved to a N2 glove box. Active layer solutions (D/A ratio 1:1.2 w/w) were prepared in CB/DCB (1:1 volume ratio) with or without DIO (polymer concentration: 6 mg mL−1; 9 mg mL−1 for PDTffBT–TVT and PDTffBT–TVTOEt, respectively). In order to completely dissolve the polymers, the solution was stirred on a hot plate at 110 °C for 3 h. Before spin coating, both the polymer solution and the ITO substrate are preheated on a hot plate at 110 °C. Active layers were spin coated from the warm polymer solution on the preheated substrate in a N2 glovebox at 1500 rpm. The polymer/fullerene films were then annealed at 80 °C for 5 min, before being transferred to the vacuum chamber of a thermal evaporator inside the same glovebox. MoO3 (10 nm) and Ag (100 nm) were deposited sequentially by thermal evaporation under 10−5 Pa. The photoactive layer area of the device was 0.04 cm2.
The J–V measurements of the devices under illumination with an AM 1.5G solar simulator (100 mW cm−2) were performed on a computer-controlled Keithley 2400 Source Measure Unit in N2. The external quantum efficiency (EQE) was measured using a Solar Cell Spectral Response Measurement System QE-R3011 (Enlitech, Taiwan). The thicknesses of the BHJ blends film were measured with a Bruker Dektak XT profilometer.
Space-charge-limited current (SCLC) measurement
The hole mobility was measured through the SCLC method by using a device architecture of ITO/PEDOT:PSS/blend film/MoO3/Ag by recording the current–voltage curves and fitting the results to a space charge limited form, where the SCLC is described by:| J = 9ε0εrμ(Vappl − Vbi)2/8L3 | (1) |
| μh = slope2 − 8L3/9ε0ε | (2) |
Acknowledgements
We acknowledge the NSFC (51303180 and 21574135), Beijing Natural Science Foundation (2162043), One Hundred Talents Program of Chinese Academy of Sciences, and University of Chinese Academy of Sciences for financial support. The DFT results described in this paper were obtained on the China Scientific Computing Grid (ScGrid).Notes and references
- G. Yu, J. Gao, J. C. Hummelen, F. Wudl and A. J. Heeger, Science, 1995, 270, 1789–1791 CAS.
- B. C. Thompson and J. M. Frechet, Angew. Chem., Int. Ed. Engl., 2008, 47, 58–77 CrossRef CAS PubMed.
- J. Chen and Y. Cao, Acc. Chem. Res., 2009, 42, 1709–1718 CrossRef CAS PubMed.
- H. Zhou, L. Yang and W. You, Macromolecules, 2012, 45, 607–632 CrossRef CAS.
- X. Guo, A. Facchetti and T. J. Marks, Chem. Rev., 2014, 114, 8943–9021 CrossRef CAS PubMed.
- R. Steim, F. R. Kogler and C. J. Brabec, J. Mater. Chem., 2010, 20, 2499 RSC.
- X. Lin, Y. Yang, L. Nian, H. Su, J. Ou, Z. Yuan, F. Xie, W. Hong, D. Yu, M. Zhang, Y. Ma and X. Chen, Nano Energy, 2016, 26, 216–223 CrossRef CAS.
- N. Adhikari, D. Khatiwada, A. Dubey and Q. Qiao, J. Photonics Energy, 2015, 5, 057207 CrossRef.
- J. Zhang, Y. Zhang, J. Fang, K. Lu, Z. Wang, W. Ma and Z. Wei, J. Am. Chem. Soc., 2015, 137, 8176–8183 CrossRef CAS PubMed.
- L. Ye, S. Zhang, L. Huo, M. Zhang and J. Hou, Acc. Chem. Res., 2014, 47, 1595–1603 CrossRef CAS PubMed.
- H. Huang, Z. Chen, R. Ponce Ortiz, C. Newman, H. Usta, S. Lou, J. Youn, Y. Y. Noh, K. J. Baeg, L. X. Chen, A. Facchetti and T. J. Marks, J. Am. Chem. Soc., 2012, 134, 10966–10973 CrossRef CAS PubMed.
- H. Huang, N. Zhou, R. P. Ortiz, Z. Chen, S. Loser, S. Zhang, X. Guo, J. Casado, J. T. López Navarrete, X. Yu, A. Facchetti and T. J. Marks, Adv. Funct. Mater., 2014, 24, 2782–2793 CrossRef CAS.
- X. Guo, Q. Liao, E. F. Manley, Z. Wu, Y. Wang, W. Wang, T. Yang, Y.-E. Shin, X. Cheng, Y. Liang, L. X. Chen, K.-J. Baeg, T. J. Marks and X. Guo, Chem. Mater., 2016, 28, 2449–2460 CrossRef CAS.
- B. Xia, K. Lu, L. Yuan, J. Zhang, L. Zhu, X. Zhu, D. Deng, H. Li and Z. Wei, Polym. Chem., 2016, 7, 1323–1329 RSC.
- W. Zhang, Z. Mao, J. Huang, D. Gao and G. Yu, Macromolecules, 2016, 49, 6401–6410 CrossRef CAS.
- S. Yum, T. K. An, X. Wang, W. Lee, M. A. Uddin, Y. J. Kim, T. L. Nguyen, S. Xu, S. Hwang, C. E. Park and H. Y. Woo, Chem. Mater., 2014, 26, 2147–2154 CrossRef CAS.
