
From molecules in solution to molecules on surfaces – using supramolecular dyads to form functional self-assembled networks on graphene†
Quentin
Fernez
a,
Shiva
Moradmand
b,
Michele
Mattera
a,
William
Djampa-Tapi
c,
Céline
Fiorini-Debuisschert
 c,
Fabrice
Charra
c,
David
Kreher
d,
Fabrice
Mathevet
a,
Imad
Arfaoui
b and
Lydia Sosa
Vargas
*a
c,
Fabrice
Charra
c,
David
Kreher
d,
Fabrice
Mathevet
a,
Imad
Arfaoui
b and
Lydia Sosa
Vargas
*a
aSorbonne Université, CNRS, Institut Parisien de Chimie Moléculaire (IPCM), F-75005, Paris, France. E-mail: lydia.sosa-vargas@sorbonne-universite.fr
bSorbonne Université, CNRS, Laboratoire de la Molécule aux Nano-Objets: Réactivité, Interactions et Spectroscopies (MONARIS), F-75005, Paris, France
cUniversité Paris-Saclay, CEA-CNRS, Service de Physique de l’état condensé (SPEC), F-91191, Gif-sur-Yvette, France
dUniversité Paris-Saclay, UVSQ-CNRS, Institut Lavoisier de Versailles (ILV), F-78035, Versailles, France
First published on 16th August 2022
Abstract
Using supramolecular chemistry to functionalise graphene for photonic applications is a challenging issue due to graphene's capacity to quench any emission from molecules adsorbed on its surface. To overcome this problem, we propose the use of molecular dyads to form ordered self-assemblies on graphene-like substrates. These dyads are designed to reduce surface quenching by positioning the emissive component out-of-the plane of the substrate. We use a zinc porphyrin and a phthalocyanine as molecular pedestals to immobilise the dyads onto the graphene thanks to a nanoporous network; and a perylenetetracarboxylic diimide, as the emissive component. This approach has been recently reported, however; we have found that the formation of these dyads is an intricate process, that requires an in-depth study of the solution phase before its study on a graphene surface. We demonstrate that two types of dyads can be formed in solution, depending on the supramolecular interactions that dominate the equilibrium, and the type of molecular pedesal used. A metal–ligand association was observed between the perylene and the porphyrin pedestal, whilst the phthalocyanine leads to a dyad formed via π–π interactions. We also conclude that scanning tunneling microscopy is not a reliable technique to characterise the on-surface assemblies, due to a strong probe–molecule interaction. Other spectroscopic techniques; such as epifluorescence micro-spectroscopy coupled with atomic force-microscopy, were investigated, however we found it is ambitious to rely solely on these techniques, to correlate observations from the nano to the micrometric scale.
 Lydia Sosa Vargas | Lydia Sosa Vargas has been a CNRS researcher at the Paris institute of molecular chemistry since 2017. Originally from Mexico, she obtained her PhD at the University of East Anglia in the UK under the supervision of professors Andrew N. Cammidge and Michael J. Cook. She carried out her first postdoctoral position in Japan, at the National Institute of Advanced Industrial Science and Technology (AIST) with Dr Yo Shimizu. In 2015, she joined the polymer chemistry team at Sorbonne University before being recruited by the french national centre of scientific research (CNRS). She is member of the Royal society of chemistry, the French chemical society and the Mexican system of researchers (SNI-1). Since 2020 she has been member of the IUPAC subcommittee of polymer terminology and polymer education. In 2019, she was an awardee of the IUPAC periodic table of young researchers. Her research focuses on the synthesis of pi-conjugated materials for applications in organic electronics and supramolecular self-assembly. |
Introduction
Graphene has captivated the focus of materials researchers, since its discovery and even more now that high-quality, micro-scale CVD graphene can be produced at competitive costs.1,2 It is easily transferable onto a variety of substrates, which makes it attractive for applications in photonics,3 electronics,4–6 and metamaterials,7,8 for example. However, being a mostly-inert, zero-bandgap semiconductor, any advanced application requires the modulation of its properties via covalent or non-covalent functionalisation.9 Between these, the non-covalent approach has gained popularity, since we can exploit graphene's crystal lattice to direct the physisorption of a variety of molecules on its surface, in a controlled manner.10,11 Significant work has been carried out on tuning its electronic properties,12–14 however its use in photonic devices remains relatively unexplored.3,15 The main reason is due to the strong Dexter energy transfer that occurs between an optically or electronically-active molecule and the graphene; which results in the quenching of the organic molecule's excited states when adsorbed onto the surface.16,17 To overcome this, the organic molecules must have a specific design where the optically active component is located at a sufficient distance to avoid the non-radiative exchange. The first example of a fluorescent molecular system on graphene employed the use of a cyclophane molecular spacer to separate the emissive ‘functional’ unit from the surface-binding pedestal.18 Other designs, with mono, bi and tripodal pyrene motifs that attach ‘functional’ components to graphene have also been used.19,20 However, in all these cases, the molecules used do not allow us to control the distance and orientation of the optically-active component from the surface, and at the same time obtain robust, long-range self-assemblies on graphenoid substrates.21 To address this problem, a strategy based on functional self-assembling molecules (dyads) that combine an optically active component with a graphene-binding pedestal was proposed, supported by preliminary observations.22 In this report, a single system composed of a zinc phthalocyanine (ZnPc) associated to a perylenetetracarboxylic diimide (PDI) chromophore was studied by STM and confocal spectroscopy, with the aim to obtain an emitting 2D-network on a graphene substrate. This work demonstrates the feasibility of the strategy through their computational studies, demonstrating that the metal–ligand interaction occurs. However, the methodology used in this paper for confirming the successful coordination, via UV-vis spectroscopy and the Job plot method has been proven to be obsolete.23,24 This, together with the inconclusive results presented when studying the surface-confined monolayer, brings into question what species are actually present on the graphene surface upon deposition. This motivated us to explore further this dyad system; and more specifically, the supramolecular interactions in solution, prior to their study on surface.In this paper, we compare two different pedestals combined with two different fluorophores based on the same model. A thorough characterization of the assemblies in solution and on-surface; by UV-vis spectroscopy, NMR spectroscopy and Scanning Tunnelling Microscopy (STM) permit to show, beyond the naïve picture, the existence of a competing supramolecular association stabilized by π–π stacking which is often the dominant one.
