
Lysosome-targeting solid state NIR emissive donor–acceptor molecules: a study on photophysical modulation through architectural distinction†
Ashish Kumar
Kushwaha
a,
Ankit Kumar
Srivastava
a,
Pradeep
Kumar
b,
Anjani
Kumar
a,
Saripella
Srikrishna
b and
Roop Shikha
Singh
 *a
*a
aDepartment of Chemistry, Institute of Science, Banaras Hindu University, Varanasi-221005, U.P., India. E-mail: roopshikha22singh@gmail.com; roopshikha.singh1@bhu.ac.in
bDepartment of Biochemistry, Institute of Science, Banaras Hindu University, Varanasi-221005, U.P., India
First published on 28th February 2024
Abstract
The prevalence of the D–A strategy in achieving red-shifted emission has been established through designing D–A molecules of D–A–D and A–D–A constructs. Architectural control over such D–A systems integrates solid state NIR emission with lysosome tracking and sets a multifarious goal of photophysical modulation in a comprehensive way. In particular, two compounds, CPM–1 (D–A–D) and CPM–2 (A–D–A), have been synthesized by introducing carbazole-based donors and difluoroboron acceptors. Lysosome targeting and imaging have been achieved through incorporation of a morpholine unit, which ultimately imparts viscosity sensitivity to the construct. The fluorophores exhibited significant emission in solution along with distinctive solvatochromism, viscochromism and TICT. A comparative account of these competitive photophysical properties revealed the superior charge transfer properties of the A–D–A construct (CPM–2), while the D–A–D molecule (CPM–1) was found to be a better molecular rotor with marked viscochromism. The solid state NIR emission has been found to be much more intense in CPM–1 relative to CPM–2, which further highlights the influence of structural aspects on photophysical behvaiour. Theoretical studies further established the distinctive characteristics of ground and excited states in these compounds. Owing to its excellent viscochromic behvaiour, CPM–1 has been successfully utilized in lysosome targeting in wild-type Drosophila fly gut tissues through co-localization studies.
Introduction
Lysosomes are membrane-bound organelles providing approximately 50 different types of acid hydrolases, which play important roles in the degradation of various biomacromolecules such as carbohydrates, nucleic acids, proteins, fats, and cellular components.1–3 Lysosomes are crucial for several cellular processes, including cholesterol in vivo balance, autophagy, plasma membrane repair, bone and tissue remodeling, pathogen defense, cell signaling and death.4–6 Due to the exclusive role of lysosomal transport during tumor invasion and metastasis, the effective monitoring of intracellular lysosome has emerged as a fascinating target for cancer diagnosis and treatment.7–9 Therefore, long term and real time visualization of lysosomes is imperative in gaining a deeper understanding of lysosomerelated cancer diagnosis and treatment. In this direction, several fluorescent probes have been illustrated in the literature and various lysotrackers are commercially available. Furthermore, cancer cells are reported to have lower pH as compared to normal cells and cancer lysosomes have higher viscosity than the lysosomes of normal cells.10–12,15,16 This fact lays down a foundation for the development of lysotrackers using pH and viscosity as potential markers leading to effective cancer cell targeting.13–15 In this context, the development of probes sensitive towards viscosity, which can be engineered to target lysosomes through incorporation of suitable amine-based anchors17–20 (viz. morpholine, piperazine etc.), is one of the most prevailing strategies. This strategy utilizes the positive membrane potential of the acidic lysosomes for accumulation of amine-modified probes. Furthermore, owing to their deep tissue penetration with minimal photodamage and low tissue autofluorescence in the NIR window of 650–900 nm, NIR flurophores have become a prudent choice for in vivo cell imaging.21–25 Recent research has seen a great surge in exploration of lysosome-specific NIR fluorescent probes and their applications in cancer cell targeting and imaging.26,27Amongst several NIR fluorescent probes based on inorganic and organic molecules, those comprising a donor–acceptor (D–A) electronic structure are particularly fascinating as these have large Stokes shifts and excellent photophysical stability.28–31 Rational design and engineering of these D–A fluorophores can be done to improve the brightness as well as the fluorescence wavelength. Steric hindrance or AIE characteristics can be employed to improve the brightness, while the incorporation of suitable fluorphores might lead to red-shifted emission. It has further been reported that the incorporation of additional donor/acceptor groups in conventional D–A molecules can lead to more red-shifted emissions.32,33 Keeping these points in mind we shifted our attention toward the modification of difluoroboron-coordinated β-diketonate (BF2bdk) compounds through suitable donors and acceptors. These compounds have become indispensible tools in bioimaging applications owing to their distinctive photophysical properties, including high molar extinction coefficients, efficient solid-state emission, two photon excited fluorescence and mechanochromism.34–42 Their ease of syntheses, high sensitivity and prompt responses have made these crucial research hotspots in recent years.43–59 In this direction, keeping our goal of developing NIR-active biocompatible fluorphores for selective targeting and imaging of lysosomes intact, we have designed D–A based β-diketonate derivatives. Herein, carbazole has been chosen as the donor (D), while a borondifluoride core acts as the acceptor (A); thus to have substantial control over intramolecular charge transfer (ICT) the D–A skeleton has been altered with the construction of D–A–D and A–D–A scaffolds. To address the unique need for lysosomal targeting and imaging, morpholine has been incorporated to engender viscosity sensitivity. In this context, the present work details the design and synthesis of two small D–A molecules, CPM–1 and CPM–2 possessing D–A–D and A–D–A architectures, respectively, wherein morpholine has been appended as a pendant group through N-substitution of the carbazole unit. This provides us with two morpholine units in CPM–1, D–A–D, and one in CPM–2, A–D–A, giving us an opportunity to evaluate viscochromism based on the ICT character of the fluorophores. The compounds have been characterized by various spectroscopic techniques, while a detailed photophysical study {solvatochromism, viscochromism, and twisted intramolecular charge transfer (TICT)} in solution as well as the solid state has been performed to assess the effect of the D–A architecture on photophysical modeling and NIR emission. The fluorophores have been found to be emissive both in solution and the solid state. The lysosomal targeting of CPM–1 has been realized in wild-type Drosophila flies (Oregon R+ strain) through LysoTracker Green co-localization studies.
Experimental methods
Reagents
Carbazole, 4-(2-chloroethyl)morpholine hydrochloride, 1,2dibromoethane, 1-phenyl-1,3-butanedione, acetylacetone and boron trifluoride diethyl etherate were purchased from Sigma Aldrich India, TCI chemicals India and Avra chemical India. Common reagents, potassium hydroxide, sodium hydroxide, n-butylamine and the solvents ethyl acetate, n-hexane, dimethylsulphoxide (DMSO), dichloromethane (DCM), dimethylformamide (DMF), methanol etc. were procured from Avra Chemicals Hyderabad, India and dried and distilled following the standard literature procedures.60 The synthetic manipulations have been performed under an oxygen-free nitrogen atmosphere and photophysical studies using spectroscopic grade solvents.General information
1H (500 MHz), and 13C (125 MHz), 11B and 19F NMR spectra have been obtained using a JEOL AL 500 FT–NMR spectrometer at room temperature using Si(CH3)4 as an internal standard. UV–vis and emission spectra have been acquired at room temperature using a Shimadzu UV–1800 and PerkinElmer LS55 fluorescence spectrometer, respectively. Electrospray ionization mass spectrometry (ESI–MS) measurements have been made using a SCIEX X500R QTOF instruments. Time-resolved fluorescence lifetime experiments have been performed using a TCSPC system from Horiba Yovin (Delta Flex). Compounds were excited at 482 nm using a pico-second diode laser (Model: delta diode). Data analysis has been performed using decay analysis software (HORIBA Scientific: EzTime). Solid state photoluminescence has been acquired at room temperature using a WITEC alpha 300 Focus Innovation by using a pulse diode laser (wavelength = 450 nm and power = 0.250 milliwatt).Syntheses
Synthesis of A1, and A2 and 9-(2-morpholinoethyl)-9H-carbazole-3-carbaldehyde (ALD–1) was done following the literature procedure.61–63 9-(2-morpholinoethyl)-9H-carbazole-3,6-dicarbaldehyde (ALD–2) was synthesized following the same synthetic procedure as for ALD–1.Synthesis of ALD–2
To DMF (2.92 g, 40 mmol) at 0 °C, phosphorus oxychloride (6 g, 40 mmol) was added dropwise. The solution was allowed to warm to room temperature, and 4-(2-(9H-carbazol-9-yl)ethyl)morpholine (1 g, 3.56 mmol) in 1,2-dichloroethane (6 mL) was added to it. The reaction mixture was heated to 90 °C and kept at this temperature for 24 h. Afterwards the reaction mixture was poured into water and extracted with chloroform. The chloroform layer was washed with water, dried over Na2SO4 and the solvent was removed in vacuo to yield a deeply colored product, which was purified on a silica gel column using n-hexane/ethyl acetate (7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3). 1H NMR (CDCl3, 500 MHz, δ ppm): 2.53 (t, J = 8 Hz, 4H), 2.82 (t, J = 13.5 Hz, 2H), 3.6 (t, J = 8.5 Hz, 4H), 4.5 (t, J = 13.5 Hz, 2H), 7.5 (d, J = 8.5 Hz, 2H), 8.08 (d, J = 8.0 Hz, 2H), 8.66 (s, 2H), 10.13 (s, 2H).13C NMR (CDCl3, 125 MHz, δ ppm):191.61, 144.93, 130.02, 128.03, 124.41, 123.41, 109.94, 66.97, 56.78, 54.20, 41.95.
