
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBuilt-in electrophilic/nucleophilic domain of nitrogen-doped carbon nanofiber-confined Ni2P/Ni3N nanoparticles for efficient urea-containing water-splitting reactions†
Jiaxin Liab,
Chun Yinac,
Shuli Wangc,
Baogang Zhang *b and
Ligang Feng*a
*b and
Ligang Feng*a
aFaculty of Materials Science and Engineering, Kunming University of Science and Technology, Kunming 650093, China. E-mail: fenglg11@gmail.com
bSchool of Water Resources and Environment, MOE Key Laboratory of Groundwater Circulation and Environmental Evolution, China University of Geosciences (Beijing), Beijing, 100083, P.R. China. E-mail: baogangzhang@cugb.edu.cn
cSchool of Chemistry and Chemical Engineering, Yangzhou University, Yangzhou 225002, P.R. China
First published on 7th August 2024
Abstract
Transferring urea-containing waste water to clean hydrogen energy has received increasing attention, while challenges are still faced in the sluggish catalytic kinetics of urea oxidation. Herein, a novel hybrid catalyst of Ni2P/Ni3N embedded in nitrogen-doped carbon nanofiber (Ni2P/Ni3N/NCNF) is developed for energy-relevant urea-containing water-splitting reactions. The built-in electrophilic/nucleophilic domain resulting from the electron transfer from Ni2P to Ni3N accelerates the formation of high-valent active Ni species and promotes favourable urea molecule adsorption. A spectral study and theoretical analysis reveal that the negatively shifted Ni d-band centre in Ni2P/Ni3N/NCNF weakens the adsorption of intermediate CO2 and facilitates its desorption, thereby improving the urea oxidation reaction kinetics. The overall reaction process is also optimized by minimizing the energy barrier of the rate-determining step. Following the stability test, the surface reconstruction of the pre-catalyst is discussed, where an amorphous layer of NiOOH as the real active phase is formed on the surface/interface of Ni2P/Ni3N for urea oxidation. Benefiting from these characteristics, a high current density of 151.11 mA cm−2 at 1.54 V vs. RHE is obtained for urea oxidation catalysed by Ni2P/Ni3N/NCNF, exceeding that of most of the similar catalysts. A low cell voltage of 1.39 V is required to reach 10 mA cm−2 for urea electrolysis, which is about 200 mV less than that of the general water electrolysis. The current work will be helpful for the development of advanced catalysts and their application in the urea-containing waste water transfer to clean hydrogen energy.
Introduction
Balancing energy supply and environmental protection is highly desired for sustainable development, and various clean energies have been exploited by utilizing natural resources in an environmentally friendly manner, such as photovoltaics, wind power, and water electrolysis, etc.1–5 Meanwhile, extracting energy through the contaminated media seems more urgent, practical, and challenging. For example, the green hydrogen generation from water splitting by coupling with anodic small organic oxidation (methanol, formic acid, glycerol, and urea, etc.) has attracted increasing attention nowadays.6–9 Among them, urea is a common environmental pollutant but with a high hydrogen content of 6.67 wt%.10,11 Moreover, the stable and non-flammable properties of urea in ambient conditions make it easy for storage and transport,12 fulfilling the expected Department of Energy (DOE) targets for hydrogen storage in transport applications. In particular, the energy extraction from urea-involved human urine is of great significance for energy utilization in the military and space field. It is reported that the concentration of urea in human urine ranges from 11 to 25 mg L−1 (0.18–0.41 M),13 and 0.33 M is the most commonly used value in research currently. In urban sewage treatment, urine only accounts for 1%, but provides about 80% of nitrogen,14 which increases operational complexity and disposal costs. Moreover, the excessive nitrogen-containing substances in domestic sewage can lead to eutrophication of surface water.15 Since urea is the main nitrogen compound in urine, developing feasible urea removal or utilization technologies such as urea electrolysis is the key to achieving source separation, by which the problems caused by long-term storage and unreasonable discharge of toilet wastewater can be avoided, and while urea degrades, hydrogen energy is generated to achieve the goal of energy production and utilization.16–19The theoretical equilibrium potential of urea electrolysis is as low as 0.37 V, far less than that of water splitting (1.23 V).20–22 As reported, 30% of energy and 36% of the cost can be saved by using the urea oxidation reaction (UOR) instead of the anodic oxygen evolution reaction (OER) in the water electrolysis technique.23,24 Therefore, UOR will be an ideal alternative for OER in water splitting to concurrently achieve the goals of energy conservation and pollutant degradation.25–27 Nevertheless, the six-electron transfer process accompanied by various intermediates intrinsically impedes the reaction kinetics of UOR (*CO(NH2)2 → *CO(NH2·NH) → *CO(NH2·N) → *CO(NH·N) → *CO(N2) → *CO(OH) → *COO).28 Thus, attention has been concentrated on the development of cost-efficient and highly stable catalysts for urea oxidation. Ni-based materials have been widely studied for UOR because of their high activity and acceptable cost.29–31 It was found that Ni even had inherently higher activity than noble metals in urea oxidation.23 In our previous study, Ni2P–C was found to be an efficient UOR catalyst, whose peak current density was 2.7 times higher than that of commercial nickel oxide catalyst.32 So far, various Ni-based catalysts and design strategies have been developed to boost UOR catalytic performance, as reported in some review reports.33–35 The construction of heterostructure catalysts becomes one of the hot spots due to the synergistic effect of the hybridized phases.36–38 The interface formed between two phases with different chemical properties will induce unique characteristics such as electron redistribution, more active site exposure, charge transfer acceleration, and binding energy optimization.39,40 Up to now, several nickel phosphide-based heterostructure catalysts such as Ni/Ni2P,41 Ni–Ni3P,42 Ni phosphate@Ni12P5,43 and NiF2/Ni2P44 have been reported for UOR.
The interstitial compound Ni3N exhibits the properties of covalent compounds, ionic crystals, and transition metals due to the integration of N atoms into the interstitial sites of Ni metal.45 The lattice parameter and d-band of the parent metal can be modified to yield good electrocatalytic behaviour.46 It was reported that the junction of Ni3N and black phosphorus (BP) had enhanced OER activity,47,48 where the Ni–P bond and Ni2P interface formed between Ni3N and BP contributed a lot due to the accelerated reaction rate and more high-valent active species. However, the UOR activity and stability are still not satisfied, and fundamentally, the catalysis mechanism induced by the specific interaction between interfaces, the variation of band structure, as well as the electron transfer direction and number, are still not clear. In addition, during the electrocatalytic reaction process, especially at the anode where positive bias is applied, surface reconstruction generally occurs on the catalyst surface, which is often accompanied by the formation of new species or structures.49 Unfortunately, the current investigation about the activity origin of heterostructure is mostly limited to the analysis of pre-catalysts, rather than the in-depth research on the reconstruction of hetero-interfaces.
Inspired by these findings, it is inferred that the direct combination of Ni3N and Ni2P in a heterostructure catalyst may synergistically give full play of their advantages to improve the electrocatalytic reaction by electron re-distribution and bandgap structure tuning. Herein, the hetero-structured Ni2P/Ni3N nanoparticles confined by N-doped carbon nanofiber (Ni2P/Ni3N/NCNF) were successfully fabricated via a facile electrospinning approach and the subsequent annealing processes under the corresponding conditions to realize the structural transformation (Fig. 1a). The spectral and theoretical analysis indicated the electron transfer between Ni2P and Ni3N induced the formation of an electrophilic/nucleophilic domain, which promoted favourable urea molecule adsorption and catalytic oxidation. Moreover, the negatively shifted Ni d-band centre in Ni2P/Ni3N/NCNF weakened the adsorption of CO2 and facilitated the reaction kinetics. As a result, the as-prepared Ni2P/Ni3N/NCNF catalyst showed the highest current density of 151.11 mA cm−2 at 1.54 V vs. RHE for urea oxidation, with excellent catalytic stability. Meanwhile, Ni2P/Ni3N/NCNF yielded the kinetic current density of 10 mA cm−2 at a small cell potential of 1.39 V for urea-assisted water splitting, significantly lowering the energy input of water electrolysis (1.59 V). As the first case of the Ni2P/Ni3N study, it will be an ideal platform to concurrently achieve the goals of energy conservation and pollutant degradation.
