
A red light-activable hetero-bimetallic [Fe(III)–Ru(II)] complex as a dual-modality PDT tool for anticancer therapy†
Abhishek
Panwar
a,
Chandi C.
Malakar
 a,
Aarti
Upadhyay
*b and
Mithun
Roy
*ac
a,
Aarti
Upadhyay
*b and
Mithun
Roy
*ac
aDepartment of Chemistry, National Institute of Technology Manipur, Langol 795004, Imphal West, Manipur, India
bDepartment of Inorganic and Physical Chemistry, Indian Institute of Science Bangalore, Bangalore 50012, Karnataka, India. E-mail: aartiu@iisc.ac.in
cDepartment of Chemistry, National Institute of Technology Agartala, Jirania, West Tripura 799046, India. E-mail: mithunroy@nitmanipur.ac.in
First published on 6th February 2025
Abstract
We developed a novel red light activable hetero-bimetallic [Fe(III)–Ru(II)] complex by combining hydroxyl radical-generating Fe(III)–catecholate as a type I PDT agent and the singlet oxygen generating Ru(II)–paracymene complex as a type II PDT agent and it potentially functions as a dual-modality PDT tool for enhanced phototherapeutic applications. 2-Amino-3-(3,4-dihydroxyphenyl)-N-(1,10-phenanthrolin-5-yl)propenamide (L2) acted as a bridging linker. The single-pot synthesis of the hetero-bimetallic [Fe(III)–Ru(II)] complex was carried out through acid–amine coupling. Various photophysical assays confirmed the photo-activated production of (˙OH) radicals and (1O2) oxygen generation upon activation of the [Fe(III)–Ru(II)] complex with red light (600–720 nm, 30 J cm−2), which resulted in enhanced cytotoxicity with a photo-index of ∼45. The complex, [Fe(III)–Ru(II)], potentially bonded to the DNA through the ruthenium moiety was responsible for minimal dark toxicity. The cytotoxic potential of the complex under red light was a result of the photo-induced accumulation of reactive oxygen species through both type I and type II photodynamic therapy (PDT) mechanisms in A549 and HeLa cells, while non-cancerous HPL1D cells remained unaffected. We probed the caspase 3/7-dependent apoptosis of the complex, [Fe(III)–Ru(II)], in vitro. Overall, the hetero-bimetallic [Fe(III)–Ru(II)] complex is an ideal example of a red light activable dual-modality next-generation PDT tool for phototherapeutic anticancer therapy.
Introduction
The introduction of light as a therapeutic modality in clinical practice heralded a significant advancement in the field of phototherapy.1–5 One of the significant advancements is photodynamic therapy (PDT), which utilizes a photosensitizer, activated by red light in the presence of oxygen to generate reactive oxygen species (ROS), which initiate apoptotic pathways in tumors leading to the killing of cancer cells. The application of light in PDT allows for meticulous regulation of the photosensitizer activation, enhancing both spatial and temporal targeting for improved selectivity.6–10 Despite the clinical success achieved with PDT, first-generation photosensitizers such as Photofrin, an FDA-approved drug effective against esophageal cancer, exhibited several limitations including issues related to solubility, bioavailability, biodistribution, skin photosensitivity, and hepatotoxicity.11–13 These limitations have driven the swift advancement of next-generation photosensitizers, though the perfect PDT drug is yet to be found. In response to these challenges, metal complexes are being extensively investigated for their potential in PDT due to their diverse physicochemical, photochemical, and photobiological properties.14–16 Key benefits include excellent water solubility, beneficial photochemical performance in the UV-visible spectrum, photostability, and effective photochemical reactions. 4d and 5d metal complexes such as Os(II), Ru(II), Ir(III), and Rh(III) have proven to be highly effective photosensitizers for photodynamic therapy (PDT), generating singlet oxygen and other reactive oxygen species through the dual PDT mechanism (type-I & type-II) against various cancer cells.17–22 For instance, ruthenium complexes with high two-photon absorption cross-sections are promising candidates for PDT applications.23,24 Recently, a ruthenium-based complex (TLD1433) has advanced to phase 2 clinical studies showing remarkable potential for PDT applications.25–27 In contrast, 3d transition metal-based complexes, such as iron(III)–catecholate, have shown considerable promise due to their red light-induced cytotoxicity mediated through hydroxyl radicals generated via photo-induced redox processes via the type I mechanism occurring during ligand-to-metal charge transfer (LMCT) transitions.28–32Recent interest has also shifted towards bimetallic systems as a promising approach for PDT.33–39 These systems offer multiple avenues for generating diverse cytotoxic agents upon light activation, thereby enhancing the cancer therapy efficiency. Examples include bimetallic complexes such as Ru(II)–Pt(IV), Mn(II)–Cu(I), Ru(II)–Co(III), and Os(II)–Pt(II), which exhibit multimodal functionality, improved efficacy, and potent photocytotoxicity compared to single-metal PDT agents.
In this context, we developed an innovative red light-activatable hetero-bimetallic complex, [Fe(III)–Ru(II)], designed to function as a dual-functional phototherapeutic agent, which functions through dual PDT mechanisms (type I & type II), providing robust anticancer effects within a cohesive molecular framework (Scheme 1). This complex leverages the distinct redox properties of iron and ruthenium centers to achieve synergistic therapeutic effects. Specifically, the iron(III) catecholate moiety generates hydroxyl radicals through the type-I PDT mechanism upon red light activation, while the ruthenium(II) paracymene moiety, known for its anticancer properties, facilitates effective DNA binding and singlet oxygen generation via the type-II PDT mechanism, thereby inducing apoptotic cancer cell death. By combining these functionalities, the [Fe(III)–Ru(II)] complex enables enhanced ROS generation upon red light activation, offering a bimodal therapeutic strategy that potentially enhances the phototherapy efficacy. The complex is conjugated using L-DOPA as a biocompatible linker.40
 | ||
| Scheme 1 Illustrative depiction of the hetero-bimetallic Fe(III)–Ru(II) complex ([Fe–Ru]), and light-triggered production of hydroxyl radicals (˙OH) and singlet oxygen (1O2). | ||
Here, we present the synthesis and characterization of the bimetallic [Fe(III)–Ru(II)] complex, along with detailed photo-physical and photo-chemical assays. These include studies on photo-activated hydroxyl radical and singlet oxygen generation, DNA binding affinity, BSA-binding interactions, cellular uptake & localization, phototoxicity in A549 and HeLa cells, ROS generation in vitro, and caspase 3/7-mediated apoptosis.