:3). 1H NMR (CDCl3, 500 MHz, δ ppm): 2.53 (t, J = 8 Hz, 4H), 2.82 (t, J = 13.5 Hz, 2H), 3.6 (t, J = 8.5 Hz, 4H), 4.5 (t, J = 13.5 Hz, 2H), 7.5 (d, J = 8.5 Hz, 2H), 8.08 (d, J = 8.0 Hz, 2H), 8.66 (s, 2H), 10.13 (s, 2H).13C NMR (CDCl3, 125 MHz, δ ppm):191.61, 144.93, 130.02, 128.03, 124.41, 123.41, 109.94, 66.97, 56.78, 54.20, 41.95.
Synthesis of CPM–1
In a 50 mL round bottom flask, 2.46 mmol of ALD–1 and 1.12 mmol of A1 were dissolved in a minimum amount of DCM, and the flask was flushed with nitrogen. n–Butylamine (0.22 eq.) was then added dropwise to the reaction mixture via a syringe and the reaction mixture was stirred overnight at room temperature. After completion of the reaction a solid separated out, which was filtered through a Buchner funnel and washed with cold ethyl acetate and diethyl ether and dried under vacuum. This yielded CPM–1 as a red colured solid (yield: ∼63%). 1H NMR (500 MHz, DMSO-D6, δ ppm): 8.76 (s, 1H), 8.31 (s, 1H), 8.24–8.20 (m, 2H), 8.01 (d, J = 8.9 Hz, 1H), 7.80 (d, J = 8.8 Hz, 1H), 7.74 (d, J = 8.5 Hz, 1H), 7.53 (s, 1H), 7.33 (t, J = 7.9 Hz, 1H), 7.26 (d, J = 15.3 Hz, 1H), 6.55 (d, J = 18.4 Hz, 1H), 4.86 (t, J = 6.0 Hz, 4H), 4.08 (t, J = 6.0 Hz, 4H), 2.77 (t, J = 7.8 Hz, 2H), 1.50 (t, J = 7.8 Hz, 2H). 13C NMR (125 MHz, DMSO-D6, δ ppm): 178.63, 147.74, 142.63, 140.73, 127.63, 126.71, 125.78, 123.15, 123.06, 122.30, 120.63, 120.40, 118.15, 110.72, 110.42, 79.18, 39.19, 39.02, 38.61, 29.08, 19.08, 13.48. 11B NMR (160 MHz, DMSO-D6, δ ppm): −0.24, −0.35, −0.47, −0.58. 19F NMR (470 MHz, DMSO-D6, δ ppm) −137.91, −137.98, −139.41, −139.52 ESI–MS calcd. for C43H43BF2N4O42+ [M + H]+ 729.33; found 729.34.Synthesis of CPM–2
In a 50 mL round bottom flask, 0.65 mmol of A2 and 0.29 mmol of ALD–2 were dissolved in a minimum amount of DCM, and the flask was flushed with nitrogen. n-Butylamine (0.22 eq.) was then added dropwise to the reaction mixture via a syringe, and the reaction mixture was stirred overnight at room temperature. After completion of the reaction a solid separated out; this was filtered through a Buchner funnel and washed with cold ethyl acetate and diethyl ether and dried under vacuum ∼73%. 1H NMR (500 MHz, DMSO-D6, δ ppm): 8.78 (s, 2H), 8.46 (d, J = 15.3 Hz, 2H), 8.20 (d, J = 7.7 Hz, 4H), 8.10 (d, J = 9.0 Hz, 2H), 7.89 (d, J = 9.0 Hz, 2H), 7.81 (t, J = 7.4 Hz, 2H), 7.68 (t, J = 7.5 Hz, 4H), 7.41 (s, 2H), 7.31 (d, J = 15.9 Hz, 2H), 4.73 (s, 2H), 3.61 (s, 4H), 2.64 (s, 2H), 2.36 (s, 4H). 13C NMR (125 MHz, DMSO-D6, δ ppm): 178.63, 147.74, 142.63, 140.73, 127.63, 125.78, 123.15, 123.06, 122.30, 120.63, 120.40, 118.15, 110.72, 110.42, 79.18, 39.19, 39.02, 38.61, 29.08, 19.08, 13.48. 11B NMR (160 MHz, DMSO-D6, δ ppm): −2.26. 19F NMR (470 MHz, DMSO-D6, δ ppm): −137.32, −148.14 ESI–MS calcd. For C40H34B2F4N2O52+ [M + H]+ 721.32; found 721.31.Results and discussion
BF2bdk complexes CPM–1 and CPM–2 have been synthesized in good yields by condensation of the corresponding ALD–1 and ALD–2 with compounds A1 and A2, following a very simple one step-reaction pathway (Scheme 1). These are air-stable, non-hygroscopic solids and possess good solubility in common organic solvents like I, DMF, and DMSO and moderate solubility in MeOH, EtOH, DCM, and EtOAc and are insoluble in water. The synthesized compounds have been well characterized by NMR (1H, 13C, 11B and 19F), ESI–MS, electronic absorption and emission spectroscopic studies and the spectral data have been given in the Experimental section and spectra shown in Fig. S1–S12 (ESI†) and Fig. 1 and 2. | ||
| Scheme 1 Synthetic routes to CPM–1 and CPM–2. | ||
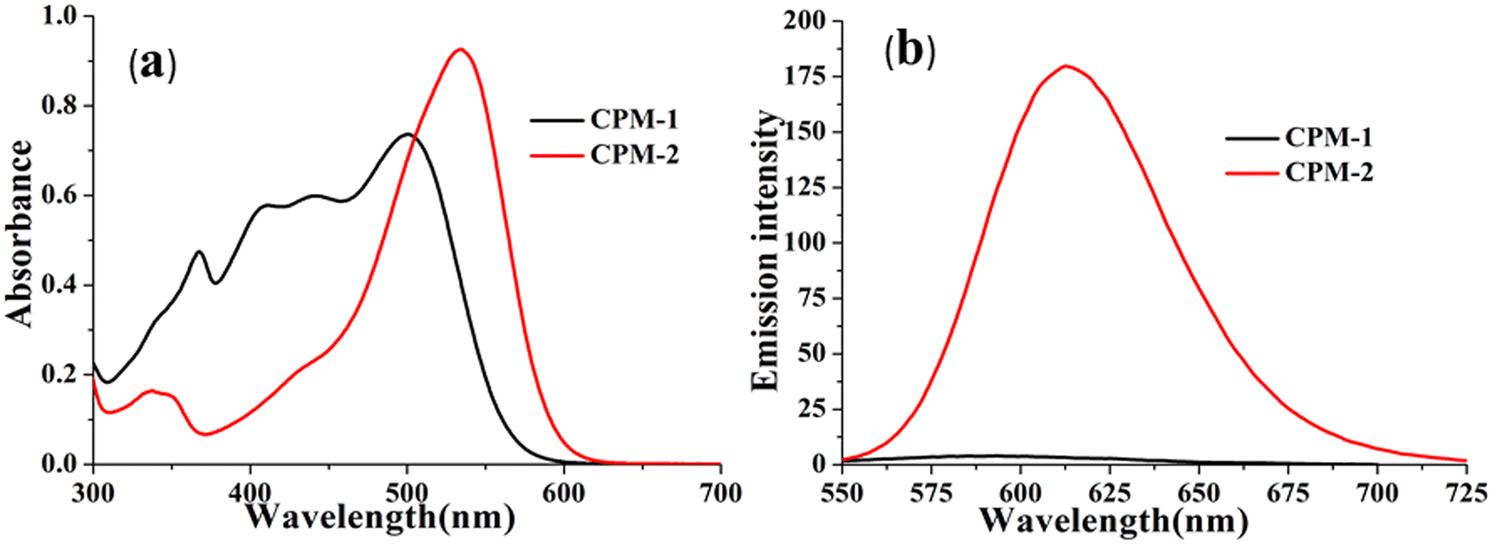 | ||
