
Glycosylation via remote activation of anomeric leaving groups: development of 2-(2-propylsulfinyl)benzyl glycosides as novel glycosyl donors†
Penghua
Shu
a,
Wang
Yao
a,
Xiong
Xiao
a,
Jiuchang
Sun
a,
Xiang
Zhao
a,
Yueqi
Zhao
a,
Yang
Xu
a,
Jinyi
Tao
a,
Guangmin
Yao
a,
Jing
Zeng
a and
Qian
Wan
*ab
aHubei Key Laboratory of Natural Medicinal Chemistry and Resource Evaluation, School of Pharmacy, Huazhong University of Science and Technology, 13 Hangkong Road, Wuhan, Hubei 430030, China. E-mail: wanqian@hust.edu.cn
bState Key Laboratory of Bio-organic and Natural Products Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, 345 Lingling Road, Shanghai 200032, China
First published on 14th December 2015
Abstract
2-(2-Propylsulfinyl)benzyl glycoside was designed as a new type of glycosyl donor which could be activated via remote mode. This glycosyl donor and its reduced form could act as active and latent donors respectively in a latent-active glycosylation strategy. Details of the discovery of this novel glycosyl donor are reported in this article.
 Qian Wan | Qian Wan received his BS degree from Central China Normal University in 1997. In 2004 he completed his PhD from University Paris 11 under the supervision of Prof. André Lubineau. He then joined the group of Prof. Samuel J. Danishefsky at the Memorial Sloan-Kettering Cancer Center as a Postdoctoral Research Fellow. From 2007 to 2011, he was a Scientist at the Department of Medicinal Chemistry at Amgen Inc. In 2012, he started his independent career as a Professor in the School of Pharmacy at the Huazhong University of Science and Technology. His current research interests focus on carbohydrate chemistry, free radical chemistry and medicinal chemistry. |
Introduction
Glycosidic bond formation plays a central role in carbohydrate chemistry.1 The past few decades have witnessed a dramatic improvement in the development of efficient chemical glycosylation strategies.2 The common activation mode of these strategies involves stimulating functional groups which are directly linked to the glycosyl donors’ anomeric position. The major used glycosyl donors include glycosyl halides,3a thioglycosides,3b–d glycosyl imidates and thioimidates,3e–h glycosyl sulfoxides,3i glycosyl phosphates,3j glycosyl phosphites,3k 1-hydroxyl sugars,3l glycosyl orthoesters,3m 1,2-anhydro sugars,3netc. Other than these donors, glycosyl donors activated by remote activation mode4 were also discovered by Fraser-Reid (n-pentenyl glycoside),5 Kim (2-(hydroxycarbonyl)benzyl glycoside),6 Hotha (propargyl glycoside)7 and Yu (glycosyl ortho-alkynylbenzoate)8et al. (Scheme 1). Despite being less investigated, glycosylation via remote activation exhibited some advantages: (1) the glycosylation conditions of these donors are normally orthogonal to those of most of the other glycosyl donors; (2) a latent-active strategy9 could be easily applied to these donors in oligosaccharide synthesis. Inspired by these pioneering studies, we have reported the development of 2-(2-propylsulfinyl)benzyl (PSB) glycosides as novel glycosyl donors which were activated via a remote mode.10 With this newly developed glycosyl donor, a natural hepatoprotective glycoside was synthesized based on a latent-active strategy. Herein, we further reported the details of our efforts in the establishment and understanding of this novel glycosylation process.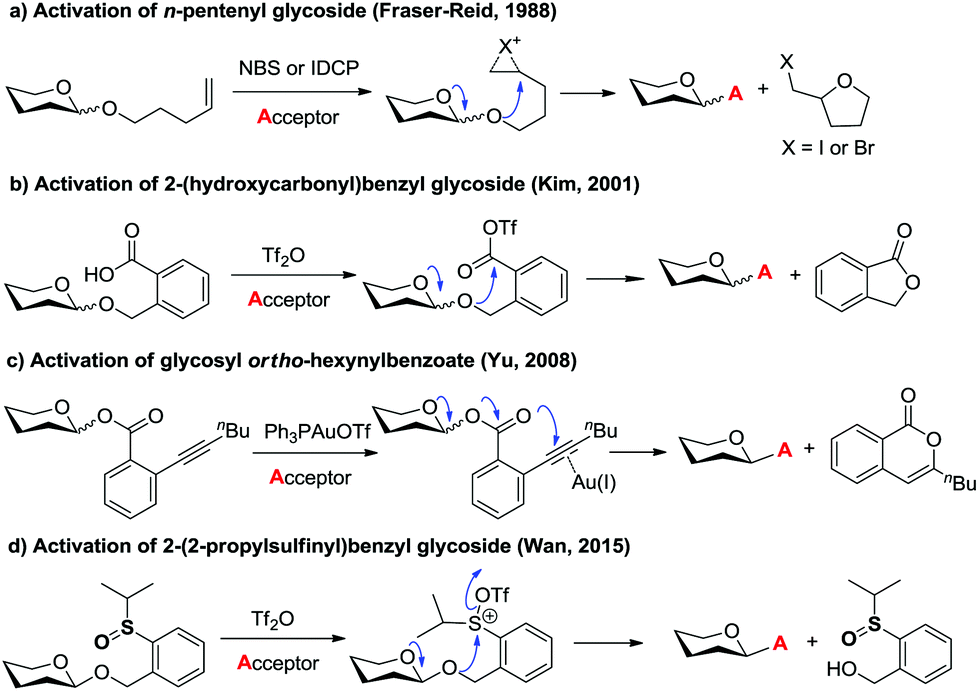 | ||
| Scheme 1 Selected examples of glycosylation via remote activation mode. | ||
Sulfoxide compounds are widely utilized in organic synthesis. It is well known that the sulfinyl group of sulfoxide could be activated by electrophiles to generate an active electrophilic species.11 This intrinsic nature of sulfoxide encouraged chemists to apply it in practical organic transformations. For example, sulfoxide has been widely used in Moffatt reaction,12a Corey–Kim oxidation,12b–d Swern oxidation12e–g and Pummerer reaction. Since the first report on the Pummerer reaction in 1909,13 a lot of related reactions have been developed for the construction of new carbon–carbon bonds and carbon–heteroatom bonds.14 The distinctive nature of sulfoxide also stimulated its application in carbohydrate chemistry. Kahne,3i Crich15 and Gin16 have made significant contributions in this area. In Kahne's and Crich's glycosylations, sulfoxide was employed as an anomeric leaving group. Activation of this leaving group by Tf2O formed a sulfonium intermediate, which instantly degraded to oxocarbenium and allowed the glycosylation to proceed smoothly.
