
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Pyrolytic lignin: a promising biorefinery feedstock for the production of fuels and valuable chemicals†
M. B.
Figueirêdo
 a,
I.
Hita
b,
P. J.
Deuss
a,
R. H.
Venderbosch
c and
H. J.
Heeres
*a
a,
I.
Hita
b,
P. J.
Deuss
a,
R. H.
Venderbosch
c and
H. J.
Heeres
*a
aDepartment of Chemical Engineering, ENTEG, University of Groningen, Nijenborgh 4, 9747 AG, Groningen, the Netherlands. E-mail: h.j.heeres@rug.nl
bMultiscale Reaction Engineering, KAUST Catalysis Center (KCC), King Abdullah University of Science and Technology (KAUST), Thuwal 23955-6900, Saudi Arabia
cBTG Biomass Technology Group BV, Josink Esweg 34, 7545 PN, Enschede, the Netherlands
First published on 6th May 2022
Abstract
Lignocellulosic biomass is a key feedstock for the sustainable production of biofuels, biobased chemicals and performance materials. Biomass can be efficiently converted into pyrolysis liquids (also known as bio-oils) by the well-established fast pyrolysis technology. Currently, there is significant interest in the application of fast pyrolysis technology as principle biomass conversion technology due to its feedstock flexibility, low cost and high energy conversion efficiency, with many emerging commercial enterprises being established around the globe. Upgrading of the bio-oils is a requisite, and is complicated by its complex and heterogeneous organic nature. Pyrolysis liquids may be further separated by a simple water fractionation, yielding an aqueous sugar-rich phase and a water-insoluble pyrolytic lignin (PL) fraction. This separation step allows the use of dedicated conversion strategies for each fraction, which can be highly advantageous due to their differences in composition and reactivity. For example, the sugar-rich fractions can be used for fermentation, while the phenolic-rich PL is a particularly promising feedstock for the production of a wide range of platform chemicals and energy-dense streams upon depolymerization. To aid the emerging use of PL, novel characterization techniques and valorization strategies are being explored. In this review, the fast pyrolysis process and PL characterization efforts are discussed in detail, followed by the state-of-the-art regarding PL processing using both oxidative and reductive (catalytic) strategies, as well as a combination thereof. Possible applications are discussed and recommendations for future research are provided.
1. Introduction
The pressing environmental concerns related to fossil fuel consumption and the ever-growing global demand for energy and products greatly encourage the development of technologies to replace petroleum with renewable sources of carbon. The Paris Agreement, a landmark treaty on climate change involving 196 countries, has an overarching goal to limit global warming to 1.5 °C above pre-industrial levels.1 This is a challenging yet achievable target, provided that all stakeholders support the development of zero-emission disruptive technologies worldwide. In this context, lignocellulosic biomass (i.e. plant-based raw material, particularly from non-edible sources) stands out as a promising alternative carbon source due to its high availability and versatility, energy content and rich chemical structure (vide infra). Accordingly, efficient valorization routes for biomass can pave the way for the decarbonization of the chemical industry, which is one of the most fossil-dependent industrial sectors, responsible for 5% of the global CO2 emissions in 2017 (i.e. 1.7 Gt of CO2).2 The use of biomass as a resource can certainly bring major positive impacts to the energy-intensive freight and long-haul transport sectors (road freight, aviation and shipping), which together were responsible for 11% of the global CO2 emissions in 2017 (i.e. 4.1 Gt of CO2).2 Unless major changes towards the use of renewables are implemented, it is expected that these sectors will account for up to 21% of energy and process emissions by 2050.Lignocellulosic biomass consists mainly of three biopolymers, namely cellulose (25–50%), hemicellulose (25–40%) and lignin (15–40%),3,4 each of them presenting a unique structure and properties (Fig. 1). The proportions of the abovementioned biopolymers, as well as moisture content and amounts of extractives and minor compounds (i.e. alkali metals and silica), vary significantly with the type of plant, climate and harvesting conditions.5 Cellulose is a linear homopolymer consisting of β-D-glucopyranose (also known as D-glucose) units, linked together by covalent 1,4-glycosidic bonds with a varying degree of polymerization (up to 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000). These elongated molecules are arranged in parallel and joined together by hydrogen bonds, being responsible for the longitudinal strength and toughness of the plant's leaves, roots and stems. Hemicellulose also consists of polysaccharides, however, its structure is branched and diverse, containing both pentoses and hexoses (primarily xylose and mannose). It binds tightly, but non-covalently, to the surface of each cellulose microfibril, contributing to the cell wall strength.6
000). These elongated molecules are arranged in parallel and joined together by hydrogen bonds, being responsible for the longitudinal strength and toughness of the plant's leaves, roots and stems. Hemicellulose also consists of polysaccharides, however, its structure is branched and diverse, containing both pentoses and hexoses (primarily xylose and mannose). It binds tightly, but non-covalently, to the surface of each cellulose microfibril, contributing to the cell wall strength.6
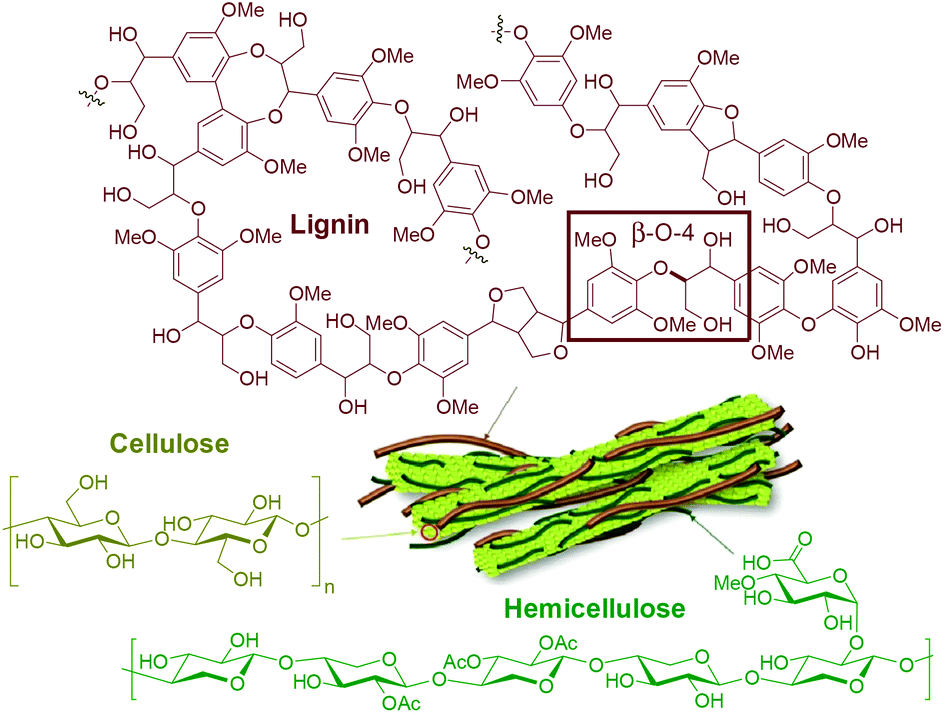 | ||
| Fig. 1 Representative chemical structures of the main biopolymers that make up lignocellulosic biomass. | ||
In contrast to cellulose and hemicellulose making up the carbohydrate part of lignocellulosic biomass, lignin is distinguished by its aromatic and hydrophobic character. It is defined as a network constituted by phenylpropanoid units connected by C–O–C and C–C bonds with a varying degree of methoxylation. Specifically, in native lignins, β-O-4 interunit linkages are dominant (45–60% in softwoods, 45–80% in hardwoods and 55–75% in monocots), typically followed in abundance by 5–5′, β-5 and β–β′ linkages.7–10 This biopolymer plays a major role in water regulation and pathogen resistance, while also providing mechanical support, strength and rigidity to the plants’ tissues.11
In detail, lignin's main biosynthetic precursors are p-coumaryl, coniferyl and sinapyl alcohols, further denominated p-hydroxyphenyl (H), guaiacyl (G) and syringyl (S) units (Fig. 2). The proportions of H, G and S units vary substantially among different biomass sources, but also with plant age and environmental conditions. In general, lignins derived from hardwood are composed of G and S units with low H units, while softwoods are mainly composed of G units (with traces of H units). Lignins derived from herbaceous plants include both G, S and H units.12 Since the structure of lignin is overall complex, variable and highly prone to condensation, it has been historically deemed unfit for dedicated valorization. Traditional biomass-processing industries such as pulp and paper and bioethanol facilities obtain high-value products from the sugar fractions to the detriment of lignin, which is heavily degraded in the process and treated as a low-value fuel and/or waste. Importantly, this view is changing with the emergence of novel technologies that aim to process lignocellulosic biomass holistically, thus treating all of its fractions as sources of valuable products.13
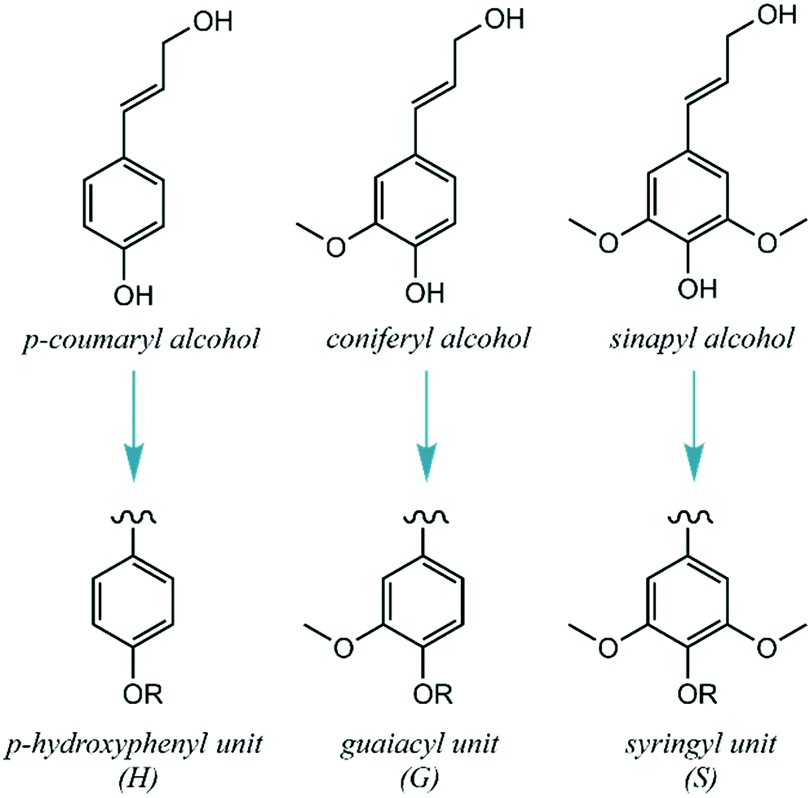 | ||
| Fig. 2 Major building blocks of native lignin. | ||
For ease of processing and effective integration in the established petro-based value chains, biomass conversion technologies producing liquid products with low oxygen content are preferred. Therefore, strategies towards using lignin as a valuable carbon source typically include depolymerization and deoxygenation steps. Due to the complexity of biobased feedstocks, associated conversion products tend to be complex as well. Fractionation/separation and funnelling strategies aiming at more homogeneous streams that can be valorized separately may be a necessity.
The chemical heterogeneity and structural complexity of biomass make the synergetic isolation and upgrading of all its fractions a major challenge. Nonetheless, to efficiently feed fossil-dependent markets with biobased counterparts, as well as to introduce new cost-competitive biobased products, biomass needs to be processed in an integrated way – the so-called biorefinery concept. Similar to the petroleum refinery, a biorefinery involves processing biomass with a minimum formation of waste and minimum energy consumption to obtain a combination of chemicals, high-end fuels and power. The typically overlooked lignin fraction then becomes an important source of aromatics and their derivatives.
Several biorefinery schemes using lignocellulosic biomass have been reported in the literature (Fig. 3), and different conversion routes and products are envisioned. One remarkable challenge in the scale-up of such schemes is their typical capital-intensive character. In this context, multiple conversion technologies can be potentially combined to reduce costs, provide energy to the process and guarantee portfolio flexibility.14 Furthermore, the development of processes able to efficiently produce fine chemicals (high value and low volumes) in addition to fuels and commodities (low value and high volumes) from waste biomass could significantly accelerate the industrialization of biorefineries. Other challenging aspects involve: (i) biomass logistics (storage, transport); (ii) continuous access to inexpensive feedstocks with standard properties; (iii) market barrier and social acceptance of new biobased products; (iv) lack of regulation and governmental incentives; (v) lack of industrial infrastructure (except for drop-in chemicals); (vi) lack of robust and comparable life cycle assessment (LCA) studies; (vii) technical and economic challenges regarding product separation. Despite these challenges, it is important to highlight that a responsible implementation of biorefineries has benefits that go way beyond lowering CO2 emissions, such as promoting energy security, economic development and overall positive environmental and social impacts.15
 | ||
| Fig. 3 Overview of lignocellulosic biomass conversions and derived products within the biorefinery concept. | ||
Among the biomass conversion possibilities highlighted above, thermochemical routes can be used to deconstruct biomass at elevated temperatures, in either oxygenic or anoxygenic atmospheres. Various thermochemical routes can be discriminated, ranging from biomass combustion to the transformation of the biomass into liquids through hydrothermal liquefaction or fast pyrolysis. Fig. 4 illustrates the main technologies used for the thermochemical upgrading of biomass, as well as their products and uses.
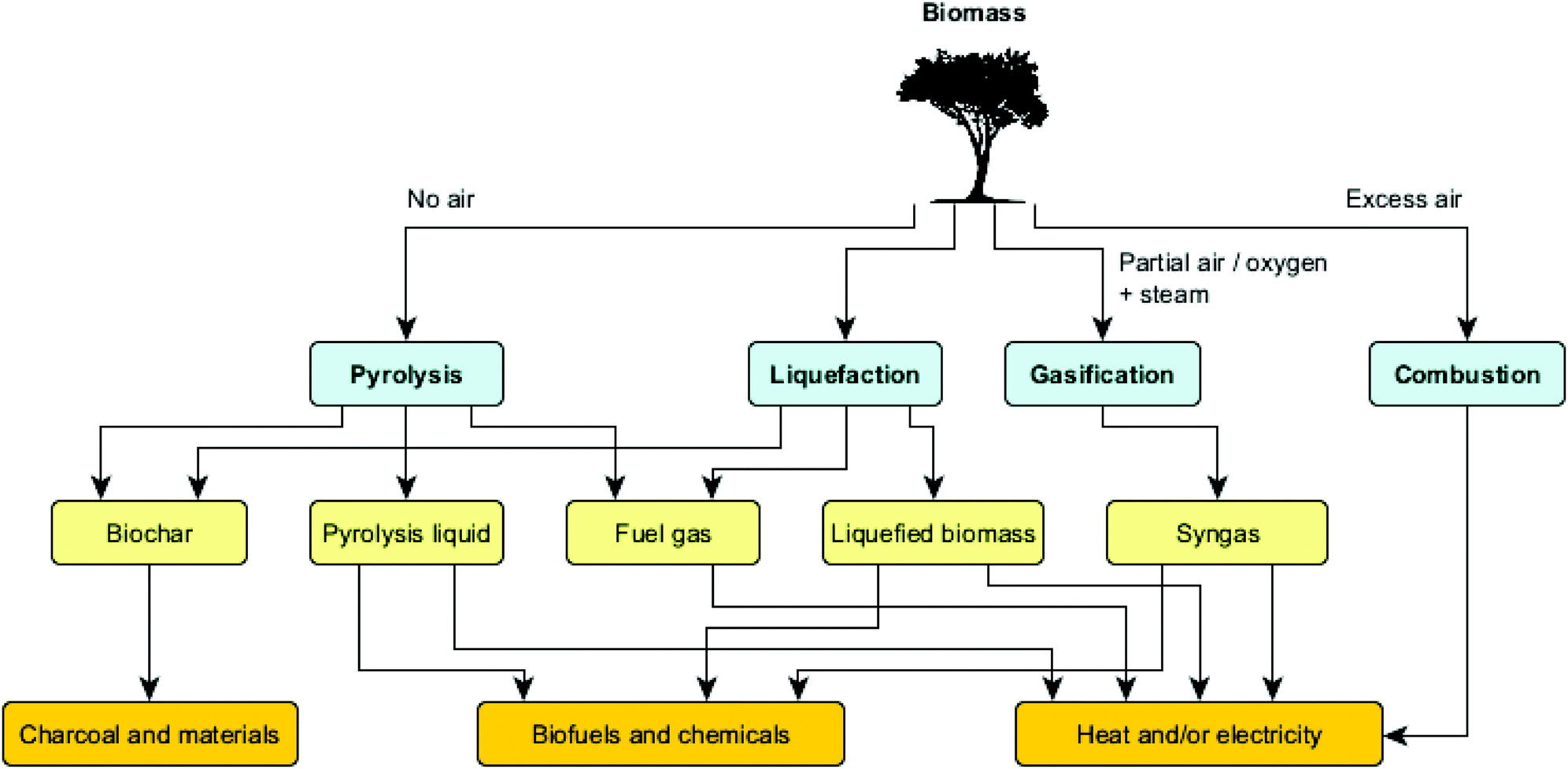 | ||
| Fig. 4 Main thermochemical routes for biomass processing. | ||
Whereas the combustion of biomass is mainly used for heat and power generation, the gasification process aims at producing so-called syngas. Gasification takes place at high temperatures (800–1100 °C) in a well-controlled environment with restricted supply of oxygen and/or steam to avoid full combustion. Syngas mainly consists of carbon monoxide, carbon dioxide, hydrogen and nitrogen, and has several applications like heat and power generation, manufacturing of liquid fuels (e.g. via Fischer–Tropsch synthesis) and production of chemicals (e.g. methanol). Gasifiers can be operated at atmospheric or pressurized conditions.16
When aiming at liquid product mixtures from biomass, two main processes stand out, namely hydrothermal liquefaction (HTL) and fast pyrolysis. HTL is a chemical reforming process in a pressurized reactor to convert wet biomass at moderate temperatures (typically 200–400 °C) and high pressures (typically 10–25 MPa), using water as the reaction medium. Since no drying step is necessary, this process is particularly suited for wet feedstocks such as food processing wastes, manure, microalgae and municipal sludge, yielding an energy-dense biocrude oil, fuel gas and limited amounts of char.17 The fast pyrolysis process involves the use of dry biomass feedstocks which are deconstructed under elevated temperatures (typically 450–600 °C) and in the absence of oxygen, resulting in a product mix consisting of solid (charcoal), liquid (bio-oil/pyrolysis liquid) and gaseous streams.18 Both the composition and product distribution vary significantly with feedstock properties and conditions applied, in ways that a desirable mix can be obtained by choosing the correct reactor configuration and adjusting main process parameters, i.e. temperature, heating rate and residence time (vide infra). Note that while ‘slow pyrolysis’ (also called carbonization) maximizes the yield of solid products, fast pyrolysis maximizes the yield of liquid products.