| Fig. 1 UV–vis absorption (a) and emission (b) spectra of CPM–1 and CPM–2 in ACN (c = 30 μM). | ||
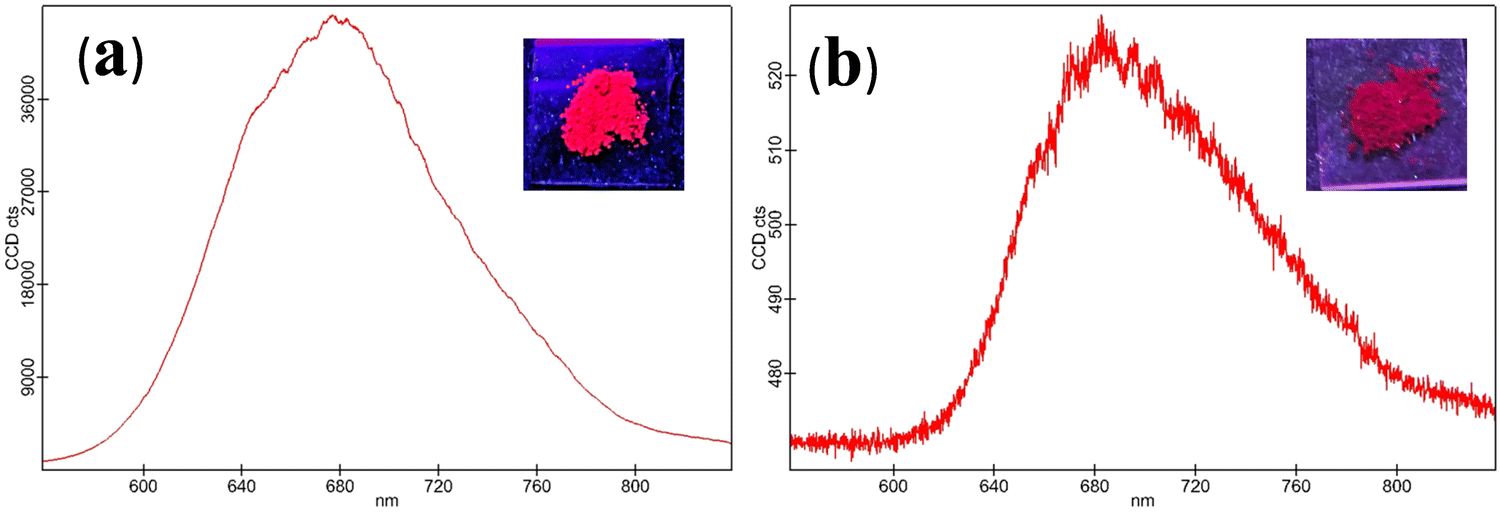 | ||
| Fig. 2 Solid state emission spectrum of CPM–1 (a) and CPM–2 (b) (λex = 502 and 535 nm, respectively). [Insets: solid state emission of CPM–1 and CPM–2 under UV light (λex = 365 nm)]. | ||
Photophysical properties
The photophysical modulation by architectural alteration of the synthesized D–A molecules has been verified through UV–vis absorption and emission studies focusing on polarity sensitivity, viscochromism and TICT behavior. The absorption spectra of CPM–1 and CPM–2 have been recorded IACN (c = 30 μM), and display low energy absorption bands at 502 and 535 nm, respectively, attributed to π–π* transitions (Fig. 1). On the other hand, the characteristic high energy absorption bands corresponding to the acetylacetonate boron difluoride unit of CPM–1 and CPM–2 were observable at 367 and 348 nm, respectively. A broad absorption band related to the morpholine-substituted carbazole units of CPM–1 and CPM–2 has been visualized at ∼ 438 nm. On the other hand in their emission profiles, CPM–1 and CPM–2 exhibited emission peaks at 594 and 613 nm upon excitation at 502 and 535 nm, respectively (Fig. 1). The Stokes’ shifts for CPM–1 and CPM–2 were measured at 3085 cm−1 and 2378 cm−1, respectively. Furthermore, the fluorescence quantum yield (Φf) of CPM–1 was found to be considerably lower (Φf = 0.3%) as compared to CPM–2 (Φf = 10%).Furthermore, solid state photoluminescence spectra of CPM–1 and CPM–2 have also been recorded (Fig. 2). CPM–1 showed a broad band (600–800 nm) with a maximum centered at ∼680 nm; on the other hand, CPM–2 displayed a similar type of band (630–800 nm) with a maximum centered at ∼685 nm. Interestingly the solid state emission intensity of CPM–1 was found to be much higher relative to CPM–2, which can further be visualized under a UV lamp (Fig. 2: inset, λex = 365 nm).
The initial indication for the effect of the altered D–A architecture can be realized by more red shifted absorption and emission of CPM–2 as well as lower Φf (solution) of CPM–1. However, a more systematic study of ICT behavior needs to be monitored to establish a comparative account in terms of photophysical attributes for A–D–A and D–A–D constructs.
In this context, the comparative ICT behavior of CPM–1 and CPM–2 has further been monitored through study of their solvatochromic behavior. In light of this, we investigated these D–A systems across a range of organic solvents with varying polarities, including benzene, toluene, 1,4-dioxane, CHCl3, THF, CH2Cl2 and CH3CN. The absorption spectra of the compounds displayed insignificant changes with different solvent polarities (Fig. S14–S15, ESI†), indicating that the ground state of the luminophores is eventually unaffected by solvent polarity. However, the emission spectra exhibited considerable changes in response to the solvent polarity, ranging from benzene to CH3CN. Specifically, for CPM–1 and CPM–2, a positive solvatochromism with bathochromic shifts of 33 nm and 47 nm, respectively, was observed, upon going from extreme non-polar to extreme polar solvents. The shift was accompanied by considerable emission quenching as well as broadening and loss of the vibronic structure of the emission spectra (Fig. 3) indicating strong ICT behavior. Furthermore, the Lippert–Mataga plots between Stokes’ shifts and orientolarizabilitybility (Δf) for CPM–1 and CPM–2 displayed linear dependency of the positive slope on Stokes’ shift, which further denotes positive solvatochromism (Fig. S13, ESI†).
