Ni(salphen)-based metal–organic framework for the synthesis of cyclic carbonates by cycloaddition of CO2 to epoxides†
Yanwei
Ren
,
Yanchao
Shi
,
Junxian
Chen
,
Shaorong
Yang
,
Chaorong
Qi
and
Huanfeng
Jiang
*
School of Chemistry and Chemical Engineering, South China University of Technology, Guangzhou 510640, China. E-mail: jianghf@scut.edu.cn; Fax: +86-20-22236337
First published on 7th December 2012
Abstract
A well-defined homogeneous molecular catalyst Ni(salphen) was introduced as a “metalloligand” in a MOF, providing an efficient and recyclable heterogeneous catalyst for the synthesis of cyclic carbonates by the cycloaddition of CO2 to epoxides under relatively mild conditions.
Introduction
Although carbon dioxide (CO2) is a nontoxic, non-flammable and abundant C1 building block, the scope of the synthetic applications of CO2 is limited because of its inert nature. The development of efficient synthetic methods for CO2 fixation is therefore an important and challenging subject.1 Cyclic carbonates derived from the coupling reactions of CO2 with epoxides are promising target molecules since they can be widely used for various applications, such as electrolytes in lithium-ion batteries, raw materials for polycarbonates, and polar aprotic solvents.2Various homogeneous metal complex catalysts, such as M(salen) complexes3 have been developed for the cycloaddition of CO2 and epoxides. Although these homogeneous catalysts usually exhibit high activity and selectivity at mild temperatures in the presence of co-catalysts, their practical applications remain limited because of catalyst instability and difficulty in catalyst/product separation. The immobilization of homogeneous catalysts can facilitate their recovery and reuse and therefore is of considerable interest to academia and industry.4 According to this strategy, various heterogeneous catalysts have been developed for the coupling reaction of CO2 with epoxides, such as functional polymers,5 ion-exchange resins,6 and quaternary ammonium or phosphonium supported on SiO2.7 Recently, based on their great potential as adsorbents/catalysts due to extremely large surface areas and well-ordered porous structures, several metal–organic framework (MOF) heterogeneous catalysts have been studied for CO2 fixation.8 However, these studies rely on the intrinsic catalytic activity (e.g., weak Lewis acidity) of the metal-connecting points. A more rational strategy than such an “opportunistic” approach is to introduce well-defined homogeneous molecular catalysts (or precatalysts) into the MOF structures.9 Until now, a few MOFs based on M(salen) ligands with additional functional groups such as carboxylates,10p-benzoic acid groups,11 and p-pyridyl groups12 in the para or meta positions to the OH group have been studied as the new generation of heterogeneous catalysts that are capable of catalyzing more advanced and efficient reactions. Surprisingly, no MOF catalyst employing this synthetic strategy for CO2 fixation has been reported so far. Herein we report a new dicarboxyl-functionalized nickel salphen complex Ni–H2L (Scheme 1) as a bridging metalloligand incorporated in a 3D MOF as a self-supported heterogeneous catalyst for the coupling reaction of CO2 with epoxides in the presence of quaternary ammonium salts. To the best of our knowledge, it is the first report of a demonstration in which a M(salen)-based MOF material is efficient both as a CO2 adsorbent and as a catalyst for its chemical fixation.