In this review, we focus on the potential of fast pyrolysis and specifically on the water-insoluble fraction of biomass-derived pyrolysis liquids, the pyrolytic lignin (PL), for the production of biobased compounds. We will start with the principles and fundamentals of fast pyrolysis followed by a discussion of different processes developed for fast pyrolysis. The concept of a pyrolysis-based biorefinery will be introduced, followed by a discussion on the fractionation of the pyrolysis liquids. We will then zoom in on the PL fraction and discuss its elemental and chemical compositions, followed by conversion technologies with an emphasis on oxidative and reductive (catalytic) strategies, as well as a combination thereof. Finally, prospective PL-derived product applications are discussed and recommendations for future research are provided.
2. Fast pyrolysis and the pyrolysis-based biorefinery
2.1. Fast pyrolysis
Fast pyrolysis is an attractive primary thermochemical process to liquefy biomass due to the flexibility of feedstock and process conditions, relatively low cost and high energy conversion efficiency.18,19 While other approaches can be sensitive to the biomass source and purity, a wide range of feedstocks (e.g. agricultural residues such as seeds, grasses, bagasse, and biomass mixtures) are suitable for pyrolysis. In addition, fast pyrolysis is a relatively simple process that allows for decentralization, meaning that small-sized pyrolysis units can be located near the biomass source to produce easily transportable products and reduce overall costs with logistics.20 This technology also allows for co-processing biomass together with other wastes, like scrap tires, sewage sludge, or plastics, further boosting its interest as a very promising conversion route.21–23During fast pyrolysis, thermal conversion of biomass takes place at elevated temperatures and in the absence of oxygen, involving very rapid heating of the biomass particles. The heating rate can be of 1000–10000 °C s−1, but to boost bio-oil production the attained peak temperature must be kept below 650 °C.24 Rapid cooling of the hot vapors (residence times <10 s) is also required to maximize the liquid yield. Through fast pyrolysis, liquid product yields up to 75 wt% can be obtained,21 with char and gaseous compounds as by-products. A precise selection and control of the process variables are required during pyrolysis, to attain the desired balance (trade-off) between the product distribution and their quality (physico-chemical properties).18
Biomass fast pyrolysis conversion units are stand-alone, not necessarily integrated within already existing refinery infrastructure, where the solid biomass is locally transformed into liquids that can be easily transported to and – likely after some additional treatment – may be handled in refineries.
As depicted in Fig. 5, fast pyrolysis can be conceived as a primary biomass depolymerization step to yield three main primary products (gas, liquid and char). The gas phase produced during fast pyrolysis consists of a mixture of primarily CO2 and CO with smaller amounts of CH4, H2 and C2–C4 compounds. Due to this composition, the most straightforward application for these gases is combustion, to produce the heat required by the process and thus aim at energetic self-sustainability. The char also offers the possibility to be combusted for heat production, or - if possibilities are developed to extract it from the process – be converted into hydrogen or syngas.25,26 Being a bio-char it can, through physical and/or chemical activation, be converted into activated carbon, which has many applications.27,28
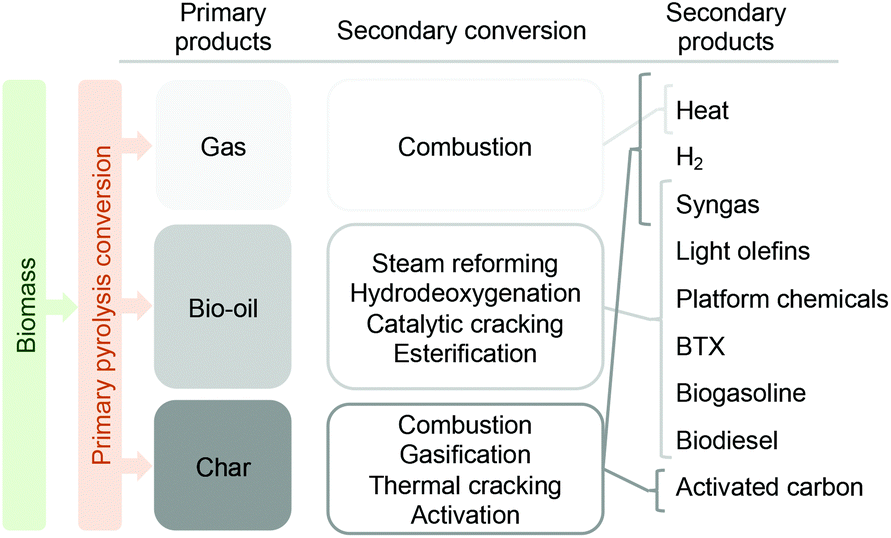 | ||
| Fig. 5 Main products derived from primary biomass fast pyrolysis, secondary conversion routes and derived products. | ||
The liquid fraction, known as bio-oil, is the most valuable of the products.29 It can be converted into biofuels and/or renewable chemicals through several thermo-catalytic routes, with the main ones being steam reforming,30 hydrodeoxygenation,31,32 catalytic cracking,33,34 or esterification.35,36 An interesting application of the liquid fraction, either as such or after upgrading, is co-feeding with refinery streams like heavy gas oils and jointly upgraded in an oil refinery for the production of diesel, gasoline or LPG products.37,38
The main objective of biomass fast pyrolysis is to obtain a liquid product in high yields, which is generally achieved at pyrolysis temperatures of ca. 500–550 °C.39,40 Liquid yields are not only a function of the reactor temperature but also affected by feedstock properties, and process aspects such as the biomass heating rate, and the rate of condensation of the vapor outlet stream. To date, several reactor technologies have been developed, and the production of pyrolysis oil is at a commercial scale. Fig. 6 displays the product distributions obtained through fast pyrolysis in different reactor configurations, as reported in the literature. Extended information on the advantages and drawbacks of the different fast pyrolysis reactor types can be found in recent overviews.41
 | ||
| Fig. 6 Product distribution reported for different types of pyrolysis reactors. Data have been collected from fast pyrolysis reactors operated at 500 °C, unless labelled otherwise. Reactor abbreviations: CSB, Conical spouted bed; FixedB, Fixed bed; FB, Fluidized bed. | ||
The composition and possible uses of the different product fractions from the primary pyrolysis process are discussed in further detail in the coming section, with a main focus on the uses and applications of pyrolysis oil.
2.2. Pyrolysis product composition and properties
The permanent or non-condensable gaseous products obtained from the pyrolysis of biomass generally consist of a mixture of predominantly CO and CO2 (70–90%) with lower amounts of CH4, H2 and C2–C5 hydrocarbons, see Table S1† for details. The low heating value (LHV) of the pyrolysis gases typically ranges between 5.5–7.0 MJ m−3 if not diluted with any inert gases.42 The simplest application for these gases is combustion to generate energy, for example, to increase the steam/electricity output.On the other hand, the solid product formed in biomass pyrolysis, known as char, is a carbon-rich material with smaller amounts of hydrogen and oxygen (see Table S2† for details). With a slightly lower H content than biomass (ca. 3–4 wt% less), its relatively high carbon content (ca. 20–30% higher than the original biomass, which contains averaged C content of 45–55 wt%) provides char with a higher high heating value (HHV) compared to its bio-oil co-product. Likewise, the calorific value of the char is also enhanced by its rather low oxygen content (ca. 35–45 wt% in biomass vs ca. 10–15 wt% in char). In practice, the char is combusted, as it is usually difficult to entrain it from the inert heat carrier materials.
| Ref. | 51 | 52 | 52 | 21 | 53 | 54 | 55 |
|---|---|---|---|---|---|---|---|
| a Reactor abbreviations: CSB, Conical spouted bed; FixedB, Fixed bed; FB, Fluidized bed. | |||||||
| Biomass source | Black poplar | Poplar | Poplar | Pinewood | Corn stover | Sugarcane bagasse | Wastewood sawdust |
| Reactor typea | CSB | FixedB | FixedB | CSB | Microwave oven | FixedB | FB |
| T (°C) | 500 | 500 | 600 | 500 | 550 | 500 | 500 |
| HHV (MJ kg−1) | 20.6 | 21.3 | 21.37 | 19.5 | — | 19.1 | 13.5 |
| pH | 2.3 | 2.57 | 2.71 | — | 3.50 | 3.02 | 3.4 |
| Water (wt%) | 49.0 | 35.9 | 54.6 | 32.0 | 68.1 | — | 37.5 |
| C (wt%) | 57.4 | 42.9 | 51.3 | 41.8 | 15.1 | 52.82 | 35.0 |
| H (wt%) | 5.9 | 6.64 | 6.3 | 8.5 | 9.0 | 7.74 | 7.5 |
| O (wt%) | 35.5 | 50.4 | 42.32 | 49.6 | 75.2 | 38.95 | 55.0 |
| N (wt%) | 1.20 | — | — | 0.1 | 0.7 | 0.49 | 2.5 |
| S (wt%) | — | 0.06 | 0.06 | 0 | 0 | — | — |
An attractive strategy for obtaining bio-oils with a reduced oxygen content and enriched in hydrocarbons such as benzene, toluene and xylenes (BTX) involves the use of acidic solid catalysts (generally zeolites), either in situ or ex situ in a reactor connected in series.47,48 Careful selection of the right type of zeolite is required as pore size and shape selectivity are known to have a great impact on catalyst performance and hence on the physicochemical properties of the end products. On the other hand, bio-oil stabilization can also be conducted through a series of physical methods like the addition of a solvent, ash and/or char removal, and emulsification.34
An increasingly popular strategy for bio-oil valorization is to conduct a water-based fractionation step, which leads to the separation of a sugar (aqueous) fraction and a water-insoluble fraction comprised mostly of the lignin-derived aromatic fragments.49,50 The latter is referred to as pyrolytic lignin (PL). In the context of a pyrolysis-based biorefinery scheme, the two fractions can be processed independently by strategies tailored to their nature and inherent properties into a wide range of valuable products.
2.3. The pyrolysis-based biorefinery and the role of pyrolytic lignin
Pyrolysis liquids are promising feedstocks in the context of biorefineries and the development of novel biobased value chains. An overview of a pyrolysis-based biorefinery concept is illustrated in Fig. 7. It involves either the direct use of the pyrolysis liquids or an alternative approach using fractionation.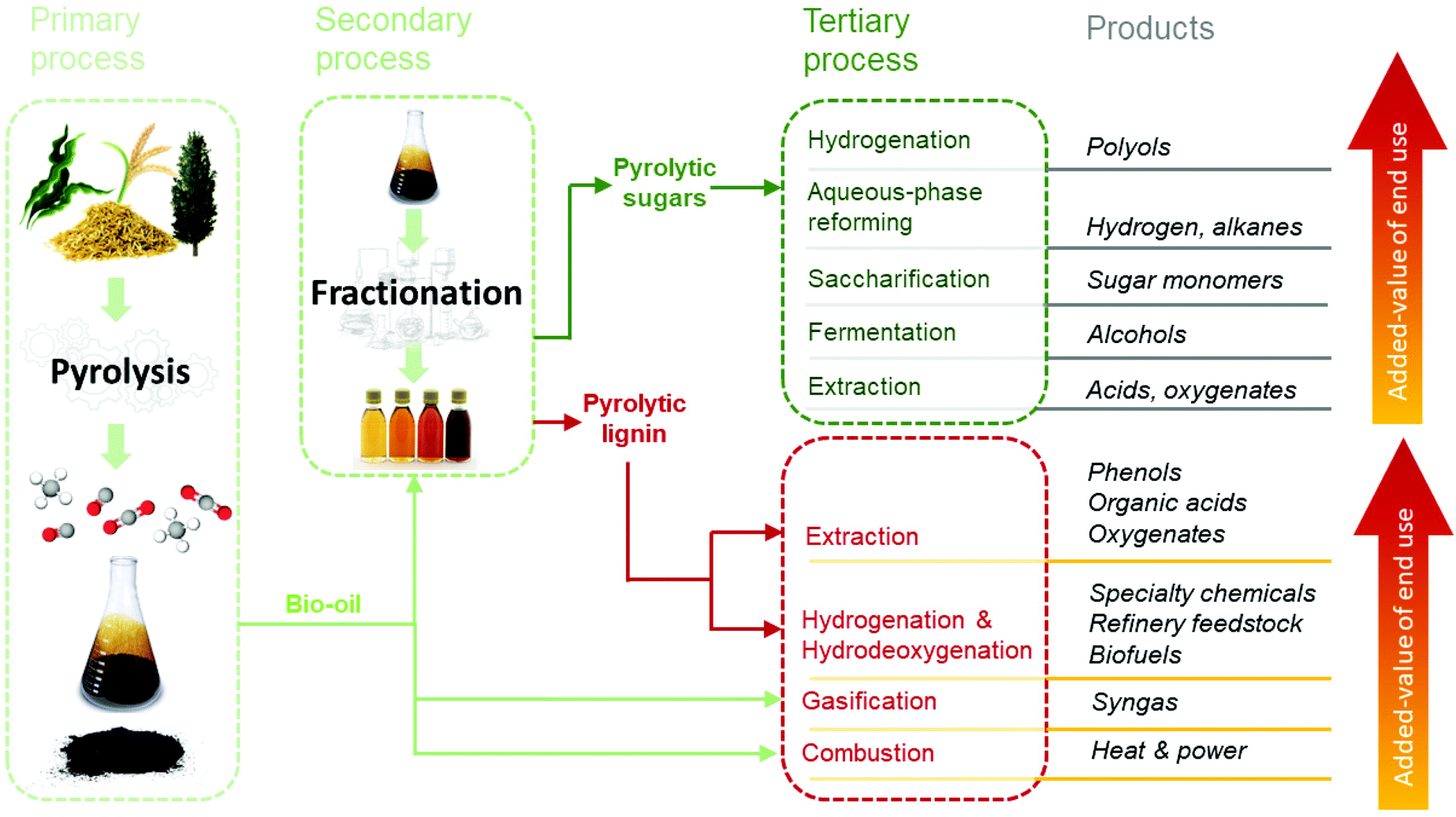 | ||
| Fig. 7 The pyrolysis-based biorefinery concept. | ||
As said in the previous paragraphs, direct upgrading of pyrolysis liquids is possible (e.g. via hydrotreatment followed by co-feeding in petro-based refineries56–58), though technology development is still at a relatively low readiness level (max TRL 5-659). Apart from downstream separation issues due to the formation of complex product mixtures, carbohydrate-derived products contribute to catalyst lifetime issues, among others by their charring tendency (e.g. humins formation).60
To circumvent these issues, fractionation of the pyrolysis oils, followed by tailored upgrading routes for the individual fractions is an attractive valorization approach. In this respect, distillation is not the preferred option due to the limited thermal stability of pyrolysis liquids, leading to substantial amounts of char. An alternative scheme involves phase separation of the oil by water addition. This leads to two main fractions, i.e. a pyrolytic sugar fraction and PL. Such water fractionation can be either applied as a simple procedure with limited amounts of water (e.g. 2.5:1 water:pyrolysis liquid w/w ratio, giving 30–40 wt% of PL obtained as a viscous oil with residual water and sugars61), or using a more extensive water wash which leads to high-purity PL (e.g. 16:1 water:pyrolysis liquid w/w ratio, 13.5–27.7 wt% of PL obtained as a fine powder62). The trade-off between water usage and lignin purity needs optimization depending on the envisioned products and techno-economical aspects. Reported yields for the sugar fraction are 60–70 wt% (wet basis).63 Overall yields are dependent on both the biomass source and water fractionation procedure.
The sugar and PL fractions are remarkably different in terms of structure and physicochemical properties. They may serve as intermediates to be processed independently in the biorefinery scheme. For instance, it was shown that the sugar fraction consists of cellulose-derived anhydrosugars (mainly levoglucosan), pentoses, organic acids and furans.63 These can be further converted by hydrolysis, fermentation, hydroprocessing and aqueous phase reforming to yield hydrocarbons, alcohols, sugar monomers and platform chemicals such as hydroxymethylfurfural (HMF).63–69 Some routes aim particularly at levoglucosan, a versatile platform molecule present in bio-oil in high amounts and that may be further concentrated in the pyrolytic sugars to >30 wt%. Derivatives from levoglucosan are glucose (by hydrolysis), gluconic acid and ketones, as well as lipids and succinates produced via metabolic pathways.70–72 In contrast to extensive research on the sugar fraction, the development of conversion processes and applications of PL are by far less mature and will be the main scope of this review.
The PL fraction can be further depolymerized and upgraded into alkylphenolics, aromatics and organic acids with various applications such as high-end fuels and additives, fine chemicals, bioactive molecules and building blocks for chemical industries.73,74 Upon an efficient oxygen removal, PL can serve as a feedstock for co-feeding in traditional refineries, i.e. a drop-in possibility that may accelerate the entry market of this feedstock.75,76 Heat and power can be produced through combustion of either the whole pyrolysis oil or the PL derived stream (which has a higher energy content than the sugar fraction) to achieve an integrated and self-sustainable process. Gaseous streams from the pyrolysis itself and PL depolymerization are also sources of energy.
The main envisioned challenges of the pyrolysis-based biorefinery scheme are related to the chemical heterogeneity of each fraction and the low concentrations of specific compounds (be it monomers, dimers or trimers), which may bring high separation costs and low yields when aiming for specific molecules with high purity (e.g. specialty chemicals). The use of heterogeneous catalysts (often noble metals) for catalytic hydrotreatment, expensive hydrogen gas and relatively harsh conditions require well-integrated processes in which the catalyst can be recycled and/or has high stability, materials can withstand the applied conditions and products achieve high quality and added-value to sustain their associated costs. The latter aspect is a bottleneck in practically any biorefinery developmentz, as it is and it will probably remain difficult to economically compete with most petro-based products. A proper societal engagement and incentives from governmental and industrial leaders are paramount to accelerate the much-needed transition towards biobased schemes.
