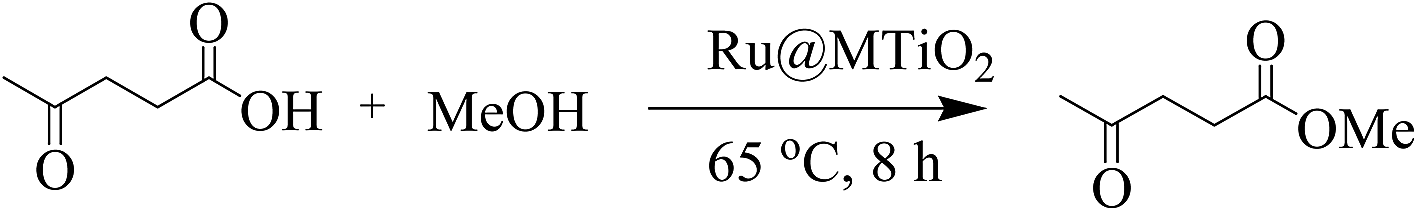DOI:
10.1039/C6RA10233F
(Paper)
RSC Adv., 2016,
6, 73440-73449
Ruthenium nanoparticles supported over mesoporous TiO2 as an efficient bifunctional nanocatalyst for esterification of biomass-derived levulinic acid and transfer-hydrogenation reactions†
Received
20th April 2016
, Accepted 25th July 2016
First published on 25th July 2016
Abstract
We have supported ruthenium nanoparticles (NPs) over a mesoporous TiO2 material (Ru@MTiO2) having nanoscale porosity and a high BET surface area. Wide-angle powder X-ray diffraction (XRD), transmission electron microscopy (TEM), energy-dispersive X-ray spectroscopy (EDX), Fourier transform infrared spectroscopy (FT-IR), ultraviolet-visible absorption spectra (UV-vis) and N2 sorption studies are used to characterize the Ru@MTiO2 material. This nanostructured ruthenium supported over a TiO2 material exhibited excellent bifunctional catalytic activity in the esterification of levulinic acid and selective transfer hydrogenation reactions of nitroarenes under mild reaction conditions. This Ru@MTiO2 catalyst can be easily recovered from the reaction mixtures and reused five times without a significant loss of its reactivity and selectivity.
Introduction
Depleting fossil resources and increasing concerns over greenhouse gas emissions have attracted a wide-scale interest in producing fuels and valuable chemicals from renewable biomass feedstocks.1 The chemical industry views biomass as a promising feedstock in the future and thus biomass conversion into value added chemicals is a matter of growing interest today. Among these routes, the synthesis of levulinate esters from levulinic acid (LA) has attracted widespread interest because LA is one of the top biomass derived platform molecules that can be made via C6 sugar carbohydrates derived from renewable lingo-cellulose.2 Levulinates such as ethyl, methyl, and n-butyl levulinate are obtained by esterification of levulinic acid (LA) or ethanolysis of furfuryl alcohol.3 These levulinate esters have several potential applications in flavour and fragrance industry. Moreover, ethyl levulinate (EL) can be used up to 5 wt% as the diesel miscible biofuel (DMB). Addition of EL to the diesel leads to considerable improvement in the fuel properties such as a clean burning fuel with high lubricity, stability in flashpoint and improvement in viscosity.4
On the other hand selective reduction or transfer hydrogenation of nitroarenes to anilines is a very important reaction both in industry and academia.5 In the past few years, several protocols for the reduction of nitroarenes employing various catalysts using a variety of metals including Cu, Ni, Fe, Pd, Ag, Ru and Rh have been investigated.6,7 But catalytic hydrogenation has two major drawbacks: the danger and inconvenience of handling highly flammable hydrogen gas and the low selectivity in the reduction of functionalized nitroarenes. Apart from hydrogen, several other stoichiometric reducing agents have been introduced that allow an efficient reduction of nitroarenes to anilines when used in combination with metal catalysts, therefore, hydrazine hydrate (N2H4·H2O) is a very suitable reagent for the reduction of nitro groups, as it generates only N2 gas as a byproduct.8 Compared to its unstable anhydrous form, hydrazine hydrate is relatively safe and easy to handle.
Nanometer-sized transition metal particles have emerged as one of the most useful and promising material because of their unique surface properties together with porosity and high surface area, which are largely different from their corresponding bulk materials.9 Their large surface area-to-volume ratio, which increases when decreasing particle size, could greatly increase their specific catalytic activity.10 Supported ruthenium catalysts have been widely used in heterogeneous catalysis, owing to their high activity and/or selectivity in many industrially important catalytic processes, including ammonia synthesis and decomposition, hydrogenation, etc.11 The effectiveness of the Ru-based catalysts in these heterogeneous reactions is strongly dependent on the metal-support interaction, which imposes significant influences on the morphology, surface and electronic structure, and the resulting catalytic performance.12 Thus, these nanoparticle based catalysts are often attached with different solid supports such as carbon nanotube, graphene, inorganic materials as SiO2, Al2O3, ZrO2, TiO2, hydrotalcite, montmorillonite clay and other materials for easier separation and presence of well-distributed Ru active sites.13 Recently, Weckhuysen et al. has employed Ru/H-ZSM-5 as a heterogeneous catalyst to directly convert LA to valeric acid (VA) and its esters but a yield of 45.8% of VA was achieved in dioxane as solvent.13h Therefore, the development of a novel catalytic system for esterification of levulinc acid with ethanol aiming for biofuel derived ethyl levulinate production in one pot with high efficiency is of great significance. Moreover TiO2 supported Ru catalyst is considered as one of the most promising catalyst support materials due to high surface area, its low cost, high photocatalytic activity, environmentally benign characteristics, high reflective index, noncorrosion, good stability.
In that context, TiO2-supported catalysts have been attracted considerable attention in many reactions of both environmental and industrial importance, because of their excellent catalytic activities compared with other supported catalysts.14 Although Ru NPs are known for their unique catalytic activity in wide range of chemical reactions15,16 and nanoporous TiO2 as a robust catalyst support,17 there are only few very recent reports on Ru NPs supported over TiO2 materials for partial hydrogenation of benzene to cyclohexene18 and photocatalytic oxidation of bezylalcohol.19 To the best of our knowledge, there are only a few reports of high surface area mesoporous TiO2 materials composed of tiny titania nanoparticles with crystalline pore walls. In recent years, with the increasing environmental restrictions and the green development of industrial processes TiO2-supported nanocatalysts offer a wide scope.20 Many research studies are focusing on fabricating various morphologies of TiO2, such as nanotubes, porous nanofibers, metal doped TiO2, etc.21 Furthermore, TiO2 was found to be a good metal oxide catalyst support due to the strong metal support interaction, chemical stability, and acid–base property.21 Herein, we wish to demonstrate the synthesis of a new and highly stable ruthenium nanoparticles embedded on mesoporous TiO2 material and its excellent catalytic activity towards the esterification of levulinc acid with ethanol aiming for biofuel derived ethyl levulinate production and also in efficient and selective reduction of nitroarene mixed in the presence of styrene by using hydrazine as reducing agent at room temperature.
Experimental
Chemicals
Ruthenium trichloride hydrate (RuCl3·nH2O) and titanium(IV) isopropoxide [Ti(OC3H7)4] were purchased from Sigma-Aldrich. Hydrochloric acid (HCl) and sodium borohydrate (NaBH4) were obtained from E-Merck, India. Sodium dodecyl sulfate (SDS) was obtained from Spectrochem, and used as anionic structure-directing agent. All other chemicals were also purchased from E-Merck, India.
Characterization techniques
Powder X-ray diffraction (XRD) patterns of the samples were investigated with a Bruker D8 Advance X-ray diffractometer using Ni-filtered Cu Kα (λ = 0.15406 nm) radiation. Transmission electron microscopy (TEM) images of the nanocomposite were analyzed to determine the particle morphology, using a JEOL JEM 2010 transmission electron microscope operating at 200 kV. A Perkin-Elmer FT-IR 783 spectrophotometer was used to record the FT-IR spectra of the samples. UV-Vis spectra were carried out using a Shimadzu UV-2401PC doubled beam spectrophotometer having an integrating sphere attachment for solid samples. N2 adsorption/desorption isotherms were obtained by using a Quantachrome autosorb-1C at 77 K. Prior to gas adsorption, the sample was degassed for 4 h at 393 K under high vacuum conditions. Thermogravimetry (TG) analysis of the samples was taken in a TGA instruments thermal analyzer Mettler Toledo TGA/DTA 851e under N2 flow. A Varian 3400 gas chromatograph equipped with a 30 m CP-SIL 8CB capillary column and a flame ionization detector was used to analyze the reaction products and a Trace DSQ II GC-MS equipped with a 60 m TR-50 MS capillary column was used to identify the products.
