
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
N-Triflylphosphoramides: highly acidic catalysts for asymmetric transformations
Guillermo
Caballero-García
 and
Jonathan M.
Goodman
*
and
Jonathan M.
Goodman
*
Yusuf Hamied Department of Chemistry, University of Cambridge, Lensfield Road, CB2 1EW, Cambridge, UK. E-mail: jmg11@cam.ac.uk
First published on 21st October 2021
Abstract
N-Triflylphosphoramides (NTPA), have become increasingly popular catalysts in the development of enantioselective transformations as they are stronger Brønsted acids than the corresponding phosphoric acids (PA). Their highly acidic, asymmetric active site can activate difficult, unreactive substrates. In this review, we present an account of asymmetric transformations using this type of catalyst that have been reported in the past ten years and we classify these reactions using the enantio-determining step as the key criterion. This compendium of NTPA-catalysed reactions is organised into the following categories: (1) cycloadditions, (2) electrocyclisations, polyene and related cyclisations, (3) addition reactions to imines, (4) electrophilic aromatic substitutions, (5) addition reactions to carbocations, (6) aldol and related reactions, (7) addition reactions to double bonds, and (8) rearrangements and desymmetrisations. We highlight the use of NTPA in total synthesis and suggest mnemonics which account for their enantioselectivity.
 Guillermo Caballero-García | Guillermo Caballero-García studied at the Autonomous University of the State of Mexico and taught chemistry in high school before coming to Cambridge to do his PhD with Jonathan Goodman. During this time, he worked on asymmetric catalysis with Brønsted acids and molecular modelling. Many experiments and calculations later, he moved to Canada to work on total synthesis with Professor Rob Britton at Simon Fraser University. |
 Jonathan M. Goodman | Jonathan Goodman is a Professor of Chemistry in the Yusuf Hamied Department of Chemistry at the University of Cambridge. He did his PhD with Professor Ian Paterson doing experiments on boron-mediated aldol reactions, and a post-doc with Professor Clark Still developing molecular modelling algorithms. He research includes the invention of the DP4 NMR analysis method, the quantitative analysis of bifurcating reaction pathways and methods of understanding asymmetric catalysis. |
1. Introduction
Making molecules with high levels of stereoselectivity is a central challenge in organic chemistry. The three-dimensional structure of a molecule is of paramount importance. Molecules with only a specific shape are biologically active. For instance, several drug compounds are administered as single enantiomers, not only to achieve the desired pharmacological treatment, but to avoid adverse side effects as well.1 In addition, more than half of the small molecules amongst the top selling drugs in 2020 contain at least one stereogenic centre.2The field of asymmetric synthesis has attracted both the attention and effort of many research groups trying to imitate nature in synthesising a wide variety of compounds with very high levels of stereocontrol and using mild reaction conditions.3 Such a task can be addressed by asymmetric organocatalysis.4–7 For example, the application of organocatalysis is widely appreciated in the medicinal chemistry area because of the high enantiomeric ratios that organocatalysts can achieve in order to supply a specific enantiomer for clinical trials.8,9
Nowadays, there is a huge repertoire of organocatalysts, with an impressive variety of molecular shapes and functionalities, designed for both general substrates and specific transformations. These catalysts can be classified into four broad categories according to their catalytic cycle and mechanism for activating substrates: Lewis bases, Lewis acids, Brønsted bases and Brønsted acids. An example of the last of these is the focus of this review.
Brønsted acids initiate their catalytic cycle either by protonating the substrate or by forming a hydrogen bond with it (Scheme 1). During the initial step in the mechanism, the electrophile is activated by lowering its LUMO energy. In addition, secondary, non-covalent interactions are likely to occur in order to stabilise intermediates or transition states.10 Amongst these interactions, ion pairing or π–π stacking of aromatic groups are important. Within the catalytic cycle, the most important stage is the enantioselectivity-determining step. Therein, the chiral information in the catalyst is transferred to favour one diastereomeric transition state over the other. This chirality-based bias is promoted by the catalyst's architecture.
 | ||
| Scheme 1 A general catalytic cycle for Brønsted acid-catalysed asymmetric transformations. | ||
Amongst the most popular chiral Brønsted acids are PA. Since the first reports from the groups of Akiyama and Terada,11–14 these catalysts have been widely used in asymmetric synthesis.15 Several reviews are devoted to PA and to the chemical transformations they catalyse. These include Mannich reactions,16 Diels–Alder reactions,17 cycloadditions,18 multicomponent reactions,19 additions to carbonyl groups,20 additions to imines21,22 and C–C bond forming reactions.23,24 Furthermore, heterogeneous Brønsted acid catalysis by PA has also been reported in an enantioselective fashion.25 However, the field of NTPA-catalysed reactions has not been reviewed exhaustively.
Since the seminal publication by the group of Yamamoto in 2006,26 reports regarding the use of NTPA for enantioselective catalysis have increased exponentially. The group of Rueping published a review of NTPA-catalysed reactions from 2006 to 2010.27 Therein, such transformations are covered in detail and will not be included in this review. In addition, the same group published another review focusing on activation modes for PA,15 in which NTPA were also taken into account, albeit without being the key topic. The group of Tsogoeva also published an account regarding NTPA and other chiral Brønsted acids.18
The NTPA-catalysed reactions published from 2006 to 2010 are summarised in Scheme 2.26,28–34,35–40
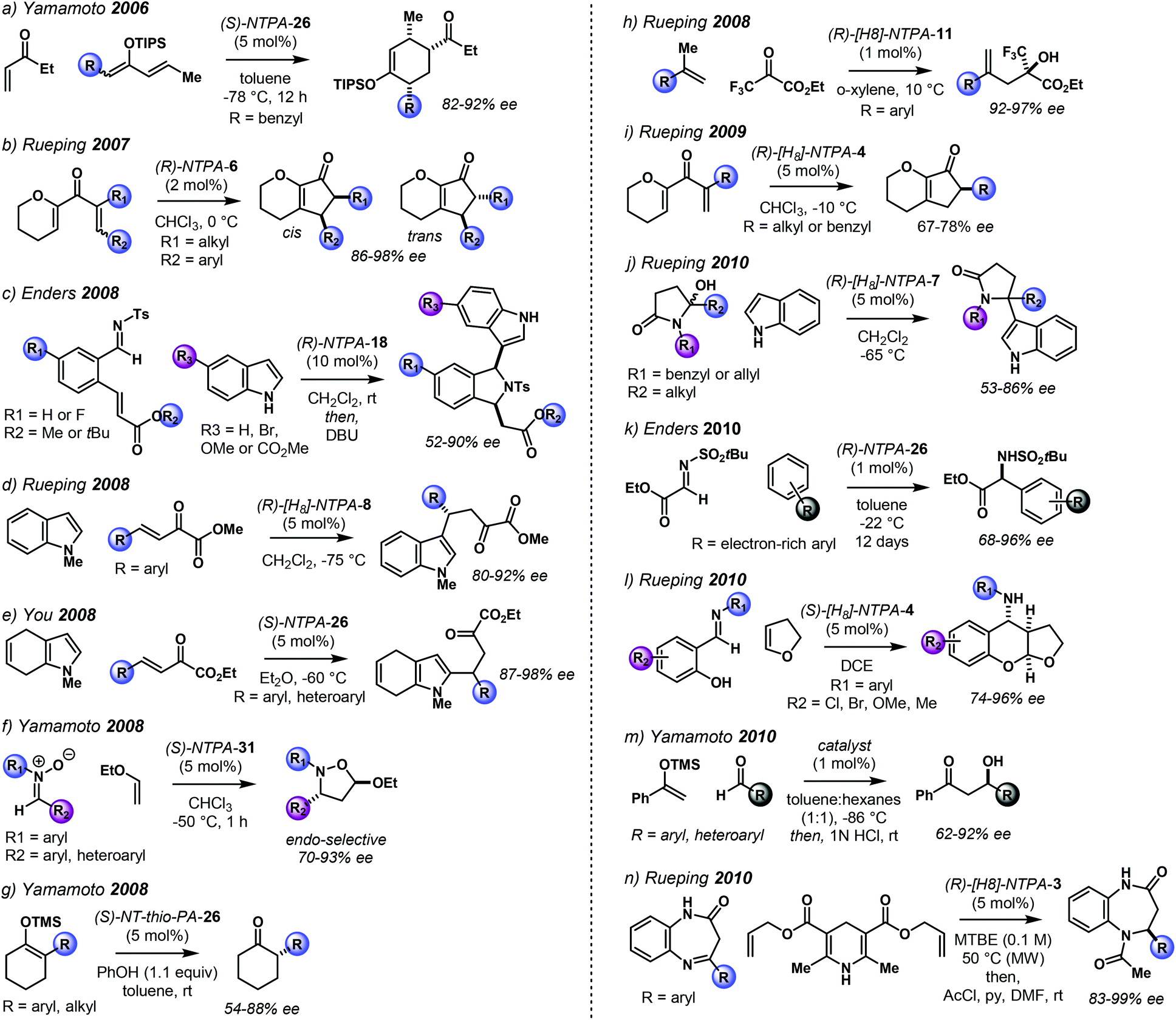 | ||
| Scheme 2 NTPA-catalysed reactions, 2006–2010 timeline. The structure of the catalysts will be discussed later in the review. | ||
1.1. Enhancing reactivity through a more acidic active site
Chiral PA have been mostly used for the activation of rather basic electrophiles, such as imines.21 Less basic substrates, like carbonyl groups, are more challenging to activate with PA. A solution to this problem is to use a more acidic catalyst. In this context, the groups of Rueping and Leito established that the reaction rate depends on the acidity of the catalyst,41 but the ability to induce chirality is dependent on the catalyst's structure.42,43Amongst the most popular chiral templates employed in NTPA are BINOL 1.1,44 [H8]-BINOL 1.2 and SPINOL 1.3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 45 frameworks (Chart 1). Each of these templates holds substituents adjacent to the N-triflylphosphoramide, and we refer to these as scaffolds because they provide a supporting framework for the enantiocontrol of these reactions. Compared to PA, not many chiral scaffolds have been utilised for these stronger Brønsted acids. Yet, depending on the substituents in the catalyst's framework, withstanding stereocontrol can be achieved. The synthesis of NTPA will be discussed in the next subsection.
45 frameworks (Chart 1). Each of these templates holds substituents adjacent to the N-triflylphosphoramide, and we refer to these as scaffolds because they provide a supporting framework for the enantiocontrol of these reactions. Compared to PA, not many chiral scaffolds have been utilised for these stronger Brønsted acids. Yet, depending on the substituents in the catalyst's framework, withstanding stereocontrol can be achieved. The synthesis of NTPA will be discussed in the next subsection.
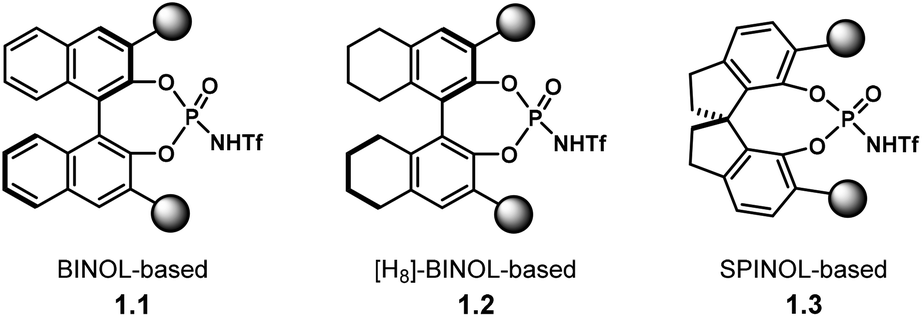 | ||
| Chart 1 Most popular chiral templates for NTPA, indicating the positions of the scaffolds which provide a supporting framework for enantiocontrol. | ||
One approach to increasing the acidity of chiral Brønsted acids is through modification of the active site (Chart 2).46 The introduction of the N-triflylphosphoramide group—1.1—increases the strength of the Brønsted acid,47 in comparison to the OH motif in 1.4. A more delocalised negative charge in the catalyst's conjugate base is expected to be one of the key elements in Brønsted acidity. Furthermore, the trifluoromethyl group contributes via inductive effects.
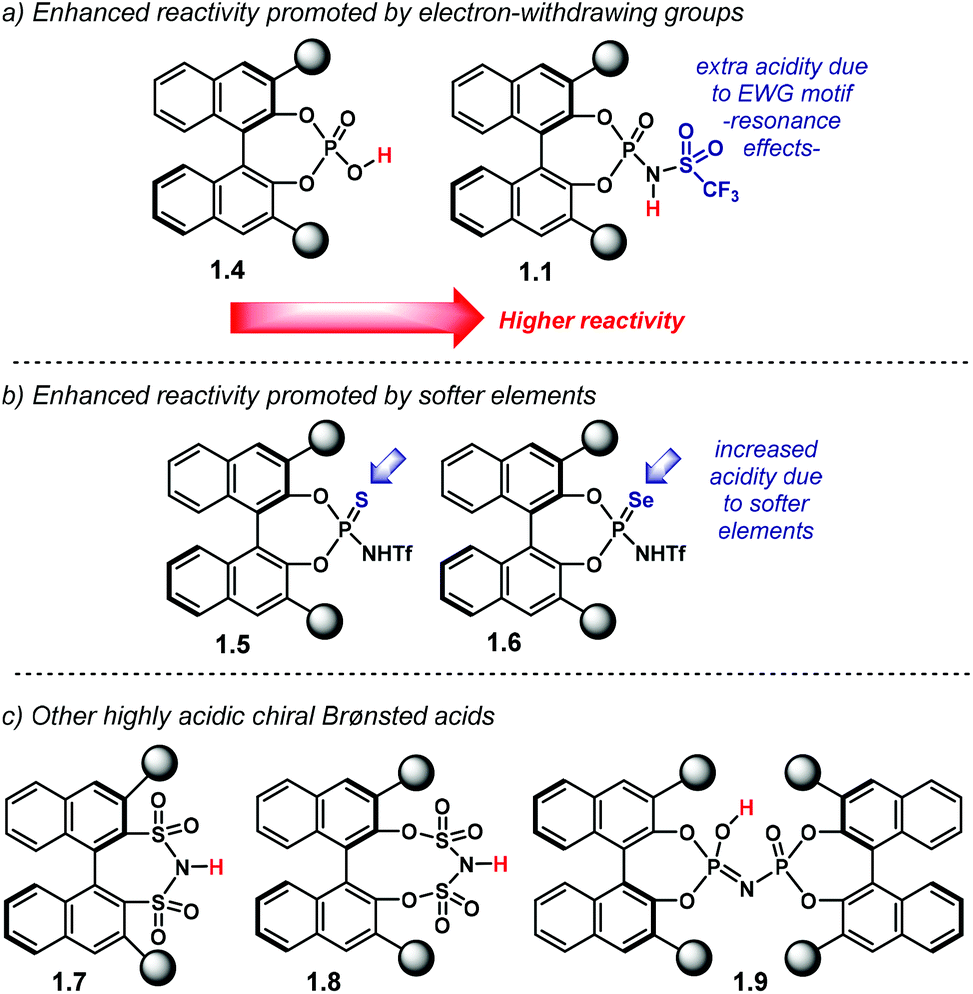 | ||
| Chart 2 (a) Increased reactivity in the catalyst's active site by introduction of electron-withdrawing motifs. (b) Increased acidity by introduction of softer elements, i.e. sulfur or selenium. (c) Stronger chiral Brønsted acids. | ||
In addition, another way in which catalyst acidity can be enhanced is by changing the phosphoryl oxygen atom in 1.1 for softer elements: sulfur or selenium (Chart 2b). These larger atoms can further stabilise the negative charge in the catalyst's counter anion. Thus, thio- and seleno-NTPA, 1.5 and 1.6, respectively, represent more acidic compounds. The mechanisms of many reactions using these catalysts depend on their ability to act as a Lewis base as well as a Brønsted acid. Changing oxygen to sulfur or selenium reduces this effect and so may lead to different activation modes. As a consequence, they perturb reaction mechanisms in complex ways and there are rather few reports of their use.
Even stronger Brønsted acids have been designed by introducing additional electron-withdrawing motifs to the active site (Chart 2c). Bis(sulfonyl)imides 1.7,48–50 bis(sulfuryl)imides 1.851 and imidodiphosphoric acids 1.952 are highly acidic catalysts for enantioselective transformations, but with very different functionalities to those shown in Chart 1. These types of catalysts, therefor, are beyond the scope of this review.
Much research in this field comprises the study of catalyst's acidity. As mentioned before, catalytic activity is intimately related to the acidity of the active site. Several research groups have developed acidity scales in order to account for reactivity.53,54 The groups of Berkessel and O'Donoghue developed one of the first experimentally determined acidity scales.55 In their work, they determined the pKa values in DMSO for a series of Brønsted acids. Albeit mostly focused on PA, they showed that the substituents in the chiral framework could also affect the acidity.
The groups of Rueping and Leito reported another acidity scale,41 determined in acetonitrile, comprising both PA and NTPA (Fig. 1a). In acetonitrile, PA cover a range of pKa values from 12.5 to 14.0, whereas, the more acidic NTPA fall into a 6.3 to 6.9 region of the scale. This shows, with experimentally measured values, the higher acidity of NTPA. In the case of PA, those bearing electron-poor substituents have a lower pKa than those with electron neutral scaffolds, for example, PA-1.1 (pKa = 12.5) against PA-1.2 (pKa = 12.7) and PA-1.3 (pKa = 13.3). The more sterically demanding PA-4 (pKa = 13.6) was shown to be less acidic. Presumably, the large substituents might be influencing the acidity of the active site somehow. In addition, the partially hydrogenated BINOL backbone in [H8]-PA-1.1 decreases the acidity by 0.7 pKa units.
 | ||
| Fig. 1 Acidity scales for Brønsted acids, pKa values are presented in parenthesis. (a) Rueping and Leito acidity scale determined in acetonitrile. (b) Acidity scale calculated by the groups of Li and Chen at the SMD(DMSO)/M06-2X/6-311++G(2df,2p)//B3LYP/6-31+G(d) level of theory. | ||
On the other hand, the electronic nature of the 3,3′ substituents in NTPA seems to have little—or even negligible—effect in the catalyst's acidity. This was observed in NTPA-2 and NTPA-2.1, for whose pKa values were 6.4. In analogy with PA, a partially hydrogenated BINOL backbone also contributed to increase the pKa value in NTPA.
Apart from experimental acidity scales, computational methods, like DFT, have become popular to assess pKa values. These offer alternative approaches when experimental data is limited or not available. In this manner, the groups of Lin and Chen developed an acidity scale derived from calculated pKa values (Fig. 1b).56,57 Therein, it was shown how acidity varies when changing heteroatoms in the catalyst's active site, i.e. replacing the P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O moiety for PS or PSe. As expected, the pKa values decreased when going from NTPA-1 (pKa = −3.40) to thioNTPA-1 (pKa = −3.83) to selenoNTPA-1 (pKa = −4.58). In addition, this computational study was helpful to observe how acidity varies when changing the chiral template as well. In that case, the SPINOL-derived NTPA turned to be more acidic than the corresponding BINOL-based one. On the other hand, the NTPA with a partially hydrogenated BINOL backbone was shown to be less acidic (pKa = −2.70). Furthermore, the values for three disubstituted NTPA were calculated. Similar to PA, it was found that scaffolds bearing electron-withdrawing groups enhance the catalyst's acidity, in comparison to electron-donating ones.
O moiety for PS or PSe. As expected, the pKa values decreased when going from NTPA-1 (pKa = −3.40) to thioNTPA-1 (pKa = −3.83) to selenoNTPA-1 (pKa = −4.58). In addition, this computational study was helpful to observe how acidity varies when changing the chiral template as well. In that case, the SPINOL-derived NTPA turned to be more acidic than the corresponding BINOL-based one. On the other hand, the NTPA with a partially hydrogenated BINOL backbone was shown to be less acidic (pKa = −2.70). Furthermore, the values for three disubstituted NTPA were calculated. Similar to PA, it was found that scaffolds bearing electron-withdrawing groups enhance the catalyst's acidity, in comparison to electron-donating ones.
1.2. Catalyst structure and synthesis of NTPA
The most important features in NTPA are presented in Fig. 2. In broad terms, the catalyst's architecture comprises a chiral template and an active site. The first of which provides the source of chiral information to be transferred during the enantio-determining step. Amongst the most popular chiral templates for NTPA, as well as for PA, is the BINOL framework (Fig. 2a).44 Herein, the 3,3′ positions are critical in catalyst engineering as they are responsible of linking the stereocontrolling scaffolds that define the chiral cavity of the catalyst—our group has conducted DFT calculations in order to understand the role of these susbtituents in stereochemical induction using BINOL-based PA.58,59 In addition, the 6,6′ positions of the template offer alternative sites for framework modifications. These include attachment of hydrocarbon chains to improve solubility, or the incorporation of electron-withdrawing groups to enhance acidity. Using a partially hydrogenated BINOL backbone might be regarded as one particular example as it provides alternative conformations to the chiral template. Besides the BINOL template, the SPINOL scaffold has also become popular in the design of NTPA (Fig. 2b)—the group of Lin is one of the pioneers in utilising this framework for chiral Brønsted acids.60 The alternative template provides a tighter chiral pocket, which can also be tuned by installing different substituents, in a similar fashion to BINOL.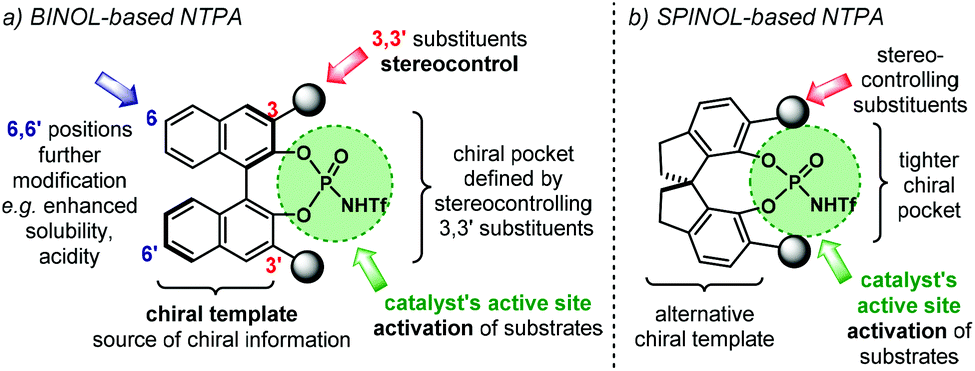 | ||
| Fig. 2 Catalyst structure in (a) BINOL-based NTPA and (b) SPINOL-based NTPA. | ||
The installation of the 3,3′ substituents in the BINOL framework, as well as in the SPINOL backbone, has been extensively covered in previous reviews,15,60,61 and will not be covered in detail here. Instead, we will focus on the available methods to install the acidic motif that constitute the active site. The general synthetic routes towards NTPA are presented on Scheme 3.