The development of novel and inexpensive catalysts as well as efficient routes and new applications for PL can greatly improve the viability of the pyrolysis biorefinery. For instance, the use of PL as an additive in formulations and materials can broaden the scope of industrial applications and add higher value to these by taking advantage of the great intrinsic properties of lignin (i.e. antioxidant, anti-UV, antimicrobial). The next section will detail the chemical and structural properties of PL, followed by possible depolymerization strategies to convert it to high-value chemicals and fuels.
3. Structure and properties of pyrolytic lignins
3.1. Molecular and chemical composition of PL
Thermal decomposition of lignin in lignocellulosic biomass involves numerous pathways, including competitive and/or consecutive reactions.77,78 Extensive cleavage of C–C and C–O–C linkages occurs. In particular, the β-O-4 linkages that make up the majority of the linkages in native lignin are extensively broken down.79,80 Cleavage of the propanoid side chains leads to the formation of considerable amounts of monomeric and oligomeric alkylated methoxyphenols, which can be further converted to alkylated phenols.78,81The biomass source and associated lignin structure determine the product composition of the pyrolysis oil and the associated PL fraction to a large extent. For example, softwood lignins consisting mostly of G units form mainly guaiacols (2-methoxyphenols), whereas hardwood lignins consisting of S and G units are decomposed in both guaiacols and syringols (2,6-dimethoxyphenols). Herbaceous biomass sources contain guaiacols, syringols and non-negligible amounts of p-hydroxyphenyl derived structures.88 Accordingly, substantial differences in elemental composition are observed when comparing values reported in the literature for PL (Table 2). It should be kept in mind that these data may also be affected by differences in water fractionation methods and pyrolysis conditions applied. This also leads to differences in the PL appearance and water content. Typically, a brown hygroscopic powder is obtained when using an elaborated water wash protocol, while a viscous dark brown oil is obtained when using a simplified fractionation procedure with lower amounts of water.50,83,85 The elemental variability is clearly visible in a Van Krevelen diagram (Fig. 8). It also shows that the elemental composition of PLs differs from typical monomeric alkylphenolics and that the O/C molar ratio of PL is considerably higher. This is due to the presence of oxygen-containing chemical functionalities other than phenolic hydroxy groups (e.g. methoxy groups), as further detailed in the following sections. The higher H/C molar ratio of some PLs might be related to higher amounts of extractives (such as fatty acids) and residual sugars.
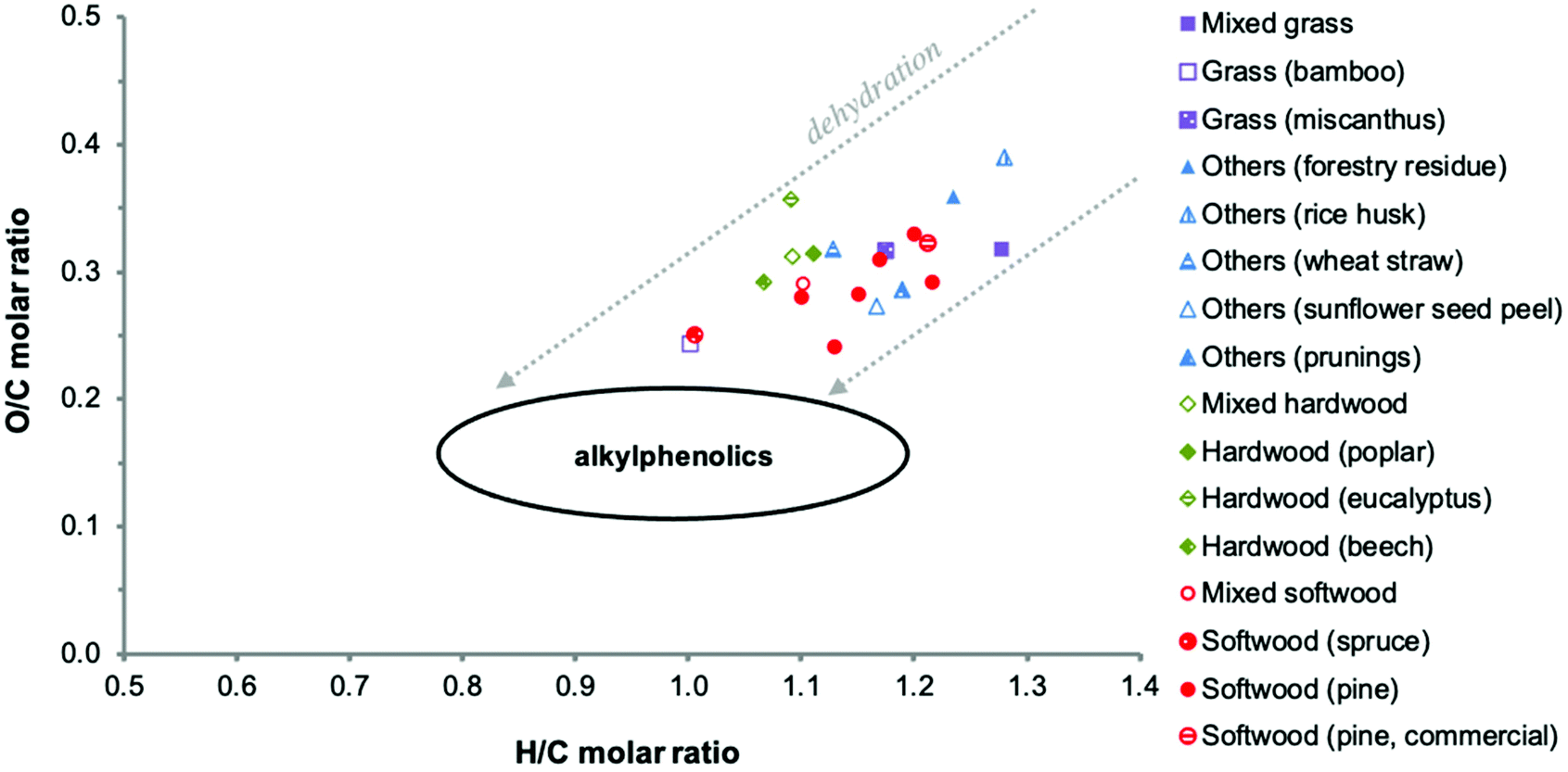 | ||
| Fig. 8 Van Krevelen plot of PLs as reported in the literature. Adapted from ref. 89 with permission from Elsevier, copyright 2020. | ||
| Ref. | Biomass source | Pyrolysis temp. (°C) | HHV (MJ kg−1) | Water (wt%) | C (wt%, dry) | H (wt%, dry) | O (wt%, dry) | N (wt%, dry) | M w (g mol−1) |
|---|---|---|---|---|---|---|---|---|---|
| 82 | Rice husk | 550–600 | 23.6 | — | 60.2 | 6.42 | 31.3 | 2.07 | — |
| 83 | Pine | — | — | — | 67.1 | 6.8 | 26.1 | — | 730 |
| 83 | Forestry residue | — | — | — | 63.2 | 6.5 | 30.3 | — | 705 |
| 84 | Beech | 470 | — | — | 67.5 | 6 | 26.3 | <0.2 | — |
| 84 | Spruce | 470 | — | — | 70.4 | 5.9 | 23.5 | <0.2 | — |
| 84 | Bamboo | 470 | — | — | 70.7 | 5.9 | 22.9 | 0.5 | — |
| 62 | Mixed hardwood | — | — | — | 66.2 | 6.02 | 27.6 | 0.23 | — |
| 62 | Mixed softwood | — | — | — | 67.4 | 6.2 | 26.1 | 0.24 | — |
| 62 | Poplar | — | — | — | 66 | 6.1 | 27.6 | 0.21 | — |
| 62 | Eucalyptus | — | — | — | 63.7 | 5.8 | 30.3 | 0.19 | — |
| 62 | Wheat straw | — | — | — | 65.2 | 6.13 | 27.6 | 1.02 | — |
| 62 | Pine | — | — | — | 70.6 | 6.6 | 22.6 | 0.15 | — |
| 85 | Pine | — | — | 4.7 | 65.9 | 6.5 | 27.5 | <0.01 | 780 |
| 61 | Pine | 500 | — | 7.5 | 67.9 | 6.5 | 25.6 | <0.1 | 741 |
| 61 | Commercial pine | 500 | — | 8.2 | 65.3 | 6.6 | 28.1 | <0.1 | 690 |
| 61 | Prunings | 500 | — | 11.2 | 66.5 | 6.6 | 25.4 | 1.5 | 662 |
| 61 | Mixed grass | 500 | — | 7.6 | 64.3 | 6.8 | 27.3 | 1.6 | 713 |
| 61 | Miscanthus | 500 | — | 7.6 | 65.7 | 6.4 | 27.7 | 0.2 | 668 |
| 61 | Sunflower seed peel | 500 | — | 7.6 | 67.5 | 6.6 | 24.5 | 1.4 | 757 |
| 86 | Pine | — | — | — | 66 | 6.5 | 27.5 | 0.1 | 650 |
| 87 | Pine | — | — | — | 68.1 | 6.3 | 25.5 | 0.1 | 725 |
| 87 | Pine | — | — | — | 64.8 | 6.5 | 28.6 | 0.1 | 616 |
Similar to other lignins, the molecular weight (Mw) distribution of PL is polydisperse, meaning that fragments of different sizes are present. However, the average Mw of PL (ca. 650–750 g mol−1, Table 2) is much lower than those of typical technical lignins (i.e. 1500–9000 g mol−1,5,89–96vide infra). This is explained by the high temperatures applied in the fast pyrolysis process, which lead to thermally-induced depolymerization. Furthermore, high Mw fragments from lignin might end up as char and not in the condensed vapor during the process. Interestingly, the Mw distribution of PL is similar to oligomeric fractions obtained by other biomass processing routes such as reductive catalytic fractionation (RCF) of pine wood (i.e. oligomers with Mw of 985 g mol−1 (ref. 97)). As a result of the oligomeric character of PL, the monomer content is relatively low. This oligomeric nature of PL also brings a challenge to its characterization. For instance, only 15 wt% of monomers were quantified in a pine-derived commercial PL using GC × GC-FID, GC × GC/TOF-MS and HPLC techniques.89 GC × GC identified mainly alkylphenolics and furans, whereas HPLC identified residual levoglucosan and organic acids such as acetic and formic acid. The GC × GC/TOF-MS chromatogram with main monomers assigned is shown is Fig. 9.
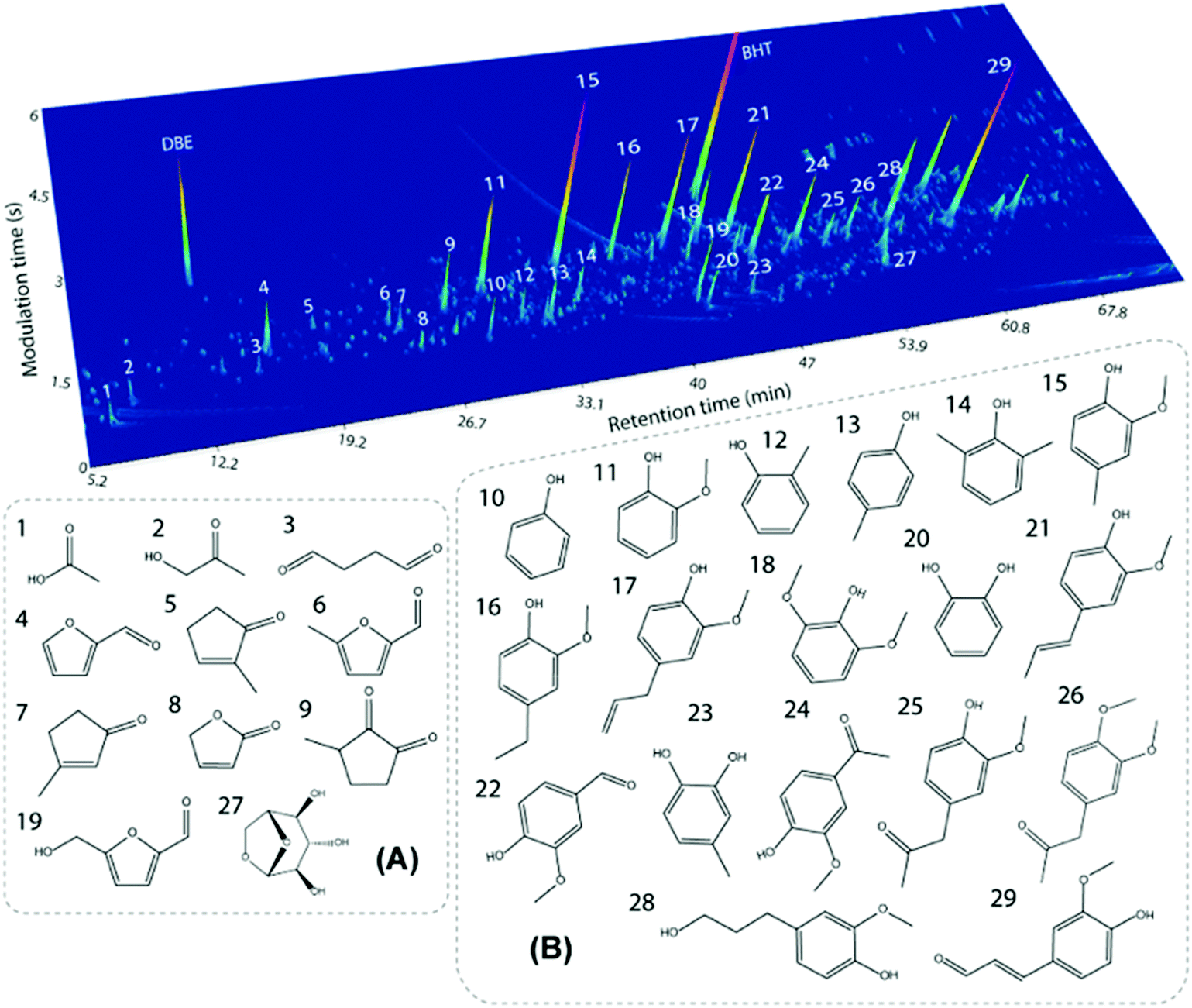 | ||
| Fig. 9 GC × GC chromatogram for a pine-derived PL with the main identified monomers in the volatile fraction. Reproduced from ref. 89 with permission from Elsevier, copyright 2020. | ||
3.2. Structural composition and formation of PL
A large fraction of the PL consists of substituted phenolic oligomers, for which two main routes of formation are proposed: (i) direct thermal ejection of lignin fragments into the gas phase;84,98 (ii) repolymerization of highly reactive monomeric species during pyrolysis and/or during condensation of the pyrolysis vapors.78,99–102 While the former route leads to the release of relatively unchanged structures existent in native lignin, the later route results in new types of inter-unit linkages. Such new linkages, formed during pyrolysis, are different from the typical alkyl-aryl-ether, being particularly carbon–carbon linkages (e.g. diphenyl, diaryl methine) and saturated aliphatic side chains.84,103–106 According to the thermal ejection theory, residual alkyl-aryl-ether linkages are present as a result of the release of “untouched” lignin oligomers.98 Such theory has been refuted by the most recent PL characterization studies using advanced techniques, in which β-O-4 linkages were not observed.84,89,102 Thermal splitting during pyrolysis is reported to generate unconjugated carbonyl groups and C–C double bonds, while the amount of methoxy groups and aliphatic hydroxy groups decreases substantially when compared to native lignin.84,105,107 Gaseous compounds (e.g. CO, CO2, H2 and CH4) and char are produced from reactions taking place during lignin pyrolysis, i.e. decarbonylation, decarboxylation, dehydration, cracking and repolymerization.77,88,101,108,109In general, lignin characterization has greatly evolved with the use of advanced tools such as NMR spectroscopy,110 DFT (density functional theory) computational modelling,102 high-resolution Fourier transform ion cyclotron resonance mass spectrometry (FT-ICR MS),111 among others. Recent studies provided important insights on the main structural features of PL, showing that it consists mainly of trimers and tetramers of HGS units, as anticipated when considering thermally-driven depolymerization reactions.84,89,103,107,112 The relative contents of aromatic, side-chain aliphatics/methoxy groups and oxygenated aliphatics reported for a range of technical lignins and a representative PL (based on 13C-NMR) are plotted in Fig. 10.86 Despite the semi-quantitative character of this analysis, it shows that PL has an overall higher amount of aliphatic side chains (from the cleavage of native aryl-ether linkages during pyrolysis) in comparison with other types of lignin. Furthermore, PL shows a much lower concentration of methoxy side groups, likely from the thermal-driven demethoxylation that occurs during pyrolysis. These observations are in line with the proposed lignin reaction pathways taking place under pyrolysis conditions (vide supra).
