
Novel reduction-sensitive pullulan-based micelles with good hemocompatibility for efficient intracellular doxorubicin delivery
Xianwu Wanga,
Jingyun Wang*ab,
Yongming Baoa,
Benhua Wangb,
Xiaohong Wanga and
Lili Chena
aSchool of Life Science and Biotechnology, Dalian University of Technology, School of Life Science and Biotechnology, No. 2 Linggong Road, Ganjingzi District, Dalian 116024, P. R. China. E-mail: wangjingyun67@dlut.edu.cn; Fax: +86 411 84706365; Tel: +86 411 84706355
bState Key Laboratory of Fine Chemicals, Dalian University of Technology, Dalian 116024, P. R. China
First published on 6th November 2014
Abstract
A novel intracellular reduction-sensitive delivery system of doxorubicin (DOX), based on pullulan–stearic acid (P-ss-SA) conjugates with disulfide bonds as reduction-sensitive bonds, was successfully developed. The conjugates could self-assemble into micelles in aqueous media and encapsulate DOX. The properties of blank and DOX-loaded micelles were studied in detail. The results showed that the mean size of the blank and DOX-loaded micelles was around 187.7 nm and 194.4 nm, respectively. The drug loading content and encapsulation efficiency of the P-ss-SA micelles were around 6.19% and 65.53%, respectively. The mean size of reduction-sensitive P-ss-SA micelles increased dramatically under reductive conditions. The drug release of P-ss-SA micelles under reductive conditions was much faster than that under non-reductive conditions. The confocal laser microscopy and flow cytometry measurements indicated that the intracellular reductive conditions broke the disulfide bonds in P-ss-SA micelles and triggered the fast release of DOX. The in vitro IC50 of the DOX-loaded P-ss-SA micelles was lower than that of DOX-loaded micelles without reduction-sensitivity against HepG2 and MCF-7 cells. The blank micelles showed negligible cytotoxicity, and possessed excellent hemocompatibility without causing undesirable hemolysis. These results indicated that the biocompatible reduction-sensitive P-ss-SA micelles can be used as potential carrier systems for the intracellular delivery of DOX.
1. Introduction
Biodegradable polymeric micelles have emerged as among the most promising nanocarrier systems for anticancer drug delivery.1–6 These biodegradable polymeric micelles can improve the bioavailability of anticancer drugs and reduce the side effects of the drugs by increasing the solubility and stability of anticancer drugs, prolonging drug blood circulation time and improving drug accumulation at tumor tissue via the enhanced permeability and retention (EPR) effect.7–9 These polymeric micelles could disassemble rapidly and give burst drug release intracellularly because of stimuli such as pH,10–14 temperature,15 ultrasound,16 enzyme,17 light,18 redox potential.19 Among these biodegradable polymeric micelles, reduction-sensitive polymeric micelles containing disulfide bonds in the main chain, at the side chain, or in the cross-linker have been intensively studied for enhanced intracellular drug release due to the great difference in the redox potential between the mildly oxidizing extracellular milieu and the reducing intracellular milieu.20–22 The significant difference in the redox potential mainly contributes to the concentration of glutathione (GSH), a thiol-containing tripeptide capable of reducing disulfide bonds, which is mM concentrations in the cell cytoplasm and even higher level in cancer cells, while only μM concentrations in blood plasma due to rapid enzymatic degradation.23,24 Based on this, a lot of reduction-responsive polymeric micelles have been designed and used as anticancer drug carrier.7,8,25–30Pullulan (P), a water-soluble and neutral liner polysaccharide produced by Aureobasidium pullulans, is formed from α-1,4-linked glucose units included in a α-1,6-linked maltotriose unit.31 P has been widely explored recently for its biomedical applications in tissue engineering, targeted drug and gene delivery due to its good biological nature such as nontoxic, non-immunogenic, non-mutagenic and non-carcinogenic nature.32–35 In addition, P has been verified that it can specifically interact with asialoglycoprotein receptor (ASGPR) present on liver parenchymal hepatocytes and it is an ideal material to prepare active liver-targeting carriers.12,35–38 Although there have been many P-based amphiphilic copolymers for drug delivery, to the best of our knowledge, few studies have focused on redox-sensitive P-based amphiphilic copolymers for doxorubicin (DOX) delivery.39
In our previous report, we synthesized stearic acid (SA) modified P derivatives with different degrees of substitution.40 The stearic acid modified P derivatives (PUSAs) can self-assembled in water and efficiently encapsulate DOX. But the DOX-loaded micelles exhibited gradual drug release over a long time and a reduced antitumor efficacy against MCF-7 cells. In this study, we for the first time introduced stearic acid (SA) into P using cystamine as bioreducible linkages to form redox-sensitive micelles (P-ss-SA) for triggered intracellular doxorubicin release (Scheme 1). Insensitive P–SA conjugates which were structurally analogous to P-ss-SA were also synthesized for comparison. The physicochemical characteristics and DOX-loading capacities of micelles were investigated by dynamic light scattering (DLS), transmission electron microscopy (TEM), and fluorescence spectroscopy. The effective disulfide bond breakage of the micelles and triggered DOX release in response to dithiothreitol (DTT) medium imitating reduction condition in human body were demonstrated by the observations of size change and in vitro DOX release study. The cytotoxicity of blank and DOX-loaded micelles was studied in HepG2 cells and MCF-7 cells by the MTT assay. The blood compatibility of blank micelles was also evaluated by hemolysis test. The behavior of the intracellular release of DOX was further studied in MCF-7 cells by confocal laser scanning microscopy (CLSM) and flow cytometry (FCM).
