
Catalytic effect of microflow space for supramolecular block co-polymerization of water-soluble porphyrins†
Haruna
Takemori
,
Chisako
Kanzaki
,
Shota
Nomura
,
Takato
Maeda
and
Munenori
Numata
 *
*
Department of Biomolecular Chemistry, Graduate School of Life and Environmental Sciences, Kyoto Prefectural University, Shimogamo, Sakyo-ku, Kyoto 606-8522, Japan. E-mail: numata@kpu.ac.jp; Fax: +81-75-703-5132
First published on 29th May 2024
Abstract
Using microflow space, a catalytic effect was achieved for supramolecular polymerization. With increasing reactivity at the polymer end, the selective connection of active monomers formed new block domains, avoiding fast homo-assembly. Binding of less-reactive monomers at the polymer end overcame steric bulkiness, affording a stable supramolecular diblock copolymer (SdiBCP).
Supramolecular polymerization is a fascinating area of research in supramolecular chemistry.1 Despite the rapid progress in this area, diversifying the structure of monomers involved in chain reactions remains challenging, especially when multiple monomer units with different reactivities are involved. The concept of supramolecular copolymerization has been developed as an analog of the well-established chain growth in covalent polymerization.2 Unlike covalent copolymerization, the combination of monomers is still restricted in supramolecular copolymerization. A comprehensive strategy that overcomes the limited monomer combinations is strongly desired for expanding the resulting supramolecular polymer (SP) structures.
Porphyrins have unique photonic and electronic properties arising from their large π-electron systems. The supramolecular polymerization of porphyrin molecules is an attractive approach for creating electro-optical materials by amplifying the effects of the inherent molecular functionality. 5,10,15,20-Tetrakis(4-sulfonatophenyl)porphyrin (H2TPPS4) and its derivatives are recognized as useful porphyrin building blocks for forming well-regulated J-aggregated fibers in aqueous media.3 Under acidic conditions, H2TPPS4 is converted to the protonated species H4TPPS4, which undergoes spontaneous slipped-stack self-assembly (forming J-aggregates), mediated by intermolecular electrostatic interactions between the sulfonate and protonated pyrrole groups. Recently, we synthesized novel H2TPPS4 derivatives by positioning two nonionic ethylene glycol (EG) units at opposite meso (trans) positions through amide groups (H2TPPS2–NHCO–EGx; x = 2, 4, 6, 8, and 18).4 These new porphyrin monomers have the advantage of being water-soluble monomers for supramolecular copolymerization because the structure differs from that of the parent H2TPPS4. As reported in our previous paper,4 these new families of porphyrins undergo proton-triggered supramolecular polymerization depending on peripheral steric repulsion. For example, H2TPPS2–NHCO–EG2 has higher aggregation ability than the parent H2TPPS4, whereas H2TPPS2–NHCO–EG8 exhibits no aggregation ability. Although supramolecular copolymerization with a combination of these monomers can prospectively create new J-aggregated materials, the strong tendency of H2TPPS2–NHCO–EG2 and –EG4 to homoaggregate and the lower reactivity of –EG8 and –EG18 arising from their steric bulkiness have hampered our attempts at their combinatorial supramolecular copolymerization.
Recently, we developed a system using microfluidics as a reaction field for supramolecular copolymerization, in which one terminus of a SP is always activated during supramolecular growth.5,6 Unlike the SP-monomer reaction governed by thermal diffusion, both the collision frequency and steric demand that are generally required to accelerate the rate of supramolecular growth were satisfied in a microflow field, realizing anisotropic supramolecular diblock copolymerization, even with less-reactive monomers. In this study, by applying the developed strategy, we demonstrate that the highly reactive monomer H2TPPS2–NHCO–EG2 and less-reactive monomer H2TPPS2–NHCO–EG6, both of which are ineffective as monomers for supramolecular copolymerization in a vial, are successfully connected at the one-terminus of the parent H4TPPS4 SP (Fig. 1).