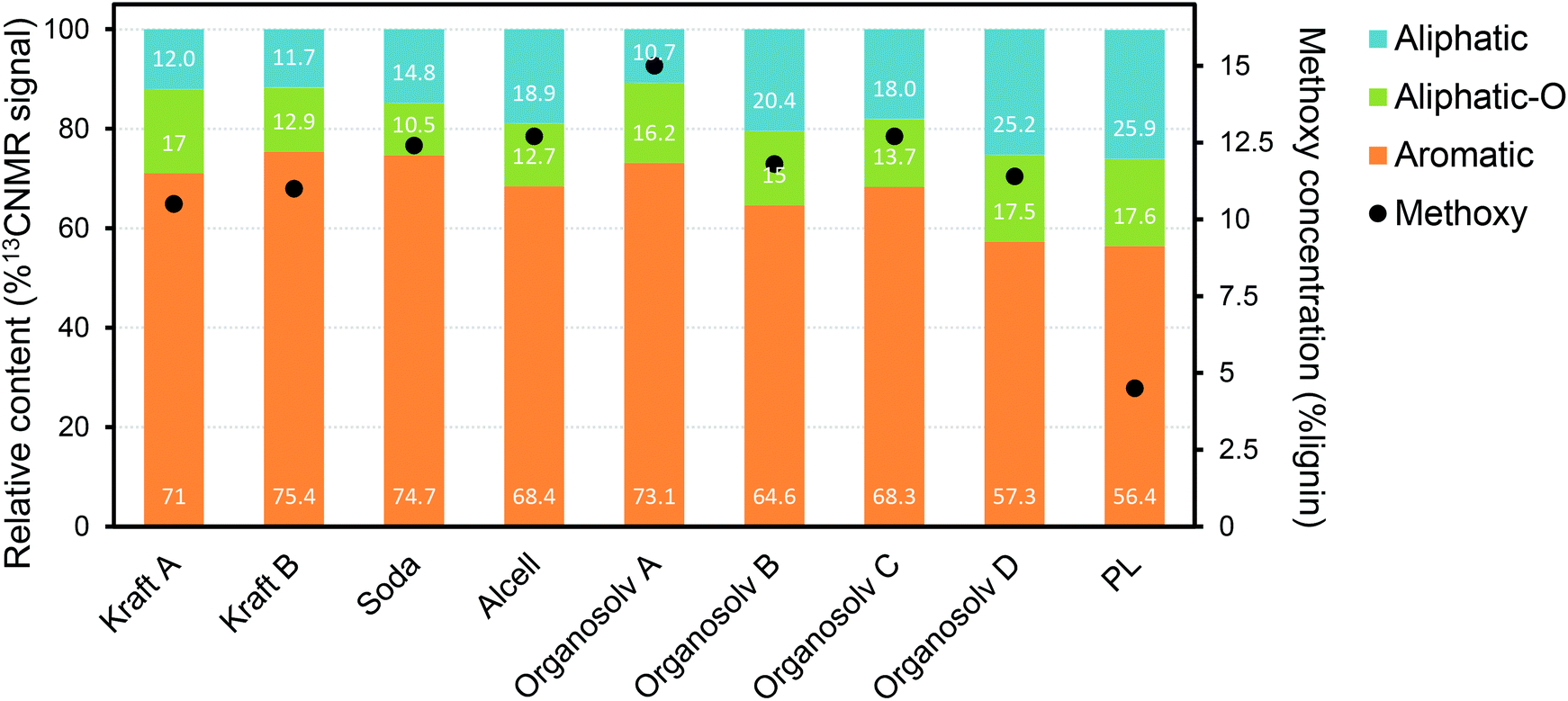 | ||
| Fig. 10 Relative contents of aromatics, side-chain aliphatics and oxygenated aliphatics in technical lignins, together with their methoxy concentrations as determined by 13C-NMR.86 | ||
An overview of the molecular structures for PL oligomers proposed so far in the literature is presented in Fig. 11. These PLs were obtained from various biomass sources and pyrolysis conditions. While there are similarities in the building blocks and C–C linkages, the proposed PL structures can be remarkably different in terms of size and the presence of β-O-4, sugars, benzofurans, stilbenes and β–β linkages. This is related to both the biomass sources and to the different analytical observations that served as base for building such models. For instance, advanced characterization tools such as HSQC and HMBC NMR are typically required for a precise fingerprinting of the complex oligomeric structures found in PL.
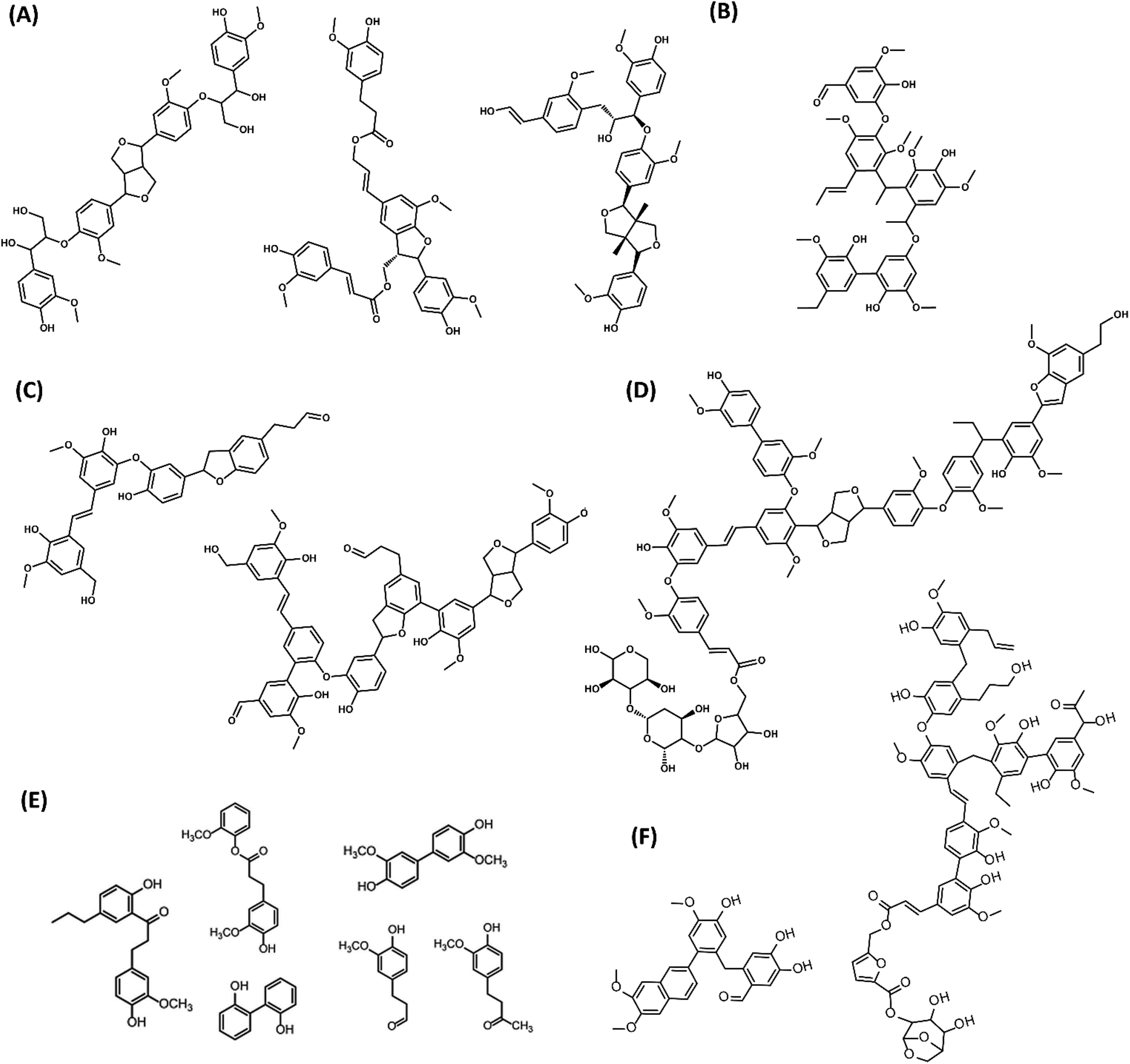 | ||
| Fig. 11 Structures of PL oligomers proposed in the literature. (A) G-based tetramers from pine sawdust PL98 (B) Pentamer from red oak PL104 (C) Oligomers from beech wood PL84 (D) Oligomer from switchgrass PL113 (E) PL dimers from rice straw, cotton stem and walnut shells102 (F) G-based oligomers from pine PL Adapted from ref. 89 with permission from Elsevier, copyright 2020. | ||
The lower molecular weight of PL compared to lignins obtained from other industrial processes is also noted by a relatively high phenolic OH content, since the native ether linkages in lignin are cleaved into phenols. By plotting the average Mw of lignins versus their phenolic OH content (calculated in mmol g−1 by 31P-NMR) as reported in the literature, a clear tendency is observed, in which lower Mw distributions lead to higher phenolic OH content (Fig. 12). While these parameters can be influenced by the isolation process, conditions applied and biomass source, the observed trend is informative. The PL is highlighted in the graph as one of the lignins with the lowest Mw and, therefore, highest phenolic OH content. This high content is particularly interesting for applications in materials such as polycarbonates and epoxy resins, in which PL could potentially replace (part of) bisphenol-A (BPA), a petroleum-based building block with well-known concerns related to endocrine disruption in humans.114–116 Other lignin applications explored in the materials literature include the partial replacement of aromatic polyols in rigid polyurethane foams and of phenol in phenol-formaldehyde resins.91,117–119 Being a particularly reactive motif, the phenol groups in PL can also serve as a handle for the engineering of advanced materials with on-demand functionalities. On the other hand, further depolymerization of PL may lead to high amounts of valuable phenolic monomers with applications as building blocks in the chemical and pharmaceutical industries, as well as energy-dense products with great potential as fuels. Such depolymerization strategies will be detailed in the next sections of this review.
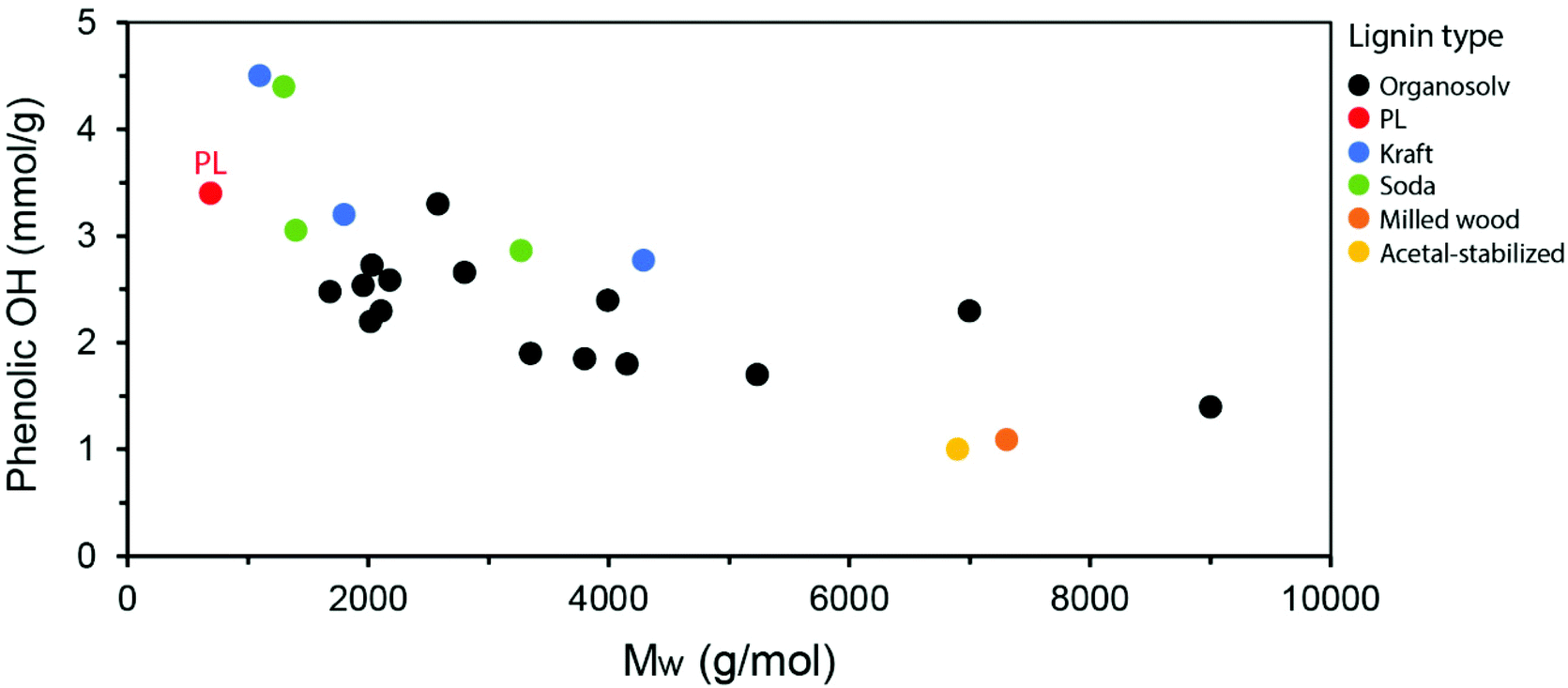 | ||
| Fig. 12 Correlation between the average Mw and the phenolic OH content for various lignin types as reported in the literature.5,89–96 | ||
4. Pyrolytic lignin valorization via depolymerization
The low amounts of monomers and chemical heterogeneity requires the PL to be further depolymerized before serving as a viable source for low molecular weight biobased chemicals and fuels.120 Similar to pyrolysis liquids, novel multistep approaches have great potential to enable efficient depolymerization of PL into valuable monomers. Yields and distribution of the desired products may be optimized and tuned by for instance process conditions, catalyst and solvent choice. An overview of the valorization possibilities for PL and possible low molecular weight products, including fields of applications, is presented in Fig. 13.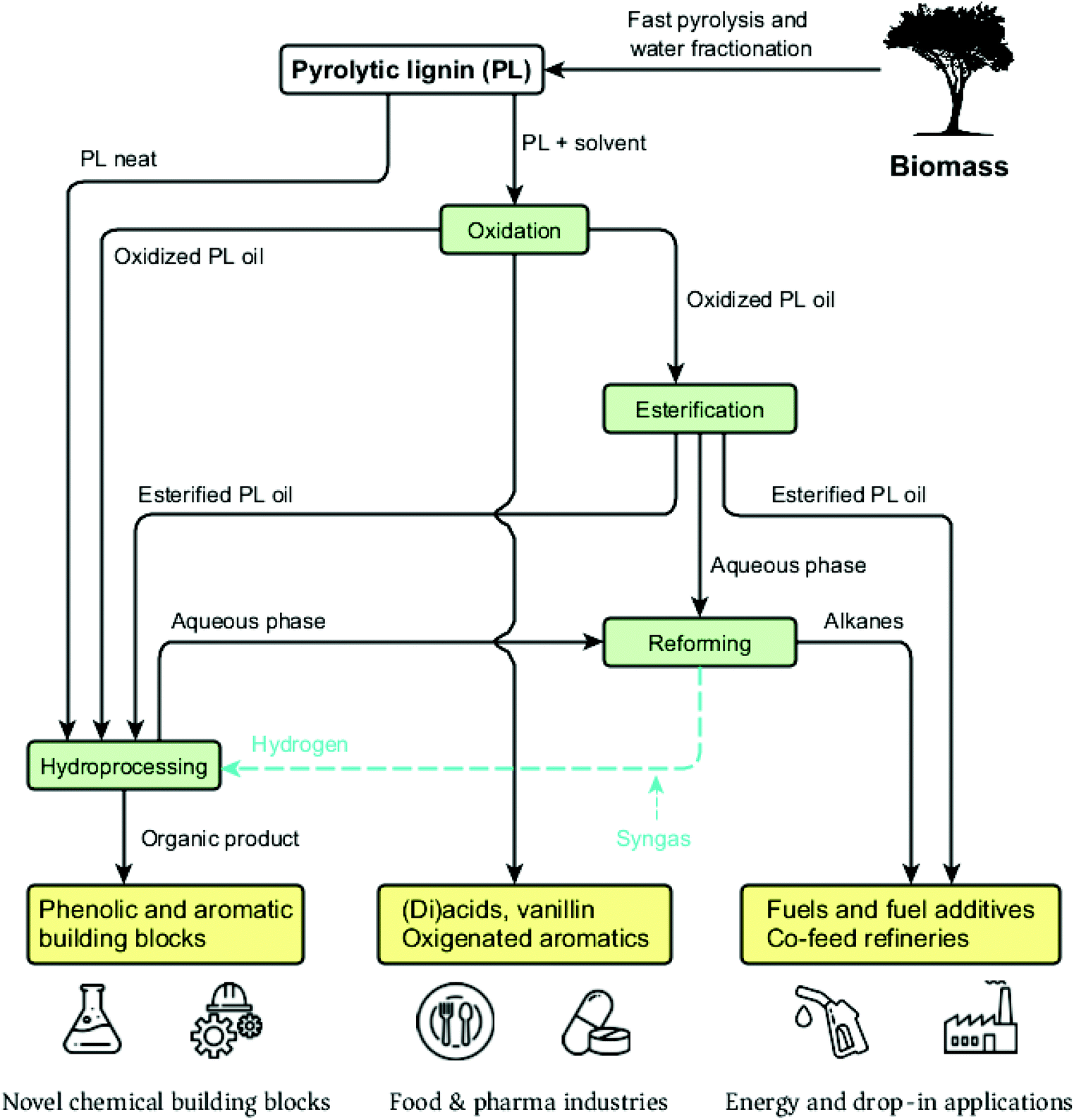 | ||
| Fig. 13 Overview of the possible PL valorization strategies discussed in this review. | ||
Typical depolymerization approaches involve reductive and/or oxidative (catalytic) conversion steps. These may lead to product mixtures with considerable amounts of phenolic and aromatic lignin building blocks that can serve as high-value platform chemicals, whereas (di)carboxylic acids formed during oxidation are of interest for applications in the polymer and food industries.74,121,122 In addition, esterified mixtures may also be targeted as biobased fuels or fuel additives.123 Recently, considerable research efforts have been undertaken to apply the PL as such (without major chemical modifications). Examples are the use of PL as filler/binder/reactive diluent in materials (e.g. polyurethanes118,124 and resins125) or to produce carbon fibers.126,127 Though such a strategy is not the core of this review, these potential applications broaden the possibilities for PL use, being complementary to the ones assessed by the depolymerization strategies here addressed.
Catalytic hydrotreatment is a reductive strategy for the upgrading of pyrolysis feedstocks (notably whole pyrolysis liquids) and has also been explored for PL depolymerization and valorization. The state of the art on PL hydrotreatment is presented below. Subsequently, potential oxidative approaches for the valorization of PL are discussed, followed by combined processes.