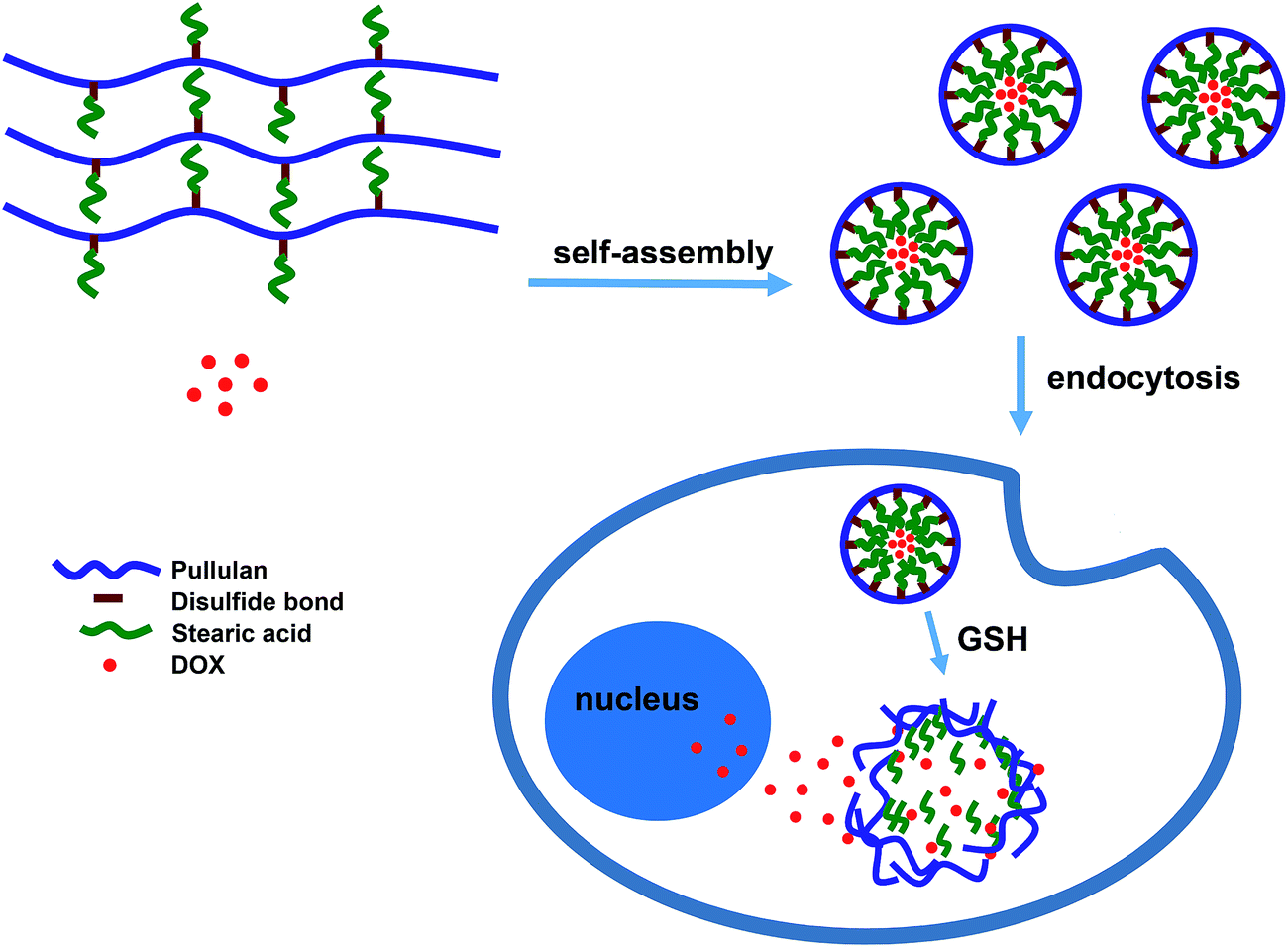 | ||
| Scheme 1 The illustration of drug release from reduction-sensitive P-ss-SA micelles. | ||
2. Materials and methods
2.1. Materials
Pullulan (P, MW: 100 kDa) was purchased from Shandong Zhongqing Biotechnology Company. 1-Ethyl-3-(3-dimethyllaminopropyl)-carbodiimide hydrochloride (EDC), N-hydroxysuccinimide (NHS), 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), trypsin, 4-dimethylaminopyridine (DMAP), pyrene, cystamine dihydrochloride, adipic acid dihydrazide, dithiothreitol (DTT), triethylamine (TEA) and other chemicals were all obtained from Sigma-Aldrich (St. Louis, USA). Doxorubicin hydrochloride (DOX·HCl) was purchased from Dalian Meilun Biotechnology Co., Ltd., China.Human hepatocellular liver carcinoma cell (HepG2) and human breast cancer cell (MCF-7) were purchased from the Institute of Biochemistry and Cell Biology, SIBS, CAS. Dulbecco's modified Eagle's medium (DMEM) and fetal bovine serum (FBS) for cell culture were obtained from Hyclone (Logan, USA).
2.2. Synthesis of P-ss-SA conjugates and P–SA conjugates
Adipic dihydrazide modified pullulan succinate (P-ADH) was carried out according to the following procedures. First, P-COOH (1.51 g, n–COOH = 3.46 mM) was dissolved in 150 mL PBS (0.2 M) and adipic dihydrazide (3.08 g, 17.3 mM) was added with agitation. The pH of the reaction mixture was adjusted to 4.75, followed by addition of EDC (672.7 mg, 3.46 mM). The reaction was carried out at 30 °C for 12 h. The pH of resulting solution was adjusted to 7.0 by addition of 0.1 M NaOH. Then the solution was dialyzed (MWCO 10000) exhaustively against 0.1 M NaCl, 25% EtOH/H2O, deionized water, each for 3 days with six exchanges. Finally, the polymer P-ADH solution was lyophilized and stored at 4 °C until further use.
2.3. Characterization of P-ss-SA conjugates and P–SA conjugates
1H NMR spectra of P-COOH, P-CYS, P-ADH, P-ss-SA conjugates and P–SA conjugates were acquired in D2O using a 400 MHz spectrometer (Varian INOVA400, USA). The degree of succinoylation of P-COOH was quantified by titration against a standard NaOH solution with phenolphthalein as an indicator. The degree of substitution (DS) of P-CYS or P-ADH which is defined as the number of free amines per 100 glucose residues of P was estimated using elemental analysis.2.4. Preparation and characterization of micelles
The morphology of micelles was observed by transmission electron microscopy (TEM, Tecnai G2 Spirit 120 kV). The samples were prepared by dropping 0.5 mg mL−1 micellar solution on the copper grid, followed by air drying and negatively stained with 2% (w/w) uranyl acetate for 2 min. The grid was dried at room temperature and then observed by TEM.
The critical micelles concentration (CMC) of amphiphilic P-ss-SA conjugates and P–SA conjugates was evaluated by fluorescence spectroscopy (LS55, PerkinElmer), using pyrene as the probe. To prepare sample solutions, a known amount of pyrene in methanol was added to each 10 mL test tubes and then methanol was evaporated. 10 mL of different concentrations of micellar solutions was added to give a final pyrene concentration of 6.0 × 10−7 M. The samples were sonicated for 40 min in an ultrasonic bath and shaken in a shaking air bath at 37 °C for 1 h. The fluorescence excitation spectra were measured at an emission wavelength of 334 nm, and the emission spectra was recorded in the range of 350–450 nm. The excitation and emission bandwidths were 5 and 2.5 nm, respectively.
2.5. Disassembly of micelles triggered by DTT
The disassemble behaviors of reduction-sensitive P-ss-SA micelles in response to 10 mM DTT (PBS; 10 mM, pH 7.4) imitating the intracellular reductive condition was monitored by DLS measurement and P-ss-SA micelles without the addition of DTT as a control. The disassembly of P–SA micelles in response to 10 mM DTT was also measured for comparison. Briefly, to a glass cell containing 3 mL solution of P-ss-SA micelles or P–SA micelles (0.5 mg mL−1), was added 10 mM DTT at predetermined time. The solution was placed in a shaking bed at 37 °C at a rotation speed of 180 rpm. The micelle size was measured by DLS through 24 h.2.6. Preparation and characterization of DOX-loaded micelles
DOX-loaded micelles were prepared by a simple dialysis method.25 Before loading DOX into the micelles, DOX·HCl was stirred overnight with twice the number of moles of TEA to obtain the DOX base. Then the DOX/DMSO solution was added into the DMSO solution of P-ss-SA conjugates or P–SA conjugates. The mixture was placed in a shaking bed at 37 °C at a rotation speed of 180 rpm for 3 h to get well mixed. The final mixture was dialyzed (MWCO 10000) against deionized water for 48 h. Afterwards, The solution was filtered through a 0.45 μm pore-sized microporous membrane and then lyophilized.The entrapment efficiency (EE) and drug-loading (DL) were obtained on the basis of the standard curve by using the UV/VIS Spectrophotometer (JASCO V-560). Lyophilized DOX-loaded polymeric micelles were dissolved in a mixture of DMSO/H2O (v/v, 9/1) to obtain clear solution, and the drug concentration was quantified by its absorbance at 485 nm. The EE and DL were calculated by the formulas as EE (%) = (weight of DOX in micelles)/(weight of DOX fed initially) × 100% and DL (%) = (weight of DOX in micelles)/(weight of polymeric micelles containing DOX) × 100%, respectively.