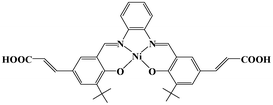 | ||
| Scheme 1 Schematic representation of Ni–H2L. | ||
Results and discussion
The Ni(salphen) ligand Ni–H2L was synthesized by a Schiff base condensation reaction between o-phenylenediamine and (E)-3-(3-tert-butyl-5-fomyl-4-hydroxyphenyl)acrylic acid and then metalated with Ni(OAc)2·4H2O. The reaction of CdCl2 and with Ni–H2L in dimethylformamide (DMF)/H2O at 80 °C for 96 h afforded brown block single crystals of [Cd2(Ni–L)2(H2O)4]·3DMF (1) in good yield. The product was stable in air and insoluble in water and common organic solvents and was formulated on the basis of elemental analysis, IR spectroscopy, and thermogravimetric analysis (TGA). The phase purity of the bulk sample was established by comparison of its observed and simulated powder X-ray diffraction (PXRD) patterns (Fig. S1, ESI†).Single-crystal X-ray diffraction studies showed that 1 adopts a 3D nanoporous framework and crystallizes in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) with two Ni–L units, two Cd ions and four H2O molecules in the asymmetric unit. The basic building blocks, dinuclear Cd2 clusters C-I and C-II (Fig. 1a) with a C2 axis passing through two bridging oxygen atoms, are clustered by two bidentate and two tridentate carboxylate groups of four Ni–L units. In the two Cd2 clusters, one of the independent Cd ions is coordinated by five oxygen atoms from three carboxylate groups, and two water molecules (in the trans-position in C-I, and the cis-position in C-II). The Ni–L units exhibit two coordination modes including the bis-bidentate chelating mode and the mode involving bis-tridentate chelating-bridging carboxylate groups (Fig. S2, ESI†). Each Ni ion is coordinated in a nearly square-planar geometry with two nitrogen atoms and two oxygen atoms from the L ligand (Ni–Oavg = 1.839 Å, Ni–Navg = 1.850 Å). Each Cd2 cluster in 1 is thus linked by four Ni–L ligands, and each Ni–L ligand is linked to two Cd2 clusters to generate a porous 2D network along the a-axis (Fig. S3, ESI†). All the thick lamellar networks are stacked together via supermolecular interactions to form a 3D nanoporous framework with 1D ellipse-shaped channels with a cross section of ∼1.0 × 1.4 nm along the b-axis, which are filled with DMF molecules (Fig. 1b). Therefore, the channel surfaces are uniformly stacked with Ni–L units with coordinatively unsaturated Ni2+ ions, and the Ni–Ni distance of adjacent Ni–L layers is about 5.128 Å, which are accessible to guest molecules and available for cooperative double-Lewis acidic activation. PLATON calculations13 reveal that 1 has 41.3% of the total volume available for guest inclusion. TGA reveals that the coordinated water and DMF molecules could be readily removed in the temperature range 30–120 °C, and the framework is stable up to 150 °C (Fig. S4, ESI†). After a sample of 1 was ground and heated at 100 °C for 5 h under vacuum to obtain activated 1, PXRD of the resultant powder showed a broad diffraction pattern similar to that of the pristine sample. This result indicates that the nanoporous framework was maintained after removal of the solvent molecules. To determine the permanent porosity of 1, 77 K N2 adsorption was carried out on the evacuated framework (Fig. S5, ESI†). A type I isotherm was observed, indicating that 1 is microporous, and the apparent surface area was calculated using the Langmuir method to be 358 m2 g−1 (245 m2 g−1 BET). The CO2 adsorption measurement of the activated 1 showed that up to 25 cm3 g−1 of CO2 uptake was achieved at 273 K and 1.0 atm (Fig. S6, ESI†), a moderate adsorption capacity of lower-pressure CO2 as compared to previously reported MOFs,14 indicating its potential application for CO2 capture and conversion.
with two Ni–L units, two Cd ions and four H2O molecules in the asymmetric unit. The basic building blocks, dinuclear Cd2 clusters C-I and C-II (Fig. 1a) with a C2 axis passing through two bridging oxygen atoms, are clustered by two bidentate and two tridentate carboxylate groups of four Ni–L units. In the two Cd2 clusters, one of the independent Cd ions is coordinated by five oxygen atoms from three carboxylate groups, and two water molecules (in the trans-position in C-I, and the cis-position in C-II). The Ni–L units exhibit two coordination modes including the bis-bidentate chelating mode and the mode involving bis-tridentate chelating-bridging carboxylate groups (Fig. S2, ESI†). Each Ni ion is coordinated in a nearly square-planar geometry with two nitrogen atoms and two oxygen atoms from the L ligand (Ni–Oavg = 1.839 Å, Ni–Navg = 1.850 Å). Each Cd2 cluster in 1 is thus linked by four Ni–L ligands, and each Ni–L ligand is linked to two Cd2 clusters to generate a porous 2D network along the a-axis (Fig. S3, ESI†). All the thick lamellar networks are stacked together via supermolecular interactions to form a 3D nanoporous framework with 1D ellipse-shaped channels with a cross section of ∼1.0 × 1.4 nm along the b-axis, which are filled with DMF molecules (Fig. 1b). Therefore, the channel surfaces are uniformly stacked with Ni–L units with coordinatively unsaturated Ni2+ ions, and the Ni–Ni distance of adjacent Ni–L layers is about 5.128 Å, which are accessible to guest molecules and available for cooperative double-Lewis acidic activation. PLATON calculations13 reveal that 1 has 41.3% of the total volume available for guest inclusion. TGA reveals that the coordinated water and DMF molecules could be readily removed in the temperature range 30–120 °C, and the framework is stable up to 150 °C (Fig. S4, ESI†). After a sample of 1 was ground and heated at 100 °C for 5 h under vacuum to obtain activated 1, PXRD of the resultant powder showed a broad diffraction pattern similar to that of the pristine sample. This result indicates that the nanoporous framework was maintained after removal of the solvent molecules. To determine the permanent porosity of 1, 77 K N2 adsorption was carried out on the evacuated framework (Fig. S5, ESI†). A type I isotherm was observed, indicating that 1 is microporous, and the apparent surface area was calculated using the Langmuir method to be 358 m2 g−1 (245 m2 g−1 BET). The CO2 adsorption measurement of the activated 1 showed that up to 25 cm3 g−1 of CO2 uptake was achieved at 273 K and 1.0 atm (Fig. S6, ESI†), a moderate adsorption capacity of lower-pressure CO2 as compared to previously reported MOFs,14 indicating its potential application for CO2 capture and conversion.
