
Asymmetric cationic lipid based non-viral vectors for an efficient nucleic acid delivery†
Rakeshchandra R. Mekaa,
Sudhakar Godeshalaa,
Srujan Marepallyb,
Ketan Thoratbc,
Hari Krishna Reddy Rachamallaa,
Ashish Dhayanibd,
Ankita Hiwaleb,
Rajkumar Banerjeea,
Arabinda Chaudhuria and
Praveen Kumar Vemula*be
aBiomaterials Group, CSIR-Indian Institute of Chemical Technology, Hyderabad 500 007, India
bInstitute for Stem Cell Biology and Regenerative Medicine (inStem), GKVK-post, Bangalore 560065, India. E-mail: praveenv@instem.res.in
cManipal University, Manipal, India
dSASTRA University, Thirumalaisamudram, Thanjavur-613401, India
eRamalingaswami Re-Entry Fellow, Dept of Biotechnology, Govt of India, India
First published on 9th August 2016
Abstract
Cationic lipids have been extensively studied for their ability to complex with nucleic acids to condense and consequently deliver them into the cells. However, developing safe and efficient cationic lipids for delivering nucleic acids is still an unmet challenge. Prior structure-activity investigations led to the path to understanding the lipid structure and its transfection efficiency. The trend in the transfection profiles of linker-based lipids is different from linker-less lipids. Influence of unsaturation in the hydrophobic chains has been investigated in linker-based lipids. However, in linker-less lipids, it remains unexplored. Herein, we demonstrate that the designed cationic lipid Lipid S-U with an asymmetric hydrophobic core having one stearyl (18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0) and one oleyl chain (18:1) showed superior transfection efficiency compared to its symmetric counterparts, Lipid S-S (hydrophobic core comprising of two stearyl chains (18:0)), and Lipid U-U (two oleyl chains (18:1)), in vitro. Mechanistic studies involving membrane fusogenicity with FACS revealed that liposomes of Lipid S-U have higher fusogenicity (89%) with B16F10 cell membrane than saturated Lipid S-S (66%) and unsaturated Lipid U-U (70%). Endosomal escape studies with confocal microscopy in HEK 293 cells revealed that lipoplexes of Lipid S-U had a higher endosomal escape and released the genetic payload in cytoplasm more efficiently than saturated Lipid S-S and unsaturated Lipid U-U. These cumulative findings support the notion that higher cellular uptake and endosomal escape resulting from fusogenic liposomes of Lipid S-U play a pivotal role in the higher transfection efficiency of asymmetric Lipid S-U.
:0) and one oleyl chain (18:1) showed superior transfection efficiency compared to its symmetric counterparts, Lipid S-S (hydrophobic core comprising of two stearyl chains (18:0)), and Lipid U-U (two oleyl chains (18:1)), in vitro. Mechanistic studies involving membrane fusogenicity with FACS revealed that liposomes of Lipid S-U have higher fusogenicity (89%) with B16F10 cell membrane than saturated Lipid S-S (66%) and unsaturated Lipid U-U (70%). Endosomal escape studies with confocal microscopy in HEK 293 cells revealed that lipoplexes of Lipid S-U had a higher endosomal escape and released the genetic payload in cytoplasm more efficiently than saturated Lipid S-S and unsaturated Lipid U-U. These cumulative findings support the notion that higher cellular uptake and endosomal escape resulting from fusogenic liposomes of Lipid S-U play a pivotal role in the higher transfection efficiency of asymmetric Lipid S-U.
1. Introduction
Translation of gene therapy from concept to clinic critically depends on the availability of safe and efficient gene transfer reagents. Gene transfer reagents are broadly classified as viral and non-viral vectors. Although viral vectors are effective in delivering nucleic acids under in vivo conditions, their applications are limited by biosafety-related concerns including immunogenic response and insertional mutagenesis through random integration into the host genome.1 Contrastingly, non-viral vectors such as cationic liposomes,2–6 cationic polymers7–9 and dendrimers10,11 hold therapeutic promise given their promising biosafety profiles, lower toxicity and moderate tissue