Since all three molecular components (Pcs, Ps, PDI's) are well-known to form on their own molecular assemblies on graphene-like substrates via π–π interactions;25–28 we purposefully introduced bulky 3,5-tert-butylphenoxy groups at the bay positions of the perylene core in order to inhibit its adsorption on the surface. This PDI (BPDI4Py) is capped at the N-positions with a pyridyl group at either end, which is known to coordinate to the zinc cation inside the Pc or DPP cavity.29–31 A second PDI (BPDIC7), without pyridil-capping groups, was also used as reference as shown in Fig. 1.
 | ||
Fig. 1 General scheme of dyad design and components: (left to right) ZnPc and ZnDPP molecular pedestals, the perylene-functional components BPDIC7 and BPDI4Py, and the resulting dyad and triads obtained via metal–ligand (M–L) or π–π associations in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 or 2:1 ratio. :1 or 2:1 ratio. | ||
In this work, we aim to give a detailed insight into the intricate process of surface functionalisation via supramolecular self-assembly. We discuss the formation and stability of the coordination dyads in chloroform and toluene, at different concentrations and using different molecular pedestals (ZnPc, ZnDPP). We report the finding of an unexpected perylene–phthalocyanine dyad, resulting from π–π stacking between the ZnPc and the two different PDIs used. Finally, these dyads were deposited onto highly oriented pyrolytic graphite (HOPG) and graphene as guest molecules within a TSB-host network. These surface-confined systems were then studied by scanning tunneling microscopy (STM), to characterise the self-assembling properties. We also used-epifluorescence micro-spectroscopy to investigate if any differences in the electronic properties can be observed when analysing the metal–ligand and the π-stacked dyad.
Results and discussion
We investigated the association of the ZnPc and ZnDPP pedestals with the BPDI4Py by performing a series of titrations using three different spectroscopic methods: absorbance, emission and nuclear magnetic resonance spectroscopy to monitor the changes in physical parameters. The absorbance titrations quickly revealed that the low solubility of the molecules studied would limit the reliability of concentrations used and therefore make it difficult to obtain accurate results. The ZnPc in particular, presented aggregation even at micromolar concentrations in chloroform (CHCl3). Fig. 2 shows the results obtained from the absorbance titration experiments. First, in Fig. 2a, we observe that during the addition of BPDI4Py to the ZnPc solution, the ZnPc's absorption at 671 nm (in black) increases from 0.25 to 0.45, and then reaches a plateau after the addition of almost 1 equivalent of BPDI4Py. This absorption remains constant until reaching ≈ 2 eq. of BPDI4Py added. We believe these regime changes can be attributed to the formation of the 2:1 and 1:1 (ZnPc–BPDI4Py) dyad. The reverse titration (ZnPc to BPDI4Py) was also performed, however no clear trends could be observed due to the predominant aggregation of ZnPc observed during this experiment (Fig. S3, ESI†).