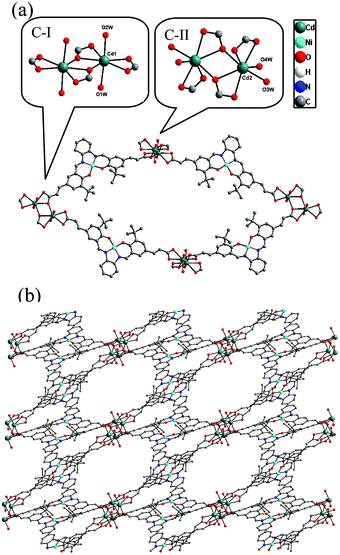 | ||
| Fig. 1 (a) Building blocks of 1 and (b) view of 3D porous structure of 1 along the b-axis. | ||
The moderate stability and uptake for CO2 of the Ni(salphen)-based MOF 1 prompted us to explore its utilization as a heterogeneous catalyst for the synthesis of cyclic carbonates by the cycloaddition of CO2 to epoxides, since M(salen) complexes are promising catalysts for the CO2 fixation reaction.3a The activity of various catalysts was tested at 80 °C and 2 MPa using the reaction of propylene oxide (PO) and CO2 to produce propylene carbonate (PC). As shown in Table 1, the catalytic performance of activated 1 depends strongly on the ammonium salt used, and highest activity was observed using NBu4Br (entries 1–4). Theoretically, iodide is the best promoter in accordance to its increased nucleophilicity, but in the presence of the microporous MOF the diffusion of the large iodide might be hampered, thus explaining the slightly reduced activity compared to the bromides. When the catalyst was not used, the yield of PC was very low (12%), indicating Lewis acid activation of PO was necessary for the reaction (entry 5). To answer the question of whether the reaction was catalyzed by the Ni–L units or the coordinatively unsaturated Cd active sites exposed in the 1D channel of activated 1, we performed test reactions under standard conditions using Ni–H2L and [Cd(bpdc)]n (H2bpdc = 4,4′-biphenyldicarboxylic acid),15 respectively as the catalyst (entries 6 and 7, the molar amounts of Ni–L and Cd were the same as in 1). The results revealed that for both the yield of PC was higher than in the test with no catalyst, but significantly lower than when using 1, clearly indicating that synergistic and/or additive activation of two kinds of Lewis acid in 1 occurred during the reaction process. Another reason for the higher activity of this MOF catalyst is that it is a porous material, which is favourable for increasing the CO2 uptake, and facilitates the access of reactants to the active sites and product diffusion through open channels. At the same time, we found the amount of NBu4Br had a great influence on the reaction. Increasing the amount of NBu4Br from 1 mol% to 3 mol% could enhance the yield of PC significantly (entries 6, 8 and 9). The PC yield was also increased by an increased reaction temperature or time (entries 10–12), but higher temperature reactions were not investigated, because this catalyst is unstable at more than 150 °C. The results of the recycling experiments showed no obvious decrease in the activity of the catalyst after being used three times (entries 13 and 14). Furthermore, the solid catalyst recovered from the catalytic reaction exhibited the same PXRD as the pristine solid MOF 1 (Fig. S1, ESI†), unambiguously supporting the stability of the MOF framework during the catalytic reactions. The good recyclability may be due to the fact that this catalyst was synthesized by the solvothermal method and therefore resisted moderate temperature and pressure. This catalytic capability of the self-supported Ni(salphen) MOF catalyst can be compared to that of other MOF based heterogenerous catalysts reported in the literature for the cycloaddition reaction of CO2 to epoxide (Table 2).