2.7. In vitro drug release from micelles triggered by DTT
The release profiles of DOX from P-ss-SA micelles and P–SA micelles were studied using a dialysis method. Lyophilized DOX-loaded P-ss-SA micelles or P–SA micelles containing 0.2 mg DOX were suspended in 2 mL of PBS (10 mM, pH 7.4) and placed in a dialysis tube (MWCO 10000). Then the tube was immersed in release media (100 mL PBS; 10 mM, pH 7.4) with or without 10 mM DTT in a shaking water bath at 37 °C. At predetermined time intervals, 3 mL of release media was withdrawn and replaced with 3 mL of the corresponding fresh buffer solution. The amount of DOX release was determined by fluorescence measurement (excitation at 485 nm).2.8. Hemolysis test
1 mL sheep blood sample was added to 2 mL PBS, and then red blood cells (RBCs) were isolated from serum by centrifugation at 2000 rpm for 5 min. The supernatant containing plasma and platelets was discarded. The washing procedure was continued with PBS until the supernatant was clear. The resultant RBC suspension (about 200 μL) was diluted to 9.8 mL of PBS, producing stock RBC solution with about 2% RBC suspensions. Herein, RBCs incubation with deionized water and PBS were used as the positive and negative controls, respectively. 1 mL of 2% RBC suspensions were added to 1 mL samples until the final concentration of P-ss-SA, P–SA and Tween 80 solutions ranged from 0.25 to 1 mg mL−1. The mixture was incubated at 37 °C for 6 h and then centrifuged at 2000 rpm for 5 min to remove intact RBCs. The absorbance of the supernatant was measured for release of hemoglobin at 545 nm.35 All measurements were performed in triplicate and the percentage of hemolysis was calculated as Hemolysis (%) = (Atest − Aneg)/(Apos − Aneg) × 100%, where Atest, Aneg, Apos are the absorbance values of the test sample, negative control (PBS) and positive control (water), respectively.2.9. MTT assay
The cytotoxicity of blank or DOX-loaded polymeric micelles was studied by the MTT assays using HepG2 cells and MCF-7 cells. Cells were seeded onto a 96-well plate at a density of 1 × 104 cells per well in 100 μL of Dulbecco's Modified Eagle medium (DMEM) containing 10% FBS and incubated for 24 h (37 °C, 5% CO2). The medium was replaced by 100 μL samples of various concentrations of blank or DOX-loaded polymeric micelles. The cells were incubated for another 48 h, and then replaced by 100 μL of MTT solution (0.5 mg mL−1). The cells were further incubated for another 4 h, and the medium was aspirated and replaced by 100 μL of DMSO to dissolve the resulting purple crystals. The absorbance was measured at 570 and 630 nm using a microplate reader (Thermo Fisher Scientific). Cell viability was expressed as a percentage of the control culture value.2.10. CLSM observation
Confocal laser scanning microscopy (CLSM) was employed to examine the intracellular distribution of DOX. MCF-7 cells were seeded on glass bottom dishes at a density of 5 × 104 cells per dish in 1 mL of DMEM containing 10% FBS and incubated for 24 h (37 °C, 5% CO2). The cells were then incubated with free DOX, DOX-loaded P–SA micelles or DOX-loaded P-ss-SA micelles at a final DOX concentration of 10 μg mL−1 in DMEM for 4 h or 8 h at 37 °C. The culture media was removed and the cells were rinsed with PBS for three times to remove micelles that were not ingested by the cells. Then the nuclei were stained with 10 μL (1 mg mL−1) of Hoechst 33342 at 37 °C for 15 min. Finally, the cells were washed three times with PBS and incubated with 1 mL DMEM. Fluorescence images of cells were obtained with OLYMPUS FV10-ASW confocal fluorescence microscope.2.11. Evaluation of cellular uptake by FCM
MCF-7 cells were seeded onto 6-well plates at a density of 1 × 105 cells per well in 1 mL of DMEM containing 10% FBS and incubated for 24 h (37 °C, 5% CO2). The cells were then incubated with free DOX, DOX-loaded P–SA micelles or DOX-loaded P-ss-SA micelles at a final DOX concentration of 10 μg mL−1 in DMEM for 4 h and 8 h at 37 °C. The culture media was removed and the cells were rinsed with PBS for three times to remove micelles that were not ingested by the cells. The cells were harvested by trypsinization and resuspended in PBS after centrifugation (1000 rpm, 5 min) and flow cytometry was done using a BD FACSCantoTM flow cytometer (FCM).2.12. Statistical analysis
Data were presented as mean ± standard deviation. Statistical analysis was conducted by using one-way analysis of variance (ANOVA) and p-values <0.05 were considered statistically significant.3. Results and discussion
3.1. Synthesis of P-ss-SA conjugates and P–SA conjugates
The synthesis schemes of P-ss-SA conjugates and P–SA conjugates were presented in Fig. 1. In order to prepare amphiphilic P-ss-SA conjugates and P–SA conjugates, P was first modified with succinic anhydride to introduce carboxyl groups. For reduction-sensitive P-ss-SA conjugate, cystamine was used as linker to couple pullulan succinate (P-COOH) and SA. The non-reduction-sensitive P–SA conjugate using adipic dihydrazide to link P-COOH and SA with both amide bonds was synthesized as control. In our preliminary experiments, stearic acid modified P derivatives with different degrees of substitution were synthesized based on our previous report and other research.40,41 We found that conjugates with degree of substitution (DS, number of SA residues per 100 glucose units in pullulan) of around 5 might be beneficial for the formation of nanomicelles which possess good physicochemical properties. Conjugates with lower DS were prone to the formation of nanomicelles with larger size and less stability. Conjugates with higher DS seemed to form nanomicelles which were too tight to show reduction-sensitivity due to the difficult access of reducing agent to the core of nanomicelles. Thus, in this paper, we presented reduction-sensitive P-ss-SA conjugates with only one kind of DS and non-reduction-sensitive P–SA conjugate was then synthesized with a similar DS for comparison. | ||
| Fig. 1 Synthetic scheme of P-ss-SA conjugate and P–SA conjugate. | ||
The chemical structures of P-ss-SA and P–SA conjugates were confirmed using 1H NMR, as shown in Fig. 2. The representative structure of pullulan (P) in D2O was shown in Fig. 2(a). The broad chemical shifts in the wide region of 3.4–4.0 ppm were mainly associated with the inner methylidyne and methylene protons (![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif)
![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) –O and 2–O) on glucose units of pullulan. The chemical shift associated with the unique methylidyne proton (O––O) of glucose units was at about 4.9 ppm.35 For P-COOH (Fig. 2(b)), the chemical shift at the range of 2.58–2.85 ppm was associated with methylene protons (C–2–2–C) of succinic acid moieties. The degree of succinoylation quantified by titration was around 48%. For P-ADH (Fig. 2(c)), the chemical shift at the range of 2.40–2.58 ppm was associated with methylene protons (CO–2–2–2–2–CO) in adipic dihydrazide. For P-CYS (Fig. 2(e)), the chemical shift at the range of 2.85–2.96 ppm was associated with methylene (NH–2–2–S) in cystamine. For P–SA (Fig. 2(d)) and P-ss-SA (Fig. 2(f)), the chemical shift at the range of 1.1–1.3 ppm was associated with the methylene and methyl protons (CO–(–CH2)16–CH3) in stearic acid. The degree of substitution (DS, defined as the number of stearic acid per 100 sugar residues in pullulan) for the P-ss-SA and P–SA micelles were 5.36 and 4.37, respectively (Table 1).