Preparation of mesoporous TiO2 nanoparticles
Synthesis of mesoporous TiO2 was carried out according to the reported procedure.17 In this process, 20 mL distilled water was acidified with 35 wt% of hydrochloric acid (1.2 g). After that, 0.01 M titanium(IV) isopropoxide (2.84 g) was added slowly into the aqueous acidic solution under vigorous stirring. The stirring was continued until a clear solution was observed. Then, in a round bottom flask this clear solution was taken and allowed to stir for 20 h at 313 K. After 20 h of stirring, milky highly dispersed nanoparticles were obtained. 20 mL distilled water was taken in another beaker and 0.12 M SDS (0.345 g) was added. After that, the prepared titania nanoparticles were slowly added to the micellar solution of SDS. The whole solution was then allowed to stir for 2 h at room temperature. Finally, this solution was transferred in a round bottom flask and allowed to stir for 3 h at 353 K. After cooling the solution at room temperature centrifugation was done to separate out the self-assembled nanoparticles. Then this was dried and calcined at 773 K for 5 h in presence of air to get the template-free self assembled mesoporous titania nanoparticles.
Synthesis of colloidal ruthenium nanoparticles (Ru NPs)
Ruthenium NPs have been synthesized through the reduction of aqueous solution of ruthenium trichloride by NaBH4 as a reducing agent. The 0.2 mM RuCl3 (0.042 g) solution was dissolved in 50 mL distilled water under vigorous stirring. In another round bottom flask, 4 mM SDS (1.15 g) was dissolved into 50 mL of water with constant stirring followed by the addition of 6 mM NaBH4 (0.23 g). Then the solution of prepared RuCl3 was slowly added into the mixture solution of NaBH4 and SDS under continuous stirring for 1 h. Then the precipitates were collected by centrifugation and washed with double distilled water for several times, yielding colloidal ruthenium nanoparticles.
Synthesis of Ru@MTiO2 nanocatalyst
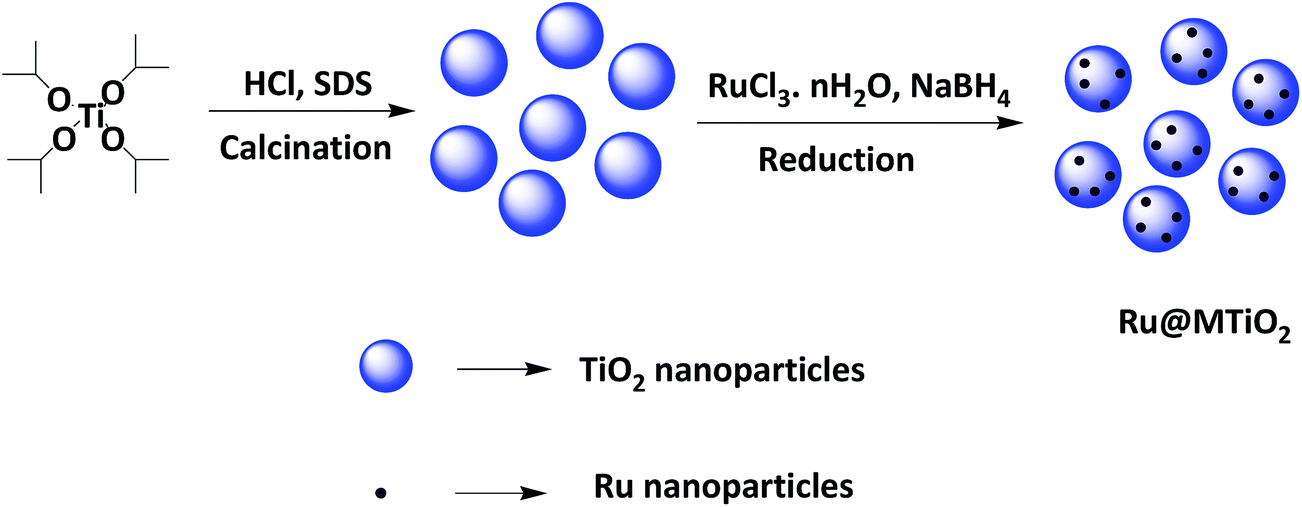
The prepared ruthenium NPs were dispersed into the methanol by sonication for 10 minutes. Then 1.0 g mesoporous TiO2 was added into the solution. The mixture was then stirred for 24 h at room temperature to obtain the homogeneous suspension. Finally, the mixture was washed with water and acetone, dried in oven and calcined at 300 °C for 6 h to obtain the Ru@MTiO2 nanocatalyst (Scheme 1).
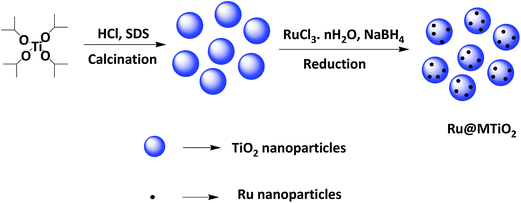 |
| | Scheme 1 Schematic diagram for the formation of Ru@MTiO2 nanocatalyst. | |
General experimental procedure for esterification of levulinic acid with alcohol using Ru@MTiO2 nanocatalyst
Esterification reaction was carried out in a 50 mL round bottom flask equipped with a reflux condenser. In a typical synthesis, levulinic acid (1.474 g, 12.7 mmol), methanol (4.1 mL, 101.6 mmol) and catalyst (40 mg) were heated to 338 K. The progress of the reaction was monitored by GC. After 8 h the solid catalysts were separated out from reaction mixture by centrifugation. Then, the obtained liquid was dried over anhydrous sodium sulphate and finally, the product was analyzed by GC-MS.
General experimental procedure for transfer-hydrogenation of nitroarenes over Ru@MTiO2 nanocatalyst
In a typical synthesis, nitrobenzene (0.5 mmol, 0.05 mL), solvent (2.0 mL) and catalyst (30 mg) were taken in a 50 mL round bottom flask. Then, hydrazine hydrate (5 mmol, 0.16 mL) was added into the reaction mixture. After completion of the reaction the catalyst was removed from the mixture by filtration. Then, the filtrate was washed with ethyl acetate and dried over anhydrous sodium sulphate. The residue obtained was purified by flash column chromatography on silica gel to afford the desired product, and then analyzed by GC-MS.