| Entry | Catalyst | Ammonium salt | Yield (%)g |
|---|---|---|---|
| a Typical reaction conditions: PO (10 mmol), ammonium salts (3 mol %), catalyst (0.05 g), CO2 pressure (2 MPa), reaction temperature (80 °C), reaction time (4 h). b 1 mol% NBu4Br was used. c 2 mol% NBu4Br was used. d Reaction temperature was 100 °C. e Reaction time was 10 h. f Reaction temperature was 100 °C and reaction time was 24 h. g The yields were determined by GC with an internal standard. | |||
| 1 | 1 | None | 0 |
| 2 | 1 | NBu4Cl | 26 |
| 3 | 1 | NBu4Br | 80 |
| 4 | 1 | NBu4I | 72 |
| 5 | None | NBu4Br | 12 |
| 6 | Ni–H2L | NBu4Br | 38 |
| 7 | [Cd(bpdc)]n | NBu4Br | 25 |
| 8b | 1 | NBu4Br | 56 |
| 9c | 1 | NBu4Br | 75 |
| 10d | 1 | NBu4Br | 85 |
| 11e | 1 | NBu4Br | 82 |
| 12f | 1 | NBu4Br | 86 |
| 13(2nd) | 1 | NBu4Br | 80 |
| 14(3rd) | 1 | NBu4Br | 78 |
| Catalyst | Co-catalyst | Pressure (MPa) | Temperature (°C) | Time (h) | Yield (%) | Ref. |
|---|---|---|---|---|---|---|
| a MIXMOF-5 represents mixed-linker MOF-5 with general formula Zn4O(BDC)x(ABDC)3−x, in which BDC is benzene-1,4-dicarboxylate, and ABDC is 2-aminobenzene-1,4-dicarboxylate. b The epoxide in this cycloaddition reaction is propylene oxide. c The epoxide in this cycloaddition reaction is chloropropylene oxide. d ZIF-8-f represents the grafting of ethylene diamine in the ZIF-8 framework. e The epoxide in this cycloaddition reaction is styrene oxide. | ||||||
| MIXMOF-5a | NEt4Br | 3.0 | 140 | 3 | 63.0b | 8a |
| MOF-5 | NBu4Br | 1.0 | 50 | 4 | 97.6b | 8b |
| Ni(salphen)-MOF | NBu4Br | 2.0 | 80 | 4 | 80.0b | Present work |
| ZIF-8 | — | 0.7 | 80 | 4 | 52.0c | 8e |
| ZIF-8-fd | — | 0.7 | 80 | 4 | 73.1c | 8e |
| Co-MOF-74 | — | 2.0 | 100 | 4 | 96.0e | 8c |
| Mg-MOF-74 | — | 2.0 | 100 | 4 | 95.0e | 8d |
| F-IRMOF-3 | — | 2.0 | 140 | 5 | 84.0e | 8f |
Table 3 summarizes the coupling reactions of CO2 with different epoxides catalyzed by the 1/NBu4Br system, and the data show that the catalytic system could convert epoxides 2a–2d to the corresponding cyclic carbonates effectively under the same conditions. The conversion for 2e is lower partly because it is bulky, reducing its diffusion rate in the 1D open channel of the solid catalyst. This result further suggests that the catalytic reaction took place in the channel not on the catalyst surface. Indeed, upon extending the reaction time to 24 h, the yield reached 82%.

Based on the above analysis, a plausible Lewis acidic activation mechanism is proposed for this CO2 fixation reaction, which is similar to that of the homogenous catalytic system with Co(salen)X complexes as catalysts and quaternary ammonium salts as co-catalysts.16 First of all, the porosity and polar nature of 1 favors CO2 uptake and further increases the local concentration of CO2 in the channel. The coupling reaction is initiated through the coordination of the coordinatively unsaturated Ni2+ (or Cd2+) in 1 to the oxygen atom of the epoxide, and this step can activate the epoxy ring. Secondly, the Br− generated from NBu4Br attacks the less-hindered carbon atom of the coordinated epoxide, followed by the ring opening step. Then, the oxygen anion of the opened epoxy ring interacts with CO2, forming an alkylcarbonate anion, which is converted into the corresponding cyclic carbonate through the ring closing step. Simultaneously, the catalyst is regenerated.