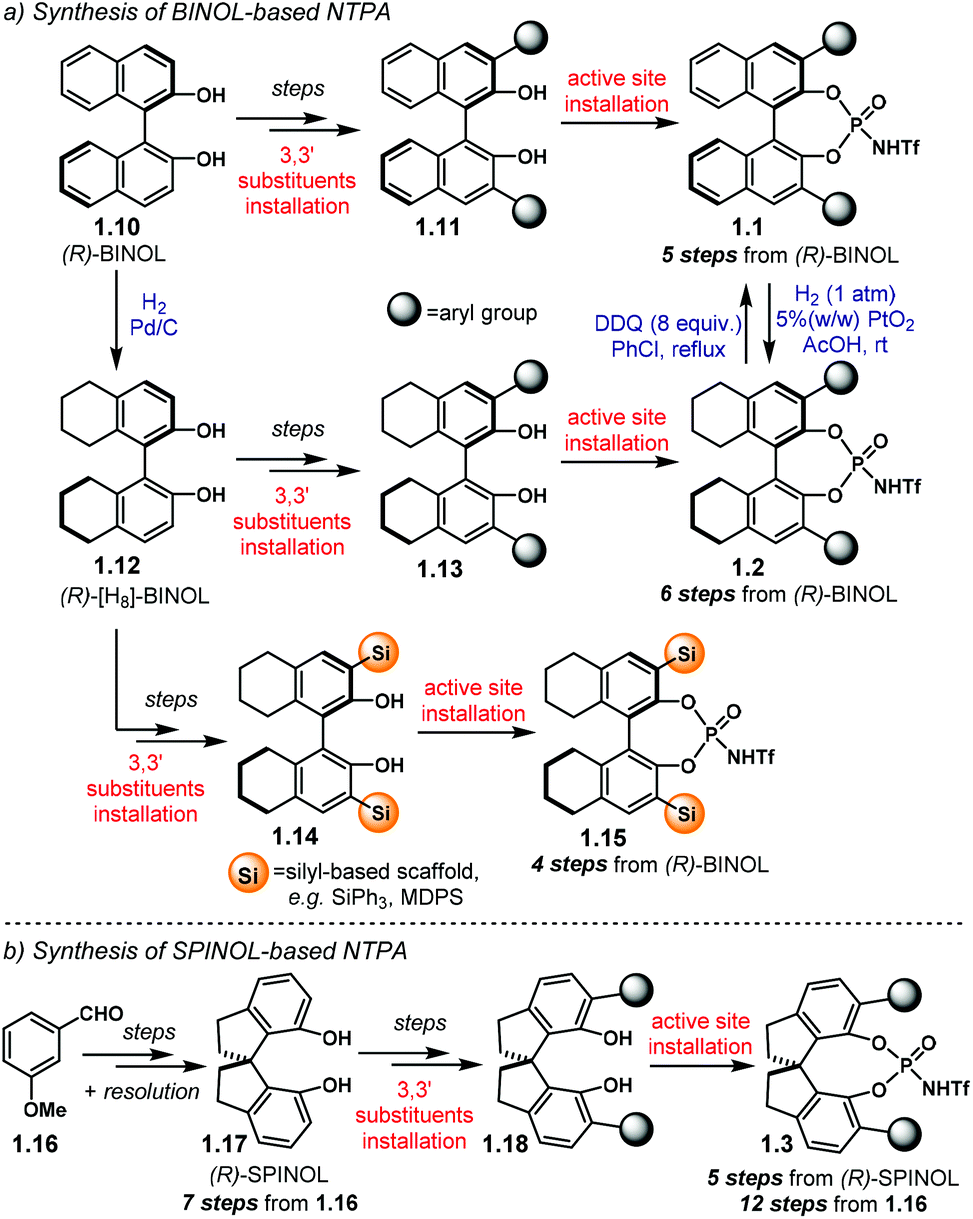 | ||
| Scheme 3 (a) Synthetic routes towards BINOL-based and [H8]-BINOL-based NTPA. (b) Synthetic route to SPINOL-based NTPA. MDPS = methyldiphenyl silyl. | ||
BINOL-based NTPA are prepared starting from commercially available (S)- or (R)-BINOL 1.10 (Scheme 3a). Several steps—comprising diol protection, installation of a cross-coupling handle in the 3,3′ positions, cross-coupling reaction of aryl substituents and diol deprotection—lead to the corresponding 3,3′-disubstituted BINOL 1.11. With such a precursor in hand, the active site can be installed to afford BINOL-based NTPA 1.1 in five steps from 1.10. The synthesis of [H8]-BINOL-based NTPA 1.2 follows a similar pathway. Initial partial hydrogenation of 1.10 provides key chiral template 1.12. After a similar series of transformations as for BINOL, 3,3′-disubstituted precursor 1.13 is ready to be converted into the corresponding NTPA 1.2 in a six step sequence. In addition, catalysts 1.1 can be partially hydrogenated using PtO2 to afford 1.2.62 Likewise, oxidation of 1.2 with DDQ offers another route to BINOL-based 1.1.
An alternative, and slightly shorter, route to [H8]-BINOL-based NTPA bearing silicon-based scaffolds on the 3,3′ positions was envisaged by the group of Rueping.61 Therein, (R)-[H8]-BINOL 1.12 is converted into the corresponding 3,3′-disubstituted compound 1.14 using a two-step sequence that involves a silyl-protection followed by a t-BuLi promoted rearrangement.63 Using this methodology, NTPA 1.15 can be obtained in four steps from 1.10.
SPINOL-based NTPA 1.3 require a longer route to be made (Scheme 3b). Starting from 3-methoxybenzaldehyde 1.16, SPINOL 1.17 is obtained after several transformations and resolution of the resulting racemate.64 Thus, this method has the advantage of providing both enantiomers of the chiral template. The stereocontrolling scaffolds are installed in a similar fashion to BINOL to furnish 1.18.60 There, the active site can now be installed. In this manner, NTPA 1.3 are afforded in twelve steps from aldehyde 1.16.
The increasing number of NTPA-catalysed transformations have rendered an impressive variety of 3,3′ substituents in the catalysts’ architecture. The most popular are aryl-based substituents, bearing diverse functionalities and substitution patterns. In addition, silicon-based scaffolds have also shown to give good levels of enantiocontrol, albeit fewer examples are reported in the literature. The catalysts used in the majority of reactions discussed throughout the review are presented on Chart 3. In order to avoid ambiguity and to keep consistency in the following sections, the labels and numbering presented in Chart 3 are used in the rest of the review. The label ‘NTPA’ is used for the BINOL-based NTPA, the ‘[H8]-NTPA’ for those catalysts with a partially hydrogenated BINOL backbone and ‘S-NTPA’ for SPINOL-derived catalysts (i.e.1.1, 1.2 and 1.3, respectively, cf.Chart 1). The (R)- and (S)-stereochemical descriptors are also included for clarity depending on the enantiomer used in a given reaction.
 | ||
| Chart 3 Catalysts used in the majority of reactions discussed throughout this review. The (R)-enantiomers are presented here for clarity. This label and numbering system are used consistently throughout the review. (a) BINOL-based NTPA. (b) [H8]-BINOL-based NTPA. (c) SPINOL-based NTPA. | ||
1.3. The N-triflylphosphoramide active site
Although the installation of the active site in NTPA is central to their preparation, it has not been discussed in detail in past reviews. Therefore, here we want to revise the available methods to do so as well as highlighting potential advantages and disadvantages inherent of each method—as a way of providing a brief and useful guide to experimentalists aiming to synthesise this type of catalyst. Amongst the literature, four main protocols to install the acidic motif in NTPA are utilised (Scheme 4). For clarity purposes, we exemplify these methods using BINOL 1.11, but they apply to [H8]-BINOL- and SPINOL-derived diols as well. | ||
| Scheme 4 Available methods to install the N-triflylphosphoramide motif. | ||
NTf) as a phosphorylating reagent—this compound can be synthesised on a relatively large scale from phosphorus(V) pentachloride and TfNH2 at 110 °C without the need of a solvent followed by fractional distillation.69,72,73 Such a method furnishes intermediate 1.20, which by subsequent hydrolysis affords the desired catalyst. The shorter reaction times, room temperature and high yields make this procedure an appealing alternative. Furthermore, other perfluorinated sulfonylamides are suitable substrates as well.74,75 The use of strict anhydrous conditions and the synthesis of Cl3PNTf might be the main disadvantages.
During the work-up stage in the aforementioned methods, the NTPA is obtained in its free, fully protonated form. However, further purification by column chromatography is often required. As it has been studied in the case of PA, this has the disadvantage of contaminating the final product with metal impurities from the silica gel, and the isolated product is generally a mixture of metal phosphates instead of the free acid.80 This can have dramatic effects in catalytic activity and enantiocontrol.81–83 Therefore, recovering the NTPA in the free acid form is paramount. An efficient method is through thorough washing with 6 N aqueous HCl after column chromatography. Recrystallization would be preferred if chromatography can be avoided.
1.4. Scope of the review
The following sections comprise a collection of NTPA-catalysed reactions published from 2011. Herein, we classify those transformations into eight different categories: (1) cycloadditions, (2) electrocyclisations, polyene and related cyclisations, (3) addition reactions to imines, (4) electrophilic aromatic substitutions, (5) addition reactions to carbocations, (6) aldol and related reactions, (7) addition reactions to double bonds, and (8) rearrangements, desymmetrisations and enantioselective protonations.Even though a given reaction can be fitted into more than one category, we took as the key criterion the enantio-determining step where enantioinduction takes place. In addition, the highlights of each transformation are discussed, whether the substrate scope, the screening of NTPA or the design of a specific type of catalyst. Throughout the sections, we also go into the detailed mechanism of the reactions, focusing on how the catalyst activates the substrates; enantiocontrol is the subject of the last section. Furthermore, where appropriate, we emphasise the use of NTPA as part of the key step in total syntheses.
In the final section, we present a mnemonic system to account for the stereochemical output of NTPA-catalysed reactions. This is a useful, qualitative guide to explain enantioselectivity without the need of sophisticated DFT calculations.
2. Pericyclic reactions I: cycloadditions
Cycloaddition reactions are amongst the most versatile methods for the rapid construction of ring systems. Those rings can bear several chiral centres. Therefore, numerous methodologies to assemble cyclic molecules with multiple stereocentres in an asymmetric fashion have been developed. Amongst these methods, catalytic processes are highly desirable.17 Over the past sixteen years, chiral Brønsted acids have shown to be effective catalysts to build up molecules with high levels of stereocontrol. BINOL-derived NTPA have outperformed their analogous PA in challenging cycloaddition reactions. The higher acidities of NTPA makes them suitable for the activation of unreactive substrates. Furthermore, their atom-rich active site can engage in several types of stabilising interactions to achieve high levels of enantiocontrol. In this section, we look at enantioselective NTPA-catalysed transformations that furnish ring systems through a cycloaddition reaction as the enantiodetermining step.2.1. (4 + 2) cycloadditions
The Diels–Alder reaction is one of the most powerful methods for the construction of six-member carbocyclic rings. Together with its variants, including the hetero Diels–Alder reaction, several catalytic methodologies have used NTPA to control the stereochemical output. Enantiocontrol can be challenging with unbiased substrates like olefins, as distinct to carbonyl groups or imines which have dipoles. In this context, NTPA are suitable catalysts for such difficult-to-activate substrates.The Nagorny group reported a challenging ionic Diels–Alder reaction of acetals 2.1 with unactivated dienes 2.2 (Scheme 5a).79 Chiral PA showed no reactivity when the cycloaddition was done in toluene. However, (S)-NTPA-21 promoted the reaction, albeit with poor stereocontrol (Scheme 5b). The enhanced reactivity was attributed to the catalyst's higher acidity. Further screening of catalysts found (S)-NTPA-26 to be the most suitable in terms of reactivity and diastereocontrol (endo/exo 5:1). Bulky substituents at the ortho and para positions in the 3,3′ aryl scaffolds were needed to enhance stereoselectivity. Solvent evaluation revealed ethyl iso-butyrate as the best reaction media for both diastereo- and enantiocontrol (endo/exo 7:1, 60% ee). Although the substrate scope offered good yields, modest enantioselectivities, up to 62% ee, were achieved, with the endo adducts being favoured.
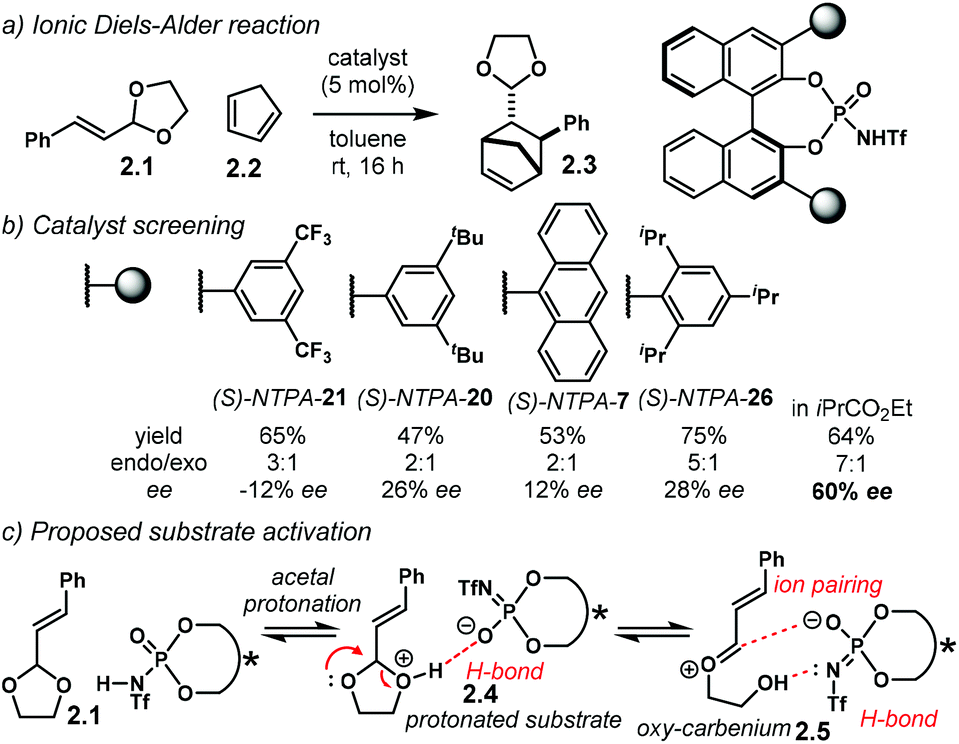 | ||
| Scheme 5 (a) Ionic Diels–Alder reaction developed by the group of Nagorny. (b) Catalyst screening, different 3,3′ substituents were tried with varying steric bulk. (c) Plausible activation of the acetal moiety by the catalyst. The open oxy-carbenium ion is proposed to be stabilised by ion pairing and H-bonding. | ||
A plausible reaction mechanism starts by protonation of acetal 2.1 (Scheme 5c). This step might be disfavoured for PA due to their low acidity. An H-bond complex 2.4 is proposed to be in equilibrium with the opened form of the acetal, 2.5. Such a complex seems to activate the dienophilic double bond through the reactive oxy-carbenium ion. This species is presumed to engage in H-bonding with the catalyst, as well as forming a tight ion pair. The low level of enantiocontrol suggests that a flexible H-bond complex, rather than a rigid ion-paired one, is most likely to be formed in the reaction—though the authors do not clarify the activation mode.
A highly enantioselective Diels–Alder reaction, generating up to three stereocentres, was developed by the group of Jiao (Scheme 6a).84 Using diene carbamates 2.6 and vinyl ketones 2.7, the reaction yields carbocycles 2.8 as single diastereoisomers with excellent enantioselectivities, up to 99% ee. The highly acidic NTPA outperformed PA, which showed no catalytic activity. Fine-tuning of the catalyst's scaffolds was found to be important to achieve high enantiocontrol (Scheme 6b). A bulky substituent in the para position, like in (R)-NTPA-26 proved to be detrimental for enantiomeric excess, which was not the case when (R)-NTPA-2 was tried. However, the extended ortho–meta π-system, anthracenyl in (R)-NTPA-7 afforded the best enantioinduction.
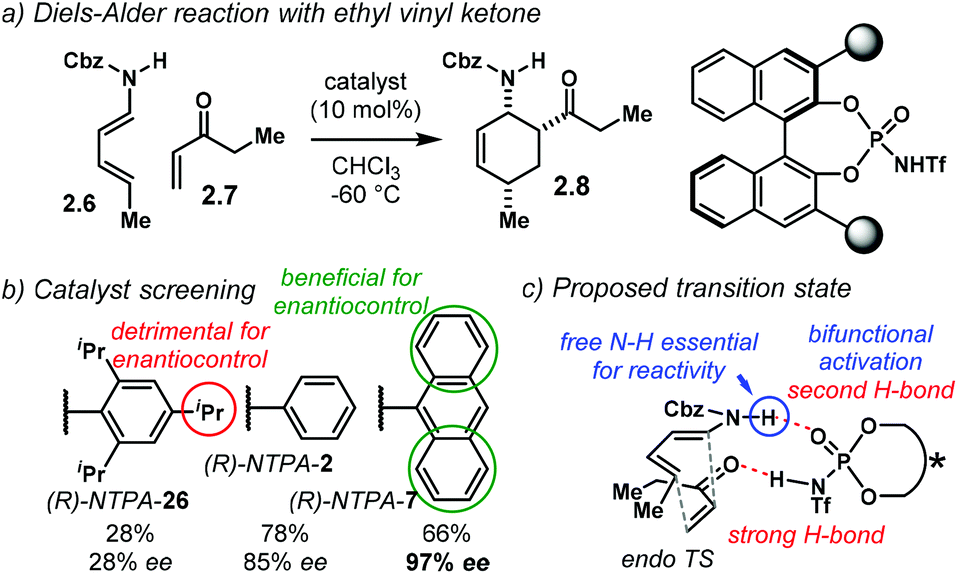 | ||
| Scheme 6 (a) Diels–Alder reaction of diene carbamates with ethyl vinyl ketone. (b) Extract from the catalyst screening. (c) Proposed endo-TS for the cycloaddition reaction. The free N–H moiety was found to be essential for reactivity. | ||
The high levels of diastereocontrol suggest a rigid transition state. Therein, the authors propose a bifunctional activation mode (Scheme 6c). In this case, the catalyst forms a strong H-bond with the oxygen atom in the ketone. At the same time, the free N–H motif in the diene engages in a second H-bond with the catalyst's PO group. Control experiments showed that the free N–H moiety is essential for reactivity. In addition, modest DFT modelling of the catalyst–substrate complex supports the proposed bifunctional activation mode.
The substrate scope in this Diels–Alder reaction is not limited to alkyl vinyl ketones. Aryl and heteroaryl substrates also worked with remarkable enantiocontrol. For the case of 2-methylphenyl vinyl ketone, the reaction fails, presumably due to unfavourable steric clash within the catalyst's active site.
The group of Gong developed an elegant methodology using a dual Au(I)/chiral-NTPA catalytic system (Scheme 7a).85 Therein, gold(I) catalyst 2.9 generates siloxadiene 2.11 as a latent intermediate, which is subsequently trapped by 2.12. The dienophile might be H-bonded to (S)-NTPA-8 to provide a tight chiral environment for the next step. The enantiodetermining Diels–Alder step is controlled by the NTPA, yielding 2.13 with remarkable enantioselectivity. For this purpose, large 1-pyrenyl substituents had to be used in the catalyst framework for efficient chirality transfer. Double bond isomerization in the last step yields the final product, 2.14.
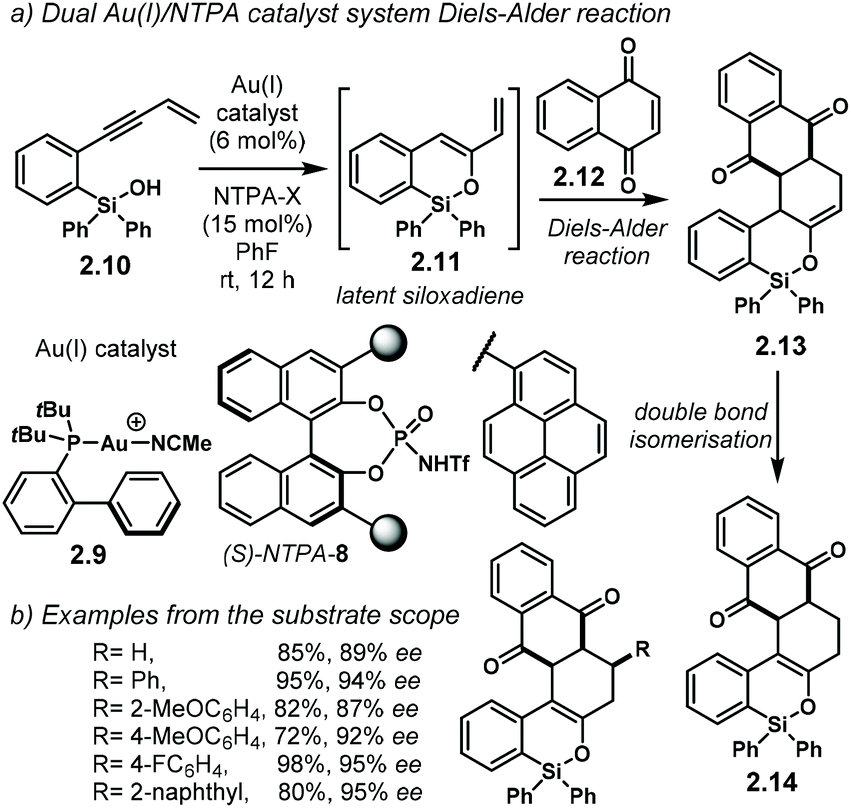 | ||
| Scheme 7 (a) Siloxylation/Diels–Alder cascade using an Au(I)/NTPA catalytic system. (b) Examples from the substrate scope. Up to three contiguous stereocentres are generated with excellent enantioselectivities. | ||
PA and NTPA have been used successfully along with gold(I)-catalysed processes as well as with other metals.86–90 However, when chiral PA were used in the latter reaction, low reactivity and low enantioselectivity were observed. Presumably, the NTPA is just acidic enough to activate dienophile 2.12. Even though the authors did not fully discuss the activation step, it is likely to go through carbonyl protonation or through a strong H-bond. This reaction shows off the high compatibility of Au(I) complexes and NTPA as Brønsted acid catalysts. The cycloaddition step can install up to three contiguous stereocentres, as shown by the group of selected examples (Scheme 7b). The reaction can tolerate both electron-withdrawing and electron-donating substituents on the substrate's aryl groups.
The group of Ishihara developed a Lewis acid-assisted and Brønsted acid-catalysed Diels–Alder reaction.91 The reaction works well using PA, achieving high yields and enantioselectivities. However, for the less-reactive diene 2.15, (R)-NTPA-26 outperformed PA both in reactivity and in stereocontrol (Scheme 8). Reaction with 2.16 afforded 2.17 with good enantioselectivity (89% ee). The enhanced reactivity of (R)-NTPA-26 can be attributed to its higher acidity. The extra iso-propyl groups in the 3,3′ substituents of the catalyst may also be playing an important role, but the authors do not discuss it.
 | ||
| Scheme 8 Lewis acid-assisted/NTPA-catalysed Diels–Alder reaction. | ||
The hetero-Diels–Alder reaction is a powerful strategy for constructing heterocyclic systems.92 In the field of Brønsted acid-catalysed reactions, imines play an important role. Due to the relatively facile protonation of the basic nitrogen atom, iminium ions are suitable dienophiles for aza-Diels–Alder reactions.93,94
The group of Hatanaka reported an asymmetric imino-Diels–Alder reaction (Scheme 9a).66 Therein, N-protected aldimines 2.18 are activated through full protonation by a Brønsted acid. The activated species readily reacts with acyclic dienes 2.19 yielding heterocyclic products 2.20 in good yields and high enantioselectivities. During catalyst screening studies (Scheme 9b), it was found that the unsubstituted (R)-PA-1 achieved modest enantioselectivity (47% ee) albeit with poor reactivity. On the other hand, the analogous (R)-NTPA-1 outperformed the PA in reactivity but with poor enantiocontrol (9% ee). Further screening of the catalyst's substituents was needed to enhance enantioinduction. Catalysts (R)-NTPA-4, (R)-NTPA-5 and (R)-NTPA-21 showed good reactivity but low enantiocontrol (<39% ee). The best results were obtained with (R)-NTPA-26, suggesting that bulky groups increased enantiodifferentiation. Despite good enantiocontrol, the reactivity of this catalyst was modest. Surprisingly, the introduction of trifluoromethyl groups at the 6,6′ positions in the backbone of the catalyst enhanced reactivity at half the catalyst loading without an appreciable decrease in enantioselectivity. This new catalyst suggests that the electronics of the BINOL backbone might also play an important role increasing the acidity of the active site.
 | ||
| Scheme 9 (a) Enantioselective aza-Diels–Alder reaction developed by the group of Hatanaka. (b) Catalyst screening: the trifluoromethyl groups at the 6,6′ positions in the backbone of the catalyst suggest that its electronic structure plays an important role. | ||
Higher enantiocontrol was achieved by changing the N-protecting group in the imine for a 2-naphthyl sulfonyl scaffold (Scheme 10a). Imines 2.21 furnished heterocycles 2.22 in higher yields and enantioselectivities. Different N-protecting groups showed to be detrimental for reactivity and enantiocontrol. The authors propose that the imine is fully protonated in active complex 2.23 (Scheme 10b). DFT calculations of such complex suggested that the linear (E)-iminium ion 2.23 might not fit in the catalyst's chiral pocket. Therefore, the (Z) isomer 2.24 reacts with the incoming diene while avoiding steric clash with the catalyst.