In view of the advantages of glycosylation via remote activation and high activity of sulfoxide, we aimed to design a new type of glycosyl donor with the sulfinyl group located at a remote site of the anomeric position. We envisaged that the activated mode would pass through the Pummerer reaction pathway: the thionium possibly induced the intramolecular attack of the anomeric oxygen atom via a 5- or 6-membered ring to form glycosyl oxocarbenium (Scheme 2).
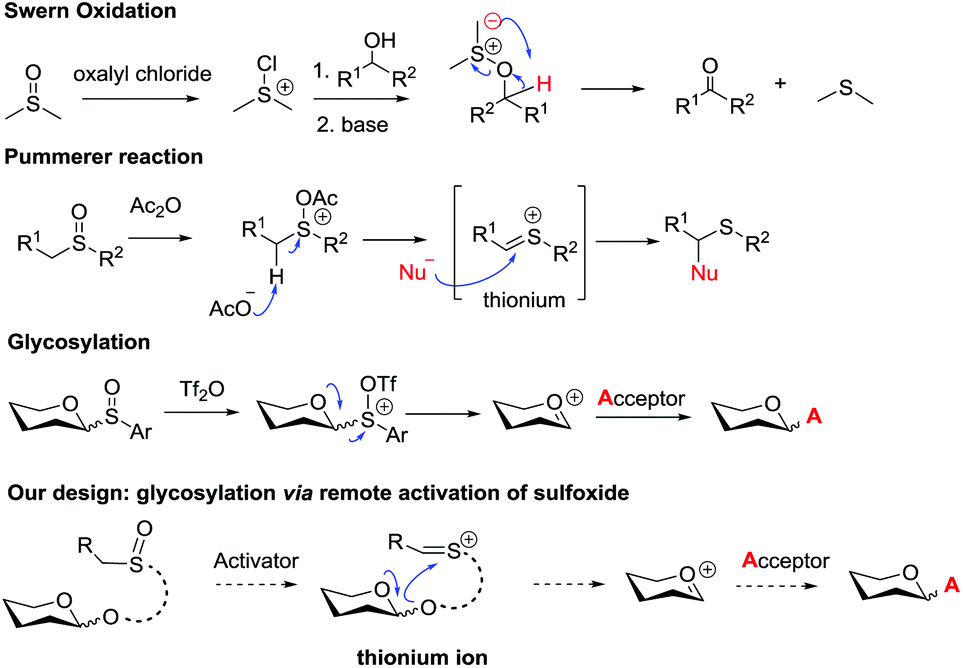 | ||
| Scheme 2 Reaction with a sulfinyl functional group. | ||
Results and discussion
Donor design
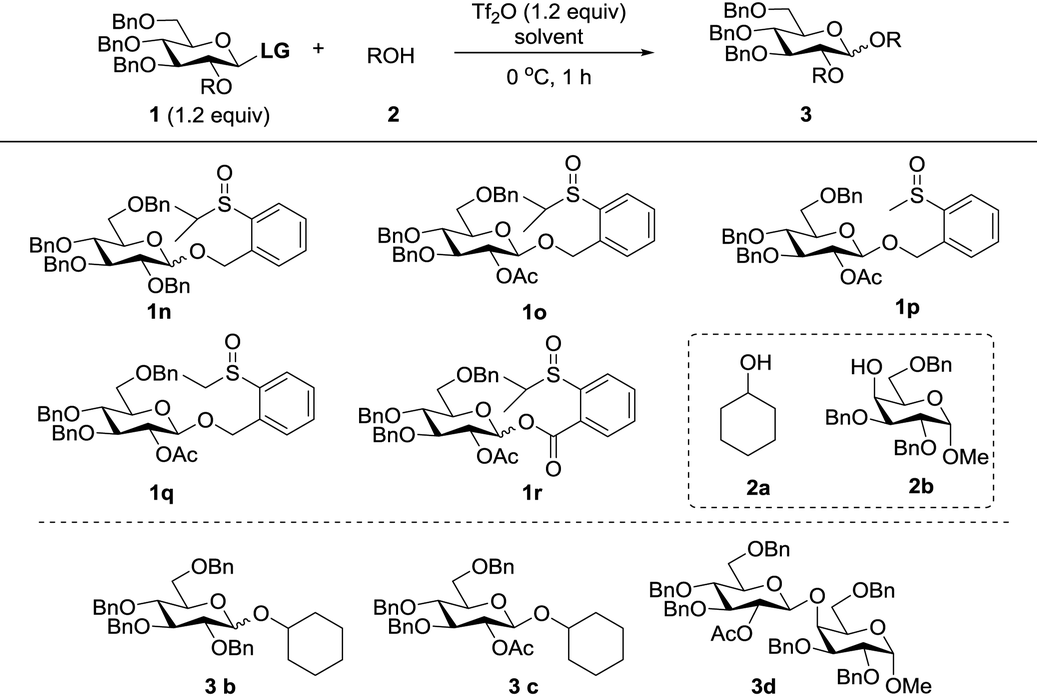
Initially, peracylated glucosides with various leaving groups possessing sulfinyl groups were chosen as glycosyl donors to conceive the idea (Table 1). Unfortunately, the commonly used activator for the Pummerer reaction such as Ac2O and trifluoroacetic anhydride (TFAA) under various conditions failed to activate the sulfinyl groups, and in most cases, only starting materials were recovered (see the ESI†). While Tf2O was used as an activator, we observed the full consumption of most of the examined donors; however, no desired product was formed and the major by-products are listed in Table 1. When 1a was used as a glycosyl donor and dimethylacetamide (DMA) as a solvent, surprisingly, the reaction afforded an aldehyde 4a in 30% yield. Reactions with donors 1d and 1e in dichloromethane (DCM), acetonitrile (CH3CN) or toluene furnished 4b and 4c in high yields respectively. However, activation of a structurally similar donor 1c was