
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Selective modification of hydroxyl groups in lignin model compounds by ruthenium-catalyzed transfer hydrogenation†
Veronika D.
Badazhkova
 ,
Risto
Savela
and
Reko
Leino
*
,
Risto
Savela
and
Reko
Leino
*
Laboratory of Molecular Science and Engineering, Åbo Akademi University, 20500 Åbo, Finland. E-mail: reko.leino@abo.fi
First published on 10th March 2022
Abstract
Selective ruthenium-catalyzed oxidation of lignin diol model compounds and lignin was accomplished by a transfer hydrogenation methodology. The developed procedure allows us to selectively oxidize benzylic secondary alcohols in model diols and spruce milled wood lignin in the presence of a commercially available Shvo catalyst under aerobic conditions. Six ketoalcohols were obtained in 70–92% yields from the model compounds, which also included lignin monomers containing 5-5′ and β-O-4 linkages. The developed method can be used as an intermediate step for the introduction of new functional groups into lignin-type structures and lignin to allow their further modifications.
Introduction
The search for innovative processes based on renewable energy sources and the development of environmentally benign methods for producing modern materials and base chemicals are significant global challenges.1 One of the potential sustainable feedstocks for fuels and materials manufacturing processes is lignin, nature's aromatic polymer. Dry wood biomass contains up to 40% of lignin, which is the second most abundant terrestrial polymer in the world after cellulose.2Structurally, lignin consists of a crosslinked network of aromatic functionalities with a surface rich in hydroxyl groups (Fig. 1). Consequently, due to its aromatic structure and high carbon content, lignin is a highly potential renewable source for the synthesis of a wide range of aromatic compounds and materials for various industrial applications.
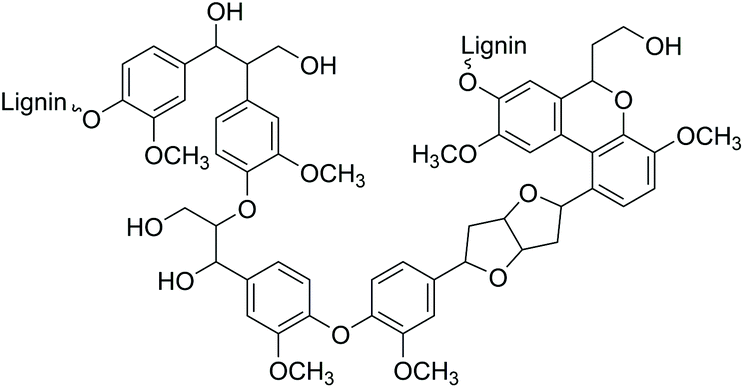 | ||
| Fig. 1 Representative lignin structure. | ||
Several reports have been published on the use of lignin and its derivatives for polymeric and carbon fibre composite materials applications.3 Moreover, lignin has been used as a commercially available starting material for the synthesis of engineering plastics and thermoplastic elastomers, polymeric foams and membranes, with end properties comparable to those of petroleum-based products.4 However, the direct use of lignin as a starting material is limited due to its low thermal and mechanical stability. For improving the stability and imparting desirable properties, lignin can be chemically modified by utilizing the functional groups in its structure. Several studies have been published on, for example, lignin alkylation, nitration, oxidation and amination.5 In different studies, it was demonstrated that the hydroxyl groups in lignin can be selectively oxidized using organocatalytic methods under an oxygen atmosphere,6 iridium catalysts under oxidant-free conditions,7 or in the presence of radical catalysts.8 Another approach for modification of lignin is based on the functionalization of the phenolic and aliphatic hydroxyl groups by hydrogen borrowing reactions.9,10 Compared to other oxidation processes, hydrogen transfer is highly selective, safe and eco-friendly and takes place in the presence of a wide range of metal catalysts.11
Fu and coworkers have reported a catalytic chemoselective dehydrogenation of aliphatic α-hydroxyl groups in lignin monomers.7 Oxidation of monomers containing only methoxy substituents in the aromatic ring resulted in the formation of C-α-ketoalcohols in high yields. However, dehydration of monomers containing phenolic hydroxyl groups led to the formation of side products, which significantly decreased the yield. Furthermore, the ruthenium and iridium catalysts used are not commercially available and require additional preparative steps.
Ruthenium-catalyzed hydrogen borrowing reactions can also be used for selective amination of alcohols.12 The reaction principle is based on the dehydrogenation of the alcohol, resulting in the formation of an intermediate carbonyl product. The carbonyl intermediate then reacts with an amine resulting in imine formation. Further hydrogenation of the imine produced leads to the formation of the desired amine (Scheme 1).
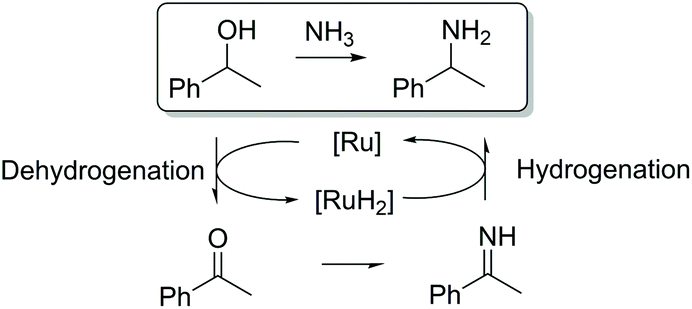 | ||
| Scheme 1 Transfer dehydrogenation–amination of benzyl alcohol. | ||
It was shown that this approach allows us to obtain various amines from bio-based alcohols in high yields. Moreover, the reactions involving ruthenium- and iridium-catalyzed amination processes can be carried out at lower temperatures and pressures compared to similar aminations in the presence of Lewis acids.13 Recently, Barta and coworkers reported the successful synthesis of benzazepines from lignin degradation products.14 Depolymerized mixtures of arylpropanols obtained after lignin fractionation were aminated in the presence of a ruthenium catalyst with further cyclization resulting in the formation of cyclic benzazepines.
In the present study, we have investigated the selective modification of monomeric lignin model compounds by a ruthenium-catalyzed hydrogen borrowing reaction methodology. Compared to other studies, with our methodology, the diol fragment can be selectively oxidized in the presence of the commercially available Shvo catalyst under aerobic conditions. The resulting ketoalcohols can potentially be used for further synthesis of new bio-based materials and biologically active compounds. The preliminary results obtained suggest the feasibility of the method also for the oxidation of native spruce milled wood lignin.
Results and discussion
The structural network of lignin contains several different monomeric units in irregular order, which complicates both the chemical modification and the analysis of lignin fractions. For preliminary screening of the chemical reactions and evaluation of the best starting parameters, model compounds were used in our study. Considering the hydroxyl groups of lignin as the main targets for modification, 3-phenyl-1,3-propanediol was selected as the simplest model compound to exclude the possible effect of substituents on the principal reaction pathway. Since lignin amination can be considered as a value-added reaction process for further synthesis of biologically active compounds from lignin-derived starting materials,15 we first aimed to screen the possibility of selective amination of the 3-phenyl-1,3-propanediol model compound using the transfer dehydrogenation–amination approach (Scheme 2). | ||
| Scheme 2 Protocol for lignin-based diol transfer dehydrogenation–amination screening. | ||
The use of primary amines as starting materials for alcohol amination typically leads to polysubstitution, resulting in a mixture of primary, secondary and tertiary amines. It has been observed, however, that the use of aniline for the amination of benzylic hydroxyl groups in the presence of ruthenium catalysts may result in a highly selective process, providing monoaminated products only.16 Based on this precedence, aniline was selected as the aminating agent also in the present study. In a set of experiments, 3-phenyl-1,3-propanediol was reacted with aniline in the presence of different ruthenium catalysts (Scheme 3). The reactions were evaluated based on the conversion of 3-phenyl-1,3-propanediol and the reaction yields (Table 1).