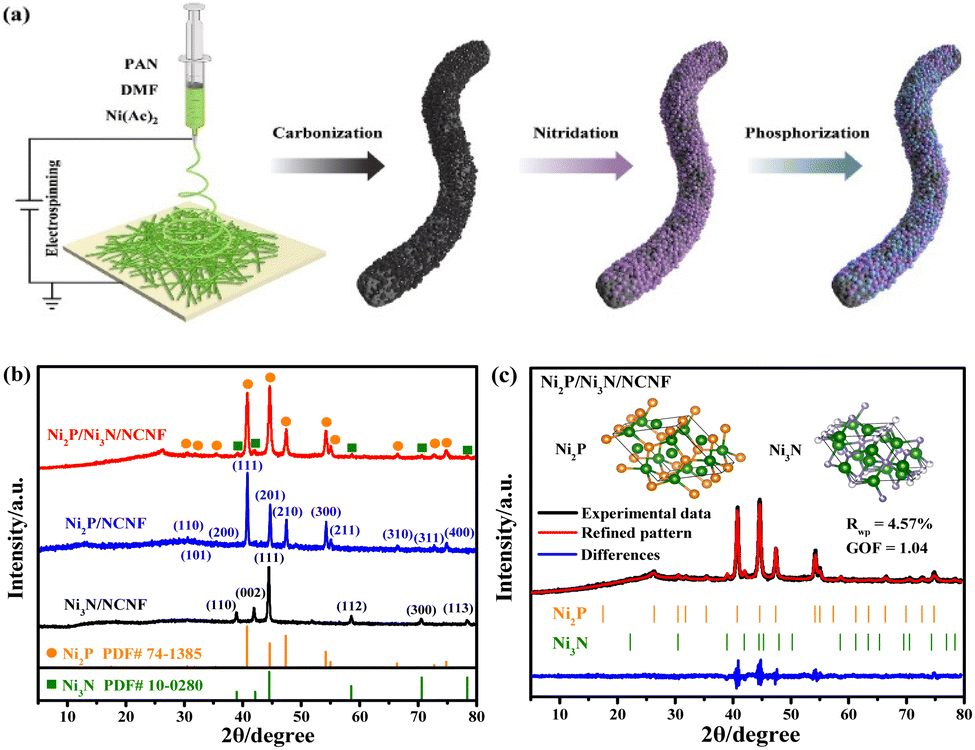 | ||
| Fig. 1 (a) Schematic illustration of the synthesis of Ni2P/Ni3N/NCNF. (b) XRD patterns of Ni3N/NCNF, Ni2P/NCNF, and Ni2P/Ni3N/NCNF. (c) Rietveld refinement of XRD pattern of Ni2P/Ni3N/NCNF. | ||
Results and discussion
Composition and microstructures
The Ni/NCNF was successfully prepared, as confirmed by the corresponding Ni diffraction peaks in the XRD patterns (Fig. S1†). The pure phase of Ni3N (JCPDS # 10-0280) and Ni2P (JCPDS # 74-1385) was obtained, respectively, as proved by the XRD patterns after the separate nitridation and phosphorization treatment of Ni/NCNF (Fig. 1b). Ni2P/Ni3N/NCNF showed both characteristic diffraction peaks of Ni2P and Ni3N crystal phases (Fig. 1b), demonstrating the successful fabrication of the hybrid catalyst. The detailed composition and structural information were analyzed by Rietveld refinement of the XRD patterns, which confirmed the hexagonal Ni2P crystal lattice with P![[6 with combining macron]](https://www.rsc.org/images/entities/char_0036_0304.gif) 2m space group, and the hexagonal Ni3N with P6322 space group (Fig. 1c). The component of Ni2P and Ni3N was calculated to be 79.8 and 20.2 wt%, respectively. Accordingly, the atomic ratio of Ni, P, and N was evaluated to be 2.70
2m space group, and the hexagonal Ni3N with P6322 space group (Fig. 1c). The component of Ni2P and Ni3N was calculated to be 79.8 and 20.2 wt%, respectively. Accordingly, the atomic ratio of Ni, P, and N was evaluated to be 2.70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.04:0.21. The cell length and cell volume of Ni2P and Ni3N in Ni2P/Ni3N/NCNF both slightly increased compared to those of standard crystal Ni2P and Ni3N, indicating a tiny lattice expansion (Table S1†).
:1.04:0.21. The cell length and cell volume of Ni2P and Ni3N in Ni2P/Ni3N/NCNF both slightly increased compared to those of standard crystal Ni2P and Ni3N, indicating a tiny lattice expansion (Table S1†).
The morphology observation by scan electron microscopy (SEM) showed a one-dimensional nanofiber structure with some small granules confined in the nanofibers for the Ni/NCNF (Fig. S2†); Ni3N/NCNF and Ni2P/NCNF showed similar nanofibrous morphology but with a rougher surface due to the nitridation and phosphorization processes, respectively (Fig. S3 and S4†). The surface of Ni2P/Ni3N/NCNF nanofiber became much rougher, and some fractures occurred because of the harsh condition of the two successive annealing treatments (Fig. 2a and S5†). In the local morphology observation by transmission electron microscopy (TEM) (Fig. 2b and c), in addition to the dark metal particles, some spherical hollows generated by the precursor Ni(Ac)2 decomposition were also observed. The coexistence of Ni2P (210) plane and Ni3N (110) plane was indicated in the high-resolution TEM (HRTEM), corresponding to the lattice fringes of 0.19 and 0.23 nm, respectively (Fig. 2d and S6†). The interface between these two phases can be clearly observed, testifying to the formation of the Ni2P/Ni3N heterostructure. The mixed crystalline phases of Ni2P and Ni3N were also evidenced by the selected area electron diffraction (SAED) (Fig. 2e), which was in line with the XRD pattern. The EDX spectroscopy detected the C, Ni, P, N, and O elements (Fig. 2f), and the atomic ratio of Ni/P/N was 2.83:1.04:3.16 (Table S2†), which shared an almost identical proportion of Ni and P with the atomic ratio estimated by XRD analysis, and the larger amount of N element would come from the N doped into the carbon nanofiber, which was not discussed in the XRD analysis. In the elemental mapping images (Fig. 2g–k), C and O were evenly distributed on the surface of the nanofiber, while Ni and P were distributed as blocks confined in the nanofiber; N was also uniformly visible throughout the nanofiber, again indicating it not only came from Ni3N but also carbon fibre in the doping form.