Results and discussion
Synthesis, characterization, and general aspects
The tetradentate ligand bis(pyridin-2-ylmethyl)glycine (L1) was prepared following a previously established protocol41 and analyzed using FT-IR and mass spectrometry (Fig. S1 and S2†). The iron(III) complex (Fe) was synthesized by following a previously published protocol31 in which a methanol solution of L1 was added to the methanol solution of [Fe(NO3)3·9H2O], following the addition of triethylamine mixed L-dopa i.e. 2-amino-3-(3,4-dihydroxyphenyl)propanoic acid to procure the complex (Fe) with a chemical notation [Fe(L1)(L-dopa)]. The prepared complex (Fe) was analyzed by infrared (IR) and mass spectroscopy (Fig. S3–S5†). The ruthenium complex (Ru) was synthesized by following a previously published protocol42 where a methanol solution of 5-amino-1,10-phenanthroline was added to the methanol solution of bis(dichloro(η6-p-cymene)ruthenium) dimer under 6 h of continuous stirring at room temperature (RT) to procure the complex (Ru) with a chemical notation of [Ru-Cl(η6-p-cymene)(phen)]+ and it was also characterized by infrared (IR) and mass spectroscopy techniques (Fig. S6–S9†). The bridging ligand 2-amino-3-(3,4-dihydroxyphenyl)-N-(1,10-phenanthrolin-5-yl)propenamide (L2) was prepared following a previously established peptide coupling protocol.43,44 At first, the amino group of Levodopa was protected with di-tert-butyl dicarbonate (Boc) to prevent the self-peptide coupling of the L-dopa ligand. Later Boc-protected L-dopa was subjected to the peptide coupling with 5-amino-1,10-phen alongside 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC), hydroxybenzotriazole (HOBt) and diisopropyl ethylamine (DIPEA). The Boc group was removed by treating the Boc-protected L2 with trifluoracetic acid (TFA). The linker or the bridging ligand (L2) [L-dopa(COO-NH2)phen] was characterized by IR, mass, and NMR (1H, 13C) spectroscopy before being used to conjugate iron and ruthenium centers together (Fig. S10–S14†). The bimetallic [Fe(III)–Ru(II)] complex was synthesized in multiple steps: (i) addition of a tetradentate ligand (L1) to the methanol solution of ferric nitrate [Fe(NO3)3] salt, (ii) incorporation of L2 (linker) dissolved in CH3OH, leading to a colour change from red to deep bluish-green, and (iv) addition of the methanolic solution of ruthenium–paracymene salt until the colour of the solution changes from bluish-green to dark greenish-violet. Slow evaporation of the mixture resulted in dark greenish-violet microcrystals of the bimetallic [Fe(III)–Ru(II)] complex (Scheme 2). The synthesized complex, [Fe–Ru] was characterized analytically and spectroscopically by FT-IR, UV-visible, and ESI mass spectrometry (Fig. S15–S17†). The molar conductance (ΛM, S (m2 mol−1)) of the [Fe–Ru] complex was found to be 64.2 in 5% DMSO–H2O at room temperature suggesting the positive nature of the heterobimetallic complex, [Fe–Ru]. The FT-IR spectra of the [Fe–Ru] complex in the KBr phase revealed distinct Ru–N bond stretching frequencies at 533 cm−1, (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Nstr) at 1573 cm−1, (CCstr) at 1485 cm−1, and (COstr) at 1685 cm−1 (Fig. S15†). These frequencies are consistent with the data reported in previous publications and provide reasonable evidence for the attachment of the phenanthroline ligand (N–N) to the Ru core and correlate with the given structure of the complex. A strong absorption at 660 nm assignable to the LMCT transitions in Fe(III)–catecholate was recorded through UV-visible assay of the complex dissolved in a 5% DMSO–PBS buffered media solution at pH 7.4 at 298 K; additionally, various electronic transitions focused on the ligand were seen in the range of (254–450 nm) (Fig. S16†). In the Q-TOF-ESI mass spectrum of the [Fe–Ru] complex in methanol, there was a fragmented molecular ion peak at m/z 460.3666, which is related to the general formula [M − Cl]2+, where M represents [Fe–Ru]. Additionally, the isotope distribution at m/z 460.3666 was further analyzed and the characteristics of the [Fe–Ru] were evaluated (Fig. S17†).
Nstr) at 1573 cm−1, (CCstr) at 1485 cm−1, and (COstr) at 1685 cm−1 (Fig. S15†). These frequencies are consistent with the data reported in previous publications and provide reasonable evidence for the attachment of the phenanthroline ligand (N–N) to the Ru core and correlate with the given structure of the complex. A strong absorption at 660 nm assignable to the LMCT transitions in Fe(III)–catecholate was recorded through UV-visible assay of the complex dissolved in a 5% DMSO–PBS buffered media solution at pH 7.4 at 298 K; additionally, various electronic transitions focused on the ligand were seen in the range of (254–450 nm) (Fig. S16†). In the Q-TOF-ESI mass spectrum of the [Fe–Ru] complex in methanol, there was a fragmented molecular ion peak at m/z 460.3666, which is related to the general formula [M − Cl]2+, where M represents [Fe–Ru]. Additionally, the isotope distribution at m/z 460.3666 was further analyzed and the characteristics of the [Fe–Ru] were evaluated (Fig. S17†).
 | ||
| Scheme 2 Synthesis scheme for the hetero-bimetallic Fe(III)–Ru(II) complex ([Fe–Ru]): (i) bis(pyridin-2-ylmethyl)glycine (L1), MeOH, dark; (ii) 2-amino-3-(3,4-dihydroxyphenyl)-N-(1,10-phenanthrolin-5-yl)propanamide (L2), MeOH, 10 min stirring in the dark; (iii) dichloro(p-cymene)ruthenium(II) dimer, MeOH, 15 min stirring in the dark. | ||
Solubility and stability studies
We examined the solubility of the [Fe–Ru] complex in a 5% DMSO–PBS buffer solution at a pH of 7.4 at 298 K, which is commonly used as a biological reference. We found that the [Fe–Ru] complex was soluble in a 5% DMSO–PBS buffer solution. The stability, chemical assays, and other photophysical studies of the [Fe–Ru] complex were examined in a 5% DMSO–PBS buffer solution with a pH of 7.4 and at room temperature using UV-visible spectroscopy. To assess the stability of the [Fe–Ru] complex in a 5% DMSO–PBS buffer solution with a pH of 7.4, the UV-visible spectra of [Fe–Ru] were recorded over 48 h in the absence of light (Fig. S18†). Minimal or insignificant variations in the UV-visible spectral patterns of [Fe–Ru] were noted and suggested that the bimetallic complex remains stable in the physiologically relevant solvent and conditions. Nevertheless, the solid-state [Fe–Ru] complex remained stable for six months when kept at temperatures between 0–4 °C and stored in the dark. The aqueous solution of the [Fe–Ru] complex had a notable impact on red light exposure, as indicated by the alterations in the UV-visible spectral traces measured in a 5% DMSO–PBS buffer medium (pH 7.4) at room temperature during the light exposure (Fig. 1).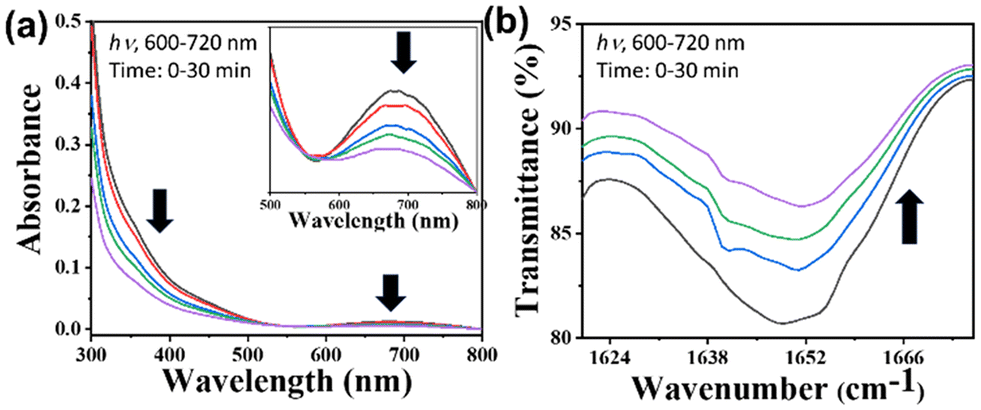 | ||
| Fig. 1 Photo-physical studies: (a) UV-visible spectral data obtained upon illuminating [Fe–Ru] (0.02 mM) with red light (600–720 nm, 10 J cm−2, 0–30 min) in 5% DMSO–PBS buffer (pH 7.4). (b) IR spectral traces show a decrease in COstr (COO of Fe-complex) upon illuminating [Fe–Ru] with red light (600–720 nm, 10 J cm−2, 0–30 min). | ||
Electrochemical studies
The electrochemical properties of the bimetallic [Fe–Ru] complex (0.03 mM) were assessed by cyclic voltammetry in a 5% DMSO–PBS buffer solution with 0.1 M KCl acting as the supporting electrolyte, with glassy carbon serving as the working electrode, Ag/AgCl as the reference electrode, and platinum as the counter electrode. The scan rate employed was 50 mV s−1. The cathodic scan of the [Fe–Ru] complex exhibited a quasi-reversible cyclic voltammetric response at E1/2 = −0.60 V, which can be attributed to the reduction of iron(III) to iron(II). Another noticeable quasi-reversible cyclic voltammetric response at E1/2 = 0.7 V was observed which matched with the reported voltammetric response of the ruthenium paracymene complex45 and was mainly attributed to the oxidation of ruthenium(II) to ruthenium(III) (Fig. S19†).Lipophilicity
The lipophilicity of pharmacological compounds, typically evaluated through the logarithm of the partition coefficient (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P), is essential for comprehending their distribution within tissues. logP represents the ratio of a compound's concentration in 1-octanol to its concentration in water, reflecting its ability to distribute between a hydrophobic solvent (like n-octanol) and water. This measure is essential for evaluating cellular absorption and drug distribution. In this study, we determined the logP of the [Fe–Ru] complex in a 5% DMSO–water solution and compared it with its logP in n-octanol, finding a value of 0.35. This positive logP indicates that the [Fe–Ru] complex has a moderate attraction to lipids and hydrophobic surroundings, which could enhance its cellular uptake. Therefore, the logP value underscores the [Fe–Ru] complex's potential for advanced exploration and advancements in biomedical applications, where effective cellular internalization is the key to clinical success.
