
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Terminal methylene phosphonium ions: precursors for transient monosubstituted phosphinocarbenes†
Pawel
Löwe
 a,
Marius A.
Wünsche
a,
Felix R. S.
Purtscher
b,
Jakob
Gamper
b,
Thomas S.
Hofer
b,
Lukas F. B.
Wilm
a,
Maike B.
Röthel
b and
Fabian
Dielmann
*ab
a,
Marius A.
Wünsche
a,
Felix R. S.
Purtscher
b,
Jakob
Gamper
b,
Thomas S.
Hofer
b,
Lukas F. B.
Wilm
a,
Maike B.
Röthel
b and
Fabian
Dielmann
*ab
aInstutut für Anorganische und Analytische Chemie Universität Münster, Corrensstraße 28/30, 48149 Münster, Germany. E-mail: fabian.dielmann@uibk.ac.at
bInstitute of General, Inorganic and Theoretical Chemistry Leopold-Franzens-Universität Innsbruck, Innrain 80–82, 6020 Innsbruck, Austria
First published on 29th June 2023
Abstract
Isolable singlet carbenes are among the most important tools in chemistry, but generally require the interaction of two substituents with the electron deficient carbon atom. We herein report a synthetic approach to monosubstituted phosphinocarbenes via deprotonation of hitherto unknown diprotic terminal methylene phosphonium ions. Two methylene phosphonium salts bearing bulky N-heterocyclic imine substituents at the phosphorus atom were isolated and fully characterized. Deprotonation studies indicate the formation of transient monosubstituted carbenes that undergo intermolecular cycloadditions or intramolecular Buchner ring expansion to afford a cycloheptatriene derivative. The reaction mechanism of the latter transformation was elucidated using DFT calculations, which reveal the ambiphilic nature of the phosphinocarbene enabling the insertion into the aromatic C–C bond. Additional computational studies on the role of substituent effects are presented.
Introduction
Carbenes are compounds of the general formula R2C featuring a divalent carbon atom with six electrons in its valence shell. While the parent carbene (H2C) has a triplet ground state and is too reactive to be isolated,1 it soon became evident that the carbene center is highly sensitive to electronic interactions with its substituents.2–6 Prompted by the pioneering work of Bertrand7 and Arduengo8 on the isolation of the first persistent singlet carbenes, countless stable carbenes with various substitution patterns have been prepared.3,9–12 Among the available heteroatom substituents, nitrogen substituents are particularly effective for the stabilization of carbenes, due to the interplay of π-donor and σ-acceptor properties.13 Indeed, N-heterocyclic carbenes (NHCs) – particularly diaminocarbenes – have evolved to be impressively useful tools in various fields of chemistry.14–17 Despite their significantly enhanced nucleophilicity and electrophilicity, several isolable NHCs bearing only one amino group and one alkyl or aryl substituent have been reported.18,19 Most remarkably, Bertrand and coworkers also succeeded in isolating the monosubstituted aminocarbene I by constructing an extremely bulky π-donor substituent to ensure its kinetic stabilization (Scheme 1a).20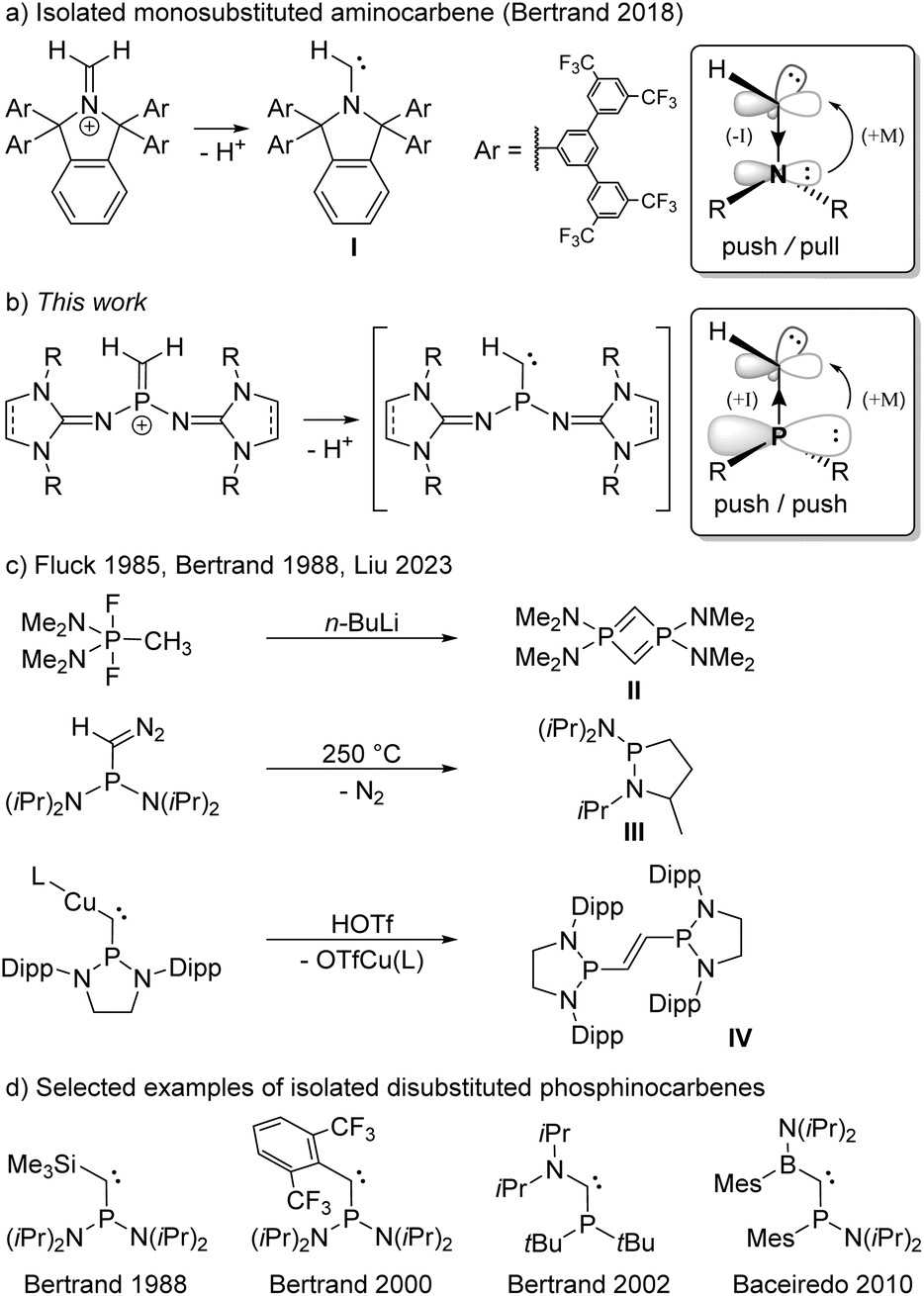 | ||
| Scheme 1 (a) Synthesis of a persistent monosubstituted aminocarbene reported by Bertrand. (b) Monosubstituted phosphinocarbenes transiently formed in this work. (c) Reactions in which phosphinocarbene intermediates have been postulated. (d) Selected examples of isolated phosphinocarbenes. L = 1,3-bis(2,6-diisopropylphenyl)imidazole-2-ylidene, Dipp = 2,6-diisopropylphenyl, OTf− = trifluoromethanesulfonate (triflate). Boxes: Mesomeric and inductive substituent effects in amino- and phosphinocarbenes. | ||
This achievement raises the question of whether substituents other than amino groups are capable of stabilizing monosubstituted carbenes. The heavier phosphino group exerts much less π-conjugation towards the carbon center than an amino group, leading to smaller singlet–triplet energy separation and a low-lying LUMO,21,22 which results in an increased electrophilicity and nucleophilicity of the carbene center. The interaction of the carbene carbon atom with a second substituent was therefore hitherto required for the formation of stable phosphinocarbenes (Scheme 1d).3,23–26 The intermediate existence of a monosubstituted phosphinocarbene was speculated by Fluck and coworkers in 1985, when they treated (Me2N)2PF2CH3 with 2 equivalents of n-butyllithium and obtained diphosphete II as the formal carbene dimer (Scheme 1c).27,28 Later, Bertrand and coworkers showed that thermolysis of (iPr2N)2PC(N2)H at 250 °C leads to heterocycle III, which formally results from a C–H insertion reaction of the corresponding phosphinocarbene (Scheme 1c).7,29 Very recently, Liu and coworkers demonstrated that protonation of the bulky copper phosphinocarbyne anion complex IV results in the formation of the corresponding R2P–CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH–PR2 carbene dimer.30 We reasoned that deprotonation of terminal methylene phosphonium ions would provide access to monosubstituted phosphinocarbenes under mild conditions. Although several methylene phosphonium salts have been reported, mainly by the groups of Appel,31 Bertrand,32 Grützmacher,33–35 Erker,36 and Baceiredo,24 stabilizing groups at both phosphorus and carbon were employed in all cases. Herein, we report a synthetic access to terminal methylene phosphonium cations and show that they are suitable precursors for transient monosubstituted phosphinocarbenes.