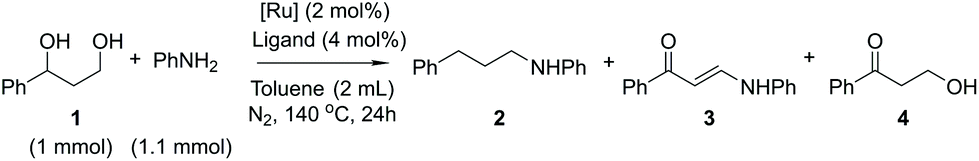 | ||
| Scheme 3 Ruthenium-catalyzed amination of the 3-phenyl-1,3-propanediol model compound. | ||
| Entry | Catalyst | Ligand | Conversion 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) e [%] e [%] |
Yield 2e [%] |
Yield 3e [%] |
Yield 4e [%] |
|---|---|---|---|---|---|---|
| All reactions were carried out using 1 mmol of compound 1, 1.1 mmol of aniline, 2 mL of toluene, 2 mol% of the Ru catalyst and 4 mol% of the ligand (entries 1–5) by stirring for 24 h at 140 °C under a N2 atmosphere.a Reactions catalyzed by dichloro(p-cymene)ruthenium(II) dimers.b Reactions catalyzed by 1-hydroxytetraphenylcyclopentadienyl(tetraphenyl-2,4-cyclopentadien-1-one)-μ-hydrotetracarbonyl-diruthenium(II) (Shvo).c 10 mol% of K2CO3 was added for catalyst activation.d According to 1H NMR and HPLC-SEC results, the diol fully oligomerizes.e Based on NMR-analysis.f The reaction residue consists of a mixture of inseparable aminated products that could not be analyzed and identified due to complexity of the mixture and small amounts of each individual side product. | ||||||
| 1 | [(p-Cym)RuCl2]2a | DPPP | 55 | 15 | 4 | 10 |
| 2 | [(p-Cym)RuCl2]2 | DPPB | 50 | 20 | 2 | 8 |
| 3 | [(p-Cym)RuCl2]2 | DPPF | 100 | 25 | 12 | — |
| 4 | [(p-Cym)RuCl2]2 | XANT | 30 | 12 | 1 | 12 |
| 5 | [(p-Cym)RuCl2]2c | DPPB | 100d | — | — | — |
| 6 | Shvob | — | 80 | — | 1 | 40f |
| 7 | Cp*RuCl(cod)c | — | 100d | — | — | — |
Five reactions were carried out with 2 mol% of dichloro(p-cymene)ruthenium(II) dimer in the presence of 4 mol% of different ligands, DPPP [1,3-bis(diphenylphosphino)propane], DPPB [1,4-bis(diphenylphosphino)butane], DPPF [1,1′-ferrocenediyl-bis(diphenylphosphine)] and XANT [4,5-bis(diphenylphosphino)-9,9-dimethylxanthene]. The products of the reaction in the presence of the DPPB ligand were purified and isolated by flash chromatography for establishing the components of the product mixture. All reactions catalyzed by the [(p-Cym)RuCl2]2–ligand system resulted in a mixture of products 2–4. Conversions of the reactions varied between 30–100% and the NMR yields of the products did not exceed 25%. In the reaction catalyzed by 2 mol% of Cp*RuCl(cod) preactivated by K2CO3, the starting material was fully oligomerized. The highest selectivity and 40% yield toward the desired product 3-hydroxypropiophenone (4), in this set of experiments, were observed in the reaction catalyzed by 2 mol% of the Shvo catalyst, indicating that the hydrogen borrowing mechanism was the main pathway for this specific reaction. Since the yield of the aminated product 3 reached only 1%, the reaction with the Shvo catalyst was repeated in the absence of any amination agent. Instead, 2-butanone was used, being well-studied previously as a hydrogen bond acceptor in transfer hydrogenation reactions (Scheme 4).17,18
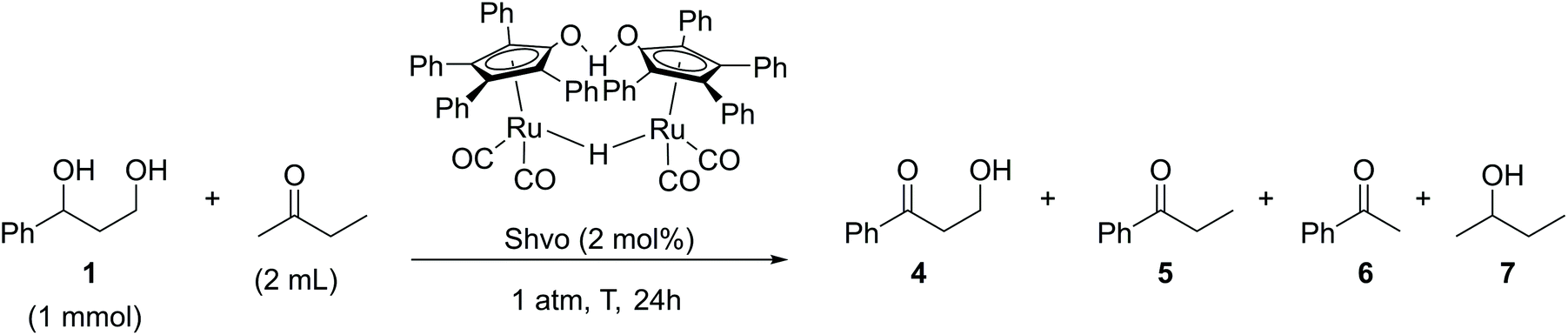 | ||
| Scheme 4 Shvo-catalyzed transfer hydrogenation reaction of the 3-phenyl-1,3-propanediol model compound. | ||
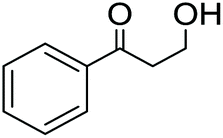
For the reaction condition screening, the experiments were carried out under different conditions (Table 2). At 140 °C the reaction resulted in a mixture of products. It was observed that an inert atmosphere is not critical for this reaction and it can also be performed under air. The best yield of the desired 3-hydroxypropiophenone (4) was reached after 24 h stirring at 60 °C in the presence of 2 mol% of the Shvo catalyst under air (Table 2).
| Entry | T (°C) | Conversion 1 [%] | Yield 4 [%] | Yield 5 [%] | Yield 6[%] |
|---|---|---|---|---|---|
| All reactions were carried out using 1 mmol of compound 1, 2 mL of 2-butanone and 2 mol% of Shvo catalyst by stirring for 24 h at the given temperature.a Reaction carried out under a N2 atmosphere.b Reaction carried out under air.c Based on NMR analysis.d Isolated yield. | |||||
| 1 | 140a | 100 | — | 30c | 10c |
| 2 | 100a | 100 | 40c | 25c | — |
| 3 | 80a | 98 | 60d | — | — |
| 4 | 60a | 87 | 82d | — | — |
| 5 | 60b | 95 | 92d | — | — |
Samples from the reaction mixtures were analyzed by 1H NMR spectroscopy and GC-MS methods without evaporation or preliminary purification. According to these analyses, temperatures higher than 100 °C led to side reactions, resulting in the formation of propiophenone (5) and acetophenone (6). When the reaction mixtures were evaporated prior to the analyses, neither propiophenone nor acetophenone was detected. At 60 °C, the starting material 3-phenyl-1,3-propanediol was fully converted to the desired 3-hydroxypropiophenone and no side products from the cleavage processes were observed. Furthermore, similar results were obtained under both oxygen and oxygen-free atmospheres. Carrying out the reaction under air allowed us to simplify the preparative process, resulting in highly selective oxidation of the benzylic hydroxyl group at 60 °C.
Amination screening with the simple model compound demonstrated a possible reaction pathway for compounds containing a 3-phenyl-1,3-propanediol fragment. The chemical structures of native lignin and lignin monomers are, however, more complicated and may significantly influence the reaction.