specificity. However, lower transfection efficiency of these vectors limits their clinical applications. Among the non-viral vectors, cationic liposomes represent the most intensively studied and employed class of vectors for delivery of nucleic acids.3Cationic amphiphiles are made of positively charged headgroup (usually tertiary or quaternary ammonium groups or polyamines) and a hydrophobic domain, usually consisting of either two long aliphatic hydrocarbon chains or a cholesterol skeleton.12 Their transfection efficiency depends on multiple parameters such as length of hydrophobic alkyl chain, nature of hydrophilic headgroup, nature of the linker functionalities and their orientation.13–18 Several attempts have been made to understand the relation between the lipid structure and its transfection efficiency.14–19 Transfection profiles of cationic amphiphiles are highly sensitive to even subtle changes such as the orientation of the linker functionalities between hydrophilic and hydrophobic domains, and asymmetry in the hydrophobic region.20,21 Koynova et al., demonstrated that the asymmetry with mono-unsaturation in the hydrophobic core of oleoyldecanoyl-ethylphosphatidylcholine (C18:1/C10-EPC) imparted 50-fold superior transfection efficiency compared to that of its structurally similar saturated symmetric counterpart stearoyldecanoyl-ethylphosphatidylcholine (C18:0/C10-EPC).21 Asymmetry in the hydrophobic core strongly influenced the biophysical properties of the liposomes by forming a pronounced non-lamellar phase which, in turn, played a dominant role in imparting high membrane fusogenicity and thereby enhanced transfection properties to C18:1/C10-EPC.21 Further, structure-activity investigations by Koynova et al., and Nantz et al., demonstrated the significance of hydrophobic domain asymmetry in modulating the gene transfer efficacies of synthetic cationic amphiphiles.22 More recently, Voshavar et al., have demonstrated the cationic lipids synthesized with naturally occurring fatty acyl chains from food coconut grade oil delivered genes >4-folds superior to their lauryl counterpart under in vivo conditions.18 These asymmetric cationic amphiphiles have ester linker functionality connecting between hydrophilic headgroup and hydrophobic tail, and studies are confined to investigating their role in cell membrane fusogenicity. In addition, unsaturation could improve the membrane fusogenicity of the liposomes. It is well documented that unsaturated helper lipids such as dioleoylphosphotidylcholine (DOPC) and dioleoylphosphotidyl ethanolamine (DOPE) could improve the transfection efficiency of the symmetric lipids, impart the membrane fusogenicity.23
Interestingly, the trend in the transfection profiles of linker-based lipids is different from linker-less lipids. Influence of unsaturation has been investigated in linker-based lipids; however, in non-linker based lipids it remains unexplored. The effect of degree of unsaturation in determining the transfection efficiency of the non-linker based cationic liposomes and their role in intra-cellular events such as endosomal escape is still elusive. To investigate this effect, being inspired by our previous work,24 we have designed and synthesized three non-linker based cationic lipids (Lipid S-S, Lipid S-U and Lipid U-U, Fig. 1A). Alkyl groups, greater than fourteen carbon chains are found to be safer than twelve carbon alkyl chains or lesser in biological applications. Hence, C18 alkyl chains were taken in these lipids to avoid cytotoxicity in interpreting the results. These lipids have the same polar headgroup and different degree of unsaturation in hydrophobic tails. Lipid S-S hydrophobic core comprising of two stearyl chains (18:0), Lipid S-U has an asymmetric hydrophobic core having one stearyl (18:0) and one oleyl chain (18:1), Lipid U-U has two oleyl chains (18:1). Gene transfection efficiency of these three lipids has been investigated in four different mammalian cells including CHO (Chinese hamster ovary), B16F10 (melanoma cells), MDA-MB 231 (triple negative breast cancer cells), SKOV3 (ovarian cancer cells), in