CH–PR2 carbene dimer.30 We reasoned that deprotonation of terminal methylene phosphonium ions would provide access to monosubstituted phosphinocarbenes under mild conditions. Although several methylene phosphonium salts have been reported, mainly by the groups of Appel,31 Bertrand,32 Grützmacher,33–35 Erker,36 and Baceiredo,24 stabilizing groups at both phosphorus and carbon were employed in all cases. Herein, we report a synthetic access to terminal methylene phosphonium cations and show that they are suitable precursors for transient monosubstituted phosphinocarbenes.
Results and discussion
Experimental results
The synthesis of the terminal diprotic methylene phosphonium ions [4a]+ and [4b]+ was realized by oxidation of the methyl-substituted phosphines to phosphonium salts and subsequent deprotonation, analogous to the original synthesis by Grützmacher (Scheme 2).37 Sterically demanding N-heterocyclic imines (NHIs) have been shown to be effective for the kinetic stabilization of low-coordinate phosphorus(V),25,38–42 and other main group cations.43 Moreover, Schoeller predicted that nitrogen-based π-donor substituents at phosphorus effectively increase the singlet–triplet gap of phosphinocarbenes.21 Hence, we chose NHIs with tert-butyl (tBu) and 2,6-diisopropylphenyl (Dipp) groups to flank the reactive PC double bond. The phosphenium salts [1a]Cl and [1b]Cl (Scheme 2) were prepared according to reported procedures by the reaction of PCl3 with NHI–SiMe3 derivatives upon elimination of ClSiMe3.25,44–46 Alkylation of the phosphenium cations with MeMgCl gave phosphines 2a and 2b as crystalline solids in very good yields. Characteristically, the phosphines show quartets (2a: 69.3 ppm, 2b: 62.0 ppm) in the 31P NMR spectrum with 2JPH coupling constants of 7–9 Hz. The solid-state structures of the bulky phosphines were determined by single-crystal X-ray diffraction (SCXRD) studies showing the expected pyramidalization of the phosphorus atoms (Fig. 1, left).
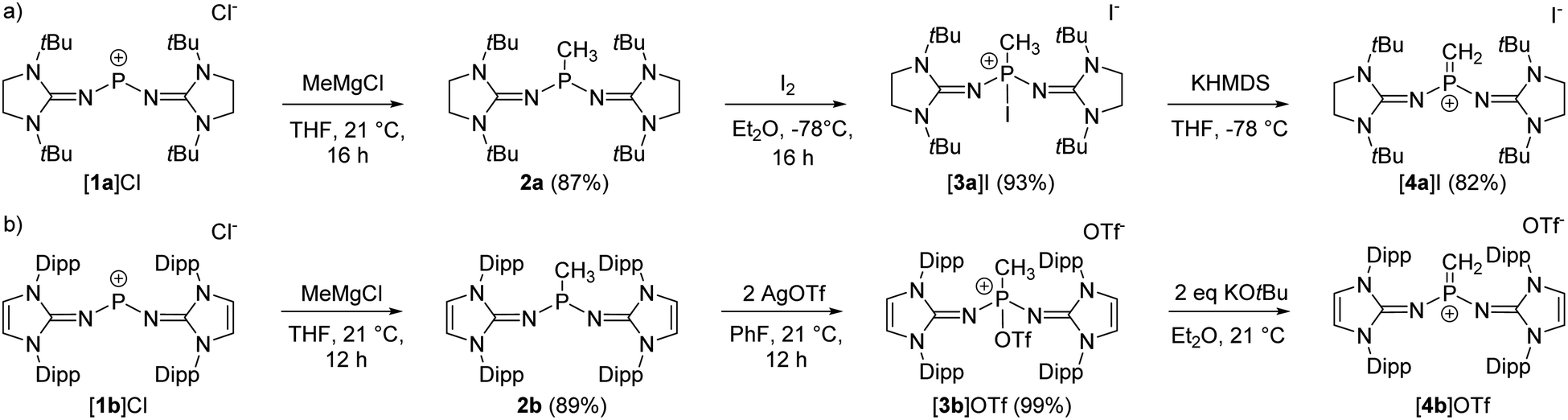 | ||
| Scheme 2 Synthesis of methylene phosphonium salts [4a]I (a) and [4b]OTf (b). | ||
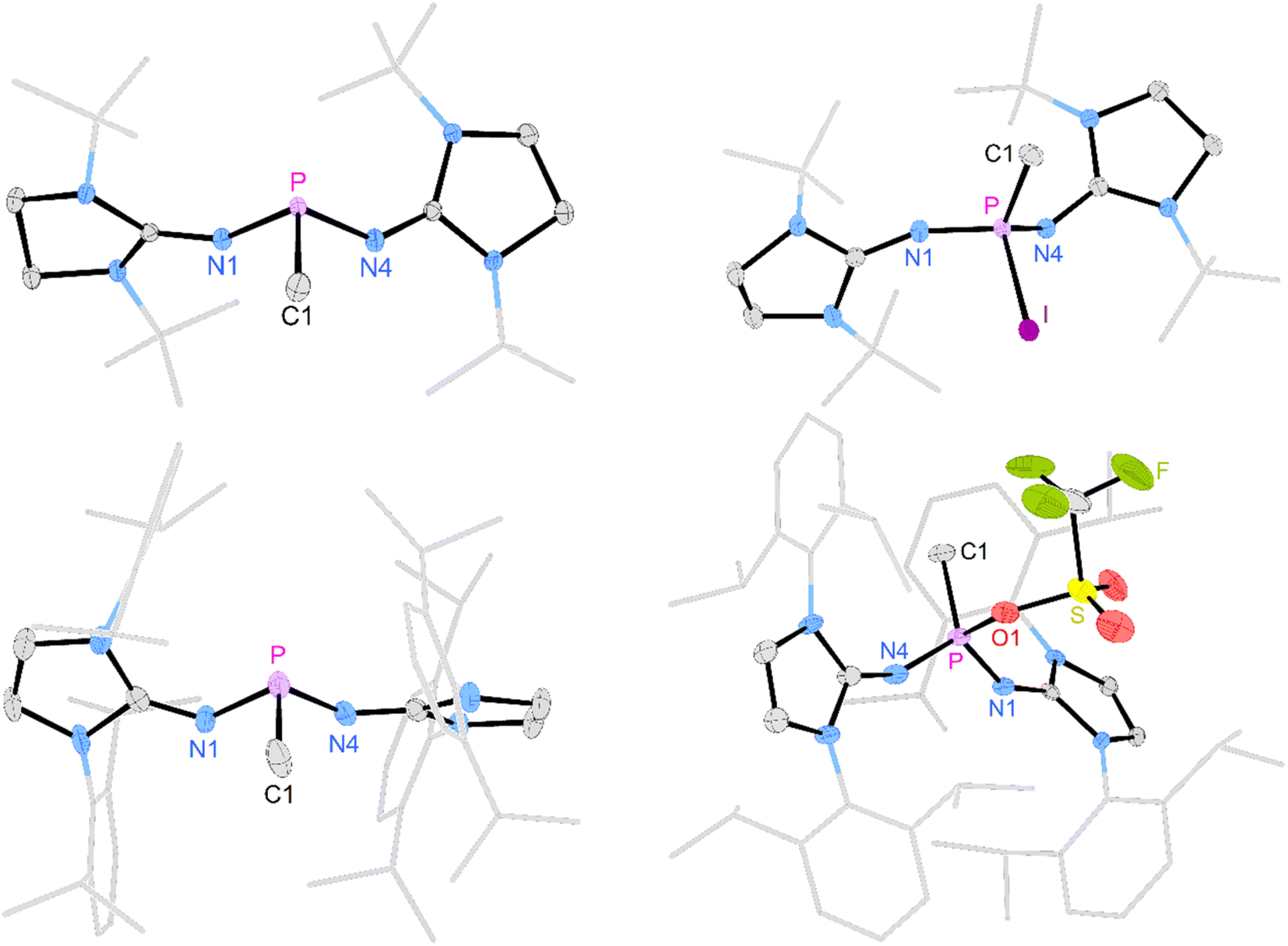 | ||
| Fig. 1 Solid-state structures of 2a (top left), 2b (bottom left), [3a]I (top right), and [3b]OTf (bottom right). In 2b and [3b]OTf, only one of the two crystallographically independent molecules is depicted. Hydrogen atoms, anions, and disordered moieties are omitted for clarity. Ellipsoids are drawn at 50% probability. tBu (2a, [3a]+) and Dipp (2b, [3b]+) groups are shown in wireframe. Selected bond lengths [Å] and angles [deg]: 2a: P–C1 1.8444(9), P–N1 1.6700(8), P–N4 1.6824(8), C1–P–N1 97.88(4), C1–P–N4 97.03(4), N1–P–N4 100.23(4). [3a]I: P–C1 1.785(2), P–N1 1.550(3), P–N4 1.570(2), P–I 2.4982(8), C1–P–N1 113.20(13), C1–P–N4 109.75(12), N1–P–N4 114.02(12), C1–P–I 99.10(9). 2b: P–C1 1.848(5), P–N1 1.702(4), P–N4 1.687(4), C1–P–N1 95.6(2), C1–P–N4 103.3(2), N1–P–N4 96.6(2). [3b]OTf: P–C1 1.787(5), P–N1 1.543(4), P–N4 1.549(4), P–O1 1.730(4), C1–P–N1 117.9(2), C1–P–N4 115.1(2), N1–P–N4 110.8(2), C1–P–O1 99.3(2). | ||