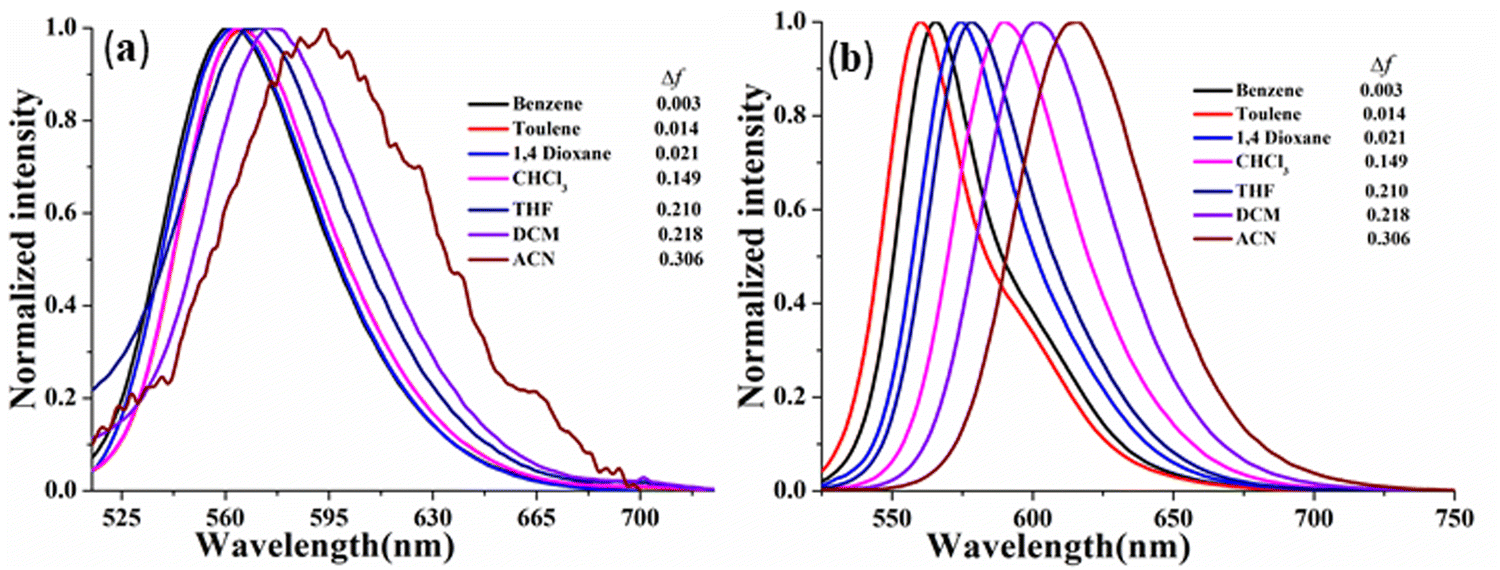 | ||
| Fig. 3 Normalized emission spectra of CPM–1 (a) and CPM–2 (b) (c = 30 μM) in solvents of varying polarity. | ||
Additionally, Φf has also been calculated in solvents of different polarity and the data have been summarized in Table S1 (ESI†). The Φf values for CPM–1 and CPM–1 were found to be the maximum in non-polar solvents, while Φf decreases rapidly with increasing solvent polarity owing to the stabilization of the transferred charge within the molecules. The solvent effect was further assessed through evaluation of excited state dynamics under the effect of differential polarity (Fig. S16, ESI†). The radiative (kr) and nonradiative (knr) decay rate constants from the excited singlet state (S1) have been calculated and the data have been compared (Table S1, ESI†). In general, the kr values decrease with increasing solvent polarity; however the comparative kr and knr values of CPM–1 and CPM–2 in CHCl3 were found to be responsible for their significant Φf in CHCl3. On the other hand the high knr and low kr can be responsible for very low quantum yields of CPM–1 and CPM–2 in CH3CN. Interestingly the effect of solvent on the kr and knr values was more pronounced in CPM–2 relative to CPM–1.
Thus, the solvatochromic study professes the superior ICT behvaiour of CPM–2 (A–D–A) relative to CPM–1 (D–A–D) and strengthens our assumption of tuning of photophysical behavior through structural modulations.
The effectiveness of the above-mentioned approach has been revealed by manifestation of the TICT state as reflected through emission spectra of CPM–1 and CPM–2 in a mixture of tetrahydrofuran (THF) and n-hexane (Fig. 4 and Fig. S17, ESI†), with increasing n-hexane fractions (fH). As expected, in accordance with the characteristics of conventional D–A molecules, both the fluorphores exhibited blue-shifted emission with gradual addition of n-hexane. However, CPM–1 displayed a hypsochromic shift of only 7 nm; however a more significant blue shift of 41 nm was observable for CPM–2 at fH = 90%. These compelling observations firmly indicated the stronger dipolar ICT character of A–D–A (CPM–2) relative to D–A–D (CPM–1).
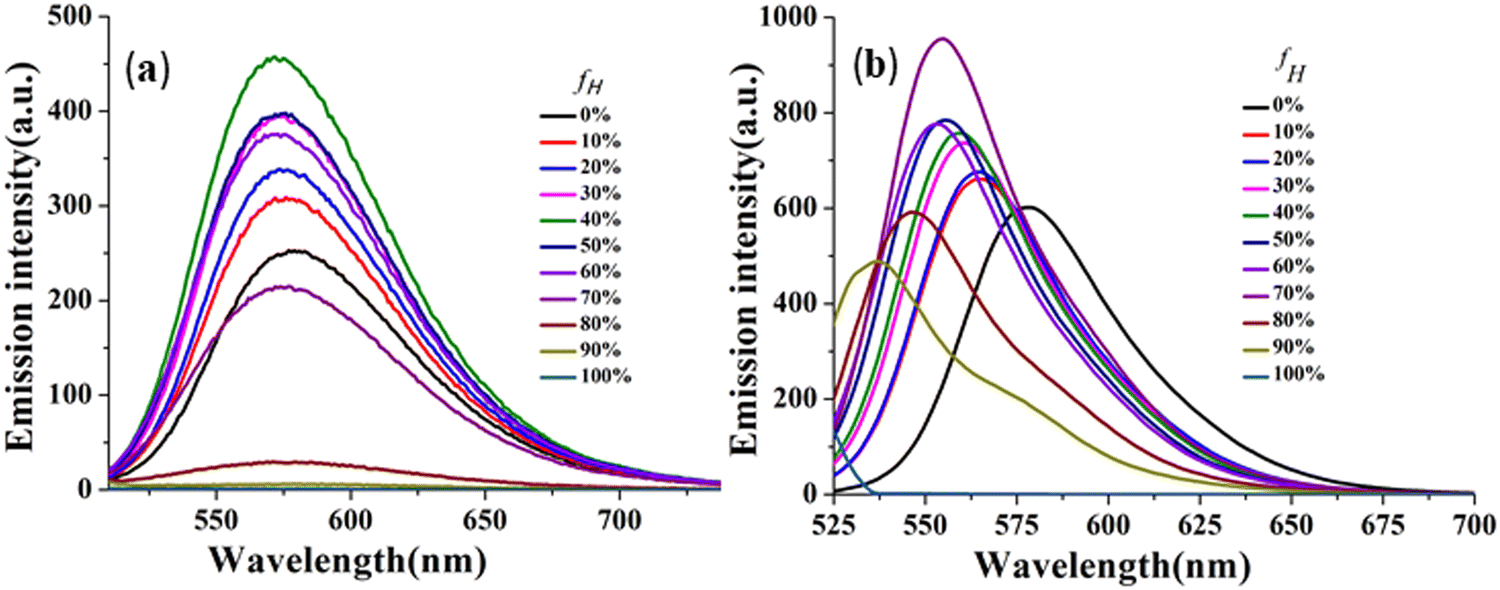 | ||
| Fig. 4 Emission spectra of CPM–1 (a) and CPM–2 (b) in THF and THF/n-hexane mixtures (c = 30 μM) with increasing fractions of n-hexane and formation of the TICT state. | ||
Viscochromism and restricted intramolecular rotation
The viscosity sensitivity of the designed probes has been verified through recording the emission spectra in glycerol–methanol mixtures by varying the percentage of glycerol (Fig. 5). CPM–1 exhibited significant emission enhancement (∼2.5 fold) upon increasing the fg from 0 to 99% for the emission maxima at 586 nm in methanol. The emission enhancement can be attributed to restriction of intramolecular rotation followed by deactivation of non-radiative channels. On the other hand, no such emission enhancement was observable for CPM–2 (Fig. S18, ESI†). In addition, the viscochromism has further been authenticated through fluorescence lifetime measurements. It was observed that the fluorescence life time increased from 0.11 ns (fg = 0%) to 0.31 ns (fg = 90%) supporting the emission enhancement. These observations cumulatively established CPM–1 as an effective molecular rotor, which can be explored for lysosomal targeting.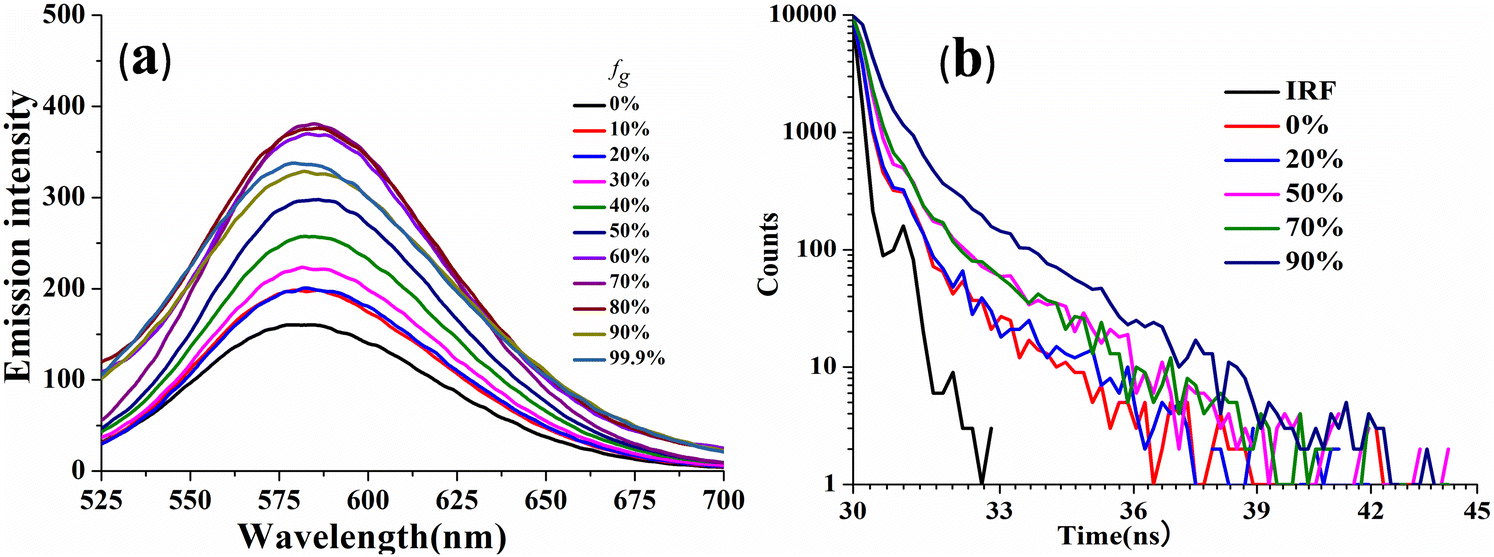 | ||
| Fig. 5 Emission spectra of CPM–1 (a) and time-resolved fluorescence decay of CPM–1 (b) in MeOH and MeOH/glycerol mixtures (c = 30 μM) with increasing fractions of glycerol. | ||
As is well known, TICT is achieved through intramolecular rotation or twisting and leads to red-shifted and quenched emission. On the other hand, blocking of intramolecular rotations in the aggregated state leads to fixed molecular conformations, which might further lead to emission enhancement. Both processes are competitive and define the emission in D–A construct-based AIEgens.64–66
One of the most compelling outcomes of the detailed photophysical study of CPM–1 and CPM–2 lies in epistemic interdependence of restriction of intramolecular rotations (RIRs) and TICT. The pragmatic aspects of evidence are demonstrated by an effective RIR but weak TICT in CPM–1, while contrasting properties are found in CPM–2, which displays a non-existent RIR and very effective TICT. These explicit evidences establish a design rationale for photophysical modulation via structural modifications.