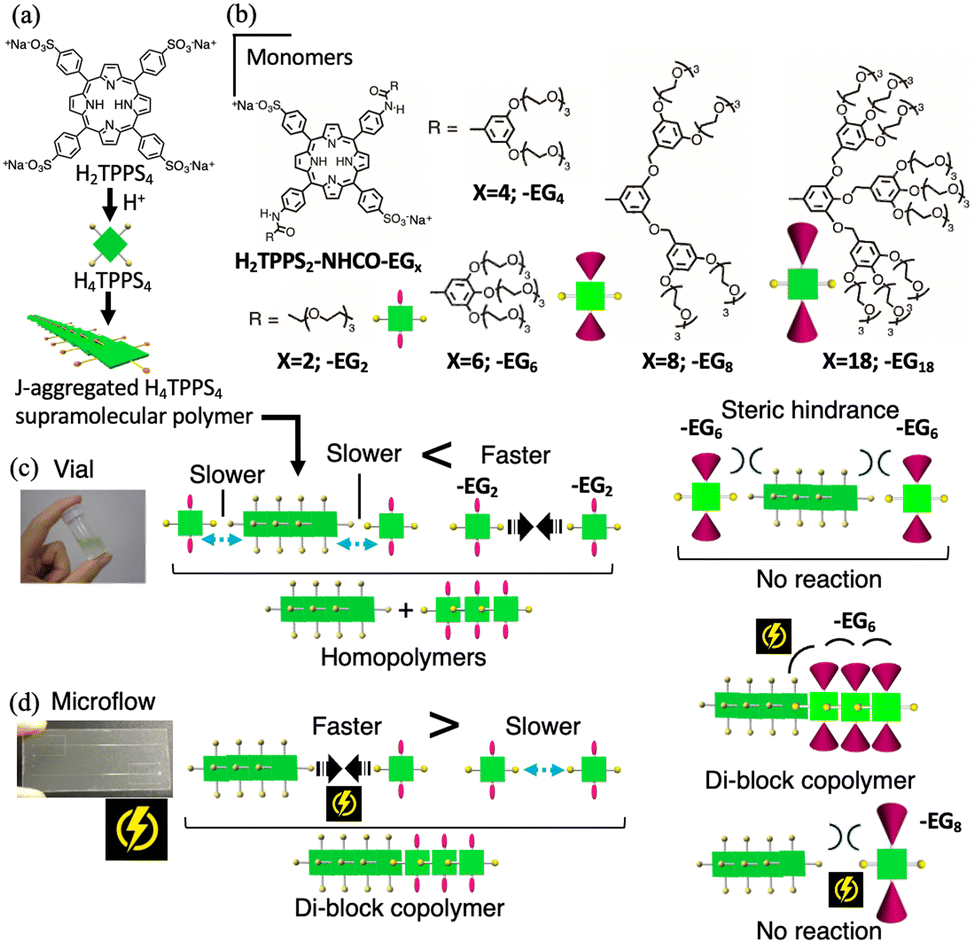 | ||
| Fig. 1 (a) Molecular structure of H2TPPS4 and illustration of its self-assembly to form J-aggregated SP; (b) monomers employed in this study: H2TPPS2–NHCO–EGx (x; 2, 4, 6, 8, 18); (c) schematic of conventional self-assembly in a vial and (d) active-assembly driven in a microflow channel. | ||
Firstly, we investigated whether H2TPPS2–NHCO–EGx reacted with the end of the H4TPPS4 SP in a vial. A solution of the H4TPPS4 SP was prepared and different concentrations of an aqueous solution of each monomer (30, 50, to 70 μM) were added (Fig. 1a and Fig. S1, ESI†). To avoid protonation and homoaggregation, the pH of the monomer solution was set to 4, and this solution was added to a more acidic H4TPPS4 SP solution (Fig. 2a). Fig. 2c summarizes the selected spectra obtained upon adding 70 μM of the monomer solution (for details, see Fig. S2 and S3, ESI†). The absorption at 490 nm (Soret band), corresponding to the J-aggregate, appeared for the monomers with smaller EG units, such as H2TPPS2–NHCO–EG2 and –EG4, suggesting that these monomers underwent supramolecular polymerization in the vial. However, it is difficult to distinguish between copolymerization at the end of the parent H4TPPS4 SP and the homoaggregated species. Thus, we focused on the changes in the Q-band. In the case of H2TPPS2–NHCO–EG2, a