The oxidation of the electron-rich phosphines 2a and 2b was carried out with elemental iodine and silver trifluoromethanesulfonate (AgOTf), respectively, to afford the corresponding phosphonium salts [3a]I and [3b]OTf as white, air-sensitive solids in excellent yield. The use of elemental iodine allows the subsequent exchange of the iodide for a weakly coordinating anion (vide infra), but entails the problem that the triiodide anion is formed with excess iodine, as confirmed by a SCXRD analysis of [3a]I3 (see the SI for details). AgOTf was therefore used as a more convenient oxidizing agent in the case of the sterically encumbered phosphine 2b. SCXRD studies reveal distorted tetrahedral bonding environments for the phosphonium cations (Fig. 1, right). The P–O bond in [3b]OTf (1.730(4) Å) is exceptionally long compared to other PV–O bonds (cf. Ph3P–O: 1.479(2) Å;47 [Ph3P–O–PPh3]2+: 1.597(3) Å,48 1.598(4) Å; [{(Ph3P)2C}P(OTf)Ph2]2+: 1.676(4) Å).49 This elongation and the planarization of the N2PC moiety (sum of angles [3b]+: 343.8°) suggests that the P–O bond is strongly activated, which is supported by the observation that [3b]OTf readily polymerizes tetrahydrofuran (THF) in solution.
The solid-state structure of [3a]I also exhibits a distorted tetrahedral geometry of the phosphorus atom. However, the N2PC unit of [3a]+ is less planarized (sum of angles: 337°) compared with that of [3b]+. There are short contacts of 3.829 Å between the phosphorus bound iodine and the iodide anion, which is not uncommon for iodophosphonium iodide salts including [iPr3I]I (3.384 Å)50 and [(Me2N)3PI]I (3.619 Å)51 (van der Waals radii for two iodine atoms: 4.3 Å).52 The 31P NMR resonances of [3a]I (−71.7 ppm) and [3b]OTf (−11.9 ppm) appear as quartets with 2JPH coupling constants of 14–16 Hz. Consistent with the solid-state structures, two distinct 19F NMR resonances are observed in solution for the phosphorus bound triflate and the free anion, respectively (−73.67 ppm and −78.77 ppm). The 127I NMR spectrum of [3a]I shows a broad signal at 62 ppm, which is in the expected range for free iodide.53 The absence of a second signal corresponding to the phosphorus-bound iodine atom is due to the quadrupolar nature (I = 5/2) of the 127I nucleus,54 since resonances with sufficiently narrow line width are only detected for iodine in highly symmetrical environments.55,56
Deprotonation of [3a]I with potassium hexamethyldisilazide (KHMDS) resulted in the formation of the desired terminal methylene phosphonium salt [4a]I, which was obtained after recrystallization as large colorless crystals in 82% yield. [4a]I is air sensitive but can be stored under inert atmosphere for months without noticeable decomposition. It is soluble in dichloromethane but sparingly soluble in THF or Et2O and decomposes in acetonitrile. Exposure to D2O leads to activation of both O–D bonds and affords [(NHI)2CH2DP–O–PCH2D(NHI)2]2+ (NHI = 1,3-di-tert-butylimidazolidin-2-ylidenamino), which is isotypic to the Hendrickson reagent [Ph3POPPh3]2+ (see the SI for details). The methylene phosphonium salt [4a]I shows a characteristic triplet at 89 ppm (2JPH = 15 Hz) in the 31P NMR spectrum. The 13C resonance of the terminal methylene group (32.2 ppm, 1JCP = 186 Hz) exhibits a significantly larger 1JPC coupling constant than [3a]I (34.3 ppm, 1JCP = 119 Hz). The broad signal at 77 ppm in the 127I NMR spectrum indicates that iodide is not bound to the cationic molecule.53 A SCXRD study confirms the ionic nature of [4a]I in the solid state with the iodine atom located 5.52 Å away from the phosphorus center (Fig. 2, top). The P atom is in a trigonal planar environment (sum of angles: 360°). The P–C bond length (1.620 Å) is slightly shorter than in the methylene phosphonium ions reported by Bacereido [Mes(iPr2N)PCH(BMes(NiPr2))]+ (1.634 Å)24 and Grützmacher [tBu2P = CPh(o-MeC6H4)]+ (1.680 Å),35 consistent with the minimum steric bulk at the methylene moiety.