Results and discussion
Characterization of catalyst
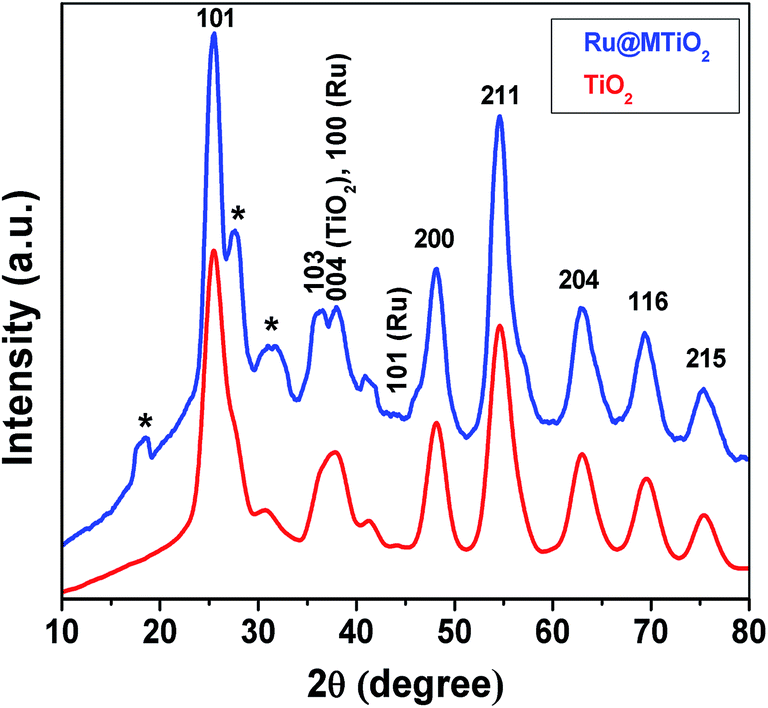
Powder XRD studies. The wide-angle powder X-ray diffraction patterns for mesoporous TiO2 and Ru@MTiO2 materials are shown in Fig. 1. The wide angle XRD patterns of the TiO2 nanoparticles showed highly crystalline planes corresponding to the anatase TiO2. The peaks at 2θ values of 25.35°, 37.77°, 48.20°, 54.65°, 62.92°, 69.52° and 75.40° agree well with the anatase (101), (004), (200), (211), (204), (116) and (215) crystal planes of TiO2 (JCPDS, CAS number 21-1272).22 Therefore, this powder XRD result indicates the successful preparation of the stable crystalline TiO2 nanoparticles. The comparison of the XRD patterns for TiO2 and Ru@MTiO2 samples shows that the characteristic diffraction peaks of titania anatase remains unchanged. This observation clearly suggests that the loss in the crystallinity or change in lattice framework of anatase titania after reduction of ruthenium(III) ions do not occurs. Furthermore, we observe two peaks for (100) and (101) crystal planes corresponding to hexagonal ruthenium NPs on titania anatase in Fig. 1 (JCPDS CAS number 89-4903).23
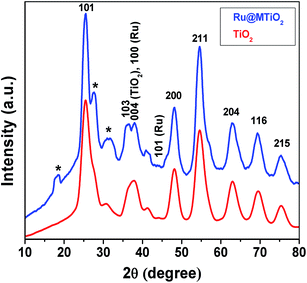 |
| | Fig. 1 Wide-angle powder XRD patterns of mesoporous TiO2 and Ru@MTiO2 nanomaterials. The asterisk sings indicate the presence of unreacted RuCl3. | |
In the Fig. 1, the peaks at 2θ values of 18.20°, 27.40° and 31.29° (indicated by asterisk signs) have come due to (011), (104) and (015) crystal planes of unreacted ruthenium trichloride, respectively (JCPDS CAS number 01-085-0528).
TEM analysis. The particle size and morphology of the samples are investigated by TEM analysis. The TEM images (Fig. 2A and B) showed the uniformly distribution of TiO2 particles of size in between 6 and 9 nm. The presence of ruthenium NPs are confirmed from EDX analysis (Fig. 2C).
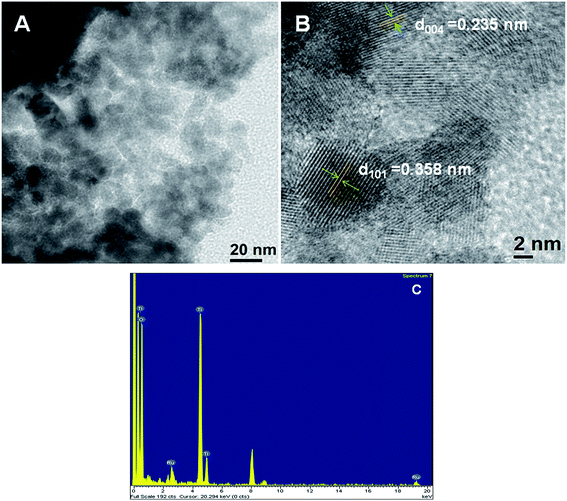 |
| | Fig. 2 TEM images (A and B) and EDX pattern (C) of Ru@MTiO2 nanocatalyst. | |
The loading Ru over TiO2 is around 0.1637 mmol g−1 from ICP-AES measurement. ICP-AES measurement has also been carried out for the reused catalyst after five consecutive catalytic cycles. It is found that, after the five catalytic cycles the Ru metal loading on TiO2 is around 0.1550 mmol g−1.
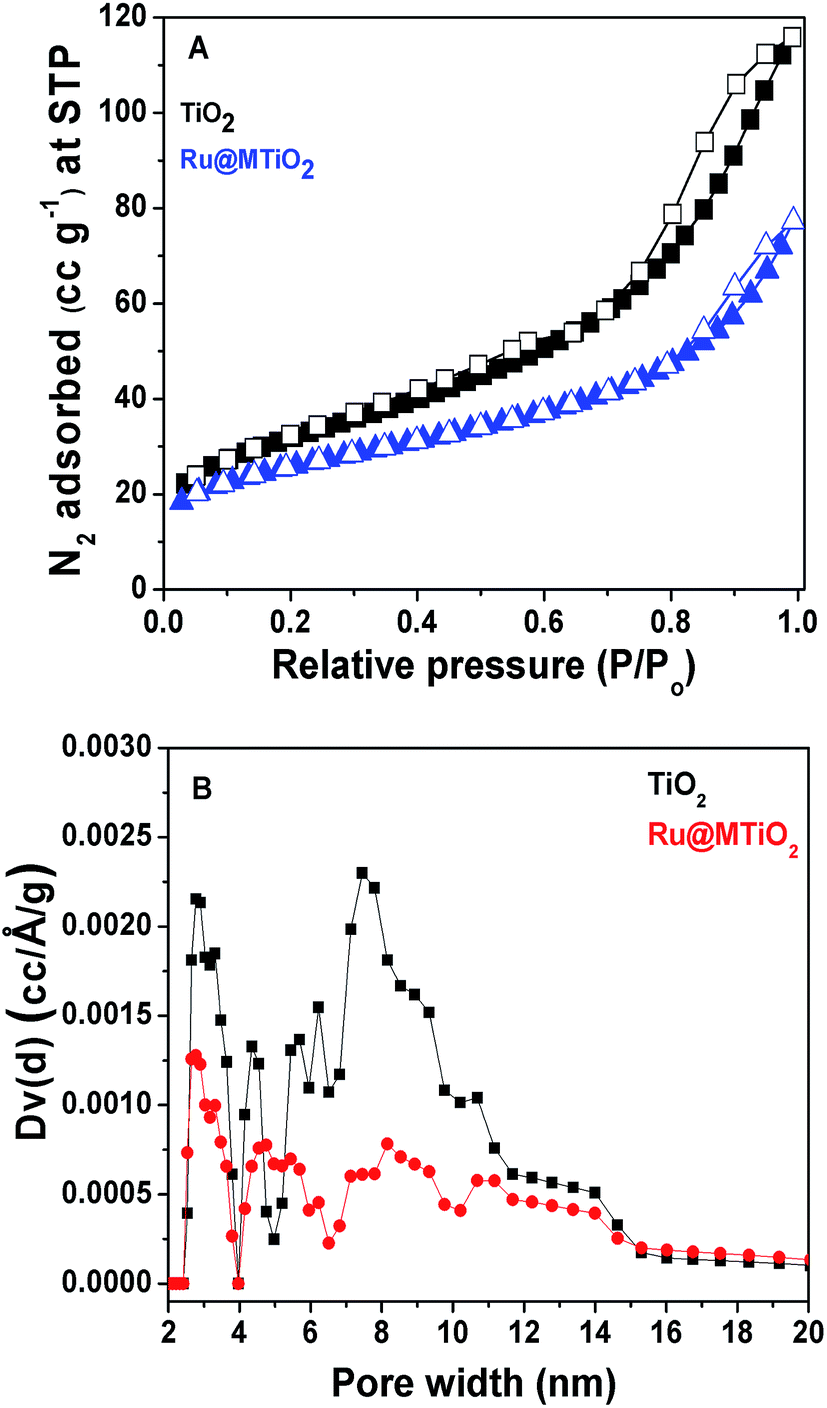
N2 adsorption–desorption analysis. The nitrogen adsorption–desorption isotherms of calcined TiO2 and Ru@MTiO2 materials describe the porosity of these materials, which is shown in Fig. 3. The BET (Brunauer–Emmett–Teller) surface areas of calcined TiO2 and Ru@MTiO2 materials are 113 m2 g−1 and 92 m2 g−1, respectively. Both isotherms can be classified as type IV isotherm corresponding to the mesoporous nature and these isotherms display H2 type hysteresis loops.22 The decrease in surface area of Ru@MTiO2 than that of calcined TiO2 indicates the successful grafting of ruthenium nanoparticles at the surface of the mesopores TiO2 material. The pore size distribution is measured by non-local density functional theory (NLDFT) method. The total pore volumes of calcined TiO2 and Ru@MTiO2 materials are 0.1796 cm3 g−1 and 0.1196 cm3 g−1, respectively.