very difficult; under the same reaction conditions, 45% of 1c was still recovered. While 1f with higher acidity of the α-proton of the sulfinyl group and better stability of the generated thionium was used as a donor, and toluene as a solvent, the reaction gave an unwanted compound 4d as the major product. In DCM, a reduced product 5f was observed as well. Donor 1g failed to give any desired product either; if a base such as triethyl amine or NaHCO3 was introduced into the reaction system, a moderate yield of the sulfinyl group reduced product 5g was isolated. An exception is donor 1b which produced the desired product 3a in 15% yield; however, attempts to further improve the reaction efficiency failed.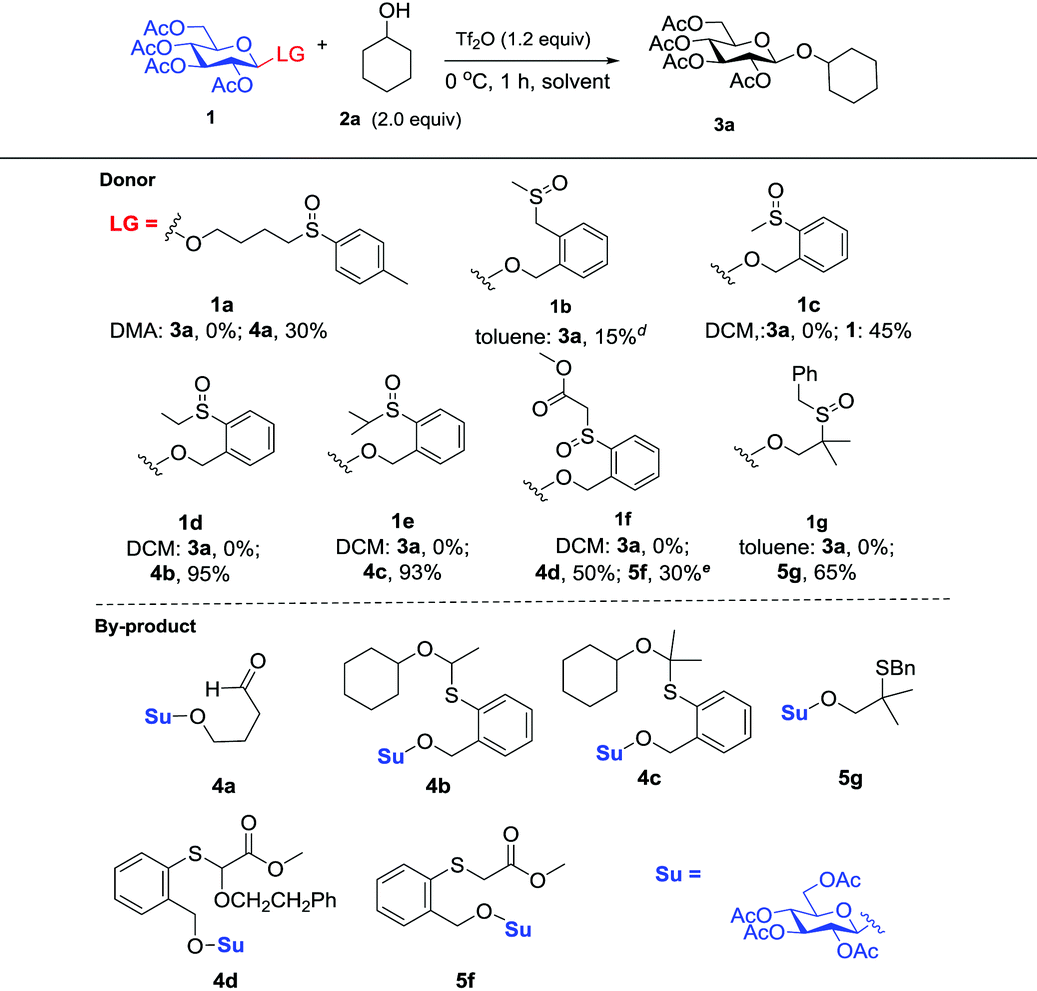
Although frustrating results were obtained for donors 1a–1g under various conditions, it is notable that the sulfinyl group was successfully activated by Tf2O. However, after activation, an intermolecular Pummerer reaction (Path A2) and/or oxidation reaction (Path B) occurred as competition reactions to our anticipated intramolecular Pummerer reaction mediated glycosylation (Path A1) (Scheme 3). These results implied that possibly the nucleophilicity of the anomeric oxygen atom was not strong enough. This speculation prompted us to further examine donors 1h–1m with ester type anomeric leaving groups. Interestingly, reactions with all of these glycosyl donors could produce the desired products albeit with low to moderate yields. But again, we could not improve the yields after screening a lot of reaction conditions (Table 2).