vitro. Transfection experiments suggest that liposome of Lipid S-U with an asymmetric hydrophobic core with mono-unsaturation has superior transfection profiles than its symmetric counterparts, saturated Lipid S-S and unsaturated Lipid U-U. The lipid/pDNA complexes (lipoplexes) of three lipids were found to be of similar size and morphology thereby ruling out any major role of different lipoplex size and shape behind variations in their gene delivery profiles. Findings in the confocal microscopic experiments in representative HEK 293 cells using lipoplexes containing green fluorescent protein encoded plasmid pDNA revealed a higher cellular uptake of asymmetric Lipid S-U complexed pDNA compared to pDNA complexes of symmetric lipids Lipid S-S and Lipid U-U. Endosomal escape study with rhodamine labeled lysotrackers and FITC labeled siRNA revealed that liposomes of asymmetric Lipid S-U escaped through early endosomes more efficiently than liposomes of symmetric Lipid S-S and Lipid U-U. Collectively, the present findings demonstrate that mono-unsaturation; imparting asymmetry in the hydrophobic core of non-linker based cationic amphiphiles can influence membrane fusogenicity, cellular uptake, endosomal escape and consequently their gene delivery efficiency.
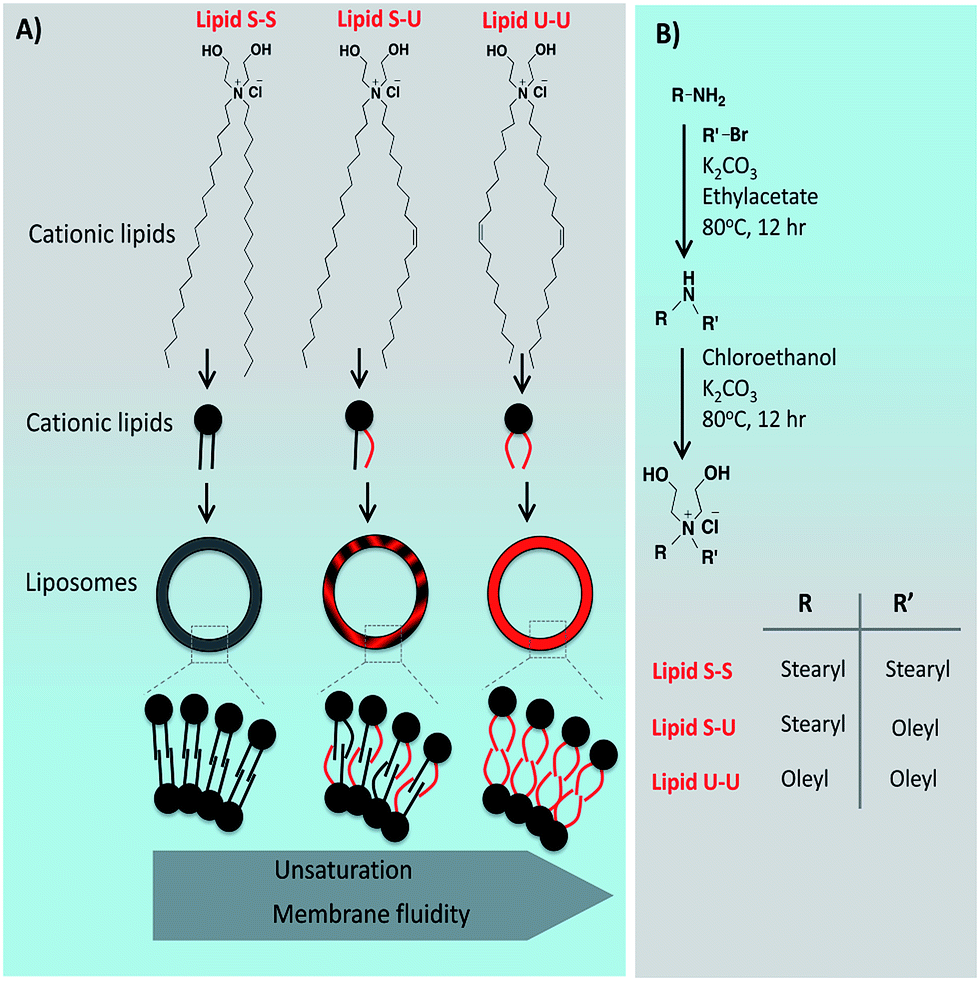 | ||
| Fig. 1 (A) Chemical structures of symmetric and asymmetric cationic lipids, and self-assembly of lipids to form cationic liposomes. (B) Synthesis of cationic lipids. | ||
2. Experimental
2.1 General procedures and reagents
Mass spectral data were acquired by using a commercial LCQ ion trap mass spectrometer (ThermoFinnigan, SanJose, CA, USA) equipped with an ESI source or micromass Quattro LC triple quadrupole mass spectrometer for ESI analysis. 1H-NMR spectra were recorded on a Varian FT 200 MHz or AV 300 MHz NMR spectrometer. Column chromatography was performed with silica gel (Acme Synthetic Chemicals, India, 60–120 mesh). The p-CMV-SPORT-β-gal plasmid was a generous gift from Dr Nalam Madhusudhana Rao of Centre for Cellular and Molecular Biology, Hyderabad, India. All the reagents were purchased from Sigma-Aldrich, St. Louis, USA unless otherwise stated. NP-40, antibiotics and agarose were procured from Hi-media, India. B16F10, MDMA-231, CHO and SKOV3 cells were procured from the National Centre for Cell Sciences (NCCS), Pune, India. Cells were grown at 37 °C either in Dulbecco's modified Eagle's medium (DMEM) with 10% FBS in a humidified atmosphere containing 5% CO2/95% air.2.2 Synthesis
Detailed procedures for synthesizing the cationic amphiphiles are given below. Structures of all synthetic intermediates were confirmed by 1H NMR and ESI-MS. The purity of the lipids was confirmed by reverse phase analytical HPLC. Synthesis of cationic lipids was shown in Fig. 1B.:95 methanol:chloroform, v/v).![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) 3–CH2–C15H31), 1.291 (m, 64H, (CH2–(C2)16–CH3))2, 2.55 (t, 4H, C2–NH). ESI-MS: calcd 523.786 (for C31H62NO5), found 523 (M+).2)–CH3), 2.0 (m, 4H, H2C–HC
3–CH2–C15H31), 1.291 (m, 64H, (CH2–(C2)16–CH3))2, 2.55 (t, 4H, C2–NH). ESI-MS: calcd 523.786 (for C31H62NO5), found 523 (M+).2)–CH3), 2.0 (m, 4H, H2C–HC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH–CH2), 3 (t, 4H, CH2–NH), 5.4 (t, 2H, HCCH). ESI-MS: m/z 520.5 [M + H]+.2)–CH3), 2.0 (m, 8H, H2C–HCCH–CH2), 3 (t, 4H, CH2–NH), 5.4 (t, 4H, HCCH). ESI-MS: m/z 518.5 [M + H]+.:95 methanol:chloroform, v/v).3–(CH2)17–N), 1.2 (m, 64H, (C2)15–CH3)2, 1.7 (m, 4H, N+(CH2–C2–CH2–)2), 3.5 (m, 4H, N+(C2–CH2–CH2–)2), 3.8 (m, 4H, (HOCH2–C2–)2), 4.2 (m, 4H, (HOC2–CH2–)2). ESI-MS: m/z 610 [M + H]+.3–(CH2)17–N), 1.2 (m, 56H, (C2)15–CH3)2, 1.7 (m, 4H, N+(CH2–C2–CH2–)2), 2.0 (m, 4H, 2C–HCCH–C2), 3.5 (m, 4H, N+(C2–CH2–CH2–)2), 3.8 (m, 4H, (HOCH2–C2–)2), 4.2 (m, 4H, (HOC2–CH2–)2), 5.4 (t, 2H, CC). ESI-MS: m/z 608 [M + H]+ ESI-MS m/z: (for C31H62NO5), 608 (M+).3–(CH2)17–N), 1.2 (m, 48H, (C2)15–CH3)2, 1.7 (m, 4H, N+(CH2–C2–CH2–)2), 2.0 (m, 8H, (2C–HCCH–C2))2, 3.5 (m, 4H, N+(C2–CH2–CH2–)2), 3.8 (m, 4H, (HOCH2–C2–)2), 4.2 (m, 4H, (HOC2–CH2–)2), 5.4 (t, 4H, (CC))2. ESI-MS: m/z 606 [M + H]+.