Density functional theory calculations
The present study focuses on the theoretical investigation of geometrical optimization for (D–A–D) and (A–D–A) systems using the B3LYP method with the 6–31G** basis set. Fig. 6 illustrates the distribution of the highest occupied molecular orbitals (HOMOs) and the lowest unoccupied molecular orbitals (LUMOs) for CPM–1 and CPM–2. It can be observed that the HOMOs are predominantly located on the respective carbazole and morpholine units, which act as the donor moieties. Conversely, the LUMOs are primarily concentrated over the BF2 core, serving as the acceptor moieties. The energy difference values between the HOMO−2, HOMO−1 and LUMO, known as the band gap, are found to be 3.44 eV for CPM–1 and 3.25 eV for CPM–2, respectively. The HOMO−2 and HOMO−1 energy levels are −5.85 eV for CPM–1 and 6.09 eV for CPM–2, respectively, while the LUMO energy levels are −2.41 eV for CPM–1 and −2.84 eV for CPM–2. The substantial spatial separation between the HOMO−2, HOMO−1 and LUMO suggests the possibility of an ICT phenomenon between the donor and acceptor moieties, corroborating the experimental observations (vide infra). Furthermore, the UV–vis spectra of CPM–1 and CPM–2 were obtained from TD–DFT calculations (Fig. S20 and Table S3, ESI†). Table S3 (ESI†) summarizes the details of the absorption wavelength, energy, oscillation strength (f), assignment, and transitions.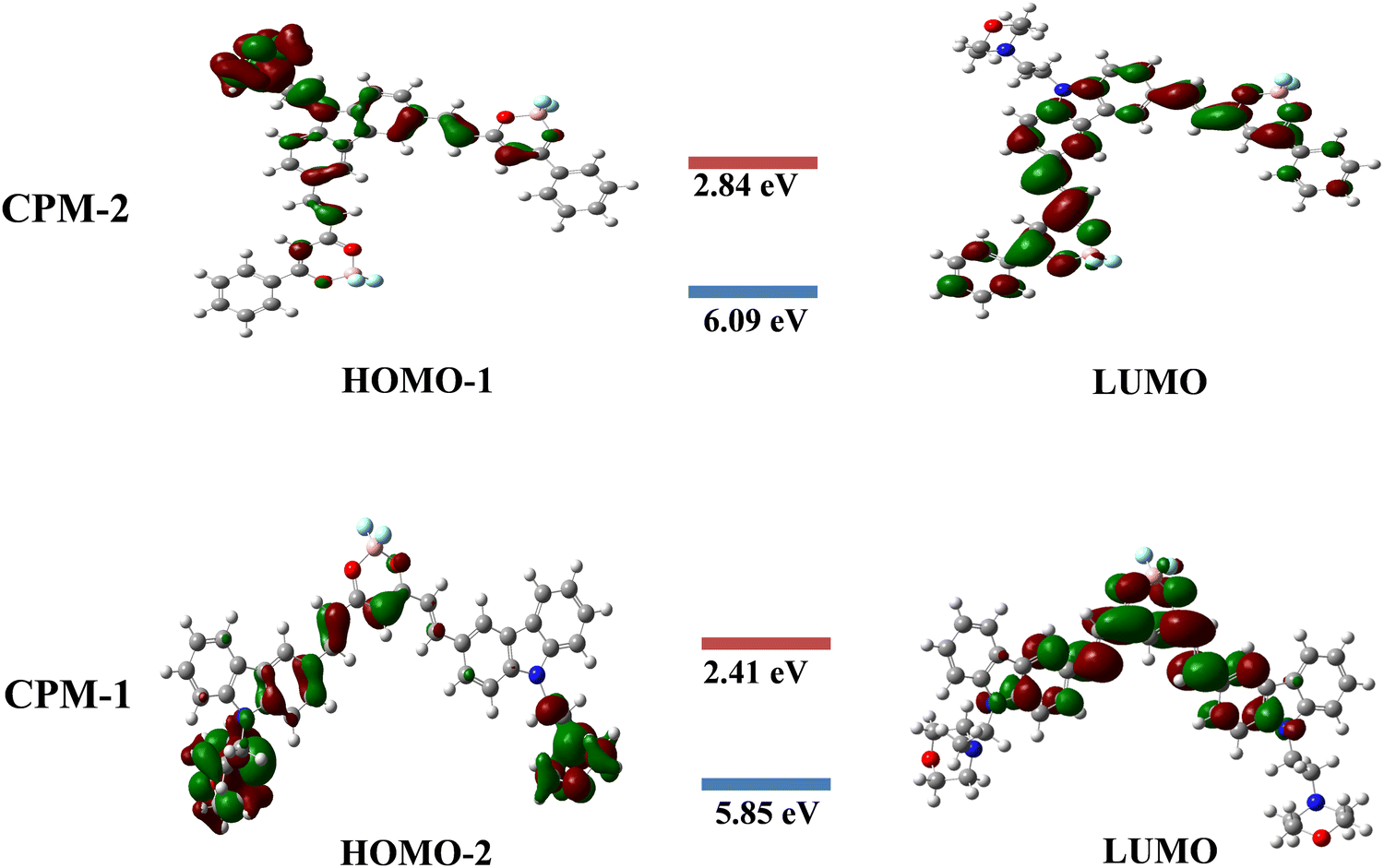 | ||
| Fig. 6 The HOMO–LUMO frontier molecular orbital (FMO) diagrams of CPM–1 and CPM–2. | ||
The TD–DFT calculations reveal that, for both CPM–1 and CPM–2, the HOMO−2 → LUMO and HOMO−1 → LUMO transitions, respectively, contribute significantly to the ICT process, accounting for 95% in CPM–1 and 97% in CPM–2. The dipole moments of CPM–1 and CPM–2 in the ground state were found to be 13.08 and 12.37 Debye, respectively, which increased to 19.05 and 18.48 in the excited state. An increase in the dipole moment was noted in the excited state, which was in good agreement with the observed solvatochromism and the anticipated charge separation. These theoretical findings provide valuable insights into the electronic structure and optical properties of the investigated D–A–D and A–D–A systems. The results not only support the experimental observations but also shed light on the potential for intramolecular charge transfer in these systems, which can have implications for various applications in optoelectronic devices.