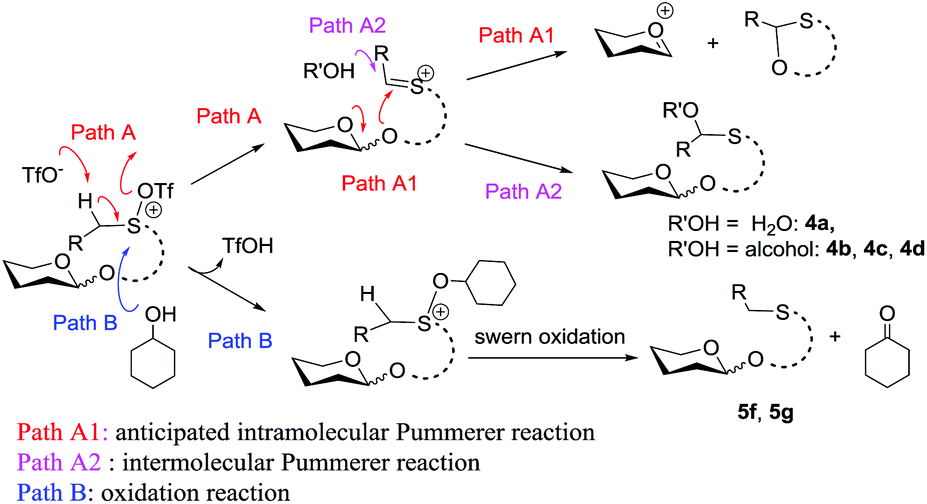 | ||
| Scheme 3 Possible competition reactions. | ||
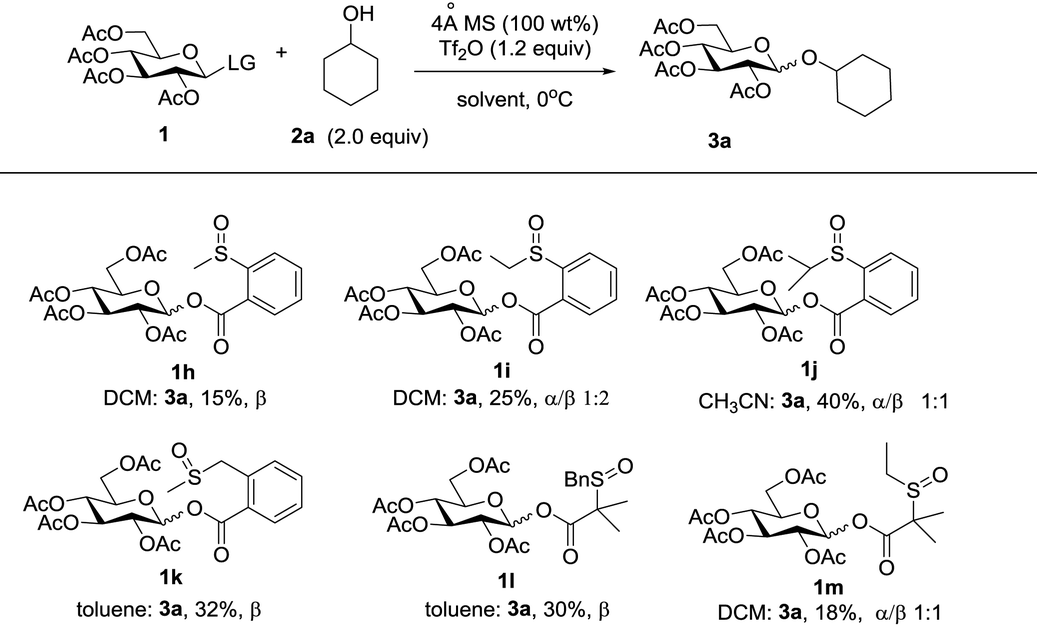
Since modification of the leaving groups could not lead to successful glycosylation, we then considered changing the disarmed protecting group of donors to the armed protecting group which definitely increased the activity of the anomeric oxygen atom (Table 3). First, the perbenzylated donor 1n was examined (entry 1). To our delight, the reaction proceeded smoothly under the activation of Tf2O in DCM at 0 °C, which generated the desired product 3a in 78% yield without any stereoselectivity. Changing the solvents led to moderate selectivity and higher yields (entries 2 and 3). To validate the concept, the superarmed donor 1o was then subjected to the reaction conditions, and the desired product 3c was obtained in excellent yield with exclusive β-selectivity (entry 4). The reaction with the 4-OH galactose acceptor 2b still provided exclusively β-disaccharide 3d in 95% yield (entry 5). Donors 1p and 1q also provided good results (entries 6 and 7); however, a small amount of acceptor 2b was recovered. Interestingly, donor 1r with the ester leaving group was less efficient and only a moderate yield of β-disaccharide was isolated (entry 8), possibly due to the instability of the donor.