CH–CH2), 3 (t, 4H, CH2–NH), 5.4 (t, 2H, HCCH). ESI-MS: m/z 520.5 [M + H]+.2)–CH3), 2.0 (m, 8H, H2C–HCCH–CH2), 3 (t, 4H, CH2–NH), 5.4 (t, 4H, HCCH). ESI-MS: m/z 518.5 [M + H]+.:95 methanol:chloroform, v/v).3–(CH2)17–N), 1.2 (m, 64H, (C2)15–CH3)2, 1.7 (m, 4H, N+(CH2–C2–CH2–)2), 3.5 (m, 4H, N+(C2–CH2–CH2–)2), 3.8 (m, 4H, (HOCH2–C2–)2), 4.2 (m, 4H, (HOC2–CH2–)2). ESI-MS: m/z 610 [M + H]+.3–(CH2)17–N), 1.2 (m, 56H, (C2)15–CH3)2, 1.7 (m, 4H, N+(CH2–C2–CH2–)2), 2.0 (m, 4H, 2C–HCCH–C2), 3.5 (m, 4H, N+(C2–CH2–CH2–)2), 3.8 (m, 4H, (HOCH2–C2–)2), 4.2 (m, 4H, (HOC2–CH2–)2), 5.4 (t, 2H, CC). ESI-MS: m/z 608 [M + H]+ ESI-MS m/z: (for C31H62NO5), 608 (M+).3–(CH2)17–N), 1.2 (m, 48H, (C2)15–CH3)2, 1.7 (m, 4H, N+(CH2–C2–CH2–)2), 2.0 (m, 8H, (2C–HCCH–C2))2, 3.5 (m, 4H, N+(C2–CH2–CH2–)2), 3.8 (m, 4H, (HOCH2–C2–)2), 4.2 (m, 4H, (HOC2–CH2–)2), 5.4 (t, 4H, (CC))2. ESI-MS: m/z 606 [M + H]+.For spectral characterization, see ESI Fig. S1–S18.†
2.3 Preparation of liposomes and plasmid pDNA
1 mM liposomes were prepared with 1:1 mole ratios of lipid and cholesterol. The cationic lipids and cholesterol in the appropriate mole ratios were dissolved in chloroform (500 μl) in a glass vial. The solvent was removed with a thin flow of moisture-free nitrogen gas and the dried lipid film was kept for drying under high vacuum for 6 h. 1 ml of sterile deionized water was added to the vacuum dried lipid films and the mixtures were allowed to swell overnight. The vials were then vortexed for 2–3 minutes at room temperature to produce multilamellar vesicles (MLVs). MLVs were then sonicated initially in a water bath followed by an ice bath until clarity using a Branson 450 sonifier at 100% duty cycle and 25 W output power to produce small unilamellar vesicles (SUVs). p-CMV-SPORT-β-gal plasmid was amplified in DH5α-strain of Escherichia coli, isolated by alkaline lysis procedure and finally purified by PEG-8000 precipitation as described previously.25 The purity of plasmid was checked by A260/A280 ratio (around 1.9) and 1% agarose gel electrophoresis.
2.4 pDNA binding assay
The pDNA binding ability of the lipids were assessed by their gel retardation assay on a 1% agarose gel (pre-stained with ethidium bromide) across the varying lipid:pDNA charge ratios of 8:1 to 1:1. pCMV-β-gal (0.30 μg) was complexed with the varying amount of cationic lipids in a total volume of 30 μl in HEPES buffer (pH 7.4) and incubated at room temperature for 20–25 minutes. 4 μl of 6× loading buffer (0.25% bromophenol blue in 40% (w/v) sucrose with sterile H2O) was added to it and from the resulting solution 30 μl was loaded on each well. The samples were electrophoresed at 80 V for 45 minutes, and the pDNA bands were visualized in the Gel documentation unit.
2.5 DNAse I sensitivity assay
Briefly, in a typical assay, 3 nM of pCMV-β-gal (1 μg) was complexed with the varying amount of cationic Lipids S-S, S-U and U-U (using indicated lipid:pDNA charge ratios) in a total volume of 30 μl in HEPES buffer (pH 7.4) and incubated at room temperature for 30 min on a rotary shaker. Subsequently, the complexes were treated with 10 μl DNAse I (at a final concentration of 1 μg ml−1 or 10 ng/3 nmol of pDNA) in the presence of 20 mM MgCl2 and incubated for 20 min at 37 °C. The reactions were then halted by adding EDTA (to a final concentration of 50 mM) and incubated at 60 °C for 10 min in a water bath. The aqueous layer was washed with 50 μl of phenol:chloroform (1:1 v/v) and centrifuged at 10000g for 5 min. The aqueous supernatants were separated, loaded (20 μl for cationic Lipids S-S, S-U and U-U on a 1% agarose gel (pre-stained with ethidium bromide) and electrophoresed at 80 V for 45 h. DNAse-I treated and untreated naked pDNA was also included in the same experiment. The binding was visualized after 45 min in the Gel documentation unit.
2.6 Zeta potential (ξ) and size measurements
The sizes and the zeta potentials (surface charges) of neat liposomes and lipoplexes with varying charge ratios (8:1–1:1) were measured by photon correlation spectroscopy and electrophoretic mobility on a Zeta sizer 3000HSA (Malvern UK). The sizes and potentials of liposomes were measured in deionised water with a sample refractive index of 1.59 and a viscosity of 0.89. Liposomes of Lipids S-S, S-U and U-U were complexed with pDNA in plain DMEM for size and potential measurements of lipoplexes. The system was validated by using the 200 nm + 5 nm polystyrene polymer (Duke Scientific Corps. Palo Alto, CA). The diameters of liposomes and lipoplexes were calculated by using the automatic mode. The zeta potential was measured using the following parameters: viscosity, 0.89 cP; dielectric constant, 79; temperature, 25 °C; F (Ka), 1.50 (Smoluchowski); maximum voltage of the current, V. Using DTS0050 standard from Malvern, UK validated the system. All the size measurements were done 10 times in triplicate with the zero field correction and values represented as the average of triplicate measurements. The potentials were measured 10 times and represented as their average values as calculated by using the Smoluchowski approximation.