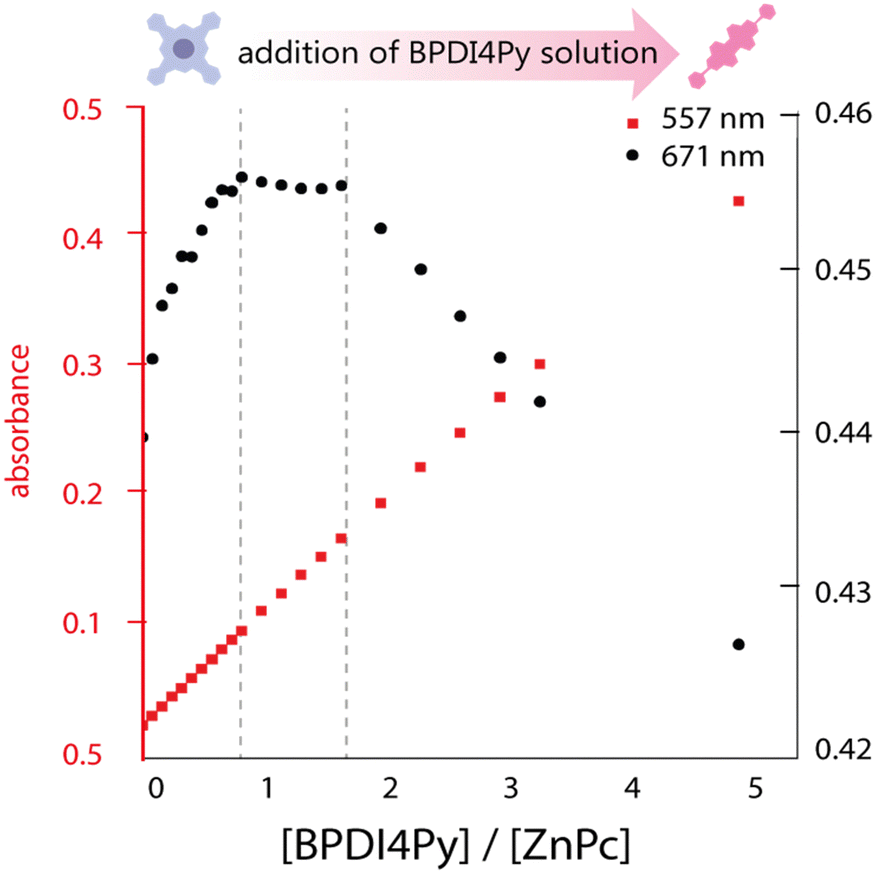 | ||
| Fig. 2 Plot of the maximum absorbance peaks of ZnPc (671 nm) and BPDI4Py (557 nm) against the ratio of BPDI4Py/ZnPc added. | ||
Next, we titrated a BPDI4Py solution with the ZnDPP pedestal. Here, we were unable to monitor the same wavelengths as with the ZnPc since the porphyrin also absorbs at the region corresponding to the BPDI4Py's absorption maxima (Fig. S2, ESI†). So, for the plot in 3 a secondary treatment of the data was performed. For each spectrum obtained during the titration, we subtracted the spectrum of a pure solution of ZnDPP. This resulted in the appearance of a new absorption peak at 404 nm which we attribute to the formation of the ZnDPP–BPDI4Py dyad (Fig. S4, ESI†). As we can see on Fig. 3, we once again observe changes in the absorption at 404 nm, after adding approximately 0.5 equivalents of BPDI4Py, which corresponds to the 2:1 triad, where two ZnDPP pedestals coordinate to a single perylene. In this experiment, we were also able to observe a clear blue-shift of 0.7 nm of the ZnDPP's Soret peak (413 nm) during the BPDI4Py addition (Fig. S5, ESI†), which is usually reported with the metal–ligand type associations.29–32
 | ||
| Fig. 3 Plot of the subtracted absorbance peaks of BPDI4Py (404 nm) and ZnDPP (413 nm) against the ratio of BPDI4Py/ZnDPP added. | ||
For the second set of experiments, we titrated both pedestals using the BPDIC7, expecting that without pyridil groups, no association with the Zn-pedestals should occur. Surprisingly, the ZnPc–BPDIC7 titration plot follows a similar trend to that of the ZnPc–BPDI4Py system (Fig. S6a, ESI†). Therefore, we believe that without the pyridyl groups, the BPDIC7 is still able to associate with the zinc pedestals. In contrast, the ZnDPP does not appear to interact with BPDIC7 as strongly or in the same manner, meaning that each pedestal possibly favours a different type of association with the PDIs. We also carried out titrations using fluorescence spectroscopy to confirm the results obtained, expecting to observe different variations in the emission maxima of the PDI signal depending on the type of association taking place. However, as with the absorbance titrations, the low solubility of the ZnPc seriously limited our ability to obtain reliable data, and the slight spectral changes observed for the ZnDPP system could mostly be attributed to dilution effects. (Fig. S7 and S8, ESI†).
Finally, we used NMR spectroscopy to investigate how the PDIs and Zn-pedestals are associating and explain the differences observed in the previous studies. In a Zn–pyridil (M–L type) association, we expect to see a pronounced change in the chemical shift of the protons located closest to the pyridil group (Ha, Hb) as the pyridil coordinates to the zinc cation.29,33 This is clearly demonstrated with the ZnDPP pedestal (Fig. 4a); with the Ha-protons shifting upfield, disappearing and then re-appearing at around 3.5 ppm. The Hb-proton also behaves in a similar manner, although with a smoother transition. With the addition of ZnPc however (Fig. 4b), both Ha, Hb-protons remain at the same position and the only variations that can be observed come from the aromatic protons located on the BPDI4Py core (Hc, Hd, He).