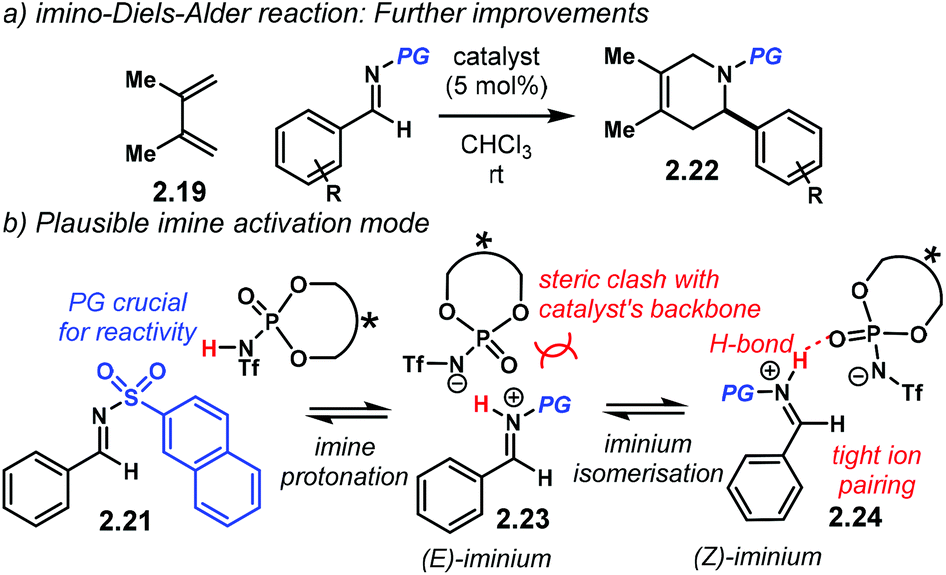 | ||
| Scheme 10 (a) imino-Diels–Alder reaction, PG = 2-naphthyl sulfonyl. (b) Plausible activation mode of the imine. A (Z)-iminium geometry seems more likely to fit into the chiral cavity of the catalyst in contrast with the extended (E)-iminium isomer. | ||
Recalling the publication from the group of Gong regarding the dual Au(I)/NTPA catalytic system (cf.Scheme 7), an hetero-Diels–Alder reaction was later envisaged by the same group (Scheme 11).95 The latent siloxadiene 2.11 obtained from 2.10 through the Au(I)-catalysed process reacts with glyoxylate 2.25 in a (4 + 2) cycloaddition. Subsequent double bond isomerisation furnishes enantioenriched oxa-heterocycles 2.26. For the hetero-Diels–Alder step, NTPA outperformed PA both in reactivity and in enantioselectivity. The bulky (S)-NTPA-28 provided access to products with good yields and enantiocontrol. Interestingly, the similar catalysts (S)-NTPA-26 and (S)-NTPA-29 were not as good as the former. This example shows that even small or subtle variations in the catalyst structure (the 3,3′ substituents in this case) can have a tremendously different output. In the majority of cases, catalyst screening is still a trial and error scenario in the development of new asymmetric reactions.
 | ||
| Scheme 11 Siloxylation/hetero-Diels–Alder cascade using an Au(I)/NTPA catalytic system. Subtle variations in the catalyst scaffolds can have meaningful effects in reactivity and enantioselectivity. | ||
The activation of glyoxylate 2.25 might be through a strong H-bond with the catalyst. Nonpolar solvents, which were found as the most suitable reaction media, point towards this. In addition, based on previous publications by the same group, the fluorenyl ester in 2.25 was needed to achieve good levels of enantiocontrol.
2.2. (3 + 2) and (4 + 3) cycloadditions
Amongst popular methods for the assembly of five and seven member rings, (3 + 2) and (4 + 3) cycloadditions play an important role. Together with the possibility of installing several chiral centres in a single step, these two types of cycloadditions are frontline candidates to develop asymmetric methodologies. The introduction of heteroatoms to any of the reaction counterparts makes them even more valuable for the synthesis of heterocycles.In the terrain of acid-catalysed (3 + 2) cycloadditions, the group of Rueping developed a method for the enantioselective construction of five-membered heterocycles 2.28 and 2.31 (Scheme 12).96 In this methodology, N-acyl hydrazones 2.27 and 2.29 react with alkenes in a NTPA-catalysed cycloadditions. PA afforded low yields despite efforts to optimise the reaction conditions. Both yield and enantioselectivity were outperformed by (R)-[H8]-NTPA-14. The enhanced reactivity is attributed to the NTPA higher acidity. Different N-acyl protecting groups on the imines were also tried. N-Benzoyl hydrazones 2.27 gave the best results for the reaction with cyclopentadiene 2.2 (Scheme 12a). The reaction showed a remarkable substrate scope. Alkyl, aryl and heteroaryl substituents worked well. Single diastereoisomers with outstanding enantiocontrol were produced with a catalyst loading as low as 2.5 mol%. This methodology was not limited to cyclopentadiene. α-Methylstyrenes 2.30 worked as well delivering excellent enantiocontrol (Scheme 12b). For this reaction, the more electron-deficient N-(4-nitrobenzoyl) hydrazones 2.29 were used. Good to excellent enantioselectivities were achieved. In addition, access to all-carbon quaternary stereocentres makes this reaction particularly valuable.
 | ||
| Scheme 12 Enantioselective (3 + 2) cycloaddition reaction with (a) cyclopentadiene and (b) α-methylstyrenes. (c) Plausible activation mode of N-acyl hydrazones; full protonation on the nitrogen atom and tight ion pairing is more likely than just an H-bond complex. | ||
The hydrazone is very likely to be protonated during the cycloaddition step (Scheme 12c). The basicity of the nitrogen atom and the remarkable diastereocontrol may be indicative of this. If a tight ion-pair, assisted by multiple H-bonds is formed, a rigid transition state can explain the efficient chirality transfer, although, the authors do not suggest an activation mechanism.
Later, the groups of Rueping and Houk investigated (3 + 2) cycloadditions of N-acyl hydrazones 2.27 in more detail (Scheme 13).97 Using ethyl vinyl thioether 2.32 and SPINOL-derived catalyst S-NTPA-11, a synthesis of enantioenriched pyrazolidines 2.33 was realised. Despite the modest yields reported, the reaction features good diastereocontrol (up to 8:1 dr in favour of the syn-diastereoisomers) and excellent enantioselectivities (up to 92% ee). Catalyst screening revealed acidity to be a crucial factor for the reaction to work. Low yields were obtained with PA and NTPA outperformed them due to their higher acidity. Interestingly, the SPINOL-derived NTPA gave the highest enantiomeric excesses, surpassing their BINOL and [H8]-BINOL analogues.
 | ||
| Scheme 13 (3 + 2) cycloaddition of N-benzoyl hydrazones with ethyl vinyl thioether. The SPINOL-derived catalyst achieved the best enantiocontrol. | ||
The reaction works well with a broad scope of alkyl substituents. The N-benzoyl protecting group gave the best enantioselectivities. The methodology also works with ethyl vinyl ether as the alkene component; however, lower yields and diminished enantiocontrol was observed, albeit with enhanced diastereoselectivities (>95:5 dr).
Through a detailed DFT computational study, the authors investigated the activation mode of N-acyl hydrazone 2.27 in this (3 + 2) cycloaddition. Full protonation on the nitrogen atom yields hydrazonium–NTPA complex 2.34 (Scheme 14a). This species is highly stabilised and locked into the active site of the catalyst through a tight ion-pair, which is further assisted by multiple H-bonding. The H-bonds are formed with the oxygen and nitrogen atoms in the phosphoryl motif of the NTPA. In the lowest-energy transition state 2.35, the H-bonds are formed with the PO and SO moieties in the active site. The sum of all these strong, non-covalent interactions provides a rigid transition state to account for the observed stereoselectivities. Given that the positive hydrazonium ion undergoes the cycloaddition step, this reaction was better described as a 1,3-monopolar (3+ + 2) cycloaddition.
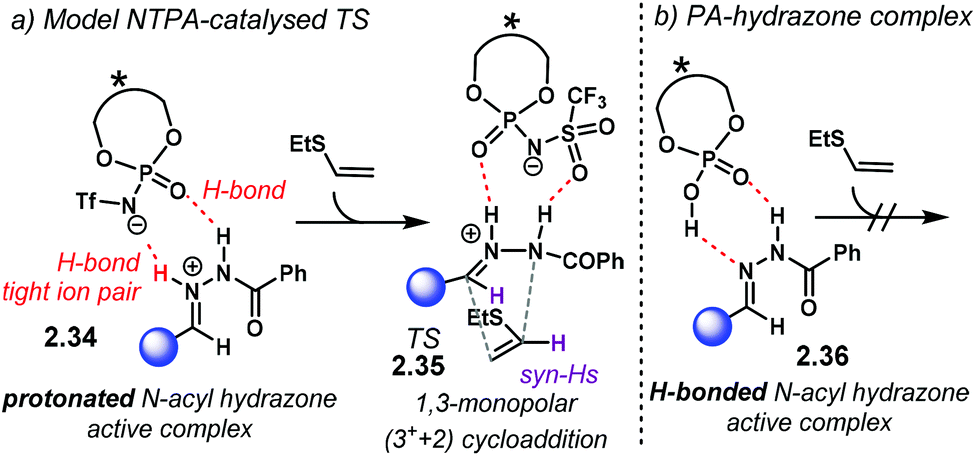 | ||
| Scheme 14 (a) Activation mode and proposed transition state for the NTPA-catalysed cycloaddition of N-acyl hydrazones. Full protonation is required to reach the transition state. The catalyst locks and activates the substrate through tight ion pairing and multiple H-bonds. These H-bonds are formed with the oxygen atoms from the phosphoryl and sulfonyl motifs. (b) PA–hydrazone model complex. Full protonation will heavily distort the H-bond complex. Such energy penalty makes PA less reactive in this reaction. | ||
In sharp contrast, PA only interact with the hydrazone through H-bonding, as shown in complex 2.36 (Scheme 14b). Yet, full protonation is needed for the reaction to occur. Through a distortion–interaction analysis, it was found that the PA–hydrazone complex 2.36 has to distort heavily in order to achieve full protonation and the ion-pair geometry required to promote the cycloaddition reaction. This work neatly accounts for the enhanced reactivity of NTPA, outperforming PA.
Further developing ionic (3 + 2) cycloadditions, the group of Shi, envisaged one such reaction using 2-indolylmethanols 2.37 as suitable carbocationic precursors for a (3 + 2) cycloaddition with hydroxystyrenes 2.38 (Scheme 15a).98 Several PA were tried unsuccessfully, affording either no reaction or low enantioselectivity. Regarding (R)-S-PA-7, (R)-[H8]-PA-7 and (R)-PA-7, it is noticeable, yet not obvious, how subtle differences in the catalyst's backbone can lead to dramatic, dissimilar outputs (Scheme 15b). With a SPINOL or an [H8]-BINOL backbone, no product 2.39 was observed in the reaction. A standard BINOL-derived PA improved reactivity and enantioselectivity, albeit modestly. The analogous (R)-NTPA-7 provided a higher yield with quite improved enantiocontrol. It is not clear, however, how changing the active site of the catalyst can boost the enantiomeric excess.
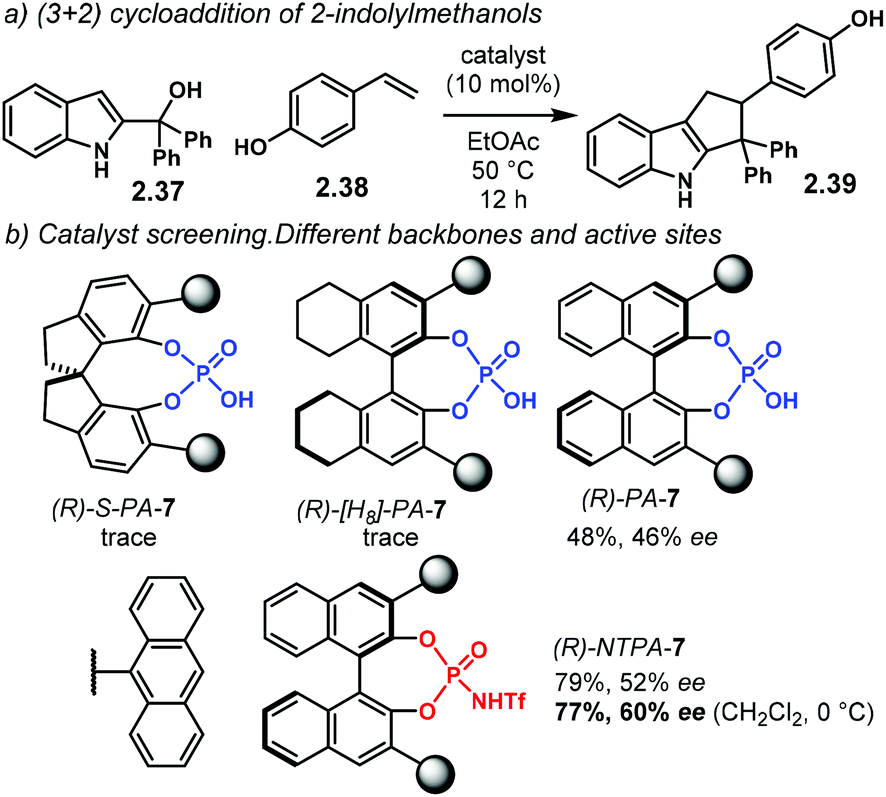 | ||
| Scheme 15 (a) Reaction developed by the group of Shi. The absolute configuration of the new chiral centre in the products was not determined. (b) Catalyst screening. Different, albeit related, chiral backbones can have important and critical effects in reactivity and stereocontrol. | ||
A proposed reaction mechanism starts with protonation of the hydroxyl group in indole 2.37 (Scheme 16). Subsequent dehydration delivers carbocationic intermediate 2.40, which may be stabilised through ion pairing or an H-bond with the NTPA anion. Addition of the hydroxystyrene component 2.38 seems to occur through a bifunctional activation mode, 2.41. The catalyst anchors both reactants through H-bonds. Control experiments showed that the free N–H and O–H moieties are crucial for reactivity. In addition, a bifunctional activation mode provides a rigid chiral environment for enantioinduction. However, the authors did not determine the absolute stereochemistry of the product.
 | ||
| Scheme 16 Proposed reaction mechanism. A bifunctional activation mode is suggested; both free N–H and O–H moieties in the substrates were shown to be essential for reactivity. | ||
The group of Vicario designed an oxidative (4 + 3) cycloaddition sequence (Scheme 17a).67 The reaction uses in situ generated oxyallyl cations from oxidation and protonation of alleneamides 2.42 and furan 2.43. Initial catalyst screening showed PA afford practically racemic products 2.44, though with modest or good yields. (R)-NTPA-10 was found the best catalyst in terms of enantioselectivity. Substrate optimisation was further required to achieve higher yields and stereocontrol. The substituents on the nitrogen atom in the alleneamide component were tuned (Scheme 17b). It was found that an extra methoxy group in the phenyl ring enhanced the enantioselectivity at expense of a slight decrease in yield (substrates 2.42a and 2.42b). On the other hand, a Boc protecting group improved the yield (2.42c and 2.42d), as well as slightly boosting the enantioselectivity. Solvent choice was another important parameter, with ethyl acetate giving the best results. This reaction is a nice example that displays how fine-tuning of the substrates can have dramatic effects in the reaction output, despite the catalyst. In fact, for most of the cases, the substrate structure plays a crucial role. Not only the catalysts have to be screened, but the starting materials as well. It again becomes a trial and error process.
 | ||
| Scheme 17 (a) Oxidative (4 + 3) cycloaddition reaction developed by the group of Vicario. (b) Fine-tuning of the substrate is still a crucial stage when developing enantioselective methodologies. (c) Proposed reaction mechanism and activation mode. Tight ion pairing and H-bonding stabilize the oxyallyl cation intermediate. | ||
Regarding the reaction mechanism (Scheme 17c), alleneamide 4.42d is readily oxidised by DMDO to yield 2.45. Subsequent protonation and ring opening affords oxyallyl cation 2.46 as the reactive intermediate. This species is possibly stabilised by the NTPA anion. A tight ion-pair, assisted by an additional H-bond, locks the conformation for efficient chirality transfer in the upcoming cycloaddition step.
2.3. Cycloaddition reactions utilising (aza)ortho-quinone methides as reactive intermediates
ortho-Quinone methides (oQM) and aza-ortho-quinone methides (aoQM) are reactive species generated from an ortho disubstituted benzene with a potential leaving group in a benzylic position (Scheme 18). In this context, a benzylic alcohol starting material 2.47 can be protonated by a Brønsted acid. Dehydration and concomitant loss of aromaticity of 2.48, assisted by an ortho oxygen or nitrogen atom, affords (aza)oQM 2.49. These intermediates are extremely reactive and can serve as suitable partners for cycloaddition reactions. Upon treatment with a suitable alkene 2.50, the corresponding hetero-Diels–Alder-like adducts 2.51 are obtained. The possibility of building up to three contiguous stereocentres makes it a powerful method for the construction of ring systems. In addition, diastereo- and enantioselective versions are highly desirable. One of the main challenges of this approach is to stabilise the latent (aza)oQM intermediate long enough to allow reaction with a π2 system. Moreover, a rigid chiral environment has to be provided if a reaction is aimed to be enantioselective. This can be achieved by tight ion pairing with the anion of a chiral Brønsted acid. Further H-bonding is desirable in order to enhance stereocontrol. | ||
| Scheme 18 (aza)oQMs as reactive intermediates in (4 + 2) cycloaddition reactions. | ||
The group of Rueping reported one of the first examples using oQM as cycloaddition partners (Scheme 19a):99 the reaction with styrenes 2.53 afforded chromanes 2.54 in high yields with excellent diastereo- and enantiocontrol (up to >49:1 dr and up to 99% ee). Several aryl alkenes were found to be suitable substrates for this reaction. NTPA were tested straightaway as chiral catalysts due to their high acidity. BINOL-backbone (R)-NTPA-4 afforded low enantioselectivity. Further screening showed (R)-[H8]-NTPA-2 as a promising catalyst (Scheme 19b). However, no explanation was given to understand how the hydrogenated backbone performs better than the standard BINOL framework. Not only catalyst screening was important, but also the optimisation of several reaction parameters had significant effects in stereocontrol. Solvent, concentration, additives and temperature had to be optimised in order to get the best results. Catalyst screening is a time-consuming process by itself, and the optimisation of reaction conditions is also a bottleneck in discovering new asymmetric methodologies. A plausible activation mechanism is presented in Scheme 19c. Initial protonation of 2.52 by the NTPA leads to 2.55, which upon dehydration affords the corresponding oQM 2.56. H-Bonding to the catalyst and a non-classic C–H⋯OP H-bond seems to stabilise and anchor the oQM. This produces a rigid transition state which may account for the high stereocontrol. The methoxy moiety in the aromatic substituent of 2.56 helps to stabilise the oQM via inductive effects. A remarkable feature of this reaction is that the catalyst activates the electrophile exclusively.
 | ||
| Scheme 19 (a) Enantioselective synthesis of chromanes using oQMs as reactive intermediates. (b) Optimisation of the reaction conditions. Such a workflow needs screening of catalysts, solvent, concentration, additives and temperatures. This is the main bottleneck in the discovery of new enantioselective reactions. (c) Suggested activation mode. The oQM is locked to the catalyst active site through H-bonding and non-classic C–H⋯OP interactions. Exclusive activation of the oQM is achieved. | ||
A related (4 + 2) cycloaddition utilising aoQMs, also called methide imines, was later reported. With o-amino benzylic alcohols 2.57, the group of Shi developed the reaction using o-hydroxystyrenes 2.58 as the alkene component (Scheme 20a).100 The reaction affords tetrahydroquinolines 2.59 with moderate yields and good enantiocontrol. The products are obtained as single syn-diastereoisomers (up to >95:5 dr). (R)-NTPA-26 gave the best results in terms of yield and enantioselectivity. The analogous PA showed similar reactivity and comparable stereocontrol. During optimisation, it was found that the free –OH moiety in 2.58 was essential to achieve high enantioselectivities. Nevertheless, styrenes lacking the hydroxyl group were also suitable substrates for the reaction, albeit with diminished enantiocontrol.
 | ||
| Scheme 20 (a) Enantioselective (4 + 2) cycloaddition reported by the group of Shi. (b) Reaction mechanism to afford stabilised aoQMs. (c) Bifunctional activation mode; the catalyst locks both substrates into the chiral cavity. | ||
For all the substrates scoped, the reaction yields remarkable diastereocontrol. This strongly points towards a concerted mechanism. A suggested mechanism starts with protonation of o-amino benzylic alcohol 2.57 (Scheme 20b). Subsequent dehydration of 2.60 renders aoQM 2.61. It is suggested that the protonated form of the NTPA stabilises the intermediate through H-bonding. For the case of o-hydroxystyrenes, a bifunctional activation mode 2.62 is very likely to be operational (Scheme 20c). Simultaneous H-bonding with both substrates establishes a rigid chiral environment to account for the high stereocontrol. On the other hand, mono activation of the aoQM occurs when the styrenes lack the free –OH motif.
The same year, the group of Rueping reported a similar reaction with amino alcohols 2.63 which does not need a free –OH group in the styrene (Scheme 21a).101 The more sterically demanding S-NTPA-11 delivered N-methyl tetrahydro-quinolines 2.64 as single diastereoisomers with excellent yields and enantiocontrol. PA, as well as BINOL and [H8]-BINOL-derived NTPA, were not effective catalysts for this reaction.