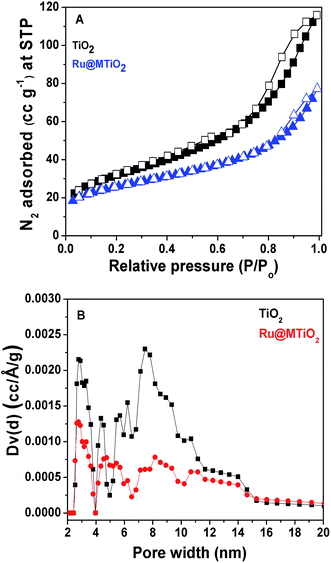 |
| | Fig. 3 N2 adsorption–desorption isotherms of TiO2 and Ru@MTiO2 materials (A) and the pore size distributions of TiO2 and Ru@MTiO2 materials (B). | |
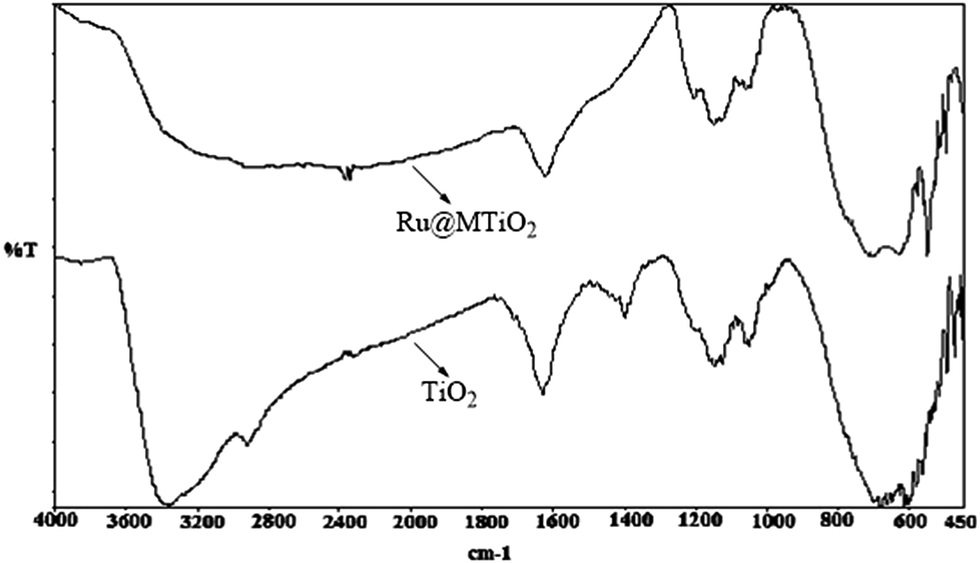
FTIR spectroscopy. The FT IR spectra of mesoporous titania and Ru@MTiO2 is shown in Fig. 4. Mesoporous titania showed a broad peak at 3300 to 3400 cm−1 which could be assigned due to the O–H stretching vibrations of the residual water present in the mesoporous titania. The mesoporous titania also shows a broad band in the 450–800 cm−1 region confirming the anatase phase of TiO2.24 The IR spectra of Ru@MTiO2 shows small shift towards lower wave number of these peaks, this may be due the interaction of ruthenium nanoparticle with anatase TiO2. Further, we have added UV-visible absorption spectroscopic data (ESI: Fig. S1†) to know the effect of the loading of Ru NPs on TiO2 matrix and TGA analysis (ESI: Fig. S2†) to understand the thermal stability of our Ru@MTiO2 catalyst.
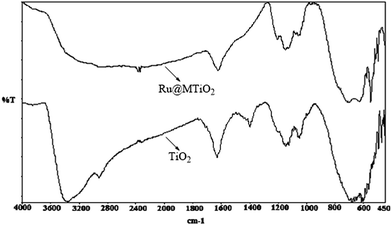 |
| | Fig. 4 FTIR spectra of mesoporous TiO2 and Ru@MTiO2 nanomaterials. | |
Catalytic activity
Recently, significant interest has been paid on designing the nanostructured materials of definite size and shape because of their various potential applications as catalysts, sensors, and optoelectronics and so on. Mesoporous materials have great demand now a day due to its high surface area, uniform pore size distribution and high catalytic activity.25 So, we have explored the catalytic activity of the mesoporous Ru@MTiO2 in the esterification of biomass derived levulinic acid and selective transfer hydrogenation reactions of nitroarenes under mild conditions.
Esterification of levulinic acid using alcohol catalyze by Ru@MTiO2 nanomaterials
The esterification of levulinic acid (LA) is an important chemical transformation to produce the biobased chemicals using environmentally friendly methods. Biofuel derived levulinate ester is widely used as a fuel in oil and gas industry, additive in some foods and fragrance in cosmetics. To explore the catalytic activity of Ru@MTiO2 nanocatalyst, esterification of levulinic acid with methanol was chosen initially as a model reaction (Scheme 2).
 |
| | Scheme 2 Esterification of levulinic acid with methanol. | |
Optimization of reaction parameters
Effect of catalyst loading. The effect of catalyst loading on LA esterification has been studied to optimize the reaction conditions. The catalyst loading was varied from 10 to 60 mg at a temperature of 338 K, reaction time of 8 h and methanol to LA molar ratio 8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. With increasing of catalyst loading overall conversion was also increased from 45 to 95% (Fig. 5). In this case, catalyst loading of 40 mg is observed to be optimum. Fig. 5 illustrated the effect of catalyst loading on LA conversion. Above 40 mg catalyst loading, the almost steady LA conversion is observed, suggested this optimum loading of Ru-catalyst necessary for the esterification reaction.
:1. With increasing of catalyst loading overall conversion was also increased from 45 to 95% (Fig. 5). In this case, catalyst loading of 40 mg is observed to be optimum. Fig. 5 illustrated the effect of catalyst loading on LA conversion. Above 40 mg catalyst loading, the almost steady LA conversion is observed, suggested this optimum loading of Ru-catalyst necessary for the esterification reaction.
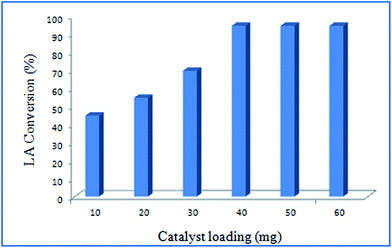 |
| | Fig. 5 Effect of catalysis loading at reaction conditions: solvent to LA ratio: 8:1, temperature: 338 K, reaction time: 8 h. | |
Effect of reaction temperature. The effect of temperature on LA esterification was studied. The reaction temperature was varied from 298 K to 378 K. The maximum conversion (95%) was obtained at 338 K, after 8 h using 40 mg catalyst and methanol to LA molar ratio 8:1. Fig. 6 displayed the effect of reaction temperature on LA conversion.