 | ||
| Fig. 4 (a) 1H NMR spectra in CDCl3 of a titration of BPDI4Py solution (0.5 mL, 5.6 mM) with increasing amounts of a solution of ZnDPP (50 μL, 5.6 mM). Protons from the pyridil group (Ha, Hb) are shaded in pink and purple respectively, whilst those from the perylene core (Hc, Hd, He) are in grey. (4b) NMR spectra in CDCl3 of a titration of BPDI4Py solution with increasing amounts of a ZnPc solution (180 μL, 0.13 mM). Increasing molar ratios of the added pedestals are indicated on the right. | ||
From these results, we deduce that no pyridyl–zinc (M–L) coordination between BPDI4Py and ZnPc occurs during this titration. Thus, the noticeable shift of the Hc, d and e protons in both cases can be explained by a face-on (π–π) interaction between the aromatic pedestals and perylene. This result confirms that with the ZnDPP, in addition to the pyridil–Zn coordination; a different association (most likely π–π) is also possible. To verify this assumption, we repeated the titrations using the BPDIC7. Fig. 5 shows that with both pedestals, the protons from the perylene core move slightly upfield, following a similar trend to that of the ZnPc–PBDI4Py experiment, confirming the face-on association between PDI and Zn-Pc via π–π interactions. By plotting the chemical shift of protons c, d and e against an increasing ratio of ZnPc or ZnDPP added, we were able to observe a significantly stronger effect on the BPDIC7's core protons upon addition of ZnPc than with ZnDPP (Fig. S9, ESI†). We assume that the smaller aromatic core and sterically-hindering phenyl groups of the porphyrin are responsible for this weaker (or non-existent) interaction at these concentrations.
 | ||
| Fig. 5 (a) 1H NMR spectra in CDCl3 of a titration of BPDIC7 solution (0.5 mL, 0.055 mM) with increasing amounts of ZnPc (180 μL, 0.13 mM) and (b) ZnDPP (400 μL, 13 mM) in CDCl3. | ||
The data obtained from the ZnDPP–BPDI4Py NMR titration experiments was fitted using different association models (1:1, 1:2, and 2:1, Fig. S10, ESI†).23 We plotted the shift of the Hb protons from the pyridyl group (BPDI4Py-host) with the concentration of guest molecule added (ZnDPP). Using the residual error plots, a reliable association constant of 1.099 × 103 ± 2.3% M−1 was obtained using the 1:2 model for the ZnDPP to BPDI4Py titration. In the opposite sense (BPDI4Py to ZnDPP), the best fit resulted in an association constant of 4.53 × 103 ± 9.5% M−1 with the 2:1 model. An association constant for the 1:1 model was also obtained with a value of 946 ± 2.5% M−1. The rates obtained are not quite comparable to the values reported with similar systems,30,33 but we know that with ZnPc case, aggregation is predominant, and thus the M–L association is not favoured. And although previous reports on similar systems report that that the metal–ligand associations quite labile in solution,27,29 these studies have given us sufficient evidence to conclude that the dyad formed are sufficiently stable to be characterised. Moreover, we were able to observe two different types of dyads and triads formed, via different supramolecular interactions; through metal–ligand coordination and surprisingly, via π–π stacking.
The titration studies using the different pedestals and fluorophores enabled us to have a better knowledge of what we have in solution, and so, we were keen to know what we have on the surface after drop casting: the self-assembling behaviour of these two different dyads and to check the integrity of the molecules after deposition.
Scanning tunneling microscopy studies on HOPG
Since the behaviour in solution of ZnDPP–BPDI4Py dyads differs from that of the ZnPc–BPDI4Py molecules, an attempt was made to observe by STM, for the first time, the layers obtained after deposition by evaporation. In the previously reported work, the STM is used as a tool for observing the molecular assemblies obtained.22 In contrast, we will mainly employ it to attempt to detect unequivocally the presence of a coordination-driven, ZnDPP–BPDI4Py dyad.The solution of the ZnDPP–BPDI4Py dyad was drop casted on the C10-TSB-patterned, highly oriented pyrolytic graphite (HOPG) surface. Fig. 6 shows the STM image collected after this process.
 | ||
| Fig. 6 STM image of a 50 × 50 nm2 area with C10-TSB and the deposited ZnDPP–BPDI4Py dyad on HOPG (bias: −0.2 V, current: 20 pA). Selected areas showing an empty TSB pore (a), the ZnDPP pedestal trapped inside a pore (b) and the ZnDPP–BPDI4Py dyad trapped by the C10-TSB network (c). | ||
Here, we observe three different types of patterning with varying contrast and which are reasonably assigned to: the C10-TSB network with empty pore (Fig. 6a); a pore filled with a ZnDPP molecule with a low contrast (Fig. 6b); and, the ZnDPP–BPDI4Py dyad trapped by the C10-TSB network appearing as a perturbated very bright spot (Fig. 6c). The STM images of C12-TSB and C10-TSB-ZnDPP assemblies on HOPG are shown in Fig. S11, ESI.† It is important to note that for the ZnDPP–BPDI4Py dyad, the feature of Fig. 5 could only be obtained at the air/solid interface. Moreover, this feature disappears when the bias is higher than 400 mV, demonstrating that the electric field (of several GV m−1) generated during STM imaging, plays an important role. Indeed, this can be seen by the presence of disturbances parallel to the scan direction on the bright spots (Fig. 6c) that have been attributed to the presence of the M–L complex with an out-of-plane BPDI4Py molecule. Thus, this could be consistent with out-of-plane, orthogonal ZnDPP–BPDI4Py dyads which interact more strongly with the STM tip (through the high electric field) than the π–π ZnDPP–BPDI4Py dyads that are expected to be more parallel to the surface. In addition to destabilizing the network of these out-of-plane dyads by increasing the electric field between the STM tip and the HOPG, we were also able to easily remove the adsorbed dyads from the host network. It is also important to note that such disturbances have never been observed in the work previously reported;22 suggesting that the PDI chromophores may not be oriented out of plane. At this point, we also refrain to confirm that this is indeed the case for our coordinated dyad, since it remains difficult to interpret observations made on STM images in an absolute way. Indeed, the apparent height in STM is mainly subjected to electronic local densities of states (LDOS) at Fermi level, rather than the physical height of the sample. For this reason, conditions such as the electric bias, the STM tip and other environment conditions can have an effect on the contrast observed.34,35 This is one of the reasons why we purposefully compared different features from the same image (highlighted in Fig. 6) based on their qualitative differences to show that they correspond to different species, rather than discussing the absolute heights.