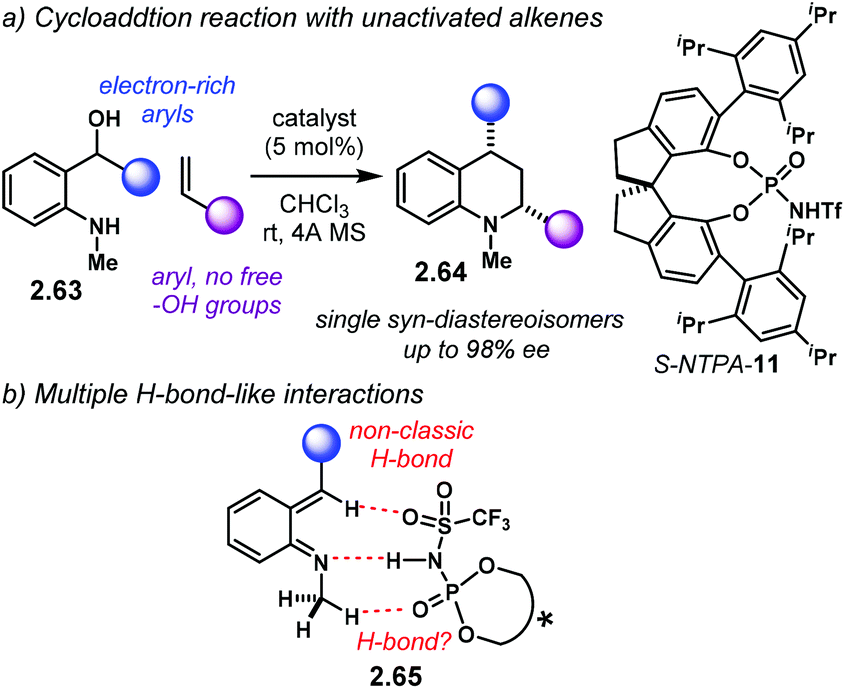 | ||
| Scheme 21 (a) (4 + 2) cycloaddition of aoQM reported by the group of Rueping. (b) Suggested activation mode of the aoQM; several H-bond and H-bond-like interactions engage to activate and stabilise the intermediate. | ||
Because only the aoQM is activated, several interactions have to be taking place in order to create a rigid chiral environment in 2.65 (Scheme 21b). H-Bonding with the N–H moiety in the catalyst's active site, as well as non-classic C–H⋯O H-bond-like interactions with the phosphoryl and sulfonyl oxygens are suggested by the authors. In addition, the electron-rich substituent in the aoQM might be playing an important role stabilising this intermediate. Altogether, these numerous interactions create a rigid chiral environment for the alkene to approach the activated aoQM. The high levels of diastereocontrol are indicative of a concerted mechanism. Presumably, the tighter chiral cavity created by the SPINOL-backbone promotes better enantiocontrol.
A complementary, intramolecular methodology to construct enantioenriched, fused heterocycles was later reported by the group of Schneider (Scheme 22).102 This reaction uses substrates 2.66 which generates aoQMs in situ. The tethered alkene to the N-substituent affords polycyclic products 2.67 as single syn-diastereoisomers in good yields and with high enantiocontrol (Scheme 22a). Moreover, vinyl ethers 2.68 grant access to hemi-aminals 2.69 in good yields, with good enantiocontrol and anti-diastereoselectivity (Scheme 22b). While PA gave low conversions, NTPA showed enhanced reactivity, with (R)-NTPA-7 achieving the best enantiocontrol.
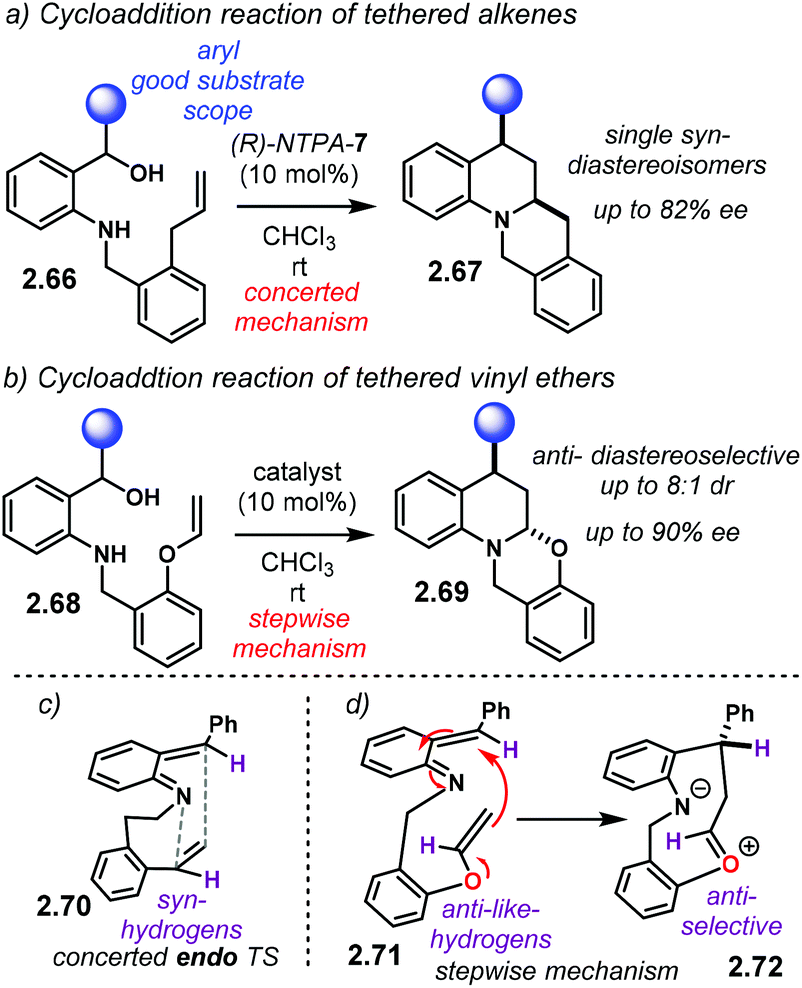 | ||
| Scheme 22 Intramolecular aza-Diels–Alder reaction using aoQMs as reactive intermediates. (a) With tethered alkenes. (b) With tethered vinyl ethers. (c) Concerted mechanism for tethered alkenes, accounting for syn-stereochemistry. (d) Stepwise mechanism for tethered vinyl ethers, accounting for anti-stereochemistry. | ||
Generation and activation of the aoQM proceeds as already discussed in Schemes 20 and 21. Multiple H-bond-like interactions might be taking place as well. It is suggested that substrates 2.66 cyclise via an endo TS 2.70 (Scheme 22c). Such a TS sits the highlighted H-atoms syn-to each other to account for relative stereochemistry. The high diastereocontrol strongly points to a concerted mechanism. On the other hand, and supported with control experiments, aoQM 2.71 reacts in a stepwise fashion (Scheme 22d). Initial attack by the enol ether delivers intermediate 2.72. Transannular attack to the oxy-carbenium ion in an anti-selective way affords anti-products 2.69. However, the authors did not account for the anti-preference in the second transition state. Yet, a pericyclic mechanism with thermodynamic epimerisation is also possible.
3. Pericyclic reactions II: Electrocyclisations, polyene and related cyclisations
Electrocyclic reactions are another powerful method for the rapid construction of ring systems, as they offer the possibility of building up complex molecules out of linear precursors. In addition, their stereospecific nature makes them suitable for developing asymmetric methodologies. Even though electrocyclisations are generally thermal reactions, Lewis or Brønsted acid catalysis can also trigger them.27,103In the past years, chiral Brønsted acids have been suitable catalysts for reactions that render cyclic products with high enantioselectivities. Amongst the most important, useful and versatile acid-catalysed electrocyclisations is the Nazarov reaction (Scheme 23a). In an oversimplified fashion, a divinyl ketone 3.1 is activated, whether through protonation or a strong H-bond to the carbonyl group, in order to access 4π system 3.2. Subsequent conrotatory electrocyclisation delivers cationic intermediate 3.3. Hydrogen abstraction leads to enol 3.4, which is then protonated to afford cyclopenten-2-ones 3.5. It is well known that the substitution pattern in the vinyl moieties of the ketone has important consequences in the overall reaction. In addition, chiral centres can be introduced either enantioselectively or diastereoselectively.
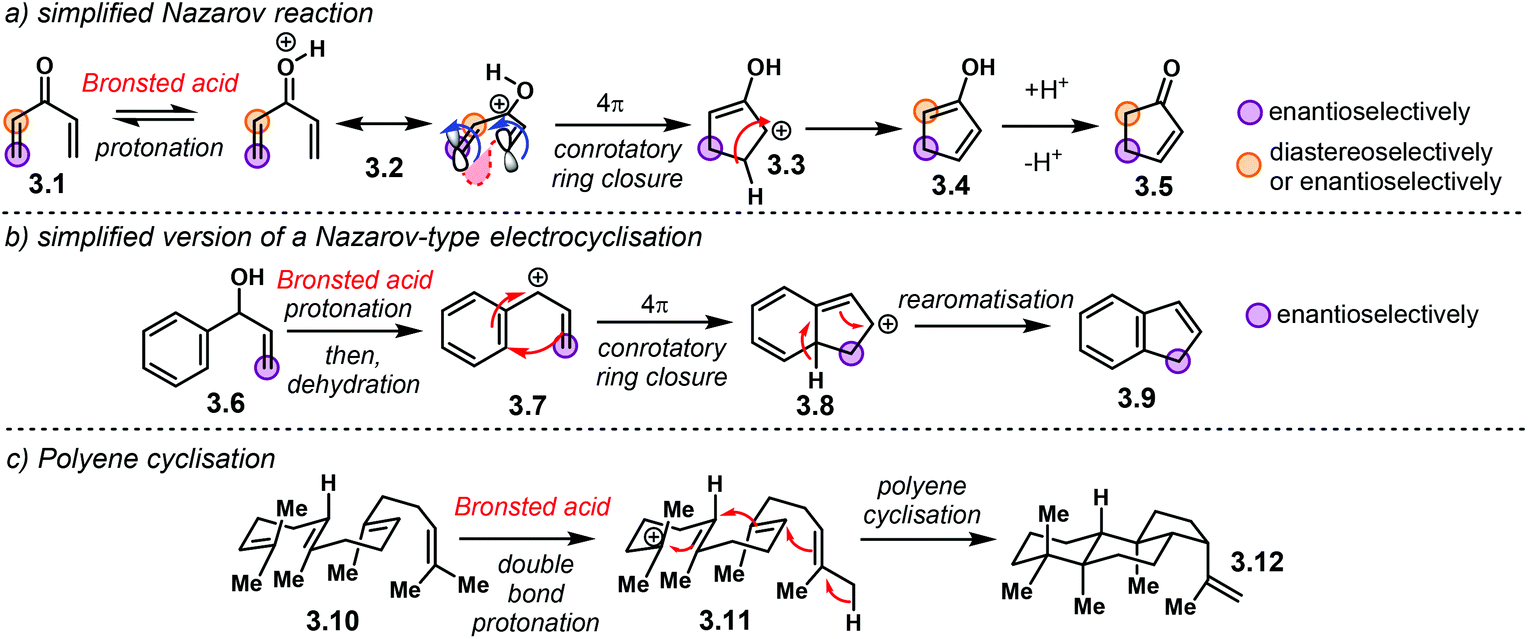 | ||
| Scheme 23 Electrocyclic reactions activated by a Brønsted acid. (a) Simplified Nazarov reaction. (b) Nazarov-type electrocyclisation. (c) Polyene cyclisation. | ||
A similar electrocyclisation process involves a protonation–dehydration sequence of a benzylic alcohol 3.6 to generate a reactive carbocation 3.7 (Scheme 23b). Conrotatory cyclisation affords intermediate 3.8, which, after hydrogen loss, furnishes the indene core structure 3.9. Electron-donating motifs in the aromatic ring are needed both to stabilize the intermediate carbocationic structures as well as to promote the electrocyclisation step.
Another useful method to synthesise multiple fused ring systems are polyene cyclisations (Scheme 23c). In an illustrative fashion, controlled protonation of a double bond in 3.10 triggers a cyclisation cascade of carbocation 3.11 to furnish complex structure 3.12. The possibility to install several stereogenic centres in a single step makes this reaction an appealing candidate for a catalytic and enantioselective version.
In the aforementioned examples, initial protonation delivers a reactive cationic intermediate that can readily cyclise. Therefore, if the conjugate base of the Brønsted acid can engage in tight ion pairing, H-bond or other locking interactions, cyclic molecules can be made enantioselectively. As we will see, several asymmetric methodologies involving these three groups of reactions have been developed in the past years, using NTPA as chiral catalysts.
Most of the computational work done to understand enantioselectivity in NTPA-catalysed transformations has been conducted in these types of reactions.103,104 Mechanistic details regarding enantioinduction models will be covered in section 10. In the current section, we summarise the advances over the past ten years using NTPA as catalysts in enantioselective electrocyclic reactions, briefly discussing the reaction mechanisms and the activation modes.
3.1. Nazarov reactions
The Brønsted acid-catalysed enantioselective Nazarov reaction has been widely studied by the group of Rueping. The seminal works on NTPA-catalysed Nazarov cyclisations date back from 2007 and 2009 (Scheme 24).28,35 These reactions have already been discussed in detail in the review comprising NTPA-catalysed reactions from 2006 to early 2011.27 In the first example, dienones 3.13 are activated through protonation or H-bond to the carbonyl group. The full Nazarov reaction afforded cis- and trans-cyclopenten-2-ones 3.14 in good yields and excellent enantioselectivities (Scheme 24a). | ||
| Scheme 24 Seminal works from the group of Rueping regarding NTPA-catalysed Nazarov reactions. | ||
After the initial publication, later, in 2009, the same group took advantage of the NTPA-catalysed Nazarov reaction to develop an electrocyclisation/enantioselective protonation sequence of dienones 3.15 (Scheme 24b). Even though the conrotatory electrocyclisation is not involved in the enantiodetermining step, decent levels of enantiocontrol were observed for cyclopenten-2-ones 3.16.
Generally, NTPA-catalysed enantioselective Nazarov cyclisations follow the reaction mechanism and activation mode outlined in Scheme 25. Dienone 3.17 is protonated by the highly acidic catalyst. Intermediate 3.18 is locked and stabilised inside the catalyst's chiral cavity through H-bonding. Further stabilisation might arise from ion pairing. 4π electrocyclisation settles carbocation 3.19, enantioselectively. This intermediate may gain extra stability by the adjacent electron-donating group. Deprotonation by the conjugate base of the NTPA regenerates the catalyst and delivers enol 3.20. Subsequent diastereoselective protonation yields cyclopenten-2-ones anti-3.21 and syn-3.21. Diastereoselection levels depend on the stereoelectronics of the substrate as well as the catalyst chiral pocket.
 | ||
| Scheme 25 Enantioselective NTPA-catalysed Nazarov cyclisation. Reaction mechanism and activation mode. | ||
Later in 2011, the group of Rueping reported an electrocyclisation–bromination sequence (Scheme 26).105 Regarding intermediate 3.20 (cf.Scheme 25) as a reactive enol, the group envisaged a way to trap it with an electrophile in a diastereoselective fashion. Starting from dienones 3.22, after the enantiodetermining electrocyclisation the enol was brominated using 3.23 as a source of bromonium ions. The reaction yields anti-3.24 as the major diastereoisomer with excellent enantiocontrol (up to 97% ee), albeit for most of the cases, the diastereoselectivities were quite low and variable (1.7:1 to 20:1 dr).
 | ||
| Scheme 26 Enantioselective Nazarov electrocyclisation – diastereoselective bromination sequence developed by the group of Rueping. | ||
The following year, the group of Rueping envisioned an enantioselective electrocyclisation of dienones 3.25 (Scheme 27a).106 The Nazarov reaction products 3.26 were subsequently treated with concentrated HCl to afford 2-hydroxy-cyclopenten-2-ones 3.27 in high yields and good enantiocontrol. The iso-butyl ether in 3.25 gave the best results for the electrocyclisation step in terms of enantioselectivity. During the development of the optimal reaction conditions, the authors performed a broad catalyst screening, using [H8]-NTPA. There are several observations regarding catalyst design, especially looking at the 3,3′ substituents (Scheme 27b). It is evident, albeit not obvious, how the stereoelectronics of the aryl substituents can have dramatic effects in enantiocontrol. For instance, catalyst [H8]-11 gave the opposite sense of enantioselectivity, even though being similar to [H8]-1, [H8]-9 and [H8]-10. These last two catalysts gave practically the same enantioselectivity (56% ee and 57% ee, respectively) even when they have different stereoelectronic properties. This observation might be indicative that a para substituent might not be that essential to improve stereocontrol. The sterically different [H8]-6, with a 3,5-disubstitution pattern, slightly improved enantioselectivity (60% ee). These observations point to ortho substituents being crucial to increase the enantiomeric excess. Such increase was indeed achieved by the more sterically demanding [H8]-3, [H8]-2 and [H8]-15. Catalyst [H8]-4 afforded the best enantioselectivity (89% ee). In summary, catalyst optimisation is still the bottleneck when developing asymmetric methodologies.
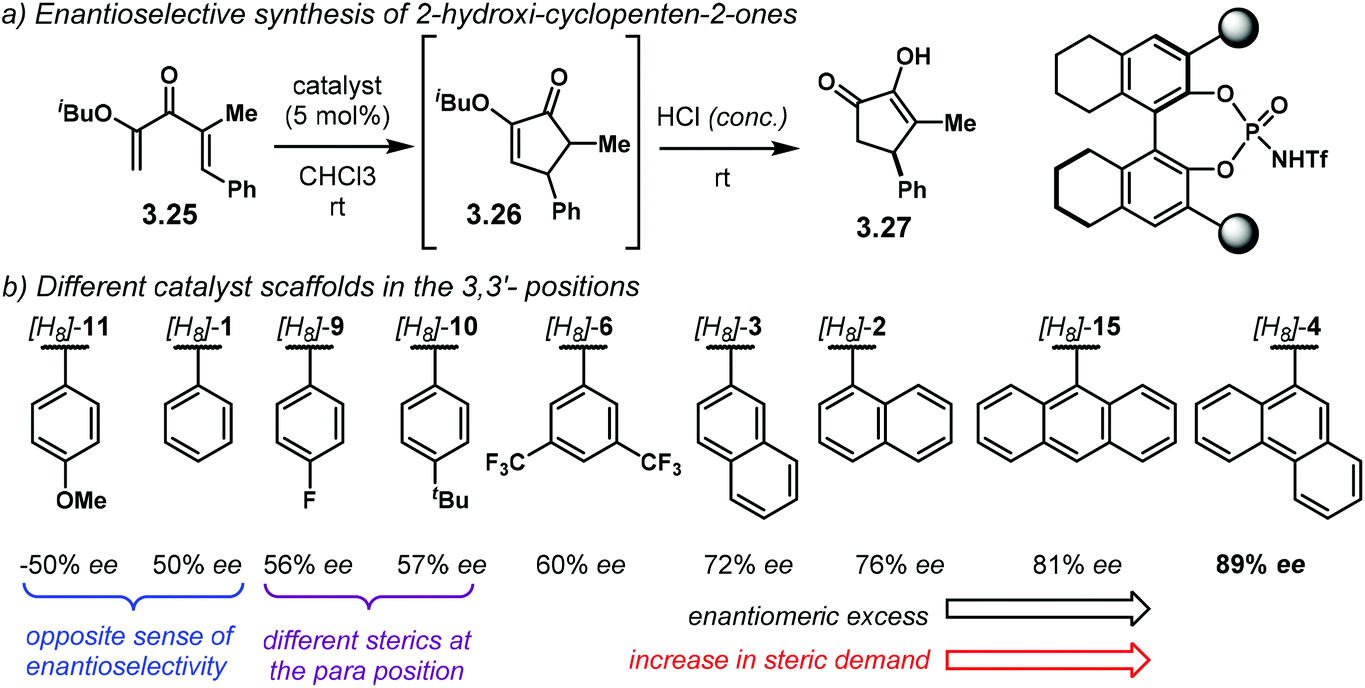 | ||
| Scheme 27 (a) Asymmetric Nazarov cyclisation. (b) Catalyst screening: fine-tuning of the catalyst scaffolds is still the bottleneck to improve enantiocontrol. | ||
Building up in the previous methodology, the group of Flynn designed an elegant, enantioselective formal synthesis of (+)-roseophilin 3.28 using an enantioselective Nazarov reaction as the key step (Scheme 28).107 Reductive coupling of fragments 3.29 and 3.40 lead to key intermediate 3.41. Nazarov cyclisation using catalyst (R)-NTPA-7 followed by acid treatment afforded advanced intermediate 2.42 in 91% yield and 82% ee. Further steps to install the macrocycle and the pyrrole motif furnished core structure 3.43 as a formal intermediate towards 3.28. This work represents a neat example of NTPA being used in enantioselective syntheses of natural products.
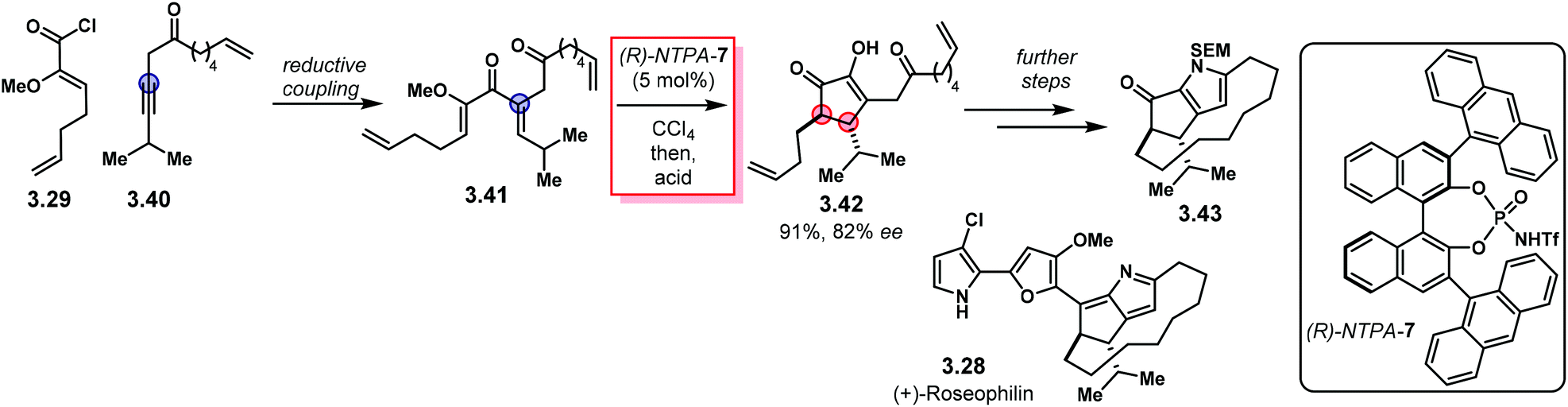 | ||
| Scheme 28 NTPA use in syntheses of natural products. Towards a formal synthesis of (+)-roseophilin. | ||
Later, on 2014, the group of Tius expanded the scope of NTPA-catalysed enantioselective Nazarov cyclisations.108 Using (R)-NTPA-14, electrocyclisation of 3.44 takes place to yield cyclopenten-2-one 3.45 as a single diastereoisomer with excellent enantiocontrol (Scheme 29a). This is a remarkable reaction, as it grants access to two contiguous all-carbon quaternary stereocentres. NTPA were needed due to their high acidity to activate the substrates. On the other hand, PA did not promote the reaction at all. The reported substrate scope showed this methodology to be effective even for the synthesis of highly congested cyclopenten-2-ones (Scheme 29b). Sterically demanding products 3.46 and 3.47 were obtained in good yields with excellent enantiocontrol (96% ee and 84% ee, respectively). Products 3.48 and 3.49, bearing a fused cyclohexane ring, were obtain with excellent enantiocontrol as well (98% ee and 94% ee, respectively). Modest enantiomeric excess was achieved for the fused seven-membered ring product 3.50 (58% ee), presumably due to its larger size.
 | ||
| Scheme 29 (a) Asymmetric Nazarov reaction yielding vicinal all-carbon quaternary stereocentres. (b) Selected examples from the substrate scope. | ||
The activation mode for substrates like 3.44 follows the mechanism proposed in Scheme 30, similar to the one previously discussed in Scheme 25. During the reaction optimization, the highest enantioselectivities were obtained in solvents with a low dielectric constant. This strongly suggests that an ion-pair 3.51 is the reactive complex in the enantiodetermining step. After the cyclisation step, 2-tert-butylphenol was required to scavenge the benzhydryl motif in 3.52.
 | ||
| Scheme 30 Proposed reaction mechanism for the enantioselective Nazarov cyclisation reported by the goup of Tius. | ||
Later, in 2017, the same group reported a full account of the enantioselective Nazarov reaction.68 Several reaction parameters, catalysts and substrate substituents were screened. Such a fully detailed study is beyond the scope of this review and we leave it to the interested reader.