Consequently, model compounds closer to real lignin containing monomeric diol structures were tested next. Native lignin contains several typical fragments connected to each other by different linkages (Fig. 2). The percentage of these linkage types depends on the lignin source, with the β-O-4 type being the most abundant.19 Consequently, also in our study, β-O-4 fragments and methoxy groups typical of lignin were introduced to the test reaction substrates. Furthermore, a lignin model monomer containing a 5-5′ linkage was prepared and tested.
 | ||
| Fig. 2 The principle for selecting the model lignin compounds containing typical lignin linkages to recreate structures with chemical properties similar to those of real lignin. | ||
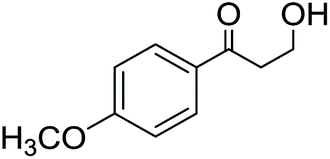
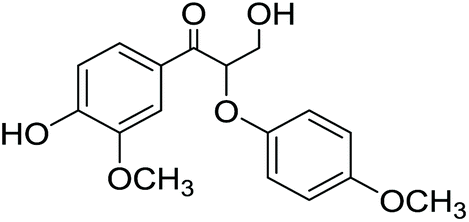
Lignin monomer model compounds 8–12, containing substituents in the aromatic ring, β-O-4 and 5-5′ linkages, were prepared from the corresponding ethyl 3-oxopropanoates as described earlier by Sheldrake and coworkers (for complete characterization details and synthesis procedures, see the ESI†).20 The obtained diols 8–12 were then applied in transfer hydrogenation reactions with 2-butanone in the presence of the Shvo catalyst (Scheme 5). For monomeric reaction products 13–16, the final isolated yields were 70–87% (Table 3). The lowest yield was observed for the bis-compound 17, which according to the 1H NMR analysis was contaminated with unreacted starting material 12.
 | ||
| Scheme 5 Shvo-catalyzed transfer hydrogenation reaction of lignin model compounds. | ||
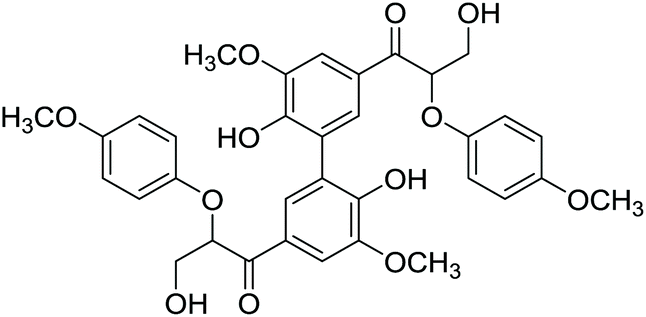
After successfully employing the transfer hydrogenation methodology to lignin model compounds, preliminary testing on native lignin was performed. As a source of lignin, milled spruce sapwood was used. As verified by the analytical methods, the sample of spruce milled wood lignin contains up to 44% β-O-4 fragments,22 being the primary target for modification. Following a literature procedure, ethanol solved lignin with high β-O-4 content was extracted from milled spruce sapwood.22 For demonstrating the presence of β-O-4 fragments, HMBC and HSQC 2D NMR spectra were recorded from the extracted product.
According to the literature, the signal at 4.90/72.9 ppm is specific for Hα in a β-O-4 fragment. The multiple signals at 4.5/83.3 ppm and 3.8/55.5 ppm correspond to Hβ and Hγ, respectively (Fig. 3).23 In addition, the HSQC NMR spectrum of the isolated lignin was compared to the HSQC NMR spectrum of the model compound 9. Signals for Hα, Hγ and Hβ in both compounds are observed in the same areas (Fig. 4), further verifying the presence of β-O-4 fragments in the lignin sample.
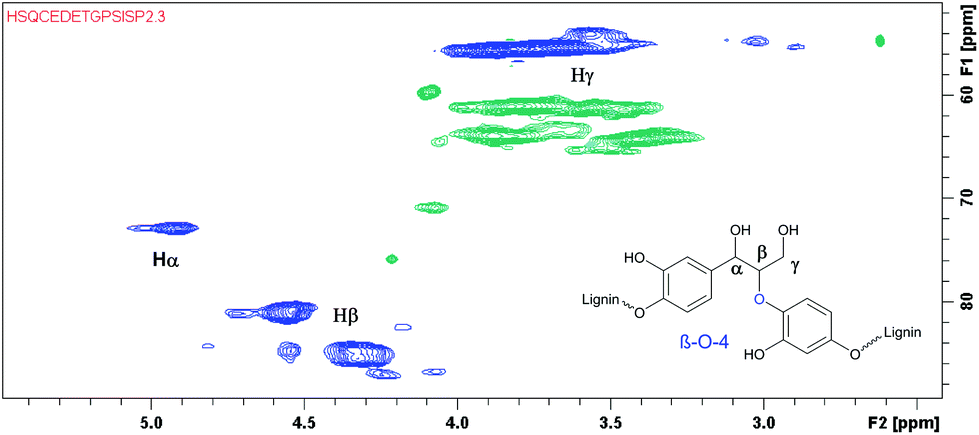 | ||
| Fig. 3 HSQC NMR spectrum of the extracted lignin with signals from the β-O-4 fragments highlighted. | ||
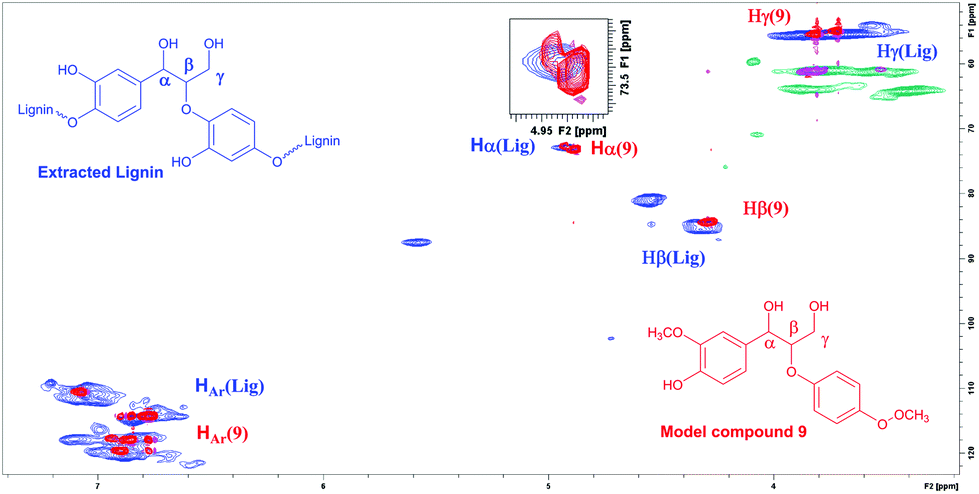 | ||
| Fig. 4 HSQC NMR spectrum of the extracted lignin and compound 9 overlaid, verifying the structural similarities and the presence of β-O-4 fragments in the lignin. | ||
For lignin modification with transfer hydrogenation methodology, the same reaction conditions were used as for the model compounds (Scheme 6). After stirring for 24 h in the presence of 2-butanone and the Shvo catalyst at 60 °C, the reaction mixture was evaporated to dryness and the resulting lignin-containing powder was analyzed by 2D NMR spectroscopy.