P), is essential for comprehending their distribution within tissues. logP represents the ratio of a compound's concentration in 1-octanol to its concentration in water, reflecting its ability to distribute between a hydrophobic solvent (like n-octanol) and water. This measure is essential for evaluating cellular absorption and drug distribution. In this study, we determined the logP of the [Fe–Ru] complex in a 5% DMSO–water solution and compared it with its logP in n-octanol, finding a value of 0.35. This positive logP indicates that the [Fe–Ru] complex has a moderate attraction to lipids and hydrophobic surroundings, which could enhance its cellular uptake. Therefore, the logP value underscores the [Fe–Ru] complex's potential for advanced exploration and advancements in biomedical applications, where effective cellular internalization is the key to clinical success.
DNA binding studies
Ruthenium–paracymene complexes are well known for their anticancer activity through DNA binding, therefore to confirm the binding of the [Fe–Ru] complex towards DNA, we have followed a previously reported protocol46–48 in which we incubated the [Fe–Ru] complex (1 mM) with a modeled DNA base, i.e. guanosine-5-monophosphate (5′-GMP) (1 mM) in (1:1) stoichiometry. The reaction was performed in a water–ethanol mixture for 24 h at 40 °C. After continuous stirring, the solvent was evaporated under vacuum and the resultant crude remnant was prepared for mass characterization. The formation of the [Fe-Ru-N7-GMP] adduct was confirmed from Q-TOF ESI mass spectra in which there was a prominent peak at m/z 641.1245 which corresponds to [M − Cl]+2 where M is [Fe-Ru-N7-GMP] (Fig. S21†). The binding of the [Fe–Ru] complex to the N7 position of GMP confirms its efficacy to act as a chemotherapeutic drug through DNA binding.
BSA binding studies
Blood serves as the main medium for the distribution of medications within the human body, with serum albumin being essential in directing drugs to their designated targets. We performed a fluorometric assay to examine the interaction between the [Fe–Ru] complex and bovine serum albumin (BSA), which acts as a representative for human serum albumin (HSA).49,50 BSA exhibits notable intrinsic fluorescence due to the existence of three luminescent amino acids: tryptophan (Trp), tyrosine (Tyr), and phenylalanine (Phe). Upon excitation at 280 nm (λex), BSA exhibits fluorescence emission at approximately 339 nm. Through the incremental addition of a [Fe–Ru] complex solution (0.001 M, stock) to an aqueous BSA solution (3.0 × 10−5 M), we noted a decrease in BSA's fluorescence intensity at 339 nm, along with a slight blue shift (Δλ = 3–7 nm). With the increasing introduction of the [Fe–Ru] complex, the luminescence of BSA consistently diminished. The binding constant, derived from Stern–Volmer and Scatchard plots, was found to be 6.2 × 103 mol−1, accompanied by a Gibbs free energy estimate of −5.13 kcal mol−1 (Fig. S22†). The outcomes demonstrate a significant affinity of the [Fe–Ru] complex for serum albumin, indicating optimal conditions for improved circulation and delivery of the complex throughout the body.Photophysical and photochemical studies
We examined the photophysical and photochemical properties of the [Fe–Ru] complex, which may be essential for evaluating its photocytotoxic effects. Notable alterations were observed in the UV-visible spectral data of the [Fe–Ru] complex in an aqueous-DMSO buffer following 30 minute red light illumination (600–720 nm, 30 W). Significant changes were noted in the LMCT band around 660 nm of the [Fe–Ru] complex, suggesting photodecarboxylation (Fig. 1a).51 Moreover, a notable reduction in the intensity of the COstr peak at 1685 cm−1 was noticed in the IR spectra of the [Fe–Ru] complex (Fig. 1b), and such reduction led us to a similar conclusion. We also observed the photodecarboxylation in the control complex Fe (Fig. S27†). In general, it can be inferred that the photodecarboxylation was caused by the photo-activated ligand to metal charge transfer (LMCT) focused on the Fe(III)–O(-carboxylate) bond.52 Photodecarboxylation in iron(III)–carboxylate complexes typically leads to the formation of (˙OH) radicals, which are toxic to cancer cells.53 We conducted a detailed investigation into the production of (˙OH) radicals by subjecting the [Fe–Ru] complex to red light exposure using UV-visible spectroscopy.