Coupling two reaction mechanisms into a single transformation serves as an elegant method to build up molecular complexity. In this context, the group of Tu developed an enantioselective Nazarov cyclisation/semipinacol rearrangement sequence (Scheme 31a).109 This method allows dienones 3.53 to afford spiro[4.4]nonane-1,6-diones 3.54 as single diastereoisomers in good yields and with excellent enantiocontrol. The reaction works well with a wide variety of electron-rich or electron-poor aryl groups. Perhaps the main drawback of this methodology is that the substrate scope is limited to the expansion of four-membered rings. Nevertheless, up to four contiguous stereocentres can be installed with remarkable stereocontrol (Scheme 31b). Tricyclic compounds 3.55 and 3.56 were obtained with good enantioselectivities (84% ee and 91% ee, respectively).
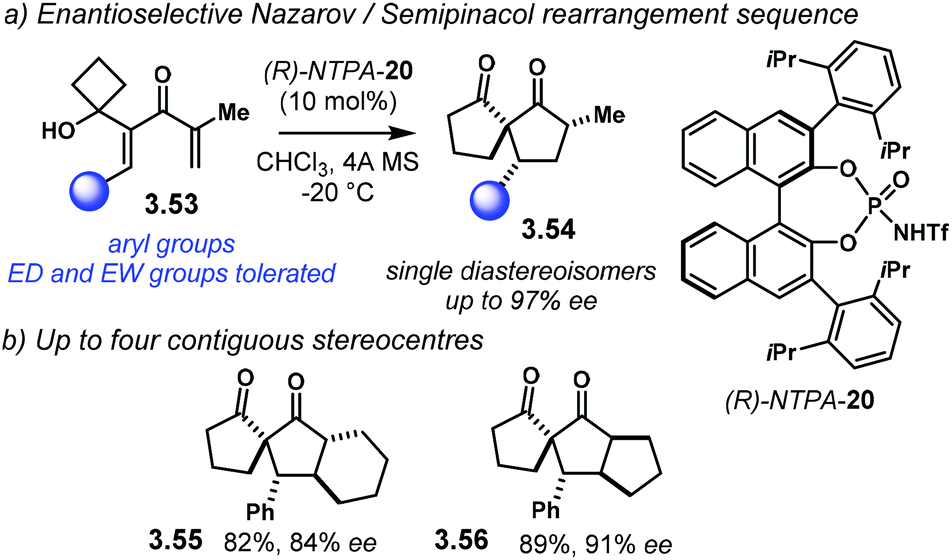 | ||
| Scheme 31 (a) Sequential enantioselective Nazarov/semipinacol rearrangement. (b) Up to four contiguous stereocentres can be obtained, one of them bearing an all-carbon quaternary one. | ||
The reaction mechanism of this sequence is summarised in Scheme 32. Protonation of 3.53 at the carbonyl group leads to cationic intermediate 3.57. The authors propose that this complex is locked within the catalyst chiral pocket through H-bonding. Enantioselective conrotatory cyclisation furnishes cation 3.58 with the first chiral centre installed. Then, the semipinacol rearrangement takes place. The chiral centre in 3.58 has the bulky substituent at the back, blocking such a face. Therefore, diastereoselective 1,2-migration occurs on the unhindered face to place the spirocyclic system in 3.59. Forward protonation delivers product 3.54.
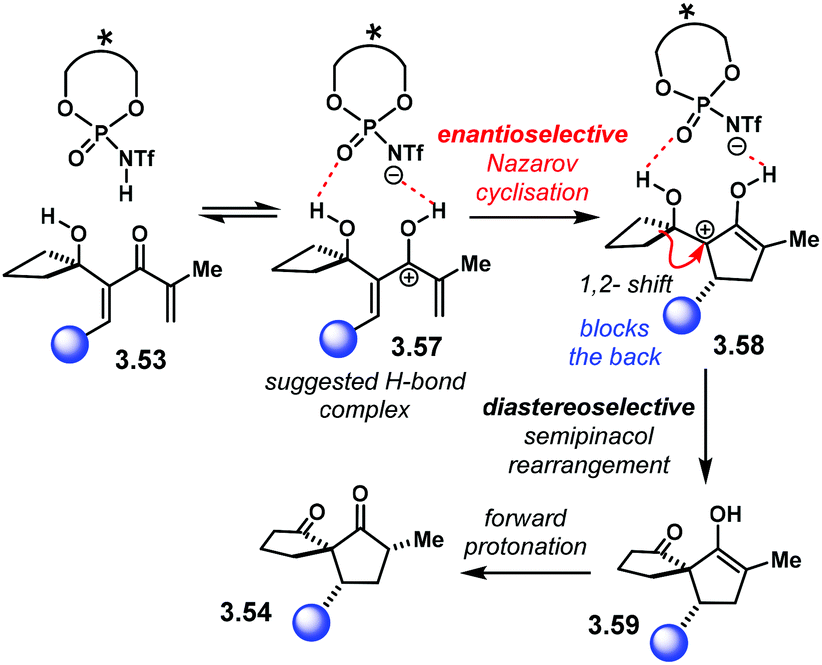 | ||
| Scheme 32 Proposed reaction mechanism and activation mode for the sequential Nazarov cyclisation/semipinacol rearrangement. | ||
DFT calculations were in agreement with the observed sense of enantioinduction.103 They also showed the electrocyclisation to be the rate-determining step. The 1,2-shift activation free energy barrier in the semipinacol rearrangement was calculated to be only 8.2 kcal mol−1, much lower than the electrocyclisation one (24.4 kcal mol−1). In addition, the free energy barrier difference between the two possible semipinacol 1,2-shifts was calculated to be 4.8 kcal mol−1. This is in agreement with the high levels of diastereocontrol.
3.2. Other 4π conrotatory electrocyclisations
Another methodology to synthesise cyclopenten-2-ones in an asymmetric fashion was developed by the group of Rueping. This protocol uses an aza-Piancatelli rearrangement of furan-2-yl methanols 3.60 and substituted anilines 3.61 to furnish products 3.62 with high stereocontrol (Scheme 33a).110(R)-[H8]-NTPA-14 gave the best results in terms of enantiocontrol. PA were suitable catalysts for the reaction, affording good yields but poor enantioselectivity. The reaction stats with protonation of 3.60 followed by dehydration (Scheme 33b). Then, aniline 3.61, possibly assisted by the conjugate base of the NTPA, attacks cationic intermediate 3.63. Ring opening of adduct 3.64 delivers 4π system 3.65, which readily undergoes enantioselective electrocyclisation towards product 3.66. The authors propose that the enantiodetermining step is triggered by mono activation of 3.65; however, a single H-bond may be too flexible to afford the high enantiomeric excesses observed. Therefore, we believe that a dual activation mode, through multiple H-bonds might be more likely. | ||
| Scheme 33 (a) Enantioselective Piancatelli rearrangement/electrocyclisation sequence. (b) Reaction mechanism and activation mode. (c) Extract from the substrate scope. | ||
As shown in Scheme 33c, this methodology is suitable for a wide variety of substrates. It is not limited for the aforementioned starting materials. Compound 3.67 was obtained with excellent enantiocontrol. Substitution in the aromatic ring from the furan component is also tolerated, both for electron withdrawing groups, like 3.68 and for electron donating groups, like in 3.69. Furthermore, products 3.70 and 3.71 bearing an all-carbon quaternary stereocentre were afforded with excellent enantioselectivity, albeit with modest diastereocontrol.
Following a protonation–dehydration strategy, the group of Chan developed a creative Nazarov-type electrocyclisation (Scheme 34a).104 In this transformation, starting materials 3.72 yield enantioenriched 1H-indenes 3.73via intermediate 3.74. When PA were tried as catalysts, the reaction did not proceed at all. The more acidic NTPA achieved the desired products and (S)-NTPA-16 was found to provide the highest enantioselectivities. The reaction worked well with a wide variety of substrates, in good yields and with good enantiocontrol, not only limited to alkynes (Scheme 34b). 1H-Indene 3.75, with an electron-deficient aryl, was obtained with excellent enantiocontrol (94.5% ee). Substitution in the alkyne motif is also tolerated. Compound 3.76 with an electron-poor aryl in the alkyne was obtained with excellent enantioselectivity (94% ee). This was also the case for the thiophene analogue 3.77 (88% ee). Substituted thiophenes were also suitable starting materials. Products 3.78 and 3.79 were obtained with very good enantiocontrol (88% ee and 89% ee, respectively).
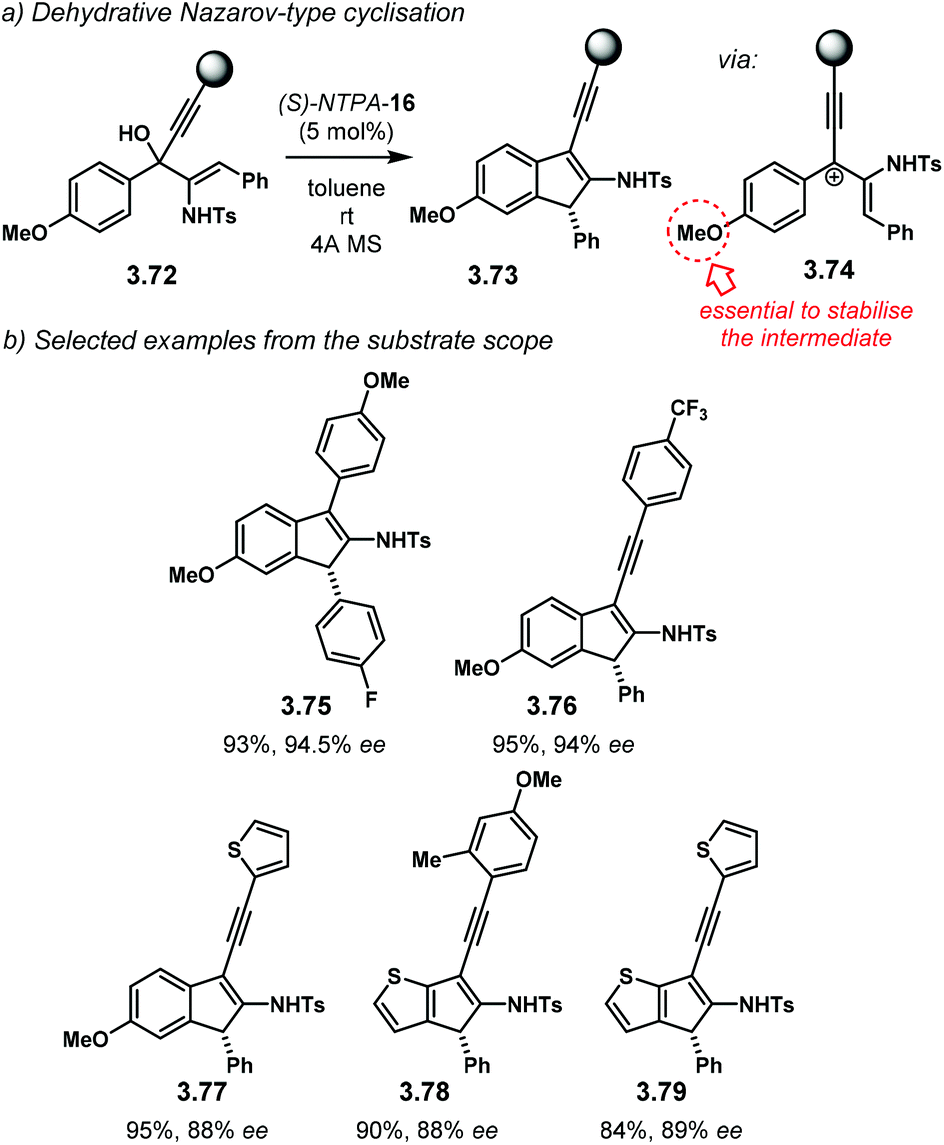 | ||
| Scheme 34 (a) Nazarov-type cyclisation developed by the group of Chan. An electron-rich substituent in the starting material was found to be essential to stabilise the carbocationic intermediate. (b) Selected examples from the substrate scope. | ||
The reaction mechanism, as well as the activation mode, were thoroughly investigated through detailed DFT calculations. In Scheme 35, we present a rather simplified but concise version. Key intermediate 3.80 is stabilised by the conjugate base of the NTPA through H-bonding and a tight ion-pair The N–H moiety was found to be essential in order to direct the catalyst to one face of the planar intermediate. Conrotatory electrocyclisation affords carbocation 3.81. This was found to be the favoured conformation as the hydrogen atom from the benzene ring points up. Such conformation allows catalyst regeneration as well as re-aromatisation to yield product 3.82.
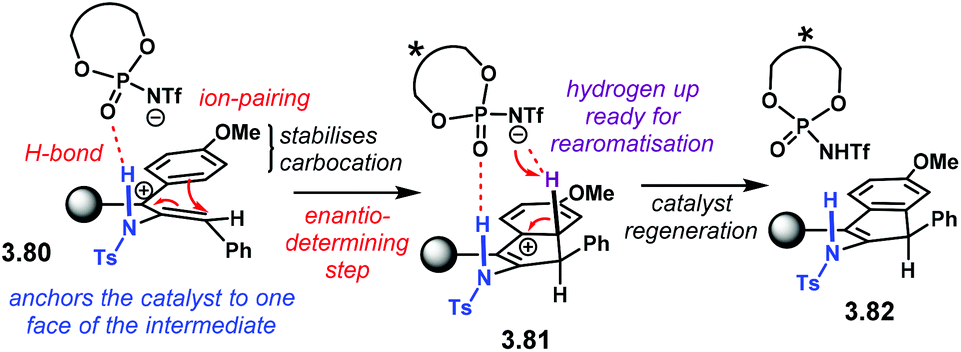 | ||
| Scheme 35 Proposed reaction mechanism and activation mode for the dehydrative Nazarov-type cyclisation. | ||
The same group extended the former methodology to include a pyrrole motif in a subsequent step (Scheme 36).111 Therein, after the NTPA-catalysed reaction, Au(I) catalysis was used to afford pyrroles 3.83 in an elegant one-pot strategy. This second step was found to proceed without erosion of enantioselectivity in the already formed chiral centre.
 | ||
| Scheme 36 One-pot, sequential synthesis of enantioenriched pyrroles. | ||
3.3. Polyene and related electrocyclisations
An exquisite method to construct fused carbocycles was reported by the group of Zhao (Scheme 37a).112 Starting from aldehydes 3.84 and tosylamine, the group developed an asymmetric Brønsted acid-catalysed cyclisation to obtain 3.85 as single diastereoisomers with excellent enantiocontrol in a single step. The reaction proceeds through the intermediate imine 3.86. PA were also tried but gave low yields and enantioselectivities. (R)-NTPA-7 showed the best for reactivity and enantiocontrol. An electron-rich aryl motif in the starting material was found to be important for reactivity. Strong electron-withdrawing groups did not promote the reaction. The authors did not discuss the reaction mechanism in detail; however, we proposed one, with its corresponding activation mode (Scheme 37b). Imine 3.87 is very likely to be protonated by the catalyst, yielding iminium ion 3.88. This iminium is electrophilic enough to trigger the cyclisation step. A mode of activation through several H-bonding is likely to be operative. This can easily provide a rigid transition state within the catalyst chiral pocket. A chair–chair conformation in the transition state with a pseudoequatorial iminium motif might explain the observed high stereocontrol. Furthermore, this conformation leaves a pseudoaxial H atom in 3.89 ready to be taken by the catalyst's counteranion in the re-aromatisation step. Catalyst regeneration in that same step affords product 3.90. A full computational study would be extremely useful in order to explain enantioselectivity, as well as to figure out how the trans-decaline system fits inside the chiral cavity. | ||
| Scheme 37 (a) Polyene cyclisation developed by the group of Zhao. (b) Plausible reaction mechanism suggested by the group of Goodman. We do not want to state that this is the reaction mechanism or activation mode. It is just a suggestion based on what we know about PA and NTPA-catalysed reactions. (c) A short total synthesis of (−)-ferruginol. | ||
In the same publication, the authors showed off the power of their newly developed methodology in an asymmetric total synthesis of (−)-ferruginol 3.93 (Scheme 37c). The key step in the synthesis highlights cyclisation of aldehyde 3.91 with tosylamine catalysed by (R)-NTPA-7. This step furnishes key intermediate 3.92 in 76% yield and 92% ee. Further steps afforded (−)-ferruginol 3.93 in 97% ee. This method uses an iminium ion as an electrophile to trigger the enantioselective step. Therefore, imines can also serve as suitable substrates for NTPA-catalysed addition reactions.
Before changing gears to other types of NTPA-catalysed reactions, we want to finalise this section with another reaction reported by the Samanta and Yamamoto. This comprises two closely related enantioselective bromonium-induced polyene cyclisations (Scheme 38).113
 | ||
| Scheme 38 Enantioselective bromocyclisation of polyenes, developed by Samanta and Yamamoto. (a) Homogeranyl benzene derivatives as substrates. (b) Geranylphenols as substrates. (c) Activation of the bromonium source and plausible activation mode. | ||
Despite the fact that the methodology does not uses an NTPA as a catalyst, a closely related thio-triflylphosphoramide was found to be the best catalyst for these reactions. In the first example (Scheme 38a), homogeranyl benzene derivatives 3.94 react with dibromo–dimethyl–hydantoine, 3.95 (DBDMH). After treatment with chlorosulfonic acid, bromocyclisation products 3.96 are obtained in good yields and excellent enantioselectivities, up to 94% ee. Presumably, excellent diastereocontrol is also involved, though not explicitly reported. Geranylphenols 3.97 (Scheme 38b) were also suitable substrates for the bromonium–polyene cyclisation cascade. This reaction provides access to complex bromo benzo-heterocycles 3.98 in good yields and remarkable enantiocontrol, up to 98% ee. These products are obtained as single diastereoisomers.
So far, we have seen NTPA functioning as highly efficient catalysts, mainly, due to its increased acidity. However, in this latter reaction, the authors suggest that the Lewis basicity of the catalyst plays an essential role. The basicity, then, was enhanced by the introduction of the softer sulfur atom into the catalyst active site. Therefore, the authors propose that Lewis basicity is important to activate the bromonium source through 3.99 (Scheme 38c). It is possible, although not fully discussed in the article, that 3.100 will be actual catalyst. In that case, bromination of the double bond in the substrate would deliver an intermediate akin to 3.101. Herein, the nucleophilic component could be directed by the catalyst counter anion. This concept of Asymmetric Counter-anion Directed Catalysis is widely evoked by the group of List.6 A chair–chair transition state, similar to 3.88 (cf.Scheme 38) may be operational in this process.
4. Addition reactions to imines
Nucleophilic additions to imines are, perhaps, the most widely exploited and studied reactions using chiral PA as Brønsted acid catalysts.21 The high basicity of the nitrogen atom in aldimines or ketimines 4.1 makes them suitable substrates to be activated by PA or NTPA (Scheme 39). Herein, the acidity of the catalyst plays a key role in substrate activation. Whether H-bonding 4.2 or full protonation to form an iminium ion 4.3 are plausible activation modes for PA (Scheme 39a). An equilibrium between the H-bond and protonated species might also be in play. However, full protonation is more likely to be the activation mode of imines if NTPA are used as catalysts (Scheme 39b, 4.4).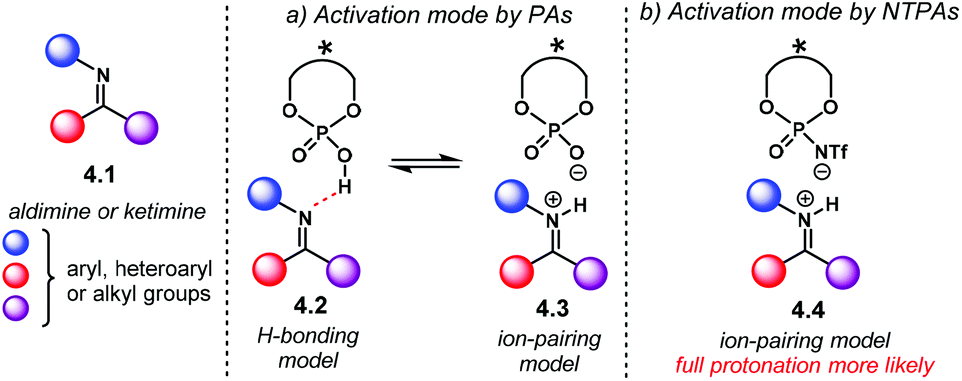 | ||
| Scheme 39 Activation modes for imines. (a) H-bonding or full protonation is likely to be operating with PA. (b) Tight ion pair model for iminium ions and NTPA. | ||
Another important feature comes regarding the incoming nucleophile in an addition reaction. If the attacking substrate can form an H-bond with the catalyst active site, a bifunctional activation mode can occur (4.5, Scheme 40). This is likely to happen for both PA and NTPA. Regarding PA, several computational studies done by our group show that a bifunctional activation mode is preferred in reactions of imines and H-nucleophiles.59,114–117 Yet, little is known about NTPA catalysing addition reactions to imines. However, a bifunctional activation mode is equally plausible, as shown in 4.6. In addition, the triflyl motif can engage in further non-covalent interactions due to its heteroatom-rich active site.
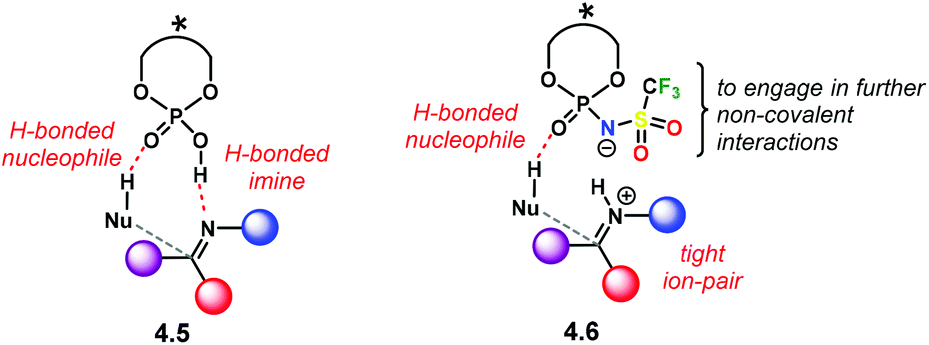 | ||
| Scheme 40 Bifunctional activation modes of chiral PAs and NTPAs. | ||
This section of the review focuses on enantioselective addition reactions to imines using NTPA as chiral Brønsted acids. Despite most of these transformations can be successfully achieved using PA, for some substrates and reactions a more acidic catalyst was required, whether to enhance reactivity or to provide higher levels of stereocontrol.