4.1. Reductive approaches for PL valorization
Catalytic hydrotreatment involves a catalyzed reaction with a hydrogen source. Typically, hydrogen gas is used but other hydrogen donors have been evaluated as well (e.g. alcohols). The process is typically carried out at rather harsh conditions (up to 450 °C and 200 bar of hydrogen pressure). Upon this treatment, hydrodeoxygenation and hydrocracking reactions occur, and valuable monomers can be obtained. Hydrodeoxygenation (HDO) is, in theory, closely related to the hydrodesulfurization (HDS) process performed in refinery industries for the removal of sulfur from petrochemical-based mixtures.128,129 Both processes use molecular hydrogen to eliminate the undesired heteroatoms, forming respectively H2O and H2S. However, despite this conceptual similarity, petrochemical-based and biobased feedstocks are extremely different in terms of structure and properties, and the oxygen content of the latter is much higher than the sulfur content in fossil fuels.During catalytic hydrotreatment, the highly complex chemical character of pyrolysis feedstocks typically leads to low product selectivity, as different starting materials combined with multiple reaction pathways occur simultaneously (Fig. 14).130 Hydrogen consumption is related to the conversion of various compound classes (acids, aldehydes, ketones, double bonds, etc.), and complex molecules can be accompanied by (often undesired) saturation.131 Ideally, the hydrogen usage during hydroprocessing should be minimized, as it is an important cost contributor, and carbon losses to the gas phase must also be suppressed to enhance overall carbon yields. Furthermore, thermally-induced repolymerization pathways must be prevented, as they ultimately lead to char formation.132
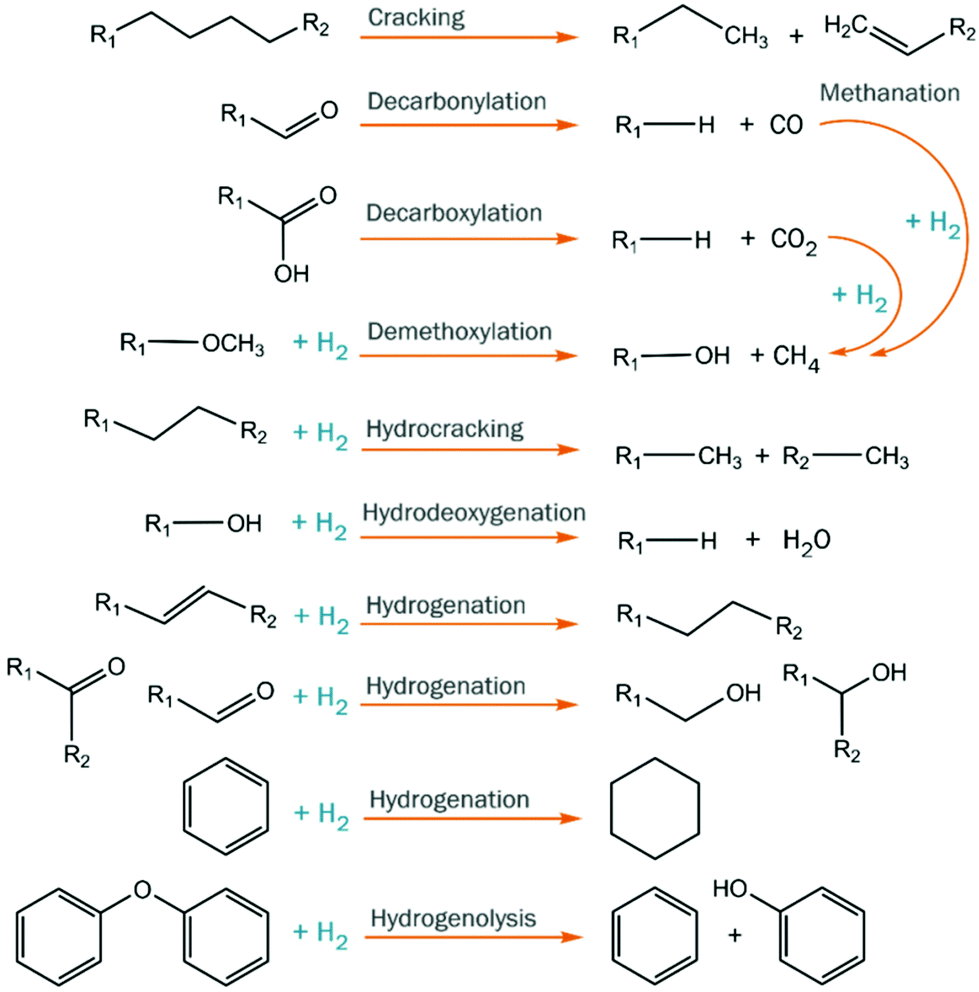 | ||
| Fig. 14 Reaction pathways involved in the catalytic hydroprocessing of pyrolysis feeds. | ||
Catalyst selection is of paramount importance. In particular, catalyst stability, selectivity and activity under the harsh conditions applied and in the reaction medium (which contains water and acidic compounds) are fundamental, yet difficult to achieve.133,134 Various catalyst deactivation mechanisms have been described in the literature, e.g. blocking of active sites, poisoning, sintering and coking.135–137 Despite these drawbacks, promising results have been reported for the catalytic hydrotreatment of bioliquids.133,138,139 Experimental studies have been extensively performed with whole pyrolysis liquids at different conditions and using a range of catalysts, which include, among others: (i) typical HDS catalysts, e.g. NiMo/Al2O3 and CoMo/Al2O3,140–144 which need an external sulfur addition to improve activity and stability;130,145 (ii) supported noble metal catalysts, e.g. Pd/C, Pt/C, Ru/C,132,138,146,147 (iii) inexpensive Ni-based supported catalysts.139,148–152 There is still great potential for further improvements, for instance by the use of tailored bimetallic and bifunctional supported catalysts.153–155
In contrast to pyrolysis liquids as a whole, the processing of the PL fraction is by far less explored. Nonetheless, interesting results were reported in studies using a range of different catalysts, set-ups, reaction conditions and solvents, see Table 3 for an overview. The vast majority of the literature regarding PL hydrotreatment consists of exploratory studies performed in batch set-ups. While these studies have given insights into reaction pathways, continuous processes suitable for scale-up need to be investigated in the future, particularly to assess catalyst stability. In some studies, the pyrolysis oil was from a technical lignin feed (e.g. Kraft, Alcell) instead of lignocellulosic biomass, and the resulting lignin oil was then hydrotreated.156,157
| PL source | T (°C) | P (bar) | Catalyst(s) | Solvent | Monomer yield (wt%) | Ref. |
|---|---|---|---|---|---|---|
| a ‘Volatile liquids’ based on PL intake, obtained from vacuum distillation of the organic product (55 °C, 170 mbar, 1 h). b ‘Organic distillate’ based on PL intake, obtained from vacuum distillation of the organic layer (200 °C, 1.7 mbar, 30 minutes). c Sum of total hydrocarbons and total phenolics as determined by GC-MS, based on PL intake. d Fraction of the organic product (≈60–65wt% of PL intake) boiling within gasoline range, estimated by simulated distillation. e Monomer yield as determined by GC × GC-FID, based on PL intake. f Monomer yield as determined by GC × GC-FID, based on the hydrotreated organic product. g Phenolic fraction separated by glycerol-assisted vacuum distillation of the pyrolysis liquid (≈10 wt% yield). h Lignin (instead of biomass) used as the pyrolysis feedstock. i Mass yield of the GC-analyzed oils after pyrolysis and hydrotreatment. | ||||||
| Water-extracted PL from pyrolysis liquid, solvent used as hydrogen donor | ||||||
| Mixed maple wood | 25–150 | 50 | Ru/TiO2 | Ethanol | 15–16.3a | 160 |
| Rice husk | 260 | 20 | Ru/ZrO2/SBA-15 | Ethanol | — | 82 |
| Red oak | 150 | 35 | Ru/C | Ethanol | — | 104 |
| Maple wood | 340–415 | 1 | HZSM-5 | Tetralin | 22.2–31.3b | 161 |
| Pine wood | 400–500 | 1 | γ-Al2O3 | Methanol | 39.5 | 159 |
| Pine wood, red oak | 300 | 1 | CuAlMgOx | Methanol | — | 162 |
| Water-extracted PL from pyrolysis liquid, H 2 used as hydrogen donor | ||||||
| Pine wood | 340 | 35 | HZSM-5, α-Al2O3, MoO3 | — | 3.1–17.1c | 163 |
| Hog fuel | 230–415 | 140 | CoMo | — | 50d | 164 |
| Forestry residue | 220–310 | 190 | Ru/C | — | — | 165 |
| Pine wood | 450 | 100 | Limonite | — | 23.4e | 86 |
| White oak | 150–400 | 69–167 | NiMo/Al2O3, Pd/C, Pt/C | — | — | 166 |
| Pine wood | 300–400 | 190–200 | Ru/C, NiMo/Al2O3 | — | — | 167 |
| Pine wood, sunflower seed peel, miscanthus, prunings, verge grass | 350–425 | 100 | Pd/C | — | 39e | 61 |
| Pine wood | 350–400 | 100 | Ru/C, Pt/C, Pd/C, Rh/C, NiMo/Al2O3, CoMo/Al2O3 | — | 33e | 85 |
| Pine wood, forestry residue | 400 | 100 | Ru/C | — | 51.3e | 83 |
| Pine wood | 350 | 120 | Ru/C, Ni/SiO2–ZrO2, Ni-Cu/SiO2–ZrO2, Ni–Pd/SiO2, Ni–Pd–Cu/SiO2, Ni–Mo–Cu/SiO2-Al2O3, Ni–Mo/SiO2–Al2O3 | — | 20–37f | 168 |
| Pyrolyzed lignin oils, solvent used as hydrogen donor | ||||||
| Rice huskg | 150–170 | 40 | Ru/SBA-15 | Isopropanol | 83–85c | 169 |
| Alcell ligninh | 350 | 100 | Ru/C | Dodecane | 26f | 156 |
| Pyrolyzed lignin oils, H2 used as hydrogen donor | ||||||
| Kraft and Organosolv ligninh | 350–400 | 100 | Ru/C, 20NiMoP/AC, CoMo/Al2O3 | — | 28.8–81.9f | 157 |
| Organocell ligninh | 400 | 1 | Fe/SiO2, Fe/AC | — | 5.7–6.1i | 170 |
The monomer yields after hydrotreatment vary substantially, and high values of >50% have been reported. A clear comparison is difficult due to differences in process conditions, and particularly in the approach to determine the yield of such monomeric products (e.g. based on input, based on product oil, based on GC detectables, etc.). Recent advances in analytical techniques such as NMR and GC × GC for bioliquids characterization have provided valuable information regarding the product composition and the amounts of alkylphenolics and aromatics obtained from PL hydroprocessing.61,83,85,86 Indeed, monomers are present in considerable amounts, and may be used in various existing applications, e.g. polymers, dyes, resins, fine chemicals, fuels and fuel additives.74,158 Remarkably, Wang et al. reported a 39.5% PL conversion into a single product (hexamethylbenzene), isolated as a high-purity (>99%) crystal.159
Overall, noble metal catalysts often favour the over-reduction of aromatic rings in PL, leading to (cyclo)alkanes.85,157 Inexpensive catalysts such as Ni-based and limonite (an iron ore) are preferred. PL is typically isolated as a low melting solid or a liquid depending on the purity. As such, it may serve as the reaction medium, and a solvent-free process may be envisioned. This prevents extra costly separation steps and other issues related to the use of solvents, such as participation in reaction pathways.
As most research is performed in batch set-ups and exploratory, further process development is required. The use of continuous set-ups enables the acquisition of intrinsic kinetic data essential to pave the way towards optimization, scale-up and ultimately industrialization of PL upgrading processes. Other important aspects to be explored are: (i) detailed studies aimed at the selection of improved catalysts regarding activity, selectivity, stability and recyclability; (ii) the biomass source, which has a direct impact on the PL structure and properties; (iii) the hydrotreatment conditions, e.g. pressure, temperature, residence time; (iv) the hydrogen donor, e.g. H2, methanol, formic acid; (v) the reactor configuration; (vi) new fractionation approaches for PL; (vii) separation technologies for the product mixtures obtained from PL; (viii) testing of PL-derived mixtures and/or specific compounds in suggested applications.
4.2. Oxidative approaches for PL valorization
In contrast to reductive processes, the oxidation of lignin has been performed industrially for many decades. A representative example is the pulp bleaching process, in which residual lignin-related chromophores are oxidized to low molecular weight, water-soluble compounds. Several oxidants are used (e.g. chlorine, oxygen, hydrogen peroxide), which react via electrophilic and radical mechanisms with the aromatic chromophores and promote delignification.171,172 An oxidative strategy similar to the one used in pulp industries has been recently proposed for the deconstruction of lignin before enzymatic digestion of cellulose, i.e. a first step able to significantly decrease biomass recalcitrance and, consequently, increase the availability of biomass-derived sugars for further processing.173–176 This approach relies on the fact that the lignin structures are highly prone to oxidation, and thus there is great potential for converting lignin into valuable products via oxidative strategies.172,177–179The oxidation of industrially available technical lignins (e.g. alkali, Alcell) as well as of lignin model compounds, has been extensively investigated with oxygen, air and hydrogen peroxide as oxidants. A broad range of catalytic systems has been reported, in which homogeneous catalysts are most often employed, e.g. TEMPO,180–182 oxovanadium complexes,183–186 metallosalen complexes187,188 and POMs.189,190 Heterogeneous catalysts (e.g. chalcopyrite,191 metal-supported,192–194 metal oxides195–197) and innovative approaches using biomimetic catalysts198–200 and ionic liquids201–203 have been studied as well. Products derived from lignin oxidation include aromatic acids and aldehydes such as vanillin,204–206 phenolic building blocks207–210 and di-carboxylic acids (DCAs)211,212 with several potential applications.
Ozone is relatively less explored as an oxidant for lignin, despite its uses in wastewater treatment as a disinfectant,213 in the paper and pulp industry as a bleaching agent,178,214 as a pretreatment to improve the enzymatic hydrolysis of biomass,173,175,215–218 and to upgrade vegetable oils.219 Ozone has a high reactivity towards both phenolic and non-phenolic nuclei at mild conditions, and neither chemical additives nor catalysts are typically needed.172 Advantageously, it can be easily generated in situ, either from oxygen or from dry air, and is relatively safe. Ozone has a short half-life of less than one hour when dissolved,220,243 thus any residual ozone in the system quickly decomposes to O2, providing an overall clean process with no need of extra separation steps. Previous works showed that ozonated solutions from biomass and/or lignin contain a range of oxygenated aromatics, quinones and carboxylic acids with potential as fuel additives,221 polyurethanes,222 surfactants223 and fine chemicals for the food and pharma industries.191,224
In contrast to lignin oxidation, studies on the oxidation of PL are nearly absent in the literature. Recently, an oxidation system with oxygen and polyoxometalates as the catalysts was reported. Here the PL showed the highest product yields (65.2 wt%) to aromatic compounds and esters compared to other lignin feeds.190 Recently, our group has for the first time studied the oxidation of PL with ozone in detail. It showed a high reactivity (mainly due to the high amounts of hydroxy and methoxy groups within the PL structure) and PL depolymerization of up to 40% upon ozone exposure at mild conditions was observed. The main products were low molecular weight (di)acids and esters, along with larger highly oxygenated aliphatics.224 Ozone was also shown to be promising to mediate the depolymerization and solvolysis of other technical lignins under ambient conditions.93
Most of the studies on lignin oxidation have been performed in (semi)batch set-ups. However, the use of continuous flow microreactors with gaseous oxidants that often bring safety issues (e.g. oxygen and ozone) is very attractive, as higher mass transfer rates from the gas to the liquid phase are attainable, much smaller volumes are used and superior control over the reaction conditions can be achieved. Accordingly, the microreactor technology is considered a sustainable solution from both safety and energy-saving aspects as it offers a substantial process intensification due to the enhanced mass and heat transfer rates, as well as the ease of upscaling by numbering-up.225,226 For lignin oxidation in general, the development of continuous flow processes in microreactors is still in its infancy, and only a few reports are currently available. Promising results were reported on the photocatalytic degradation of lignin model substrates,227 ultrafast hydrothermal228 and copper chloride mediated oxidative229 depolymerization of Kraft lignin, and supercritical extraction of Kraft lignin oxidation products.230 Our group has recently demonstrated the potential of microreactors for the ozone oxidation of PL at ambient conditions, and the system showed good performance at lower residence times compared to a semi-batch set-up.231
Similar to the reductive approaches, the development of feasible systems that minimize reagents consumption while maximizing product yields is fundamental for future oxidative upgrading processes for PL. This includes tuning process conditions according to the characteristics of the feedstock and targeted products, optimizing the process set-up and, in the case of catalytic systems, improving the catalyst stability and selectivity. While the focus of this review is on depolymerization, it is important to mention that lignin oxidation is also interesting as a functionalization step, as the OH groups incorporated in the structure can be further used as polymerization sites in the development of novel biobased materials.223
New oxidation approaches not discussed in detail in this paper but worthwhile mentioning (as they will be subject to future investigations) are electrochemical processes to depolymerize lignin, e.g. to aromatic fine chemicals such as vanillin and syringaldehyde.232
4.3. Combined oxidative-reductive approaches for PL valorization
The previous sections showed that reductive and oxidative routes have good potential for PL valorization. In addition to that, a combination of both an oxidative and reductive step may be of interest to improve product yields. This approach has been, for example, explored for the controlled depolymerization of lignins rich in β-O-4 linkage into aromatic monomers.233 While in PL the aliphatic C–C double bonds, i.e. stilbene linkages, are readily cleaved during oxidation, these are hydrogenated to stable C–C bonds during catalytic hydrotreatment (which are difficult to cleave). Furthermore, even though aromaticity can be lost upon harsh oxidation, a controlled and mild oxidative step can greatly improve lignin's accessibility at the expense of some aromaticity, still leading to good yields of aromatic and phenolic monomers.Successful combined oxidative-reductive approaches were demonstrated for whole pyrolysis liquids.234–236 For example, a sequence of an oxidation reaction based on H2O2/ozone and a subsequent hydrotreatment ultimately gave a product enriched in hydrocarbons. Compared to the direct hydrotreatment, products from the two-step approach were obtained in higher yields and had improved HHVs, as well as lower acid values, char and oxygen contents. A follow-up study achieved similar promising results using syngas (from biomass gasification) instead of pure hydrogen in the hydrotreatment step.123 While combined approaches for PL depolymerization are nearly absent, the use of a straightforward oxidative pre-treatment step to reduce the molecular weight of PL before hydrotreatment was shown to have a positive effect on monomeric product yields and overall depolymerization, yielding products of higher volatility and improved calorific values.231,236
Esterification of acidic oxidized pyrolysis liquids has been also explored to stabilize them and to prevent aging and corrosion issues due to the presence of the organic acids. In addition, as boiling points of esters are lower than those of their parent acids, separation through reactive distillation is possible.237–239 Various catalysts have been investigated for this purpose, i.e. ion-exchange resins,240 ionic liquids,241 zeolites,242,243 homogeneous/solid acids244–247 and mixed oxides.248 Previous studies showed that aldehydes can negatively affect the conversion of acids into esters and promote repolymerization pathways during esterification. As such, the oxidation of aldehydes in the preceding step is a convenient solution to improve product properties.239,249 Accordingly, oxidation-esterification systems using both ozone and H2O2/ozone for the first step were reported and showed potential to upgrade pyrolysis liquids into fuels.239,250
4.4. Other potential approaches for PL valorization
To the best of our knowledge, the approaches addressed below have not yet been applied to PL. Nonetheless, it is worth mentioning current trends in the broad lignin transformation space, which can also be used for PL and likely expand the possibilities for PL valorization. For instance, biological valorization routes using enzymes and microorganisms are receiving great attention due to their milder operational conditions and low energy consumption. Ligninolytic enzymes, mainly peroxidases, were shown to catalyze the depolymerization of technical lignins (e.g. Kraft lignin, alkali lignin) and lignin model compounds.251–253 Lignin degradation by bacteria has been also investigated as a way to produce mixtures of low Mw fragments, and several reports are available on the bacterial conversion of technical lignins and different lignocellulosic biomasses.254,255 Such biological biorefining concepts typically involve a first conversion step that generates lignin-derived intermediates which are further upgraded into platform chemicals through subsequent funnelling steps. Accordingly, metabolic engineering strategies have been explored for the production of value-added chemicals from lignin, such as vanillin, catechol, vanillic acid, adipic acid, muconic acid, bioactive polyphenols, among others.254 A recent work specifically highlighted the potential of pyrolysis products as a carbon source for the microbial biosynthesis of lipids, which are known important platform molecules for fuels, food and feed applications.256While such biological routes in theory provide a promising technological platform for lignin, several technical barriers and challenges still need to be overcome. The main drawbacks are related to the high costs, difficulties on obtaining enzymes on a large scale, low product yields, lignin heterogeneity and low enzyme stability due to their overall high sensitivity. The latest point is crucial when considering PL as the feed. Given that lignin-derived streams are often water-insoluble and toxic to microbes, industrial strains can only be used at low feedstock concentrations. Therefore, the development of processes more tolerant to lignin monomers/small fragments is of paramount importance to increase the feasibility of biological upgrading steps when combined with the pyrolysis biorefinery. Finally, as the knowledge of lignin degrading metabolic pathways is still very limited, there is a remarkable opportunity in place for the development of systems biology approaches (e.g. ‘omics’ technologies) able to leverage microbial engineering for lignin valorization.257,258 The reader is referred to dedicated reviews that cover this topic in detail.254,255,259,260
Another rapidly growing topic in the lignin field is the synthesis and use of lignin nanoparticles (LNPs), for which various methods have been investigated (e.g. solvent exchange, acid precipitation, ultrasonication).261,262 LNP formation is related to the naturally amphiphilic character of lignin, comprised by a hydrophobic aromatic backbone decorated with hydrophilic motifs (e.g. hydroxyl and carboxyl groups, vide supra). The properties of LNPs can be tuned to a high extent, as they are related to the production method and conditions (such as temperature and pH)263,264 as well as very dependent on the lignin source and its properties (i.e. hydroxy content and distribution, molecular weight, S/G unit ratio). Overall, LNPs have the advantages of being an abundant, biocompatible and inexpensive nanomaterial with very interesting properties. Several applications of LNPs have been consolidated, such as their use in thermoplastics, nanocomposites, dispersants, coatings, foams, bactericides and products that require protection from UV radiation.262,265 While mostly explored for technical lignins of larger Mw (e.g. Kraft and organosolv lignins), it has been shown that LNPs can also be produced using low Mw lignin fractions with properties similar to those of PL.264 This valorization strategy doesn't necessarily involve depolymerization (our focus in this review), but it can be certainly envisioned in a pyrolysis-based biorefinery as a way to produce valuable surface-active materials and bioactive ingredients from PL.