Photo-activated hydroxyl radical (˙OH) generation
The main outcome of the photo-Fenton-type reaction involving iron-phenolato/carboxylato complexes, molecular oxygen (O2), and light is the formation of reactive oxygen species (ROS) like superoxide ions (O2˙−), hydrogen peroxide (H2O2), or hydroxyl radicals (˙OH).54 We examined the emergence of (˙OH) radicals from a [Fe–Ru] complex (0.3 mM) using a 30 W red LED light source for photoactivation. We also examined the generation of hydroxyl radicals through control complex Fe (Fig. S26†). The assay was probed by employing UV-visible spectroscopy with benzoic acid (0.2 mM) and externally added Fe(NO3)3·9H2O. Upon red light illumination (600–720 nm, 30 W), the iron(III) moiety in the [Fe–Ru] complex generated hydroxyl radicals which then interacted with benzoic acid to produce salicylaldehyde. Salicaldehyde (sal) formed a complex ([Fe(sal)3]3−) with the externally added iron(III) ions which was characterized by strong and broad LMCT bands of [Fe(sal)3]3 at around 520 nm. Formation of [Fe(sal)3]3− suggested the formation of hydroxyl radicals (˙OH) on photo-activation of [Fe–Ru] (Fig. 2a).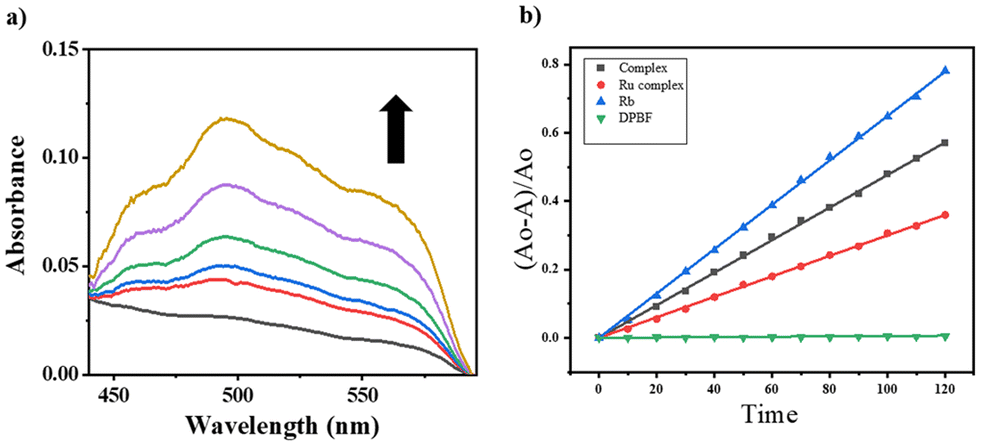 | ||
| Fig. 2 (a) UV-visible spectral traces showing the formation of hydroxyl radicals (˙OH) on photo-activation of [Fe–Ru] (0.3 mM) with red light (600–720 nm, 30 W, 0–30 min) in 5% DMSO–PBS buffer (pH 7.4) in the presence of benzoic acid. The enhancement in the absorbance at ∼520 nm suggested the formation of an Fe(III)–salicylate complex. (b) Comparative plots of (A0 − A)/A0 against exposure time (sec.), where A represents the value of A417 at a certain time (t), and A0 represents the value of A417 at t = 0 in the DBPF assay (Table S28†) showing the changes in the absorbance (A417) of DPBF (50 μM), examined with Rose Bengal (5 μM) (blue), [Fe–Ru] (0.2 mM) (black) and the Ru-complex (0.2 mM) (red), relative to LED red light (30 W, 600–720 nm) in DMSO at 298 K. | ||
Photo-activated singlet oxygen generation
Ruthenium complexes are recognized in terms of their capacity to produce singlet oxygen upon illumination with light.55–57 This study investigates the capability of the [Fe–Ru] complex to produce singlet oxygen upon activation with red light. We utilized a 1,3-diphenylisobenzofuran (DPBF) degradation assay to assess the singlet oxygen production of the [Fe–Ru] complex. DPBF exhibits a UV-visible absorption peak at 417 nm and demonstrates sensitivity to singlet oxygen. The absorbance of DPBF at this wavelength diminishes with singlet oxygen, and the degree of this reduction in A417 can reflect the quantity of singlet oxygen generated by the [Fe–Ru] complex upon illumination with red light (Fig. S23†).To determine the singlet oxygen quantum yield, we plotted (A0 − A)/A0 against exposure time (Fig. 2b), where A represents the A417 value at a specific time (t), and A0 denotes the A417 value at t = 0. Rose Bengal, recognized for its singlet oxygen quantum yield of 0.76, served as a reference. The [Fe–Ru] complex demonstrated a singlet oxygen quantum yield of 0.46, marginally surpassing that of the control ruthenium paracymene-phen complex (Ru), which showed a quantum yield of 0.39. This indicated that the bimetallic [Fe–Ru] system demonstrated enhanced efficiency in generating reactive oxygen species when activated with red light.
Cellular uptake and localization
The intake of the [Fe–Ru] complex in A549 cells was investigated by utilizing flow cytometry. The A549 cells were treated with [Fe–Ru] at a concentration of 2.5 μM and incubated for 2 and 4 h. Subsequent flow cytometry and fluorescence-assisted cell sorting analyses were conducted, revealing a significant increase in fluorescence emission in the treated cells compared to the untreated ones, indicating significant uptake of [Fe–Ru] (Fig. 3a). The complex exhibited blue fluorescence with an emission wavelength (λem) of 438 nm (λex, 300 nm) (Fig. S20†), and the intracellular distribution of the [Fe–Ru] complex in A549 cells was probed through confocal microscopy. The A549 cells were cultured with [Fe–Ru] for 4 h, and treated with mitotracker red (MTR) for mitochondria staining. Confocal images (Fig. 3b) showed that the [Fe–Ru] complex localized in the mitochondria, with Pearson's value of 0.64. The localization of the [Fe–Ru] complex in mitochondria may be due to the formal positive charge on the complex as mitochondria are a negatively charged membrane. | ||
| Fig. 3 (a) Fluorescence-assisted cell-sorting analysis revealing the uptake of [Fe–Ru] (2.5 μM) in A549 cells incubated for 2 and 4 h. (b) Confocal imaging of A549 cells incubated with [Fe–Ru] (2.5 μM) and mitotracker red showing mitochondrial localization of [Fe–Ru] in A549 cells. | ||
Cell viability assays
The cell viability assays of the iron(III)–catecholate complex (Fe), the ruthenium(II)–paracymene phen complex (Ru), and the mixed-metal complex, [Fe–Ru] were probed in adenocarcinoma human alveolar basal epithelial cells (A549) and HeLa cells under dark conditions and with red light (600–720 nm, 30 J cm−2). In addition, the cytotoxicity of the [Fe–Ru] complex, (Fe) complex, and (Ru) complex was investigated in immortalized peripheral lung epithelial (HPL1D) cells which may be regarded as normal cells. We inoculated approximately 30000 cells on a 96-well plate and incubated them with the Fe, Ru, and [Fe–Ru] complexes (1–100 μM). Following exposure to red light (600–720 nm, 30 J cm−2) for 30 min, the viability of the treated cells was assessed using the MTT assay. An analogous experiment was conducted while ensuring the absence of light. A graph was created to show the relationship between cell viability and the logarithm of concentration. Nonlinear regression analysis was conducted to determine the IC50 values. The results are shown in Fig. 4, Fig. S24, S25† and Table 1. The complex (Fe) demonstrated photocytotoxicity against A549 and HeLa cells in red light, with an IC50 value of (25.5 ± 1.4) μM and (35.6 ± 1.5) μM respectively. However, in the absence of light, the complex (Fe) showed no toxicity, with an IC50 value >100 μM in A549 and HeLa cells (PI > 6.0). The (Ru) complex exhibited photocytotoxicity against A549 and HeLa cells in red light, with an IC50 value of (18.8 ± 1.1) μM and (26.8 ± 1.6) μM respectively. However, in the absence of light, the (Ru) complex was shown to be relatively less toxic with an IC50 value of 72.9 ± 2.5 μM in A549 cells and 82.4 ± 2.7 μM in HeLa cells (PI, 6.0). The impact of red light (600–720 nm, 30 J cm−2) on the toxicity of the [Fe–Ru] complex in A549 cells was significant with an IC50 value of (1.4 ± 0.7) μM and (2.9 ± 0.8) μM in HeLa cells, while under dark conditions, the IC50 value of the mixed-metal complex, [Fe–Ru], was determined to be 86.1 ± 3.2 μM in A549 and 95.9 ± 4.1 μM in HeLa cells. The photo-index (PI) in this case was remarkable and the value was ∼35.0. The photo-cytotoxic effectiveness of the [Fe–Ru] (PI: 35.0) compound was even more noticeable than that of (Fe) (PI: >6.0) and (Ru) (PI: ∼6.0) complexes in A549 and HeLa cells. This may be due to the combined influence of hydroxyl radicals (˙OH) and singlet oxygen (1O2) of the bimetallic complex, [Fe–Ru] on red light activation. The differential photocytotoxic behavior of [Fe–Ru] was investigated in the HPL1D cells. Although the complex was benign to HPL1D cells (IC50 > 100 μM) in the dark, we observed reduced cell viability (IC50, 53.3 ± 3.4 μM) upon red light exposure. The complex, [Fe–Ru] was considerably less toxic in HPL1D cells compared to the A549 and HeLa cells by a factor of ∼8.0. This could be attributed to the less intake of [Fe–Ru] in HPL1D cells, which explains the distinct cytotoxic behavior of the [Fe–Ru] complex. Although PDT mostly relies on the concentration of molecular oxygen in the intracellular matrix, the reported bimetallic complex offered scope for a dual-modality type I and type II PDT mode for anticancer therapy.