shoulder band clearly appeared at 720–740 nm. Because the bands at approximately 732 nm could be reasonably assigned to supramolecular homopolymers,4 the Q band was deconvoluted into four spectra (Fig. 2d and Fig. S4, ESI†). The intensity of the deconvoluted peak at 732 nm increased with increasing monomer concentration. Namely, homoaggregates were predominant, and the connection at the end of the H4TPPS4 SP was suppressed in the vial. A similar trend was observed for H2TPPS2–NHCO–EG4 (Fig. S5, ESI†). Thus, for H2TPPS2–NHCO–EG2 and –EG4, the rate of homoaggregation was faster than that at the end of the parent J-aggregate. In contrast, for H2TPPS2–NHCO–EG8 and –EG18, no changes in the Q-band were observed, suggesting that homoaggregation was suppressed due to steric hindrance. For H2TPPS2–NHCO–EG6, although the intensity of the peak at 732 nm increased slightly, no clear concentration dependence was observed, indicating no polymerization ability (Fig. 2e and Fig. S6, ESI†). Based on these results, we conclude that in the vial, H2TPPS2–NHCO–EG2 and –EG4 underwent self-aggregation without reacting at the end of the H4TPPS4 SP. On the other hand, H2TPPS2–NHCO–EG6 –EG8, and –EG18 did not undergo homo- or copolymerization because of steric hindrance (Fig. 1c).
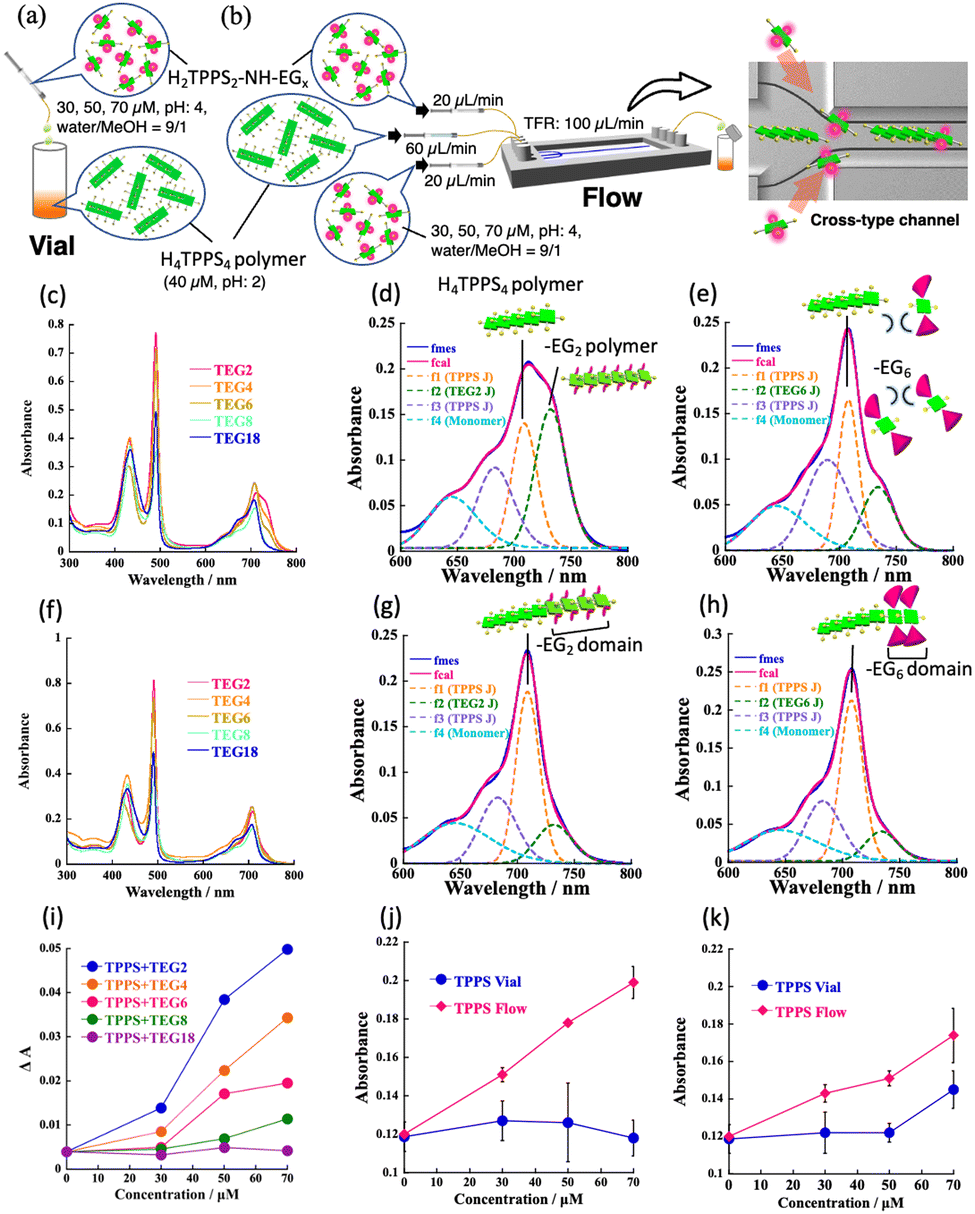 | ||