–O and 2–O) on glucose units of pullulan. The chemical shift associated with the unique methylidyne proton (O––O) of glucose units was at about 4.9 ppm.35 For P-COOH (Fig. 2(b)), the chemical shift at the range of 2.58–2.85 ppm was associated with methylene protons (C–2–2–C) of succinic acid moieties. The degree of succinoylation quantified by titration was around 48%. For P-ADH (Fig. 2(c)), the chemical shift at the range of 2.40–2.58 ppm was associated with methylene protons (CO–2–2–2–2–CO) in adipic dihydrazide. For P-CYS (Fig. 2(e)), the chemical shift at the range of 2.85–2.96 ppm was associated with methylene (NH–2–2–S) in cystamine. For P–SA (Fig. 2(d)) and P-ss-SA (Fig. 2(f)), the chemical shift at the range of 1.1–1.3 ppm was associated with the methylene and methyl protons (CO–(–CH2)16–CH3) in stearic acid. The degree of substitution (DS, defined as the number of stearic acid per 100 sugar residues in pullulan) for the P-ss-SA and P–SA micelles were 5.36 and 4.37, respectively (Table 1).
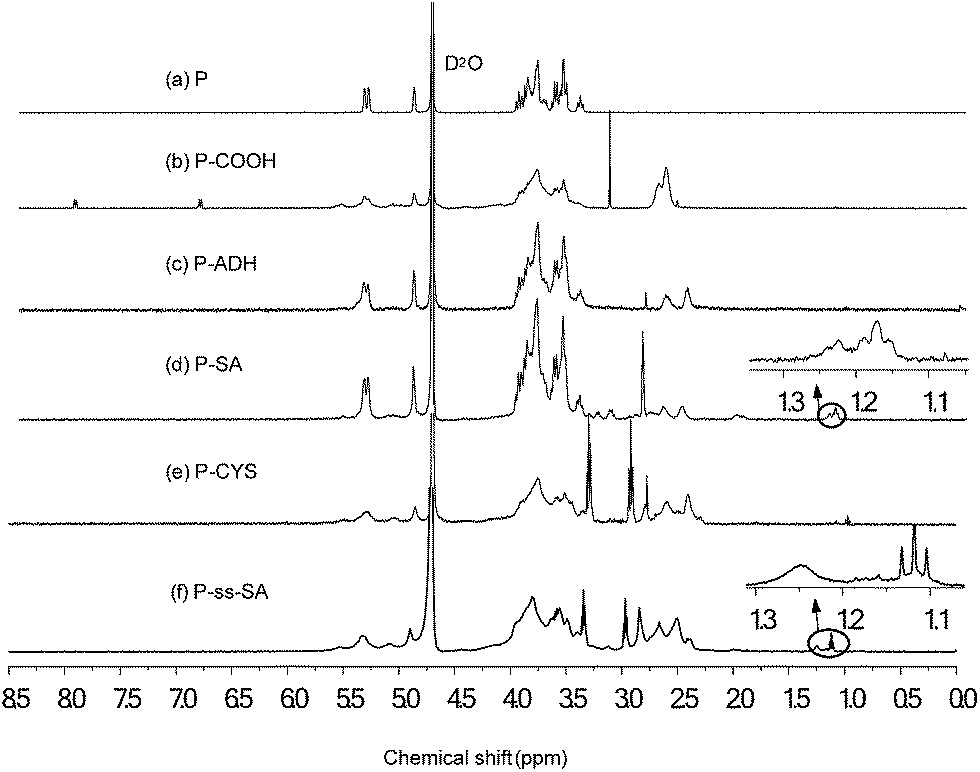 | ||
| Fig. 2 1H NMR spectra of P (a), P-COOH (b), P-ADH (c), P–SA (d), P-CYS (e), and P-ss-SA (f). | ||
| Sample | DSa | CMCb (mg L−1) | Sizec (nm) | PDIc | Zeta potentialc (mV) |
|---|---|---|---|---|---|
| a Degree of substitution of stearic acid.b Measured using pyrene as a fluorescence probe.c Micelle size and zeta potential was measured by DLS. | |||||
| P–SA | 4.37 | 88.23 | 194.8 ± 2.2 | 0.227 ± 0.002 | −16.1 ± 2.2 |
| P-ss-SA | 5.36 | 51.24 | 187.7 ± 7.8 | 0.228 ± 0.013 | −25.9 ± 4.2 |
3.2. Preparation and characterization of micelles
The reduction-sensitive P-ss-SA and non-reduction-sensitive P–SA micelles were prepared by a simple dialysis method. The particle size of P-ss-SA and P–SA micelles measured by dynamic light scattering (DLS) were 187.7 ± 7.8 nm and 194.8 ± 2.2 nm, respectively (Table 1). The morphology of P-ss-SA and P–SA micelles was observed by TEM, and as shown in Fig. 3, both P-ss-SA and P–SA micelles were approximate spherical in shape and the mean size was 26.9 ± 8.2 nm and 29.7 ± 10.9 nm, respectively. The size measured from TEM was smaller than that from DLS. This difference probably was because the samples for TEM undergo a shrinkage due to water evaporation under air-drying, while size determination by DLS was conducted under aqueous condition.27,29,30 Additionally, because only part of the carboxyl group of P-COOH was modified with cystamine or adipic dihydrazide,27,29 the zeta potential of P-ss-SA and P–SA micelles were around −25.9 mV and −16.1 mV, respectively (Table 1). The negatively-charged surface provides a repelling force between the micelles and increases the stability of the micelles. No precipitation was observed in P-ss-SA or P–SA micelles solution after stewing at 4 °C for 1 month and the particle size and zeta potential didn't change much measured by DLS (Fig. 4(c)). The critical micelles concentration (CMC) of amphiphilic P-ss-SA conjugates and P–SA conjugates evaluated by fluorescence spectroscopy using pyrene as a probe was 51.24 mg L−1 and 88.23 mg L−1, respectively (Table 1). Such low CMC values suggested that the micellar structure would retain intact under highly diluted conditions after administration, which were advantageous to prolong blood circulation time.27 These results suggested that the P-ss-SA micelles possess suitable properties to be drug carriers. | ||
| Fig. 3 TEM images of blank micelles and DOX-loaded micelles. | ||
 | ||
| Fig. 4 The size change of P-ss-SA micelles (a) and P–SA micelles (b) in response to 10 mM DTT and the stability of P-ss-SA micelles (c) in PBS (10 mM, pH 7.4) determined by DLS measurement. | ||
3.3. Disassembly of micelles triggered by DTT
To investigate reduction triggered disassemble behaviors of P-ss-SA micelles, DTT which is a water-soluble reducing agents was utilized to simulate the intracellular reductive condition. P-ss-SA micelles were treated with 10 mM DTT, a reductive environment analogous to that of the intracellular compartments such as cytosol and the cell nucleus.42 At predetermined time, the size of micelles was determined by DLS. As shown in Fig. 4(a), the mean size of P-ss-SA micelles without DTT treatment didn't change much after 24 h, by contrast, the mean size of P-ss-SA micelles increased continuously from about 188 nm to 716 nm after adding DTT for 24 h. The disulfide linkages in the side chain in the core of the P-ss-SA micelles were reduced into free thiols after adding reducing agent DTT, which made hydrophobic stearic acid groups detach from hydrophilic pullulan main chain and thus resulted in the dissociation of micelles to enlarge the mean size. But the pullulan main chain were very long chain, when the conjugates self-assembled into nano-micelles, the pullulan polysaccharide backbones coiled to form hydrophilic shells, they couldn't get completely decomposed in 24 h. Such trend was also observed in other amphiphilic copolymers.7,29,42,43 On the other hand, for P–SA micelles with or without DTT treatment (Fig. 4(b)), the mean micelle size didn't change much.3.4. Preparation and characterization of DOX-loaded micelles