Conclusions
In summary, we introduced a well-defined homogeneous Ni(salphen) catalyst as a “metalloligand” in a porous MOF. The Ni(salphen) units and coordinatively unsaturated Cd active sites accessible via the open MOF channels were utilized to generate an efficient heterogeneous catalyst for the coupling reactions of CO2 with epoxides under relatively mild conditions. The MOF catalyst features a high local density of coorperative layer Ni(salphen) motifs, exhibiting improved catalytic performance relative to the monomeric homogeneous catalyst. This solid catalyst can be easily recycled and reused without any apparent loss of catalytic activity after being used three times. This work establishes a new strategy in the rational design of effective self-supported MOF catalysts for CO2 absorption and in situ fixation based on functional metallosalens or metalloporphyrins.Acknowledgements
We are grateful to the National Natural Science Foundation of China (No. 20932002 and 21172076), the National Basic Research Program of China (973 Program) (No. 2011CB808600), the Natural Science Foundation of Guangdong Province, China (No. 10351064101000000), and the Fundamental Research Funds for the Central Universities (No. 2011ZM0044) for the financial support.Notes and references
- R. L. Paddock and S. B. T. Nguyen, J. Am. Chem. Soc., 2001, 123, 11498 CrossRef CAS.
- (a) W. Leitner, Coord. Chem. Rev., 1996, 155, 257 CrossRef CAS; (b) D. J. Darensbourg and M. W. Holtcamp, Coord. Chem. Rev., 1996, 153, 155 CrossRef CAS; (c) A. G. Shaikh and S. Sivaram, Chem. Rev., 1996, 96, 951 CrossRef CAS.
- (a) R. M. Haak, A. Decortes, E. C. Escudero-Adán, M. M. Belmonte, E. Martin, J. Benet-buchholz and A. W. Kleij, Inorg. Chem., 2011, 50, 7934 CAS; (b) A. Decortes, M. M. Belmonte, J. Benet-buchholz and A. W. Kleij, Chem. Commun., 2010, 46, 4580 RSC; (c) A. Decortes and A. M, Angew. Chem., Int. Ed., 2010, 49, 9822 CrossRef CAS; (d) R. M. Haak, S. J. Wezenberga and A. W. Kleij, Chem. Commun., 2010, 46, 2713 RSC; (e) H. W. Jing, S. K. Edulji, J. M. Gibbs, C. L. Stern, H. Y. Zhou and S. T. Nguyen, Inorg. Chem., 2004, 43, 4315 CrossRef.
- Z. Wang, G. Chen and K. Ding, Chem. Rev., 2009, 109, 322 CrossRef CAS.
- Y. Xie, Z. Zhang, T. Jiang, J. He, B. Han, T. Wu and K. Ding, Angew. Chem., Int. Ed., 2007, 46, 7255 CrossRef CAS.
- Y. Du, F. Cai, D. L. Kong and L. N. He, Green Chem., 2005, 7, 518 RSC.
- (a) J. Q. Wang, D. L. Kong, J. Y. Chen, F. Cai and L. N. He, J. Mol. Catal. A: Chem., 2006, 249, 143 CrossRef CAS; (b) T. Takahashi, T. Watahiki, S. Kitazume, H. Yasuda and T. Sakakura, Chem. Commun., 2006, 1664 RSC; (c) T. Sakai, Y. Tsutsumi and T. Ema, Green Chem., 2008, 10, 337 RSC.
- (a) W. Kleist, F. Jutz, M. Maciejewski and A. Baiker, Eur. J. Inorg. Chem., 2009, 3552 CrossRef CAS; (b) J. Song, Z. Zhang, S. Hu, T. Wu, T. Jiang and B. Han, Green Chem., 2009, 11, 1031 RSC; (c) H. Y. Choa, D. A. Yanga, J. Kima, S. Y. Jeongb and W. S. Ahn, Catal. Today, 2012, 185, 35 Search PubMed; (d) D. A. Yang, H. Y. Cho, J. Kim, S. T. Yang and W. S. Ahn, Energy Environ. Sci., 2012, 5, 6465 RSC; (e) C. M. Miralda, E. E. Macias, M. Zhu, P. Ratnasamy and M. A. Carreon, ACS Catal., 2012, 2, 180 Search PubMed; (f) X. Zhou, Y. Zhang, X. Yang, L. Zhao and G. Wang, J. Mol. Catal. A: Chem., 2012, 361-362, 12 Search PubMed.
- L. Ma, C. Abney and W. Lin, Chem. Soc. Rev., 2009, 38, 1248 RSC.