 | ||
| Fig. 2 (a) SEM image, (b and c) TEM images, (d) HRTEM image, (e) SAED pattern, (f) EDX pattern, (g–k) STEM and elemental mapping images of Ni2P/Ni3N/NCNF. | ||
It is known that high hydrophilicity can accelerate the penetration of electrolytes to active sites and weaken the adhesion force of gas bubbles, thereby improving the catalytic performance.50 The hydrophilicity of the as-prepared catalysts was probed by the contact angle. Compared with the pristine Ni/NCNF, the modified catalysts with phosphorization and nitridation both helped reduce the contact angle, and Ni2P/Ni3N/NCNF showed the smallest contact angle of 30.0° for its superior hydrophilicity (Fig. 3a and b). The surface chemical states of these samples were investigated by X-ray photoelectron spectroscopy (XPS). All the concerned elements of C, Ni, P, N, and O were detected in the survey spectrum (Fig. S7a†). The whole binding energies were calibrated against the C 1s peak at 284.60 eV (Fig. S7b†). The XPS fine spectrum of Ni 2p can be fitted by metallic Ni, oxidized Ni, and satellite peaks (Fig. 3c and Table S3†). In Ni/NCNF, the peaks of metallic Ni (Ni0) were located at 852.35 and 869.95 eV,51 while the binding energy of Ni0 for Ni3N/NCNF and Ni2P/NCNF shifted to a higher direction by about 0.10 and 0.79 eV, respectively, deriving from the electron transfer from Ni to N and P. Notably, the metallic Ni sat at even much higher binding energies of 853.24 and 870.84 eV for Ni2P/Ni3N/NCNF, which indicated that in addition to the electron transfer within the single compound, the electron transfers also occurred between the Ni2P and Ni3N. In the XPS spectra of P 2p for Ni2P/NCNF and Ni2P/Ni3N/NCNF (Fig. S7c†), apart from the P–O peak derived from the surface oxidation, two peaks of P 2p3/2 and P 2p1/2 were also observed, demonstrating the formation of Ni–P bond (Table S4†).52 Whereas, the N 1s spectra of all the samples showed the peaks of pyridinic-N (398.80 eV), pyrrolic-N (400.10 eV), and graphitic-N (401.20 eV), confirming the doping of N into carbon nanofibers (Fig. 3d and Table S5†).47,53 Notably, the Ni–N bond of Ni2P/Ni3N/NCNF was located at a lower binding energy of 397.62 eV than that of Ni3N/NCNF (397.85 eV), revealing that N attracted some electrons from neighbour atoms. The dopant N came from N,N-dimethylformamide, which was commonly used as a nitrogen source to synthesize N-doped carbon nanofiber during electrospinning.54,55 Doping N into carbon nanofiber is a versatile strategy for tailoring their properties in catalysis and energy-related fields by influencing the surface chemistry, electronic structure, and conductivity. In specific, N-doping can introduce heteroatoms into the carbon lattice, facilitating electron transfer processes and enhancing the kinetics. The O 1s spectra were further analysed (Fig. S7d and Table S6†). The signal peaks of Ni–O, C–O, and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O could be fitted for all the samples.56 In addition, the P–O peak was indicated in Ni2P/NCNF and Ni2P/Ni3N/NCNF,57 which was consistent with surface oxidation indicated in the spectra of P 2p. Compared to the Ni/NCNF and Ni3N/NCNF, the formation of Ni2P in the system would reduce the surface oxidation as low intensity of Ni–O was found in the Ni2P/Ni3N/NCNF and Ni2P/NCNF.
O could be fitted for all the samples.56 In addition, the P–O peak was indicated in Ni2P/NCNF and Ni2P/Ni3N/NCNF,57 which was consistent with surface oxidation indicated in the spectra of P 2p. Compared to the Ni/NCNF and Ni3N/NCNF, the formation of Ni2P in the system would reduce the surface oxidation as low intensity of Ni–O was found in the Ni2P/Ni3N/NCNF and Ni2P/NCNF.
 | ||
| Fig. 3 (a) Contact angle of Ni2P/Ni3N/NCNF, Ni2P/NCNF, Ni3N/NCNF, and Ni/NCNF. (b) The illustration of the contact angle; XPS spectra of Ni 2p region (c) and N 1s region (d) for Ni/NCNF, Ni3N/NCNF, Ni2P/NCNF, and Ni2P/Ni3N/NCNF. | ||
Water and urea oxidation study
Considering the competition reaction of water oxidation and urea oxidation in urea-containing wastewater handling, the electrochemical water oxidation was first studied in alkaline electrolytes of 1 M KOH at 5 mV s−1. The benchmark current density of 10 mA cm−2, the predicted current density for 10% competent solar to fuel conversion devices under 1 sun illumination,58,59 was employed for the performance comparisons. The Ni2P/Ni3N/NCNF catalyst showed the lowest overpotential of 283 mV to reach the benchmark current density of 10 mA cm−2, showing superior OER activity (Fig. 4a). This performance was also comparable to that of the state-of-the-art commercial RuO2 catalyst, but with the advantage of low cost. The largely improved activity could be due to the facile Ni species oxidation as indicated by the Ni oxidation peaks (inset of Fig. 4a). The Ni/NCNF showed the onset potential and peak potential at 1.332 and 1.379 V for redox of Ni2+/Ni3+ (Table S7†), while these potentials were reduced by forming the Ni2P, Ni3N, and their hybrids. Specifically, the Ni2P/Ni3N/NCNF catalyst showed the onset potential and peak potential at 1.316 and 1.362 V, respectively, which indicated the much easier Ni oxidative species formation, a significant intermediate for water and urea oxidation.60 The kinetics for water oxidation were evaluated by Tafel slopes and the charge transfer resistance. The Ni2P/Ni3N/NCNF electrode showed the Tafel slope of 91 mV dec−1 (Fig. 4b), smaller than other electrodes, signifying faster catalytic kinetics. The charge transfer ability of these electrodes was also probed by EIS (Fig. 4c and S8†). By fitting the Nyquist plots, the Ni2P/Ni3N/NCNF exhibited the smallest charge transfer resistance (Rct) of 64.37 Ω, indicating