2.7 Transfection assay
Cells were seeded at a density of 15000 cells (SKOV3, CHO, MDA-MB-231, B16F10) per well in a 96-well plate 12–18 h before the transfection. 0.3 μg of plasmid pDNA was complexed with varying amounts of lipids (0.9–7.2 nmol) in plain DMEM/MEM medium (total volume made up to 100 μl) for 30 minutes. The lipid:pDNA (+/−) charge ratios were from 8:1 to 1:1 over these ranges of the lipids. The complexes were then added to the cells. After 3 h of incubation, DMEM was removed, 10% complete medium was added to the cells. The reporter gene activity was estimated between 36 and 48 h. The cells were washed with PBS (2 × 100 μl) and lysed with 50 μl lysis buffer [0.25 M Tris–HCl pH 8.0, 0.5% NP40]. Care was taken to ensure complete lysis. The β-galactosidase activity per well was estimated by adding 50 μl of 2×-substrate solution [1.33 mg ml−1 of ONPG, 0.2 M sodium phosphate (pH 7.3) and 2 mM magnesium chloride] to the lysate in a 96-well plate. Absorption at 405 nm was converted to β-galactosidase units using a calibration curve constructed with a pure commercial β-galactosidase enzyme. The values of β-galactosidase units in triplicate experiments assayed on the same day varied by less than 10%. The transfection experiment carried in duplicate, and the transfection efficiency values shown are the average of triplicate experiments performed on the same day. The day-to-day variation in average transfection efficiency was found to be within 2-fold. The transfection profiles obtained on different days were identical. The transfection experiments repeated 3 times.
2.8 Cytotoxicity assay
The cytotoxicities of DODEAC analogs were evaluated in representative CHO (Chinese hamster ovary) cells across the lipid:pDNA charge ratios of 8:1–1:1 using MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) based reduction assay as described earlier.25 The cytotoxicity assay was performed in 96-well plates by maintaining the same ratio of number of cells to the amount of cationic lipid, as used in the previously described transfection experiments. Briefly, 4 h after the addition of lipoplexes, MTT (5 mg ml−1 in PBS) was added to cells and incubated for 4 h at 37 °C. Results were expressed as percent viability = [A540 (treated cells)-background/A540 (untreated cells)-background] × 100. Assay was performed in triplicate manner and repeated for three times.
2.9 Cellular uptake and expression studies by epifluorescence microscopy
For fluorescence microscopy experiments, 10000 cells were seeded in each well of a 96-well plate (Corning Inc., Corning, NY) 12 h in 500 μl of growth medium such that the well became 30–50% confluent at the time of transfection. Rhodamine-PE labeled Lipids S-S, S-U and U-U were complexed with pCMV-SPORT-β-gal (0.3 μg per well) at 2:1 lipid:pDNA charge ratio in a total volume of 100 μl DMEM for 15–20 min. The complexes were then added to the cells. After 4 h incubation, cells were washed with PBS (2 × 100 μl) and fixed with 3.8% paraformaldehyde in PBS at room temperature for 10 min. The red fluorescent cells were detected under an inverted fluorescence microscope (Nikon, Japan).
For a5GFP pDNA expression experiment, 4 × 104 per well cells were seeded in 24-well plates (Corning Inc., Corning, NY) 12 h in 500 ml of growth medium such that the well became 30 × 1050% confluent at the time of transfection. Lipids S-S, S-U and U-U were complexed with GFP expressing pDNA (pα5GFP, 0.9 mg per well) at 2:1 lipid:DNA charge ratio in plain DMEM (total volume made up to 100 ml) for 15–20 min. The complexes were then diluted with 300 ml DMEM and added to the cells. After 4 h of incubation, DMEM was removed and cells were supplemented with complete medium. The cells were allowed for 24 h incubation. Cells were washed with PBS (100 ml) and fixed with 3.8% paraformaldehyde in PBS at room temperature for 10 min. The green fluorescent cells expressing GFP were detected under an inverted fluorescence microscope (Nikon, Japan).
2.10 Cellular uptake of liposomes by FACS analysis
Approximately 300000 B16F10 cells were seeded per well in a 6 well plate (Corning Inc., Corning, NY) and incubated for 12 h in 2 ml of growth medium prior to the experiment. The seeded cells were then transfected with Rhodamine-PE labeled liposomes made of Lipids S-S, S-U and U-U, in serum free medium. After 6 h the cells were tripsinised, centrifuged, and washed with ice-cold PBS at least three times before analysis using a BD FACS Caliber flow cytometer. The cells were gated using forward versus side scatter to exclude debris and dead cells before analyzing in FACS with 10000 cell counts. The data were analyzed with BD Cell Quest Pro software.
2.11 Cellular uptake study with lysotracker
HEK 293 cells were seeded on a cover slip placed into a six well plate 12 h prior to transfection. The seeded cells were then transfected with Rhodamine-PE labeled liposomes made of Lipids S-S, S-U and U-U, in serum free medium. After 2 h, media was removed and the cells were treated with 50 nM green lysotracker (Molecular Probes, Invitrogen) solution in serum free medium. After 1 h, media was removed; the cells were washed twice with PBS, fixed with 4% para-formaldehyde in PBS for 20 min and permeabilized with 0.1% TritonX-100 in PBS for 5 min. The cover slip was finally mounted on a slide using mounting medium (VectaShield) and imaged under confocal microscope (Olympus, FV1000).3. Results and discussion
3.1 Physiochemical characterization
Self-assembly of cationic liposomes produced unilamellar vesicles that were characterized using the transmission electron microscope (TEM) and dynamic light scattering (DLS). TEM images of liposomal formulations confirmed the presence of spherical unilamellar vesicles (Fig. 2). DLS data suggests that all liposomal formulations showed hydrodynamic diameter in the range of 110 to 140 nm (ESI Table TS1†), while liposome–pDNA complexes (lipoplexes) showed increasing trend from 170 nm to 500 nm with changing lipid/pDNA charge ratio from 8:1 to 1:1 (ESI Table TS2†). As expected, zeta potential was decreased with reducing the fraction of cationic lipids from 8:1 to 1:1 lipoplexes (ESI Table TS2†). Interestingly, the ratio of 2:1 which showed maximum transfection efficiency for all formulations showed size in the range of 300 to 370 nm and zeta potential of +2 to +7 meV, suggesting the possibility that efficient transfection requires only slight positive charge instead of high positive charge.