 | ||
| Scheme 6 Shvo-catalyzed transfer hydrogenation of lignin. | ||
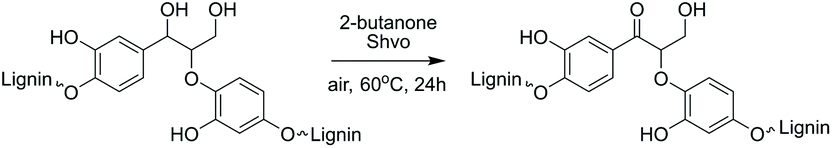
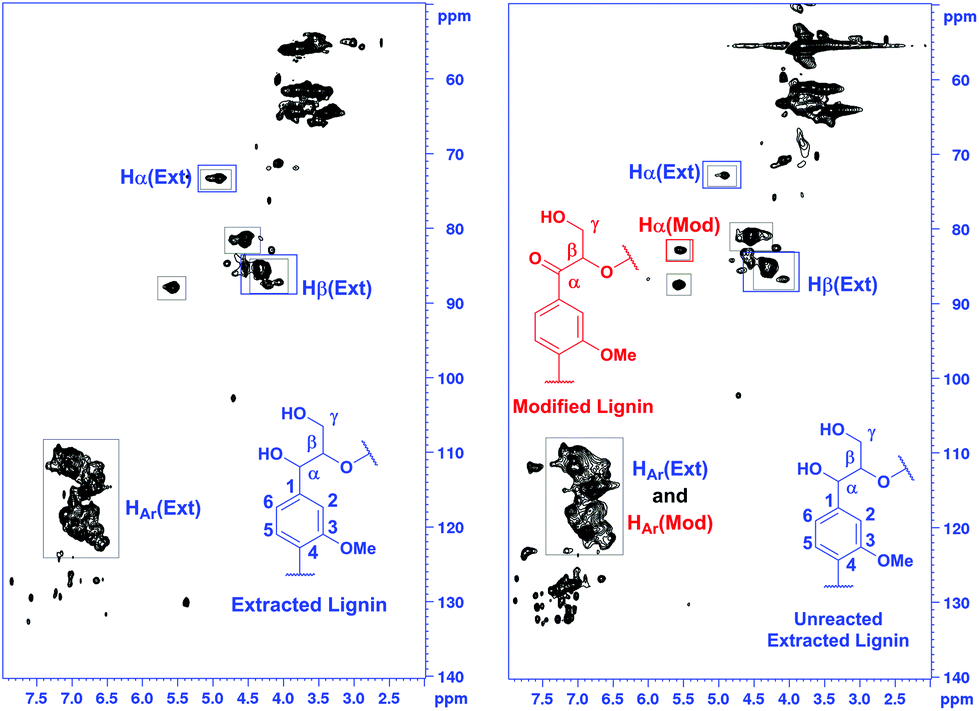
For estimating the outcome of this reaction, the HMBC and HSQC 2D NMR spectra of the resulting product were compared to the spectra of model compound 9 and the corresponding transfer hydrogenation product 14, and further to native lignin prior to the modification. In ketoalcohol 14, the formation of the new α-keto group results in the disappearance of the Hα signal and shifting of the Hβ, H2 and H6 signals to a weaker field in the HSQC spectrum, compared to the starting material 9. In comparison with the HSQC NMR spectrum of extracted lignin, the spectrum of modified lignin shows a new signal in the same area as Hβ in the model compound 14 and new signals in the aromatic region with similar chemical shifts as observed for H2 and H6 in compound 14. The signals for Hα did not fully disappear in modified lignin while decreasing in intensity (Fig. 5). Apparently, the reaction was not fully completed in 24 hours due to the complexity of the lignin structure.
 | ||
| Fig. 5 Comparison of the HSQC NMR spectra of the starting material 9 and the corresponding transfer hydrogenation product 14 (top) and the extracted and modified lignin (bottom), demonstrating the efficiency of the reaction. | ||
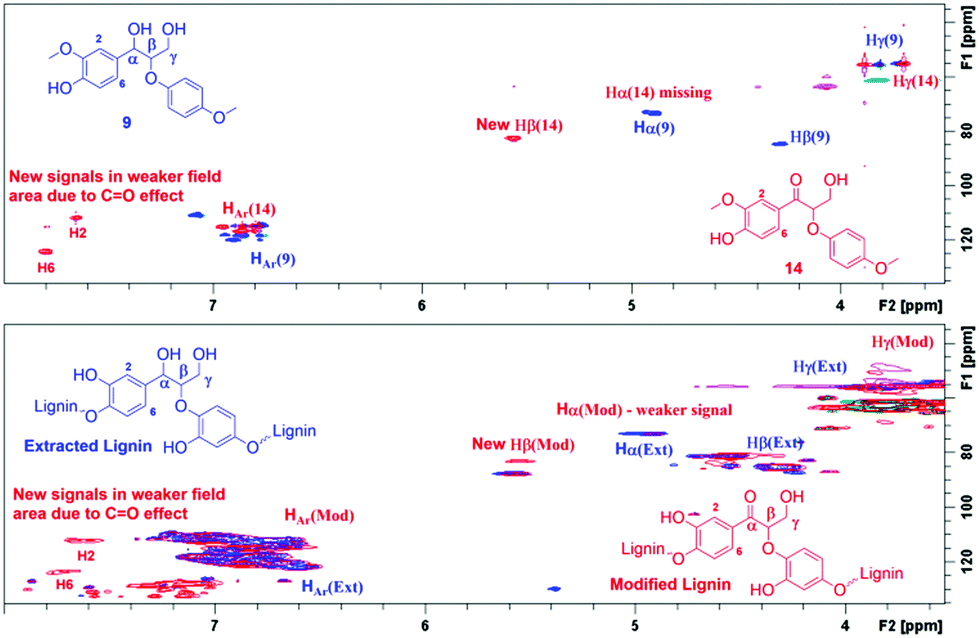
The same correlations were also observed in the HMBC NMR spectra. In the case of ketoalcohol 14, the Hα signal disappeared and the Hβ, H2 and H6 signals were shifted toward weaker fields compared to compound 9. Furthermore, in the HMBC it is possible to observe the specific signal for C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O at 195.8 ppm. In the HMBC spectrum for modified lignin, the Hα signal had almost disappeared, although it was not possible to observe the signal for the new Hβ. This could possibly be explained by the complexity of the polymeric structure. Finally, it was possible to observe new signals specific for a benzylic carbonyl group at 196.4 ppm and the corresponding correlations from H2 and H6 (Fig. 6).
O at 195.8 ppm. In the HMBC spectrum for modified lignin, the Hα signal had almost disappeared, although it was not possible to observe the signal for the new Hβ. This could possibly be explained by the complexity of the polymeric structure. Finally, it was possible to observe new signals specific for a benzylic carbonyl group at 196.4 ppm and the corresponding correlations from H2 and H6 (Fig. 6).
 | ||
| Fig. 6 Comparison of the HMBC NMR spectra of the starting material 9 and the corresponding transfer hydrogenation product 14 (top) and the extracted and modified lignin (bottom), demonstrating the efficiency of the reaction. | ||
In addition, since the analyzed modified lignin was contaminated with minor amounts of the Shvo catalyst, the 2D NMR spectrum of the resulting lignin product was compared with the 2D NMR spectrum of the Shvo ligand. In both the HMBC and HSQC NMR spectra, signals from the ligand appear in different areas than the new signals from modified lignin (Fig. 7), further verifying that the new signals result from the structural changes in lignin itself, instead of possible contamination with the Shvo catalyst.