its fastest charge transfer ability (Table S8†). The stability of Ni2P/Ni3N/NCNF for water oxidation was probed by recording 1000 CV cycles at the scan rate of 150 mV s−1 and a long-term operation to offer the kinetic current density of 10 mA cm−2 for 10 hours. After the accelerated attenuation test, good dynamic stability was observed by comparing the CV curves before and after 1000 CV cycles running, where no serious performance decay was observed (Fig. 4d); in the steady state stability evaluated by the CA test (inset of Fig. 4d), good performance was also obtained where the kinetic current density of 10 mA cm−2 was steadily kept for 10 h at 1.52 V. The efficiency for water oxidation to oxygen was evaluated by comparing the experimentally generated oxygen volume to the theoretical amount of O2 (Fig. S9†), and a high current efficiency close to 100% was found for Ni2P/Ni3N/NCNF. Thus, Ni2P/Ni3N/NCNF would be a proper catalyst for urea-containing water oxidation.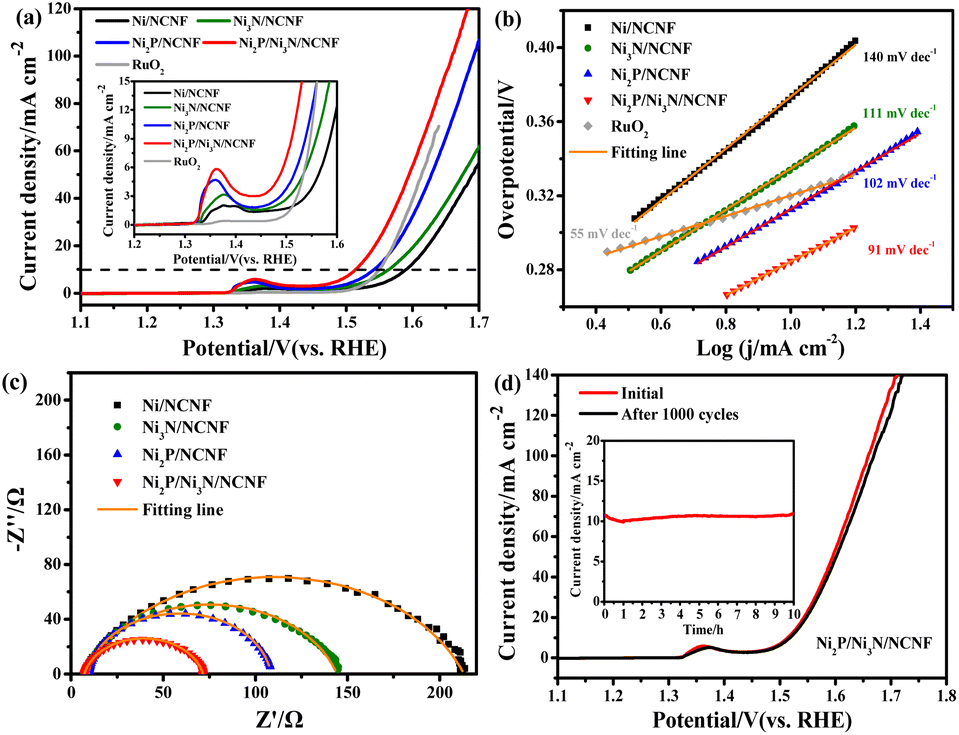 | ||
| Fig. 4 (a) LSV curves and (b) Tafel slopes of all the samples. (c) EIS curves of Ni/NCNF, Ni3N/NCNF, Ni2P/NCNF, and Ni2P/Ni3N/NCNF. (d) LSV curves before and after 1000 CV cycles and the CA curve (inset in (d)) of Ni2P/Ni3N/NCNF in 1 M KOH solution. | ||
Then we carefully investigated the UOR catalytic activity and kinetics catalysed by these electrodes in 1 M KOH and 0.33 M urea electrolyte.61 The Ni2P/Ni3N/NCNF electrode still achieved the highest current density of 151.11 mA cm−2 at 1.54 V, much higher than that of Ni2P/NCNF (82.57 mA cm−2), Ni3N/NCNF (50.66 mA cm−2), and Ni/NCNF (35.48 mA cm−2) (Fig. 5a). Note that the catalytic ability of this electrode was also much better than the counterparts and many other similar catalysts reported in recent years (Table S9†). The competition of water oxidation and urea oxidation was compared for the Ni2P/Ni3N/NCNF electrode by the LSV curve (Fig. 5b). Ni2P/Ni3N/NCNF can deliver 10 mA cm−2 at 1.337 V for UOR, which was negatively shifted by 176 mV than that of water oxidation, showing its advantage of energy saving. Notably, the oxidation of Ni2+ to Ni3+ at ca. 1.36 V was close to the onset potential of UOR, which verified the role of high-valent Ni as an active phase in UOR.30 The Ni oxidation peak was not observed during UOR; this was because the electrochemically formed Ni3+ quickly chemically oxidized urea molecules and continually catalysed urea oxidation under the control of an electrochemical–chemical mechanism.62 By plotting the current density under different scan rates (Fig. S10†), a linear relationship was obtained on the peak current density vs. the square root of the scan rate (Fig. S11†), indicating a diffusion-controlled process.63 The slope of the linear fitting was proportional to the electron transfer coefficient in the rate-determining step;64 thus, the largest slope of Ni2P/Ni3N/NCNF confirmed its superior catalytic kinetics. Consistently, the Ni2P/Ni3N/NCNF showed the fastest reaction kinetics for UOR, indicated by the smallest Tafel slope of 29 mV dec−1 (Fig. 5c). The charge transfer resistance was evaluated by impedance measured at 0.34 V for UOR, and Ni2P/Ni3N/NCNF showed the smallest arc diameter, meaning the lowest charge transfer resistance (Fig. 5d); and the smallest charge transfer resistance (Rct) of 45.24 Ω was obtained by fitting and analysing the Nyquist plots with an equivalent circuit model consisting of resistance, constant phase element, and capacitance (Fig. S12† and Table S10†). The improved kinetics could be attributed to the interface between Ni2P and Ni3N, which efficiently reduced the charge transfer resistance and accelerated the electron transfer.65
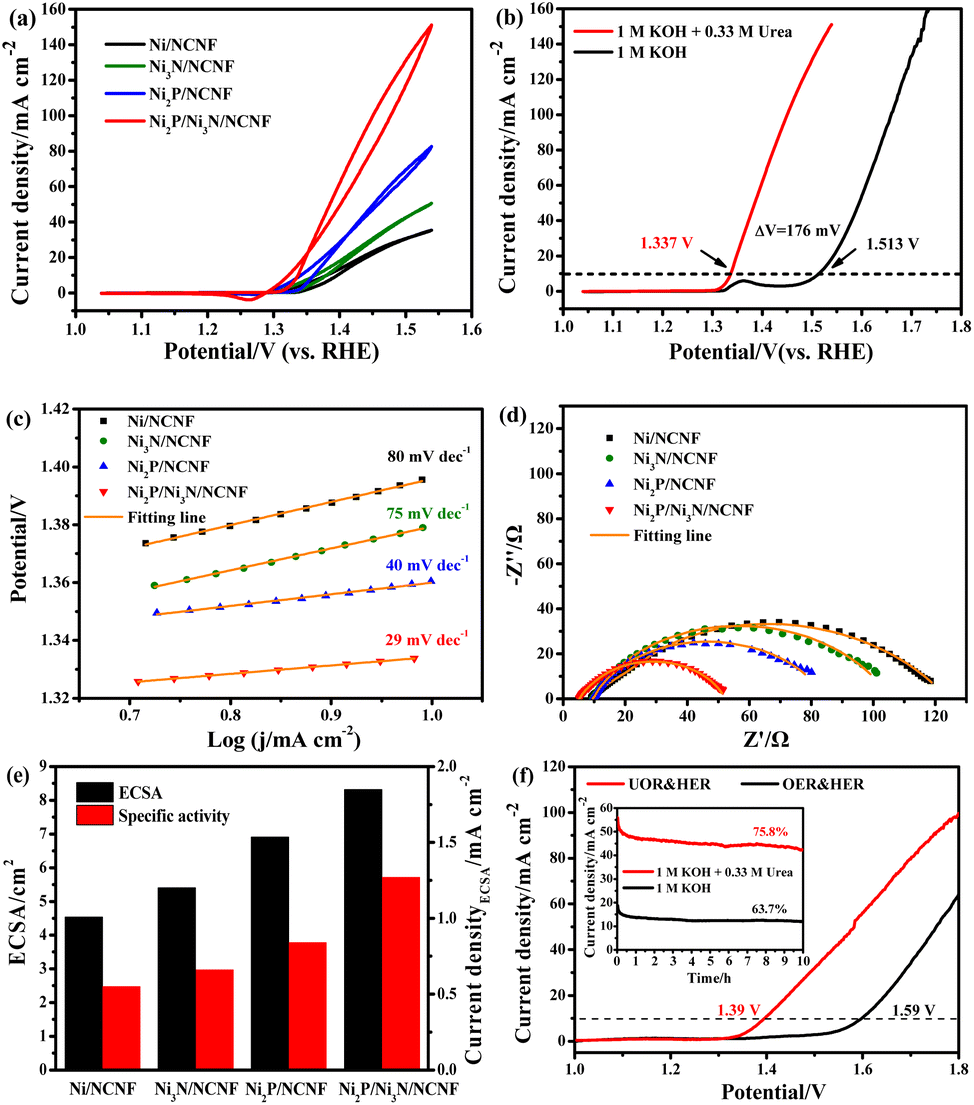 | ||
| Fig. 5 (a) CV curves measured in 1 M KOH with 0.33 M urea at 10 mV s−1. (b) LSV curves of Ni2P/Ni3N/NCNF in 1 M KOH with and without 0.33 M urea. (c) Tafel slopes, (d) EIS, (e) ECSA and specific activity for Ni/NCNF, Ni3N/NCNF, Ni2P/NCNF, and Ni2P/Ni3N/NCNF. (f) LSV plots of urea electrolysis and overall water splitting of the Ni2P/Ni3N/NCNF‖Pt/C electrode with (inset) durability tests at 1.6 V. | ||
The electrochemical surface area (ECSA) was then estimated by integrating the reduction peak of Ni species in the CV curves conducted in the background KOH solution (Fig. S13†). The results indicated that Ni2P/Ni3N/NCNF had the biggest ECSA of 8.32 cm2, larger than that of Ni2P/NCNF (6.91 cm2), Ni3N/NCNF (5.40 cm2), and Ni/NCNF (4.54 cm2), showing its rougher surface and more available active sites. We also calculated the ECSA from the double-layer capacitance (Cdl) by measuring CV at various scan rates in a non-faradaic region (Fig. S14†). The ECSA obtained for these samples was higher than the corresponding values mentioned above (Table S11†). This was due to the excess capacitive current contribution from the carbon materials in the catalyst.34 In this work, the specific activity was compared by normalizing the current to the ECSA calculated from the Ni reduction peak to get a fair active site efficiency (Fig. 5e and S15†). Ni2P/Ni3N/NCNF still displayed the largest specific activity of 1.27 mA cm−2 at 1.54 V, revealing the effectively enhanced intrinsic activity and efficiency for UOR. Furthermore, the turnover frequency (TOF) of all the samples was compared to evaluate the catalytic efficiency (details see the ESI†). Ni/NCNF exhibited the lowest TOF of 0.005 s−1, and Ni2P/Ni3N/NCNF showed the highest TOF value of 0.026 s−1 at 1.54 V, which was nearly two times of Ni2P/NCNF (0.015 s−1) and three times of Ni3N/NCNF (0.008 s−1). TOF results demonstrated that Ni2P/Ni3N/NCNF can provide the most efficient active sites among all the samples.