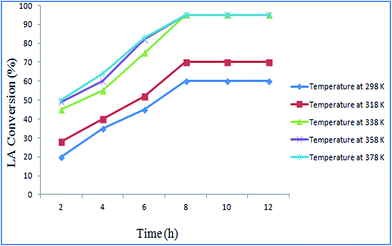 |
| | Fig. 6 Effect of temperature at reaction conditions: solvent to LA ratio: 8:1, catalyst: 40 mg, reaction time: 8 h. | |
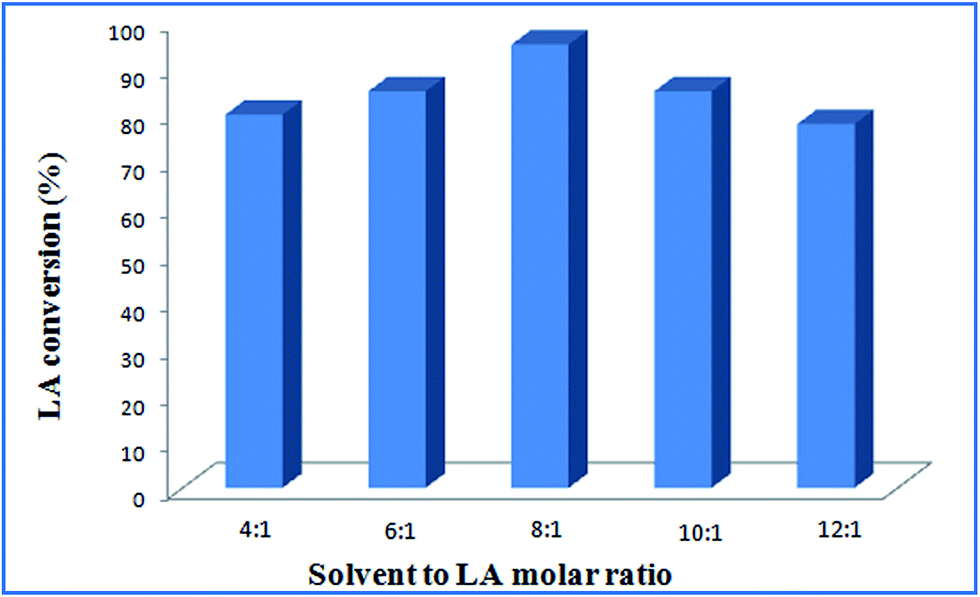
Effect of molar ratio of solvent to levulinic acid. The molar ratio of solvent to levulinic acid on LA esterification was investigated. The methanol to LA molar ratio was varied between 4:1 and 10:1 over Ru@MTiO2 catalyst at 338 K for 8 h using 40 mg catalyst. Fig. 7 illustrated the effect of molar ratio of solvent on LA conversion. LA conversion increased with increase in ethanol to LA molar ratio from 4:1 to 8:1. Fig. 7 showed that the maximum conversion of 95% was observed when methanol to LA molar ratio was 8:1. However, further increase in molar ratio to 12:1, LA conversion was decreased from 95% to 78%. This is because of mass transfer limitation, as more methanols may dilute the reactant and lowering LA conversion. In present case, molar ratio of methanol to LA 8:1 is observed to be optimum.
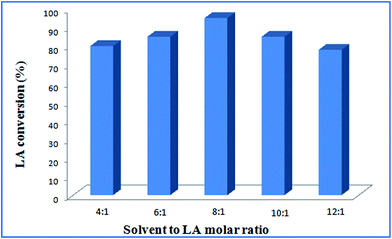 |
| | Fig. 7 Effect of solvent to LA molar ratio at reaction conditions: catalyst: 40 mg, temperature: 338 K, reaction time: 8 h. | |
Esterification of levulinic acid over Ru@MTiO2
To explore the activity of Ru@MTiO2 catalyst for the esterification of LA in the presence of different alcohols, methanol, ethanol, iso-propanol and 2-butanol have also been investigated. As seen from Table 1, conversions of levulinic acid to corresponding alkyl levulinate in the presence of different alcohols were studied under optimized reaction conditions. Esterification of levulinic acid with methanol and ethanol afforded the methyl levulinate and ethyl levulinate in high yields of 94% and 95% respectively (Table 1, entry 1 and 2). However, alcohols with bulkier groups slowed down the LA esterification. Thus, iso-propanol and 2-butanol gave the esterification product in 85% and 80% respectively (Table 1, entry 3 and 4).
Table 1 Levulinic acid conversion using different solvents over Ru@MTiO2 nanomateriala
| Entry |
Reactants |
Solvents |
Conversionb (%) |
| Reaction conditions: solvent to LA ratio: 8:1, temperature: 338 K, catalyst: 40 mg, reaction time: 8 h. The products were confirmed by GC-MS, the yields was determined by GC. 1H NMR spectra of methyl and ethyl levulinates are provided in the ESI. |
| 1 |
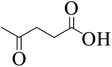 |
Methanol |
95 |
| 2 |
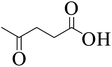 |
Ethanol |
94 |
| 3 |
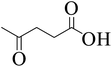 |
Iso-propanol |
85 |
| 4 |
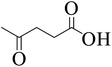 |
2-Butanol |
80 |
Esterification of levulinic acid over different TiO2 and Ru@TiO2 materials
We have performed the esterification of levulinic acid with methanol in the presence of titanium dioxide (TiO2) and Ru@TiO2 as the catalysts and the results are summarized in Table 2. When we have carried out the esterification of levulinic acid by using bulk (TiO2 and Ru@TiO2) and mesoporous TiO2 catalysts, then the conversion of the desired product are very low compared to the mesoporous Ru@MTiO2 material (Table 2, entry 4). High catalytic activity of mesoporous Ru@MTiO2 material for the production of methyl levulinate in high yields as seen from the Table 2 could be attributed to its high surface area, porosity and self-assembled nanostructure.
Table 2 Levulinic acid conversion to methyl levulinate in presence of different catalystsa
| Entry |
Catalyst |
Conversionb (%) |
| Reaction conditions: methanol to LA ratio: 8:1, temperature: 338 K, catalyst: 40 mg, reaction time: 8 h. The products were confirmed by GC-MS, the yields was determined by GC. |
| 1 |
Bulk TiO2 |
30 |
| 2 |
Bulk Ru@TiO2 |
38 |
| 3 |
Mesoporous TiO2 |
55 |
| 4 |
Mesoporous Ru@MTiO2 |
95 |
| 5 |
Without catalyst |
No reaction |
Transfer hydrogenation of nitrobenzene over Ru@MTiO2 nanomaterial. Catalytic activity of Ru@MTiO2 nanocatalyst was tested in the selective transfer hydrogenation of various nitroarenes. For this purpose, we have choosen transfer hydrogenation of nitrobenzene in the presence of styrene as model reaction (Scheme 3) where, nitrobenzenes only selectively reduces to the corresponding anilines.
 |
| | Scheme 3 Selective transfer hydrogenation of nitrobenzene mixed with styrene. | |
Selective transfer hydrogenation of nitrobenzene in the presence of styrene has been carried out to understand the optimized reaction conditions over Ru@MTiO2 nanocatalyst. The reaction is governed by number of factors such as solvent, amount of catalyst, reaction time. Firstly, transfer hydrogenation of nitrobenzene with different solvents such as MeOH, EtOH, H2O, CH3CN and PEG-400 were investigated. All the results are summarized in Table 2. In the presence of MeOH at room temperature, after 2 h >99% aniline (Table 2, entry 1) was formed. Using ethanol as solvent 98% product (Table 3, entry 2) was obtained after 3 h of reaction. When water, CH3CN and PEG-400 were used as solvent the yield of anilines were 95%, 90% and 88% respectively (Table 3, entries 3–5) but it took longer time to give the desired reduced product. Therefore, the best result was obtained when MeOH was used as solvent at room temperature under atmospheric pressure. The effect of catalyst amount for the transfer hydrogenation of nitrobenzene was also investigated. It was observed that using 30 mg of catalyst gave the higher product (Table 3, entry 1). When we decrease the catalyst amount from 20 mg to 10 mg lower conversion of aniline was obtained (Table 3, entries 6, 7). Using mesoporous TiO2 as catalyst only 35% yield was observed after 12 h. Without using the catalyst 10% anilines was observed (Table 3, entry 9). So, the best result observed for the transfer hydrogenation of nitrobenzene when MeOH was used as solvent for 2 h reaction using 30 mg Ru@MTiO2 catalyst at room temperature.