5. How far are we from the implementation of a pyrolysis-based biorefinery?
In the previous sections, we have showcased the pyrolysis biorefinery concept with a focus on valorization strategies for depolymerizing PL into fuels and chemicals. Most of the routes aforementioned are still very exploratory and academic in scope, and therefore not close to implementation. The pyrolysis technology itself is available commercially, but the obtained pyrolysis liquid is currently mainly used for energy generation purposes.266While proven to be a promising source of biobased aromatic molecules, several challenges have to be overcome in order to bring the PL fraction closer to the high-value applications that could enable the industrialization of an integrated pyrolysis-based biorefinery. These challenges are related to technological, economic and regulatory aspects, which are intertwined. To highlight some points addressed earlier, it is fundamental to achieve a good catalyst stability/recyclability in the PL depolymerization, to develop efficient product separation steps and/or further funnelling steps toward mixtures of lower complexity and high value, as well as to achieve a performance as good (or superior to) fossil-based counterparts. On this note, a particularly important point is the need for more applied research on PL valorization, in which specific applications/properties are targeted and industrial benchmarks are used for a relevant comparison that should ideally include technical and economic analyses (TEA). A good example from the recent literature is a TEA and a LCA of the integrated utilization of bio-oil from cotton straw for the production of levoglucosan (i.e. the main component of the pyrolytic sugar fraction), phenol-formaldehyde resins and noncorrosive road deicers. The results imply that this scheme is likely commercially attractive (particularly for the first two products) if high product yields are achieved and the use of non-renewable reagents and energy is minimized.267
Besides technical aspects, the implementation of more advanced biorefining schemes is a multidisciplinary challenge including supply chain, sustainability and regulatory aspects. A robust model must therefore consider, among others, feedstock selection (as biomass properties and costs vary and are subject to location and seasonality, as well as impacted by natural events); logistics aspects such as feedstock transportation and storage/stability; fluctuations and sourcing of energy, reagents and materials; LCA/sustainability aspects such as recyclability, biobased content and biodegradability of products; product portfolio flexibility; heat integration and waste management. Along with this comes the regulatory framework, which is not fully established for many novel lignin-derived products, delaying market entry and increasing the risk of new technologies. Finally, governmental support as in funding schemes for bioeconomy and environmentally-sound policies (e.g. emissions taxation, tighter regulations) will play a pivotal role in the development of biorefineries – a space also highly influenced by societal pressure and global politics.268 It is clear that a new era of biobased industries, in which biomass is valorized as a whole by cascading upgrading steps of its components, is in full-speed development. Differently from the traditional “biomass to biofuels” models, such schemes unlock new types of businesses, often decentralized and suitable for more complex industries that aim at higher-value products and diversified markets.269 The integrated pyrolysis-based biorefinery (here explored by “zooming in” the PL fraction) is part of such movement, and its scale-up and implementation will largely depend on both disruptive scientific/technology developments as well as said external factors.
6. Conclusions and future recommendations
Fast pyrolysis is a well-established technology to thermochemically convert lignocellulosic biomass into pyrolysis liquids (also known as bio-oils). Pyrolysis liquids are easily fractionated into a sugar-rich aqueous stream and the water-insoluble PL, two fractions that can be efficiently upgraded via tailored routes in a pyrolysis-based biorefinery. PL is mostly comprised of lignin fragments and, as this review details, holds great potential as a biobased source of valuable compounds to replace fossil-based counterparts. While its complex and chemically heterogeneous structure may represent a challenge, increasing research efforts highlight the potential of PL to yield aromatics, phenolics and aliphatic monomers such as DCAs and esters through (catalytic) reductive, oxidative and combined valorization strategies.Despite the promising results reported in the literature, research on PL valorization is still in its infancy, and further investigations are necessary to advance the field. For instance, the range of biomass feedstocks can be greatly expanded beyond wood, particularly towards agricultural wastes with a local approach that can bring social developments and accelerate scale-up and implementation. Detailed studies will be required to further elucidate reaction pathways and support the selection of improved and viable catalysts concerning activity, selectivity, stability and recyclability. Studies under continuous operation are of great interest to better understand the kinetics and scale-up possibilities. Importantly, the downstream processing of lignin-derived mixtures to individual compounds or specific compound classes is still an overlooked topic that requires further investigation. Finally, product research to use PL or derived products as reactive-diluent/filler in materials (phenol-formaldehyde resins, polyurethanes, epoxy resins, carbon fibers) and functional ingredients needs further attention. Identification of promising end uses for (upgraded) PL will accelerate the introduction and implementation of feasible pyrolysis-based refineries with a low carbon footprint. Accordingly, such biorefining schemes will largely benefit from robust TEA and LCA models supported by environmentally-sound regulations to speed up the market entry of novel products from biomass.
Author contributions
Monique B. Figueirêdo: conceptualization, investigation, writing – original draft, visualization, funding acquisition. Idoia Hita: conceptualization, investigation, writing – original Draft, visualization. Peter J. Deuss: conceptualization, writing – review & editing, supervision. Robbie Venderbosch: writing – review & editing. Erik Heeres: conceptualization, writing – review & editing, supervision.Conflicts of interest
The authors declare the following competing financial interest(s): R. H. V. is a senior researcher at BTG Biomass Technology Group, which explores commercial opportunities for pyrolysis oils and its fractions.Acknowledgements
Financial support from the Science without Borders program (CNPq, Brazil) on the PhD project of MBF is gratefully acknowledged.References
- J. Jacquet and D. Jamieson, Nat. Clim. Change, 2016, 6, 643–646 CrossRef.
- International Renewable Energy Agency (IRENA), Reaching zero with renewables: Eliminating CO2 emissions from industry and transport in line with the 1.5 °C climate goal, Abu Dhabi, 2020 Search PubMed.
- E. Novaes, M. Kirst, V. Chiang, H. Winter-Sederoff and R. Sederoff, Plant Physiol., 2010, 154, 555–561 CrossRef CAS PubMed.
- A. Kumar, A. Gautam and D. Dutt, Adv. Biosci. Biotechnol., 2016, 7, 149–168 CrossRef.
- S. Bertella and J. S. Luterbacher, Trends Chem., 2020, 2, 440–453 CrossRef CAS.
- Y. Pu, F. Hu, F. Huang, B. H. Davison and A. J. Ragauskas, Biotechnol. Biofuels, 2013, 6, 15 CrossRef CAS PubMed.
- J. Ralph, C. Lapierre and W. Boerjan, Curr. Opin. Biotechnol., 2019, 56, 240–249 CrossRef CAS PubMed.
- M. T. Amiri, S. Bertella, Y. M. Questell-Santiago and J. S. Luterbacher, Chem. Sci., 2019, 10, 8135–8142 RSC.
- C. W. Lahive, P. C. J. Kamer, C. S. Lancefield and P. J. Deuss, ChemSusChem, 2020, 13, 4238–4265 CrossRef CAS PubMed.
- M. Bergs, X. T. Do, J. Rumpf, P. Kusch, Y. Monakhova, C. Konow, G. Völkering, R. Pude and M. Schulze, RSC Adv., 2020, 10, 10740–10751 RSC.
- M. P. Pandey and C. S. Kim, Chem. Eng. Technol., 2011, 34, 29–41 CrossRef CAS.
- S. Nikafshar, J. Wang, K. Dunne, P. Sangthonganotai and M. Nejad, ChemSusChem, 2021, 14, 1184–1195 CrossRef CAS PubMed.
- Y. Liao, S.-F. Koelewijn, G. V. den Bossche, J. V. Aelst, S. V. den Bosch, T. Renders, K. Navare, T. Nicolaï, K. V. Aelst, M. Maesen, H. Matsushima, J. M. Thevelein, K. V. Acker, B. Lagrain, D. Verboekend and B. F. Sels, Science, 2020, 367, 1385–1390 CrossRef CAS PubMed.
- G. G. Zaimes, N. Vora, S. S. Chopra, A. E. Landis and V. Khanna, Processes, 2015, 3, 634–663 CrossRef CAS.
- J. K. Kurian, G. R. Nair, A. Hussain and G. S. V. Raghavan, Renewable Sustainable Energy Rev., 2013, 25, 205–219 CrossRef.
- A. Molino, S. Chianese and D. Musmarra, J. Energy Chem., 2016, 25, 10–25 CrossRef.
- Y. Zhang and W.-T. Chen, Direct Thermochemical Liquefaction for Energy Applications, ed. L. Rosendahl, Woodhead Publishing, 2018, pp. 127–168 Search PubMed.
- A. V. Bridgwater, Biomass Bioenergy, 2012, 38, 68–94 CrossRef CAS.
- T. Dickerson and J. Soria, Energies, 2013, 6, 514–538 CrossRef CAS.
- X. Chen, H. Zhang and R. Xiao, Energy Fuels, 2018, 32, 4178–4188 CrossRef CAS.
- J. Alvarez, M. Amutio, G. Lopez, L. Santamaria, J. Bilbao and M. Olazar, Waste Manage., 2019, 85, 385–395 CrossRef CAS PubMed.
- S. Deng, H. Tan, X. Wang, F. Yang, R. Cao, Z. Wang and R. Ruan, Bioresour. Technol., 2017, 239, 302–310 CrossRef CAS PubMed.
- J. Yang, J. Rizkiana, W. B. Widayatno, S. Karnjanakom, M. Kaewpanha, X. Hao, A. Abudula and G. Guan, Energy Convers. Manage., 2016, 120, 422–429 CrossRef CAS.
- R. W. Nachenius, F. Ronsse, R. H. Venderbosch and W. Prins, Advances in Chemical Engineering, ed. D. Y. Murzin, Academic Press, 2013, vol. 42, pp. 75–139 Search PubMed.
- Y. A. Situmorang, Z. Zhao, N. Chaihad, C. Wang, A. Anniwaer, Y. Kasai, A. Abudula and G. Guan, Int. J. Hydrogen Energy, 2021, 46, 3640–3650 CrossRef CAS.
- A. Pattiya, in Direct Thermochemical Liquefaction for Energy Applications, ed. L. Rosendahl, Woodhead Publishing, 2018, pp. 3–28 Search PubMed.
- M. Balajii and S. Niju, Environ. Chem. Lett., 2019, 17, 1447–1469 CrossRef CAS.
- S. Anto, M. P. Sudhakar, T. S. Ahamed, M. S. Samuel, T. Mathimani, K. Brindhadevi and A. Pugazhendhi, Fuel, 2021, 285, 119205 CrossRef CAS.
- J. G. Rogers and J. G. Brammer, Biomass Bioenergy, 2012, 36, 208–217 CrossRef CAS.
- H. D. Setiabudi, M. A. A. Aziz, S. Abdullah, L. P. Teh and R. Jusoh, Int. J. Hydrogen Energy, 2020, 45, 18376–18397 CrossRef CAS.
- I. Hita, T. Cordero-Lanzac, G. Bonura, C. Cannilla, J. M. Arandes, F. Frusteri and J. Bilbao, J. Ind. Eng. Chem., 2019, 80, 392–400 CrossRef CAS.
- C. Zerva, S. A. Karakoulia, K. G. Kalogiannis, A. Margellou, E. F. Iliopoulou, A. A. Lappas, N. Papayannakos and K. S. Triantafyllidis, Catal. Today, 2021, 366, 57–67 CrossRef CAS.
- Á. Ibarra, I. Hita, J. M. Arandes and J. Bilbao, Energy Fuels, 2019, 33, 7458–7465 CrossRef.
- I. Hita, J. M. Arandes and J. Bilbao, in Chemical Catalysts for Biomass Upgrading, John Wiley & Sons, Ltd, 2020, pp. 61–96 Search PubMed.
- Y. Y. Chong, S. Thangalazhy-Gopakumar, S. Gan, L. Y. Lee and H. K. Ng, Bioresour. Technol. Rep., 2020, 12, 100560 CrossRef.
- L. Li, B. Yan, H. Li, S. Yu and X. Ge, Renewable Energy, 2020, 146, 643–650 CrossRef CAS.
- Á. Ibarra, I. Hita, J. M. Arandes and J. Bilbao, Catalysts, 2020, 10, 1157 CrossRef.
- A. de R. Pinho, M. B. B. de Almeida, F. L. Mendes, L. C. Casavechia, M. S. Talmadge, C. M. Kinchin and H. L. Chum, Fuel, 2017, 188, 462–473 CrossRef CAS.
- M. Amutio, G. Lopez, M. Artetxe, G. Elordi, M. Olazar and J. Bilbao, Resour., Conserv. Recycl., 2012, 59, 23–31 CrossRef.
- T. Aysu and M. M. Küçük, Energy, 2014, 64, 1002–1025 CrossRef CAS.
- A. Oasmaa, J. Lehto, Y. Solantausta and S. Kallio, Energy Fuels, 2021, 35, 5683–5695 CrossRef CAS.
- A. M. Shoaib, R. A. El-Adly, M. H. M. Hassanean, A. Youssry and A. A. Bhran, Egypt. J. Pet., 2018, 27, 1305–1311 CrossRef.
- J. Piskorz, D. S. Scott and D. Radlein, ACS Symp. Ser., 1988, 16, 167–178 CrossRef.
- R. H. Venderbosch and H. J. Heeres, Pyrolysis Oil Stabilisation by Catalytic Hydrotreatment, IntechOpen, 2011 Search PubMed.
- M. E. Boucher, A. Chaala, H. Pakdel and C. Roy, Biomass Bioenergy, 2000, 19, 351–361 CrossRef CAS.
- E. J. Soltes and T. Elder, Org. Chem. Biomass, 1981, 1, 63–99 Search PubMed.
- H. Shafaghat, H. W. Lee, Y. F. Tsang, D. Oh, J. Jae, S.-C. Jung, C. H. Ko, S. S. Lam and Y.-K. Park, Chem. Eng. J., 2019, 366, 330–338 CrossRef CAS.
- S. Ryu, H. W. Lee, Y.-M. Kim, J. Jae, S.-C. Jung, J.-M. Ha and Y.-K. Park, Appl. Surf. Sci., 2020, 511, 145521 CrossRef.
- X. Zhang, H. Ma, S. Wu, W. Jiang, W. Wei and M. Lei, Fuel, 2019, 242, 587–595 CrossRef CAS.
- M. Zhang and H. Wu, Fuel, 2019, 242, 580–586 CrossRef CAS.
- I. Hita, T. Cordero-Lanzac, T. Kekäläinen, O. Okafor, J. Rodríguez-Mirasol, T. Cordero, J. Bilbao, J. Jänis and P. Castano, ACS Sustainable Chem. Eng., 2020, 8, 18433–18445 CrossRef CAS.
- Z. E. Zadeh, A. Abdulkhani and B. Saha, Energy, 2021, 214, 118930 CrossRef.
- A. E. M. Fodah, M. K. Ghosal and D. Behera, J. Energy Inst., 2021, 94, 242–251 CrossRef.
- N. Ahmed, M. Zeeshan, N. Iqbal, M. Z. Farooq and S. A. Shah, J. Cleaner Prod., 2018, 196, 927–934 CrossRef CAS.
- E. Salehi, J. Abedi and T. Harding, Energy Fuels, 2011, 25, 4145–4154 CrossRef CAS.
- P. Manara, S. Bezergianni and U. Pfisterer, Energy Convers. Manage., 2018, 165, 304–315 CrossRef CAS.
- A. Dimitriadis, L. P. Chrysikou, G. Meletidis, G. Terzis, M. Auersvald, D. Kubička and S. Bezergianni, Renewable Energy, 2021, 168, 593–605 CrossRef CAS.