Though the UOR is measured below the potential of 1.55 V, the competitive reaction of UOR and OER still occurs. The faradaic efficiency of UOR was further probed by gas chromatographic analysis by collecting the gas product during an 8 h CA test at 1.39 V in an electrode system (Fig. S16 and S17†). The faradaic efficiency was calculated to be ca. 95.07% using the electron numbers consumed for N2 generation for urea oxidation, indicating the process is mainly for UOR. The UOR ability for 10 hours was probed by the chronoamperometry (CA) measurement (Fig. S18†). Ni2P/Ni3N/NCNF delivered the largest final current density of 47.72 mA cm−2 with the highest retention rate of 82.21%, demonstrating good stability. Though the catalytic stability can be improved by pure phosphorization or nitridation, the most significant improvement came from the Ni2P/Ni3N/NCNF heterostructure owing to the more exposed active sites with high catalytic ability and the synergistic effect between Ni2P and Ni3N.
The potential application of the Ni2P/Ni3N/NCNF for water and urea oxidation in the two-electrode system was finally compared in 1 M KOH with/without 0.33 M urea (Fig. 5f). Obviously, Ni2P/Ni3N/NCNF achieved a current density of 99.63 mA cm−2 at 1.8 V in urea containing alkaline electrolyte; however, it was 64.29 mA cm−2 in pure KOH solution. Most importantly, it only needed 1.39 V to offer the benchmark current density of 10 mA cm−2 in urea electrolysis, about 200 mV lower than that for the water-splitting reaction. This performance was comparable to some Ni-based catalysts reported recently (Table S12†). In addition, a higher current density and retention ability were also obtained in urea electrolysis during the 10 h durability test (inset of Fig. 5f). Therefore, by assisting the water splitting with urea oxidation, the energy input for hydrogen generation can be significantly reduced, which was beneficial for the practical application.
Catalytic mechanism discussion
The UOR catalysed by Ni generally follows the electrochemical–chemical (EC) mechanism (eqn (1) and (2)).62Electrochemical:
| 6Ni(OH)2 + 6OH− → 6NiOOH + 6H2O + 6e− | (1) |
Chemical:
| 6NiOOH + CO(NH2)2 + H2O → 6Ni(OH)2 + N2 + CO2 | (2) |
The high-valent Ni species were believed to be the active phase for urea decomposition. Taking Ni(OH)2 as an example, Ni(OH)2 is electrochemically oxidized to NiOOH with the increase of applied voltage at first, then the formed NiOOH chemically oxidized urea with its reduction to Ni(OH)2. Therefore, the surface reconstruction will occur to form the active Ni phase with high valence states to chemically catalyse the process.66 To explore the surface reconstruction, the morphology, phase composition, and surface chemical state of Ni2P/Ni3N/NCNF were thus probed after the stability test in the urea-assisted water-splitting reaction. Due to the harsh alkaline electrolyte and the strong oxidative environment, the surface of the nanofibrous catalyst became much rougher and showed more cracks owing to the surface reconstruction and the dissolution of phosphate species (Fig. 6a). Some particles were still observed in the carbon nanofiber, while some voids were also found due to the metal species leaching (Fig. 6b). The HRTEM images still showed some crystalline lattice fringes of 0.19 nm and 0.24 nm corresponding to Ni2P (210) and Ni3N (110) crystal faces, and their interface was still visible (Fig. 6c). Significantly, an amorphous layer around the surface and interface of Ni2P/Ni3N was found, which was attributed to the active phase of Ni oxyhydroxide. The XRD patterns showed the reduced diffraction peaks intensity of Ni3N and Ni2P (Fig. 6d), and consistently, the very weak diffraction rings of Ni2P (210) and Ni3N (110) were observed in the SAED pattern (Fig. 6e). The variation of the element composition of Ni2P/Ni3N/NCNF was investigated by EDX analysis (Fig. 6f and Table S13†). It was obvious that the content of O element increased a lot (from 6.36 to 11.03 wt%), indicating the partial surface oxidation and the formation of metal oxyhydroxide. Meanwhile, the decreased P element meant some P was leached out during the urea oxidation (from 2.33 to 0.95 wt%). The content of Ni and N was nearly kept constant. The details of the element distribution were illustrated in the element mapping images (Fig. 6g–k).