 | ||
| Fig. 2 Transmission electron microscopic images of liposomes (scale 0.2 μm). | ||
Gel retardation assay was employed to study electrostatic complexation of pDNA with cationic lipids through varying lipid:pDNA ratio from 0.5:1 to 8:1. Results from gel retardation assay showed that pDNA was completely complexed with the liposomes from lipid/pDNA charge ratios, 2:1, 4:1 and 8:1, whereas at ratios 1:1 and 0.5:1 liposomes are inadequate to complex entire pDNA (Fig. 3). Although we have seen slight DNA in the well at charge ratio 2:1 (white arrows in Fig. 3), interestingly, at charge ratios 4:1 and 8:1, we did not observe any staining. This is not surprising, as often, a tight complex of liposome/pDNA prevents the access to ethidium bromide to intercalate with DNA, and thus signal is not seen. When assayed for protection from DNase I mediated degradation, at charge ratio 8:1 from all three liposomes protected pDNA from the enzyme, and lower charge ratios of these three lipids have decreased ability to protect the pDNA from the degradation (ESI Fig. S19†).
 | ||
| Fig. 3 Plasmid DNA binding assay symmetric and asymmetric lipids with varying lipid/DNA charge ratio. White arrow indicates the weak signal from complexed/condensed pDNA. | ||
3.2 Cytotoxicity and transfection efficiency
Cytotoxic effect of lipid–pDNA complexes was studied with B16F10 and CHO as representative cell lines using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT)-based cell viability assay at varying charge ratios of lipid/pDNA. The most effective charge ratio of lipid/pDNA (2:1) showed cell viability of ∼85–95% in both cell lines (ESI Fig. S20 and S21†). Given the negligible cytotoxic profile of three lipids could be used potential transfection agents.
Transfection efficiency of lipid formulations was evaluated in vitro in four different cell lines namely SKOV3 (ovarian cancer), CHO (Chinese hamster ovary cells), MDA-MB 231 (human breast cancer) and B16F10 (murine melanoma) varying lipid/pDNA charge ratio from 8:1 to 1:1 using β-galactosidase expressing p-CMV-SPORT-β-gal as reporter plasmid. In all four-cell lines, the asymmetric Lipid S-U showed maximum transfection efficiency with a lipid/pDNA charge ratio of 2:1 (Fig. 4). The observed gene delivery efficiency of Lipid S-U (with 2:1 charge ratio) was either equal or moderately higher than commercially available transfection reagent, LipofectAmine2000 in all four-cell lines (Fig. 5).
 | ||
| Fig. 4 Transfection efficiency of lipid formulations evaluated in SKOV3, CHO, MDA-MB 231 and B16F10 cells with varying lipid/DNA charge ratio from 8:1 to 1:1 using β-galactosidase expressing p-CMV-SPORT-β-gal as a reporter plasmid. | ||
 | ||
| Fig. 5 (A) Transfection efficiency of lipid formulations at lipid/pDNA charge ratio of 2:1 compared to commercially available transfection agent lipofectamine. (B) Epifluorescence microscopic images of representative SKOV3 cells transfected with lipoplexes of Lipids S-S, S-U and U-U with GPF expressing plasmid pα5GFP keeping the lipid/DNA charge ratio constant at 2:1; after 24 h of transfection (scale bar: 50 μm). Inset shows the quantification of total fluorescence. | ||
We further studied cell transfection ability of lipid/pDNA complexes by transfecting SKOV3 cells with Lipid S-S, Lipid S-U and Lipid U-U formulations complexed with GPF-expressing plasmid pα5GFP keeping the lipid/pDNA charge ratio constant at 2:1 followed by evaluation using epifluorescence imaging. Fluorescence images in Fig. 5B suggest that although the number of GFP-expressing cells are comparable, total GFP expression is higher for Lipid S-U compared to Lipid S-S and Lipid U-U (inset of Fig. 5B). These results are consistent with the previous results that cationic amphiphiles with hydrophobic chain asymmetry have superior transfection profiles than their symmetric analogs,22 the transfection efficiencies of Lipid S-U were found to be higher than those of their symmetric saturated, Lipid S-S and unsaturated Lipid U-U, respectively (Fig. 4 and 5). These findings convincingly demonstrate that asymmetry imparting mono-unsaturation in the hydrophobic domain is sufficient than their symmetric di-unsaturation.