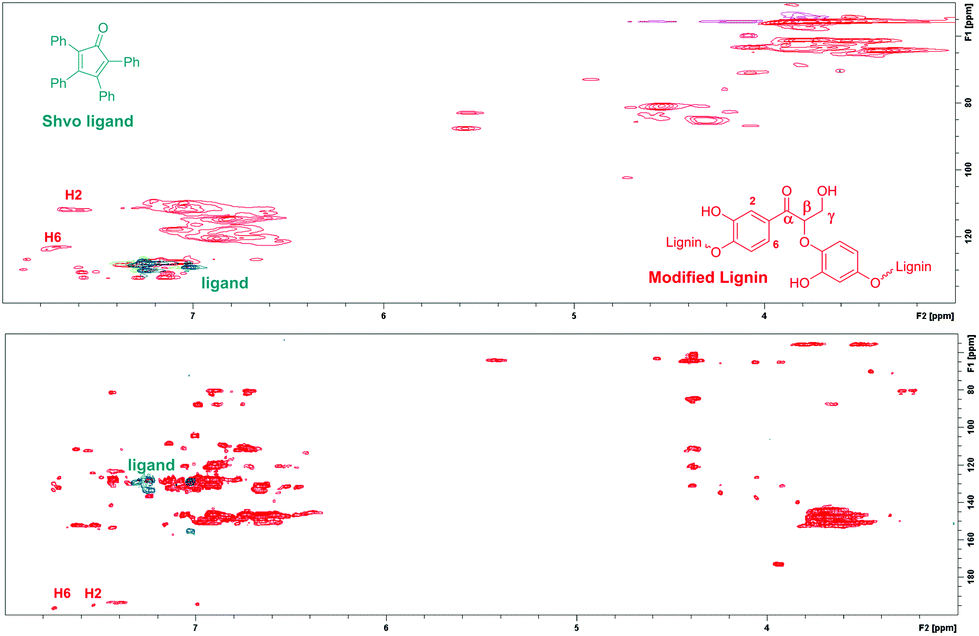 | ||
| Fig. 7 Comparison of the HMBC NMR spectra of the Shvo ligand and the modified lignin. | ||
In addition, based on the HSQC spectra recorded, a semi-quantitative analysis of the extracted and modified lignin fractions was carried out by following a literature protocol.22 For the calculations, correlation peaks from the three aromatic C–H groups were used as the reference, considered to represent the total amount of aromatic units. Then, the amount of linkages in the samples was calculated as parts per 100 aromatic units. Based on the literature22 and the NMR data obtained, ethanol-solved and modified lignin contain β-O-4 linkages with specific signals in their 2D NMR HSQC spectra (Fig. 8).
 | ||
| Fig. 8 Integration of the corresponding specific signals of extracted and modified lignin from the HSQC spectra. | ||
The results of the integration are presented in Table 4. Based on this semi-quantitative integration method, the content of the diol containing β-O-4 fragments is approximately 11 parts per 100 aromatic units prior to the modification. After modification, the content of unmodified (unreacted) β-O-4 diol fragments equals to 6 parts per 100 aromatic units. Consequently, the number of oxidized units in the modified lignin corresponds to 5 units and approximately 45% conversion of the benzylic hydroxyl groups to ketone.
| Linkage | Parts per 100 aromatic units | ||
|---|---|---|---|
| Extracted | Modified | ||
| β-O-4 | α (A) | 11 | 6 |
| β-CO (Aox) | 5 | ||
| Conversion of α-OH | 45% | ||
The data acquired in this work demonstrate that the transfer hydrogenation methodology can be applied for the oxidation of the benzylic hydroxyl groups in β-O-4 fragments in native lignin. However, as evidenced by the HSQC NMR spectrum, the reaction was not fully completed in 24 hours. This could be due to the structural complexity of lignin or the presence of fragments that resonate in the same area in the NMR spectrum but do not participate in the transfer hydrogenation process.
Finally, while one of the initial aims of this study was to obtain aminated products from the lignin model compounds, as illustrated in Scheme 3, the ruthenium-catalyzed amination procedure resulted in a mixture of products. A modification of the originally tested procedure, carried out as a two-step solvent replacement sequence, provided, however, the desired 1-phenyl-3-(phenylamino)propan-1-one 18 from the model compound 1 in 40% yield (Scheme 7). In the first step of this sequence, compound 1 was subjected to the conventional Shvo-catalyzed transfer hydrogenation procedure. Next, instead of purification, the reaction mixture was evaporated and the resulting oil, containing both the ruthenium catalyst and the product diol, was dissolved in acidic methanol, which is followed by the addition of aniline. After stirring, the produced precipitate was filtered, washed and analyzed by NMR-spectroscopy, indicating that instead of the expected imine formation, aniline had reacted with the primary hydroxyl group, forming the product 18. Furthermore, by applying the same two-step procedure to the model compound 8, the corresponding product 1-(4-methoxyphenyl-3-(phenylamino)propan-1-one (19) was obtained in 50% yield.
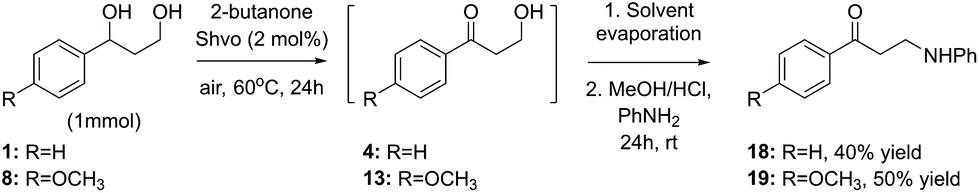 | ||
| Scheme 7 Two-step diol amination procedure involving solvent replacement. | ||
It has been reported previously that 4-hydroxybutan-2-one reacts with different aromatic amines by forming β-aminoketones, which are important intermediates in the synthesis of several biologically active compounds.24 From this point of view, the synthesis of lignin-based β-aminoketones could be considered as a prospective way for lignin valorization. Optimization of this two-step amination protocol and its application to lignin model compounds containing β-O-4 linkages will be investigated in more detail in our future work.
Summary and conclusions
In summary, we have developed a feasible methodology for selective functionalization of diol fragments in lignin model compounds using a ruthenium-catalyzed hydrogen borrowing procedure. It was demonstrated that benzylic hydroxyl groups in several model compounds could be selectively oxidized in the presence of the Shvo catalyst under aerobic conditions via the transfer hydrogenation pathway. After screening of the model compounds, this methodology was applied to native lignin, resulting in selective modification of the benzylic hydroxyl groups. Furthermore, preliminary studies demonstrated the possibility of amination of the obtained ketoalcohols to form aminoketones. Expansion of the developed methodology will be pursued in our ongoing work.Experimental section
General considerations
All chemicals were purchased from TCI, ACBR or Sigma-Aldrich and used without further purification unless otherwise indicated. For reactions under a protective atmosphere, solvents and liquid reagents used in the experiments involving glass reactors were dried and degassed and stored in a glovebox. The NMR spectra were recorded using 500 and 600 MHz NMR spectrometers. The measured NMR spectra were calibrated against the residual solvent signal that is used as the internal standard.25 The NMR signal assignments were based on 2D NMR (NOESY, DEPT, COSY, HSQC and HMBC). High resolution mass spectroscopy (HRMS) was carried out with a micrOTOF spectrometer (electrospray (ESI) equipped with a time of flight (TOF) mass analyzer. The product distribution was monitored by both GC/MS and GC/FID. The GC/MS instrument was equipped with an MS detector (EI) and a HP-5MS column (30 m × 250 μm × 0.25 μm) and He was used as the carrier gas with the following temperature program: injector 250 °C, oven Tinitial = 50 °C (4 min), rate 10 °C min−1, Tfinal = 300 °C, and hold 5 min. The GC/FID instrument was equipped with a HP-1 column (30 m × 320 μm × 0.25 μm), He was used as the carrier gas, and the following temperature program was followed: injector 220 °C, oven Tinitial = 50 °C (2 min), rate 10 °C min−1, Tfinal = 300 °C, and hold 2 min. Flash column chromatography was carried out by using an automated purification system using pre-packed columns with 20–40 µm particle size. The lignin sample used was extracted according to a previously published procedure21 from milled spruce sapwood (particle size approx. below 0.5 mm).General procedure for the anaerobic [(p-Cym)RuCl2]2 catalyzed amination reaction