|
|
||||
|---|---|---|---|---|
| Entry | Donor | Acceptor | Solvent | Yield (α/β)b |
| a 1 (0.12 mmol, 1.2 equiv.), 2 (0.1 mmol, 1.0 equiv.), Tf2O (0.12 mmol, 1.2 equiv.), solvent (2.0 mL), 4 Å MS (100 wt%). b Isolated yield. c 13% of 2b recovered. d 11% of 2b recovered. e 21% of 2b recovered. | ||||
| 1 | 1n | 2a | DCM |
3b, 78% (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) :1) |
| 2 | 1n | 2a | Et2O |
3b, 82% (5:1) |
| 3 | 1n | 2a | CH3CN |
3b, 84% (1:5) |
| 4 | 1o | 2a | DCM | 3c, 87%, β only |
| 5 | 1o | 2b | DCM | 3d, 95%, β only |
| 6 | 1p | 2b | DCM | 3d,79%, β onlyc |
| 7 | 1q | 2b | DCM | 3d, 82%, β onlyd |
| 8 | 1r | 2b | DCM | 3d, 68%, β onlye |
With the discovery of 2-(2-propylsulfinyl)benzyl (PSB) glycosides as novel glycosyl donors and the optimized reaction conditions in hand, we have examined this new glycosylation strategy by employing various glycosyl acceptors, and these results have been previously reported.10
Latent-active
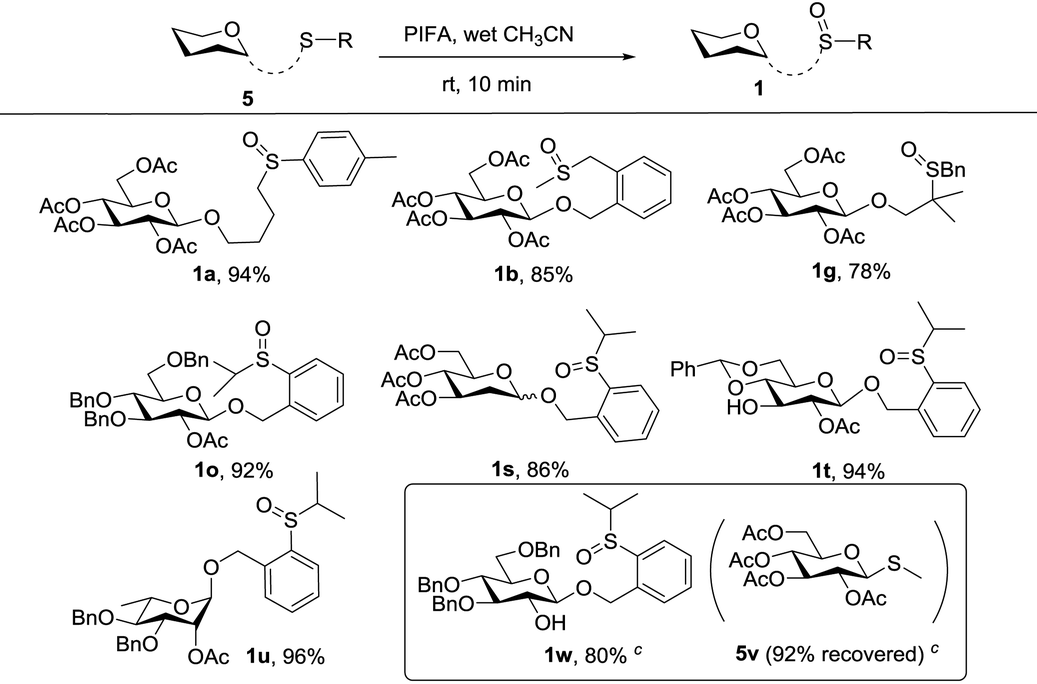
We next considered the possibility of merging this remote activation glycosylation reaction with a latent-active strategy in the synthesis of oligosaccharides. Two basic requirements of a successful latent-active strategy are: (1) a latent glycosyl donor should be easily converted to an active glycosyl donor; (2) a latent glycosyl donor must be stable enough under the glycosylation conditions with an active glycosyl donor. In our case, we envisioned that the 2-(2-propylsulfinyl)benzyl (PSB) glycoside could act as an active donor, while its reduced form 2-(2-propylthiol)benzyl (PTB) glycoside which was readily obtained could act as a latent donor.First, we investigated the oxidative conditions of the transforming PTB donor 5o to the PSB donor 1o. Although a lot of oxidants could oxidize thioether to sulfoxide, most of the conditions required careful control of the reaction conditions even though over-oxidation to sulfone was inevitable. After screening of several conditions,17 we found that the best condition was the usage of PIFA (bis(trifluoroacetoxy)iodobenzene, 1.2 equiv.) in wet CH3CN (containing ca. 5% water). Under this condition, 92% yield of 1o was obtained from the corresponding thioether (5o) without any over-oxidation product even extending the reaction time.18 This condition was also successfully applied to oxidize other thioethers to form sulfoxides as depicted in Table 4. Most interestingly, when 5w (corresponding thioether of 1w) was mixed with 1.2 equiv. of 5v and subjected to 1.2 equiv. of PIFA in anhydrous DCM, after aqueous workup of the reaction mixture, 5w was selectively oxidized to 1w in 80% yield with only a trace amount of 1v formed.
Having established the effective transformation from the latent donor to the active donor, we next investigated the tolerance of the PTB donor under the conditions of the activating PSB donor. First, the PTB containing acceptor 2c was reacted with the PSB donor 1o under our remote activation conditions, and the desired disaccharide 3e was isolated in 76% yield without affecting the PTB group. The stability was also demonstrated by the high yield of 3f from the PTB donor 5t and PSB donor 1u (Scheme 4).