Application to lysosome tracking in vivo
The applicability of the developed fluorophores in biological systems has been realized through lysosome tracking experiments. In order to assess the toxic effects of CPM–1 on Drosophila, flies were administered with concentrations of 5 μM, 50 μM, and 100 μM.The results revealed that CPM–1 at concentrations of 5 μM and 50 μM exhibited lower toxicity, which is non-significant, as evidenced by the successful eclosion of 92% and 87% of the adult flies compared to the control group (Fig. 7A). However, at a concentration of 100 μM, CPM–1 was found to be toxic to fly development, with a reduced eclosion rate; hence, the 50 μM concentration is the safe dose for oral administration. The cell viability of larval gut tissue treated with the CPM–1 formulation was assessed using the MTT (3-[4,5-dimethylthiazol-2-yl]-2,5 diphenyl tetrazolium bromide) assay. The results demonstrated that larvae treated with CPM–1of 5 μM and 25 μM concentrations presented 95.5% and 73.7% viability, respectively. However, at a concentration of 50 μM, the viability decreased to only 65% (Fig. 7B). These findings indicate that the viability of the larval gut tissue is dose-dependent, with higher concentrations of CPM–1 leading to reduced cell viability.
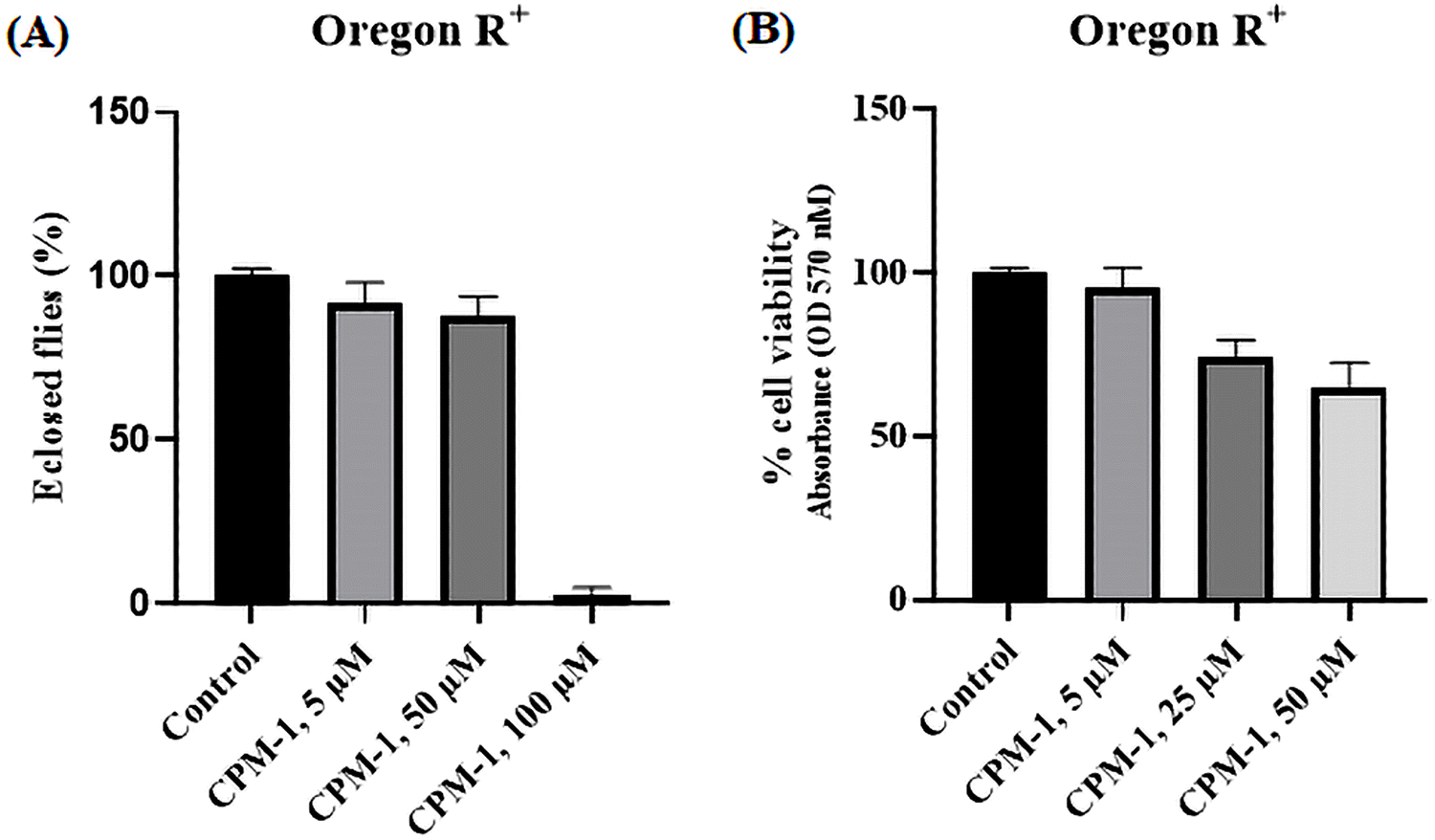 | ||
| Fig. 7 In histogram (A), the percentage of flies that eclosed during the toxicity assay after CPM–1 treatment is presented. Histogram (B) illustrates the percentage of cell viability following the MTT assay of CPM–1-treated larval gut tissue. | ||
CPM–1 colocalization with LysoTracker Green
The colocalization experiment performed in the gut tissue of 3rd instar Drosophila larvae involved comparing the fluorescence patterns of CPM–1 and LumiTracker Lyso Green to assess the specificity of CPM–1 for tracking the lysosomes. The results showed that the red fluorescence emitted by CPM–1 (Fig. 8a and d) colocalized with the green fluorescence emitted by LumiTracker Lyso Green (Fig. 8b and e) and the merged images (Fig. 8c and f) showed absolute colocalization in the corresponding lysosomal regions. The colocalization of CPM–1 (red fluorescence) and LumiTracker Lyso Green (green fluorescence) indicated that CPM–1 specifically targets and localizes to lysosomes in the gut tissue of Drosophila larvae. The magnitude of the Pearson coefficient is 0.741, indicating a high correlation between the variables (Fig. 8g, h and i). The coincidence of the fluorescence signal suggests that CPM–1 is a reliable marker for visualizing and studying lysosomes in this experimental in vivo model.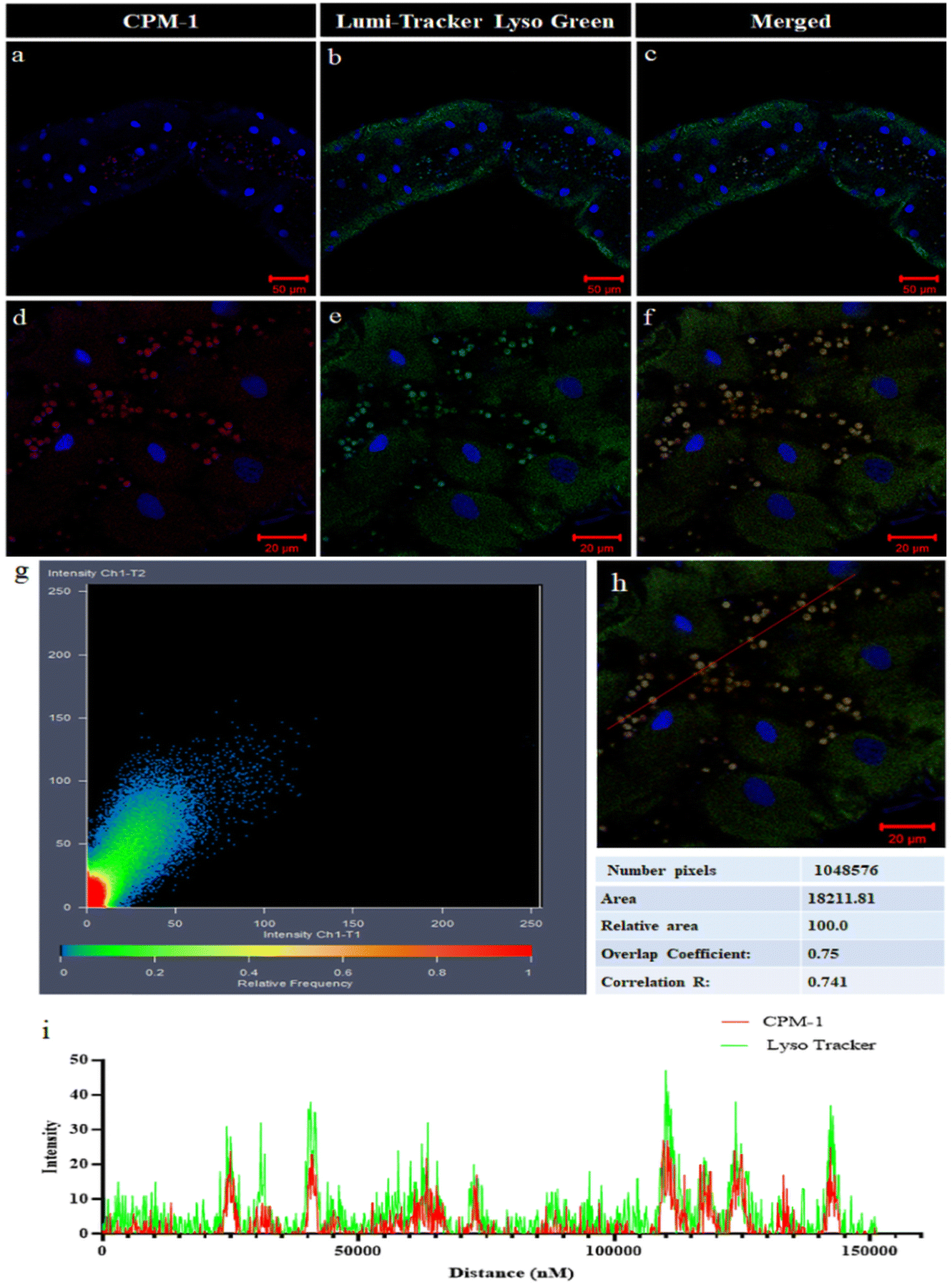 | ||
| Fig. 8 Confocal micrographs of 1 μM CPM–1 (panels a and d) and LumiTracker Lyso Green at 1 μM (panels b and e). (Panels c and f) represent the merged images, showing the co-localization of CPM–1 and LumiTracker Lyso Green. Panel (g) shows the correlation scatter plot of panel (f) and graph (i) the relative intensity correlation of panel (h). | ||
Conclusions
In summary, we present an empirical strategy that prioritizes the architectural alterations to achieve desired photophysical properties through the development of unconventional D–A molecules CPM–1 and CPM–2. The meticulously designed D–A molecules bearing D–A–D and A–D–A constructs displayed distinct photophysical properties. The study of the competitive RIR and TICT behavior reflects a weaker ICT with a more effective RIR in CPM–1, while a stronger TICT and non-existent RIR in CPM–2. These contrasting properties have been accompanied by strong NIR emission in the solid state by both the molecules, which is reminiscent of their inherent D–A constructs. Owing to its intricate structural attributes and excellent viscosity sensitivity, CPM–1 has been explored for its lysosome tracking ability wherein it displayed selective lysosomal localization in larval gut tissue of wild-type Drosophila flies. The proposed hypothesis of architectural control can be used to provide more in-depth understanding of competitive photophysical properties of D–A molecules and get the desired applications.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
The authors gratefully acknowledge the Science and Engineering Research Board (SERB), New Delhi, India (Scheme ECR/2018/001810) and BHU IOE cell (Seed/Incentive Grant) for providing financial assistance.References