 | ||
| Fig. 4 (a) Cell viability plots determining the IC50 (μM) values of the complex ([Fe–Ru]) in A549 cells under dark conditions (black line) and with red light (600–720 nm, 30 J cm−2) for 30 min (red line). (b) Cell viability plots determining the IC50 (μM) values of the complex ([Fe–Ru]) in HeLa cells under dark conditions (black line) and under the red light illumination (600–720 nm, 30 J cm−2) for 30 min (red line). | ||
| Complex | A549 cells, IC50/(μM) | HeLa cells, IC50/(μM) | HPL1D cells, IC50/(μM) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Darkb | P value | Lighta | P value | Darkb | P value | Lighta | P value | Darkb | P value | Lighta | P value | |
| a 24 hours of incubation in the dark. b 4.0 h of incubation in the dark, followed by 15 min of exposure to red light (600–720 nm, 30 J cm−2) and then 19 h of post-incubation. | ||||||||||||
| Fe | >100 | <0.0001 | 25.5 ± 1.4 | 0.0034 | >100 | <0.0001 | 35.6 ± 1.5 | 0.0032 | >100 | <0.0001 | 87.2 ± 3.8 | <0.0001 |
| Ru | 72.9 ± 2.5 | <0.0001 | 18.8 ± 1.1 | <0.0001 | 82.4 ± 2.7 | <0.0001 | 26.8 ± 1.6 | <0.0001 | >100 | <0.0001 | 61 ± 4.1 | <0.0001 |
| [Fe–Ru] | 86.1 ± 3.2 | 0.0035 | 1.4 ± 0.7 | <0.0001 | 95.9 ± 4.1 | 0.0037 | 2.9 ± 0.8 | <0.0001 | >100 | <0.0001 | 53.3 ± 3.4 | <0.0001 |
Intracellular ROS generation
The [Fe–Ru] complex exhibited the capability to generate hydroxyl radicals (˙OH), as confirmed by previous UV-visible assays. The production of reactive oxygen species through photo-induced processes may significantly contribute to the cytotoxic effects observed under light activation. The production of ROS by the [Fe–Ru] complex, induced by red light, was investigated in vitro utilizing the 2′,7′-dichlorodihydrofluorescein diacetate (2′,7′-DCFDA) assay and assessed via flow cytometry.58 A549 cells were subjected to treatment with the [Fe–Ru] complex at a concentration of 2.5 μM for 4 h, subsequently followed by exposure to red light (600–720 nm, 30 J cm−2) for 15 min. Following this, the cells were incubated with DCFDA (5 μM) and subsequently analyzed using flow cytometry. The assay was performed under dark conditions to evaluate ROS generation in the absence of light activation. Upon photo-activation, DCFDA undergoes oxidation with reactive oxygen species (ROS), resulting in the formation of the fluorescent compound 2′,7′-dichlorodihydrofluorescein (2,7-DCF), which emits at 525 nm and is excited at 488 nm. Flow cytometry facilitated the qualitative detection of DCF, demonstrating an increase in fluorescence intensity in the treated A549 cells, which indicates elevated ROS production. The data indicated an increased fluorescence intensity in the red light-activated [Fe–Ru] complex relative to the dark conditions, highlighting significant ROS generation by the complex (Fig. 5). | ||
| Fig. 5 Fluorescence-assisted cell-sorting DCFDA analysis revealing ROS generation by [Fe–Ru] (2.5 μM) in A549 cells under dark conditions and upon red light exposure (600–720 nm, 30 J cm−2) for 30 min. | ||
Annexin-V-FITC/propidium iodide (PI) assay
Reactive oxygen species can contribute to cell death, with apoptosis being the preferred mode over necrosis in cancer therapy due to its non-inflammatory characteristics. We conducted an investigation into the nature of cell death induced by the bimetallic complex [Fe–Ru] upon activation with red light, utilizing the Annexin-V-FITC/PI assay alongside fluorescence-assisted cell sorting (FACS).59 This assay detects early apoptosis through the presence of phosphatidylserine on the outer surface of the cell membrane, as evidenced by green fluorescence from Annexin-V-FITC. The observation of blue staining from PI signifies disruption of the cell membrane. Late apoptosis is characterized by the presence of both Annexin-V-FITC and PI staining, whereas necrosis is indicated by the exclusive presence of PI staining. A549 cells underwent treatment with [Fe–Ru] at a concentration of 2.5 μM for 4 h, followed by exposure to red light (600–720 nm) at a dose of 30 J cm−2 for 30 min. The findings indicated that the [Fe–Ru] complex resulted in a distribution of 50.8% healthy cells, 0.16% early apoptotic cells, 41.0% late apoptotic cells, and 8.64% necrotic cells when exposed to red light. In the absence of light, the distribution was 86.90% healthy cells, 3.61% early apoptotic cells, 6.61% late apoptotic cells, and 2.88% necrotic cells (Fig. 6). The findings suggest that photo-activated [Fe–Ru] produces ROS, leading to oxidative stress in A549 cells and facilitating apoptosis, probably via intrinsic pathways. | ||
| Fig. 6 Annexin-V-FITC/PI assay confirming the apoptosis of the A549 cells incubated with [Fe–Ru] (2.5 μM) for 4 h accompanied by red light exposure (L). “D” indicates dark conditions. | ||
Caspase 3/7 mediated apoptosis
The impact of a chemotherapeutic medication on the activity of caspase-3/7 can be analyzed by utilizing the Caspase-Glo assay kit.60 The Caspase-Glo assay kit was used to measure the mechanism of apoptotic cell death caused by the [Fe–Ru] complex upon red light photoexcitation. A549 cells were subjected to treatment with the [Fe–Ru] complex (2.5 μM), followed by a 4 hour incubation, and red light illumination (600–720 nm, 30 J cm−2). This treatment resulted in a remarkable three-fold rise in caspase 3/7 activity (Fig. 7), in contrast to the minimal effect observed on cellular caspase 3/7 activity in the absence of light.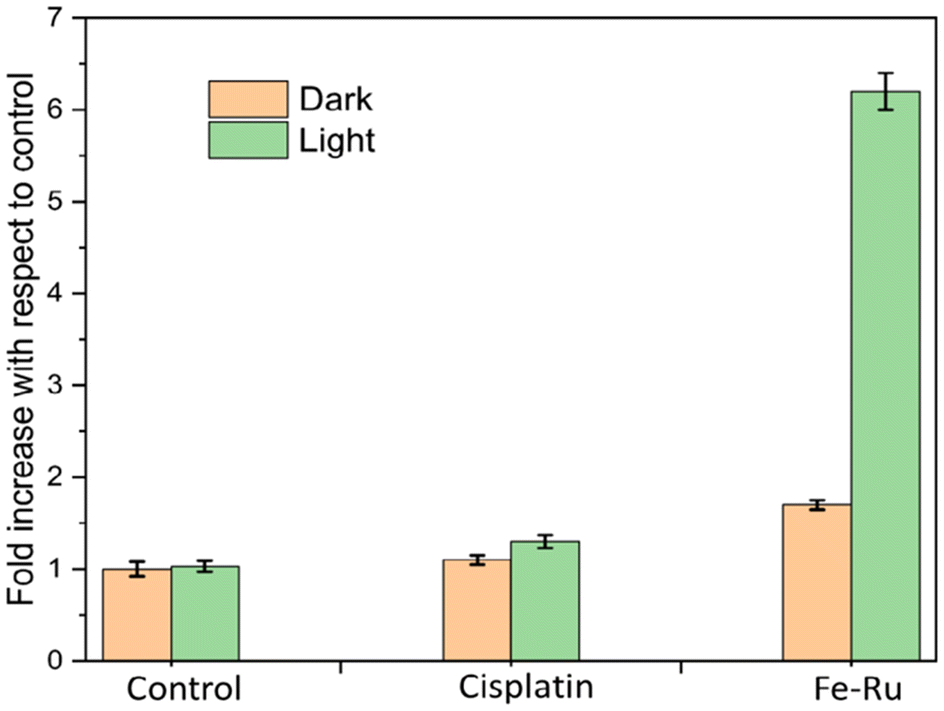 | ||
| Fig. 7 Caspase 3/7 assay: assessment of caspase 3/7 activation in A549 cells pre-treated with culture medium (negative control, NC), 1% DMSO culture medium (DMSO), cisplatin (25 μM), and [Fe–Ru] (2.5 μM) complex, evaluated before (orange) and after red light exposure (600–720 nm, 30 J cm−2, 30 min) (green). In cisplatin- and [Fe–Ru]-treated cells, non-overlapping error bars between light and dark conditions indicate a statistically significant difference in fold activation. Conversely, overlapping error bars in control groups suggest no statistically significant difference. | ||
Furthermore, cells treated with cisplatin exhibited an elevation in caspase 3/7 activity. However, the [Fe–Ru] compound exhibited a notably elevated caspase 3/7 activity compared to the untreated cells or cells treated with cisplatin when exposed to red light irradiation. The findings of this study suggested that the cell death caused by [Fe–Ru] was mediated through caspase-dependent pathways. In general, the light-induced generation of ROS from the bimetallic complex, [Fe–Ru], is potentially responsible for the remarkable increase in oxidative stress and serves as the catalyst for apoptosis through caspase-3/7 activation.