3.3 Cellular uptake of lipoplexes
Lipid carriers were used for enhanced delivery of nucleic acids due to their unique ability to fuse with the cell membrane. Therefore, we studied the cell internalization of lipoplexes in a detailed manner using Rho-PE labeled lipoplexes of lipid formulations in B16F10 cell line. Quantification has been done using Fluorescence Activated Cell Sorter (FACS) and imaging epifluorescence microscopy. Asymmetric Lipid S-U showed higher cell fusogenicity with ∼89% fluorescence positive cells than their symmetric counterparts, Lipid S-S (66%) and Lipid U-U (70%) which was confirmed qualitatively using epifluorescence images (Fig. 6). Thus, the cellular uptake likely to play a dominant role in determining the transfection profiles of the cationic amphiphiles. These results were consistent with the previous reports by Koynova et al. that liposomes of glycerol based asymmetric lipids exhibit more membrane fusogenicities due to their nonlamellar structures.21 | ||
| Fig. 6 (A) Schematic presentation of dye-labeled liposome fusion with cell membrane to quantify the cell fusogenicity of liposomes. Cell fusogenicity of Rho-PE labeled (B) Lipid S-S, (C) Lipid S-U and (D) Lipid U-U in B16F10 cells, evaluated using FACS and epifluorescence microscopy (inset, scale bar = 50 μm). | ||
3.4 Endosomal escape and cellular localization
Transfection efficiency of lipoplexes critically depends on two cellular events: firstly, their ability to internalize into the intracellular compartments such as endosomes. Secondly, the endosomal escape of lipoplexes into cytoplasm before lysosomal degradation. Cellular uptake experiments suggest that these lipids can internalize into cells efficiently, and asymmetric Lipid S-U has higher internalizing ability than its counter symmetric lipids (Fig. 6). To gain insights into their endosomal escape, we performed transfection experiment using FITC labeled siRNA and with liposomes of Lipids S-S, S-U and U-U. FITC tagged siRNA was trailed using lysotracker red for pre-lysosomal escape.Confocal images in HEK 293 cells demonstrated that the superior part of lipoplexes of Lipids S-S (35.7 ± 3.1%) and U-U (39.7 ± 2.1%) were co-localized in lysosomal compartment. On the contrary, only a few of lipoplexes of Lipid S-U (21.0 ± 3.4%) were found in the lysosomes. Arrows in Fig. 7 indicate a greater degree of siRNA delivery into the cytoplasm with Lipid S-U when compared with Lipids S-S and U-U (Fig. 7). Further, we confirmed the endosomal escape results with N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl)-1,2-dihexadecanoyl-sn-glycero-3 phosphoethanolamine (NBD-PE) labeled liposomes evident from Fig. S22.† These results suggest that the difference in the endosomal escaping ability of lipids could be influencing their transfection efficiency.
 | ||
| Fig. 7 Confocal laser scanning microscope images for endosomal escape of lipoplexes with siRNA tagged with FITC (green) and lysosomes trailed with lysotracker red (arrow points localization in the nucleus) in HEK 293 cells (see ESI Fig. S22† for more images). | ||
Cationic liposome mediated gene transfer requires: (a) cellular uptake of the lipid/pDNA complex (lipoplex), (b) release of pDNA from the resulting endosomes into the cell cytoplasm, and (c) nuclear transport of the released pDNA followed by transcription and gene expression. Improved membrane fusogenicity of cationic liposomes is critical for enhanced cellular uptake (step-a) and efficient endosomal escape (step-b) of the cationic liposome associated pDNA. Typically, membrane fusogenic ability of given liposome determines the transfection efficiency. Given that asymmetric Lipid S-U exhibited enhanced transfection efficiency than symmetric counterparts Lipid S-S and Lipid U-U, we examined the membrane fusogenic properties of all three liposomes independently.
4. Conclusions
In the present investigation, toward addressing the influence of asymmetry in the hydrophobic region and degree of unsaturation in determining the transfection efficiency of cationic amphiphiles, we designed and synthesized three non-linker based cationic lipids, bearing the same polar head group and varying the unsaturation in hydrophobic chains. Mono-unsaturated, asymmetric Lipid S-U showed higher transfection efficiency to deliver reporter genes into multiple cultured mammalian cells including CHO, B16F10, MDAMB-231, and SKOV3, than symmetric, saturated Lipid S-S and unsaturated Lipid U-U. Epifluorescence microscopic studies and FACS studies using rhodamine labeled liposomes demonstrated a higher cellular uptake of liposomes of Lipid S-U compared to liposomes of Lipid S-S and Lipid U-U. Cumulative results suggest that liposomes of asymmetric Lipid S-U have higher membrane fusogenicity than its symmetric counterparts, and lipoplexes of asymmetric Lipid S-U have higher endosomal escape ability than its symmetric counterparts. Thus, Lipid S-U lipid exhibited enhanced transfection ability than symmetric Lipid S-S and Lipid U-U. Collectively, the present findings further enriched our understanding of the lipid asymmetry and its superior influence on the cellular events including plasma membrane fusion and endosomal escape consequently determining the gene transfer efficacies over unsaturation. Thus, we will explore the possibility of using this lipid for in vivo gene delivery in future research.Acknowledgements
GS and RHR thank the Council of Scientific and Industrial Research, India, for the doctoral research fellowship. AD thanks the Indian University Grant Commission for Junior Research Fellowship. PKV thanks Ramalingaswami Re-Entry fellowship, Department of Biotechnology, India. PKV thanks Dept. of Science and Technology for funding support (INT/SWISS/SNSFP-51/2015). MS thanks SERB, Department of Science &Technology, New Delhi for the financial assistance under Fast Track Scheme for Young Scientist (SB/FT/CS-198/2013). We thank Central Imaging Facility at inStem/NCBS, Bangalore and Renu for helping in TEM imaging.References
- E. Check, Nature, 2003, 423, 573–574 CAS
.