| Fig. 2 (a, b) Illustration of supramolecular copolymerization through (a) vial mixing and (b) separate injection using a cross-type microflow channel; (c)–(h) UV-vis spectra for –EGx; x = 2–18 (c, f) and selected deconvoluted spectra for –EG2 (d, g) and –EG6 (e, h) monomer; (c)–(e) vial and (f)–(h) flow samples obtained upon adding 70 μM of monomers (other spectra (for –EG2, –EG4, –EG6) recorded with different monomer concentrations, see Fig. S4–S6, ESI†) [1.0 mm cell, r.t.]; (i) plots of ΔA at 490 nm with respect to monomer (–EGx; x = 2–18) concentration; (j) and (k) plots of absorbance at 708 nm versus monomer concentration (j; –EG2, k; –EG6, blue line; vial, red line; flow sample) (for plots at 732 nm, see Fig. S7, ESI†). | ||
The possibility of copolymerization in a microflow field was evaluated. A cross-type microflow system was employed, in which the parent H4TPPS4 SP solution was injected into the central flow, and the monomer solution was injected from two lateral inlets (Fig. 2b). According to our previous work, the injected H4TPPS4 SP was aligned in the microflow channel, which makes one terminus of the SP active for supramolecular polymerization.5 The spatially separated injection, therefore, has the benefit of suppressing unfavorable homoaggregation, especially in the case of H2TPPS2–NHCO–EG2, because the monomers meet at the activated SP end, concurrent with protonation.