Experimental
Materials and methods
[Fe(NO3)3·9H2O], bis(dichloro(η6-p-cymene)ruthenium), bis(pyridin-2-ylmethyl)amine, bromoacetic acid, L-dopa, 1,10-phenanthroline-5-amine, (3-dimethylamino-propyl)-ethyl-carbodiimide (EDC·HCl), HOBt(1-hydroxybenzotriazole), N,N-diisopropylethylamine (DIPEA), MTT, BSA, and sodium sulphate were procured from Sigma-Aldrich. All chemicals and solvents were sourced from TCI Chemicals, Alfa Aesar, SRL Chemical Company (India), and HI-MEDIA, and were used as received without further purification. Measurements were conducted using a PerkinElmer UV-vis spectrophotometer for UV-vis analysis, a UATR Two FT-IR instrument for FT-IR measurements, and a Hitachi F-7000 fluorescence spectrophotometer for photoluminescence assessments. Mass spectra were obtained utilizing a Bruker Esquire 3000 Q-TOF-ESI system. The absorbance values during the MTT assay were quantified utilizing a Molecular Devices Versa Max tunable microplate reader. ChemDraw Professional 15 was employed to ascertain the IUPAC names and produce structural diagrams of the compounds.Synthesis
The tetradentate ligand L1i.e. bis(pyridin-2-ylmethyl)glycine (L1) was prepared according to a previously established protocol41 and analyzed by FT-IR and mass spectroscopy techniques (Fig. S1 and S2†).Synthesis of Fe
The complex (Fe) with a chemical notation of [Fe(L1)L-dopa] was synthesized in accordance with a previously published protocol31 where a methanolic solution of L1 (0.19 g, 0.49 mmol) was mixed with the methanolic solution of [Fe(NO3)3·9H2O] (0.18 g, 0.44 mmol) giving a deep red color solution and kept for stirring for 1 hour. To this solution, triethylamine mixed L-dopa (0.45 ml, 4.2 mmol) was added dropwise until the color was changed to a dark greenish-purple color. The whole process was performed under an inert atmosphere and dark conditions at room temperature for a period of 4 h, and then the resulting solution was kept for slow evaporation which resulted in a dark greenish precipitate that was carefully rinsed with hexane and diethyl ether to obtain a greenish-purple microcrystalline solid product. Yield 0.08 g (40%).
Fe: UV-visible spectroscopy in 5% DMSO/H2O ([λmax, nm]): 255 nm, 282 nm, and 640 nm. FT-IR solid phase (cm−1): 3362 (N–H stretching), 3213 (O–H stretching), 2925 (C–H stretching), and 1650 (CO stretching). Q-TOF ESI mass analysis in DMF: m/z 530.0456, [M + Na]+ (Fig. S3–S5†).
Synthesis of Ru
The complex (Ru) with a molecular formula of [Ru-Cl(η6-p-cymene)(phen)]+ was synthesized according to a previously published protocol42 where a methanolic solution of 5-amino 1,10-phenanthroline (0.5 mmol) was slowly added dropwise to a methanolic solution containing the bis(dichloro(η6-p-cymene)ruthenium) dimer (0.25 mmol) in an inert atmosphere and under dark conditions for 6 h. After continuous stirring, the solvent was evaporated under vacuum and the resultant material was rinsed with n-hexane (50 ml) and dried under vacuum to obtain the yellowish-orange product. Yield: 0.072 g (67%).
Ru: UV-visible spectroscopy in 5% DMSO/H2O ([λmax, nm]): 290 nm and 330 nm. FT-IR solid phase (cm−1): 3450, 3350 and 3250 (N–H stretching), 2970 (C–H stretching), 1648 (CN stretching) and 1464 (CC stretching). Q-TOF ESI mass analysis in methanol: m/z 466.0685 [M]+ (Fig. S6–S9†).
Synthesis of the bridging linker L2
The bridging linker L2i.e 2-amino-3-(3,4-dihydroxyphenyl)-N-(1,10-phenanthrolin-5-yl)propenamide (L2) was synthesized by following a previously published peptide coupling procedure,43,44 where the first step was to protect the amino group of Levodopa through di-tert-butyl dicarbonate (Boc2O). The Boc-protection of L-dopa was done by following a previously reported protocol. After that, to a solution of Boc-protected L-dopa (0.09 g, 0.19 mmole, 1 eq.) in anhydrous DMF at 0° C, (N,N-diisopropylethylamine) DIPEA (0.35 mmol, 2 equiv.), hydroxybenzotriazole (HOBt) (0.150 g, 0.21 m mol, 1.5 eq.) and ((dimethylamino-propyl)-ethyl-carbodiimide) EDC (0.180 g, 0.20 mmol, 1.5 equiv.) were introduced, accompanied by 5-amino 1,10-phen(0.11 g, 0.20 mmol, 1.1 equiv.), and left for continous stirring overnight at room temperature. After continuous stirring for 3 days, the solvent was evaporated under vacuum and the resultant material was isolated with ethyl acetate and water. The ethyl acetate part was then removed under low pressure and left to dry in a vacuum to obtain a brown color viscous oil product. After getting the Boc-protected peptide product, it was subjected to deprotection by adding trifluoracetic acid (TFA) to a methylene chloride solution containing the Boc-protected peptide product i.e. tert-butyl (1-((1,10-phenanthrolin-5-yl)amino)-3-(3,4-dihydroxyphenyl)-1-oxopropan-2-yl)carbamate to obtain a brownish-orange color solid having a free amino group. Yield: 0.132 g (67%).