- Z. Guo, Z. Jin, J. Wang and J. Pei, Chem. Commun., 2014, 50, 6088–6090 RSC
.
- Q. Zhao, H. Xiao, F. Yang, Y. Wang, P. Li and Z. Zheng, Pharmacol. Res. Commun., 2023, 188, 106627 CrossRef PubMed
- J. Cheng, Biomolecules, 2015, 5, 1099–1121 CrossRef PubMed
- Z. Zhang, P. Yue, T. Lu, Y. Wang, Y. Wei and X. Wei, J. Hematol. Oncol., 2021, 79 CrossRef PubMed
- K. K. Mahapatra, S. R. Mishra, B. P. Behera, S. Patil, D. A. Gewirtz and S. K. Bhutia, CMLS, 2021, 78, 7435–7449 CrossRef CAS PubMed
- S. R. Bonam, F. Wang and S. Muller, Nat. Rev. Drug Discovery, 2019, 18, 923–948 CrossRef CAS PubMed
- K. Pal, P. Kumar and A. P. Koner, J. Photochem. Photobiol., B, 2019, 206, 111848 CrossRef PubMed
- B. Jana, S. Jin, E. M. Go, Y. Cho, D. Kim, S. Kim, S. K. Kwak and J. Ryu, J. Am. Chem. Soc., 2023, 145, 18414–18431 CrossRef CAS PubMed
- S. Rafiq, S. L. McKenna, S. Muller, M. P. Tschan and M. Humbert, Leukemia, 2021, 35, 2759–2770 CrossRef PubMed
- C. Kong, Y. Li, Z. Liu, J. Ye, Z. Wang, L. Zhang, W. Kong, H. Liu, H. Pang, Z. Hu, J. Gao and F. Qian, ACS Nano, 2019, 13, 4049–4063 CrossRef CAS PubMed
- Y. Song, H. Zhang, X. Wang, X. Geng, Y. Sun, J. Liu and Z. Li, Anal. Chem., 2021, 93, 1786–1791 CrossRef CAS PubMed
- Y. Wei, X. Weng, X. Sha, R. Sun, Y. Xu and J. Ge, Sens. Actuators, B, 2021, 326, 128954 CrossRef CAS
- L. Wang, Y. Xiao, W. Tian and L. Deng, J. Am. Chem. Soc., 2013, 135, 2903–2906 CrossRef CAS PubMed
- A. Tantipanjaporn, K. K. Y. Kung, W. C. Chan, J. R. Deng, B. C. B. Ko and M. K. Wong, Sens. Actuators, B, 2022, 367, 132003 CrossRef CAS
- Y. H. Zhan, X. J. Li, R. Sun, Y. Xu and J. Ge, Anal. Chim. Acta, 2016, 933, 175–181 CrossRef CAS PubMed
- J. Zhang, Q. Chen, Y. Fan, H. Qiu, Z. Ni, Y. Li and S. Yin, Dyes Pigm., 2021, 193, 109500 CrossRef CAS
- B. Feng, Y. Ma, F. Zheng, Y. Huang, X. Feng, K. Zhang, L. Liu and W. Zeng, J. Chem. Eng., 2023, 464, 142554 CrossRef CAS
- N. Choi, J. Lee, E. Park, J. Lee and J. Lee, Molecules, 2021, 26, 217 CrossRef CAS PubMed
- J. Hong, Q. Li, Q. Xia and G. Feng, Anal. Chem., 2021, 93, 16956–16964 CrossRef CAS PubMed
- Y. Song, H. Zhang, X. Wang, X. Geng, Y. Sun, J. Liu and L. Zhaohui, Anal. Chem., 2021, 93, 1786–1791 CrossRef CAS PubMed
- Q. Liu, C. Liu, X. Jiao, S. Cai, S. He, L. Zhao, X. Zeng and T. Wang, Dyes Pigm., 2012, 190, 109293 CrossRef
- J. Li, X. Li, J. Jia, X. Chen, Y. Lv, Y. Gua and J. Li, Dyes Pigm., 2019, 166, 433–442 CrossRef CAS
- M. Grossi, M. Morgunova, S. Cheung, D. Scholz, E. Conroy, M. Terrile, A. Panarella, J. C. Simpson, W. M. Gallagher and D. F. O. Shea, Nat. Commun., 2016, 7, 10855 CrossRef CAS PubMed
- J. M. Li, F. F. Xiang, D. H. Zhou, J. X. Xu, H. Zhang, Y. Z. Liu, Q. Q. Kong, X. Q. Yu and K. Li, Chem. Biomed. Imaging, 2023, 2, 126–134 CrossRef
- Z. Li, Y. Pei, Y. Wang, Z. Lu, Y. Dai, Y. Duan, Y. Ma and H. Guo, J. Org. Chem., 2019, 84, 13364–13373 CrossRef CAS PubMed
- R. Mengji, C. Acharya, V. Vangala and A. Jana, Chem. Commun., 2019, 55, 14182 RSC
- Q. Liu, C. Lib, X. Jiao, S. Cai, S. Hb, L. Zhao, X. Zeng and T. Wang, Dyes Pigm., 2021, 190, 109293 CrossRef CAS
- P. Cheng, X. Du, S. Chen, K. Chen, Y. Yuan, J. Shao, Q. Shen, P. Sun and Q. Fan, ACS Appl. Nano Mater., 2023, 6, 10736–10745 CrossRef CAS
- J. Li, A. Ji, M. Lei, L. Xuan, R. Song, X. Feng, H. Lin and H. Chen, J. Med. Chem., 2023, 66, 7880–7893 CrossRef CAS PubMed
- T. Han, Y. Wang, J. Xu, N. Zhu, L. Bai, X. Liu, B. Sun, C. Yu, Q. Meng, J. Wang, Q. Su, Q. Cai, K. S. Hettie, Y. Zhang, S. Zhu and B. Yang, Chem. Sci., 2022, 13, 13201–13211 RSC
- X. Yang, C. Li, L. Liu, H. Zhang, H. T. Feng, Y. Li, G. Jiang and J. Wang, New J. Chem., 2022, 46, 9819–9824 RSC
- Y. Sugihara, N. Inai, M. Taki, T. Baumgartner, R. Kawakami, T. Saitou, T. Imamura, T. Yanai and S. Yamaguchi, Chem. Sci., 2021, 12, 6333–6341 RSC
- T. Kim, W. Kim, H. Mori, A. Osuka and D. Kim, J. Phys. Chem. C, 2018, 122, 19409–19415 CrossRef CAS
- A. K. Gupta, A. Kumar, R. Singh, M. Devi, A. Dhir and C. P. Pradeep, ACS Omega, 2018, 3, 14341–14348 CrossRef CAS PubMed
- J. Shaya, P. R. Corridon, B. Al-Omari, A. Aoudi, A. Shunnar, M. I. H. Mohindeen, A. Qurashi, B. Y. Michel and A. Burger, J. Photochem. Photobiol., C, 2022, 52, 100529 CrossRef CAS