Table 3 Optimization of reaction conditions for the selective transfer hydrogenation of nitrobenzene using Ru@MTiO2 nanomateriala
| Entry |
Catalyst (mg) |
Solvent |
Reaction time (h) |
Yieldb (%) |
| Reaction conditions: nitrobenzene (0.5 mmol), styrene (0.5 mmol), 30 mg catalyst, solvent (2 mL) and N2H4 (5 mmol), at room temperature. The products were confirmed by GC-MS and the yields were determined by GC. |
| 1 |
Ru@MTiO2 (30) |
MeOH |
2 |
>99 |
| 2 |
Ru@MTiO2 (30) |
EtOH |
3 |
98 |
| 3 |
Ru@MTiO2 (30) |
H2O |
7 |
95 |
| 4 |
Ru@MTiO2 (30) |
CH3CN |
10 |
90 |
| 5 |
Ru@MTiO2 (30) |
PEG-400 |
12 |
88 |
| 6 |
Ru@MTiO2 (20) |
MeOH |
2 |
75 |
| 7 |
Ru@MTiO2 (10) |
MeOH |
2 |
58 |
| 8 |
MTiO2 (50) |
MeOH |
12 |
35 |
| 9 |
— |
MeOH |
12 |
10 |
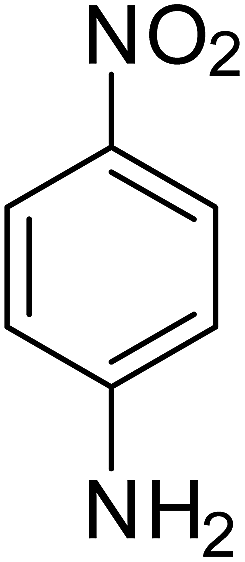
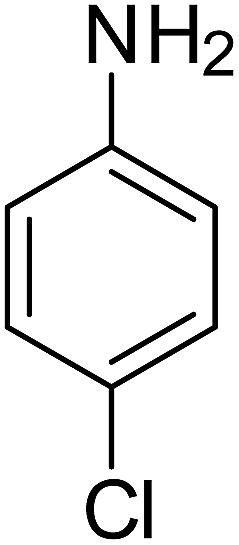
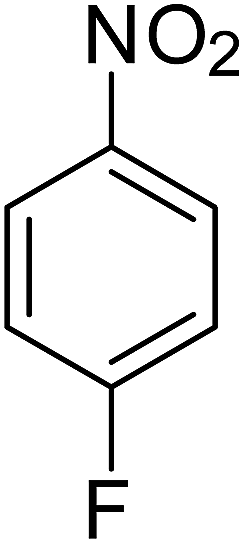
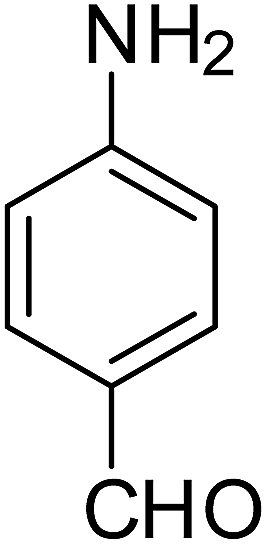
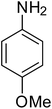
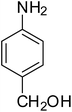
After optimization of the reaction conditions, selective transfer hydrogenation of other nitroarenes were carried out by using Ru@MTiO2 nanocatalyst and MeOH at room temperature under air. It was seen from the Table 4 that, the transfer hydrogenation of nitroarenes took place very smoothly and chemoselectively to produce the desired anilines in high yields. Nitrobenzene gave the aniline in high yield of >99% (Table 4, entry 1). Substituents like –CH3, –OCH3, –CH2OH, –OH were successfully reduced to obtain the product in good yield (Table 4, entries 2–5). The nitroaniline reduction produced phenyl diamine in 90% yield (Table 4, entry 6). The reduction of halogen-substituted nitrobenzenes afforded the corresponding amines in high yields 91–95% (Table 2, entries 7 and 8). Nitrobenzaldehyde and nitrobenzoic acid gave the respective amines in 90–94% yield without reducing the –CHO and –COOH groups (Table 4, entries 9 and 10). This result suggested that Ru@MTiO2 is highly selective in this transfer hydrogenation reaction.
Table 4 Transfer hydrogenation of nitrobenzene catalysed by Ru@MTiO2 nanomateriala
| Entry |
Nitroarene |
Product |
Yieldb (%) |
| Reaction conditions: nitroarene (0.5 mmol), N2H4 (5 mmol), 30 mg catalyst and MeOH (2 mL) at room temperature for 2 h. The products were confirmed by GC-MS, the yields were determined by GC. |
| 1 |
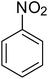 |
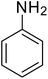 |
>99 |
| 2 |
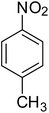 |
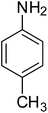 |
95 |
| 3 |
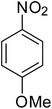 |
 |
92 |
| 4 |
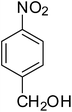 |
 |
92 |
| 5 |
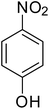 |
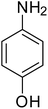 |
88 |
| 6 |
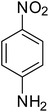 |
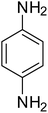 |
90 |
| 7 |
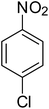 |
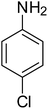 |
95 |
| 8 |
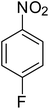 |
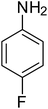 |
91 |
| 9 |
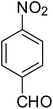 |
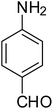 |
94 |
| 10 |
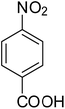 |
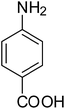 |
90 |
Recyclability of Ru@MTiO2 nanomaterial
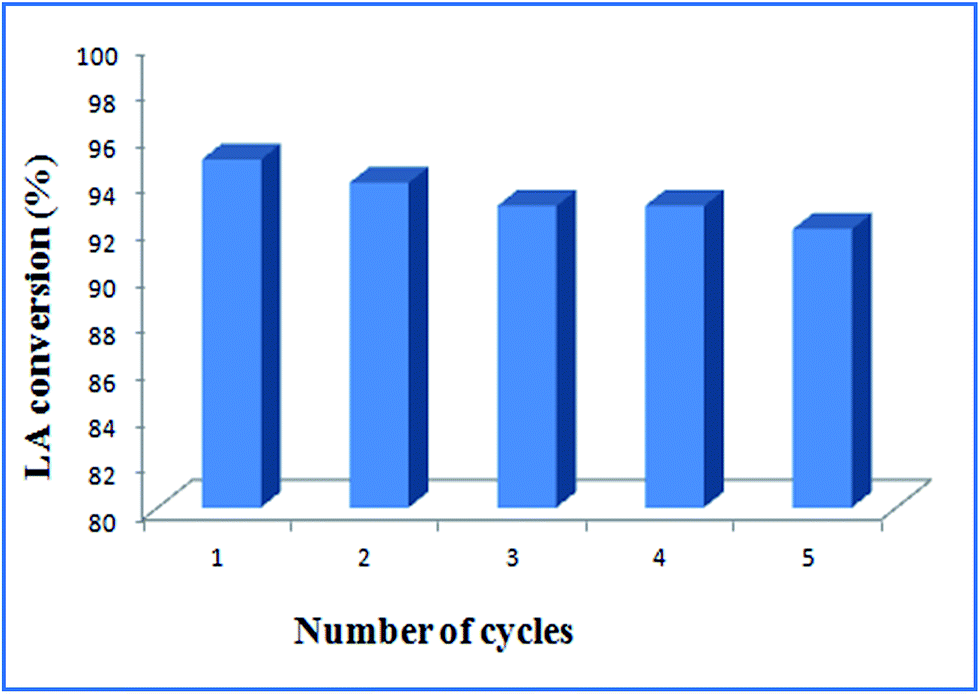
The recycling-efficiency of ruthenium nanocatalyst has been investigated to understand its potential for reuse as this is one of the most advantageous features for a heterogeneous catalyst. For this purpose, esterification of levulinic acid with methanol has been carried out over Ru@MTiO2 catalyst under optimized reaction conditions. After the completion of the reaction, solid catalyst was separated by centrifugation, washed with water and acetone. Then dried in 100 °C and used in the next catalytic run. As seen from Fig. 8 that the catalyst can be efficiently recycled and reused for five times without any appreciable loss in its product yields.
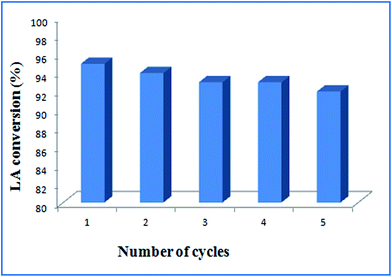 |
| | Fig. 8 Recycling efficiency of Ru@MTiO2 in the esterification of levulinic acid. | |
Heterogeneity test
Hot filtration test. An experiment for heterogeneity test was carried out in the esterification of levulinic acid with methanol with our Ru@MTiO2 nanocatalyst. The catalyst was filtered from the reaction mixture after 3 h reaction at 65 °C and the filtrate was allowed to react further. We observed that no change in the conversion of levulinic acid after 6 h reaction time. This result suggests that our Ru@MTiO2 catalyst is heterogeneous in nature. It is also observed that ruthenium was not leached out or decomposed from the mesoporous support during the catalytic reaction as no ruthenium was observed in the filtrate of the reaction mixture in the atomic absorption spectrometric analysis. Thus, we can conclude from this hot filtration test that ruthenium nanoparticles are strongly bound at the mesoporous TiO2 support and Ru@MTiO2 is a robust catalyst for carrying out liquid phase catalytic reactions.