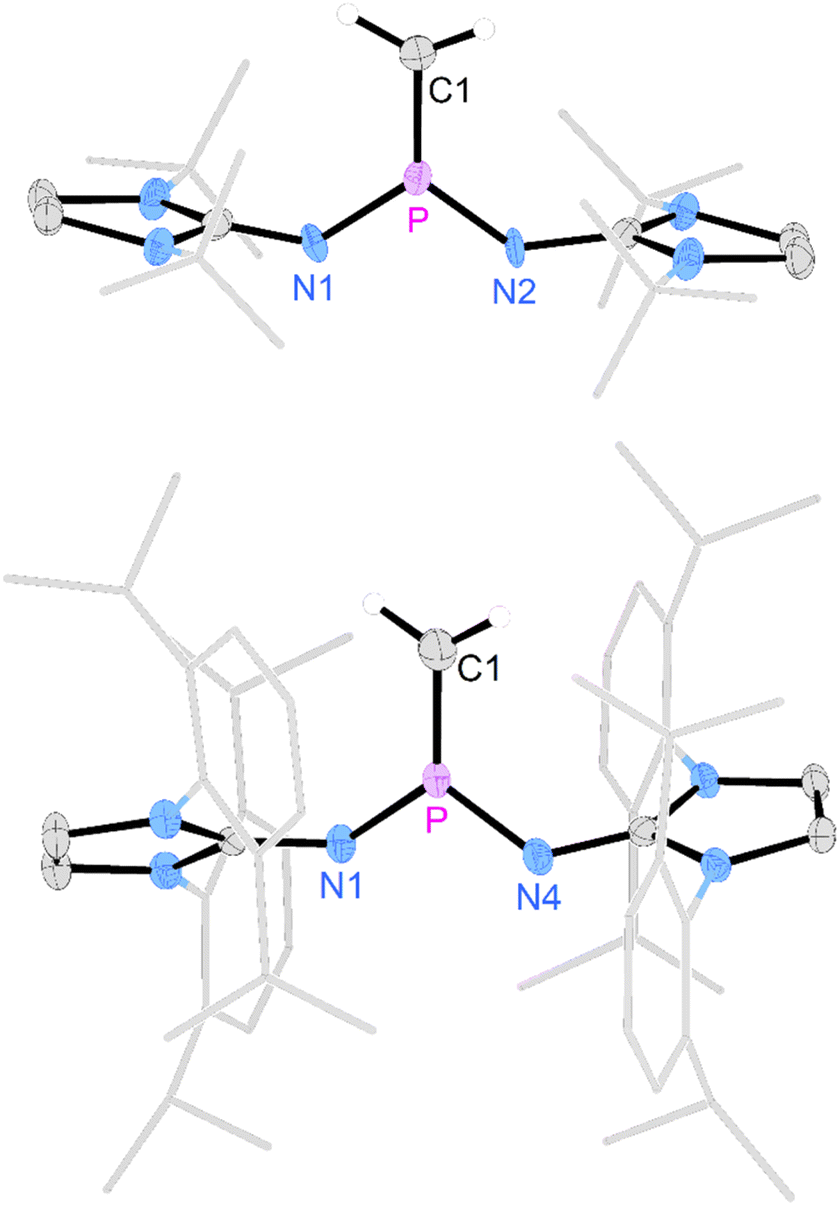 | ||
| Fig. 2 Solid-state structure of [4a]I (top) and [4b]OTf (bottom). Hydrogen atoms (except C1 hydrogen atoms) and anions are omitted for clarity. Ellipsoids are drawn at 50% probability. tBu ([4a]+) and Dipp ([4b]+) groups are shown in wireframe. Only one of two crystallographically independent molecules of [4b]OTf is depicted. Selected bond lengths [Å] and angles [deg]: [4a]I: P–C1 1.621(5), P–N1 1.569(7), P–N2 1.523(9), C1–P–N1 123.7(4), C1–P–N2 125.9(3), N1–P–N2 110.4(5). [4b]OTf: P–C1 1.619(3), P–N1 1.566(2), P–N4 1.565(2), C1–P–N1 125.9(2), C1–P–N4 123.29(13), N1–P–N4 110.69(9). | ||
The deprotonation of the sterically encumbered phosphonium salt [3b]OTf turned out to be unselective regardless of the type of base. Treatment with lithium organyls such as MeLi, nBuLi and PhLi led to the reduction to the phosphine 2b in more than 70% yield according to 31P NMR analysis. The fact that the lithium organyls act as reducing agents57,58 rather than as bases can be attributed to the effective steric shielding of the methyl protons by the Dipp substituents, which is also evident in the solid-state structure of [3b]OTf (see Fig. S97 and S98† for steric maps of [3a]+ and [3b]+). NBO charges of 1.842e and 2.238e were calculated for the phosphorus atoms of [3a]+ and [3b]+, respectively, making an electron transfer to the electrophilic P atom a plausible alternative pathway when deprotonation is hampered by bulky substituents. The attempted deprotonation of [3b]OTf with stoichiometric amounts of KHMDS or KOtBu did not afford the reduced phosphine 2b, but gave several phosphorus species. However, when using two equivalents of KOtBu, the 31P NMR spectrum of the reaction mixture showed the characteristic triplet of the methylene phosphonium cation at 102.6 ppm (2JPH = 13 Hz) as the major species along with a resonance at 0.0 ppm, which we putatively assign to the phosphine oxide.59,60 The formation of the latter can be explained by a nucleophilic attack of the base at the sulfur atom of the triflate. A detailed study on the reactivity of the triflate-containing phosphonium ion [{(Ph3P)2C}P(OTf)Ph2]2+ with nucleophiles by Weigand and co-workers showed that both the P atom and the S atom are prone to nucleophilic attack.49 Substitution at the latter leads to the elimination of the phosphine oxide. Although we were not able to isolate [4b]OTf on preparative scale, a SCXRD of a single crystal grown from the reaction mixture confirmed its molecular structure, which exhibits geometric parameters analogous to [4a]I (Fig. 2, bottom).
We next performed deprotonation studies to generate the desired monosubstituted phosphinocarbene (Scheme 3). For this purpose, [4a]BArF24 (BArF24 = [2,6-bis(trifluoromethyl)phenyl]borate) was prepared from the reaction of [4a]I with Na(BArF24) to increase the solubility of the methylene phosphonium ion in ether solvents. Treatment of [4a]BArF24 with various bases gave the diphosphete salt [7]BArF24 as the major product (>80% yield using KHMDS) along with other unidentified products (see the ESI† for details), which leads to the conclusion that the free carbene reacts readily with the methylene phosphonium precursor. The composition of [7]BArF24 was confirmed by high resolution mass spectrometry and it was characterized using multinuclear 1D and 2D NMR spectroscopy (see the SI for details). The 31P resonance at −39.7 ppm is split into a triplet (2JPH = 18 Hz) due to coupling with the CH2 ring protons. In agreement with the isostructural phosphete ion [(NMe2)2PCH2CHP(NMe2)2]+, the 2JPH coupling constant with the CH bridge is not resolved.61 In the 13C{1H} NMR spectrum of [7]BArF24, characteristic triplets are observed for both the CH bridge (63.1 ppm, 1JCP = 134 Hz) and the CH2 bridge (51.7 ppm, 1JCP = 84 Hz).
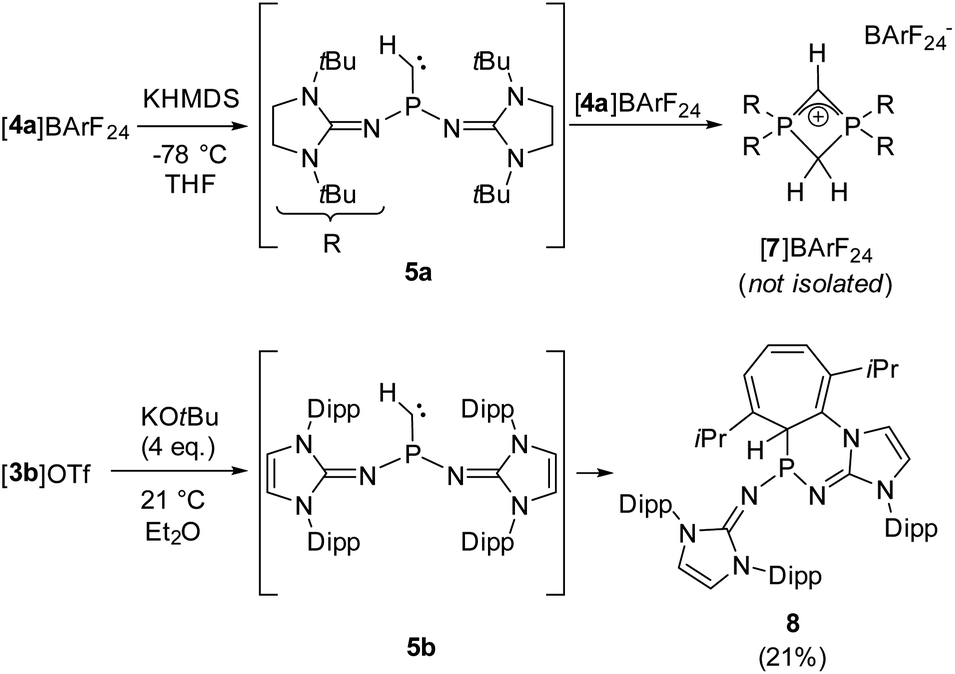 | ||
| Scheme 3 Reactivity of [4a]BArF24 with KHMDS and reactivity of [3b]OTf with an excess of KOtBu. | ||
Aiming at intercepting the generated phosphinocarbene, the deprotonation of [4a]BArF24 was performed in the presence of various trapping reagents including cyclohexene, diphenylacetylene, 1-propynylbenzene, elemental selenium and [Rh(COD)Cl]2 (COD = 1,5-cyclooctadiene). However, the formation of [7]BArF24 did not appear to be affected by the trapping reagents, which we attribute to the fact that both 5a and [4a]BArF24 are highly reactive. In addition, the basic carbene atom is expected to bind to Lewis acids after deprotonation, which hampers carbene-type reactivity but does not prevent the adjacent phosphino group from initiating the cyclization with the electrophilic methylene phosphonium cation.