- (a) C. Zhu, G. Yuan, X. Chen, Z. Yang and Y. Cui, J. Am. Chem. Soc., 2012, 134, 8058 CrossRef CAS; (b) J. M. Falkowski, C. wang, S. Liu and W. Lin, Angew. Chem., Int. Ed., 2011, 50, 8674 CrossRef CAS; (c) P. W. Roesky, A. Bhunia, Y. Lan, A. K. Powell and S. Kureti, Chem. Commun., 2011, 47, 2035 RSC; (d) A. Bhunia, Y. lan, V. Mereacre, M. T. Gamer, A. K. Powell and P. W. Roesky, Inorg. Chem., 2011, 50, 12697 Search PubMed; (e) S. Jung, W. Cho, H. J. Lee and M. Oh, Angew. Chem., Int. Ed., 2009, 48, 1459 CrossRef CAS; (f) G. Yuan, C. Zhu, W. Xuan and Y. Cui, Chem.–Eur. J., 2009, 15, 6428 CrossRef CAS; (g) A. Bhunia, P. W. Roesky, Y. Lan, G. E. Kostakis and A. K. Powell, Inorg. Chem., 2009, 48, 10483 CrossRef CAS; (h) S. Jung and M. Oh, Angew. Chem., Int. Ed., 2008, 47, 2049 CrossRef CAS; (i) Y. Jeon, G. S. Armatas, J. Heo, M. G. Kanatzidis and C. A. Mirkin, Adv. Mater., 2008, 20, 2105 CrossRef CAS; (j) J. Heo, Y. Jeon and C. A. Mirkin, J. Am. Chem. Soc., 2007, 129, 7712 CrossRef CAS; (k) M. Oh and C. A. Mirkin, Angew. Chem., Int. Ed., 2006, 45, 5492 CrossRef CAS; (l) M. Oh and C. A. Mirkin, Nature, 2005, 438, 651 CrossRef CAS; (m) R. Kitaura, G. Onoyama, H. Sakamoto, R. Matsuda, S. Noro and S. Kitagawa, Angew. Chem., Int. Ed., 2004, 43, 2684 CrossRef CAS.
- (a) F. Song, C. Wang and W. Lin, Chem. Commun., 2011, 47, 8256 RSC; (b) F. Song, C. Wang, J. M. Falkowski, L. Ma and W. Lin, J. Am. Chem. Soc., 2010, 132, 15390 CrossRef; (c) Y. Jeon, J. Heo and C. A. Mirkin, J. Am. Chem. Soc., 2007, 129, 7480 CrossRef CAS.
- (a) J. M. Falkowski, S. Liu, C. Wang and W. Lin, Chem. Commun., 2012, 48, 6508 RSC; (b) Y. Huang, T. Liu, J. Lin, Jian Lü, Z. Lin and R. Cao, Inorg. Chem., 2011, 50, 2191 CrossRef CAS; (c) G. Li, C. Zhu, X. Xi and Y. Cui, Chem. Commun., 2009, 2118 RSC; (d) B. Chen, X. Zhao, A. Putkham, K. Hong, E. B. Lobkovsky, E. J. Hurtado, A. J. Fletcher and K. M. Thomas, J. Am. Chem. Soc., 2008, 130, 6411 CrossRef CAS; (e) S. Cho, B. Ma, T. S. Nguyen, J. T. Hupp and T. E. lbrecht-Schmitt, Chem. Commun., 2006, 2563 RSC.
- P. Van der Sluis and A. L. Spek, Acta Crystallogr., Sect. A: Found. Crystallogr., 1990, A46, 194.
- K. Sumida, D. L. Rogow, J. A. Mason, T. M. McDonald, E. D. Bloch, Z. R. Herm, T. Bae and J. R. Long, Chem. Rev., 2012, 112, 724 CrossRef CAS.
- W. Tao, J. Liu, Y. Zheng and C. Sun, Chin. J. Inorg. Chem., 2011, 27, 2419 Search PubMed.
- X. B. Lu, B. Liang, Y. J. Zhang, Y. Z. Tian, Y. M. Wang, C. X. Bai, H. Wang and R. Zhang, J. Am. Chem. Soc., 2004, 126, 3732 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis and characterization of Ni-H2L ligand and MOF 1, X-ray crystallography, PXRD data, TGA data, nitrogen and carbon dioxide adsorption isotherms, and catalytic reaction. CCDC 900477. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/C2RA22550F |
| This journal is © The Royal Society of Chemistry 2013 |