DOX was chosen because it is one of the most commonly used chemotherapeutic drugs and it is easily examined for its inherent fluorescence. In this study, DOX was loaded into micelles by a simple dialysis method.11,25,30 Firstly, DOX·HCl was stirred with twice the number of moles of TEA in DMSO to detach HCl and render the drug hydrophobic. Then DOX/DMSO solution was added to P-ss-SA or P–SA in DMSO at different drug-to-carrier ratio. Then, the mixture solutions were transferred into dialysis bag (MWCO 10000) and dialyzed against deionized water for 48 h. As DMSO went out and water came in, the hydrophobic stearic acid groups aggregated to form many hydrophobic cores and the pullulan backbones coiled to form hydrophilic shells outside these hydrophobic cores, resulting in the formation of nano-micelles with a core–shell structure. Meanwhile, DOX was entrapped into the hydrophobic cores of micelles through its hydrophobic interactions with stearic acid groups during the formation of nano-micelles.The properties of DOX-loaded micelles were summarized in Table 2. DOX loading capacity increased from 3.51% to 7.56% with the weight ratio of DOX to P-ss-SA increasing from 1/20 to 1/5, but entrapment efficiency decreased from 72.81% to 40.88% at the same time. Moreover, with the drug feed ratio increased, the particle size increased slightly because of the increase in the inner core volume. The DOX-loaded P–SA micelles showed the same trend with DOX-loaded P-ss-SA micelles. Thus, the optimal weight ratio 1/10 based on the drug loading properties was selected for further investigation.
| Sample | Drug/carrier | EEa (%) | DLa (%) | Sizeb (nm) | PDIb | Zeta potentialb (mV) |
|---|---|---|---|---|---|---|
| a EE and DL are short for entrapment efficiency and drug-loading, respectively.b Micelle size and zeta potential was measured by DLS. | ||||||
| P–SA | 1/5 | 45.36 ± 1.60 | 8.31 ± 0.27 | 228.8 ± 7.8 | 0.106 ± 0.005 | −16.0 ± 6.7 |
| 1/10 | 73.31 ± 2.50 | 6.83 ± 0.22 | 214.6 ± 2.5 | 0.089 ± 0.008 | −14.9 ± 3.3 | |
| 1/20 | 77.58 ± 3.78 | 3.73 ± 0.18 | 205.6 ± 0.6 | 0.175 ± 0.004 | −13.0 ± 0.4 | |
| P-ss-SA | 1/5 | 40.88 ± 1.33 | 7.56 ± 0.23 | 220.8 ± 5.4 | 0.223 ± 0.005 | −26.4 ± 5.9 |
| 1/10 | 65.53 ± 0.78 | 6.19 ± 0.06 | 194.4 ± 0.4 | 0.213 ± 0.010 | −24.2 ± 1.9 | |
| 1/20 | 72.81 ± 6.79 | 3.51 ± 0.32 | 190.1 ± 5.8 | 0.249 ± 0.001 | −23.2 ± 1.6 | |
The morphology of DOX-loaded micelles was also observed by TEM (Fig. 3). The mean size of DOX-loaded P-ss-SA micelles (P-ss-SA/DOX) and DOX-loaded P–SA micelles (P–SA/DOX) was 29.2 ± 4.7 nm and 30.6 ± 13.6 nm, respectively. The size measured from TEM (around 30 nm) was smaller than that from DLS (around 200 nm) because of different sample preparation technologies.27,29,30
3.5. In vitro drug release from micelles triggered by DTT
In order to evaluate the reduction triggered DOX release behavior, 10 mM DTT was used to mimic the intracellular reductive condition with reduction-sensitive DOX-loaded P-ss-SA micelles and non-reduction-sensitive DOX-loaded P–SA micelles. As shown in Fig. 5(a), the release of DOX from reduction-sensitive P-ss-SA micelles was significantly accelerated by adding DTT to the release media (p < 0.05). The P-ss-SA micelles released more than 80% DOX in the first 5 h under 10 mM DTT. However, in the case without DTT, less than 60% DOX in P-ss-SA micelles was released within the same period (p < 0.05). By contrast, the release rate and level of DOX from P–SA micelles revealed negligible change after the addition of 10 mM DTT (Fig. 5(b)). These results were coincident with the outcomes illustrated in Fig. 4, the mean size of reduction-sensitive P-ss-SA micelles increased continuously under reductive condition due to the reductive cleavage of the disulfide bonds, and the encapsulated DOX released much faster and more due to the disassembly of the micelles. Therefore, compared with reduction-insensitive P–SA micelles, reduction-sensitive P-ss-SA micelles showed preferable stability and reduction sensitivity and may achieve tumor site-specific DOX delivery under reducing environment. The similar results were also reported for other reduction-sensitive micelles.25,28,43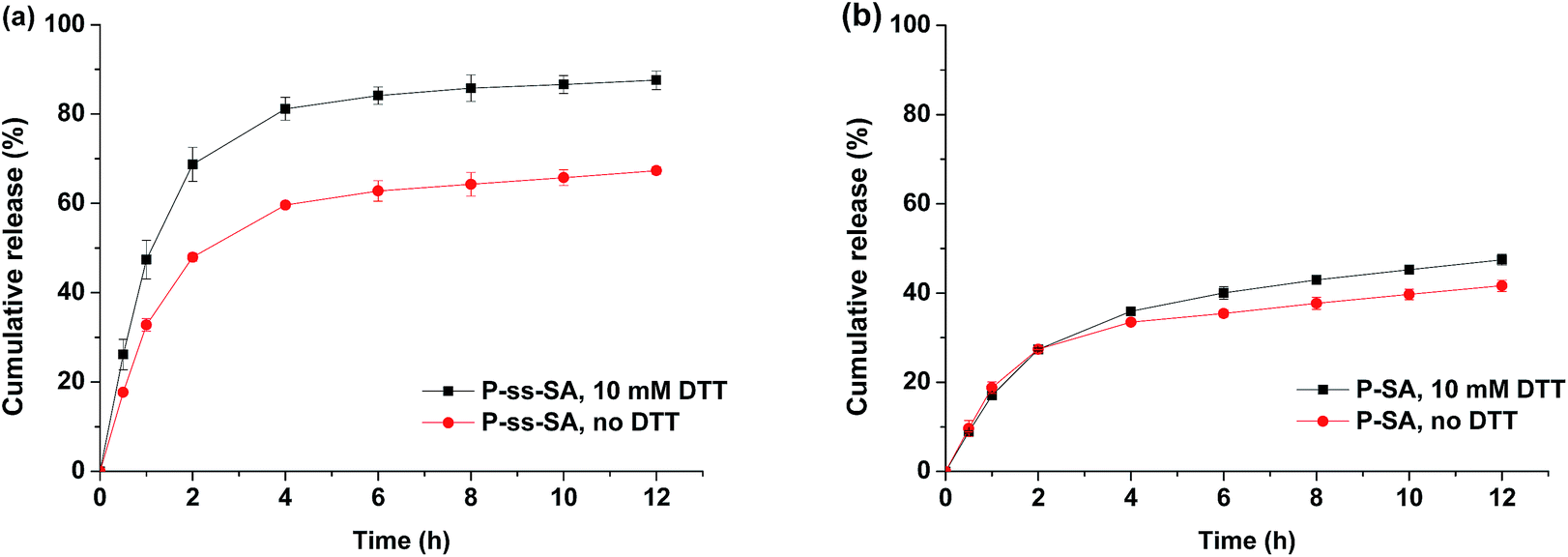 | ||
| Fig. 5 Reduction-triggered release of DOX from P-ss-SA micelles (a) and P–SA micelles used as a reduction insensitive control (b). Error bars represent the standard deviation of three measurements (mean ± SD, n = 3). | ||
3.6. Hemolysis test
In order to evaluate the blood compatibility of P–SA and P-ss-SA micelles, the hemolysis test was performed. The level of hemolysis of P–SA and P-ss-SA micelles was compared with that of Tween 80, which can cause hemolysis. As shown in Fig. 6, the hemolysis induced by Tween 80 increased dramatically from 17.51 ± 0.47% to 89.34 ± 1.45% with the concentration increasing from 0.25 mg mL−1 to 1 mg mL−1, while the hemolysis caused by P–SA or P-ss-SA micelles were all below 2% at the same concentration range, which was significantly different from Tween 80 (p < 0.05). It was reported that up to 5% hemolysis is permissible for biomaterials.35 Thus, both P–SA and P-ss-SA micelles have shown good blood compatibility. | ||