The deconvoluted Q-band spectra provided evidence that in the case of the H2TPPS2–NHCO–EG2 and –EG4, the desired copolymerization occurred with the suppression of homo-polymerization. As shown in Fig. 2f, g, and Fig. S5 (ESI†), the increase in the absorbance at 732 nm was suppressed compared to that of the vial sample. In contrast, the absorption band at 708 nm was predominant, particularly for H2TPPS2–NHCO–EG2. This peak at 708 nm can be assigned to supramolecular copolymers (SCPs). The trend showed a clear concentration dependence (Fig. S4, ESI†). For these monomers, an increase in the absorbance at 490 nm was also observed (Fig. 2f). Combined with the findings from the Q-band analysis, unlike the vial sample, the change in the Soret band was responsible for the formation of the SCP in the flow sample. Therefore, to quantitatively evaluate the degree of copolymerization, the change in the absorption band (ΔA = Aeluted solution − Ainjected solution) at 490 nm was considered. Fig. 2i summarizes the plots of ΔA against the monomer concentration. The clear concentration dependence of the reaction of the H2TPPS2–NHCO–EG2, –EG4, and –EG6 monomers suggests that these monomers react at the end of the H4TPPS4 SP. In the case of H2TPPS2–NHCO–EG8 and –EG18, no significant concentration dependence was observed, confirming that neither homoaggregation nor copolymerization occurred, even in the microflow. Importantly, H2TPPS2–NHCO–EG6, which has low propensity for self-assembly in a vial, successfully reacted at the end of the parent H4TPPS4 SP (Fig. 2h and Fig. S6, ESI;† for the self-assembly ability of monomer in a microflow channel; see Fig. S13 and S14, ESI†). To gain further insight into the reaction, the absorption changes at 732 nm in the deconvoluted spectra were plotted against the monomer concentration (Fig. S7, ESI†). Remarkably, the absorption changes were limited even for the H2TPPS2–NHCO–EG2 and –EG4 monomers, indicating that their homopolymerization was suppressed in the microflow channel. Similar plots based on the absorption changes at 708 nm (Fig. 2j and Fig. S5g, ESI†) indicated that supramolecular copolymerization proceeded with increasing monomer concentration, the trend of which was in good accordance with that confirmed by the Soret band analysis. Furthermore, for H2TPPS2–NHCO–EG6, a similar concentration dependence was confirmed (Fig. 2k), suggesting that –EG6 reacted at the end of the H4TPPS4 SP, overcoming steric hindrance. For H2TPPS2–NHCO–EG8 and –EG18, no spectral changes were observed in the Q-band range, indicating that these sterically hindered monomers remained unreacted, even in the microflow channel (Fig. S3g–j, ESI†).
The AFM images provided evidence that one terminus of the H4TPPS4 SP was activated for the reaction with the monomers, thereby generating SdiBCPs. The average length of the initial H4TPPS4 SP in the injected solution was approximately 0.85 μm (Fig. S1, ESI†). The AFM images confirmed that the SP length increased with increasing monomer concentration. This trend was conspicuous for the H2TPPS2–NHCO–EG2 (Fig. 3a–d). The length distribution measured from the AFM images revealed that the SP extension rate increased with increasing monomer concentration (Fig. S8, ESI†). A similar trend was also confirmed with the H2TPPS2–NHCO–EG4 and –EG6 (Fig. S9 and S10, ESI;† for –EG8 and –EG18; Fig. S11 and S12, ESI†). The DLS data for the eluted solution further supported this view; that is, the H2TPPS2–NHCO–EG2, –EG4, and –EG6 monomers extended from the end of the H4TPPS4 SP while passing through the microflow channel (Fig. 3h and i). Advantageously, for H2TPPS2–NHCO–EG4 and –EG6, the extended block domain was visually confirmed by AFM; that is, as shown in Fig. 3e and f, two domains with different heights were directly connected in a single SP. The height of the thicker domain for the H2TPPS2–NHCO–EG4 monomer was estimated to be ca. 4 nm, which is slightly higher than that of the H4TPPS4 SP domain (ca. 2 nm). Importantly, even in the case of the H2TPPS2–NHCO–EG6 monomer, the creation of a SdiBCP was confirmed, indicating that during passage through the microflow channel, the inert monomer in the vial reacted to make form a block domain, overcoming the steric bulkiness. The effect of the flow rate on the degree of SP extension confirmed that the dynamic motion of the microsolution played an essential role in creating the block domain (Fig. 3k–m; for detail data; Fig. S15–S21, ESI†); that is, the domain length tended to increase at higher flow rates (Fig. 3n). No decomposition of the resultant SdiBCPs was observed after elution, suggesting that once the monomer was connected at the end of the H4TPPS4 SP, the peripheral –EG4 or –EG6 units contributed to stabilizing the created domain.