Therefore, if we compare the patterning of the ZnDPP–BPDI4Py dyads with the ZnPc-chromophore dyads from Kim et al.,22 we cannot completely exclude that the brightest spots (Fig. 6 and c) could also correspond to the presence of the π–π driven ZnDPP–BPDI4Py complex, evidenced in the solution studies. However, in Fig. 6, we observe a different and surprising patterning where the ZnDPP–BPDI4Py dyads appear to fill only one out of three pores, with the lone ZnDPP pedestal filling most of the remaining ones. We propose this could be due to steric repulsions between the perylene units which we assume are orthogonal to the surface. In contrast, in the case of ZnPc-chromophore dyads reported, most of the pores are filled.22 From all these STM observations, it is very difficult to claim that the pedestal-chromophore associations, π–π or M–L, observed in solution are fully transferable to the surface after drop casting. In order to go further than the STM study presented by Kim et al., and to force the π–π ZnPc-PDI association in solution, we studied the ZnPc dyad with the BPDIC7 (Fig. 1). Lacking pyridyl groups, the M–L association with this PDI is not possible either in solution or on the surface. The solution of ZnPc–BPDIC7 molecules was drop casted on the C12-TSB-patterned HOPG surface (Fig. 7). As for ZnDPP–BPDI4Py dyad, we observe three different types of patterning with different contrast and which could be assigned to: the C12-TSB network with empty pore (Fig. 7a); a pore filled with a ZnPc molecule with a low contrast (Fig. 7b); and, the ZnPc–BPDIC7 dyad trapped by the C12-TSB network appearing as a homogeneous very bright spot (Fig. 7c). Comparing Fig. 6 and 7, and assuming that the brightest spots of STM images, respectively, correspond to the presence of the dyads, we first notice that the coverage rate of the ZnPc–BPDIC7 dyads is almost three times higher than that of the ZnDPP–BPDI4Py dyads. Then, in the case of ZnDPP–BPDI4Py, the bright spots are not homogeneous, and are easily disturbed while those corresponding to ZnPc–BPDIC7 dyads are homogeneous and well defined. These two characteristics, coverage rate and aspect ratio of the bright spots, would then allow us to distinguish qualitatively, between the two types of associations, π–π and M–L, after deposition on a surface from a solution. In this case, this would call into question the interpretation of the STM results of the previous work.
 | ||
| Fig. 7 STM image of a 50 × 50 nm2 area with ZnPc–BPDIC7 deposited on C12-TSB. As with the ZnDPP–BPDI4Py dyad, we can observe similar regions of (a) empty TSB cavities, (b) ZnPc pedestals only and (c) the brightly contrasted ZnPc–BPDIC7 dyad. | ||
Note that without a pedestal, the BPDI4Py and BPDIC7 molecules are not able to insert themselves into the TSB cavities (Fig. S13, ESI†).