| Fig. 6 Hemolysis test results of P–SA, P-ss-SA and Tween 80 at different concentrations. Error bars represent the standard deviation of three measurements (mean ± SD, n = 3, *p < 0.05). | ||
3.7. In vitro cytotoxicity studies
In order to investigate the cytotoxicity of blank micelles and DOX-loaded micelles, MTT assay was conducted against HepG2 and MCF-7 cells. As shown in Fig. 7, with the concentrations of blank micelles ranged from 25 to 200 μg mL−1, the cell viability of HepG2 and MCF-7 cells against P–SA or P-ss-SA micelles after 48 h were all around 90%, which indicated that the two micelles were non-toxic to HepG2 and MCF-7 cells and fairly safe to be used as drug carriers. | ||
| Fig. 7 Cytotoxicity of P-ss-SA micelles and P–SA micelles in HepG2 cells (a) and MCF-7 cells (b) after 48 h. Error bars represent the standard deviation of six measurements (mean ± SD, n = 6). | ||
The in vitro cytotoxicity of DOX-loaded micelles was characterized by the half maximal inhibitory concentration (IC50) to HepG2 and MCF-7 cells. The results were presented in Fig. 8. The IC50 of DOX-loaded P-ss-SA micelles, DOX-loaded P–SA micelles and free DOX to HepG2 cells were 0.740 ± 0.108 μg mL−1, 12.181 ± 0.589 μg mL−1 and 1.123 ± 0.437 μg mL−1. Compared with DOX-loaded P–SA micelles, DOX-loaded P-ss-SA micelles exhibited significantly higher cytotoxicity to HepG2 cells (p < 0.05). This finding can be explained as the much faster release of DOX from reduction-sensitive P-ss-SA micelles by cleavage of the disulfide bond in the intracellular redox potential. The IC50 of DOX-loaded P-ss-SA micelles, DOX-loaded P–SA micelles and free DOX to MCF-7 cells were 0.949 ± 0.184 μg mL−1, 8.493 ± 3.997 μg mL−1 and 1.181 ± 0.179 μg mL−1. Reduction-sensitive DOX-loaded P-ss-SA micelles also exhibited significantly superior anti-tumor activity compared with DOX-loaded P–SA micelles due to fast DOX release under the intracellular reductive conditions in MCF-7 cells. However, both DOX-loaded P-ss-SA micelles and DOX-loaded P–SA micelles didn't exhibit significantly higher cytotoxicity to HepG2 cells than to MCF-7 cells, it has to be noted that extensive chemical modification of the native polysaccharide may greatly influence its affinity for the liver.38,44
 | ||
| Fig. 8 Cytotoxicity of P-ss-SA/DOX micelles, P–SA/DOX micelles and free DOX in HepG2 cells (a) and MCF-7 cells (b) after 48 h. Error bars represent the standard deviation of six measurements (mean ± SD, n = 6). | ||
3.8. In vitro cellular uptake of DOX-loaded micelles
In order to investigate the cellular uptake of DOX-loaded micelles, CLSM and FCM were used to study the cellular uptake of the two DOX-loaded micelles. CLSM images were shown in Fig. 9. Both DOX-loaded micelles could be effectively internalized in MCF-7 cells after 4 h. Free DOX presented red fluorescence in both cytoplasm and nuclei, this can be explained that DOX is a small molecule and it can quickly diffuse into cells and enter nuclei by passive diffusion.13,24 DOX-loaded P–SA micelles presented red fluorescence only in cytoplasm, but DOX-loaded P-ss-SA micelles presented red fluorescence not only in cytoplasm but also weak red fluorescence in nuclei. With further incubation for 8 h, Cells incubated with DOX-loaded P-ss-SA micelles presented stronger DOX fluorescence in the cytoplasm and nuclei because of more DOX released from P-ss-SA micelles under reducing environment. By contrast, almost no DOX fluorescence was observed in nuclei of the cells incubated with DOX-loaded P–SA micelles, which may be caused by the lower release rate and level of DOX from DOX-loaded P–SA micelles. Because it has been reported that at the same concentration of DOX, DOX fluorescence loaded in micelles was lower compared to that of free DOX due to the self-quenching effect of DOX in confined environment.25 Therefore, the images in Fig. 9 indicated the time-dependent cellular uptake pathways of the DOX-loaded micelles and rapid DOX release from reduction-sensitive P-ss-SA micelles under reductive intracellular condition because of reductive cleavage of the disulfide bonds and disassembly of the micelles. | ||
| Fig. 9 CLSM images of MCF-7 cells treated with DOX loaded micelles for 4 h and 8 h (DOX dosage was 10 μg mL−1). | ||
The quantitative fluorescence intensity was further measured by FCM. The results were shown in Fig. 10. It was clear that the fluorescence intensity of MCF-7 cells treated with DOX-loaded P-ss-SA micelles was significant higher than that of cells treated with DOX-loaded P–SA micelles at both 4 and 8 h. Pullulan-based nanomicelles have been reported to enter cancer cells by endocytosis and then the loaded drug was subsequently released.11,13 The fluorescence signals are associated with the DOX release quantity from the micelles. Therefore, the results revealed that DOX released much faster from the reduction-sensitive P-ss-SA micelles inside the cells, which is well in accordance with the CLSM observation. Hence, the reduction-sensitive P-ss-SA micelles might be used as a suitable anticancer drug carrier.