Gratifyingly, unequivocal evidence for the formation of the transient phosphinocarbene was provided by deprotonation of [3b]OTf using an excess of KOtBu, which gave the formal ring insertion product 8 (Scheme 3). 31P NMR analysis of the reaction mixture indicates 62% conversion to the polycyclic phosphine 8 (Fig. S79†), which was isolated after extraction with n-hexane and recrystallization as yellow crystalline solid in 21% yield. Phosphine 8 shows a diagnostic doublet at 73.2 ppm in the 31P NMR spectrum with a 2JPH coupling constant of 11 Hz. The molecular structure was established by a SCXRD study and reveals that the CH carbene moiety was inserted into a CC bond of a former Dipp substituent (Fig. 3). The cycloheptatriene ring of the resulting tricyclic structure shows alternating CC double bonds (C4–C10 1.366 Å, C6–C7 1.358 Å, C8–C9 1.366 Å) and C–C single bonds (C4–C5 1.510 Å, C5–C6 1.510 Å, C7–C8 1.433 Å, C9–C10 1.438 Å).
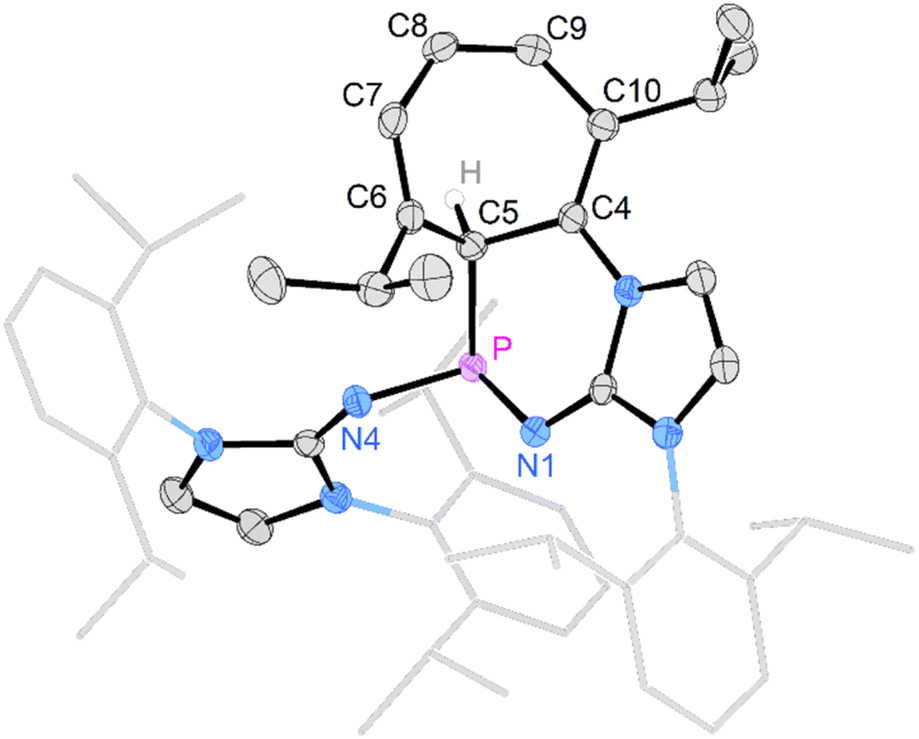 | ||
| Fig. 3 Solid-state structure of 8. Hydrogen atoms (except C5 hydrogen) are omitted for clarity. Ellipsoids are drawn at 50% probability. Dipp groups are shown in wireframe. Selected bond lengths [Å] and angles [deg]: P–C5 1.866(2), P–N1 1.7017(14), P–N4 1.693(2), C5–C6 1.510(3), C6–C7 1.358(2), C7–C8 1.433(3), C8–C9 1.366(3), C9–C10 1.438(3), C10–C4 1.366(2) C4–C5 1.510(3), N1–P–N4 199.67(7), N1–P–C5 103.78(7), C4–P–C5 100.84(7). | ||
The formation of compound 8 is rationalized by the intermediate generation of the phosphinocarbene 5b which undergoes an intramolecular cyclopropanation with an adjacent Dipp substituent followed by a divinylcyclopropane–cycloheptadiene rearrangement. The analogous Buchner ring expansion reactions, involving benzene derivatives and transient carbenes or complexes thereof, has been widely used to introduce cycloheptatriene rings into natural products and materials.62–69 Similar intramolecular ring expansions were reported for other transient main-group carbene analogues, such as silylenes,70–76 and phosphinidenes.77 Attempts to trap the transient carbene by performing the deprotonation at −40 °C in fluorobenzene in the presence of 2 bar dihydrogen or carbon monoxide, both known to react with electrophilic carbenes,78–80 were unsuccessful.