A mixture of solid 3-phenyl-1,3-propanediol 1 (152 mg, 1 mmol), Ru catalyst (0.02 mmol, 2 mol%) and ligand (0.04 mmol, 4 mol%) was loaded into a Schlenk reactor, aniline (0.1 mL, 1.1 mmol) was loaded into a 4 mL vial, both vessels were degassed three times under vacuum, refilled with nitrogen and then transferred into a glovebox. Next, 2 ml of degassed toluene was added to the aniline and the resulting mixture was transferred into the reactor under nitrogen. The reactor was sealed with a Teflon plug and heated at 140 °C for 24 h. After 24 h of stirring, the reaction was cooled down and 10 mol% of ethyl 3,5-dinitrobenzoate was added to the reaction mixture as an internal standard for 1H NMR analysis. After 5 min of stirring, 0.2 mL of the reaction mixture was evaporated under vacuum, dissolved in 0.6 mL of CDCl3 and analyzed by 1H NMR spectroscopy. Only one reaction carried out with the DPPB ligand was purified by flash chromatography and the 1H NMR spectra of the fractionated products were obtained.General procedure for the anaerobic Ru-catalyzed amination reaction with catalyst activation
A mixture of solid 3-phenyl-1,3-propanediol 1 (152 mg, 1 mmol) and ligand (0.04 mmol, 4 mol%) was loaded into a Schlenk reactor, Ru catalyst (0.02 mmol, 2 mol%) was separately loaded into a 4 mL vial, aniline (0.1 mL, 1.1 mmol) was loaded into a 4 mL vial, and all vessels were degassed three times under vacuum, refilled with nitrogen and transferred into the glovebox. Next, 10 mol% of anhydrous K2CO3 and 2 mL of degassed toluene were added to the catalyst and stirred for 10 minutes. The resulting mixture and aniline were transferred into a Schlenk reactor. The reactor was sealed with a Teflon plug under nitrogen and heated at 140 °C for 24 h. After 24 h of stirring, the reaction was cooled down and 10 mol% ethyl 3,5-dinitrobenzoate was added to the reaction mixture as an internal standard for 1H NMR analysis. After 5 min of stirring, 0.2 mL of the reaction mixture was evaporated under vacuum, dissolved in 0.6 mL of CDCl3 and analyzed by 1H NMR spectroscopy.General procedure for the anaerobic Shvo-catalyzed amination reaction
A mixture of solid 3-phenyl-1,3-propanediol 1 (152 mg, 1 mmol) and Shvo catalyst (0.02 mmol, 2 mol%) was loaded into a Schlenk reactor and aniline (0.1 mL, 1.1 mmol) was loaded into a 4 mL vial. Then, both vessels were degassed three times under vacuum, refilled with nitrogen and transferred into a glovebox. Next, 2 mL of degassed toluene was added to aniline and the resulting mixture was transferred into the reactor under nitrogen. The reactor was sealed with a Teflon plug and heated at 140 °C for 24 h. After 24 h of stirring, the reaction was cooled down and 10 mol% of ethyl 3,5-dinitrobenzoate was added to the reaction mixture as an internal standard for 1H NMR analysis. After 5 min of stirring, 0.2 mL of the reaction mixture was evaporated under vacuum, dissolved in 0.6 mL of CDCl3 and analyzed by 1H NMR spectroscopy.General procedure for the anaerobic Shvo-catalyzed transfer hydrogenation reaction
A mixture of solid 3-phenyl-1,3-propanediol 1 (152 mg, 1 mmol) and the Shvo catalyst (0.02 mmol, 2 mol%) was loaded into a Schlenk reactor, degassed three times under vacuum, refilled with nitrogen and transferred into a glovebox. Then 2 mL of degassed 2-butanone was added into the reactor under nitrogen. Then the reactor was sealed with a Teflon plug and heated at 60–140 °C for 24 h. After 24 h of stirring, the reaction was cooled down and 10 mol% of ethyl 3,5-dinitrobenzoate was added to the reaction mixture as an internal standard for 1H NMR analysis. After 5 min of stirring, 0.2 mL of the reaction mixture was evaporated under vacuum, dissolved in 0.6 mL of CDCl3 and analyzed by 1H NMR spectroscopy.General procedures for the aerobic Shvo-catalyzed transfer hydrogenation reaction
General procedure for the synthesis of lignin model diols from ethyl 3-aryl-2-(4-methoxyphenoxy)-3-oxopropanoates20
:EtOAc) and dried under vacuum. For compounds 9 and 12, additional Pd/H2 reduction was carried out for deprotection of the 4-OH group by dissolving the protected compound (0.01 mol) in THF (50 mL). To this mixture, palladium on carbon 10% (280 mg) was added. The flask was transferred into a hydrogenation reactor. The reaction mixture was stirred under a hydrogen atmosphere (4 bars) for 3 hours. Next, the reaction mixture was filtered and evaporated. The resulting oil was purified by flash chromatography (gradient: 20 → 100; hexane:EtOAc) and dried under vacuum.
General procedure for the Shvo-catalyzed transfer hydrogenation reaction for selective oxidation of lignin model compounds
:EtOAc).
Procedure for Shvo-catalyzed transfer hydrogenation reaction for selective oxidation of spruce milled wood lignin
The extracted lignin (380 mg), Shvo catalyst (0.02 mmol, 2 mol%) and 2-butanone (2 mL) were loaded into a Schlenk reactor. The reactor was sealed with a Teflon plug and heated at 60 °C for 24 h. Next, the reaction was cooled down, the solvents were evaporated and the resulting powder was dried under vacuum. Then, 50 mg of the lignin powder was dissolved in 0.6 mL of acetone-d6 and analyzed by 2D NMR spectroscopy.Two-step amination procedure for ketoamine synthesis
The first step for diol (1 mmol) oxidation was carried out according to the general method C, followed by evaporation of the solvents under vacuum. The resulting oily residue was dissolved in MeOH (2 mL) containing concentrated HCl (0.2 mL). To this, aniline (1 mmol) was added and the reaction mixture was stirred at room temperature. After 24 hours, a white precipitate was formed. The precipitate was filtered, washed with K2CO3, water and hexane and dried under air.1-(4-Hydroxy-3-methoxyphenyl)-2-(4-methoxyphenoxy)propane-1,3-diol (9) was obtained according to the General method B in 2.5 mmol scale. The yield was 0.7 g, 58%. 1H NMR (CDCl3 with 0.05% v/v TMS, 500 MHz): δH 6.78–6.90 (3H, m, H2, H5 and H6), 6.68–6.78 (4H, H2′, H3′, H5′ and H6′), 5.54–5.61 (1H, m, ArOH), 4.85–4.96 (1H, m, Hα), 4.11–4.20 (1H, m, Hβ), 3.75–3.88 (5H, m, overlapping signals from Hγ and OCH33), 3.67–3.70 (3H, multiple overlapping singlets OCH34′ from diastereoisomers), 2.68–2.83 (1H, br, OHα), 2.13–2.25, (1H, br, OHγ). 13C (CDCl3, 500 MHz): δC 154.78 (C4′), 151.67 (C1′), 146.62 (C3), 145.31 (C4), 132.34 (C1), 119.29 (C6), 118.33, 118.13 (C2′ and C6′), 114.75, 114.84 (C3′ and C5′), 114.36, 114.32 (C5), 108.82 (C2), 83.51 (Cβ), 73.94 (Cα), 61.43 (Cγ), 55.99 (OCH33), 55.69 (OCH34′). [M + Na]+ calculated 343.1158 found 343.1148.
1-(3,4-Dimethoxyphenyl)-2-(4-methoxyphenoxy)propane-1,3-diol (10) was obtained according to the General method B in 0.014 mol scale. The yield was 2.5 g, 53%. 1H NMR (CDCl3 with 0.05% v/v TMS, 500 MHz): δH 6.92–6.99 (2H, m), 6.77–6.88 (5H, m), 5.02 (1H, dd, J = 3.38 Hz, 5.42 Hz), 4.23–4.28 (1H, q, J = 5.07 Hz), 3.88 (3H, s), 3.87 (3H, s), 3.83–3.98 (2H, m), 3.77 (3H, s), 2.81–2.87 (1H, m), 2.24–2.31 (1H, m). The NMR spectrum obtained is consistent with the literature data.26 [M + Na]+ calculated 357.1314 found 357.1322.