 | ||
| Fig. 6 (a) SEM image, (b) TEM image, (c) HRTEM image, (d) XRD pattern, (e) SAED pattern, (f) EDX pattern, (g–k) STEM and elemental mapping images of Ni2P/Ni3N/NCNF after urea electrolysis. | ||
The surface chemical state change after the stability test also confirmed the surface oxidation. Typically for the Ni 2p spectrum, the peaks of metallic Ni disappeared, and only the peak for oxidized Ni with the valence of Ni3+ at 857.13 and 874.73 eV was required for the peak fitting (Fig. 7a). Consistently, the intensity of Ni–P and Ni–N peaks fitted in the P 2p and N 1s spectra were almost invisible (Fig. S19a and b†). Correspondingly, the adsorbed water and increased intensity of the Ni–O bond in the O 1s spectrum were observed compared to the fresh sample, confirming the increased oxygen-containing Ni species on the catalyst surface (Fig. S19c†).67 This phenomenon was consistent with the above-mentioned E-C mechanism for urea oxidation, where the high-valent Ni3+ active species generated from the Ni2P/Ni3N/NCNF catalysed the reaction. Herein, Ni2P/Ni3N/NCNF facilitated the formation of high-valent Ni3+ active species, as confirmed by the surface catalyst. Similar results were also reported on FeNi–FeNiO/CNS and NiS2–MoS2 catalysts.68,69
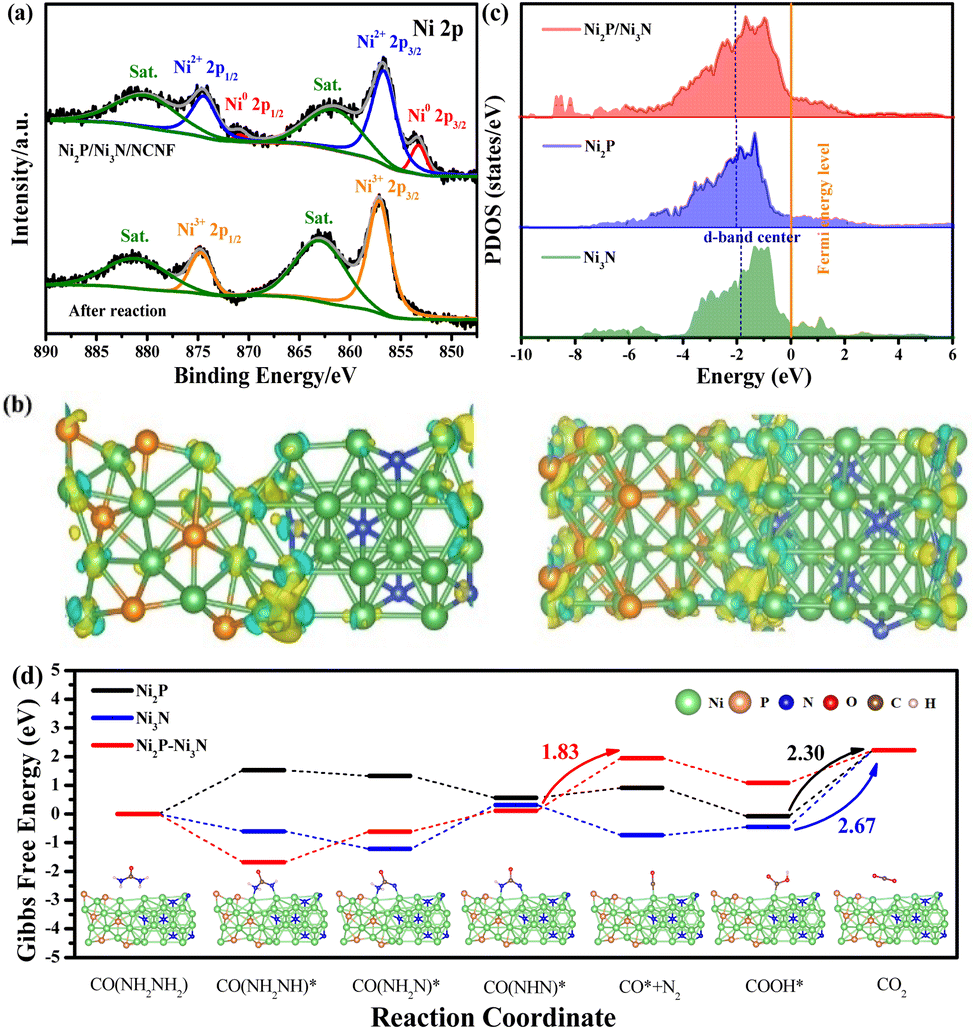 | ||
| Fig. 7 (a) The XPS spectra of Ni 2p of Ni2P/Ni3N/NCNF before and after urea electrolysis. (b) The differential charge density of the Ni2P/Ni3N heterostructure. (c) d density of states of Ni2P, Ni3N, and Ni2P/Ni3N. The Fermi level is set to zero, and the vertical short dash lines represent the d-band centre. (d) The adsorption models of Ni2P/Ni3N with the intermediate products and the Gibbs free energy in each elementary reaction step of UOR under the catalysis of Ni2P/Ni3N. | ||
In order to theoretically study the mechanism of the enhanced UOR catalytic activity of the Ni2P/Ni3N catalyst, we built the corresponding Ni3N (110) surface, Ni2P (210) surface, and the heterojunction of Ni3N (110) and Ni2P (210) surface as models to probe the in-depth reason (Fig. S20†). A prominent electron transfer from Ni2P to Ni3N was demonstrated by the differential charge density and Bader analysis in the hybrid catalyst of Ni2P/Ni3N (Fig. 7b). The electrons are strongly disturbed at the interface of Ni2P and Ni3N, where the charge density increases (yellow) and decreases (blue) are observed. To quantitatively identify the electron transfer number, Bader analysis was employed; there was 2.41 e transferred from Ni2P to Ni3N, verifying the electronic effect in the Ni2P/Ni3N interface. Furthermore, by calculating the total density of states (TDOS) (Fig. S21†), the metallic property of all the samples was revealed because their Fermi levels passed through the conduction band.70 Especially in the d density of states of Ni2P, Ni3N, and Ni2P/Ni3N (Fig. 7c), the peaks of Ni 3d in Ni2P/Ni3N negatively shifted to lower energy near the Fermi level compared to those of Ni2P and Ni3N. Specifically, the centre of the Ni 3d orbital peak moved from −1.79 eV (Ni3N) to −2.03 eV (Ni2P) and then to −2.06 eV (Ni2P/Ni3N). The distinct d-band centre of Ni2P/Ni3N from the other two samples convinced the manipulated electronic configuration of heterostructure. From the perspective of molecular functional groups, a urea molecule consists of one electron-withdrawing carbonyl group (CO) and two electron-donating amino groups (–NH2). For the sample of Ni3N, the N atoms interstitially occupy the octahedral interstices of the hcp-structured Ni, increasing the metal-like properties, which promote the urea molecule adsorption via the –NH2 and CO groups. Likewise, the phosphorization of Ni produced Ni2P, which could also provide double sites for the adsorption of –NH2 and CO groups, and the interaction of the urea molecular to the Ni sites can be enhanced through electronic effects offered by phosphorus atoms.71 Both Ni sites in the Ni3N and Ni2P can be transferred into high-valent Ni species through the self-oxidation during the electrocatalytic process, and the loss of N and P into the electrolyte could further enhance the surface area and increase surface active site exposure. However, the high occupied orbital energy of Ni in both Ni3N and Ni2P was still not good at the urea molecular activation, and it cannot be simply realized by a single-phase catalyst that lacks synergistic active sites for multiple reaction intermediate adsorption/desorption during the electrocatalytic process. In this work, the electron transfer between the Ni2P/Ni3N resulted in the formation of nucleophilic Ni3N and electrophilic Ni2P, which was beneficial for selectively adsorbing the electron-withdrawing carbonyl group and the electron-donating amino groups,33 thus accelerating the decomposition of urea molecules and lowering the reaction barriers. On the other hand, the downshift of the d-band centre was reported to increase the electron filling degree in the antibonding orbital and led to weaker binding strength between metal and intermediates.72 The CO2 desorption was a difficult step during the UOR due to its large free energy change,73 and the lower d-band centre was believed to weaken the adsorption of CO2,74 facilitating the reaction kinetics and catalytic efficiency. We calculated the Gibbs free energy between Ni2P/Ni3N and the intermediate products in each elementary reaction step of the UOR process (Fig. 7d). Results indicated that the rate-determining step (RDS) of Ni2P and Ni3N was the step of CO2 formation. This was generally observed in the Ni sample, like Ce–Ni3N@CC.75
Surprisingly, the RDS of heterostructure Ni2P/Ni3N was changed from the traditional CO2 desorption to N2 production. This phenomenon verified the role of hybrid catalysts in promoting carbon dioxide desorption, consistent with the conclusion of d-band centre analysis. In addition, the much easier CO2 desorption was beneficial for releasing Ni3+ active sites in time, leading to enhanced long-term stability. More importantly, the Gibbs free energy change in RDS of Ni2P/Ni3N (1.83 eV) was smaller than those of Ni2P (2.30 eV) and Ni3N (2.67 eV). This much lower value indicated better performance of Ni2P/Ni3N for stabilizing intermediates, which reduced the energy barrier and improved the intrinsic reactivity and catalytic kinetics. This meant that urea oxidation could be achieved at a lower potential input under the catalysis of Ni2P/Ni3N heterostructure.