- A. Dimitriadis, D. Liakos, U. Pfisterer, M. Moustaka-Gouni, D. Karonis and S. Bezergianni, Biomass Bioenergy, 2021, 151, 106171 CrossRef CAS.
- Q. Zhang, J. Chang, T. Wang and Y. Xu, Energy Convers. Manage., 2007, 48, 87–92 CrossRef CAS.
- H. Wang and Y. Wang, Top. Catal., 2016, 59, 65–72 CrossRef CAS.
- M. B. Figueirêdo, P. J. Deuss, R. H. Venderbosch and H. J. Heeres, Biomass Bioenergy, 2020, 134, 105484 CrossRef.
- B. Scholze and D. Meier, J. Anal. Appl. Pyrolysis, 2001, 60, 41–54 CrossRef CAS.
- Y. Yu, Y. W. Chua and H. Wu, Energy Fuels, 2016, 30, 4145–4149 CrossRef CAS.
- N. M. Bennett, S. S. Helle and S. J. B. Duff, Bioresour. Technol., 2009, 100, 6059–6063 CrossRef CAS PubMed.
- R. M. Abdilla, C. B. Rasrendra and H. J. Heeres, Ind. Eng. Chem. Res., 2018, 57, 3204–3214 CrossRef CAS PubMed.
- Z. Chi, M. Rover, E. Jun, M. Deaton, P. Johnston, R. C. Brown, Z. Wen and L. R. Jarboe, Bioresour. Technol., 2013, 150, 220–227 CrossRef CAS PubMed.
- H. Wang, D. Livingston, R. Srinivasan, Q. Li, P. Steele and F. Yu, Appl. Biochem. Biotechnol., 2012, 168, 1568–1583 CrossRef CAS PubMed.
- W. Yin, R. H. Venderbosch, G. Bottari, K. K. Krawzcyk, K. Barta and H. J. Heeres, Appl. Catal., B, 2015, 166–167, 56–65 CrossRef CAS.
- R.-J. van Putten, J. C. van der Waal, E. de Jong, C. B. Rasrendra, H. J. Heeres and J. G. de Vries, Chem. Rev., 2013, 113, 1499–1597 CrossRef CAS PubMed.
- H. Zhou, M. A. Akhtar, Y. Wan, K. Ding, R. Ruan, H. Zhang and S. Zhang, J. Anal. Appl. Pyrolysis, 2020, 152, 104973 CrossRef CAS.
- D. Santhanaraj, M. R. Rover, D. E. Resasco, R. C. Brown and S. Crossley, ChemSusChem, 2014, 7, 3132–3137 CrossRef CAS PubMed.
- X. Xiong, J. Lian, X. Yu, M. Garcia-Perez and S. Chen, J. Ind. Microbiol. Biotechnol., 2016, 43, 1551–1560 CrossRef CAS PubMed.
- Z. Sun, B. Fridrich, A. de Santi, S. Elangovan and K. Barta, Chem. Rev., 2018, 118, 614–678 CrossRef CAS PubMed.
- J. Zakzeski, P. C. A. Bruijnincx, A. L. Jongerius and B. M. Weckhuysen, Chem. Rev., 2010, 110, 3552–3599 CrossRef CAS PubMed.
- S. D. Stefanidis, K. G. Kalogiannis and A. A. Lappas, Wiley Interdiscip. Rev.: Energy Environ., 2018, 7, e281 Search PubMed.
- F. de Miguel Mercader, M. J. Groeneveld, S. R. A. Kersten, N. W. J. Way, C. J. Schaverien and J. A. Hogendoorn, Appl. Catal., B, 2010, 96, 57–66 CrossRef CAS.
- H. Yang, R. Yan, H. Chen, D. H. Lee and C. Zheng, Fuel, 2007, 86, 1781–1788 CrossRef CAS.
- H. Kawamoto, J. Wood Sci., 2017, 63, 117–132 CrossRef CAS.
- W. Boerjan, J. Ralph and M. Baucher, Annu. Rev. Plant Biol., 2003, 54, 519–546 CrossRef CAS PubMed.
- O. Derkacheva and D. Sukhov, Macromol. Symp., 2008, 265, 61–68 CrossRef CAS.
- C. Liu, J. Hu, H. Zhang and R. Xiao, Fuel, 2016, 182, 864–870 CrossRef CAS.
- Z. Tang, Y. Zhang and Q. Guo, Ind. Eng. Chem. Res., 2010, 49, 2040–2046 CrossRef CAS.
- A. Kloekhorst, J. Wildschut and H. J. Heeres, Catal. Sci. Technol., 2014, 4, 2367–2377 RSC.
- R. Bayerbach and D. Meier, J. Anal. Appl. Pyrolysis, 2009, 85, 98–107 CrossRef CAS.
- M. B. Figueirêdo, Z. Jotic, P. J. Deuss, R. H. Venderbosch and H. J. Heeres, Fuel Process. Technol., 2019, 189, 28–38 CrossRef.
- I. Hita, H. J. Heeres and P. J. Deuss, Bioresour. Technol., 2018, 267, 93–101 CrossRef CAS PubMed.
- P. S. Marathe, R. J. M. Westerhof and S. R. A. Kersten, Appl. Energy, 2019, 236, 1125–1137 CrossRef CAS.
- Q. Liu, S. Wang, Y. Zheng, Z. Luo and K. Cen, J. Anal. Appl. Pyrolysis, 2008, 82, 170–177 CrossRef CAS.
- M. B. Figueirêdo, R. H. Venderbosch, H. J. Heeres and P. J. Deuss, J. Anal. Appl. Pyrolysis, 2020, 149, 104837 CrossRef.
- I. Hita, P. J. Deuss, G. Bonura, F. Frusteri and H. J. Heeres, Fuel Process. Technol., 2018, 179, 143–153 CrossRef CAS.
- B. Pang, S. Yang, W. Fang, T.-Q. Yuan, D. S. Argyropoulos and R.-C. Sun, Ind. Crops Prod., 2017, 108, 316–326 CrossRef CAS.
- S. Bertella and J. S. Luterbacher, Green Chem., 2021, 23, 3459–3467 RSC.
- M. B. Figueirêdo, H. J. Heeres and P. J. Deuss, Sustainable Energy Fuels, 2020, 4, 265–276 RSC.
- S. Constant, H. L. J. Wienk, A. E. Frissen, P. de Peinder, R. Boelens, D. S. van Es, R. J. H. Grisel, B. M. Weckhuysen, W. J. J. Huijgen, R. J. A. Gosselink and P. C. A. Bruijnincx, Green Chem., 2016, 18, 2651–2665 RSC.
- F. Huang, P. M. Singh and A. J. Ragauskas, J. Agric. Food Chem., 2011, 59, 12910–12916 CrossRef CAS PubMed.
- N.-E. E. Mansouri and J. Salvadó, Ind. Crops Prod., 2006, 24, 8–16 CrossRef.
- K. V. Aelst, E. V. Sinay, T. Vangeel, Y. Zhang, T. Renders, S. V. den Bosch, J. V. Aelst and B. F. Sels, Chem. Commun., 2021, 57, 5642–5645 RSC.
- E. Fratini, M. Bonini, A. Oasmaa, Y. Solantausta, J. Teixeira and P. Baglioni, Langmuir, 2006, 22, 306–312 CrossRef CAS PubMed.
- X. Bai, K. H. Kim, R. C. Brown, E. Dalluge, C. Hutchinson, Y. J. Lee and D. Dalluge, Fuel, 2014, 128, 170–179 CrossRef CAS.
- A. I. Afifi, J. P. Hindermann, E. Chornet and R. P. Overend, Fuel, 1989, 68, 498–504 CrossRef CAS.
- P. R. Patwardhan, R. C. Brown and B. H. Shanks, ChemSusChem, 2011, 4, 1629–1636 CrossRef CAS PubMed.
- S. Li, Z. Luo, W. Wang, K. Lu, Y. Yang and X. Liang, Fuel, 2020, 262, 116516 CrossRef CAS.
- B. Scholze, C. Hanser and D. Meier, J. Anal. Appl. Pyrolysis, 2001, 58–59, 387–400 CrossRef CAS.
- D. J. McClelland, A. H. Motagamwala, Y. Li, M. R. Rover, A. M. Wittrig, C. Wu, J. S. Buchanan, R. C. Brown, J. Ralph, J. A. Dumesic and G. W. Huber, Green Chem., 2017, 19, 1378–1389 RSC.
- C. A. Mullen and A. A. Boateng, J. Anal. Appl. Pyrolysis, 2011, 90, 197–203 CrossRef CAS.
- R. Y. Nsimba, C. A. Mullen, N. M. West and A. A. Boateng, ACS Sustainable Chem. Eng., 2013, 1, 260–267 CrossRef CAS.
- S. Wang, H. Lin, B. Ru, W. Sun, Y. Wang and Z. Luo, J. Anal. Appl. Pyrolysis, 2014, 108, 78–85 CrossRef CAS.
- W. Mu, H. Ben, A. Ragauskas and Y. Deng, Bioenergy Res., 2013, 6, 1183–1204 CrossRef CAS.
- V. Strezov, M. Patterson, V. Zymla, K. Fisher, T. J. Evans and P. F. Nelson, J. Anal. Appl. Pyrolysis, 2007, 79, 91–100 CrossRef CAS.
- E. A. Capanema, M. Y. Balakshin and J. F. Kadla, J. Agric. Food Chem., 2004, 52, 1850–1860 CrossRef CAS PubMed.
- V. Echavarri-Bravo, M. Tinzl, W. Kew, F. Cruickshank, C. L. Mackay, D. J. Clarke and L. E. Horsfall, New Biotechnol., 2019, 52, 1–8 CrossRef CAS PubMed.
- F. Leng, Y. Wang, J. Chen, S. Wang, J. Zhou and Z. Luo, Chin. J. Chem. Eng., 2017, 25, 324–329 CrossRef CAS.
- M. Fortin, M. M. Beromi, A. Lai, P. C. Tarves, C. A. Mullen, A. A. Boateng and N. M. West, Energy Fuels, 2015, 29, 8017–8026 CrossRef CAS.
- D. Tiwari, J. Kamble, S. Chilgunde, P. Patil, G. Maru, D. Kawle, U. Bhartiya, L. Joseph and G. Vanage, Mutat. Res. – Genet. Toxicol. Environ. Mutagen., 2012, 743, 83–90 CrossRef CAS PubMed.
- E. Feghali, D. J. van de Pas, A. J. Parrott and K. M. Torr, ACS Macro Lett., 2020, 9, 1155–1160 CrossRef CAS.
- E. Feghali, D. J. van de Pas and K. M. Torr, Biomacromolecules, 2020, 21, 1548–1559 CrossRef CAS PubMed.
- J. E. Q. Quinsaat, E. Feghali, D. J. van de Pas, R. Vendamme and K. M. Torr, ACS Appl. Polym. Mater., 2021, 3, 5845–5856 CrossRef CAS.
- T. Saffar, H. Bouafif, F. L. Braghiroli, S. Magdouli, A. Langlois and A. Koubaa, Waste Biomass Valorization, 2020, 11, 6411–6427 CrossRef CAS.
- L.-Y. Liu, M. A. Karaaslan, Q. Hua, M. Cho, S. Chen and S. Renneckar, Ind. Eng. Chem. Res., 2021, 60, 11882–11892 CrossRef CAS.
- F. Cherubini, Energy Convers. Manage., 2010, 51, 1412–1421 CrossRef CAS.
- J. G. Zeikus, M. K. Jain and P. Elankovan, Appl. Microbiol. Biotechnol., 1999, 51, 545–552 CrossRef CAS.
- T. Polen, M. Spelberg and M. Bott, J. Biotechnol., 2013, 167, 75–84 CrossRef CAS PubMed.
- S. K. Tanneru and P. H. Steele, Renewable Energy, 2015, 80, 251–258 CrossRef CAS.
- J. Gharib, S. Pang and D. Holland, Eur. Polym. J., 2020, 133, 109725 CrossRef CAS.
- B. Sukhbaatar, P. H. Steele, L. I. Ingram and M. G. Kim, BioResources, 2009, 4, 789–804 CAS.
- W. Qu and X. Bai, J. Appl. Polym. Sci., 2020, 137, 48843 CrossRef CAS.
- H. Shi, Q. Ouyang, J. Wang, J. Hao and X. Huang, Macromol. Mater. Eng., 2020, 305, 1–8 CrossRef.
- R. Prins, V. H. J. De Beer and G. A. Somorjai, Catal. Rev., 1989, 31, 1–41 CrossRef CAS.
- A. R. Ardiyanti, A. Gutierrez, M. L. Honkela, A. O. I. Krause and H. J. Heeres, Appl. Catal., A, 2011, 407, 56–66 CrossRef CAS.
- P. M. Mortensen, J.-D. Grunwaldt, P. A. Jensen, K. Knudsen and A. D. Jensen, Appl. Catal., A, 2011, 407, 1–19 CrossRef CAS.
- M. Badawi, S. Cristol, J.-F. Paul and E. Payen, C. R. Chim., 2009, 12, 754–761 CrossRef CAS.
- R. H. Venderbosch, A. R. Ardiyanti, J. Wildschut, A. Oasmaa and H. J. Heeres, J. Chem. Technol. Biotechnol., 2010, 85, 674–686 CrossRef CAS.
- J. Wildschut, I. Melián-Cabrera and H. J. Heeres, Appl. Catal., B, 2010, 99, 298–306 CrossRef CAS.
- D. C. Elliott, Energy Fuels, 2007, 21, 1792–1815 CrossRef CAS.
- E. Furimsky and F. E. Massoth, Catal. Today, 1999, 52, 381–495 CrossRef CAS.
- E. Laurent and B. Delmon, J. Catal., 1994, 146, 281–291 CrossRef CAS.
- S. Kadarwati, X. Hu, R. Gunawan, R. Westerhof, M. Gholizadeh, M. D. M. Hasan and C.-Z. Li, Fuel Process. Technol., 2017, 155, 261–268 CrossRef CAS.
- H. Wang, J. Male and Y. Wang, ACS Catal., 2013, 3, 1047–1070 CrossRef CAS.
- C. Boscagli, C. Yang, A. Welle, W. Wang, S. Behrens, K. Raffelt and J.-D. Grunwaldt, Appl. Catal., A, 2017, 544, 161–172 CrossRef CAS.
- E. Furimsky, Appl. Catal., A, 2000, 199, 147–190 CrossRef CAS.
- D. C. Elliott and G. F. Schiefelbein, Am. Chem. Soc., 1989, 34, 1160 CAS.
- S. Zhang, Y. Yan, T. Li and Z. Ren, Bioresour.Technol., 2005, 96, 545–550 CrossRef CAS PubMed.
- N. Priharto, F. Ronsse, W. Prins, I. Hita, P. J. Deuss and H. J. Heeres, Biomass Bioenergy, 2019, 126, 84–93 CrossRef CAS.
- D. C. Elliott, H. Wang, M. Rover, L. Whitmer, R. Smith and R. Brown, ACS Sustainable Chem. Eng., 2015, 3, 892–902 CrossRef CAS.
- O. İ. Şenol, E.-M. Ryymin, T.-R. Viljava and A. O. I. Krause, J. Mol. Catal. A: Chem., 2007, 277, 107–112 CrossRef.
- J. Wildschut, M. Iqbal, F. H. Mahfud, I. M. Cabrera, R. H. Venderbosch and H. J. Heeres, Energy Environ. Sci., 2010, 3, 962–970 RSC.
- J. Wildschut, F. H. Mahfud, R. H. Venderbosch and H. J. Heeres, Ind. Eng. Chem. Res., 2009, 48, 10324–10334 CrossRef CAS.
- A. R. Ardiyanti, S. A. Khromova, R. H. Venderbosch, V. A. Yakovlev and H. J. Heeres, Appl. Catal., B, 2012, 117–118, 105–117 CrossRef CAS.
- A. R. Ardiyanti, M. V. Bykova, S. A. Khromova, W. Yin, R. H. Venderbosch, V. A. Yakovlev and H. J. Heeres, Energy Fuels, 2016, 30, 1544–1554 CrossRef CAS.
- W. Laosiripojana, W. Kiatkittipong, C. Sakdaronnarong, S. Assabumrungrat and N. Laosiripojana, Renewable Energy, 2019, 135, 1048–1055 CrossRef CAS.
- C. Boscagli, K. Raffelt, T. A. Zevaco, W. Olbrich, T. N. Otto, J. Sauer and J.-D. Grunwaldt, Biomass Bioenergy, 2015, 83, 525–538 CrossRef CAS.
- V. A. Yakovlev, M. V. Bykova and S. A. Khromova, Catal. Ind., 2012, 4, 324–339 CrossRef.
- M. Breysse, P. Afanasiev, C. Geantet and M. Vrinat, Catal. Today, 2003, 86, 5–16 CrossRef CAS.
- D. A. Ruddy, J. A. Schaidle, J. R. F. Iii, J. Wang, L. Moens and J. E. Hensley, Green Chem., 2014, 16, 454–490 RSC.
- N. Arun, R. V. Sharma and A. K. Dalai, Renewable Sustainable Energy Rev., 2015, 48, 240–255 CrossRef CAS.
- P. de Wild, R. Van der Laan, A. Kloekhorst and E. Heeres, Environ. Prog. Sustainable Energy, 2009, 28, 461–469 CrossRef CAS.
- P. J. de Wild, W. J. J. Huijgen, A. Kloekhorst, R. K. Chowdari and H. J. Heeres, Bioresour. Technol., 2017, 229, 160–168 CrossRef CAS PubMed.
- S. Gillet, M. Aguedo, L. Petitjean, A. R. C. Morais, A. M. da C. Lopes, R. M. Łukasik and P. T. Anastas, Green Chem., 2017, 19, 4200–4233 RSC.
- C. Wang, M. Li and Y. Fang, Green Chem., 2019, 21, 1000–1005 RSC.
- W. Chen, D. J. McClelland, A. Azarpira, J. Ralph, Z. Luo and G. W. Huber, Green Chem., 2016, 18, 271–281 RSC.
- R. K. Sharma and N. N. Bakhshi, Fuel Process. Technol., 1993, 35, 201–218 CrossRef CAS.
- J. Li, P. H. Galebach, J. K. Johnson, T. Fredriksen, A. Wittrig, X. Bai, H. Yang and G. W. Huber, Green Chem., 2020, 22, 8403–8413 RSC.