1-(3,4,6-Trimethoxyphenyl)-2-(4-methoxyphenoxy)propane-1,3-diol (11) was obtained according to the General method B in 2.5 mmol scale. The yield was 0.55 g, 61%. 1H NMR (CDCl3 with 0.05% v/v TMS, 500 MHz): δH 6.69–6.89 (4H, m, H2′, H3′, H5′ and H6′), 6.53–6.63 (2H, multiple singlets from diastereoisomers, H2 and H6), 4.87–4.95 (1H, m, Hα), 4.15–4.22 (1H, m, Hβ), 3.72–3.92 (11H, m, H2γ, OCH33, OCH34 and OCH35), 3.67–3.72 (3H, multiple singlets from diastereoisomers, OCH34′), 2.65–2.85 (1H, br, OHα), 2.04–2.17, 1.68–1.82 (1H, two broad peacks from diastereoisomers, OHγ). 13C (CDCl3, 500 MHz): 154.86 (C4′), 153.39, 153.35 (C3, C4 and C5), 151.62 (C1′), 135.99 (C1), 118.35, 118.09 (diastereoisomers, C2′ and C6′), 114.84, 114.75 (diastereoisomers, C3′ and C5′), 103.87, 103.28 (diastereoisomers, C2 and C6), 83.45 (Cβ), 74.09 (Cα), 61.48, 61.15 (Cγ), 60.89 (OCH34), 56.19 (OCH33 and OCH35), 55.71, 55.69 (OCH34′). [M + Na]+ calculated 387.1415 found 387.1403.
5-5-Bis-1-(4-hydroxy-3-methoxyphenyl)-2-(4-methoxyphenoxy)propane-1,3-diol (12) was obtained according to the General method B in 0.5 mmol scale. The yield was 0.30 g, 31%. 1H NMR (CDCl3 with 0.05% v/v TMS, 500 MHz): δH 6.64–6.96 (12H, m, Ar), 4.84–4.93 (2H, m, Hα), 4.11–4.20 (2H, m, Hβ), 3.70–3.90 (4H, m, H2γ), 3.76–3.83 (6H, m, OCH33), 3.62–3.69 (6H, m, OCH34′). 13C (CDCl3, 500 MHz): δC 154.64 (C4′), 151.78 (C1′), 147.29 (C3), 142.32 (C4), 132.39 (C1), 124.01 (C5), 121.43 (C6), 118.30, 118.27, 118.12, 118.10 (C2′ and C6′), 114.80, 114.74 (C3′ and C5′), 108.56 (C2), 83.30 (Cβ), 73.97 (Cα), 61.55 (Cγ), 56.18 (OCH33), 55.69 (OCH34′). [M + Na]+ calculated 661.2255 found 661.2293.
3-Hydroxy-1-(4-hydroxy-3-methoxyphenyl)-2-(4-methoxyphenoxy)propan-1-one (14) was obtained according to the General method C in 1 mmol scale. The yield is 277 mg, 87%. 1H NMR (CDCl3 with 0.05% v/v TMS, 500 MHz): δH 7.56–7.62 (1H, dd, J = 2.0 Hz, 8.5 Hz, H6), 7.45–7.48 (1H, d, J = 1.8 Hz, H2), 6.80–6.85 (1H, d, J = 8.3 Hz, H5), 6.62–6.77 (4H, m, H2′, H3′, H5′ and H6′), 5.33–5.37 (1H, dd, J = 4.1 Hz, 6.45, Hβ), 3.95–4.07 (2H, m, H2γ), 3.75 (3H, s, OCH33), 3.61 (3H, s, OCH34′), 2.72–3.18 (1H, br, OHγ). 13C (CDCl3, 500 MHz): 195.79 (Cα), 154.66 (C4′), 151.51 (C4), 151.48 (C1′), 147.55 (C3), 127.73 (C1), 124.20 (C6), 116.57 (C2′ and C6′), 114.27 (C3′ and C5′), 111.79 (C2), 81.97 (Cβ), 60.52 (Cγ), 55.99 (OCH33), 55.64 (OCH34′). [M + Na]+ calculated 341.1001 found 341.1027.
1-(3,4-Dimethoxyphenyl)-3-hydroxy-2-(4-methoxyphenoxy)propan-1-one (15) was obtained according to the General method C in 1 mmol scale. The yield was 235 mg, 70%. 1H NMR (CDCl3 with 0.05% v/v TMS, 500 MHz): δH 7.67–7.69 (1H, dd, J = 2.0 Hz, 8.5 Hz, H6), 7.49–7.50 (1H, d, J = 2.0 Hz, H2), 6.70–7.02 (5H, m, H5, H2′, H3′, H5′ and H6′), 5.35–5.37 (1H, dd, J = 4.2 Hz, 6.4 Hz, Hβ), 3.99–4.08 (2H, m, H2γ), 3.88 (3H, s, OCH33), 3.82 (3H, s, OCH34), 3.67 (3H, s, OCH34′), 1.90–2.52 (1H, br, OHγ). 13C (CDCl3, 500 MHz): 195.28 (Cα), 154.67 (C4′), 154.13 (C4), 151.45 (C1′), 149.29 (C3), 127.91 (C1), 123.58 (C6), 116.57 (C2′ and C6′), 114.83 (C3′ and C5′), 110.86 (C2), 110.15 (C5), 81.98 (Cβ), 63.61 (Cγ), 56.16 (OCH33), 55.99 (OCH34), 55.67 (OCH34′). [M + Na]+ calculated 355.1158 found 355.1122.
1-(3,4,5-Trimethoxyphenyl)-3-hydroxy-2-(4-methoxyphenoxy)propan-1-one (16) was obtained according to the General method C in 1 mmol scale. The yield was 260 mg, 78%. 1H NMR (CDCl3 with 0.05% v/v TMS, 500 MHz): δH 7.23 (2H, s, H2 and H6), 6.73–6.78 (2H, m, H2′, and H6′), 6.65–6.71 (2H, H3′ and H5′), 5.27–5.32 (1H, dd, J = 4.33 Hz, 3.11 Hz, Hβ), 3.98–4.08 (1H, br, OHγ). 13C (CDCl3, 500 MHz): 194.95 (Cα), 153.65 (C4′), 152.06 (C3 and C5), 150.47 (C1′) 142.33 (C3), 128.80 (C1), 115.54 (C2′ and C6′), 113.81 (C3′ and C5′), 105.44 (C2 and C6), 81.31 (Cβ), 62.39 (Cγ), 59.91 (OCH34), 55.21 (OCH33 and OCH35), 54.60 (OCH34′). [M + Na]+ calculated 385.1263 found 385.1222.
5-5-Bis-3-hydroxy-1-(4-hydroxy-3-methoxyphenyl)-2-(4-methoxyphenoxy)propan-1-one (17) was obtained according to the General method C in 0.5 mmol scale. The yield was 250 mg, 60%. NMR-purity: 70%, contaminated with starting material 12b. 1H NMR (CDCl3 with 0.05% v/v TMS, 500 MHz): δH 7.65–7.85 (2H, m, H6), 7.40–7.53 (2H, m, H2), 6.60–7.00 (8H, m, H2′, H3′, H5′ and H6′), 5.25–5.45 (2H, br, Hβ), 3.68–4.20 (10H, m, OCH33 and H2γ), 3.55–3.67 (6H, m, OCH34′). 13C (CDCl3, 500 MHz): 195.29 (Cα), 154.59 (C4′), 151.50 (C1′), 151.58 (C4), 148.75 (C4), 147.39 (C3), 126.87 (C1), 126.29 (C6), 122.29 (C5), 116.71 (C2′ and C6′), 114.80 (C3′ and C5′), 109.90 (C2), 82.14 (Cβ), 63.77 (Cγ), 56.24 (OCH33), 55.66 (OCH34′). [M + Na]+ calculated 657.1948 found 657.1910.