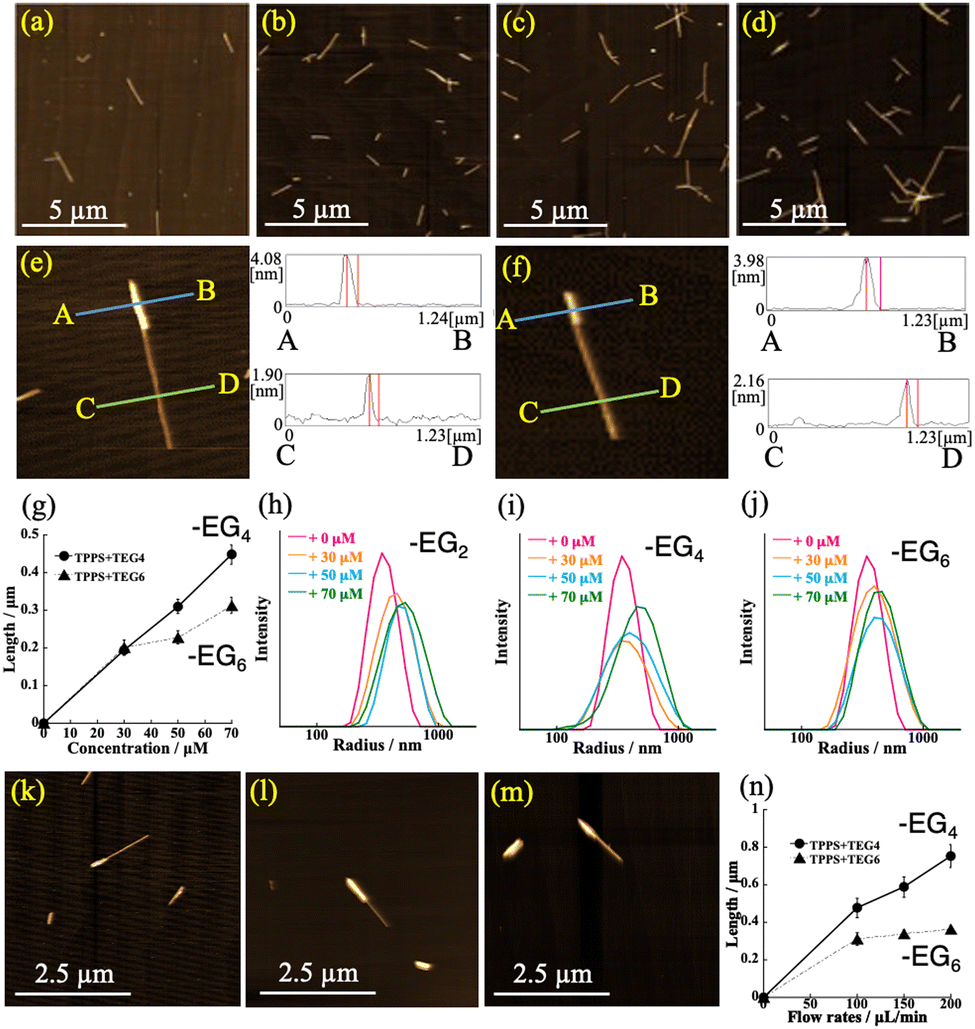 | ||
| Fig. 3 (a)–(d) AFM images of H4TPPS4/–EG2 SdiBCP; (a) vial sample prepared in the presence of 70 μM –EG2; (b)–(d) eluted solutions obtained through injection of (b) 30, (c) 50, (d) 70 μM −EG2; (e) and (f) enlarged AFM images and height profiles of (e) H4TPPS4/–EG4 and (f) –EG6 SdiBCP; (g) plots of the degree of extension against monomer concentration, estimated from AFM images (solid line; –EG4, dashed line; –EG6 domain); (h)–(j) concentration-dependent DLS data of (h) H4TPPS4/–EG2, (i) /–EG4, and (j) /–EG6; (k)–(m) AFM images showing effect of flow rate of H4TPPS4/–EG6 (k: 100, l: 150, m: 200 μL min−1); (n) plots of extension length against flow rate (solid line; –EG4, dashed line; –EG6 domain). | ||
With the advantage of directly visualizing the –EG4 and –EG6 block domain, we evaluated the extended domain length by counting 300 SPs in the AFM images. As shown in Fig. 3g, for H2TPPS2–NHCO–EG4, the domain extended linearly with increasing monomer concentration, whereas for the –EG6 domain, the extension ratio tended to saturate, even at higher concentrations. In other words, the domain length of the –EG6 unit is shorter (0.31 μm) than that of –EG4 (0.45 μm) at 70 μM. Similarly, as shown in Fig. 3n, the length of the –EG4 domain increased with increasing flow rate (0.75 μm at 200 μL min−1), whereas in the case of the –EG6 domain, a saturation profile was again observed (0.37 μm at the same flow rate). These findings indicate that the steric bulkiness of the –EG6 unit strongly influences the extension rate, even under flow-driven conditions. In contrast, the total number of SdiBCPs increased linearly with increasing monomer concentration or flow rate, regardless of the steric bulkiness (Fig. S21, ESI†). Combined with the finding of a shorter extended domain for H2TPPS2–NHCO–EG6 to avoid steric bulkiness, the reaction progressed predominantly at the end of the H4TPPS4 SP (i.e., the H4TPPS4 end) rather than the H2TPPS2–NHCO–EG6 end in the growing block domain. In