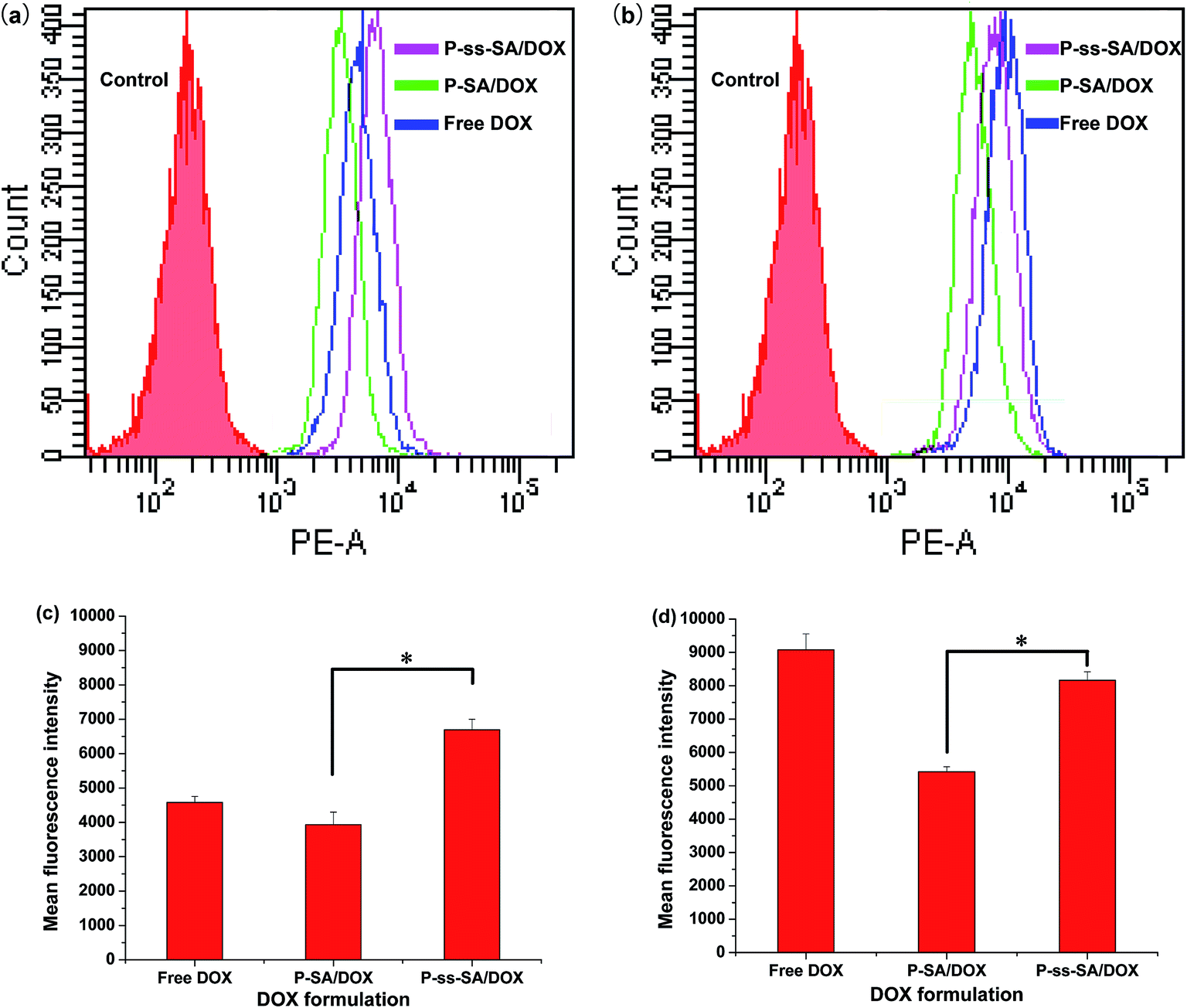 | ||
| Fig. 10 FCM results of MCF-7 cells treated with DOX loaded micelles for 4 h ((a) and (c)) and 8 h ((b) and (d)). DOX dosage was 10 μg mL−1. | ||
4. Conclusion
In this study, a novel reduction-sensitive amphiphilic polymer P-ss-SA was successfully synthesized by introducing stearic acid into pullulan with a reduction-sensitive disulfide bond. The chemical structure of P-ss-SA was confirmed by 1H NMR. The polymer can easily self-assemble into micelles in aqueous media and encapsulate DOX. The results measured by DLS showed the mean size of blank and DOX-loaded micelles was around 190 nm, and the mean size of reduction-sensitive P-ss-SA micelles increased dramatically under reductive conditions. In vitro release of DOX from reduction-sensitive P-ss-SA micelles showed a reduction-triggered drug release under reducing environment. The confocal laser microscopy and flow cytometry measurements indicated that the intracellular reductive conditions broke the disulfide bonds in P-ss-SA micelles and triggered the fast release of DOX. The in vitro IC50 of the DOX-loaded P-ss-SA micelles was lower than that of DOX-loaded P–SA to both HepG2 and MCF-7 cells. The blank micelles showed negligible cytotoxicity and possessed excellent hemocompatibility. Therefore, the biocompatible reduction-sensitive P-ss-SA micelles can be used as potential carrier systems for the intracellular delivery of DOX and enhance the anticancer efficacy.Acknowledgements
The authors are grateful to the financial support of the NSF of China (grant no. 21376039).References
- R. Haag and F. Kratz, Angew. Chem., Int. Ed., 2006, 45, 1198–1215 CrossRef CAS PubMed.
- M. E. Davis, Z. G. Chen and D. M. Shin, Nat. Rev. Drug Discovery, 2008, 7, 771–782 CrossRef CAS PubMed.
- A. Schroeder, D. A. Heller, M. M. Winslow, J. E. Dahlman, G. W. Pratt, R. Langer, T. Jacks and D. G. Anderson, Nat. Rev. Cancer, 2012, 12, 39–50 CrossRef CAS PubMed.
- C. Deng, Y. Jiang, R. Cheng, F. Meng and Z. Zhong, Nano Today, 2012, 7, 467–480 CrossRef CAS PubMed.
- S. Mura, J. Nicolas and P. Couvreur, Nat. Mater., 2013, 12, 991–1003 CrossRef CAS PubMed.
- Q. Zhang, N. R. Ko and J. K. Oh, Chem. Commun., 2012, 48, 7542–7552 RSC.
- W. Chen, Y. Zou, F. Meng, R. Cheng, C. Deng, J. Feijen and Z. Zhong, Biomacromolecules, 2014, 15, 900–907 CrossRef CAS PubMed.