L2: FT-IR solid phase (cm−1): 3438 (O–H stretching), 3321 (N–H stretching), 2985 (C–H stretching), 1661 (CO stretching) and 1516 (CN stretching). 1H NMR (500 MHz, DMSO-d6; δ ppm): 9.05 (1H, d, 1.3 Hz), 9.04 (1H, d, 1.3 Hz), 8.03 (1 H, d, 4 Hz), 8.02 (1 H, d, 4 Hz), 7.35–7.33 (1 H, m), 7.33–7.30 (1 H, m), 6.88 (1 H, s), 6.68 (1 H, d), 6.51 (1 H, s), 6.49 (1 H, d), 6.06 (1 H, s), 4.14 (1 H, m), 3.23 (1 H, dd, 3.1 Hz), 2.98 (1 H, dd, 3.3 Hz), 2.71 (1H, s) and 2.66 (1 H, s), (s, singlet; d, doublet; m, multiplet). 13C NMR (126 MHz, DMSO-d6; δ ppm): 171.17, 149.15, 146.11, 145.06, 144.68, 143.88, 142.50, 140.50, 132.52, 130.63, 130.42, 128.16, 122.99, 121.85, 121.74, 119.80, 116.62, 115.36, 101.77, 55.73, 36.35. Q-TOF ESI mass analysis in methanol: m/z 413.1604 [M + K]+ (Fig. S10–S14†).
Synthesis of [Fe–Ru]
To synthesize the bimetallic [Fe(III)–Ru(II)] complex we have followed a series of subsequent steps under dark conditions, beginning with the addition of the tetradentate ligand (L1) (0.03 mmol, 1 eq.) to a methanol solution of ferric nitrate [Fe(NO3)3] salt (0.035 mmol, 1 eq.) (i), accompanied by the addition of L2 (0.45 mmol 1 eq.) dissolved in CH3OH, leading to a colour change from red to deep bluish-green which changes the color of the solution from red to deep bluish-green (ii). After continuous stirring for 30 min ruthenium–paracymene salt (0.21 mmol, 0.5 eq.) in a methanolic solution was gradually added to the mixture, leading to a color change from bluish-green to dark greenish-violet (iii). The reaction flask was wrapped in aluminum foil and left for continuous stirring for 4 h and kept for slow evaporation for another 24 h; the resultant crude residue was redissolved in excess diethyl ether (50 mL) and kept for slow evaporation under ice-cold conditions to obtain the [Fe(III)–Ru(II)] complex as a dark greenish-violet precipitate (Scheme 2). Yield 0.062 g (22%)[Fe–Ru]: UV-visible spectroscopy in 5% DMSO/H2O ([λmax, nm]): 290 nm, 332 nm, and 660 nm. FT-IR solid phase (cm−1): 3445 (O−H stretching), 3149 (N−H stretching), 1685 (CO stretching) and 1573 (CN stretching). Q-TOF-ESI MS in CH3OH: m/z 460.0666, [M − Cl]+2 (Fig. S15–S17†).
Conclusions
Here, we presented the design, synthesis, characterization, photochemical and photophysical roles of a novel red light-activable hetero-bimetallic [Fe(III)–Ru(II)] complex as the potential tool for dual-modality PDT. The remarkable ability of the [Fe(III)–Ru(II)] in generating reactive oxygen species (ROS: ˙OH and 1O2) in red light resulted in caspase 3/7-dependent apoptosis due to the enhanced oxidative stress in cancerous A549 and HeLa cells with significantly high photo-indices of ∼45 and ∼42 respectively in red light. The photo-cytotoxic potential of [Fe(III)–Ru(II)] was differential with minimum effects on the non-cancerous HPL1D cells. Overall, the hetero-bimetallic [Fe(III)–Ru(II)] complex has emerged as a next-generation and dual-modality PDT tool for anticancer therapy.Author contributions
Abhishek Panwar: led the project execution, synthesis, characterization, and photophysical studies and played a key role in drafting the original document. Chandi C. Malakar: characterization of ligands. Aarti Upadhyay: conducted in vitro assays and played a role in drafting the manuscript. Mithun Roy: directed the conceptualization of the project, obtained funding, oversaw the research process, and played a key role in writing and editing the original draft.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors wish to acknowledge the financial support received from the Board of Research in Nuclear Sciences (BRNS), Mumbai (37(2)/14/18/2017-BRNS), and the Indian Council of Medical Research (ICMR) (R.11.13/42/2021-GIA/HR, ICMR). They express their gratitude to NIT Manipur. We extend our sincere gratitude to Prof. Akhil R. Chakravarty from IISc, Bangalore for his invaluable support with the cell culture laboratory facilities. We also acknowledge the contributions of Punjab University and IIT Mandi for their assistance with various characterization methods.References
- J.-K. Yang, H. Kwon and S. Kim, J. Mater. Chem. B, 2024, 12, 2650–2669 RSC.
- S. Li, R. Liu, X. Jiang, Y. Qiu, X. Song, G. Huang, N. Fu, L. Lin, J. Song, X. Chen and H. Yang, ACS Nano, 2019, 13, 2103–2113 CAS.
- Y. Yang, S. Chen, L. Liu, S. Li, Q. Zeng, X. Zhao, H. Li, Z. Zhang, L.-S. Bouchard, M. Liu and X. Zhou, ACS Appl. Mater. Interfaces, 2017, 9, 23400–23408 CrossRef CAS PubMed.
- F. Sozmen, M. Kucukoflaz, M. Ergul and Z. D. S. Inan, RSC Adv., 2021, 11, 2383–2389 RSC.
- X. Luan, Y. Pan, Y. Gao and Y. Song, J. Mater. Chem. B, 2021, 9, 7076–7099 RSC.
- O. J. Stacey and S. J. A. Pope, RSC Adv., 2013, 3, 25550 RSC.
- A. Escudero, C. Carrillo-Carrión, M. C. Castillejos, E. Romero-Ben, C. Rosales-Barrios and N. Khiar, Mater. Chem. Front., 2021, 5, 3788–3812 RSC.
- A. G. Niculescu and A. M. Grumezescu, Appl. Sci., 2021, 11, 3626 CrossRef CAS.
- L. Huang, S. Zhao, J. Wu, L. Yu, N. Singh, K. Yang, M. Lan, P. Wang and J. S. Kim, Coord. Chem. Rev., 2021, 438, 213888 CrossRef CAS.
- Y. Yang, S. Jiang, S. Stanciu, H. Peng, A. Wu and F. Yang, Mater. Horiz., 2024, 11, 5815–5842 RSC.
- M. R. Hamblin, Photochem. Photobiol., 2019, 96, 506–516 CrossRef PubMed.
- D. Kessel, Photochem. Photobiol., 2022, 99, 199–203 CrossRef PubMed.
- I. S. Mfouo-Tynga, L. D. Dias, N. M. Inada and C. Kurachi, Photodiagn. Photodyn. Ther., 2021, 4, 102091 CrossRef PubMed.
- Dr. J. Karges, Angew. Chem., Int. Ed., 2022, 61, e202112236 CrossRef PubMed.
- L. B. Josefsen and R. W. Boyle, Met.-Based Drugs, 2007, 2008, 1–24 Search PubMed.
- L. K. McKenzie, H. E. Bryant and J. A. Weinstein, Coord. Chem. Rev., 2019, 379, 2–29 CrossRef CAS.
- L. Conti, A. Bencini, C. Ferrante, C. Gellini, P. Paoli, M. Parri, G. Pietraperzia, B. Valtancoli and C. Giorgi, Chem. – Eur. J., 2019, 25, 10606–10615 CrossRef CAS PubMed.
- A. Mani, T. Feng, A. Gandioso, R. Vinck, A. Notaro, L. Gourdon, P. Burckel, B. Saubaméa, O. Blacque, K. Cariou, J. Belgaied, H. Chao and G. Gasser, Angew. Chem., 2023, 135, e202218347 CrossRef.
- W. Su, Z. Luo, S. Dong, X. Chen, J.-A. Xiao, B. Peng and P. Li, Photodiagn. Photodyn. Ther., 2019, 26, 448–454 CrossRef CAS PubMed.
- H. Huang, S. Banerjee and P. J. Sadler, ChemBioChem, 2018, 19, 1574–1589 CrossRef CAS PubMed.