Computational studies
To assess the electronic properties of the transient phosphinocarbene and elucidate the mechanism of the ring insertion reaction, theoretical calculations at the B3LYP-GD3BJ/def2-TZVP level of theory81–83 were performed. The HOMO/LUMO energies in the respective singlet state along with the associated singlet–triplet energy difference (ΔEST) have been calculated for simplified model compounds (Fig. 4, E and F) and compared to prototype aliphatic and heteroatom-substituted (N, P) carbenes (Fig. 4, A–D). While the non-heteroatom substituted carbenes A and B are predicted to have a triplet ground state, the singlet state is energetically favored in case of the heteroatom substituted carbenes (C–F) in agreement with previous studies.84,85 Furthermore, substitution of the nitrogen atom in compound C by a phosphorus atom (D) leads to a much smaller ΔEST. Although the gap increases by the employment of NHI substituents (E, F), it is still much lower compared to the ones from NHCs and CAACs (>200 kJ mol−1).86,87 Moreover, the calculated ΔEST of E and F are lower than that of the acyclic amino(alkyl)carbene iPr2N–C–tBu (112.5 kJ mol−1)20 but slightly higher than that of the persistent amino(silyl)carbene iPr2N–C–SiPh2tBu (84.9 kJ mol−1).88 Among the carbenes considered, E and F show the highest HOMO energy, in the same range like iPr2N–C–tBu (−4.69 eV),20 indicating that they are stronger σ-donors compared to carbenes A–D. In contrast, the HOMO–LUMO gap of E and F (4.57 eV and 4.87 eV) is comparable to the other monosubstituted carbenes B–D.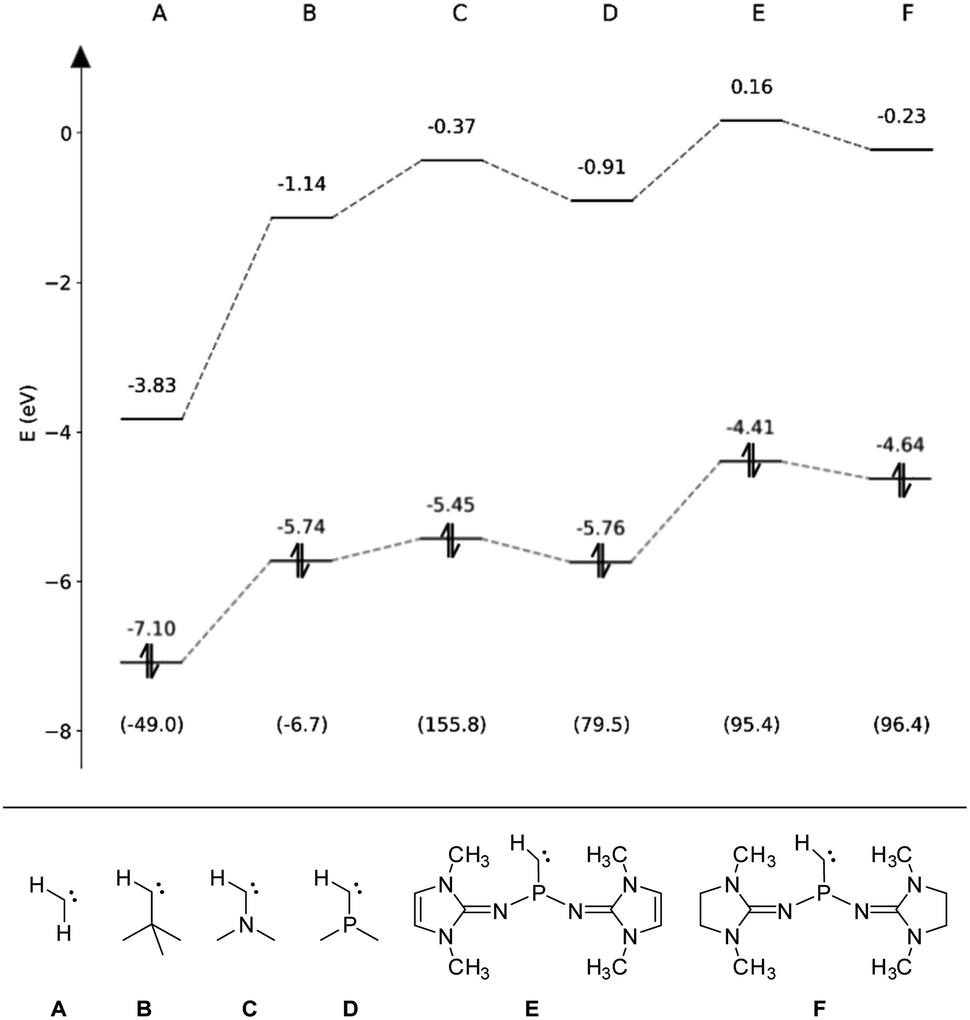 | ||
| Fig. 4 Eigenenergies of the HOMO and LUMO for carbenes A–F obtained at the B3LYP-GD3BJ/def2-TZVP level. The respective singlet–triplet energy differences (ΔEST) are given in kJ mol−1 in parentheses. | ||
In addition, the reaction path of the ring insertion of the phosphinocarbene has been characterized for a simplified model system (Fig. 5, G) by DFT calculations, considering both a vacuum environment and implicit solvation in diethyl ether. Although the targeted analog of model compound G could not be isolated experimentally, the theoretical calculations confirm the proposed reaction mechanism of a carbene formation and subsequent ring insertion via an initial [1 + 2] cycloaddition reaction step in both vacuum as well as implicit solvation. Two different transition states for the reaction G → H could be identified, which display a comparable activation energy. However, close inspection of the associated P–Ccarbene bond distance rPC and the angular sum around the P atom reveal that the respective reactions are fundamentally different: Phosphinocarbene G exhibits a PC double bond of about 1.61 Å and a planar P atom (sum of angles: 359°). While this structural pattern is very well retained for transition state GH‡2, the bond distance and angular sum changed to 1.69 Å and 330° in case of GH‡1. The latter implies a conversion of the double bond towards a P–C single bond along with the pyramidalization of the P atom, which is associated with an electrophilic addition of the carbene to the phenyl ring. On the other hand, the unchanged geometry observed for GH‡2 implies a nucleophilic attack of the carbene at the aromatic moiety.
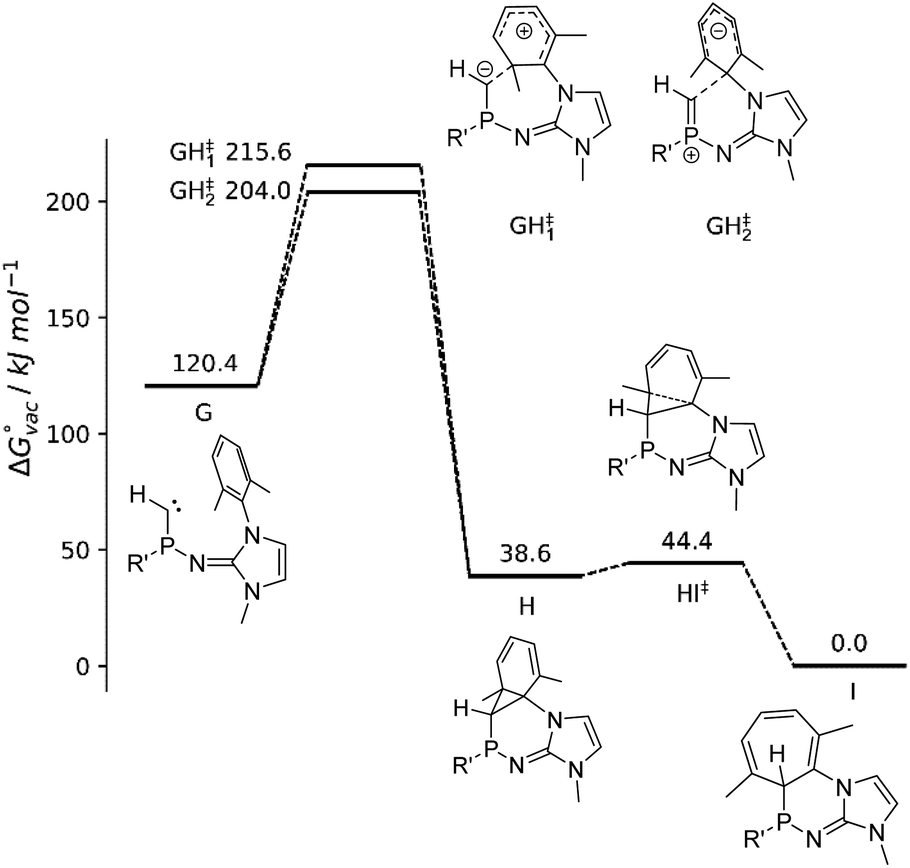 | ||
| Fig. 5 Free energy profile of the reaction path G → I including the identified transition states GH‡1–2 and HI‡ obtained at B3LYP-GD3BJ/def2-TZVP level of theory in the gas phase. R′ = 1,3-dimethylimidazolin-2-ylidenamino. | ||
From the intermediate state H onward, the geometrical properties correspond to that of a P–C single bond in the range of 1.85 to 1.86 Å with the corresponding angular sums being close to 300° as expected for NHI-substituted phosphines (2b: 295°).
The isolation of the intermediate H appears not possible due to the small activation barrier of 9.7 kJ mol−1 and 9.2 kJ mol−1 in vacuum and implicit solvent, respectively, corresponding to an effectively instantaneous reaction to the final product I. The reaction barriers associated with the two transitions states GH‡1 and GH‡2 are in the range of 83.6 to 95.2 kJ mol−1, which implies that the phosphinocarbene should be sufficiently long-lived to be trapped by a suitable reagent. However, the basic phosphinocarbene is expected to interact with Lewis acids such as K+ in the reaction mixture, which might significantly facilitate the electrophilic addition mechanism. Similarly, the electrophilic addition of a transient phosphinidene to a mesityl ring was promoted by its coordination to AuCl.77
Conclusions
We report the synthesis and full characterization of the first terminal methylene phosphonium ions and demonstrate that they are suitable precursors for the generation of monosubstituted phosphinocarbenes. The latter are highly reactive and, depending on the steric bulk of the NHI substituents, either undergo intermolecular [2 + 2] cycloadditions to form P2C2 four-membered rings or insert intramolecularly into the CC bond of an adjacent aryl substituent, leading to a Buchner ring expansion and the formation of a cycloheptatriene derivative. DFT calculations reveal similar reaction barriers for both the electrophilic and the nucleophilic attack at the phenyl ring, corroborating the ambiphilic character of the phosphinocarbene. Although we were not able to isolate the phosphinocarbenes, the present study shows that NHI substituents effectively increase the singlet triplet energy gap. Thus, the challenge in designing isolable monosubstituted phosphinocarbenes appears to narrow down to the design of appropriate substituents that can withstand the ambiphilic carbon center, which is the subject of ongoing research in our laboratories.