- B. Fu, C.-Y. Wang, B. D. Rose, Y. Jiang, M. Chang, P.-H. Chu, Z. Yuan, C. Fuentes-Hernandez, B. Kippelen, J.-L. Brédas, D. M. Collard and E. Reichmanis, Chem. Mater., 2015, 27, 2928–2937 CrossRef CAS.
- S. C. Price, A. C. Stuart, L. Yang, H. Zhou and W. You, J. Am. Chem. Soc., 2011, 133, 4625–4631 CrossRef CAS PubMed.
- H. Zhou, L. Yang, A. C. Stuart, S. C. Price, S. Liu and W. You, Angew. Chem., Int. Ed. Engl., 2011, 50, 2995–2998 CrossRef CAS PubMed.
- A. C. Stuart, J. R. Tumbleston, H. Zhou, W. Li, S. Liu, H. Ade and W. You, J. Am. Chem. Soc., 2013, 135, 1806–1815 CrossRef CAS PubMed.
- Y. Liu, J. Zhao, Z. Li, C. Mu, W. Ma, H. Hu, K. Jiang, H. Lin, H. Ade and H. Yan, Nat. Commun., 2014, 5, 5293 CrossRef CAS PubMed.
- J. Zhao, Y. Li, G. Yang, K. Jiang, H. Lin, H. Ade, W. Ma and H. Yan, Nat. Energy, 2016, 1, 15027 CrossRef CAS.
- N. Wang, Z. Chen, W. Wei and Z. Jiang, J. Am. Chem. Soc., 2013, 135, 17060–17068 CrossRef CAS PubMed.
- Z. Chen, P. Cai, J. Chen, X. Liu, L. Zhang, L. Lan, J. Peng, Y. Ma and Y. Cao, Adv. Mater., 2014, 26, 2586–2591 CrossRef CAS PubMed.
- H. Hu, K. Jiang, G. Yang, J. Liu, Z. Li, H. Lin, Y. Liu, J. Zhao, J. Zhang, F. Huang, Y. Qu, W. Ma and H. Yan, J. Am. Chem. Soc., 2015, 137, 14149–14157 CrossRef CAS PubMed.
- J. Zhao, Y. Li, A. Hunt, J. Zhang, H. Yao, Z. Li, J. Zhang, F. Huang, H. Ade and H. Yan, Adv. Mater., 2016, 28, 1868–1873 CrossRef CAS PubMed.
- J. Ren, Y. Zhang, F. Liu, Y. Yan, M. Qiu, V. A. L. Roy, H. Zheng, M. Sun and R. Yang, RSC Adv., 2016, 6, 68049–68057 RSC.
- J. Ren, W. Chen, Y. Zhang, M. Qiu, J. Ren, T. Zhu, F. Liu, M. Sun and R. Yang, J. Mater. Chem. C, 2016, 4, 11088–11095 RSC.
- R. Kim, P. S. K. Amegadze, I. Kang, H.-J. Yun, Y.-Y. Noh, S.-K. Kwon and Y.-H. Kim, Adv. Funct. Mater., 2013, 23, 5719–5727 CrossRef CAS.
- H. Huang, J. Youn, R. Ponce Ortiz, Y. Zheng, A. Facchetti and T. Marks, Chem. Mater., 2011, 23, 2185–2200 CrossRef CAS.
- M. Leclerc, C. Roux and J. Y. Bergeron, Synth. Met., 1993, 55, 287–292 CrossRef CAS.
- H. Sirringhaus, T. Kawase, R. H. Friend, T. Shimoda, M. Inbasekaran, W. Wu and E. P. Woo, Science, 2000, 290, 2123–2126 CrossRef CAS PubMed.
- E. M. Breitung, C.-F. Shu and R. J. McMahon, J. Am. Chem. Soc., 2000, 122, 1154–1160 CrossRef CAS.
- A. Campos, N. Oxtoby, S. Galindo, R. Pfattner, J. Veciana, S. T. Bromley, C. Rovira and M. Mas-Torrent, CrystEngComm, 2016, 18, 6149–6152 RSC.
- E. F. Oliveira and F. C. Lavarda, Polymer, 2016, 99, 105–111 CrossRef CAS.
- G. R. Hutchison, M. A. Ratner and T. J. Marks, J. Am. Chem. Soc., 2005, 127, 2339–2350 CrossRef CAS PubMed.
- X. Wang, H. Wang, W. Huang and J. Yu, Org. Electron., 2014, 15, 3000–3005 CrossRef CAS.
- L. Huo, T. Liu, X. Sun, Y. Cai, A. J. Heeger and Y. Sun, Adv. Mater., 2015, 27, 2938–2944 CrossRef CAS PubMed.
- P. Schilinsky, C. Waldauf and C. J. Brabec, Appl. Phys. Lett., 2002, 81, 3885–3887 CrossRef CAS.
- G. Lu, H. Usta, C. Risko, L. Wang, A. Facchetti, M. A. Ratner and T. J. Marks, J. Am. Chem. Soc., 2008, 130, 7670–7685 CrossRef CAS PubMed.
- M. Lenes, M. Morana, C. J. Brabec and P. W. M. Blom, Adv. Funct. Mater., 2009, 19, 1106–1111 CrossRef CAS.
- Y. Lin, J. Wang, Z. G. Zhang, H. Bai, Y. Li, D. Zhu and X. Zhan, Adv. Mater., 2015, 27, 1170–1174 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6qm00296j |
| ‡ These authors contributed equally to this work. |
| This journal is © the Partner Organisations 2017 |