- B. Yuan, J. Liu, R. Guan, C. Jin, L. Ji and H. Chao, Dalton Trans., 2019, 48, 6408–6415 RSC.
- M. Obata, S. Hirohara, R. Tanaka, I. Kinoshita, K. Ohkubo, S. Fukuzumi, M. Tanihara and S. Yano, J. Med. Chem., 2009, 52, 2747–2753 CrossRef CAS PubMed.
- J. Liu, Y. Chen, G. Li, P. Zhang, C. Jin, L. Zeng, L. Ji and H. Chao, Biomaterials, 2015, 56, 140–153 CrossRef CAS PubMed.
- X. Wei, W.-B. Cui, G.-Y. Qin, X.-E. Zhang, F.-Y. Sun, H. Li, J.-F. Guo and A.-M. Ren, J. Med. Chem., 2023, 66, 4167–4178 CrossRef CAS PubMed.
- M. A. Munegowda, A. Manalac, M. Weersink, S. A. McFarland and L. Lilge, Coord. Chem. Rev., 2022, 470, 214712 CrossRef PubMed.
- J. Karges, S. Kuang, F. Maschietto, O. Blacque, I. Ciofini, H. Chao and G. Gasser, Nat. Commun., 2020, 11, 1–13 CrossRef PubMed.
- Q. Chen, V. Ramu, Y. Aydar, A. Groenewoud, X.-Q. Zhou, M. J. Jager, H. Cole, C. G. Cameron, S. A. McFarland, S. Bonnet and B. E. Snaar-Jagalska, Cancers, 2020, 12, 587 CrossRef CAS PubMed.
- U. Basu, I. Pant, I. Khan, A. Hussain, P. Kondaiah and A. R. Chakravarty, Chem. – Asian J., 2014, 9, 2494–2504 CrossRef CAS PubMed.
- A. Garai, U. Basu, I. Khan, I. Pant, A. Hussain, P. Kondaiah and A. R. Chakravarty, Polyhedron, 2014, 73, 124–132 CrossRef CAS.
- A. Garai, A. Gandhi, V. Ramu, M. K. Raza, P. Kondaiah and A. R. Chakravarty, ACS Omega, 2018, 3, 9333–9338 CrossRef CAS PubMed.
- U. Basu, I. Pant, P. Kondaiah and A. R. Chakravarty, Eur. J. Inorg. Chem., 2016, 2016, 1002–1012 CrossRef CAS.
- U. Basu, I. Khan, A. Hussain, P. Kondaiah and A. R. Chakravarty, Angew. Chem., Int. Ed., 2012, 51, 2658–2661 CrossRef CAS PubMed.
- Y. Wang, P. S. Felder, P. Mesdom, O. Blacque, T. L. Mindt, K. Cariou and G. Gasser, ChemBioChem, 2023, 24, e202300467 CrossRef CAS PubMed.
- L. Ma, L. Li and G. Zhu, Inorg. Chem. Front., 2022, 9, 2424–2453 RSC.
- A. Van Niekerk, P. Chellan and S. F. Mapolie, Eur. J. Inorg. Chem., 2019, 2019, 3432–3455 CrossRef CAS.
- X.-R. Ma, J.-J. Lu, B. Huang, X.-Y. Lu, R.-T. Li and R.-R. Ye, J. Inorg. Biochem., 2023, 240, 112090 CrossRef CAS PubMed.
- N. Roy, S. Shanavas, B. Kar, L. T. Babu, U. Das, S. Vardhan, S. K. Sahoo, B. Bose, V. Rajagopalan and P. Paira, ACS Omega, 2023, 8, 12283–12297 CrossRef CAS PubMed.
- S. L. H. Higgins, T. A. White, B. S. J. Winkel and K. J. Brewer, Inorg. Chem., 2010, 50, 463–470 CrossRef PubMed.
- Z. Zhou, J. Liu, T. W. Rees, H. Wang, X. Li, H. Chao and P. J. Stang, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 5664–5669 CrossRef CAS PubMed.
- S. Ovallath and B. Sulthana, Ann. Indian Acad. Neurol., 2017, 20, 185 CrossRef PubMed.
- Y.-H. Chiu and J. W. Canary, Inorg. Chem., 2003, 42, 5107–5116 CrossRef CAS PubMed.
- H. G. Miserachs, M. Cipriani, J. Grau, M. Vilaseca, J. Lorenzo, A. Medeiros, M. A. Comini, D. Gambino, L. Otero and V. Moreno, J. Inorg. Biochem., 2015, 150, 38–47 CrossRef CAS PubMed.
- R. Larsson, N. Blanco, M. Johansson and O. Sterner, Tetrahedron Lett., 2012, 53, 4966–4970 Search PubMed.
- J.-S. Hwang, J.-M. An, H. Cho, S. H. Lee, J.-H. Park and I.-O. Han, Eur. J. Pharmacol., 2015, 746, 41–49 CrossRef CAS PubMed.
- T. Rüther, C. P. Woodward, T. W. Jones, C. I. Coghlan, Y. Hebting, R. L. Cordiner, R. E. Dawson, D. E. J. E. Robinson and G. J. Wilson, J. Organomet. Chem., 2016, 823, 136–146 CrossRef.
- F. Wang, J. Xu, A. Habtemariam, J. Bella and P. J. Sadler, J. Am. Chem. Soc., 2005, 127, 17734–17743 CrossRef CAS PubMed.
- A. R. Simović, R. Masnikosa, I. Bratsos and E. Alessio, Coord. Chem. Rev., 2019, 398, 113011 CrossRef.
- S. Liu, A. Liang, K. Wu, W. Zeng, Q. Luo and F. Wang, Int. J. Mol. Sci., 2018, 19, 2137 Search PubMed.
- J. Shao, W.-G. Bao, H. Tian, B. Li, X.-F. Zhao, X. Qiao and J.-Y. Xu, Dalton Trans., 2014, 43, 1663–1671 RSC.
- K. Phopin, W. Ruankham, S. Prachayasittikul, V. Prachayasittikul and T. Tantimongcolwat, Int. J. Mol. Sci., 2019, 21, 249 CrossRef PubMed.
- R. Bhowmik, A. Upadhyay, M. Pal, A. Bera and M. Roy, New J. Chem., 2024, 48, 5465–5474 RSC.
- A. Panwar, M. Pal and M. Roy, J. Inorg. Biochem., 2023, 238, 112055 CrossRef CAS PubMed.
- U. Basu, M. Roy and A. R. Chakravarty, Coord. Chem. Rev., 2020, 417, 213339 CrossRef CAS.
- S. Gazi and R. Ananthakrishnan, Curr. Anal. Chem., 2012, 8, 143–149 CrossRef CAS.
- A. A. Abdel-Shafi, D. R. Worrall and A. Y. Ershov, Dalton Trans., 2004, 30–36 RSC.
- H. G. Miserachs, M. Cipriani, J. Grau, M. Vilaseca, J. Lorenzo, A. Medeiros, M. A. Comini, D. Gambino, L. Otero and V. Moreno, J. Inorg. Biochem., 2015, 150, 38–47 Search PubMed.
- N. E. Aksakal, H. H. Kazan, E. T. Eçik and F. Yuksel, New J. Chem., 2018, 42, 17538–17545 RSC.
- D. Musib, M. Pal, M. K. Raza and M. Roy, Dalton Trans., 2020, 49, 10786–10798 RSC.
- F. Wallberg, T. Tenev and P. Meier, Cold Spring Harbor Protoc., 2016, 4, 1559–6095 Search PubMed.
- B. Peña, S. Saha, R. Barhoumi, R. C. Burghardt and K. R. Dunbar, Inorg. Chem., 2018, 57, 12777–12786 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4dt03433c |
| This journal is © The Royal Society of Chemistry 2025 |