Data availability
Further details of the experimental procedures, the computational studies, and the charactization data for the new compounds are available in the ESI.†Author contributions
P. L. and M. A. W. synthesized the compounds and performed the experimental studies. M. B. R. helped with experiments and obtained characterization data. P. L., L. F. B. W. and F. D. performed the SCXRD studies. F. R. S. P., J. G and T. S. H. performed the computational studies. P. L., T. S. H. and F. D. wrote the manuscript. F. D. directed the investigation. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
P. L. and M. A. W. gratefully acknowledge financial support from the DFG (IRTG 2027). L. W. thanks the German Academic Scholarship Foundation for a PhD fellowship. We thank Dr Tim Witteler for his assistance in chemical synthesis and Dr Alexander Hepp for performing the NMR experiments and for his help with assigning the data.Notes and references
- J. M. Hollis, P. R. Jewell and F. J. Lovas, Astrophys. J., 1995, 438, 259 CrossRef CAS.
- J. Vignolle, X. Cattoën and D. Bourissou, Chem. Rev., 2009, 109, 3333–3384 CrossRef CAS PubMed.
- D. Bourissou, O. Guerret, F. P. Gabbaï and G. Bertrand, Chem. Rev., 2000, 100, 39–92 CrossRef CAS PubMed.
- K. Öfele, J. Organomet. Chem., 1968, 12, P42–P43 CrossRef.
- H.-W. Wanzlick and E. Schikora, Chem. Ber., 1961, 94, 2389–2393 CrossRef CAS.
- R. Breslow, J. Am. Chem. Soc., 1958, 80, 3719–3726 CrossRef CAS.
- A. Igau, H. Grutzmacher, A. Baceiredo and G. Bertrand, J. Am. Chem. Soc., 1988, 110, 6463–6466 CrossRef CAS.
- A. J. Arduengo, R. L. Harlow and M. Kline, J. Am. Chem. Soc., 1991, 113, 361–363 CrossRef CAS.
- P. de Frémont, N. Marion and S. P. Nolan, Coord. Chem. Rev., 2009, 253, 862–892 CrossRef.
- S. C. Sau, P. K. Hota, S. K. Mandal, M. Soleilhavoup and G. Bertrand, Chem. Soc. Rev., 2020, 49, 1233–1252 RSC.
- M. Soleilhavoup and G. Bertrand, Chem, 2020, 6, 1275–1282 CAS.
- Á. Vivancos, C. Segarra and M. Albrecht, Chem. Rev., 2018, 118, 9493–9586 CrossRef PubMed.
- R. R. Sauers, Arkivoc, 2014, 2014, 376–383 Search PubMed.
- M. N. Hopkinson, C. Richter, M. Schedler and F. Glorius, Nature, 2014, 510, 485–496 CrossRef CAS PubMed.
- S. Kumar Kushvaha, A. Mishra, H. W. Roesky and K. Chandra Mondal, Chem.–Asian J., 2022, 17, e202101301 CrossRef CAS.
- U. S. D. Paul and U. Radius, Ber. Dtsch. Chem. Ges., 2017, 2017, 3362–3375 CAS.
- C. A. Smith, M. R. Narouz, P. A. Lummis, I. Singh, A. Nazemi, C.-H. Li and C. M. Crudden, Chem. Rev., 2019, 119, 4986–5056 CrossRef CAS PubMed.
- V. Lavallo, J. Mafhouz, Y. Canac, B. Donnadieu, W. W. Schoeller and G. Bertrand, J. Am. Chem. Soc., 2004, 126, 8670–8671 CrossRef CAS PubMed.
- M. Soleilhavoup and G. Bertrand, Acc. Chem. Res., 2015, 48, 256–266 CrossRef CAS PubMed.
- R. Nakano, R. Jazzar and G. Bertrand, Nat. Chem., 2018, 10, 1196–1200 CrossRef CAS PubMed.
- W. W. Schoeller, Ber. Dtsch. Chem. Ges., 2000, 2000, 369–374 Search PubMed.
- M. Z. Kassaee, F. A. Shakib, M. R. Momeni, M. Ghambarian and S. M. Musavi, J. Org. Chem., 2010, 75, 2539–2545 CrossRef CAS PubMed.
- G. Bertrand and R. Reed, Coord. Chem. Rev., 1994, 137, 323–355 CrossRef CAS.
- F. Lavigne, E. Maerten, G. Alcaraz, N. Saffon-Merceron, C. Acosta-Silva, V. Branchadell and A. Baceiredo, J. Am. Chem. Soc., 2010, 132, 8864–8865 CrossRef CAS.
- P. Löwe and F. Dielmann, Chem. Commun., 2022, 58, 11831–11834 RSC.
- N. Merceron, K. Miqueu, A. Baceiredo and G. Bertrand, J. Am. Chem. Soc., 2002, 124, 6806–6807 CrossRef CAS PubMed.
- J. Svara, E. Fluck and H. Riffel, Z. Naturforsch., B: J. Chem. Sci., 1985, 40, 1258–1263 CrossRef.
- W. Plass, M. Spahn, G. Heckmann and E. Fluck, Z. Naturforsch., B: J. Chem. Sci., 1992, 47, 947–951 CrossRef CAS.
- A. Baceiredo and G. Bertrand, Phosphorus Sulfur Relat. Elem., 1986, 26, 57–62 CrossRef CAS.
- R. Wei, X.-F. Wang, C. Hu and L. L. Liu, Nat. Synth., 2023, 2, 357–363 CrossRef.
- R. Appel and R. Schmitz, Chem. Ber., 1983, 116, 3521–3523 CrossRef CAS.
- A. Igau, A. Baceiredo, H. Gruetzmacher, H. Pritzkow and G. Bertrand, J. Am. Chem. Soc., 1989, 111, 6853–6854 CrossRef CAS.
- H. Grützmacher, U. Heim, H. Schönberg and H. Pritzkow, Phosphorus, Sulfur Silicon Relat. Elem., 1993, 76, 21–24 CrossRef.
- H. Grützmacher and H. Pritzkow, Angew. Chem., 1991, 103, 721–723 CrossRef.
- U. Heim, H. Pritzkow, H. Schönberg and H. Grützmacher, Chem. Commun., 1993, 673–674 RSC.
- Y. Hasegawa, G. Kehr, S. Ehrlich, S. Grimme, C. G. Daniliuc and G. Erker, Chem. Sci., 2014, 5, 797–803 RSC.
- H. Grützmacher and H. Pritzkow, Angew. Chem., Int. Ed., 1991, 30, 709–710 CrossRef.
- M. A. Wünsche, T. Witteler and F. Dielmann, Angew. Chem., Int. Ed., 2018, 57, 7234–7239 CrossRef PubMed.
- P. Löwe, T. Witteler and F. Dielmann, Chem. Commun., 2021, 57, 5043–5046 RSC.
- P. Mehlmann, T. Witteler, L. F. B. Wilm and F. Dielmann, Nat. Chem., 2019, 11, 1139–1143 CrossRef CAS PubMed.
- F. Dielmann, O. Back, M. Henry-Ellinger, P. Jerabek, G. Frenking and G. Bertrand, Science, 2012, 337, 1526–1528 CrossRef CAS PubMed.