contrast, once the first H2TPPS2–NHCO–EG4 monomer was attached to the end of the H4TPPS4 SP, the extension reaction from the end occurred in a cooperative manner because of the inherently strong aggregation ability. Similarly, although we could not directly observe the extension behavior by AFM, a cooperative extension mechanism was reasonably proposed for the H2TPPS2–NHCO–EG2 domain. From the statistical data obtained from the AFM images, the overall yield of the H4TPPS4/EG6 SdiBCPs was estimated to be 3% in the vial, but increased to 14% in the microflow channel.
The mechanism by which the highly active H2TPPS2–NHCO–EG2 or less-active –EG6 monomer selectively reacted at one terminus of the H4TPPS4 SP was reasonably explained by the enhanced collision frequency at the polymer end and favored steric effects during the extension reaction (Fig. 4a). The Hagen–Poiseuille flow restricted the SPs’ motion; that is, the orientation of the H4TPPS4 SPs was fixed, and the SPs were linearly transported along a streamline. In contrast, the monomers diffused freely across the streamline with a parabolic velocity distribution, which made monomers’ motion active. Thereby, the monomers met the fixed SP ends with a relatively greater velocity compared to those in the vial, thereby increasing the rate of reaction between the monomer and the SP end. Moreover, because of the parabolic velocity distribution, a monomer arrives from a slower layer and reacts with the terminus of the SP facing forward with a greater differential velocity, making the terminus facing forward more reactive (Fig. 4b). The enhanced collision frequency at the SP end was further supported by the heating effect of monomer layer (Fig. S22–S24, ESI†). Based on this mechanism, which activates the SP end by microflow, the reaction rate of H2TPPS2–NHCO–EG2 at the SP end was greater than that of the homoaggregate. When the monomer impacts the SP end, sufficient kinetic energy is provided to form a heterojunction, overcoming the energetically unfavorable steric hindrance.