Fluorescence microscopy studies on graphene
We attempted to study the optical properties of the 2D-assemblies resulting from the different dyads (M–L and π–π), in order to evidence the different surface de-coupling effects. We investigated the emission of the 2D-network of the ZnPc–PDI4Py dyad deposited on a (10 × 10 μm) graphene monolayer (previously patterned with a C12-TSB network) using simultaneous and correlated AFM and fluorescence microspectroscopy and imaging (see Fig. 8).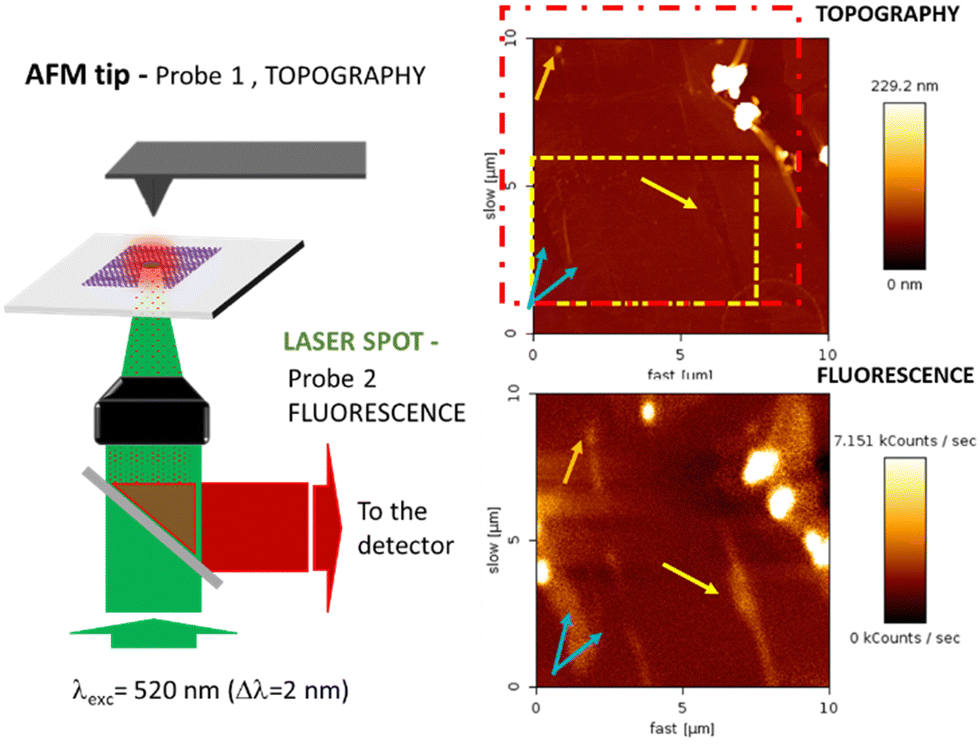 | ||
| Fig. 8 (left) Combined Fluorescence and AFM microscopy set-up with an inverted microscope (Olympus IX71) coupled to a cantilever type AFM platform (NanoWizard III, JPK) and associated to a focused laser excitation (SuperK EVO NKT Photonics supercontinuum laser, associated to an adjustable band pass filtering (SuperK Varia), here l = 520 nm (Dl = 2 nm) focused through a 100× oil immersion microscope objective). Topography and fluorescence mapping of a ZnPc–BPDI4Py, TSB system deposited on a graphene monolayer (right). | ||
The red dotted square indicates the slightly shifted fluorescence acquisition frame relative to the AFM map following a not fully-perfect tip/laser adjustment. In order to correlate topographic and optical measurements, the requirement for this experiment was the preliminary alignment of the AFM tip with the laser spot at the focus, the sample being then raster scanned enabling simultaneous topography (tapping mode) and fluorescence recording. Important to note also is that the two correlated images are obtained through two probes of different sizes (AFM tip: ≈ 1–10 nm and laser spot ≈ 300 nm) which explains the difference in resolution of these correlated photon and topography images. The excitation intensity was chosen to be very small (≈ 90 μW average power), in order to avoid any photochemical degradation of the sample. The fluorescence emission is collected through the same microscope objective and separated from the incident light by a dichroic mirror (SemRock FF735-Di670-25x36). This signal is then sent to a channel plate multiplier working in the photon counting mode (PerkinElmer MP-993-CL). As indicated by the different sets of arrows, beyond the two large clusters that can be easily identified in the upper right corner of the figure, quite large signal variations can also be observed from different areas. These can either be associated to dips or peaks in the topography, illustrating the non-homogeneous nature of the self-assembled monolayer deposition at the scale of the optical measurements. The dashed rectangle in yellow indicates the cropped area which topography is shown in more detail in Fig. S14 (ESI†).
With this specific set-up, we hoped to determine the characteristic signal of the BPDI4Py component of the dyad when confined to the graphene via the ZnPc pedestal. However, AFM evidenced the presence of many defects; such as tears, ripples and folds in the graphene monolayer. This means that any fluorescence signals observed at this scale cannot be attributed solely to the dyad assemblies, and are most likely the result of microaggregates present in the defects observed. From this experience, we can affirm that attempting to study this nanoscale system at a larger scale by just simply extrapolating the molecular organization observed through STM imaging is not possible.
Experimental
Synthesis of dyad components The ZnDPP was synthesized following reported procedures. The PDI4Py and the PDIC7 were obtained from commercially-available perylene dianhydride (PDA) using a modified synthetic pathway, and the ZnPc used purchased from Merck® (97%, sublimated). (Please refer to the ESI† for detailed synthetic procedures and characterisation data).Preparation of dyads: for each dyad, stoichiometric amounts of the pedestal units (ZnDPP, ZnPc) were mixed with the perylene counterparts (BPDI4Py, BPDIC7) in a 1:1 ratio, and dissolved in chloroform. The mixture was stirred at 40 °C during 30 minutes, after which the solvent is evaporated under reduced pressure.
Preparation of monolayers on HOPG
:1 ratio of the two species. The solvent was removed by in vacuo slow evaporation, to obtain a purple powder of the ZnPc–BPDI4Py dyad. 0.5 mg of this powder was dissolved into 500 μL of toluene. From this solution (0.65 μM), 50 μL were taken and added to 250 μL of toluene. This solution will be referred as BDPI-4-ZnPc-dil1 (0.13 μM).