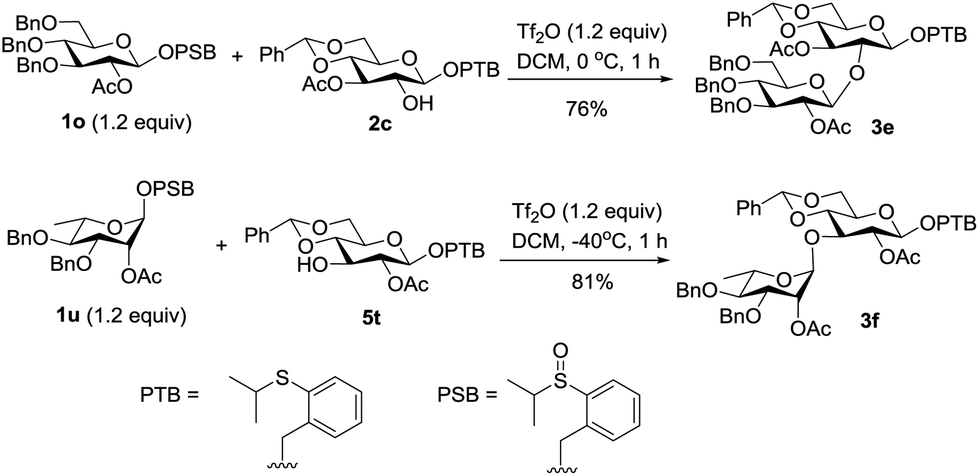 | ||
| Scheme 4 A latent-active strategy. | ||
In addition, the ground applicability of a latent-active strategy to our developed new glycosyl donors was finally proved by the successful total synthesis of leonoside F as previously reported,10 and will be further established by the synthesis of complex naturally occurring polysaccharides.
Orthogonality
The orthogonality of PTB and PSB glycosides to other glycosyl donors was further discovered. Normally, O-alkyl glycosides are rather stable towards the activation conditions of most glycosyl donors. PTB glycosides could be prepared from glycosyl acetates, glycosyl imidates and glycosyl orthoesters.10 Undoubtedly, the activation conditions of these donors will not affect the PTB group. Since PTB glycosides contained a thioether group, we then examined its stability towards the conditions of activating thioglycosides by reaction of the PTB containing acceptor 5w with thioglycoside 5v which is a less active disarmed glycosyl donor. The commonly used NIS in combination with TfOH for glycosylation with thioglycosides performed very well in this reaction, which furnished disaccharide 6 in 92% yield. MeOTf also did not affect the PTB functional group and activated 5v to produce 6 in 65% yield, with 25% PTB glycoside 5w recovered. These results clearly indicated that PTB glycoside is stable enough to tolerate various activation conditions for most commonly used glycosyl donors (Scheme 5).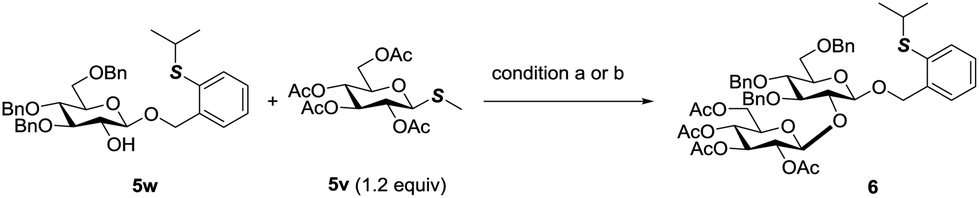 | ||
| Scheme 5 Condition a: NIS (2.4 equiv.), TfOH (0.24 equiv.), DCM, 0 °C, 92%; b: MeOTf (1.2 equiv.), DCM, 0 °C, rt, 24 h, 65%, with 25% of 5w recovered. NIS = N-iodosuccinimide. | ||
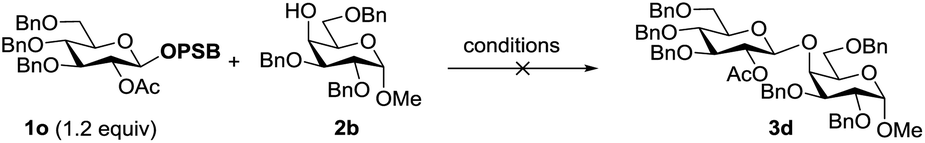
The stability of the PSB donors was tested by reaction of 1o with 2b towards various strongly acidic activators, including TfOH or TfOH in combination with NIS, even MeOTf. In each case, 1o and 2b were recovered perfectly, implying the extreme stability of PSB donors to acidic conditions (Table 5).