- F. Dielmann, C. E. Moore, A. L. Rheingold and G. Bertrand, J. Am. Chem. Soc., 2013, 135, 14071–14073 CrossRef CAS PubMed.
- T. Ochiai, D. Franz and S. Inoue, Chem. Soc. Rev., 2016, 45, 6327–6344 RSC.
- N. Kuhn, R. Fawzi, M. Steimann and J. Wiethoff, Chem. Ber., 1996, 129, 479–482 CrossRef CAS.
- M. A. Wünsche, P. Mehlmann, T. Witteler, F. Buß, P. Rathmann and F. Dielmann, Angew. Chem., Int. Ed., 2015, 54, 11857–11860 CrossRef PubMed.
- M. D. Böhme, T. Eder, M. B. Röthel, P. D. Dutschke, L. F. B. Wilm, F. E. Hahn and F. Dielmann, Angew. Chem., Int. Ed., 2022, 61, e202202190 CrossRef PubMed.
- K. A. Al-Farhan, J. Crystallogr. Spectrosc. Res., 1992, 22, 687–689 CrossRef CAS.
- S.-L. You, H. Razavi and J. W. Kelly, Angew. Chem., Int. Ed., 2003, 42, 83–85 CrossRef CAS PubMed.
- S. Yogendra, F. Hennersdorf, A. Bauzá, A. Frontera, R. Fischer and J. J. Weigand, Chem. Commun., 2017, 53, 2954–2957 RSC.
- F. Ruthe, P. G. Jones, W.-W. Du Mont, P. Deplano and M. L. Mercuri, Z. Anorg. Allg. Chem., 2000, 626, 1105–1111 CrossRef CAS.
- N. A. Barnes, S. M. Godfrey, R. T. A. Halton, I. Mushtaq and R. G. Pritchard, Dalton Trans., 2008, 1346–1354 RSC.
- S. S. Batsanov, Inorg. Mater., 2001, 37, 871–885 CrossRef CAS.
- N. Kagawa, M. Suzuki, N. Kogure and K. Toume, Tetrahedron Lett., 2015, 56, 5795–5798 CrossRef CAS.
- T. Drakenberg and S. Forsén, in The Multinuclear Approach to NMR Spectroscopy, ed. J. B. Lambert and F. G. Riddell, Springer Netherlands, Dordrecht, 1983, pp. 405–444 Search PubMed.
- M. F. A. Dove, J. C. P. Sanders and E. H. Appelman, Magn. Reson. Chem., 1995, 33, 44–58 CrossRef CAS.
- J. F. Lehmann, G. J. Schrobilgen, K. O. Christe, A. Kornath and R. J. Suontamo, Inorg. Chem., 2004, 43, 6905–6921 CrossRef CAS PubMed.
- E. C. Ashby and T. N. Pham, J. Org. Chem., 1987, 52, 1291–1300 CrossRef CAS.
- G. A. Russell and D. W. Lamson, J. Am. Chem. Soc., 1969, 91, 3967–3968 CrossRef CAS.
- P. Mehlmann, C. Mück-Lichtenfeld, T. T. Y. Tan and F. Dielmann, Chem.–Eur. J., 2017, 23, 5929–5933 CrossRef CAS.
- F. Buß, M. B. Röthel, J. A. Werra, P. Rotering, L. F. B. Wilm, C. G. Daniliuc, P. Löwe and F. Dielmann, Chem.–Eur. J., 2022, 28, e202104021 CrossRef PubMed.
- E. Fluck, G. Heckmann and K. Lange, Z. Naturforsch., B: J. Chem. Sci., 1993, 48, 1149–1150 CrossRef CAS.
- E. Buchner and T. Curtius, Ber. Dtsch. Chem. Ges., 1885, 18, 2377–2379 CrossRef.
- E. Buchner and T. Curtius, Ber. Dtsch. Chem. Ges., 1885, 18, 2371–2377 CrossRef.
- T. Ye and M. A. McKervey, Chem. Rev., 1994, 94, 1091–1160 CrossRef CAS.
- S. Reisman, R. Nani and S. Levin, Synlett, 2011, 2011, 2437–2442 CrossRef.
- L. Chan and S. A. Matlin, Tetrahedron Lett., 1981, 22, 4025–4028 CrossRef CAS.
- A. L. Crombie, J. L. Kane, K. M. Shea and R. L. Danheiser, J. Org. Chem., 2004, 69, 8652–8667 CrossRef CAS PubMed.
- B. Frey, A. P. Wells, D. H. Rogers and L. N. Mander, J. Am. Chem. Soc., 1998, 120, 1914–1915 CrossRef CAS.
- J. L. Kane, K. M. Shea, A. L. Crombie and R. L. Danheiser, Org. Lett., 2001, 3, 1081–1084 CrossRef CAS PubMed.
- M. Kira, S. Ishida, T. Iwamoto and C. Kabuto, J. Am. Chem. Soc., 2002, 124, 3830–3831 CrossRef CAS PubMed.
- T. Kosai, S. Ishida and T. Iwamoto, Chem. Commun., 2015, 51, 10707–10709 RSC.
- H. Suzuki, N. Tokitoh and R. Okazaki, J. Am. Chem. Soc., 1994, 116, 11572–11573 CrossRef CAS.
- D. Wendel, A. Porzelt, F. A. D. Herz, D. Sarkar, C. Jandl, S. Inoue and B. Rieger, J. Am. Chem. Soc., 2017, 139, 8134–8137 CrossRef CAS PubMed.
- C. Xu, Z. Ye, L. Xiang, S. Yang, Q. Peng, X. Leng and Y. Chen, Angew. Chem., Int. Ed., 2021, 60, 3189–3195 CrossRef CAS PubMed.
- L. Zhu, J. Zhang and C. Cui, Inorg. Chem., 2019, 58, 12007–12010 CrossRef CAS PubMed.
- H. Zhu, A. Kostenko, D. Franz, F. Hanusch and S. Inoue, J. Am. Chem. Soc., 2023, 145, 1011–1021 CrossRef CAS PubMed.
- L. L. Liu, J. Zhou, L. L. Cao, R. Andrews, R. L. Falconer, C. A. Russell and D. W. Stephan, J. Am. Chem. Soc., 2018, 140, 147–150 CrossRef CAS PubMed.
- G. D. Frey, V. Lavallo, B. Donnadieu, W. W. Schoeller and G. Bertrand, Science, 2007, 316, 439–441 CrossRef CAS PubMed.
- T. W. Hudnall and C. W. Bielawski, J. Am. Chem. Soc., 2009, 131, 16039–16041 CrossRef CAS PubMed.
- U. Siemeling, C. Färber, C. Bruhn, M. Leibold, D. Selent, W. Baumann, M. von Hopffgarten, C. Goedecke and G. Frenking, Chem. Sci., 2010, 1, 697 RSC.
- A. D. Becke, J. Chem. Phys., 1993, 98, 1372–1377 CrossRef CAS.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- S. Gronert, J. R. Keeffe and R. A. More O'Ferrall, J. Am. Chem. Soc., 2011, 133, 3381–3389 CrossRef CAS PubMed.
- H. M. Sulzbach, E. Bolton, D. Lenoir, P. v. R. Schleyer and H. F. Schaefer, J. Am. Chem. Soc., 1996, 118, 9908–9914 CrossRef CAS.
- J. C. Bernhammer, G. Frison and H. V. Huynh, Chem.–Eur. J., 2013, 19, 12892–12905 CrossRef CAS PubMed.
- B. Rao, H. Tang, X. Zeng, L. L. Liu, M. Melaimi and G. Bertrand, Angew. Chem., Int. Ed., 2015, 54, 14915–14919 CrossRef CAS PubMed.
- Y. Canac, S. Conejero, B. Donnadieu, W. W. Schoeller and G. Bertrand, J. Am. Chem. Soc., 2005, 127, 7312–7313 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures, NMR spectra, mass spectrometry data, crystallographic data, and computational details. CCDC 2244191–2244199. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc02899b |
| This journal is © The Royal Society of Chemistry 2023 |