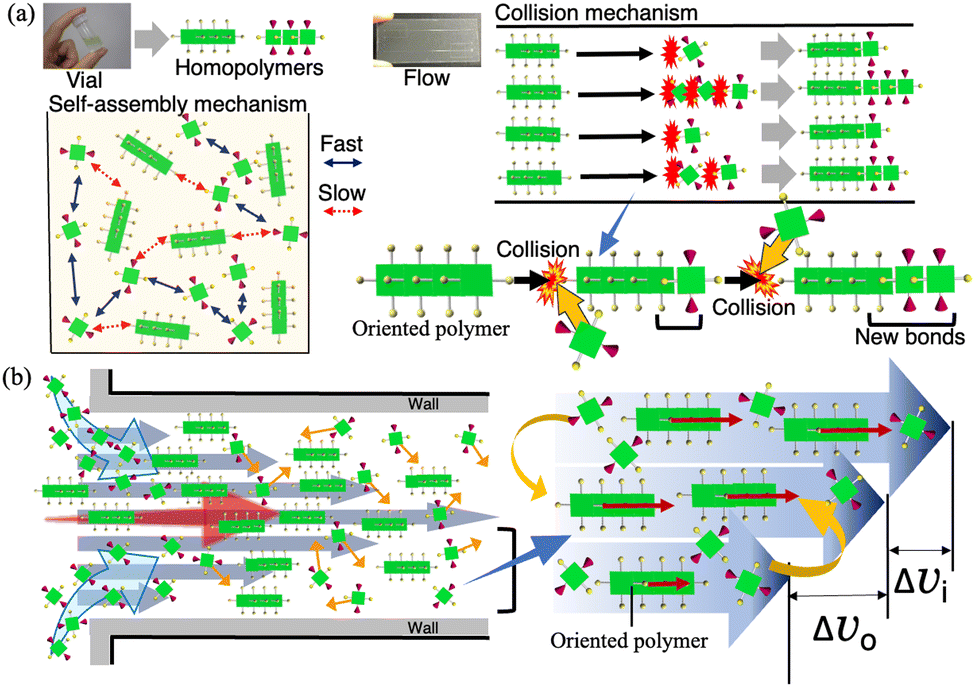 | ||
| Fig. 4 (a) Illustration of conventional self-assembly in a vial and the present flow-driven assembly; (b) schematic of mechanism by which motion of microflow is converted to molecular impact at the end of SP. | ||
In conclusion, a relatively slow supramolecular reaction can become predominant in a microflow channel. Liner solution’s motion in the microflow space was the energetic origin to make the unfavorable non-covalent bonds. Once monomers were connected to the SP end, the resultant domain was stable. Thus, the microflow space acts as a catalyst and opens up a new kinetic pathway to make new non-covalent bonds.
This study was supported by the Asahi Glass Foundation and JSPS KAKENHI (grant no. 22K19070 and 22H02065).
Conflicts of interest
There are no conflicts to declare.Notes and references
-
(a) D. Zhao and J. S. Moore, Org. Biomol. Chem., 2003, 1, 3471 RSC
; (b) R. Dobraw and F. Würthner, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 4981 CrossRef
-
(a) S. K. Yang,
et al.
, Chem. Soc. Rev., 2011, 40, 129 RSC
-
(a) P. J. Collings,
et al.
, J. Phys. Chem. B, 1999, 103, 8474 CrossRef CAS
-
(a) C. Kanzaki,
et al.
, RSC Adv., 2022, 12, 30670 RSC
- S. Matoba,
et al.
, J. Am. Chem. Soc., 2021, 143, 8731 CrossRef CAS PubMed
-
(a) M. Pumera, Chem. Commun., 2011, 47, 5671 RSC
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cc02003k |
| This journal is © The Royal Society of Chemistry 2024 |