1-Phenyl-3-(phenylamino)-1-propanone (18) was obtained according to the two-step amination procedure in 1 mmol scale. The yield was 90 mg, 40%. 1H NMR (CDCl3 with 0.05% v/v TMS, 400 MHz): δH 7.93–8.03 (2H, d, J = 7.5 Hz), 7.55–7.66 (1H, t, J = 7.2 Hz), 7.44–7.54 (2H, t, J = 7.4 Hz), 7.16–7.24 (2H, t, J = 7.7 Hz), 6.70–6.78 (1H, t, J = 7.46 Hz), 6.62–6.70 (2H, d, J = 6.7 Hz), 4.06–4.18 (1H, br), 3.60–3.70 (2H, q, J = 6.1 Hz), 3.27–3.36 (2H, t, J = 6.0). The 1H NMR spectrum is consistent with the literature data.27
1-(4-Methoxyphenyl)-3-(phenylamino)-1-propanone (19) was obtained according to the two-step amination procedure in 1 mmol scale. The yield was 128 mg, 50%. 1H NMR (CDCl3 with 0.05% v/v TMS, 500 MHz): δH 7.82–7.88 (2H, d, J = 8.8 Hz, H2 and H6), 7.06–7.15 (2H, t, J = 7.7 Hz, H3′ and H5′), 6.83–6.89 (2H, d, J = 8.9 Hz, H3 and H5), 6.60–6.65 (1H, t, J = 7.5 Hz, H4′), 6.54–6.59 (2H, d, J = 7.8 Hz, H2′ and H6′), 3.98–4.15 (1H, br, NH), 3.79 (3H, s, 4-OCH3), 3.50–3.54 (2H, t, J = 6.18 Hz, CH2γ), 3.12–3.18 (2H, t, J = 6.51 Hz, CH2β). 13C (CDCl3, 500 MHz): 197.84 (Cα), 163.69 (C4), 147.82 (C1′), 130.35 (C2 and C6), 129.91 (C1), 129.35 (C3′ and C5′), 117.53 (C2′ and C6′), 113.81 (C3 and C5), 113.07 (C4′), 55.52 (4-OCH3), 38.93 (Cβ), 37.31 (Cγ).
Detailed procedures for the synthesis of lignin model compounds, their characterization and NMR spectra of all the new compounds are provided in the ESI.†
Author contributions
The manuscript was written through the contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the Finnish Natural Resources Research Foundation (grant #20210053) for financial support. Dr Jani Rahkila and Lucas Lagerquist are thanked for their assistance with the analytical work.Notes and references
- A. J. Ragauskas, G. T. Beckhammary, M. J. Biddy, R. Chandra, F. Chen, M. F. Davis, B. H. Davison, R. A. Dixon, P. Gilna, M. Keller, P. Langan, A. K. Naskar, J. N. Saddler, T. J. Tschaplinski, G. A. Tuskan and C. E. Wyman, Science, 2014, 344, 6185 CrossRef PubMed.
- K. Barta and P. C. Ford, Acc. Chem. Res., 2014, 47(5), 1503–1512 CrossRef CAS PubMed.
- N. Meek, D. Penumadu, O. Hosseinaei, D. Harper, S. Young and T. Rials, Compos. Sci. Technol., 2016, 137, 60–68 CrossRef CAS.
- H. Chung and N. R. Washburn, Green Mater., 2013, 1, 137–160 CrossRef.
- S. Laurichesse and L. Avérous, Prog. Polym. Sci., 2014, 39, 1266–1290 CrossRef CAS.
- A. Rahimi, A. Azarpira, H. Kim, J. Ralph and S. S. Stahl, J. Am. Chem. Soc., 2013, 135, 6415–6418 CrossRef CAS PubMed.
- R. Zhu, B. Wang, M. Cui, J. Deng, X. Li, Y. Ma and Y. Fu, Green Chem., 2016, 18, 2029–2036 RSC.
- N. D. Patil and N. Yan, Catal. Commun., 2016, 84, 155–158 CrossRef CAS.
- Z. Zhang, C. W. Lahive, D. S. Zijlstra, Z. Wang and P. J. Deuss, ACS Sustainable Chem. Eng., 2019, 7, 12105–12116 CAS.
- R. Ghosh, N. Jana, C. S. Panda and B. Bagh, ACS Sustainable Chem. Eng., 2021, 9, 4903–4914 CrossRef CAS.
- R. Karvembu, R. Prabhakaran, K. Senthilkumar, P. Viswanathamurthi and K. Natarajan, React. Kinet. Catal. Lett., 2005, 86, 211–216 CrossRef CAS.
- W. Baumann, A. Spannenberg, J. Pfeffer, T. Haas, A. Köckritz, A. Martin and J. Deutsch, Chem. – Eur. J., 2013, 19, 17702–17706 CrossRef CAS PubMed.
- M. Pera-Titus and F. Shi, ChemSusChem, 2014, 7, 720–722 CrossRef CAS PubMed.
- S. Elangovan, A. Afanasenko, J. Haupenthal, Z. Sun, Y. Liu, A. K. Hirsch and K. Barta, ACS Cent. Sci., 2019, 5(10), 1707–1716 CrossRef CAS PubMed.
- Y. Rong, N. Ji, Z. Yu, X. Diao, H. Li, Y. Lei, X. Lu and A. Fukuoka, Green Chem., 2021, 23, 6761–6788 RSC.
- S. Bähn, S. Imm, L. Neubert, M. Zhang, H. Neumann and M. Beller, ChemCatChem, 2011, 3, 1853–1864 CrossRef.
- B. S. Akpa, C. D'Agostino, L. F. Gladden, K. Hindle, H. Manyar, J. McGregor, R. Li, M. Neurock, N. Sinha, E. H. Stitt, D. Weber, J. A. Zeitler and D. W. Rooney, J. Catal., 2012, 289, 30–41 CrossRef CAS.
- G. Szöllösi and M. Bartók, Catal. Lett., 1999, 59, 179–185 CrossRef.
- C. W. Lahive, P. C. Kamer, C. S. Lancefield and P. J. Deuss, ChemSusChem, 2020, 13, 4238 CrossRef CAS PubMed.
- W. G. Forsythe, M. D. Garrett, C. Hardacre, M. Nieuwenhuyzen and G. N. Sheldrake, Green Chem., 2013, 15, 3031–3038 RSC.
- M. Balakshin, E. A. Capanema, X. Zhu, I. Sulaeva, A. Potthast, T. Rosenau and O. J. Rojas, Green Chem., 2020, 22, 3985–4001 RSC.
- D. S. Zijlstra, A. de Santi, B. Oldenburger, J. de Vries, K. Barta and P. J. Deuss, J. Visualized Exp., 2019, 143, 1–12 Search PubMed.
- J. L. Wen, S. L. Sun, B. L. Xue and R. C. Sun, Materials, 2013, 6, 359–391 CrossRef PubMed.
- C. Miao, L. Jiang, L. Ren, Q. Xue, F. Yan, W. Shi, X. Li, J. Sheng and S. Kai, Tetrahedron, 2019, 75, 2215–2228 CrossRef CAS.
- G. R. Fulmer, A. J. Miller, N. H. Sherden, H. E. Gottlieb, A. Nudelman, B. M. Stoltz and K. I. Goldberg, Organometallics, 2010, 29, 2176–2179 CrossRef CAS.
- S. G. Yao, M. S. Meier, R. B. Pace III and M. Crocker, RSC Adv., 2016, 6, 104742–104753 RSC.
- E. Haak, Eur. J. Org. Chem., 2007, 2815–2824 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures and characterization data for ethyl 3-oxopropanoate starting materials and the NMR spectra of lignin material. See DOI: 10.1039/d2dt00267a |
| This journal is © The Royal Society of Chemistry 2022 |