- X. Zhang, Q. Chen, Q. Zhang, C. Wang, L. Ma and Y. Xu, J. Anal. Appl. Pyrolysis, 2018, 135, 60–66 CrossRef CAS.
- J. Piskorz, P. Majerski, D. Radlein and D. S. Scott, Energy Fuels, 1989, 3, 723–726 CrossRef CAS.
- F. de M. Mercader, M. J. Groeneveld, S. R. A. Kersten, C. Geantet, G. Toussaint, N. W. J. Way, C. J. Schaverien and K. J. A. Hogendoorn, Energy Environ. Sci., 2011, 4, 985–997 RSC.
- R. J. French, S. K. Black, M. Myers, J. Stunkel, E. Gjersing and K. Iisa, Energy Fuels, 2015, 29, 7985–7992 CrossRef CAS.
- S. Kadarwati, S. Oudenhoven, M. Schagen, X. Hu, M. Garcia-Perez, S. Kersten, C.-Z. Li and R. Westerhof, J. Anal. Appl. Pyrolysis, 2016, 118, 136–143 CrossRef CAS.
- W. Yin, M. V. Alekseeva, R. H. Venderbosch, V. A. Yakovlev and H. J. Heeres, Energies, 2020, 13, 285 CrossRef CAS.
- J. Guo, R. Ruan and Y. Zhang, Ind. Eng. Chem. Res., 2012, 51, 6599–6604 CrossRef CAS.
- R. N. Olcese, G. Lardier, M. Bettahar, J. Ghanbaja, S. Fontana, V. Carré, F. Aubriet, D. Petitjean and A. Dufour, ChemSusChem, 2013, 6, 1490–1499 CrossRef CAS PubMed.
- S. M. Ghoreishi and M. R. Haghighi, Chem. Eng. J., 2007, 127, 59–70 CrossRef CAS.
- R. Ma, Y. Xu and X. Zhang, ChemSusChem, 2015, 8, 24–51 CrossRef CAS PubMed.
- M. T. García-Cubero, G. González-Benito, I. Indacoechea, M. Coca and S. Bolado, Bioresour.Technol., 2009, 100, 1608–1613 CrossRef PubMed.
- A. Panneerselvam, R. R. Sharma-Shivappa, P. Kolar, T. Ranney and S. Peretti, Bioresour.Technol., 2013, 148, 242–248 CrossRef CAS PubMed.
- R. Travaini, M. D. M. Otero, M. Coca, R. Da-Silva and S. Bolado, Bioresour.Technol., 2013, 133, 332–339 CrossRef CAS PubMed.
- P. Sannigrahi, F. Hu, Y. Pu and A. Ragauskas, J. Wood Chem. Technol., 2012, 32, 361–375 CrossRef CAS.
- C. Cheng, J. Wang, D. Shen, J. Xue, S. Guan, S. Gu and K. H. Luo, Polymers, 2017, 9, 240 CrossRef PubMed.
- C. Liu, S. Wu, H. Zhang and R. Xiao, Fuel Process. Technol., 2019, 191, 181–201 CrossRef CAS.
- H. Lange, S. Decina and C. Crestini, Eur. Polym. J., 2013, 49, 1151–1173 CrossRef CAS.
- S. Gharehkhani, Y. Zhang and P. Fatehi, Prog. Energy Combust. Sci., 2019, 72, 59–89 CrossRef.
- S. Dabral, J. G. Hernández, P. C. J. Kamer and C. Bolm, ChemSusChem, 2017, 10, 2707–2713 CrossRef CAS PubMed.
- A. Rahimi, A. Azarpira, H. Kim, J. Ralph and S. S. Stahl, J. Am. Chem. Soc., 2013, 135, 6415–6418 CrossRef CAS PubMed.
- S. K. Hanson, R. Wu and L. A. “Pete” Silks, Angew. Chem., 2012, 124, 3466–3469 CrossRef.
- S. K. Hanson, R. T. Baker, J. C. Gordon, B. L. Scott and D. L. Thorn, Inorg. Chem., 2010, 49, 5611–5618 CrossRef CAS PubMed.
- Y. Ma, Z. Du, J. Liu, F. Xia and J. Xu, Green Chem., 2015, 17, 4968–4973 RSC.
- B. Sedai, C. Díaz-Urrutia, R. T. Baker, R. Wu, L. P. Silks and S. K. Hanson, ACS Catal., 2011, 1, 794–804 CrossRef CAS.
- B. Biannic and J. J. Bozell, Org. Lett., 2013, 15, 2730–2733 CrossRef CAS PubMed.
- B. Biannic, J. J. Bozell and T. Elder, Green Chem., 2014, 16, 3635–3642 RSC.
- T. Voitl and P. Rudolf von Rohr, ChemSusChem, 2008, 1, 763–769 CrossRef CAS PubMed.
- Y. Zhao, Q. Xu, T. Pan, Y. Zuo, Y. Fu and Q.-X. Guo, Appl. Catal., A, 2013, 467, 504–508 CrossRef CAS.
- R. Ma, M. Guo and X. Zhang, ChemSusChem, 2014, 7, 412–415 CrossRef CAS PubMed.
- S. Bhargava, H. Jani, J. Tardio, D. Akolekar and M. Hoang, Ind. Eng. Chem. Res., 2007, 46, 8652–8656 CrossRef CAS.
- J. Zakzeski, A. Dębczak, P. C. A. Bruijnincx and B. M. Weckhuysen, Appl. Catal., A, 2011, 394, 79–85 CrossRef CAS.
- W. Deng, H. Zhang, X. Wu, R. Li, Q. Zhang and Y. Wang, Green Chem., 2015, 17, 5009–5018 RSC.
- V. R. Mate, M. Shirai and C. V. Rode, Catal. Commun., 2013, 33, 66–69 CrossRef CAS.
- H. Deng, L. Lin, Y. Sun, C. Pang, J. Zhuang, P. Ouyang, J. Li and S. Liu, Energy Fuels, 2009, 23, 19–24 CrossRef CAS.
- A. Jha, K. R. Patil and C. V. Rode, ChemPlusChem, 2013, 78, 1384–1392 CrossRef CAS PubMed.
- C. Zhu, W. Ding, T. Shen, C. Tang, C. Sun, S. Xu, Y. Chen, J. Wu and H. Ying, ChemSusChem, 2015, 8, 1768–1778 CrossRef CAS PubMed.
- C. Crestini, R. Saladino, P. Tagliatesta and T. Boschi, Bioorg. Med. Chem., 1999, 7, 1897–1905 CrossRef CAS PubMed.
- C. Crestini, A. Pastorini and P. Tagliatesta, J. Mol. Catal. A: Chem., 2004, 208, 195–202 CrossRef CAS.
- R. Prado, A. Brandt, X. Erdocia, J. Hallet, T. Welton and J. Labidi, Green Chem., 2016, 18, 834–841 RSC.
- G. Chatel and R. D. Rogers, ACS Sustainable Chem. Eng., 2014, 2, 322–339 CrossRef CAS.
- J. Zakzeski, A. L. Jongerius and B. M. Weckhuysen, Green Chem., 2010, 12, 1225–1236 RSC.
- M. Fache, B. Boutevin and S. Caillol, Eur. Polym. J., 2015, 68, 488–502 CrossRef CAS.
- M. Fache, B. Boutevin and S. Caillol, ACS Sustainable Chem. Eng., 2016, 4, 35–46 CrossRef CAS.
- T. Rinesch, J. Mottweiler, M. Puche, P. Concepción, A. Corma and C. Bolm, ACS Sustainable Chem. Eng., 2017, 5, 9818–9825 CrossRef CAS.
- W. Schutyser, J. S. Kruger, A. M. Robinson, R. Katahira, D. G. Brandner, N. S. Cleveland, A. Mittal, D. J. Peterson, R. Meilan, Y. Román-Leshkov and G. T. Beckham, Green Chem., 2018, 20, 3828–3844 RSC.
- M. Wang, J. Lu, X. Zhang, L. Li, H. Li, N. Luo and F. Wang, ACS Catal., 2016, 6, 6086–6090 CrossRef CAS.
- C. S. Lancefield, O. S. Ojo, F. Tran and N. J. Westwood, Angew. Chem., 2015, 127, 260–264 CrossRef.
- R. Ma, M. Guo, K. Lin, V. R. Hebert, J. Zhang, M. P. Wolcott, M. Quintero, K. K. Ramasamy, X. Chen and X. Zhang, Chem. – Eur. J., 2016, 22, 10884–10891 CrossRef CAS PubMed.
- Z. Bi, Z. Li and L. Yan, Green Process. Synth., 2018, 7, 306–315 CAS.
- D. J. Cronin, X. Zhang, J. Bartley and W. O. S. Doherty, ACS Sustainable Chem. Eng., 2017, 5, 6253–6260 CrossRef CAS.
- W. De los Santos Ramos, T. Poznyak, I. Chairez and I. Córdova R., J. Hazard. Mater., 2009, 169, 428–434 CrossRef CAS PubMed.
- Y. Ni, A. R. P. van Heiningen, J. Lora, L. Magdzinski and E. K. Pye, J. Wood Chem. Technol., 1996, 16, 367–380 CrossRef CAS.
- R. Travaini, J. Martín-Juárez, A. Lorenzo-Hernando and S. Bolado-Rodríguez, Bioresour. Technol., 2016, 199, 2–12 CrossRef CAS PubMed.
- Y. Rosen, H. Mamane and Y. Gerchman, Bioenerg. Res., 2019, 12, 292–301 CrossRef CAS.
- T. Miura, S.-H. Lee, S. Inoue and T. Endo, Bioresour. Technol., 2012, 126, 182–186 CrossRef CAS PubMed.
- I. Barrera-Martínez, N. Guzmán, E. Peña, T. Vázquez, R. Cerón-Camacho, J. Folch, J. A. H. Salazar and J. Aburto, Biomass Bioenergy, 2016, 94, 167–172 CrossRef.
- J. Sadowska, B. Johansson, E. Johannessen, R. Friman, L. Broniarz-Press and J. B. Rosenholm, Chem. Phys. Lipids, 2008, 151, 85–91 CrossRef CAS PubMed.
- S. T. OYAMA, Catal. Rev., 2000, 42, 279–322 CrossRef CAS.
- C. J. Chuck, H. J. Parker, R. W. Jenkins and J. Donnelly, Bioresour. Technol., 2013, 143, 549–554 CrossRef CAS PubMed.
- Y. Zhang, R. Yan, T. Ngo, Q. Zhao, J. Duan, X. Du, Y. Wang, B. Liu, Z. Sun, W. Hu and H. Xie, Eur. Polym. J., 2019, 117, 114–122 CrossRef CAS.
- C. Shi, S. Zhang, W. Wang, R. J. Linhardt and A. J. Ragauskas, ACS Sustainable Chem. Eng., 2020, 8, 22–28 CrossRef CAS.
- M. B. Figueirêdo, P. J. Deuss, R. H. Venderbosch and H. J. Heeres, ACS Sustainable Chem. Eng., 2019, 7, 4755–4765 CrossRef.
- H. P. L. Gemoets, Y. Su, M. Shang, V. Hessel, R. Luque and T. Noël, Chem. Soc. Rev., 2016, 45, 83–117 RSC.
- B. Gutmann, D. Cantillo and C. O. Kappe, Angew. Chem., Int. Ed., 2015, 54, 6688–6728 CrossRef CAS PubMed.
- J. D. Nguyen, B. S. Matsuura and C. R. J. Stephenson, J. Am. Chem. Soc., 2014, 136, 1218–1221 CrossRef CAS PubMed.
- N. Abad-Fernández, E. Pérez and M. J. Cocero, Green Chem., 2019, 21, 1351–1360 RSC.
- H. Werhan, N. Assmann and P. Rudolf von Rohr, Chem. Eng. Process., 2013, 73, 29–37 CrossRef CAS.
- N. Assmann, H. Werhan, A. Ładosz and P. Rudolf von Rohr, Chem. Eng. Sci., 2013, 99, 177–183 CrossRef CAS.
- M. B. Figueirêdo, F. W. Keij, A. Hommes, P. J. Deuss, R. H. Venderbosch, J. Yue and H. J. Heeres, ACS Sustainable Chem. Eng., 2019, 7, 18384–18394 CrossRef.
- M. Breiner, M. Zirbes and S. R. Waldvogel, Green Chem., 2021, 23, 6449–6455 RSC.
- Y. Cui, S. L. Goes and S. S. Stahl, in Advances in Inorganic Chemistry, ed. P. C. Ford and R. van Eldik, Academic Press, 2021, vol. 77, pp. 99–136 Search PubMed.
- S. K. Tanneru and P. H. Steele, Fuel, 2014, 133, 326–331 CrossRef CAS.
- S. K. Tanneru and P. H. Steele, Fuel, 2015, 154, 268–274 CrossRef CAS.
- Y. Luo, E. B. Hassan, V. Guda, R. Wijayapala and P. H. Steele, Energy Convers. Manage., 2016, 115, 159–166 CrossRef CAS.
- X. Junming, J. Jianchun, S. Yunjuan and L. Yanju, Biomass Bioenergy, 2008, 32, 1056–1061 CrossRef.
- L. Ciddor, J. A. Bennett, J. A. Hunns, K. Wilson and A. F. Lee, J. Chem. Technol. Biotechnol., 2015, 90, 780–795 CrossRef CAS.
- J. Xu, J. Jiang, W. Dai, T. Zhang and Y. Xu, Energy Fuels, 2011, 25, 1798–1801 CrossRef CAS.
- J.-J. Wang, J. Chang and J. Fan, Energy Fuels, 2010, 24, 3251–3255 CrossRef CAS.
- W.-M. Xiong, M.-Z. Zhu, L. Deng, Y. Fu and Q.-X. Guo, Energy Fuels, 2009, 23, 2278–2283 CrossRef CAS.
- Z. Tang, Q. Lu, Y. Zhang, X. Zhu and Q. Guo, Ind. Eng. Chem. Res., 2009, 48, 6923–6929 CrossRef CAS.
- M. Milina, S. Mitchell and J. Pérez-Ramírez, Catal. Today, 2014, 235, 176–183 CrossRef CAS.
- Q. Zhang, J. Chang, T. Wang and Y. Xu, Energy Fuels, 2006, 20, 2717–2720 CrossRef CAS.
- T. Sundqvist, A. Oasmaa and A. Koskinen, Energy Fuels, 2015, 29, 2527–2534 CrossRef CAS.
- J. Lu, S. Guo, Y. Fu and J. Chang, Fuel Process. Technol., 2017, 161, 193–198 CrossRef CAS.
- E. Reyhanitash, M. Tymchyshyn, Z. Yuan, K. Albion, G. van Rossum and C. Xu, Fuel, 2016, 179, 45–51 CrossRef CAS.
- Y. Liu, Z. Li, J. J. Leahy and W. Kwapinski, Energy Fuels, 2015, 29, 3691–3698 CrossRef CAS.
- N. Lohitharn and B. H. Shanks, Catal. Commun., 2009, 11, 96–99 CrossRef CAS.
- S. K. Tanneru, D. R. Parapati and P. H. Steele, Energy, 2014, 73, 214–220 CrossRef CAS.
- M. Ahmad, J. N. Roberts, E. M. Hardiman, R. Singh, L. D. Eltis and T. D. H. Bugg, Biochemistry, 2011, 50, 5096–5107 CrossRef CAS PubMed.
- R. R. Pour, A. Ehibhatiomhan, Y. Huang, B. Ashley, G. M. Rashid, S. Mendel-Williams and T. D. H. Bugg, Enzyme Microb. Technol., 2019, 123, 21–29 CrossRef PubMed.
- R. Rahmanpour and T. D. H. Bugg, Arch. Biochem. Biophys., 2015, 574, 93–98 CrossRef CAS PubMed.
- G. W. Park, G. Gong, J. C. Joo, J. Song, J. Lee, J.-P. Lee, H. T. Kim, M. H. Ryu, R. Sirohi, X. Zhuang and K. Min, Renewable Sustainable Energy Rev., 2022, 157, 112025 CrossRef CAS.
- Z. Chen and C. Wan, Renewable Sustainable Energy Rev., 2017, 73, 610–621 CrossRef CAS.
- M. A. Palazzolo and M. Garcia-Perez, Biotechnol. Adv., 2022, 54, 107791 CrossRef CAS PubMed.
- G. N. Ijoma, S. M. Heri, T. S. Matambo and M. Tekere, J. Fungi, 2021, 7, 700 CrossRef CAS PubMed.
- E. C. Moraes, T. M. Alvarez, G. F. Persinoti, G. Tomazetto, L. B. Brenelli, D. A. A. Paixão, G. C. Ematsu, J. A. Aricetti, C. Caldana, N. Dixon, T. D. H. Bugg and F. M. Squina, Biotechnol. Biofuels, 2018, 11, 75 CrossRef PubMed.
- Z.-H. Liu, R. K. Le, M. Kosa, B. Yang, J. Yuan and A. J. Ragauskas, Renewable Sustainable Energy Rev., 2019, 105, 349–362 CrossRef CAS.
- N. L. Radhika, S. Sachdeva and M. Kumar, Fuel, 2022, 312, 122935 CrossRef CAS.
- Q. Tang, Y. Qian, D. Yang, X. Qiu, Y. Qin and M. Zhou, Polymers, 2020, 12, 2471 CrossRef CAS PubMed.
- P. S. Chauhan, Bioresour. Technol. Rep., 2020, 9, 100374 CrossRef.
- P. K. Mishra and A. Ekielski, Nanomaterials, 2019, 9, 243 CrossRef PubMed.
- I. V. Pylypchuk, A. Riazanova, M. E. Lindström and O. Sevastyanova, Green Chem., 2021, 23, 3061–3072 RSC.
- M. Österberg, M. H. Sipponen, B. D. Mattos and O. J. Rojas, Green Chem., 2020, 22, 2712–2733 RSC.
- R. Kaur and S. P. Singh, in Zero Waste Biorefinery, ed. Y. K. Nandabalan, V. K. Garg, N. K. Labhsetwar and A. Singh, Springer, Singapore, 2022, pp. 489–514 Search PubMed.
- J.-L. Zheng, Y.-H. Zhu, M.-Q. Zhu, G.-T. Sun and R.-C. Sun, Green Chem., 2018, 20, 3287–3301 RSC.
- P. B. V. Scholten and M. B. Figueirêdo, Macromol. Chem. Phys., 2022, 2200017 CrossRef.
- L. Lange, Frontiers in Sustainability 20223 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2gc00302c |
| This journal is © The Royal Society of Chemistry 2022 |