4.1. Aza-Darzens reactions
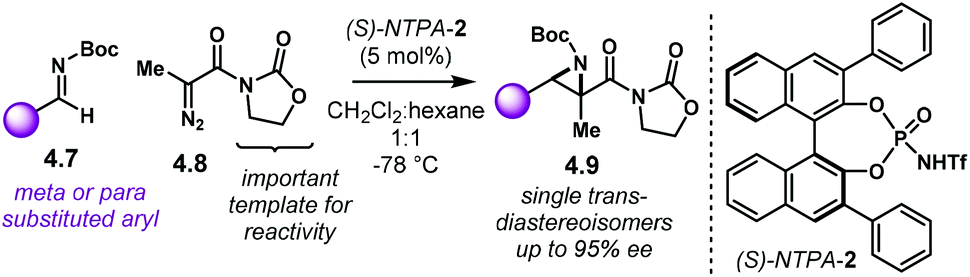
Aziridines are an important family of chiral building blocks, mostly used in the synthesis of enantioenriched amines.118 Therefore, asymmetric methodologies to obtain these three-membered heterocycles are highly desirable.119–121 In this context, the aza-Darzens reaction utilises imines as starting materials to build up the aziridine core.The group of Maruoka reported a versatile enantioselective aza-Darzens reaction (Scheme 41).122 Starting from aldimines 4.7, the reaction with α-diazo compounds 4.8 yields chiral trisubstituted aziridines 4.9 as single trans-diastereoisomers with good enantiocontrol. (S)-NTPA-2 was selected as catalyst for this transformation. The high levels of diastereo- and enantiocontrol achieved in this reaction are remarkable for a catalyst that does not bear heavily bulky substituents in the 3,3′ positions. The reaction works well for N-Boc aryl aldimines bearing meta and para substituents. The oxazolidinone motif in the α-diazo compound was found to serve as an important template to enhance reactivity.
 | ||
| Scheme 41 Enantioselective synthesis of aziridines from aldimines and α-diazo compounds, developed by the group of Maruoka. | ||
PA did not work for this transformation, which plausibly points to the need of the fully protonated iminium ion 4.10 for the reaction to occur (Scheme 42). Given that the α-diazo compound cannot engage in H-bonding with the counter anion of the NTPA, monoactivation of the iminium ion is very likely to be happening. In addition, the use of low polarity reaction media strongly suggests a tight ion-pair as the reactive species.
 | ||
| Scheme 42 Monoactivation mode for iminium ions. | ||
In addition, a complementary route to tertiary substituted aziridines was also included in the same publication (Scheme 43). Using highly reactive ketimino esters 4.11 and α-unsubstituted diazo compound 4.12, aziridines 4.13 are obtained as single trans-diastereoisomers with high enantiocontrol. Extensive tuning of the substrates had to be done to achieve the best reaction conditions. For instance, the N-Boc, tert-butyl ester and oxazolidinones motifs were found to be crucial. However, no detailed explanation was given. Catalyst (S)-NTPA-2 was also suitable for this transformation.
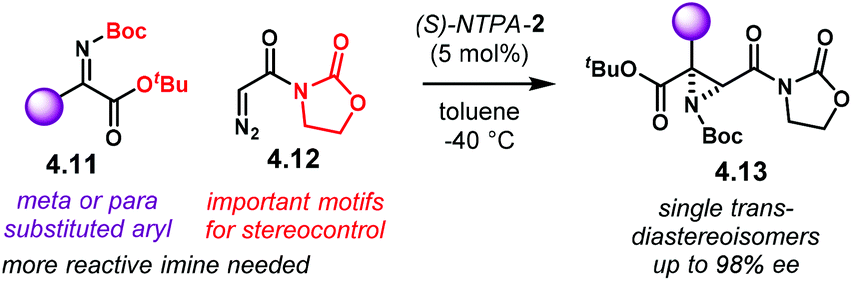 | ||
| Scheme 43 Complementary enantioselective synthesis of aziridines from ketimino esters and α-unsubstituted diazo compounds, developed by the group of Maruoka. | ||
The group of Bew developed another enantioselective aza-Darzens reaction (Scheme 44).123 As in the previously discussed aza-Darzens reaction, PA did not promote the addition at all. The more acidic (S)-NTPA-7 was found to be the best catalyst in terms of reactivity and enantiocontrol. Therein, imines 4.14 react with α-diazo ester 4.15 to furnish cis-aziridines 4.16 as single diastereoisomers with excellent enantiocontrol. The reaction tolerates a wide variety of aryl and heteroaryl aldimines. The bulky 2-tert-butoxyphenyl N-protecting group was necessary to achieve withstanding enantioselectivities. The low catalyst loading, 1 mol%, is also remarkable.
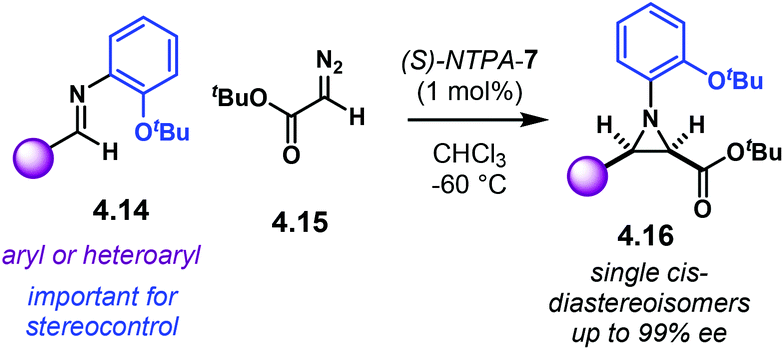 | ||
| Scheme 44 Enantioselective synthesis of aziridines from aldimines and α-diazo compounds, developed by the group of Bew. | ||
Mechanistically, imine 4.14 is protonated by the catalyst (Scheme 45). A tight ion-pair between iminium 4.17 and the NTPA anion is likely to be formed. Moreover, an intramolecular H-bond is formed with the tert-butoxy motif in the N-protecting group. This H-bond might also lock the conformation of the iminium ion and block one of the faces. This is important as the 1,2-addition step is both diastereo- and enantiodetermining, which has to occur through a monoactivation mode. With intermediate 4.18 formed, nitrogen extrusion, also possibly assisted by the NTPA anion, delivers product 4.16. The two bulky tert-butyl groups in 4.18 have to be as far away from each other to avoid steric clash in the addition step. This, as suggested by the authors, accounts for the high diastereoselectivity observed. It is, however, not entirely clear, how enantioinduction occurs.
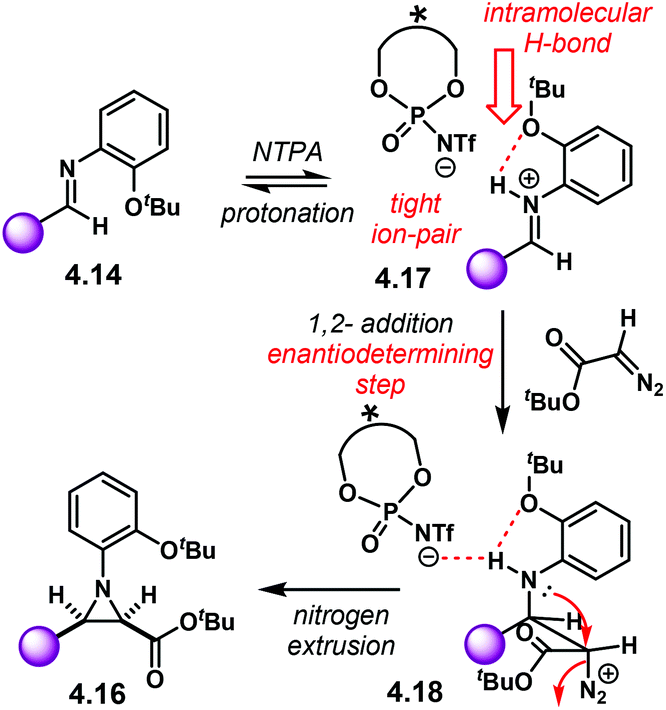 | ||
| Scheme 45 Mechanism and activation mode for the asymmetric aza-Darzens reaction reported by the group of Bew. | ||
A detailed computational study of this enantioselective aza-Darzens reaction (cf.Scheme 44) as well as the one reported by the group of Maruoka (cf.Scheme 41) would be highly desirable, both to understand how enantioselection arises, as well as to establish a model for NTPA-catalysed addition reactions to imines.
This methodology can be performed with preformed imines or in a one-pot two step sequence. This is highlighted with several examples given in the publication as well as with a short and enantioselective synthesis of (+)-chloramphenicol 4.19 (Scheme 46). The synthesis uses this new methodology as the key step to set up both of the stereocentres in the molecule. Starting with p-nitrobenzaldehyde 4.20 and aniline 4.21, the corresponding imine is generated in situ. Then, addition of 4.15 affords the key N-protected aziridine 4.22 in 99% yield and 96% ee. Further steps yield (+)-chloramphenicol 4.19.
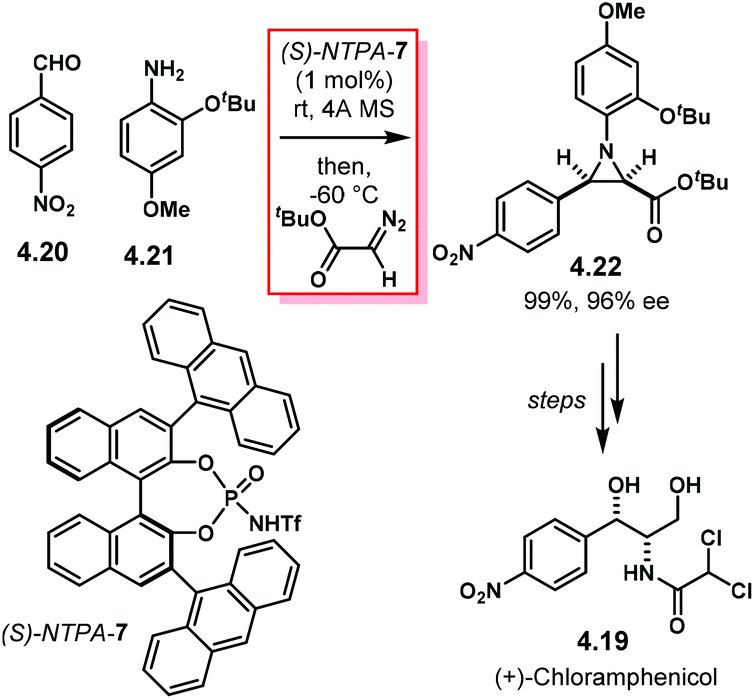 | ||
| Scheme 46 A short, enantioselective synthesis of (+)-chloramphenicol. | ||
4.2. Reduction reactions
The field of asymmetric reductions of imines is mainly dominated by PA catalysis. Such catalysts are acidic enough to activate the substrates and achieve remarkable levels of enantiocontrol. Therefore, limited examples of catalytic reduction reactions using NTPA have been reported to date.The group of Enders reported one example of an NTPA-catalysed reduction of imines (Scheme 47a).124 Therein, methyl ketimines 4.23 react with catecholborane to afford secondary amines 4.24 in modest enantioselectivities. Several parameters of this reaction were studied in detail. The benzyl N-protecting group in the imine was found to get the best reactivity-enantiocontrol balance. The substrate scope is, however, limited to aryl methyl imines. Catalyst (R)-NTPA-26 achieved the highest enantiomeric ratios. PA did catalyse the reaction affording comparable yields but with diminished enantiocontrol.
 | ||
| Scheme 47 (a) Asymmetric reduction of ketimines using catecholborane. (b) Proposed bifunctional activation mode. Herein, the NTPA–borane complex serves as the catalytic species. | ||
This fact suggests that an activation mode different from protonation and ion-pairing might be happening. The authors propose that the actual active catalyst is an NTPA–borane complex (4.25, Scheme 47b). Such complex might then serve as a Lewis acid to activate the imine. Bifunctional activation of both the imine and the reducing agent triggers the reaction. The authors also suggest that the (E)/(Z) ratio of the imine might affect the observed enantioselectivity, but this was not clarified.
4.3. Mannich–Mukaiyama reactions
The Mannich reaction is a useful method to make C–C bonds while installing an amine motif.16 The use of iminium ions in this transformation makes it suitable for an asymmetric NTPA-catalysed version. The groups of Kano and Maruoka reported an enantioselective Mannich–Mukaiyama reaction (Scheme 48a).125 This transformation also features an (E)-selective double bond isomerisation. The reaction presents (Z)-substrates 4.26 as key Mannich precursors and Mukaiyama reagents 4.27 to afford products 4.28 with excellent enantiocontrol and up to 1:20 (Z)/(E) ratios.
 | ||
| Scheme 48 (a) Asymmetric Mannich-Mukaiyama reaction reported by the groups of Kano and Maruoka. (b) Possible mechanism and activation mode. | ||
In the reaction mechanism, precursor 4.26 is protonated by the NTPA (Scheme 48b). Elimination of a BocNH2 motif in 4.29 might be assisted by H-bonding to deliver iminium ion 4.30. Isomerisation of the double bond is likely to occur at this stage of the reaction. Enantioselective 1,2-addition to the iminium ion with subsequent loss of the TMS group gets the desired product.
Zhou and Yamamoto developed an enantioselective Mannich–Mukaiyama reaction (Scheme 49).126 Using catalyst (S)-NTPA-34, they design a reaction of N-phenyl imines 4.31 and ketene silyl ether 4.32 to furnish anilines 4.33 in good yields and up to 95% ee. This transformation features a carefully designed catalyst. The nitro groups in the 3,3′ aryl substituents provide higher acidity to the catalyst's active site. Moreover, the para-methyl group serves as a steric deadlock to keep the nitro moieties orthogonal to the aryl ring. This forces the oxygen atoms in the nitro groups to point towards the active site, increasing the acidity through an intramolecular H-bond. Furthermore, such oxygen atoms may serve as extra coordinating groups to stabilise any intermediate. A computational study of this reaction would be helpful to show how the iminium ion fits into the catalyst's chiral pocket and establish a model of enantioinduction.
 | ||
| Scheme 49 Asymmetric Mannich-Mukaiyama reaction using highly acidic NTPA. | ||
4.4. Other Mannich-like reactions
The group of Jiang developed an asymmetric aza-Piancatelli/Friedel–Crafts cascade cyclisation (Scheme 50a).127 However, the enantiodetermining step features a Mannich-like addition. The reaction uses furans 4.34 and anilines 4.35 to furnish seven-membered heterocycles 4.36 with excellent diastereo- and enantioselectivity (>20:1 dr and up to 97% ee). Furthermore, the final product features three contiguous stereocentres. A wide variety of substituted furans reacted to achieve excellent stereocontrol. Substitution in the aniline component is also tolerated. (R)-[H8]-NTPA-26 was the best catalyst for this transformation. PA did not promote the reaction at all.
 | ||
| Scheme 50 (a) Asymmetric aza-Piancatelli/Friedel–Crafts cascade cyclisation developed by the group of Jiang. (b) Proposed reaction mechanism and activation mode by the Goodman group. | ||
Mechanistically, the authors did not account for the observed diastereocontrol. We suggest a mechanism as well as a plausible activation mode of the reacting species in Scheme 50b. Initially, protonation and subsequent dehydration of 4.34 yields cationic intermediate 4.37, possibly stabilised by ion pairing. Addition of aniline 4.35 furnishes hemi-aminal 4.38. Ring opening, presumably facilitated by protonation, leads to enol–iminium ion 4.39. Double H-bonding to the catalyst's active site would place the substituents in the reacting faces anti-to each other. In this case, the chiral pocket has to be tight enough to lock this conformation, accounting for the remarkable enantiocontrol and the anti-relationship. After this Mannich-like ring closure, the catalyst might still be locking intermediate 4.40, which resembles an activated α,β-unsaturated carbonyl compound. Moreover, from such conformation, it can be accounted the other observed syn-relationship. Subsequent attack of the pyrrole motif triggers the Friedel–Crafts stage of the cascade in a highly diastereoselective fashion, further leading to product 4.36.
To finalise this section we want to go into a metal NTPA-catalysed Mannich reaction, reported by the group of Smith (Scheme 51).65 This reaction uses starting material 4.41 and aldehydes 4.42 to furnish enantioenriched quinolones 4.43 as single trans-diastereoisomers. PA were tried and low reactivity was observed. On the other hand, the reaction gave higher yields, albeit with modest enantiocontrol, when the copper(II) salt of (R)-NTPA-26 was used. The dimethylamido motif on 4.41 was found to be essential for reactivity and stereocontrol, presumably, due to its coordination ability to the copper(II) centre. The reaction worked reasonably well for several aldehydes. Aryl aldehydes gave modest or low enantioselectivities, regardless the substitution pattern on the phenyl ring. Alkyl aldehydes with bulky groups were shown to improve enantiocontrol. In addition, the low catalyst loading, 1 mol%, is remarkable.
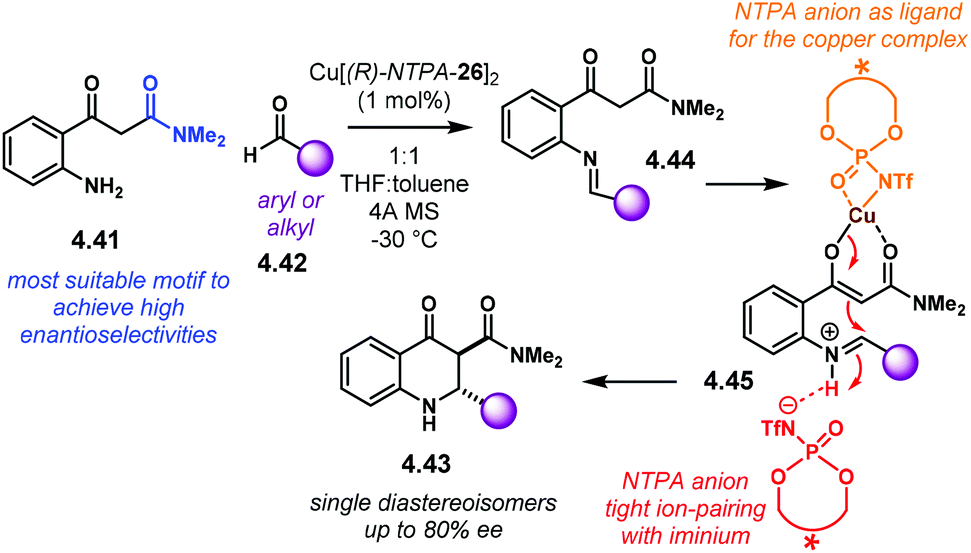 | ||
| Scheme 51 Enantioselective copper(II)–NTPA-catalysed Mannich reaction. | ||
This reaction uses the actual copper(II) N-triflylphosphorimidate, therefore, the reaction mechanism is expected to be different from that one of NTPA in the free acid form. Initially, imine 4.44 is formed from the corresponding starting materials—a Knoevenagel condensation was shown not to be operational. Then, as suggested by the authors, deprotonation of 4.44 generates copper(II) enolate 4.45. Herein, one of the NTPA anions serves as a chiral ligand for the copper centre. At the same time, the other anion activates the imine through iminium-tight ion pairing in a Brønsted acid-like fashion. Subsequent cyclisation and epimerisation to the more thermodynamically stable trans-diastereoisomer, yields product 4.43.
5. Electrophilic aromatic substitution: Friedel–Crafts and related reactions
Electrophilic aromatic substitution is another class of reactions in which NTPA catalysis can be advantageous. If a Brønsted acid activates an electrophile, an aromatic or heteroaromatic ring can attack it. Scheme 52 pictures the general Friedel–Crafts reaction pathway that display most of the transformations included in this section. Mechanistically, an electrophile 5.1 is activated by the NTPA (e.g. H-bonding or full protonation). Attack of an electron rich aryl or heteroaryl ring in the enantiodetermining step delivers Wheland intermediate 5.2 with the defined chiral centre. Subsequent re-aromatisation, possibly assisted by the counter anion of the catalyst, furnishes enantioenriched product 5.3. These reactions can be either intermolecular or intramolecular, with the latter allowing the construction of polycarbocyclic or polyheterocyclic systems.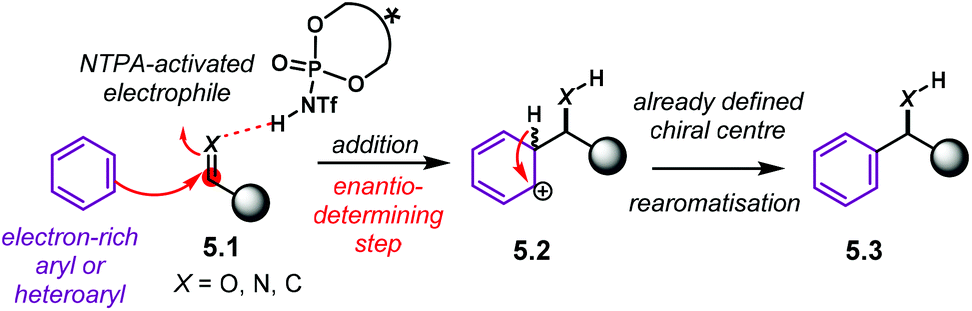 | ||
| Scheme 52 General picture of an enantioselective Friedel–Crafts reaction. In this section, we considered those reactions in which the addition step is enantiodetermining. | ||
In this section, we include those reactions in which the enantiodetermining step is the addition, delivering a chiral Wheland intermediate like 5.2, and yielding, overall, a Friedel–Crafts process. The following subsections are organised by the activated electrophilic species that participates in the addition step. These being iminium ions, α,β-unsaturated carbonyl compounds and oxy-carbenium ions.
5.1. Iminium ions as electrophiles: aza-Friedel–Crafts reactions
In the last section, we discussed how iminium ions, generated from protonation of imines, serve as suitable electrophiles for enantioselective 1,2 addition reactions. In this context, for this subsection of the review, we consider iminium ions that are involved in the enantiodetermining step of a Friedel–Crafts reaction—namely, the 1,2 addition step. In most of the cases, the precursor imines are generated in situ or pre-formed from the corresponding amines and carbonyl compounds.One reaction that uses an iminium ion in an intramolecular Friedel–Crafts sequence is the Pictet–Spengler reaction. The group of Zhou reported an enantioselective variant (Scheme 53a).128 In this transformation, aniline 5.4 condenses with aryl aldehydes 5.5 to give the corresponding imine. This imine is readily protonated by the chiral Brønsted acid to furnish reactive iminium ion 5.6. Then, enantioselective 1,2 addition of the pyrrole motif delivers Wheland intermediate 5.7. Herein, the chiral centre of the final product is already defined. To complete the Friedel–Crafts sequence, re-aromatisation takes place, possibly by NTPA-assisted deprotonation to regenerate the catalyst. Additional treatment with acetyl chloride delivers final product 5.8.
 | ||
| Scheme 53 (a) Asymmetric Pictet–Spengler-like reaction reported by the group of Zhou. (b) Extract from the catalyst screening. Reaction conditions: aryl = phenyl; 10 mol% catalyst loading; CH2Cl2, 20 °C. Numbers in red correspond to yields and enantioselectivities when NTPA were used as catalyst. Numbers in blue correspond to PA used instead. | ||
Pyrrolo-fused heterocycles are obtained in good yields albeit with modest enantioselectivities (up to 89% ee). The relatively low enantiocontrol might be due to the lack of substantial non-covalent interactions in the 1,2-addition step.
During catalyst screening, both PA and NTPA showed good reactivity, but higher enantiomeric excesses were obtained with NTPA (Scheme 53b). Catalyst (R)-[H8]-NTPA-4 provided the best enantiocontrol. As we have discussed throughout the review, catalyst tuning is still a cryptic process. As an example, it is not obvious how to explain the enantioselectivity increases when adding an extra aryl ring to the catalyst's substituents, comparing (R)-[H8]-NTPA-3 (17% ee) and (R)-[H8]-NTPA-4 (51% ee). As usual, further variables like temperature and solvent must be screened in order to attain the highest enantioselectivities.