- Y. Zhong, W. Yang, H. Sun, R. Cheng, F. Meng, C. Deng and Z. Zhong, Biomacromolecules, 2013, 14, 3723–3730 CrossRef CAS PubMed.
- J. Fang, H. Nakamura and H. Maeda, Adv. Drug Delivery Rev., 2011, 63, 136–151 CrossRef CAS PubMed.
- J. Cao, T. Su, L. Zhang, R. Liu, G. Wang, B. He and Z. Gu, Int. J. Pharm., 2014, 471, 28–36 CrossRef CAS PubMed.
- H. Guo, Y. Liu, Y. Wang, J. Wu, X. Yang, R. Li, Y. Wang and N. Zhang, Carbohydr. Polym., 2014, 111, 908–917 CrossRef CAS PubMed.
- Y. Wang, H. Chen, Y. Liu, J. Wu, P. Zhou, Y. Wang, R. Li, X. Yang and N. Zhang, Biomaterials, 2013, 34, 7181–7190 CrossRef CAS PubMed.
- Y. Wang, Y. Liu, Y. Liu, Y. Wang, J. Wu, R. Li, J. Yang and N. Zhang, Polym. Chem., 2014, 5, 423–432 RSC.
- L. Sun, X. Zhang, J. An, C. Su, Q. Guo and C. Li, RSC Adv., 2014, 4, 20208–20215 RSC.
- R. Banerjee and D. Dhara, Langmuir, 2014, 30, 4137–4146 CrossRef CAS PubMed.
- P. Yang, D. Li, S. Jin, J. Ding, J. Guo, W. Shi and C. Wang, Biomaterials, 2014, 35, 2079–2088 CrossRef CAS PubMed.
- S. H. Medina, M. V. Chevliakov, G. Tiruchinapally, Y. Y. Durmaz, S. P. Kuruvilla and M. E. Elsayed, Biomaterials, 2013, 34, 4655–4666 CrossRef CAS PubMed.
- Q. Yuan, Y. Zhang, T. Chen, D. Lu, Z. Zhao, X. Zhang, Z. Li, C.-H. Yan and W. Tan, ACS Nano, 2012, 6, 6337–6344 CrossRef CAS PubMed.
- J. Wang, G. Yang, X. Guo, Z. Tang, Z. Zhong and S. Zhou, Biomaterials, 2014, 35, 3080–3090 CrossRef CAS PubMed.
- F. Meng, W. E. Hennink and Z. Zhong, Biomaterials, 2009, 30, 2180–2198 CrossRef CAS PubMed.
- H. Wei, R.-X. Zhuo and X.-Z. Zhang, Prog. Polym. Sci., 2013, 38, 503–535 CrossRef CAS PubMed.
- A. Jhaveri, P. Deshpande and V. Torchilin, J. Controlled Release, 2014, 190, 352–370 CrossRef CAS PubMed.
- R. Cheng, F. Feng, F. Meng, C. Deng, J. Feijen and Z. Zhong, J. Controlled Release, 2011, 152, 2–12 CrossRef CAS PubMed.
- X. Zhang, K. Achazi, D. Steinhilber, F. Kratz, J. Dernedde and R. Haag, J. Controlled Release, 2014, 174, 209–216 CrossRef CAS PubMed.
- C. Cui, Y. N. Xue, M. Wu, Y. Zhang, P. Yu, L. Liu, R. X. Zhuo and S. W. Huang, Biomaterials, 2013, 34, 3858–3869 CrossRef CAS PubMed.
- H. Wang, L. Tang, C. Tu, Z. Song, Q. Yin, L. Yin, Z. Zhang and J. Cheng, Biomacromolecules, 2013, 14, 3706–3712 CrossRef CAS PubMed.
- C. Yu, C. Gao, S. Lü, C. Chen, Y. Huang and M. Liu, Chem. Eng. J., 2013, 228, 290–299 CrossRef CAS PubMed.
- J. Yang, Y. Huang, C. Gao, M. Liu and X. Zhang, Colloids Surf., B, 2014, 115, 368–376 CrossRef CAS PubMed.
- J. Li, M. Huo, J. Wang, J. Zhou, J. M. Mohammad, Y. Zhang, Q. Zhu, A. Y. Waddad and Q. Zhang, Biomaterials, 2012, 33, 2310–2320 CrossRef CAS PubMed.
- P. Sun, D. Zhou and Z. Gan, J. Controlled Release, 2011, 155, 96–103 CrossRef CAS PubMed.
- J. Wang, B. Dou and Y. Bao, Mater. Sci. Eng., C, 2014, 34, 98–109 CrossRef CAS PubMed.
- V. D. Prajapati, G. K. Jani and S. M. Khanda, Carbohydr. Polym., 2013, 95, 540–549 CrossRef CAS PubMed.
- J. C. Fricain, S. Schlaubitz, C. Le Visage, I. Arnault, S. M. Derkaoui, R. Siadous, S. Catros, C. Lalande, R. Bareille, M. Renard, T. Fabre, S. Cornet, M. Durand, A. Leonard, N. Sahraoui, D. Letourneur and J. Amedee, Biomaterials, 2013, 34, 2947–2959 CrossRef CAS PubMed.
- H. Zhang, F. Li, J. Yi, C. Gu, L. Fan, Y. Qiao, Y. Tao, C. Cheng and H. Wu, Eur. J. Pharm. Sci., 2011, 42, 517–526 CrossRef CAS PubMed.
- X. C. Yang, Y. L. Niu, N. N. Zhao, C. Mao and F. J. Xu, Biomaterials, 2014, 35, 3873–3884 CrossRef CAS PubMed.
- M. R. Rekha and C. P. Sharma, Acta Biomater., 2011, 7, 370–379 CrossRef CAS PubMed.
- H. Li, S. Bian, Y. Huang, J. Liang, Y. Fan and X. Zhang, J. Biomed. Mater. Res., Part A, 2013, 102, 150–159 CrossRef PubMed.
- M. R. Rekha and C. P. Sharma, Biomaterials, 2009, 30, 6655–6664 CrossRef CAS PubMed.
- H. Li, Y. Cui, J. Liu, S. Bian, J. Liang, Y. Fan and X. Zhang, J. Mater. Chem. B, 2014, 2, 3500–3510 RSC.
- J. Wang, D. Song and Y. Bao, Acta Chim. Sin., 2012, 70, 1193–1200 CrossRef CAS.
- Y.-Z. Du, Q. Weng, H. Yuan and F.-Q. Hu, ACS Nano, 2010, 4, 6894–6902 CrossRef CAS PubMed.
- Q. Guo, P. Luo, Y. Luo, F. Du, W. Lu, S. Liu, J. Huang and J. Yu, Colloids Surf., B, 2012, 100, 138–145 CrossRef CAS PubMed.
- L.-Y. Tang, Y.-C. Wang, Y. Li, J.-Z. Du and J. Wang, Bioconjugate Chem., 2009, 20, 1095–1099 CrossRef CAS PubMed.
- K. Raemdonck, T. F. Martens, K. Braeckmans, J. Demeester and S. C. De Smedt, Adv. Drug Delivery Rev., 2013, 65, 1123–1147 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2014 |