- Z. Xiang, Z. Y. Wang, T. B. Ren, W. Xu, Y. Liu, X. Zhang, P. Wu, L. Yuan and X. Zhang, Chem. Commun., 2019, 55, 11462–11465 RSC
- D. B. Biernacka, S. G. Mucha, L. Firlej, F. Formalik, J. L. Bantignies, E. Anglaret, M. Samoc and K. Matczyszyn, ACS Appl. Mater. Interfaces., 2023, 15, 32717–32731 CrossRef PubMed
- E. Horak, M. Robic, A. Simanovic, V. Mandic, R. Vianello, M. Hranjec and I. M. Steinberg, Dyes Pigm., 2019, 162, 688–696 CrossRef CAS
- R. Singh, A. K. Gupta and C. P. Pradeep, Cryst. Growth Des., 2021, 21, 1062–1076 CrossRef CAS
- Z. Lia, Y. Hub, K. Zhang, Y. Zhang, Q. Q. Hu, X. J. Zhang, X. K. Zhang and Y. P. Zhu, Dyes Pigm., 2020, 182, 108686 CrossRef
- Z. Li, Y. Pei, Y. Wang, Z. Lu, Y. Dai, Y. Duan, Y. Ma and H. Guo, J. Org. Chem., 2019, 84, 13364–13373 CrossRef CAS PubMed
- A. D'Aléo, A. Felouat, V. Heresanu, A. Ranguis, D. Chaudanson, A. Karapetyan, M. Giorgic and F. Fages, J. Mater. Chem. C, 2014, 2, 5208–5215 RSC
- C. A. DeRosa, S. Hiroto and C. L. Fraser, J. Phys. Chem. C, 2019, 123, 20488–20496 CrossRef CAS
- T. Butler, M. Zhuang and C. L. Fraser, J. Phys. Chem. C, 2018, 122, 19090–19099 CrossRef CAS
- T. Butler, W. A. Morris, J. S. Kosicka and C. L. Fraser, ACS Appl. Mater. Interfaces, 2016, 8, 1242–1251 CrossRef CAS PubMed
- X. Zhang, Y. Tian, H. Zhang, A. Kavishwar, M. Lynes, A. L. Brownell, H. Sun, Y. H. Tseng, A. Moore and C. Ran, Sci. Rep., 2015, 5, 13116 CrossRef CAS PubMed
- X. Zhang, Y. Tian, Z. Li, X. Tian, H. Sun, H. Liu, A. Moore and C. Ran, J. Am. Chem. Soc., 2013, 135, 16397–16409 CrossRef CAS PubMed
- A. Chaicham, S. Kulchat, G. Tumcharern, T. Tuntulani and B. Tomapatanaget, Tetrahedron, 2010, 66, 6217–6223 CrossRef CAS
- G. Zhang, S. H. Kim, R. E. Evans, B. H. Kim, J. N. Demas and C. L. Fraser, J. Fluoresc., 2009, 19, 881–889 CrossRef CAS PubMed
- S. B. Ebrahimi, D. Samanta and C. A. Mirkin, J. Am. Chem. Soc., 2020, 142, 11343–11356 CrossRef CAS PubMed
- H. Cheng, X. Cao, S. Zhang, K. Zhang, Y. Cheng, J. Zhao, L. Zhou, X. Liang and J. Yoon, Adv. Mater., 2022, 35, 2207546 CrossRef PubMed
- M. Grossi, M. Morgunova, S. Cheung, D. Scholz, E. Conroy, M. Terrile, A. Panarella, J. C. Simpson, W. M. Gallagher and D. F. Shea, Nat. Commun., 2016, 7, 1–13 Search PubMed
- P. C. Saha, T. Chatterjee, R. Pattanayak, R. S. Das, A. Mukherjee, M. Bhattacharyya and S. Guha, ACS Omega, 2019, 4, 14579–14588 CrossRef CAS PubMed
- V. N. Nguyen and H. Li, Recent Development of Lysosome-Targeted Organic Fluorescent Probes for Reactive Oxygen Species, Molecules, 2023, 28, 6650 CrossRef CAS PubMed
- P. Z. Chen, L. Niu, Y. Chen and Q. Yang, Coord. Chem. Rev., 2017, 350, 196–216 CrossRef CAS
- W. Liu, Y. Wang, G. Ge, L. Ma, L. Ren and Y. Zhang, Dyes Pigm., 2019, 171, 107704 CrossRef CAS
- N. Liu, P. Z. Chen, J. Wang, L. Niu and Q. Yang, Chin. Chem. Lett., 2019, 30, 1939–1941 CrossRef CAS
- H. Rai, S. Gupta, S. Kumar, J. Yang, S. K. Singh, C. Ran and G. Modi, J. Med. Chem., 2022, 65, 8550–8595 CrossRef CAS PubMed
- K. K. Laali, A. T. Zwarycz, S. D. Bunge, G. L. Borosky, M. Nukaya and G. D. Kennedy, ChemMedChem, 2019, 14, 1173–1184 CrossRef CAS PubMed
-
D. D. Perrin, W. L. F. Armango and D. R. Perrin, Purification of laboratory Chemicals, Pergamon, Oxford, UK, 1986 Search PubMed
- A. D’Aleó, M. H. Sazzad, D. H. Kim, E. Y. Choi, J. W. Wu, G. Canard, F. Fages, J. C. Ribierre and C. Adachi, Chem. Commun., 2017, 53, 7003–7006 RSC
- T. Kim, W. Kim, H. Mori, A. Osuka and D. Kim, J. Phys. Chem. C, 2018, 122, 19409–19415 CrossRef CAS
- Z. F. Fang, F. W. Pei, Z. J. Chao, L. S. Hui, Q. X. Ying and W. S. Xiang, Indian J. Chem., 2016, 55, 713–717 Search PubMed
- R. S. Singh, S. Mukhopadhyay, A. Biswas and D. S. Pandey, Chem. – Eur. J., 2016, 22, 753–763 CrossRef CAS PubMed
- H. Li, Y. Guo, G. Li, H. Xiao, Y. Lei, X. Huang, J. Chen, H. Wu, J. Ding and Y. Cheng, J. Phys. Chem. C, 2015, 119, 6737–6748 CrossRef CAS
- N. Zhao, Z. Yang, J. W. Y. Lam, H. H. Y. Sung, N. Xie, S. Chen, H. Su, M. Gao, I. D. Williams, K. S. Wong and B. Z. Tang, Chem. Commun., 2012, 48, 8637–8639 RSC
Footnote |
| † Electronic supplementary information (ESI) available: 1H, 13C, 11B, and 19F NMR, ESI–MS, Uv-vis, and fluorescence spectra, plots and tables are provided. See DOI: https://doi.org/10.1039/d4nj00295d |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2024 |