Leaving group recovery
In the performance of these remote activation glycosylation reactions, a polar compound other than the glycoside products was always isolated in high yield. For example, reaction of 1o with 2b furnished compound 7 in 92% yield. This compound was characterized as exactly the leaving group of the PSB donor (Scheme 6). The recovery of the leaving group prompted us to test its recyclability and regenerability.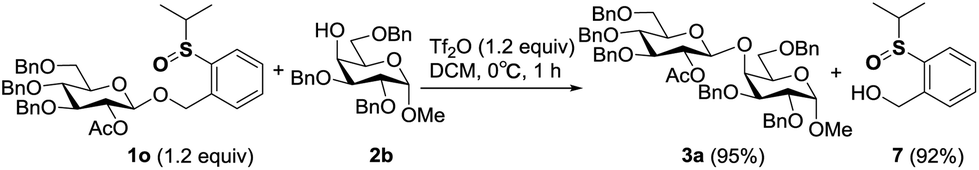 | ||
| Scheme 6 Recovery of the leaving group. | ||
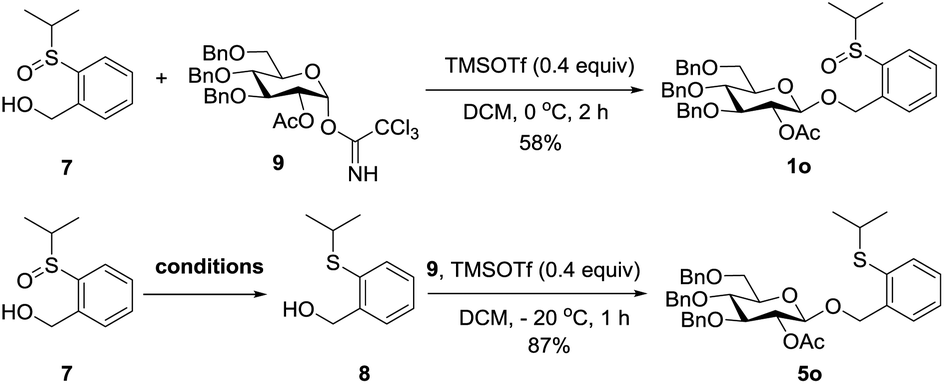
First, compound 7 was directly coupled with glycosyl imidate 9 to generate the PSB donor 1o, albeit a moderate yield was obtained due to the poor solubility of 7, which suggested that the leaving group could be recycled. We then tried to reduce compound 7 to its thioether form which was the leaving group of PTB donors. Several reported reducing conditions were screened for this reaction. Among them, AcSH/I2 was inefficient and most of the starting material was recovered;19 NaBH4/I220 and Mg21 produced better results. The best reducing agents are thiosalicylic acid in combination with catalytic amounts of NBS or I2,22 which generated the reduced PTB-OH 8 in 90% and 95% yields, respectively. Obviously, this regenerated PTB-OH coupled with 9 efficiently to provide the latent donor 5o (Table 6).
|
|
||
|---|---|---|
| Entry | Conditions | Yielda |
| a Isolated yield, yield in parentheses was recovered 7. AcSH = thioacetic acid. TSA = thiosalicylic acid. NBS = N-bromosuccinimide. | ||
| 1 | AcSH (2.2 equiv.), I2 (0.01 equiv.), CH3CN, rt, 12 h | 10% (75%) |
| 2 | NaBH4 (1.0 equiv.), I2 (2.0 equiv.), THF, rt, 0.5 h | 74% (8%) |
| 3 | Mg (20.0 equiv.), MeOH, rt, 4 h | 71% (15%) |
| 4 | TSA (3.0 equiv.), NBS (0.1 equiv.), DCM, rt, 16 h | 90% |
| 5 | TSA (3.0 equiv.), I2 (0.1 equiv.), DCM, rt, 6 h | 95% |
Mechanism
The high recovery yield of compound 7 prompted us to reconsider the reaction mechanism. In our initial design, we envisaged that the reaction could pass through an intramolecular Pummerer reaction pathway. In this pathway, after activation of the sulfinyl group, the abstraction of the α-proton led to a thionium ion B that allowed the anomeric oxygen atom to attack the α-carbon position to form an oxocarbenium C and an oxathiane 10. Therefore, in the reaction mixture, we should obtain compound 10 or its hydrolyzed product. However, in all of the reactions we carried out, neither of them was observed. Instead, compound 7 was always isolated in high yield. This phenomenon implied that the reaction should pass through a different way. Possibly, the thionium ion was not formed due to the weak basicity of the triflate anion, instead, the anomeric oxygen atom prior to directly attacking the sulfur cation of A formed oxocarbenium C and a sulfonium ion D. The sulfonium ion further hydrolyzed to form 7 during aqueous workup. The latter pathway is actually an intramolecular interrupted Pummerer reaction.23 In addition, since we have proved that the PSB glycosides are quite stable under strongly acidic conditions, the possibility of formation of 7 by direct cleavage of the anomeric C–O bond by TfOH should be excluded (Scheme 7).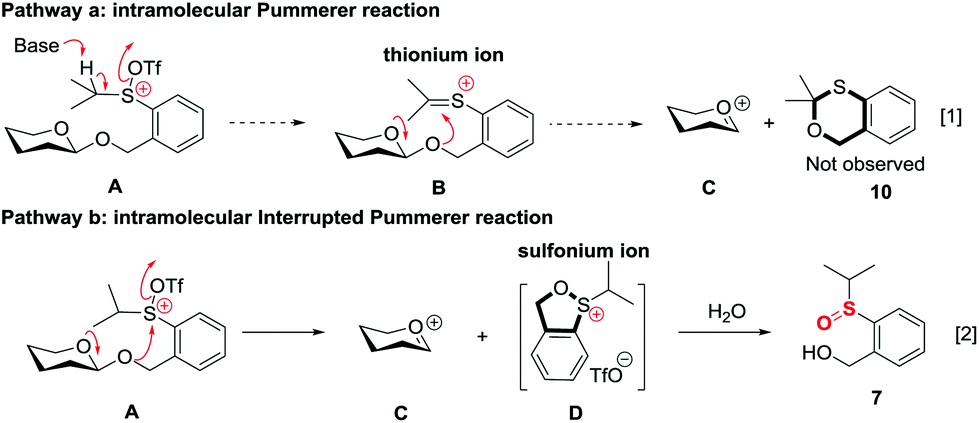 | ||
| Scheme 7 Proposed mechanism. | ||
Conclusion