Scanning tunneling microscopy experiments
Scanning tunneling microscopy (STM) was used to study the deposition of the different molecules, on the modified HOPG surface. All the experiments were performed using Keysight STM scanner working with 5100 AFM/SPM microscope system. The tip-sample bias voltage and tunneling current set points were kept between –1000 mV and −300 mV and between 11 pA and 15 pA, respectively. STM images were recorded in constant current mode, at room temperature in air or at liquid (1 – phenyl octane) – _solid (HOPG) interface using mechanically cut Pt/Ir (80/20) tips (GoodFellow, UK). All experiments were repeated several times with different tips, at different spots on the sample, and the results presented in this work are representative and consistent with the more comprehensive data set. The graphical contrast of each STM image is adjusted independently using the scale of the chosen pseudo-color palette for the best readability. We do not discuss absolute heights of different images here; the chosen pseudo-color scale has been added to provide the reader with complete experimental data.Conclusions
Taking advantage of supramolecular interactions, we were able to prepare a series of dyads composed of zinc-metallated aromatic macrocycles (ZnPc, ZnDPP) as pedestal and functionalised perylenediimides (BPDI4Py, BPDIC7) as chromophore. By studying these dyads in solution, we observed that depending on the zinc pedestal chosen, it is possible to obtain dyads where firstly the BPDI4Py is associated to the porphyrin through the expected metal–ligand coordination and, unexpectedly and exclusively through a π–π association with the ZnPc. These dyads were then deposited by a simple drop-cast deposition on an HOPG substrate an immobilized through a nanoporous network (TSB). From the STM observations, we can confirm the presence of two different dyad geometries. In the first case, with the ZnPc–BPDI4Py (evidenced as π–π association in solution) we observe a close packed, network similar to that previously reported by Kim et al.22 In contrast, the ZnDPP–BPDI4Py dyad, which in solution shows predominantly a metal–ligand association, presents a less compact network where the dyad is filling 1 out of 3 cavities. This leads us to conclude that either in solution or on surface, each pedestal favours a different type of association. Our main results, demonstrate that our dyads are able to form organised monolayers on sp2 carbon substrates like HOPG and graphene, and we can control the positioning of the emissive PDI component in relation to the surface. STM enabled us to confirm that these dyads self-assembled well when deposited on HOPG, and show that the patterning observed is very different depending on the association that holds these dyads together. However, when attempting to measure the photonic properties, we find that the situation is much more complicated when measuring at the micron scale, given that small PDI aggregates located on the various defects present in the graphene monolayer are usually the source of the emission signals observed. Thanks to these experiments we have now optimised our deposition method and will explore in the near future other microspectroscopic techniques that will enable us to study the photonic properties in a more accurate manner.Finally, although we encountered some limitations when studying the de-coupling of our supramolecular dyads; we believe this accessible method of functionalisation, combined with the versatility of the molecular design of the building blocks, make it an attractive and practical approach for graphene functionalisation for photonic applications.
Author contributions
Q. F. performed the synthesis, characterisation and titration experiments of the studied molecules; S. M. and M. M. carried out the self-assembly studies by STM; and W. D. W. D.-T.-T. and C. F.-D. the AFM-fluorescence microscopy experiments. C. F.-D., F. C., I. A. and L. S. V. verified the analytical methods and contributed to the interpretation of the results. D. K., F. M., C. F.-D., I. A. and L. S. V. were responsible for the supervision of Q. F., M. M. S. M. and W. D.-T. data visualisation was prepared by Q. F., S. M., C. F.-D., I. A. and L. S. V. The presented idea was conceived by L. S. V. Lastly, L. S. V. and I. A. wrote the manuscript with contributions from C. F.-D. and F. C.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Labex MiChem inititative. The authors would like to thank the FR2769 NMR platform for the NMR experiments performed.References
- A. K. Geim and K. S. Novoselov, Nat. Mater., 2007, 6, 183–191 CrossRef CAS PubMed.
- N. Petrone, C. R. Dean, I. Meric, A. M. van der Zande, P. Y. Huang, L. Wang, D. Muller, K. L. Shepard and J. Hone, Nano Lett., 2012, 12, 2751–2756 CrossRef CAS PubMed.
- F. Bonaccorso, Z. Sun, T. Hasan and A. C. Ferrari, Nat. Photonics, 2010, 4, 611–622 CrossRef CAS.
- S. Bae, H. Kim, Y. Lee, X. Xu, J.-S. Park, Y. Zheng, J. Balakrishnan, T. Lei, H. Ri Kim, Y. I. Song, Y.-J. Kim, K. S. Kim, B. Özyilmaz, J.-H. Ahn, B. H. Hong and S. Iijima, Nat. Nanotechnol., 2010, 5, 574–578 CrossRef CAS PubMed.
- J. Wu, M. Agrawal, H. A. Becerril, Z. Bao, Z. Liu, Y. Chen and P. Peumans, ACS Nano, 2010, 4, 43–48 CrossRef CAS PubMed.
- N. O. Weiss, H. Zhou, L. Liao, Y. Liu, S. Jiang, Y. Huang and X. Duan, Adv. Mater., 2012, 24, 5782–5825 CrossRef CAS PubMed.
- B. Wang, X. Zhang, F. J. García-Vidal, X. Yuan and J. Teng, Phys. Rev. Lett., 2012, 109, 073901 CrossRef PubMed.