The group of You developed a double Friedel–Crafts strategy for an asymmetric synthesis of fluorenes using an iminium ion in the enantiodetermining step (Scheme 54a).129 In the first (not enantioselective) Friedel–Crafts reaction, indole 5.9 reacts with aldehyde 5.10, which presumably is activated by H-bonding through the carbonyl group, to give secondary alcohol 5.11. Then, this alcohol is protonated by the NTPA and dehydrates to yield a vinylogous iminium ion 5.12. Such iminium is likely to be locked to the catalyst's active site through H-bonding and ion pairing. In the subsequent step, the dimethoxy phenyl ring attacks the electrophile in a 1,4 addition fashion. This is the enantiodetermining step of the reaction. Both the free N–H moiety and the 2-methyl group in the iminium ion were found to be essential for enantiocontrol. Moreover, an electron-rich, nucleophilic aryl is crucial for reactivity. After the addition step, NTPA-assisted re-aromatisation regenerates the catalyst and delivers enantioenriched fluorenes 5.14 in good yields and up to 92% ee.
 | ||
| Scheme 54 (a) Enantioselective synthesis of fluorenes via a double Friedel–Crafts reaction. (b) Selected examples from the catalyst screening; R = H; reactions done in toluene. | ||
During catalyst screening, briefly presented in Scheme 54b, (S)-NTPA-7 achieved the highest enantioselectivity (89% ee). The rather similar phenanthryl substituents in (S)-NTPA-6 showed lower enantiocontrol (66% ee). A more extended π system, like pyranyl in (S)-NTPA-8 resulted in even lower enantiocontrol (58% ee). Larger groups are not always better. On the other hand, 4-biphenyl 3,3′ substituents, (S)-NTPA-16, manage to get only 39% ee. Thus, the right balance in the catalyst's substituents size has to be found. This is another example to the list on how fine-tuning of the catalyst can be quite tricky.
This last reaction also worked well when PA were used as catalysts. However, the opposite sense of enantioinduction was observed. This is remarkable because the authors use the same (S)-BINOL-backbone for both the PA and the NTPA. Thus, it is not clear how a different active site can lead to enantioinversion. A detailed computational study might provide a clearer insight, highlighting the importance and utility of DFT calculations.
The group of Lete developed an elegant method for the construction of fused N-heterocycles.130 In this two-step methodology, shown in Scheme 55, imide 5.15 reacts with organolithium reagents, methyl lithium or n-butyl lithium in the scheme, to furnish hydroxy amides 5.16. Upon treatment with catalyst (R)-NTPA-7, the hydroxyl moiety is protonated. Subsequent dehydration furnishes reactive N-acyl iminium ion 5.17. N-Acyl iminium ions have proven to be suitable substrates in Brønsted acid-catalysed enantioselective transformations. Intramolecular 1,2 addition of the electron-rich aryl substituent in the enantiodetermining step yields intermediate 5.18. After that, re-aromatisation delivers enantioenriched heterocycles 5.19 in acceptable yields, albeit with modest enantioselectivities (up to 67% ee).
 | ||
| Scheme 55 Asymmetric synthesis of fused isoquinolines developed by the group of Lete. | ||
Regarding the activation mode, the authors proposed that the 1,2 addition step happens through a bifunctional/dual activation mode. N-Acyl iminium 5.17 engages in tight ion pairing with the NTPA anion. At the same time, H-bonding from one of the hydroxy groups to the catalyst's active site directs the attack inside the chiral cavity, accounting for enantioinduction. PA were also tried as catalysts; however, lower reactivity was achieved, presumably due to their lower acidity.
Despite the success of NTPA over PA, there remains the question if only acidity might be what makes NTPA more efficient catalysts. In this context, and to mention an example, the group of Piersanti reported an aza-Friedel–Crafts/lactonisation sequence using N-acyl iminium ions (Scheme 56).131 Therein, the highly acidic NTPA was beaten by a less reactive PA. For this reaction, 5.20 reacts with N-acyl iminium 5.23, generated by in situ tautomerisation of enamine 5.21. The Friedel–Crafts/lactonisation cascade delivers enantioenriched lactones 5.22. It is not clear in the publication, however, why a more acidic NTPA was not a suitable catalyst, outperformed by the corresponding PA.
 | ||
| Scheme 56 Enantioselective Friedel–Crafts/lactonisation sequence developed by the group of Piersanti. Is only acidity what makes NTPA good catalysts?. | ||
aoQMs (cf. section 2) can be regarded as a special kind of iminium ions for 1,4 Michael-like additions. The group of Rueping showed this in an enantioselective synthesis of triarylmethanes (Scheme 57).132o-Amino benzylic alcohols 5.24 undergo a protonation–dehydration sequence to deliver aoQM 5.26. This intermediate can be regarded as an α,β-unsaturated iminium ion, stabilised by an electron-rich substituent and activated by the catalyst through H-bonding. Then, 1,4 addition of indole 5.25 delivers Wheland intermediate 5.27, which, upon re-aromatisation, furnishes 5.28.
 | ||
| Scheme 57 aoQMs as potential Michael-like acceptors in aza-Friedel–Crafts reactions. | ||
The reaction features a broad substrate scope for C2-substituted indoles (Scheme 58a). 2-Phenyl indoles, however, were found to be the most suitable substrates in terms of enantioselectivity. Product 5.29a was obtained with excellent enantiocontrol (94% ee). In addition, 5.29b, bearing a 4-fluorophenyl substituent yielded excellent enantioselectivity as well (99% ee). C2-Indole substitution was also tolerated, as in the case of a methyl group in 5.29c and a 4-chloro phenyl group, 5.29d, both with remarkable enantiocontrol (90% ee and 92% ee, respectively). In addition, C3-substituted indoles provided access to the corresponding regioisomeric products 5.30a–5.30d, with excellent yields and enantioselectivities, up to 96% ee (Scheme 58b). The authors did not suggest an explanation that accounts for using free N–H-3-substituted indoles and N-methyl-2-substituted indoles in these complementary methodologies.
 | ||
| Scheme 58 (a) Substrate scope with C2-substitutes indoles, in this case, N-methyl indoles were used. (b) Substrate scope with C3-substituted indoles, in this case free N–H indoles were used. | ||
5.2. 1,4 additions to α,β-unsaturated carbonyl compounds
In the previous subsection, we regarded aoQMs as Michael-like acceptors, which can undergo a 1,4 addition. Herein, we focus on actual Michael acceptors, specifically α,β-unsaturated carbonyl compounds, activated by an NTPA. These can serve as suitable electrophiles for Friedel–Crafts alkylation reactions. Activation of carbonyl groups is not as straightforward as it is for imines. Therefore, highly acidic NTPA, rather than PA, are required in most of the cases.We start this subsection with an enantioselective intermolecular Friedel–Crafts alkylation developed by the group of You.133 The reaction comprises a Michael-like addition of pyrrole 5.31 to β-keto ester 5.32 (Scheme 59a). The authors did not suggest a mechanism or activation mode for the Friedel–Crafts step, apart from evoking an H-bond with the β-keto moiety. However, we would like to suggest that it seems possible that both starting materials are activated via a bifunctional mode, like in 5.33. In this way, the β-keto ester is H-bonded to the catalyst's proton. Simultaneously, another H-bond from the N–H moiety in the pyrrole to the catalyst seems equally feasible. Such a bifunctional activation mode can provide a conformationally locked, rigid transition state inside the catalyst chiral cavity. This would be in accordance with the excellent levels of enantiocontrol observed. After the 1,4-addition step, re-aromatisation of intermediate 5.34 delivers the alkylation product 5.35.
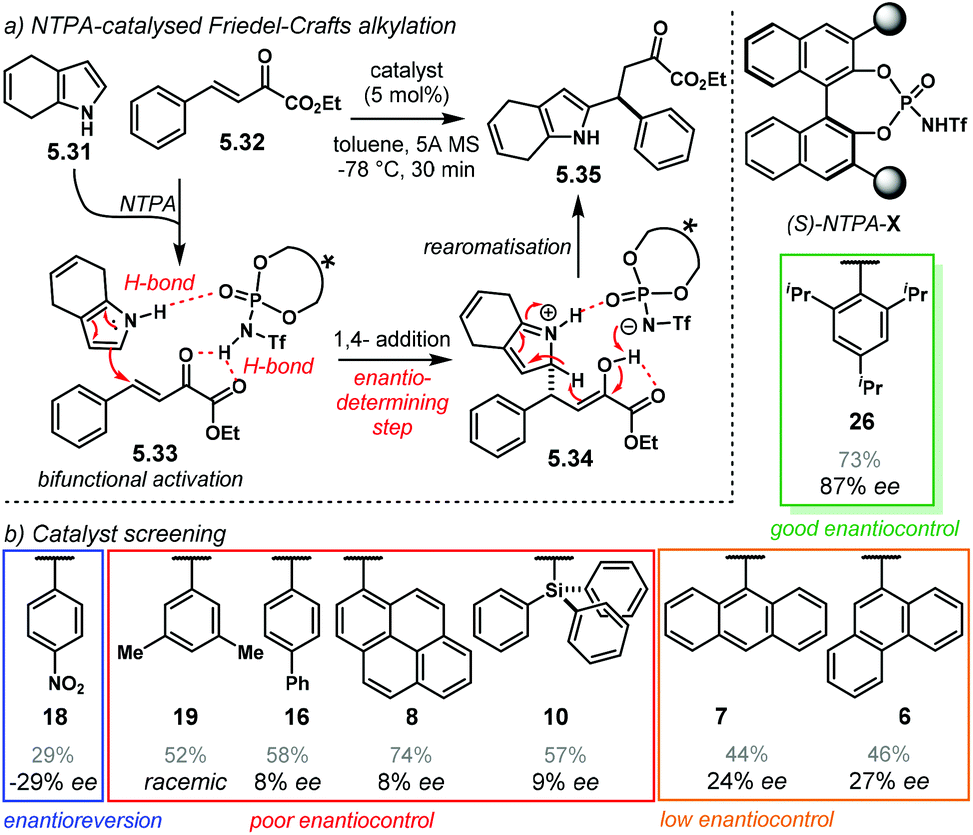 | ||
| Scheme 59 (a) Enantioselective Friedel–Crafts alkylation developed by the group of You. (b) Catalyst screening: yields obtained are greyed out for clarity in order to highlight the trend in enantioselectivity values. | ||
During the reaction optimisation, the authors performed an intriguing screening of catalysts (Scheme 59b). Several 3,3′ substituents provided poor or low enantioselectivities. Catalysts (S)-NTPA-19, (S)-NTPA-16, (S)-NTPA-8 and (S)-NTPA-10 yielded practically racemic products, even when the substituents are diverse in steric bulk. (S)-NTPA-18, highlighted in blue, got the opposite sense of enantioinduction. A slightly improved, yet low enantiocontrol (highlighted in orange), was achieved by (S)-NTPA-7 and (S)-NTPA-6. (S)-NTPA-26 gave an impressive result (87% ee), which is more than double the values obtained with the other catalysts. This screening is another example that shows that catalyst optimisation is far from being a trivial process. Herein, the development of new steric parameters or ‘easy-to-understand’ qualitative models would be desirable. Steric parameters have been developed in our group, although for PA.58 Therefore, reactions like this one provides an area of opportunity for computational chemists to develop such models that can accelerate the catalyst selection process.
In a short communication, the same group reported a similar Friedel–Crafts alkylation in an intramolecular fashion.134 Shown in Scheme 60, reaction of 5.36 with (S)-NTPA-7 leads to the activated complex 5.38. Because the reaction does not require the free N–H moiety in the indole, it is believed that this is a mono-activation mode. A strong H-bond to the carbonyl is therefore triggering the enantiodetermining 1,4-addition step to get 5.38. Thenceforth, re-aromatisation furnishes indole-fused heterocycles 5.39 with remarkable enantioselectivities (up to 98% ee). However, the methodology is limited to aryl ketones. Attempts to use alkyl ketones resulted in reduced enantiocontrol (e.g. 16% ee for the methyl ketone).
 | ||
| Scheme 60 Enantioselective intramolecular Friedel–Crafts reaction reported by the group of You. | ||
This reaction is an example in which NTPA perform equally well in reactions where PA already yielded excellent results. The same group reported the same reaction back in 2009 using chiral PA.135 Comparable yields and enantioselectivities are achieved with PA and NTPA. In addition, the same enantiomers of products 5.39 are obtained. This suggest that a similar mechanism and activation mode is in operation for this reaction.
Continuing with enantioselective intramolecular Friedel–Crafts reactions, the group of Jiang developed a method for the facile construction of seven-membered heterocycles.136 This reaction is a useful one because of the difficulty of installing chiral centres in seven-membered cycles through ring closure. It is suggested that substrates 5.40 (Scheme 61) are activated by the catalyst through H-bonding. Active complex 5.41, via mono-activation, undergoes the enantiodetermining Friedel–Crafts addition step. Intermediate 5.42 re-aromatises en route to enantioenriched heterocycles 5.43. (R)-[H8]-NTPA-14 afforded the best reactivity and enantioselectivities. PA were tried as well. However, unsuccessful outcomes were obtained, with either low reactivity or low enantioinduction. Substitution was tolerated in the aryl motif in the Michael acceptor. A broad substrate scope furnished good enantiocontrol (up to 92% ee). Substitution in the N-aryl group was tolerated as well, yet with diminished enantioselectivities.
 | ||
| Scheme 61 Enantioselective synthesis of seven membered rings developed by the group of Jiang. | ||
The groups of Fustero and del Pozo developed a similar enantioselective intramolecular Friedel–Crafts reaction using pyrrole as the nucleophile.137 This time to build up both six- and seven-membered heterocycles (Scheme 62a). Starting materials 5.44 were obtained by double alkene metathesis.
 | ||
| Scheme 62 (a) Cycloaromatisation/enantioselective Friedel–Crafts reaction. (b) Extract of examples from the substrate scope. | ||
Treatment of 5.44 with a Brønsted acid triggers a cycloaromatisation reaction, a Paal–Knorr-type pyrrole synthesis, to yield pyrroles 5.45. The NTPA activates the Michael acceptor through H-bonding. Enantioselective 1,4-addition of the pyrrole motif in this mono-activated reactant delivers intermediate 5.46, which upon re-aromatisation and tautomerisation furnishes enantioenriched heterocycles 5.47. (R)-NTPA-26 was the best catalyst for this tandem process, both in terms of yield and enantioselectivity. Weaker Brønsted acids managed to furnish pyrrole 5.45 but its reactivity towards the Friedel–Crafts was low. In general, the methodology is rather limited to short alkyl chains (Scheme 62b), for example, in products 5.48a and 5.48b, and the enantioselectivities are modest (60% ee and 66% ee, respectively).
A phenyl substituent is tolerated, achieving better enantiocontrol (5.48c 72% ee). Enantioinduction diminishes if the ring size is increased (5.48d, 57% ee), or with the introduction of heteroatoms, like oxygen (5.48e, 30% ee) or sulfur (5.48f, 46% ee). In seven-membered rings, enantioinduction was improved for the case of pyrrolo-azepinone 5.48g (68% ee).
5.3. Other Friedel–Crafts-like reactions
To finalise this section devoted to Friedel–Crafts and related reactions, we include herein an example in which the electrophilic species could be regarded as a bis-vinylogous iminium ion, or perhaps, as some sort of indole-benzylic-type carbocation. Nevertheless, the enantiodetermining step of the reaction is still the addition step in the Friedel–Crafts.The group of Han reported an enantioselective synthesis of diindoylarylmethanes.138 In this reaction, shown in Scheme 63, compound 5.49 is activated through a protonation–dehydration sequence. The resulting intermediate can be regarded either as a bis-vinylogous-iminium-like ion 5.51-A or as a benzylic carbocation 5.51-B. Either of these resonant structures are tightly paired with the NTPA counter anion. Due to the lack of H-bond donors in the substrate, ion pairing is the most likely activation mode. Addition of indole 5.50 leads to intermediate 5.52. Subsequent re-aromatisation completes the Friedel–Crafts sequence to deliver products 5.53 in excellent yields and enantioselectivties. PA were not suitable catalysts for this transformation. Surprisingly, (R)-NTPA-1 gave the best results in both reactivity and enantioinduction. Substitution at the 3,3′ positions of the catalyst framework was detrimental for enantioselectivity. This reaction is a unique example where a Brønsted acid with an unsubstituted BINOL-based backbone achieves remarkable levels of enantiocontrol. The authors do not provide a mechanism that accounts for the unsubstituted NTPA providing high enantio-differentiation; however, it is likely that two molecules of the catalyst might be involved in the enantiodetermining step.
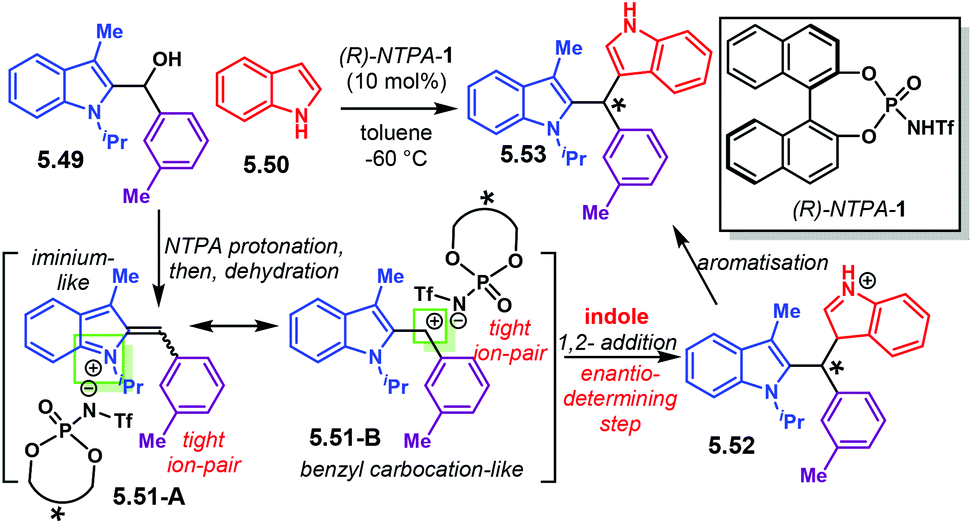 | ||
| Scheme 63 Enantioselective Friedel–Crafts reaction reported by the group of Han. The absolute configuration of the products was not determined. | ||
Several aryl groups, N-protecting groups and substitution patterns were tolerated. An extract of examples from the substrate scope is presented in Scheme 64. Products 5.54 are obtained in excellent yields with remarkable enantiocontrol.
 | ||
| Scheme 64 Substrate scope. Selected examples from the reaction published by the group of Han. The absolute configuration of the products was not determined. | ||
This last example paves the way to the next section of our review, which comprises addition reactions to carbocations.
6. Addition reactions to carbocations
Carbocations are ubiquitous reactive species. They can be attacked by nucleophiles to form new C–C or C-heteroatom bonds. In addition, their trigonal planar geometry provides them with two enantiotopic faces. This gives the possibility of installing a new chiral centre. Therefore, asymmetric catalysis has used them as suitable intermediates to construct molecules in an enantioselective fashion.In this section, we account for those reactions that involve a carbocation as the reactive electrophile. However, in order to construct this section, we considered those reactions in which the enantiodetermining step involves the attack of a nucleophile to the carbocation, thus, delivering a chirality element in such an addition step.
One of the most utilised methods to generate carbocations in Brønsted acid-catalysed reactions is protonation–dehydration. This sequence is outlined in a simplified way in Scheme 65. Alcohols 6.1 can be in equilibrium with its protonated form 6.2. The need of a highly acidic catalyst is therefore reasonable. Dehydration yields the corresponding carbocation 6.3. The presence of stabilising groups is desirable. Such groups can be electron-donating or any other motif that stabilises the intermediate. Benzylic 6.4 or allylic 6.5 carbocations can be generated in this manner. Most of the reactions discussed in this section follow this general outline as a mode of substrate activation or carbocation generation. In addition, oxy-carbenium ions 6.6 are also included herein,139 as well as carbocations generated from protonation of double bonds.
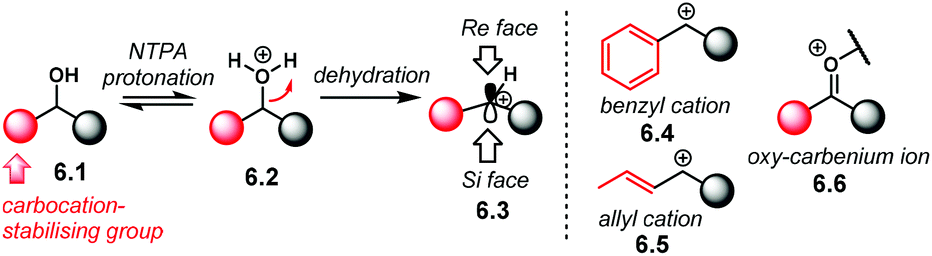 | ||
| Scheme 65 General outline to produce carbocations in a protonation–dehydration sequence. The dehydration step works best if electron-rich groups are present. | ||
6.1. Addition to allyl and benzyl carbocations
We left section 5 with an example in which a benzyl–indoyl cation was used as the electrophilic species in a Friedel–Crafts reaction (cf.Scheme 63, 5.51-B). In 2015, the group of Han used the same kind of intermediates in an addition reaction with enamides as the nucleophilic counterpart (Scheme 66a).140 Indol-2-yl carbinols 6.7 react with enamides 6.8. The asymmetric NTPA-catalysed addition delivers, presumably, intermediate imine 6.9, which is not isolated but hydrolysed with HBr to yield indoles 6.10 in excellent yields and enantioselectivities (up to 99% ee). Different substitution patterns and groups were tolerated (Scheme 66b), in both the carbinol component (6.10a, 6.10b, 6.10c) and the enamide (6.10d, 6.10e). Electron-rich aryl motifs in the carbinol were required to stabilise the carbocationic intermediate. In addition, different N-protecting groups were tested in the enamide; the benzyl group was found to be the best one for enantioselectivity. Other groups resulted in diminished enantiocontrol. It was also found that using p-nitrobenzoic acid as an additive improved the reaction yields, presumably assisting catalyst turnover. | ||
| Scheme 66 (a) Enantioselective addition of enamides to indol-2-yl carbinols developed by the group of Han. (b) Extract from the substrate scope. | ||
The authors propose two alternative bifunctional activation modes for the addition step in that reaction (Scheme 67). In 6.11, a protonation–dehydration sequence yields benzyl–indolyl carbocation which ion-pairs with the NTPA counteranion. At the same time, the enamide H-bonds to the catalyst's active site. Tertiary enamides proved to be ineffective, indicating that the free N–H moiety, as in the secondary enamide, is essential for reactivity; this is consistent with a bifunctional activation mode. On the other hand, activation mode 6.12 constitutes a doubly H-bonded transition state. In this case, the carbinol starting material binds to the catalyst through the hydroxyl moiety. Due to the high levels of enantioinduction observed, the former bifunctional activation mode seems more likely. A trigonal planar carbocation would provide better enantio-differentiation when it is inside the chiral cavity.
 | ||
| Scheme 67 Proposed bifunctional activation modes. | ||
In a remarkable piece of work, the group of Terada reported a challenging reaction featuring the induction of two contiguous stereocentres,76 both quaternary and congested. In the reaction, presented in Scheme 68a, unactivated alkenes 6.13 react with azlactones 6.14 to yield Markovnikov addition products 6.15. These were obtained in good yields, with excellent syn-diastereocontrol albeit with modest enantioselectivities. Nevertheless, the reaction is outstanding given the simplicity of the substrates. In the reaction mechanism, the alkene is protonated in a Markovnikov fashion to deliver a benzylic tertiary carbocation (6.16, Scheme 68a). Such cation has to ion pair tightly to the catalyst's counterion to be locked within the chiral cavity. Moreover, it is likely that the azlactone nucleophile engages through H-bonding. This can provide an extra bonus for enantioinduction.
 | ||
| Scheme 68 (a) Enantioselective addition of azlactones to styrene-derived carbocations developed by the group of Tereada. Although the publication does not propose an activation mode, we suggest a two point, bifunctional ion-pairing/H-bonding activation. (b) Extract from the substrate scope. | ||
Despite the modest levels of enantiocontrol, the reaction shows a broad substrate scope (Scheme 68b). Substitution in the styrene component is tolerated, as shown in 6.15a and 6.15b. In addition, indene-like products 6.15c and 6.15d were obtained with fair enantioselectivity. A lower temperature was needed for these last two examples. Substitution in the azlactone component was also tolerated and found to improve enantioselectivity. Products 6.15e and 6.15f were obtained with 73% ee and 93% ee, respectively.