In conclusion, we further reported the details of the discovery of 2-(2-propylsulfinyl)benzyl (PSB) glycoside as a novel glycosyl donor which could be efficiently activated via a remote mode. With this type of glycosyl donor, the latent-active strategy was successfully applied by utilizing 2-(2-propylsulfinyl)benzyl (PSB) glycoside as an active glycosyl donor and 2-(2-propylthiol)benzyl (PTB) glycoside as a latent glycosyl donor. The orthogonality and stability of donors were examined and the observations suggested their practical applicability in combination with other known glycosyl donors in oligosaccharide synthesis. Further extension of this strategy to disarmed glycosyl donors as well as a new application to naturally occurring oligosaccharide synthesis is under way.Acknowledgements
We thank the National Natural Science Foundation of China (21272082, 21402055, and 21472054), the State Key Laboratory of Bio-organic and Natural Products Chemistry (SKLBNPC13425), the Natural Science Funds of Hubei Province for Distinguished Young Scholars (2015CFA035), the Specialized Research Fund for the Doctoral Program of Higher Education (20120142120092), “Thousand Talents Program” Young Investigator Award, and the Huazhong University of Science and Technology (2014ZZGH015) for support.Notes and references
-
(a)
A. V. Demchenko, in Handbook of Chemical Glycosylation: Advances in Stereoselectivity and Therapeutic Relevance, ed. A. V. Demchenko, Wiley-VCH, Weinheim, Germany, 2008, pp. 1–27 Search PubMed
; (b) K. Toshima and K. Sasaki, in Comprehensive Glycoscience, ed. J. P. Karmerling, G.-J. Boons, Y. C. Lee, A. Suzuki, N. Taniguchi and A. G. J. Voragen, Elsevier, Oxford, UK, 2007, pp. 261–311 Search PubMed
-
(a) G.-J. Boons, Tetrahedron, 1996, 52, 1095–1121 CrossRef CAS
-
(a) M. Shimizu, H. Togo and M. Yokoyama, Synthesis, 1998, 799–822 CrossRef CAS
- For reviews, see: S. C. Ranade and A. V. Demchenko, J. Carbohydr. Chem., 2013, 32, 1–43 CrossRef CAS
-
(a) D. R. Mootoo, V. Date and B. Fraser-Reid, J. Am. Chem. Soc., 1988, 110, 2662–2663 CrossRef CAS
- K. S. Kim, J. H. Kim, Y. J. Lee, Y. J. Lee and J. Park, J. Am. Chem. Soc., 2001, 123, 8477–8481 CrossRef CAS PubMed
- S. Hotha and S. Kashyap, J. Am. Chem. Soc., 2006, 128, 9620–9621 CrossRef CAS PubMed
-
(a) Y. Li, Y. Yang and B. Yu, Tetrahedron Lett., 2008, 49, 3604–3608 CrossRef CAS
- For recent reviews, see:
(a) T. C. Shiao and R. Roy, Top. Curr. Chem., 2011, 301, 69–108 CrossRef CAS PubMed
- P. Shu, X. Xiao, Y. Zhao, Y. Xu, W. Yao, J. Tao, H. Wang, G. Yao, Z. Lu, J. Zeng and Q. Wan, Angew. Chem., Int. Ed., 2015, 54, 14432–14436 CrossRef CAS PubMed
-
R. J. Cremlyn, in An Introduction to Organosulfur Chemistry, John Wiley & Sons, Chichester, England, 1996, pp. 63–79 Search PubMed
-
(a) K. E. Pfitzner and J. G. Moffatt, J. Am. Chem. Soc., 1965, 87, 5661–5670 CrossRef CAS
- R. Pummerer, Ber. Dtsch. Chem. Ges., 1909, 42, 2282–2291 CrossRef CAS
- Selected reviews:
(a) S. C. Bur and A. Padwa, Chem. Rev., 2004, 104, 2401–2432 CrossRef CAS PubMed
-
(a) D. Crich and S. Sun, J. Org. Chem., 1996, 61, 4506–4507 CrossRef CAS PubMed
-
(a) V. Di Bussolo, Y.-J. Kim and D. Y. Gin, J. Am. Chem. Soc., 1998, 120, 13515–13516 CrossRef CAS
- Urea hydrogen peroxide (UHP) adduct and cyanuric chloride (CC) were used to oxidize thioethers in our previous report, see the ESI.†.
- Oxidation of sulfides by hypervalent iodine:
(a) V. G. Shukla, P. D. Salgaonkar and K. G. Akamanchi, J. Org. Chem., 2003, 68, 5422–5425 CrossRef CAS PubMed
- A. Jabbari, M. Zarei and A. Jamaleddini, J. Sulfur Chem., 2012, 33, 413–418 CrossRef CAS
- B. Karimi and D. Zareyee, Synthesis, 2003, 335–336 CrossRef CAS
- J. M. Khurana, V. Sharma and S. A. Chacko, Tetrahedron, 2007, 63, 966–969 CrossRef CAS
- B. Karimi and D. Zareyee, Synthesis, 2003, 1875–1877 CrossRef CAS
- For selected examples, see:
(a) L.-C. Chen, H.-M. Wang and I.-J. Kang, Heterocycles, 1999, 51, 1437–1441 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5qo00359h |
| This journal is © the Partner Organisations 2016 |