Conclusions
In summary, we demonstrated an efficient Ni2P/Ni3N/NCNF catalyst and its insight into the structure–activity relationship for urea-containing wastewater oxidation in hydrogen generation. The created built-in electrophilic/nucleophilic domains in the heterointerface between Ni2P and Ni3N generated lattice expansion and electronic effect, which modified the electronic configuration, lowered the Ni d-band centre, and changed the RDS of urea oxidation. These characteristics promoted the adsorption and decomposition of urea molecules, largely exposed the active sites, accelerated the desorption of CO2, and lowered the energy barrier, thereby improving the catalytic activity and kinetics. The spectral study and theoretical analysis also revealed the facile formation of high-valent active Ni species and the high catalytic efficiency of their active sites. The Ni2P/Ni3N/NCNF catalyst delivered the highest current density of 151.11 mA cm−2 at 1.54 V, accompanied by fast kinetics and good stability. When evaluated in the urea-containing water for hydrogen generation as anode catalysts, the Ni2P/Ni3N/NCNF‖Pt/C electrode only needed the cell voltage of 1.39 V to offer 10 mA cm−2, 200 mV less than that of pure water splitting, largely reducing the energy input. The findings would be helpful for insight into the advanced catalyst development and their application in the urea-containing waste water transfer to clean hydrogen energy.Data availability
Data supporting the findings of this study are available within the article ESI.†Author contributions
Jiaxin Li: conceptualization, methodology, investigation, validation, writing – original draft; Chun Yin: methodology, investigation, and formal analysis; Shuli Wang: methodology, investigation, and formal analysis; Baogang Zhang: writing – review and editing; Ligang Feng: conceptualization, writing – review and editing, resources, funding acquisition.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (22272148, 21972124).References
- Y. Ge, Z. Lyu, M. Marcos Hernández and D. Villagrán, Chem. Sci., 2022, 13, 8597–8604 RSC.
- Y. Kuang, W. Qiao, F. Yang and L. Feng, J. Energy Chem., 2023, 85, 447–454 CrossRef CAS.
- Z. Miao, G. Wu, Q. Wang, J. Yang, Z. Wang, P. Yan, P. Sun, Y. Lei, Z. Mo and H. Xu, Mater. Rep.: Energy, 2023, 3, 100235 CAS.
- P. Kuang, Z. Ni, J. Yu and J. Low, Mater. Rep.: Energy, 2022, 2, 100081 CAS.
- C. Wang, L. Yu, F. Yang and L. Feng, J. Energy Chem., 2023, 87, 144–152 CrossRef CAS.
- Y. Kuang, W. Qiao, S. Wang, F. Yang and L. Feng, ACS Mater. Lett., 2024, 6, 1722–1731 CrossRef CAS.
- Y. Liu, Y. Wang, B. Liu, M. Amer and K. Yan, Acta Phys.-Chim. Sin., 2023, 39, 2205028 Search PubMed.
- J. White, L. Peters, D. Martín-Yerga, I. Terekhina, A. Anil, H. Lundberg, M. Johnsson, G. Salazar-Alvarez, G. Henriksson and A. Cornell, J. Electrochem. Soc., 2023, 170, 086504 CrossRef.
- M. Li, F. Yang, J. Chang, A. Schechter and L. Feng, Acta Phys.-Chim. Sin., 2023, 39, 2301005 Search PubMed.
- J. Hong, S. Huang, H. Wu, N. Yao, X. Luo and Z. Zheng, Sci. Total Environ., 2023, 891, 164507 CrossRef CAS PubMed.
- A. N. Rollinson, J. Jones, V. Dupont and M. V. Twigg, Energy Environ. Sci., 2011, 4, 1216–1224 RSC.
- R. K. Singh, K. Rajavelu, M. Montag and A. Schechter, Energy Technol., 2021, 9, 2100017 CrossRef CAS.
- R. F. Madsen, J. R. Thomassen, D. Vial and R. A. Binot, Desalination, 1991, 83, 123–136 CrossRef CAS.
- T. A. Larsen, A. C. Alder, R. I. L. Eggen, M. Maurer and J. Lienert, Environ. Sci. Technol., 2009, 43, 6121–6125 CrossRef CAS PubMed.
- L. Du, Y. Sun and B. You, Mater. Rep.: Energy, 2021, 1, 100004 CAS.
- J. Li, Y. Li, Q. Xue, Y. Gao and Y. Ma, Chin. J. Struct. Chem., 2022, 41, 2207035–2207039 CAS.
- M. Luo, J. Yang, X. Li, M. Eguchi, Y. Yamauchi and Z. Wang, Chem. Sci., 2023, 14, 3400–3414 RSC.
- L. Gu, M. Kuang, J. Chen and J. Yang, Chin. J. Struct. Chem., 2023, 42, 100067 CrossRef CAS.
- Z. Yin, R. He, Y. Zhang, L. Feng, X. Wu, T. Wågberg and G. Hu, J. Energy Chem., 2022, 69, 585–592 CrossRef CAS.
- L. Baldinelli, G. M. Rodriguez, I. D'Ambrosio, A. M. Grigoras, R. Vivani, L. Latterini, A. Macchioni, F. De Angelis and G. Bistoni, Chem. Sci., 2024, 15, 1348–1363 RSC.
- F. Abdelghafar, X. Xu, S. P. Jiang and Z. Shao, Mater. Rep.: Energy, 2022, 2, 100144 CAS.
- H. Sun, H. Kim, S. Song and W. Jung, Mater. Rep.: Energy, 2022, 2, 100092 CAS.
- B. K. Boggs, R. L. King and G. G. Botte, Chem. Commun., 2009, 4859–4861 RSC.
- J. Zhang, T. He, M. Wang, R. Qi, Y. Yan, Z. Dong, H. Liu, H. Wang and B. Y. Xia, Nano Energy, 2019, 60, 894–902 CrossRef CAS.
- L. Yu, X. Pang, Z. Tian, S. Wang and L. Feng, Electrochim. Acta, 2023, 440, 141724 CrossRef CAS.
- X. Huang, R. He, S. Wang, Y. Yang and L. Feng, Inorg. Chem., 2022, 61, 18318–18324 CrossRef CAS PubMed.
- P. Hao, X. Dong, H. Wen, R. Xu, J. Xie, Q. Wang, G. Cui, J. Tian and B. Tang, Chin. Chem. Lett., 2023, 34, 107843 CrossRef CAS.
- D. Zhu, H. Zhang, J. Miao, F. Hu, L. Wang, Y. Tang, M. Qiao and C. Guo, J. Mater. Chem. A, 2022, 10, 3296–3313 RSC.
- L. Wang, Y. Zhu, Y. Wen, S. Li, C. Cui, F. Ni, Y. Liu, H. Lin, Y. Li, H. Peng and B. Zhang, Angew. Chem., Int. Ed., 2021, 60, 10577–10582 CrossRef CAS PubMed.
- C. Yin, F. Yang, S. Wang and L. Feng, Chin. J. Catal., 2023, 51, 225–236 CrossRef CAS.
- C. Liu, F. Yang, A. Schechter and L. Feng, Adv. Sens. Energy Mater., 2023, 2, 100055 CrossRef.
- D. Yang, Y. Gu, X. Yu, Z. Lin, H. Xue and L. Feng, ChemElectroChem, 2018, 5, 659–664 CrossRef CAS.
- J. Li, S. Wang, S. Sun, X. Wu, B. Zhang and L. Feng, J. Mater. Chem. A, 2022, 10, 9308–9326 RSC.
- J. Li, S. Wang, J. Chang and L. Feng, Adv. Powder Mater., 2022, 1, 100030 CrossRef.