Comparison of catalytic activity of Ru@MTiO2 for the esterification of levulinic acid with other reported systems
We have studied the esterification of levulinic acid with alcohols over a variety of catalysts. In Table 5, a comparative results for the esterification of levulinic acid is shown for different catalysts to understand the catalytic activity of our present catalytic system with those reported in the literature.26–29 It is clear from the Table 5, that our Ru-catalyst showed higher yields in the esterification reaction compared to the other reported systems.26–29
Table 5 Catalytic conversion of levulinic acid with other reported catalysts
| Catalysts |
Reaction conditions |
LA conversion (%) |
Reference |
| 20 wt% H4SiW12O40–SiO2 |
Levulinic acid: 205 mg, ethanol: 2 mL, cyclohexane: 5 mL, temp: 75 °C, time: 6 h, catalyst: 104 mg |
75 |
26 |
| 15% (w/w) DTPA/DH-ZSM-597 |
LA to ethanol molar ratio: 1:8, catalyst to LA ratio: 0.25, temperature: 351 K, reaction time: 4 h |
94 |
27 |
| H/BEA zeolite |
LA to ethanol molar ratio = 1:6, temp: 78 °C, time: 5 h, catalyst loading: 20 wt% of LA |
40 |
28 |
| 40-WD-S |
LA: 2 mmol, ethanol: 128 mmol, temp: 78 °C, time: 10 h, catalyst: 0.01 mmol |
76 |
29 |
| Ru@MTiO2 nanomaterial |
LA to ethanol molar ratio: 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :8, temp: 338 K, time: 8 h, catalyst loading: 40 mg :8, temp: 338 K, time: 8 h, catalyst loading: 40 mg |
95 |
This work |
Comparison of catalytic activity of Ru@MTiO2 for the transfer hydrogenation of nitrobenzene with other reported system
Table 6 showed the comparison data for the transfer hydrogenation of nitrobenzene with our catalyst and other reported catalytic system.30–33 The catalytic activity of our catalyst showed higher conversion than other reported catalysts in the transfer hydrogenation of nitrobenzene.
Table 6 Catalytic conversion of transfer hydrogenation with other reported catalysts
| Catalysts |
Reaction conditions |
Nitrobenzene conversion (%) |
Reference |
| Cu(Oac)2 |
Nitrobenzene (1 mmol), ethylene glycol (10 mL), N2H4 (5 mmol), catalyst (50 mg), 125 °C, 3.5 min, CMUI |
97 |
30 |
| Zinc phthalocyanine |
Nitrobenzene (5 mmol), PEG 400, N2H4 (4 equiv.), catalyst (1 mol%), 100 °C, 8 h |
96 |
31 |
| Ru0–montmorillonite clay |
Nitrobenzene (0.2 mmol), NaOH (0.5 mmol), isopropanol (20 mL), catalyst (50 mg), 85 °C, 12 h |
97 |
32 |
| Iron(0) nanoparticles |
Nitrobenzene (1 mmol), water, catalyst (3 mmol), RT, 2–3 h |
95 |
33 |
| Ru@MTiO2 nanocatalyst |
Nitrobenzene (0.5 mmol), MeOH (2 mL), N2H4 (5 mmol), catalyst (30 mg), RT, 2 h |
99 |
This work |
Conclusions
In summary, we can conclude that a new Ru-nanocatalyst has been prepared by grafting of Ru NPs on the surface of mesoporous TiO2. This Ru-nanocatalyst has been successfully used as a highly efficient recyclable heterogeneous catalytic system for the esterification of levulinic acid and transfer hydrogenation of nitroarenes under mild reaction conditions. Furthermore, the Ru@MTiO2 nanomaterial could be easily separated from the reaction mixture by simple filtration and reused several times with minimal loss of its activity and selectivity. The catalytic process is green, economically advantageous and environment friendly. Thus, green and cost effective bifunctional catalytic pathways reported over mesoporous Ru@MTiO2 nanomaterial suggested a sustainable future and large scale application for the synthesis of a wide range of value added fine chemicals.
Acknowledgements
S. M. I. acknowledges the Department of Science and Technology (DST-SERB), New Delhi, India, University Grant Commission (UGC), New Delhi, India, Department of Science and Technology, West Bengal (DST-W.B) for funding. UM is thankful to the UGC, New Delhi and SKK is also thankful to the CSIR, New Delhi, India for their senior research fellowship. NS acknowledges the Science and Engineering Research Board (SERB), DST, Govt. Of India, for his National Post-Doctoral Fellowship Scheme (File No. PDF/2015/000460). AB wishes to thank DST SERB for funding. AB wishes to thanks DST for funding through DST-UKIERI research grant. We gratefully acknowledge the DST and UGC, New Delhi, Govt. of India, for award of grant under FIST, PURSE and SAP program to the Department of Chemistry, University of Kalyani.
Notes and references
- R. Luque, L. Herrero-Davila, J. M. Campelo, J. H. Clark, J. M. Hidalgo, D. Luna, J. M. Marinas and A. A. Romero, Energy Environ. Sci., 2008, 1, 542–564 CAS.
- D. W. Rackemann and W. O. S. Doherty, Biofuels, Bioprod. Biorefin., 2011, 5, 198–214 CrossRef CAS.
-
(a) J. J. Bozell, Science, 2010, 329, 522–523 CrossRef CAS PubMed;
(b) Z. Li, R. Wnetrzak, W. Kwapinski and J. J. Leahy, ACS Appl. Mater. Interfaces, 2012, 4, 4499–4505 CrossRef CAS PubMed.
-
(a) C. Chang, G. Xu and X. Jiang, Bioresour. Technol., 2012, 121, 93–99 CrossRef CAS PubMed;
(b) D. R. Fernandes, A. S. Rocha, E. F. Mai, C. J. A. Mota and V. T. da Silva, Appl. Catal., A, 2012, 425–426, 199–204 CrossRef CAS;
(c) P. Sun, G. Gao, Z. Zhao, C. Xia and F. Li, ACS Catal., 2014, 4, 4136–4142 CrossRef CAS.
-
(a) R. S. Downing, P. J. Kunkeler and H. Bekkum, Catal. Today, 1997, 37, 121 CrossRef CAS;
(b) N. Ono, The Nitro Group in Organic Synthesis, Wiley-VCH, New York, 2001 CrossRef.
-
(a) A. Corma, P. Concepción and P. Serna, Angew. Chem., Int. Ed., 2007, 46, 7266 CrossRef CAS PubMed;
(b) T. Joseph, K. V. Kumar, A. V. Ramaswamy and S. B. Halligudi, Catal. Commun., 2007, 8, 629 CrossRef CAS.
-
(a) Y. Liu, Y. Lu, M. Prashad, O. Repič and T. J. Blacklock, Adv. Synth. Catal., 2005, 347, 217 CrossRef CAS;
(b) R. Dhanda and M. Kidwai, J. Mater. Chem. A, 2015, 3, 19563–19574 RSC;
(c) R. V. Jagadeesh, D. Banerjee, P. B. Arockiam, H. Junge, K. Junge, M. M. Pohl, J. Radnik, A. Brückner and M. Beller, Chem. Commun., 2011, 47, 3601–3603 RSC;
(d) W. Gao, W. Li, Z. Xue, M. Pal, Y. Liu, C. Wang, J. Wang, S. Wang, X. Wan, Y. Liu and D. Zhao, New J. Chem., 2016, 40, 4200–4205 RSC;
(e) D. Cantillo, M. Mirhosseini Moghaddam and C. Oliver Kappe, J. Org. Chem., 2013, 78, 4530–4542 CrossRef CAS PubMed;
(f) P. P. Sarmah and D. P. Dutta, Green Chem., 2012, 1086–1093 RSC.
- A. Furst, R. C. Berlo and S. Hooton, Chem. Rev., 1965, 65, 51–68 CrossRef CAS.
-
(a) K. Mori and H. Yamashita, Phys. Chem. Chem. Phys., 2010, 12, 14420–14432 RSC;
(b) Nanoparticles, ed. G. Schmidt, Wiley-VCH, Weinheim, 2004 Search PubMed.