- H. Lin, B. C. P. Sturmberg, K.-T. Lin, Y. Yang, X. Zheng, T. K. Chong, C. M. de Sterke and B. Jia, Nat. Photonics, 2019, 13, 270–276 CrossRef CAS.
- C. Anichini and P. Samorì, Small, 2021, 17, 2100514 CrossRef CAS PubMed.
- J. M. MacLeod and F. Rosei, Small, 2014, 10, 1038–1049 CrossRef CAS PubMed.
- V. Georgakilas, M. Otyepka, A. B. Bourlinos, V. Chandra, N. Kim, K. C. Kemp, P. Hobza, R. Zboril and K. S. Kim, Chem. Rev., 2012, 112, 6156–6214 CrossRef CAS PubMed.
- X. Dong, D. Fu, W. Fang, Y. Shi, P. Chen and L.-J. Li, Small, 2009, 5, 1422–1426 CrossRef CAS PubMed.
- R. Phillipson, C. J. L. de la Rosa, J. Teyssandier, P. Walke, D. Waghray, Y. Fujita, J. Adisoejoso, K. S. Mali, I. Asselberghs, C. Huyghebaert, H. Uji-i, S. D. Gendt and S. D. Feyter, Nanoscale, 2016, 8, 20017–20026 RSC.
- H. Lee, K. Paeng and I. S. Kim, Synth. Met., 2018, 244, 36–47 CrossRef CAS.
- A. N. Grigorenko, M. Polini and K. S. Novoselov, Nat. Photonics, 2012, 6, 749–758 CrossRef CAS.
- P. Anger, P. Bharadwaj and L. Novotny, Phys. Rev. Lett., 2006, 96, 113002 CrossRef PubMed.
- A. Kasry, A. A. Ardakani, G. S. Tulevski, B. Menges, M. Copel and L. Vyklicky, J. Phys. Chem. C, 2012, 116, 2858–2862 CrossRef CAS.
- S. Le Liepvre, P. Du, D. Kreher, F. Mathevet, A.-J. Attias, C. Fiorini-Debuisschert, L. Douillard and F. Charra, ACS Photonics, 2016, 3, 2291–2296 CrossRef CAS.
- J. A. Mann and W. R. Dichtel, ACS Nano, 2013, 7, 7193–7199 CrossRef CAS PubMed.
- M. Garrido, J. Calbo, L. Rodríguez-Pérez, J. Aragó, E. Ortí, M. Á. Herranz and N. Martín, Chem. Commun., 2017, 53, 12402–12405 RSC.
- L. Sosa-Vargas, E. Kim and A.-J. Attias, Mater. Horiz., 2017, 4, 570–583 RSC.
- B. Kim, C. Cho, I. Arfaoui, C. Paris, C. Petit, T. L. Bahers, E. Kim and A.-J. Attias, Mater. Horiz., 2020, 7, 2741–2748 RSC.
- P. Thordarson, Chem. Soc. Rev., 2011, 40, 1305–1323 RSC.
- D. B. Hibbert and P. Thordarson, Chem. Commun., 2016, 52, 12792–12805 RSC.
- J. M. Gottfried, Surf. Sci. Rep., 2015, 70, 259–379 CrossRef CAS.
- Y. Qian, B. Liu, W. Duan and Q. Zeng, J. Porphyrins Phthalocyanines, 2018, 22, 717–725 CrossRef CAS.
- F. Würthner, C. R. Saha-Möller, B. Fimmel, S. Ogi, P. Leowanawat and D. Schmidt, Chem. Rev., 2016, 116, 962–1052 CrossRef PubMed.
- S. Yoshimoto and N. Kobayashi, in Functional Phthalocyanine Molecular Materials, ed. J. Jiang, Springer, Berlin, Heidelberg, 2010, pp. 137–167 Search PubMed.
- B. Gao, Y. Li, J. Su and H. Tian, Supramol. Chem., 2007, 19, 207–210 CrossRef CAS.
- M. E. El-Khouly, A. M. Gutiérrez, Á. Sastre-Santos, F. Fernández-Lázaro and S. Fukuzumi, Phys. Chem. Chem. Phys., 2012, 14, 3612–3621 RSC.
- M. Lederer, U. Hahn, J. Fernández-Ariza, O. Trukhina, M. S. Rodríguez-Morgade, C. Dammann, T. Drewello, T. Torres and D. M. Guldi, Chem. – Eur. J., 2015, 21, 5916–5925 CrossRef CAS PubMed.
- M. E. El-Khouly and S. Fukuzumi, Photochem. Photobiol. Sci., 2016, 15, 1340–1346 CrossRef CAS PubMed.
- J. Fernández-Ariza, R. M. Krick Calderón, M. S. Rodríguez-Morgade, D. M. Guldi and T. Torres, J. Am. Chem. Soc., 2016, 138, 12963–12974 CrossRef PubMed.
- P. K. Hansma and J. Tersoff, J. Appl. Phys., 1987, 61, R1–R24 CrossRef CAS.
- J. K. Gimzewski and R. Möller, Phys. Rev. B: Condens. Matter Mater. Phys., 1987, 36, 1284–1287 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2tc01331b |
| This journal is © The Royal Society of Chemistry 2022 |