- C. Yin, J.-X. Li and L.-G. Feng, Chin. J. Appl. Chem., 2023, 40, 1158–1174 CAS.
- H. An, Y. Hu, N. Song, T. Mu, S. Bai, Y. Peng, L. Liu and Y. Tang, Chem. Sci., 2022, 13, 3035–3044 RSC.
- X.-Q. Qiao, W. Chen, C. Li, Z. Wang, D. Hou, B. Sun and D.-S. Li, Mater. Rep.: Energy, 2023, 3, 100234 CAS.
- R. He, C. Wang and L. Feng, Chin. Chem. Lett., 2023, 34, 107241 CrossRef CAS.
- G. Zhang, B. Wang, L. Li, S. Yang, J. Liu and S. Yang, J. Mater. Chem. A, 2020, 8, 18945–18954 RSC.
- M. Li and L. Feng, Chin. J. Struct. Chem., 2022, 41, 2201019–2201024 CAS.
- J. Li, S. Sun, Y. Yang, Y. Dai, B. Zhang and L. Feng, Chem. Commun., 2022, 58, 9552–9555 RSC.
- G. Li, J. Wang, J. Yu, H. Liu, Q. Cao, J. Du, L. Zhao, J. Jia, H. Liu and W. Zhou, Appl. Catal., B, 2020, 261, 118147 CrossRef CAS.
- X. Xu, P. Du, T. Guo, B. Zhao, H. Wang and M. Huang, ACS Sustain. Chem. Eng., 2020, 8, 7463–7471 CrossRef CAS.
- H. Liu, Z. Liu and L. Feng, Nanoscale, 2019, 11, 16017–16025 RSC.
- H. Wang, J. Li, K. Li, Y. Lin, J. Chen, L. Gao, V. Nicolosi, X. Xiao and J.-M. Lee, Chem. Soc. Rev., 2021, 50, 1354–1390 RSC.
- S. Dong, X. Chen, X. Zhang and G. Cui, Coord. Chem. Rev., 2013, 257, 1946–1956 CrossRef CAS.
- X. Wang, Q. Li, P. Shi, J. Fan, Y. Min and Q. Xu, Small, 2019, 15, 1901530 CrossRef CAS PubMed.
- T. Wu, S. Zhang, K. Bu, W. Zhao, Q. Bi, T. Lin, J. Huang, Y. Li and F. Huang, J. Mater. Chem. A, 2019, 7, 22063–22069 RSC.
- T. Tang, X. Bai, Z. Wang and J. Guan, Chem. Sci., 2024, 15, 5082–5112 RSC.
- Y. Wei, C. H. Shin, E. B. Tetteh, B. J. Lee and J. S. Yu, ACS Appl. Energy Mater., 2020, 3, 822–830 CrossRef CAS.
- L. Peng, C. Wang, Q. Wang, R. Shi, T. Zhang and G. I. N. Waterhouse, Adv. Energy Sustainability Res., 2021, 2, 2100078 CrossRef CAS.
- H. Sun, Y. Min, W. Yang, Y. Lian, L. Lin, K. Feng, Z. Deng, M. Chen, J. Zhong, L. Xu and Y. Peng, ACS Catal., 2019, 9, 8882–8892 CrossRef CAS.
- M. Q. Wang, C. Ye, H. Liu, M. Xu and S. J. Bao, Angew. Chem., Int. Ed., 2018, 57, 1963–1967 CrossRef CAS PubMed.
- X. Chen, S. Yan, S. Wen, J. Chen, J. Xu, C. Wang and X. Lu, J. Colloid Interface Sci., 2023, 641, 782–790 CrossRef CAS PubMed.
- X. Li, G. Guan, B. Cheng, X. Zhang, K. Zhang and J. Xiang, J. Mater. Chem. C, 2024, 12, 2894–2902 RSC.
- C. Wang, W. Chen, D. Yuan, S. Qian, D. Cai, J. Jiang and S. Zhang, Nano Energy, 2020, 69, 104453 CrossRef CAS.
- P. Bhanja, B. Mohanty, S. Chongdar, A. Bhaumik, B. K. Jena and S. Basu, ACS Appl. Energy Mater., 2021, 4, 12827–12835 CrossRef CAS.
- J. Luo, D. A. Vermaas, D. Bi, A. Hagfeldt, W. A. Smith and M. Grätzel, Adv. Energy Mater., 2016, 6, 1600100 CrossRef.
- J. Zhu, L. Hu, P. Zhao, L. Y. S. Lee and K. Y. Wong, Chem. Rev., 2020, 120, 851–918 CrossRef CAS PubMed.
- W. Hu, M. Zheng, H. Duan, W. Zhu, Y. Wei, Y. Zhang, K. Pan and H. Pang, Chin. Chem. Lett., 2022, 33, 1412–1416 CrossRef CAS.
- F. Yang, J. Li and L. Feng, Univ. Chem., 2023, 38(9), 242–251 CAS.
- V. Vedharathinam and G. G. Botte, Electrochim. Acta, 2013, 108, 660–665 CrossRef CAS.
- C. Tang, R. Zhang, W. Lu, Z. Wang, D. Liu, S. Hao, G. Du, A. M. Asiri and X. Sun, Angew. Chem., Int. Ed., 2017, 56, 842–846 CrossRef CAS PubMed.
- J. Li, Y. Chang, D. Li, L. Feng and B. Zhang, Chem. Commun., 2021, 57, 7035–7038 RSC.
- W. Z. Chen, M. Zhang, L. Zhang, J. L. He, Z. L. Liu and Y. Q. Wang, Electrochim. Acta, 2023, 441, 141868 CrossRef CAS.
- L. Zhang, L. Wang, H. Lin, Y. Liu, J. Ye, Y. Wen, A. Chen, L. Wang, F. Ni, Z. Zhou, S. Sun, Y. Li, B. Zhang and H. Peng, Angew. Chem., Int. Ed., 2019, 58, 16820–16825 CrossRef CAS PubMed.
- Y. Li, Y. Wang, J. Lin, Y. Shi, K. Zhu, Y. Xing, X. Li, Y. Jia and X. Zhang, Front. Energy Res., 2022, 10 DOI:10.3389/fenrg.2022.924515.
- Z. Liu, B. Tang, X. Gu, H. Liu and L. Feng, Chem. Eng. J., 2020, 395, 125170 CrossRef CAS.
- S. Wang, L. Zhao, J. Li, X. Tian, X. Wu and L. Feng, J. Energy Chem., 2022, 66, 483–492 CrossRef CAS.
- C. Hou, J. Wang, W. Du, J. Wang, Y. Du, C. Liu, J. Zhang, H. Hou, F. Dang, L. Zhao and Z. Guo, J. Mater. Chem. A, 2019, 7, 13460–13472 RSC.
- Y. Li, Z. Dong and L. Jiao, Adv. Energy Mater., 2020, 10, 1902104 CrossRef CAS.
- P. Li, Y. Huang, Q. Huang, R. Chen, J. Li and S. Tian, Appl. Catal., B, 2022, 313, 121444 CrossRef CAS.
- D. A. Daramola, D. Singh and G. G. Botte, J. Phys. Chem. A, 2010, 114, 11513–11521 CrossRef CAS PubMed.
- S. Lu, M. Hummel, S. Kang, R. Pathak, W. He, X. Qi and Z. Gu, ACS Omega, 2021, 6, 14648–14654 CrossRef CAS PubMed.
- M. Li, X. Wu, K. Liu, Y. Zhang, X. Jiang, D. Sun, Y. Tang, K. Huang and G. Fu, J. Energy Chem., 2022, 69, 506–515 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc01862a |
| This journal is © The Royal Society of Chemistry 2024 |