-
(a) R. Narayanan and M. El-Sayed, Top. Catal., 2008, 47, 15–21 CrossRef CAS;
(b) P. Puthiaraj and W. S. Ahn, Catal. Sci. Technol., 2016, 6, 1701–1709 RSC;
(c) Y. Yamada, S. Shikano, T. Akita and S. Fukuzumi, Catal. Sci. Technol., 2015, 979–988 RSC.
-
(a) X. Liu, C. G. Meng and Y. Han, Nanoscale, 2012, 4, 2288–2295 RSC;
(b) M. A. Soria, C. Mateos-Pedrero, I. Rodriguez-Ramos and A. Guerrero-Ruiz, Catal. Today, 2011, 171, 126–131 CrossRef CAS;
(c) S. Anantharaj, M. Jayachandran and S. Kundu, Chem. Sci., 2016, 7, 3188–3205 RSC.
- A. X. Yin, W. C. Liu, J. Ke, W. Zhu, J. Gu, Y. W. Zhang and C. H. Yan, J. Am. Chem. Soc., 2012, 134, 20479–20489 CrossRef CAS PubMed.
-
(a) X. M. Yang, X. N. Wang and J. S. Qiu, Appl. Catal., A, 2010, 382, 131–137 CrossRef CAS;
(b) M. Gopiraman, S. G. Babu, Z. Khatri, W. Kai, Y. A. Kim, M. Endo, R. Karvembu and I. S. Kim, J. Phys. Chem. C, 2013, 117, 23582–23596 CrossRef CAS;
(c) Y. H. Kim, S. K. Hwang, J. W. Kim and Y. S. Lee, Ind. Eng. Chem. Res., 2014, 53, 12548–12552 CrossRef CAS;
(d) P. Kumar, S. Varma and S. L. Jain, J. Mater. Chem. A, 2014, 2, 4514–4519 RSC;
(e) S. H. Lee and D. J. Moon, Catal. Today, 2011, 174, 10–16 CrossRef CAS;
(f) P. P. Sarmah and D. K. Dutta, Green Chem., 2012, 14, 1086 RSC;
(g) N. Salam, S. K. Kundu, A. S. Roy, P. Mondal, K. Ghosh, A. Bhaumik and S. M. Islam, Dalton Trans., 2014, 43, 7057–7068 RSC;
(h) W. H. Luo, U. Deka, A. M. Beale, E. R. H. van Eck, P. C. A. Bruijnincx and B. M. Weckhuysen, J. Catal., 2013, 301, 175 CrossRef CAS.
-
(a) G. M. Dhar, B. N. Srinivas, M. S. Rana, M. Kumar and S. K. Maity, Catal. Today, 2003, 86, 45 CrossRef CAS;
(b) M. Chen and D. W. Goodman, Chem. Soc. Rev., 2008, 37, 1860 RSC;
(c) M. Chen and D. W. Goodman, Chem. Soc. Rev., 2008, 37, 1860 RSC;
(d) B. D. Chandler, S. Kendell, H. Doan, R. Korkosz, L. C. Grabow and C. J. Pursell, ACS Catal., 2012, 2, 684–694 CrossRef CAS.
- A. Dhakshinamoorthy and H. Garcia, Chem. Soc. Rev., 2012, 41, 5262–5284 RSC.
- H. Can and O. Metin, Appl. Catal., B, 2012, 125, 304–310 CrossRef CAS.
- J. H. Yang, H. Piao, A. Vinu, A. A. Elzatahry, S. M. Paek and J. H. Choy, RSC Adv., 2015, 5, 8210–8215 RSC.
- G. B. Zhou, R. F. Dou, H. Z. Bi, S. H. Xie, Y. Pei, K. N. Fan, M. H. Qiao, B. Sun and B. N. Zong, J. Catal., 2015, 332, 119–126 CrossRef CAS.
- J. Tian, J. Li, N. Wei, X. H. Xu, H. Z. Cui and H. Liu, Ceram. Int., 2016, 42, 1611–1617 CrossRef CAS.
-
(a) X. Liu, Y. Chen, C. Cao, J. Xu, Q. Qian, Y. Luo, H. Xue, L. Xiao, Y. Chenac and Q. Chen, New J. Chem., 2015, 39, 6944–6950 RSC;
(b) I. Fechete, Y. Wang and J. C. Védrine, Catal. Today, 2012, 189, 2 CrossRef CAS.
-
(a) L. Lang, D. Wu and Z. Xu, Chem.–Eur. J., 2012, 18, 10661–10668 CrossRef CAS PubMed;
(b) J. Li, H. Qiao, Y. Du, C. Chen, X. Li, J. Cui, D. Kumar and Q. Wei, Sci. World J., 2012, 2012, 154939 Search PubMed;
(c) A. Mezzi, S. Kaciulis, I. Cacciotti, A. Bianco, G. Gusmano, F. R. Lamastra and M. E. Fragalà, Surf. Interface Anal., 2010, 42, 572–575 CrossRef CAS;
(d) C. Zhang, Y. Li, Y. Wang and H. He, Environ. Sci. Technol., 2014, 48, 5816–5822 CrossRef CAS PubMed;
(e) B. D. Chandler, S. Kendell, H. Doan, R. Korkosz, L. C. Grabow and C. J. Pursell, ACS Catal., 2012, 2, 684–694 CrossRef CAS;
(f) P. Kumar, S. Varma and S. L. Jain, J. Mater. Chem. A, 2014, 2, 4514–4519 RSC.
- S. K. Das, M. K. Bhunia and A. Bhaumik, Dalton Trans., 2010, 39, 4382–4390 RSC.
- R. B. N. Baig, M. N. Nadagouda and R. S. Varma, Green Chem., 2014, 16, 2122–2127 RSC.
- K. Bhattacharyya, S. Varma, A. K. Tripathi, S. R. Bharadwaj and A. K. Tyagi, J. Phys. Chem. C, 2008, 112, 19102–19112 CAS.
-
(a) U. Mandi, A. S. Roy, B. Banerjee and S. M. Islam, RSC Adv., 2014, 4, 42670–42681 RSC;
(b) U. Mandi, M. Pramanik, A. S. Roy, N. Salam, A. Bhaumik and S. M. Islam, RSC Adv., 2014, 4, 15431–15440 RSC;
(c) K. Ghosh, M. A. Iqubal, R. A. Molla, A. Mishra, Kamaluddin and S. M. Islam, Catal. Sci. Technol., 2015, 5, 1606–1622 CAS;
(d) N. Salam, S. K. Kundu, A. S. Roy, P. Mondal, K. Ghosh, A. Bhaumik and S. M. Islam, Dalton Trans., 2014, 43, 7057–7068 RSC;
(e) S. Mondal, J. Mondal and A. Bhaumik, ChemCatChem, 2015, 7, 3570–3578 CrossRef CAS.
- K. Yan, G. Wu, J. Wen and A. Chen, Catal. Commun., 2013, 34, 58–63 CrossRef CAS.
- K. Y. Nandiwale, S. K. Sonar, P. S. Niphadkar, P. N. Joshi, S. S. Deshpande, V. S. Patil and V. V. Bokade, Appl. Catal., A, 2013, 460–461, 90–98 CrossRef CAS.
- C. R. Patil, P. S. Niphadkar, V. V. Bokade and P. N. Joshi, Catal. Commun., 2014, 43, 188–191 CrossRef CAS.
- G. Pasquale, P. Vazquez, G. Romanelli and G. Baronetti, Catal. Commun., 2012, 18, 115–120 CrossRef CAS.
- H. Feng, Y. Li, S. Lin, E. V. V. Eycken and G. Song, Sustainable Chem. Processes, 2014, 2, 14 CrossRef.
- U. Sharma, N. Kumar, P. K. Verma, V. Kumar and B. Singh, Green Chem., 2012, 14, 2289–2293 RSC.
- P. P. Sarmah and D. K. Dutta, Green Chem., 2012, 14, 1086–1093 RSC.
- R. Dey, N. Mukherjee, S. Ahammed and B. C. Ranu, Chem. Commun., 2012, 48, 7882–7984 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra10233f |
|
| This journal is © The Royal Society of Chemistry 2016 |