- R. Srinivas, P. P. Karmali, D. Pramanik, A. Garu, Y. V. Mahidhar, B. K. Majeti, S. Ramakrishna, G. Srinivas and A. Chaudhuri, J. Med. Chem., 2010, 53, 1387–1391 CrossRef CAS PubMed
- D. Pezzoli, A. Kajaste-Rudnitski, R. Chiesa and G. Candiani, Methods Mol. Biol., 2013, 1025, 269–279 CAS
- B. S. Reddy and R. Banerjee, Angew. Chem., Int. Ed. Engl., 2005, 44, 6723–6727 CrossRef CAS PubMed
- V. Gopal, J. Xavier, M. Z. Kamal, S. Govindarajan, M. Takafuji, S. Soga, T. Ueno, H. Ihara and N. M. Rao, Bioconjugate Chem., 2011, 22, 2244–2254 CrossRef CAS PubMed
- S. Bhattacharya and A. Bajaj, Chem. Commun., 2009, 4632–4656 RSC
- A. Bajaj, P. Kondaiah and S. Bhattacharya, Bioconjugate Chem., 2008, 19, 1640–1651 CrossRef CAS PubMed
- J. E. Dahlman, C. Barnes, O. F. Khan, A. Thiriot, S. Jhunjunwala, T. E. Shaw, Y. Xing, H. B. Sager, G. Sahay, L. Speciner, A. Bader, R. L. Bogorad, H. Yin, T. Racie, Y. Dong, S. Jiang, D. Seedorf, A. Dave, K. Singh Sandhu, M. J. Webber, T. Novobrantseva, V. M. Ruda, A. K. Lytton-Jean, C. G. Levins, B. Kalish, D. K. Mudge, M. Perez, L. Abezgauz, P. Dutta, L. Smith, K. Charisse, M. W. Kieran, K. Fitzgerald, M. Nahrendorf, D. Danino, R. M. Tuder, U. H. von Andrian, A. Akinc, D. Panigrahy, A. Schroeder, V. Koteliansky, R. Langer and D. G. Anderson, Nat. Nanotechnol., 2014, 9, 648–655 CrossRef CAS PubMed
- C. Goncalves, M. Berchel, M. P. Gosselin, V. Malard, H. Cheradame, P. A. Jaffres, P. Guegan, C. Pichon and P. Midoux, Int. J. Pharm., 2014, 460, 264–272 CrossRef CAS PubMed
- A. Tschiche, S. Malhotra and R. Haag, Nanomedicine, 2014, 9, 667–693 CrossRef CAS PubMed
- M. Wang and Y. Cheng, Biomaterials, 2014, 35, 6603–6613 CrossRef CAS PubMed
- P. P. Karmali and A. Chaudhuri, Med. Res. Rev., 2007, 27, 696–722 CrossRef CAS PubMed
- S. Bhattacharya and V. Praveen Kumar, Langmuir, 2005, 21, 71–78 CrossRef CAS PubMed
- J. A. Heyes, D. Niculescu-Duvaz, R. G. Cooper and C. J. Springer, J. Med. Chem., 2002, 45, 99–114 CrossRef CAS PubMed
- J. H. Felgner, R. Kumar, C. N. Sridhar, C. J. Wheeler, Y. J. Tsai, R. Border, P. Ramsey, M. Martin and P. L. Felgner, J. Biol. Chem., 1994, 269, 2550–2561 CAS
- R. Srinivas, S. Samanta and A. Chaudhuri, Chem. Soc. Rev., 2009, 38, 3326–3338 RSC
- R. Mukthavaram, S. Marepally, M. Y. Venkata, G. N. Vegi, R. Sistla and A. Chaudhuri, Biomaterials, 2009, 30, 2369–2384 CrossRef CAS PubMed
- V. Chandrashekhar, M. Srujan, R. Prabhakar, R. C. Reddy, B. Sreedhar, K. K. Rentam, S. Kanjilal and A. Chaudhuri, Bioconjugate Chem., 2011, 22, 497–509 CrossRef CAS PubMed
- A. Bajaj, P. Kondaiah and S. Bhattacharya, J. Med. Chem., 2008, 51, 2533–2540 CrossRef CAS PubMed
- M. Rajesh, J. Sen, M. Srujan, K. Mukherjee, B. Sreedhar and A. Chaudhuri, J. Am. Chem. Soc., 2007, 129, 11408–11420 CrossRef CAS PubMed
- R. Koynova, L. Wang and R. C. MacDonald, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 14373–14378 CrossRef CAS PubMed
- R. Koynova, B. Tenchov, L. Wang and R. C. Macdonald, Mol. Pharm., 2009, 6, 951–958 CrossRef CAS PubMed
- D. Simberg, D. Danino, Y. Talmon, A. Minsky, M. E. Ferrari, C. J. Wheeler and Y. Barenholz, J. Biol. Chem., 2001, 276, 47453–47459 CrossRef CAS PubMed
- R. Banerjee, P. K. Das, G. V. Srilakshmi, A. Chaudhuri and N. M. Rao, J. Med. Chem., 1999, 42, 4292–4299 CrossRef CAS PubMed
- M. Srujan, V. Chandrashekhar, R. C. Reddy, R. Prabhakar, B. Sreedhar and A. Chaudhuri, Biomaterials, 2011, 32, 5231–5240 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: 1H NMR, ESI-MS and HRMS mass spectral characterizations for cationic amphiphiles as well as for their secondary amine precursors (Fig. S1–S15), reverse phase HPLC chromatograms and HPLC conditions in two mobile phases (Fig. S16–S18). Gel retardation assay (Fig. S19), cytotoxicity assays (Fig. S20–S21) and confocal images (Fig. S22). Size and surface potential of liposomes data (Tables S1 and S2). See DOI: 10.1039/c6ra07256a |
| This journal is © The Royal Society of Chemistry 2016 |