The most distinctive feature of this reaction is the unprecedented perfluorinated BINOL-backbone of the catalyst. Catalyst (R)-[F10]-NTPA-8 was found to achieve the best enantiocontrol, and, more importantly, the best reactivity. However, some trends are still arguable (Scheme 69). Reactivity enhances when an NTPA is used instead of the corresponding PA, for example, in the case of (R)-CPA-2 and (R)-NTPA-2 (4% and 38% yields, respectively). The same applies for (R)-[F10]-PA-2 and (R)-[F10]-NTPA-2 (54% and 75% yield, respectively). However, this trend breaks if we look at (R)-NTPA-2 and (R)-[F10]-PA-2 (38% and 54%). According to the pKa values, (R)-NTPA-2 (pKa = −3.36) should enhance reactivity more than (R)-[F10]-PA-2 (pKa = 1.03). That is not the case. Therefore, the fluorinated backbone has to be playing an important role regarding reactivity, and thus, substrate activation. These examples show that acidity on its own, which is directly related to the pKa values, may not be telling the full story. Therefore, complementary physical chemical descriptors associated to acidity, such as surface electrostatic potential,141,142 could be beneficial in the catalyst optimisation process.
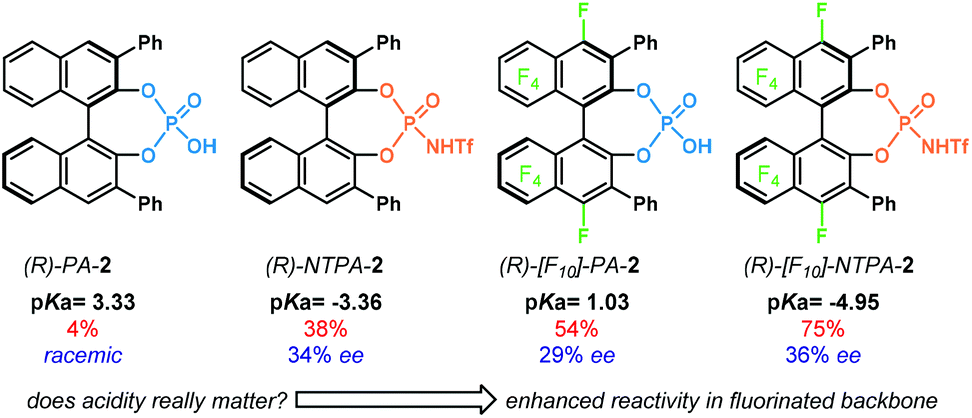 | ||
| Scheme 69 Catalyst screening. Enhanced reactivity with fluorinated backbone. Yields and enantioselectivities correspond to the reaction that gets product 6.15a. Reaction conditions: 5 mol% catalyst loading; CH2Cl2; 0 °C; ees correspond to the major syn-enantiomer, diastereoisomeric ratios omitted for clarity. | ||
Changing focus, we will now look at reactions with allylic carbocations. In 2014, the group of Du reported an intermolecular allylic amination.143 Allylic alcohols 6.17 reacted with tosyl amine to yield allylic N-tosyl amines 6.18 (Scheme 70a). A limited number of products are obtained in good yields but with modest enantioselectivities. Furthermore, enantiocontrol was sensible to the substitution pattern in the aryl motifs in the starting materials. para-Substitution achieved modest enantiocontrol (66% to 85% ee), followed by meta-substitution, albeit slightly decreased (62% ee). ortho-Substituents were detrimental for enantiocontrol (<25% ee). In addition, solvent effect was significant. The reaction can also be done with unsymmetrical substrates. The electronic effect was found to be the most important factor controlling regioselectivity. Yet, enantiocontrol was also modest or low.
 | ||
| Scheme 70 (a) Enantioselective allylic amination developed by the group of Du. (b) Plausible activation mode via bifunctional ion-pairing/H-bonding. | ||
The authors propose that a chiral contact ion pair could be the activation mode (Scheme 70b). Moreover, it is likely that tosyl amine might be engaging in an H-bond with the active site of catalyst. This would provide a relatively rigid bifunctional model. In addition, internal interactions might provide further rigidity. Yet, it is not quite clear how the carbocation is anchored to the catalyst's chiral pocket. Such interactions could be an opportunity for computational chemists.
A related intramolecular reaction, using an allylic/benzylic carbocation, was reported in 2018 by the group of Xie.144 As shown in Scheme 71, alcohols 6.20 deliver enantioenriched N-heterocycles 6.21 in good yields and with up to 93% ee. Mechanistically, the reaction proceeds in the usual NTPA protonation–dehydration sequence to furnish carbocation 6.22. The authors suggest a bifunctional, two-interaction, mode of activation. A tight ion pair locks the reactive carbocationic electrophile while an H-bond to the N–H moiety in the aniline delivers the nucleophile in an enantioselective fashion.
 | ||
| Scheme 71 Intramolecular enantioselective amination reported by the group of Xie. | ||
Several features of this reaction are noticeable. For instance, (R)-[H8]-NTPA-1 was found to be the best catalyst in terms of enantioinduction, despite not bearing particularly bulky scaffolds. Nonetheless, the Fmoc protecting group, which gave the best enantioselectivities after screening other protecting groups, is as large as the main substrate itself; accounting, perhaps for the catalyst choice. Other NTPA were equally reactive but achieved lower enantiomeric excesses. PA fell behind in both reactivity and enantiocontrol. The reaction also features an uncommon but interesting choice of solvent, dichloromethane:benzene, 1:1. Although not discussed in the text, such a solvent system might be tampering with both solubility and the dielectric constant of the reaction medium, the latter to achieve proper ion pairing. Finally, the reaction works well with a broad range of electron-rich, electron-neutral and halo-substituted aryls.
Another related intramolecular reaction, using the same relatively unhindered catalyst, (R)-[H8]-NTPA-1, was reported by the group of Rueping (Scheme 72).145 Alcohols 6.23 are protonated and dehydrated to render an allylic carbocation, which, upon cyclisation, delivers enantioenriched 2H-chromenes 6.24 with excellent enantiocontrol (up to 96% ee). Several catalyst were screened. It was found that enantioselectivity was highly dependent on the catalyst's substituents. The best enantiocontrol was achieved by (R)-[H8]-NTPA-1. PA were tried but were outperformed by the more acidic NTPA.
 | ||
| Scheme 72 Enantioselective allylic substitution reported by the group of Rueping. | ||
The authors propose an activation mode via a chiral contact ion pair 6.25. After the dehydration step in the mechanism, the carbocation is locked in the catalyst's chiral pocket through electrostatic interactions. The use of toluene as solvent might be indicative of such activation mode to account for the high enantiocontrol. In addition, it is likely that an H-bond from the –OH moiety in the intermediate is formed with the NTPA anion. This interaction would provide extra rigidity to the system for an efficient chirality transfer.
6.2. ortho-Quinone and para-quinone methides as a case of stabilised carbocations
oQMs are highly reactive species. In section 2, we discussed how these intermediates could be suitable partners for cycloaddition reactions (cf.Scheme 18). Additionally, in section 5, we presented aoQMs as suitable electrophilic species in Friedel–Crafts transformations (cf.Scheme 57). Building on that, in this section we regard (aza)oQMs as a special type of highly stabilised carbocations, mostly stabilised by resonance. We note that they are not “pure” carbocations as defined by Rueping.145 In addition, para-quinone methides (pQM) are also featured in this section as highly resonance-stabilised carbocations. These species can serve as electrophiles for addition reactions. Furthermore, due to the π systems that accompany these structures, conjugated additions are headliner reactions.We begin this subsection with a 1,4 addition–cyclisation cascade reported by the group of Rueping.146Scheme 73a shows the NTPA-catalysed reaction of alcohols 6.26 with cyclohexane-1,3-dione 6.27 to afford enantioenriched 2H-chromenes 6.28. Initially, starting material 6.26 undergoes an NTPA-protonation/dehydration sequence to deliver oQM 6.29. This intermediate is stabilised by its electron-donating motif as well as by the catalyst. Through H-bonding, the tautomer of diketone 6.27 is activated in a similar fashion.
 | ||
| Scheme 73 (a) Enantioselective synthesis of 2H-chromenes via a 1,4-addition – cyclisation cascade, developed by the group of Rueping. (b) Extract from the substrate scope. (c) Enantioselective addition followed by desymmetrisation-ring closure. (d) Bifunctional activation mode. (e) Diastereoselective cyclisation step also undergoing a bifunctional activation. | ||
The authors suggest that such a bifunctional activation mode provides a rigid chiral environment inside the catalyst pocket, in order to account for the high enantioselectivities. 1,4-Addition of the enol to the oQM installs the stereogenic centre in an enantioselective fashion to yield 6.30. This intermediate readily cyclises. That cyclisation step might be assisted by a two-point contact interaction with the NTPA. Finally, dehydration of 6.31 delivers the desired products. NTPA were used straightaway during the catalyst screening stage, likely due to their high reactivity to promote the protonation–dehydration step. Several catalysts worked well, in both reactivity and enantiocontrol. (R)-NTPA-5 afforded the highest enantioselectivities and was used in the reaction scope.
This reaction tolerates a fair variety of substitution, although an electron-donating group seems to be essential. Products 6.28a, 6.28b and 6.28c are obtained in good yields with excellent enantiocontrol (Scheme 73b). In addition, other electron-rich moieties, like in 6.28d, can be used without detriment in enantioselectivity.
In the same publication, the methodology was extended to include an enantioselective 1,4 addition–cyclisation–desymmetrisation cascade. This was achieved when meso-5-substituted cyclohexane-1,3-diones 6.32 were used (Scheme 73c). The same alcohol substrates, 6.26, were suitable reaction partners. Bicyclic hemiacetals 6.33 are obtained. In the same pot, dehydration with FeCl3 delivers the desired products 6.34 with excellent enantioselectivities (up to 93% ee) and outstanding diastereocontrol, practically as single diastereoisomers (up to >98:2 dr). For these reactions, the catalyst screening had to be done again, finding (R)-NTPA-26 as the most suitable one.
Mechanistically, the 1,4 addition step proceeds in a similar fashion as with the unsubstituted diketones (Scheme 73d). A bifunctional activation mode, 6.35, anchors both substrates and locks them inside the catalyst's chiral cavity. This step is also enantiodetermining. Different from the other reactions, the ring closure step now becomes diastereoselective (Scheme 73e). Intermediate 6.36 desymmetrises in order to yield the corresponding products with excellent diastereocontrol. In addition, a two-point contact bifunctional activation seems likely to be driving the reaction. This is a remarkable transformation as it accounts for the installation of a remote stereocentre.
In 2017, the group of Sun reported an enantioselective synthesis of tetrasubstituted allenes (Scheme 74a).147 Reaction of 6.37 with a Brønsted acid forms a pQM which is attacked by thioacetic acid in order to afford tetrasubstituted allenes 6.38 with excellent enantioselectivities. Chiral catalyst (R)-S-NTPA-11, which has an SPINOL-backbone, was found to deliver the best enantiocontrol for this transformation. The reaction offers a broad substrate scope in the alkyne component. Products 6.38 are obtained in high yields and with excellent enantiocontrol regardless the wide variety of substituents (Scheme 74b), making it a general methodology. The reaction works well with aryl (6.38a, 94% ee) and heteroaryl substituents (6.38b, 90% ee). Alkyne protecting groups are also tolerated (6.38c, 92% ee). Alkyl chains were also suitable substrates without detriment in enantiocontrol (6.38d, 93% ee and 6.38e, 92% ee).
 | ||
| Scheme 74 (a) Enantioselective synthesis of tetrasubstituted allenes via an oQM, reported by the group of Sun. (b) Extract from the substrate scope. The reaction tolerates a wide variety of substituents. | ||
In addition, the methodology also works with 1,3 dicarbonyl compounds as the nucleophilic partner (Scheme 75a). Reaction of 6.39 with 6.40 delivers enantioenriched allenes 6.41. These compounds are obtained with excellent enantioselectivity. Moreover, this transformation features the installation of a vicinal all-carbon quaternary stereocentre with good levels of diastereocontrol (up to 21:1 dr). Chiral Brønsted acid (R)-NTPA-26 was found to be the best catalyst. The substrate scope also tolerates a wide variety of substitution. Aryl and heteroaryl scaffolds in the alkyne worked well (6.41a and 6.41b). Different electron-donating groups, like thiomethyl 6.41c, gave good results too. The 1,3 dicarbonyl component can also be modified, albeit with a slight decrease in yield but still with excellent enantiocontrol (6.41d).
 | ||
| Scheme 75 (a) Enantioselective synthesis of tetrasubstituted allenes with a vicinal quaternary stereocentre, reported by the group of Sun. (b) Extract from the substrate scope. The reaction tolerates a wide variety of substituents. | ||
These transformations, although closely related, are mechanistically different (Scheme 76). According to the authors and series of control experiments, if the substrate has a free –OH moiety in the aromatic ring, a pQM is formed (Scheme 76a, 6.42). The catalyst locks both reactants through bifunctional activation via H-bonding. The nucleophile, then, attacks in an enantioselective 1,8 addition fashion. On the other hand, if the –OH moiety is not available, a carbocation 6.43 (Scheme 76b) is likely to be the reactive intermediate. Herein, a bifunctional activation mode via ion-pairing/H-bonding is likely to take place. Both types of bifunctional activation might create a rigid chiral environment inside the catalyst pocket; this accounts for the high levels of enantiocontrol.
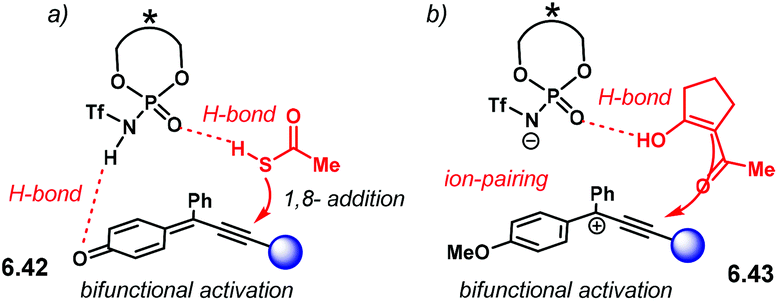 | ||
| Scheme 76 Bifunctional activation mode via (a) a pQM intermediate and (b) via a stabilised carbocation. | ||
Aza-para-quinone methides (apQM) are also suitable electrophiles for 1,8-addition reactions. The groups of Li and Li reported an interesting example using this class of intermediates.148 Propargylic alcohols 6.44 react with heterocycles 6.45 to furnish tetrasubstituted allenes 6.46, featuring both axial and centro chirality with remarkable enantiocontrol and as single diastereoisomers (Scheme 77).
 | ||
| Scheme 77 Diastereo- and enantioselective synthesis of tetrasubstituted allenes via a 1,8 addition to an apQM. | ||
(S)-NTPA-7 was the most suitable catalyst for this transformation. PA achieved good yield and comparable high diastereocontrol, but affording products in a practically racemic fashion.
The reaction mechanism features the well-known protonation–dehydration sequence to deliver intermediate 6.47. This is proposed by the authors as an apQM 6.48. Such intermediate is locked to the catalyst via an H-bond from the N–H amide moiety, the adamantyl motif achieved both the best diastereo and enantiocontrol, presumably due to steric bulk. The nucleophilic counterpart also binds to the catalyst through H-bonding via its enol tautomer. This bifunctional activation mode is likely to create a rigid chiral environment to account for the impressive stereocontrol. The construction of the allene happens via the enantioselective 1,8 addition to the nucleophile. Regioselectivity is also a remarkable feature of this transformation.
To finish this subsection on quinone methides as reactive intermediates, we include a similar reaction, also featuring an apQM as the electrophilic species. The group of Maruoka developed an asymmetric alkenylation of propargyl alcohols.149 The reaction is presented in Scheme 78a. Propargylic alcohols 6.49 react with boroxines 6.50 in the presence of a Brønsted acid, and alkenylated products 6.51 are obtained in good yields and excellent enantiocontrol. This reaction features a unique mechanism in comparison with all the other reactions discussed so far (Scheme 78b). The authors suggest that a protonation–dehydration sequence takes place to generate an apQM as key intermediate. At the same time, the NTPA forms a covalently bound complex with the boroxine, 6.52.
 | ||
| Scheme 78 (a) Enantioselective alkenylation using boroxines, developed by the group of Maruoka. (b) Bifunctional H-bonding/covalent activation mode. The authors propose an NTPA-boroxine complex as the reactive species. | ||
Complex 6.52 delivers the alkenyl substituent from the boroxine in an enantioselective fashion. In sharp contrast, 1,6 regioselectivity is observed. Given that the nucleophile is covalently bound to the catalyst, a 1,8 addition might be too far for the nucleophile to be delivered. This bifunctional H-bond/covalent activation mode should be rigid enough to account for the remarkable enantiocontrol.
In addition, the reaction works with a wide variety of substrates (Scheme 79). Aryl and heteroaryl substituents in the propargyl alcohol component affords products 6.51a, 6.51b and 6.51c with excellent enantiocontrol. Furthermore, different groups in the boroxine component are also tolerated, like heteroaryl 6.51d. Also, alkyl substituted propargylic alcohols afford the corresponding products, e.g.6.51e, without decrease in enantioselectivity.
 | ||
| Scheme 79 Extract from the substrate scope for the enantioselective alkenylation developed by the group of Maruoka. | ||
6.3. Addition to oxy-carbenium ions
Oxy-carbenium ions (cf.6.6, Scheme 65) are also suitable electrophiles for asymmetric addition reactions. However, not so many reactions are reported in this area. The group of Floreancig reported one of these transformations.150 In that reaction, shown in Scheme 80, oxy-carbenium ions are generated through oxidative C–H bond cleavage. Starting materials 6.53 deliver oxy-carbenium ion 6.54 through treatment with DDQ. Presumably, 6.54 forms a tight ion pair with the counter anion of (R)-NTPA-26. Then, addition of 6.55 affords product 6.56 in modest yield and enantioselectivity. | ||
| Scheme 80 Asymmetric allylation of oxycarbenium ions generated via oxidative C–H bond cleavage. | ||
PA delivered practically racemic products. Both yield and enantiocontrol was highly dependent on the reaction conditions and allylating reagents used. Allyl silane 6.55-A afforded the desired product in 62% ee; however, a stoichiometric amount of the chiral Brønsted acid was needed. At a lower temperature and using stannane 6.55-B the catalyst loading could be lowered to 20 mol% in order to obtain the allylated product in 66% yield and 81% ee. A slight improvement in enantiocontrol was achieved with silane 6-55C, albeit with diminished yield (86% ee, 56%, respectively). With lower catalyst loadings, the background reaction becomes competitive. The reaction shows a broad substrate scope for the racemic version, but in an asymmetric fashion, only substrate 6.53 was tried.
To finish this section, we include a reaction reported by the groups of Wang and Goeke.151 Although the reaction does not strictly involves an oxy-carbenium ion in the enantiodetermining step, it serves as a suitable example to bridge to the next section, which accounts for aldol reactions.
The groups reported an enantioselective Prins bicyclisation (Scheme 81a). Using aldehydes 6.57 and glyoxylates 6.58, the reaction affords dioxabicycles 6.59 in good yields as single endo-diastereoisomers with impressive enantiocontrol. The authors propose a transition state similar to 6.60. To account for the remarkable stereocontrol, it is plausible that a bifunctional activation mode is operating. Activation of a carbonyl group through full protonation, or a strong H-bond, with a simultaneous C–H⋯OP non-classical H-bond with the formyl moiety in the glyoxylate, might be responsible for creating a rigid structure within the catalyst's chiral pocket. After that step, intermediate 6.61 undergoes a second cyclisation to afford the desired products. PA did not promote the reaction at all. In addition, solvent polarity was a crucial factor. Enantioselectivity was higher in non-polar solvents, which suggests a tight ion pair in the reaction mechanism.
 | ||
| Scheme 81 (a) Enantioselective Prins bicyclisation reported by the groups of Wang and Goeke. (b) Extract from the substrate scope in the enantioselective Prins bicyclisation reaction. | ||
The reaction shows a reasonable substrate scope. Different glyoxylates and aldehydes were suitable (Scheme 81b). Ethyl, benzyl and isopropyl glyoxylates afforded the corresponding products 6.59a, 6.59b and 6.59c, respectively, with good enantioselectivities (86%, 87% and 88% ee). Substitution in the aldehyde was also tolerated, as in 6.59d (91% ee). Product 6.59e was obtained with diminished enantioinduction (43% ee).
7. Aldol and related reactions
The aldol reaction is another powerful transformation in the organic chemist toolbox. It can also be exploited in an asymmetric fashion. Although several enantioselective aldol reactions have been developed using Lewis acid catalysis, NTPA can render good levels of enantiocontrol via Brønsted acid catalysis.In a creative methodology, the group of Gong developed an enantioselective hydrosiloxylation/Mukaiyama aldol cascade (Scheme 82a).152 Under Au(I) catalysis, silanols 7.1 furnish silyl enol ethers 7.2. Then, it enters the NTPA catalytic cycle. Using (S)-NTPA-26 and fluorenyl glyoxylate 2.25, the reaction delivers aldol adduct 7.3 with excellent stereocontrol. After rearrangement, products 7.4 are obtained in high yields with good diastereoselectivity (up to 11:1 dr) and excellent enantioselectivity (up to 94% ee). PA afforded low yields of the final product. NTPA catalysed the aldol step more effectively, probably due to their higher acidity.
 | ||
| Scheme 82 (a) Enantioselective hydrosiloxylation/Mukaiyama aldol cascade developed by the group of Gong. (b) Plausible activation mode. | ||
A plausible activation mode suggests a dual activation of the glyoxylate, as shown in 7.5 in Scheme 82b. Such double H-bond would lock the glyoxylate inside the catalyst chiral cavity for the silyl enol ether to attack one of the two enantiotopic faces of the carbonyl. The bulky fluorenyl motif was needed to achieve high stereocontrol.
This methodology has a broad substrate scope. Several aryl groups in the alkyne motif of the substrate were tolerated. However, the real power of the aldol reaction, in this case, the Mukaiyama aldol, is demonstrated in the total synthesis of natural products. In the same publication, the asymmetric Mukaiyama aldol proved to be highly valuable in the synthesis of a potential precursor towards metasequirin B, 7.11 (Scheme 83a).
 | ||
| Scheme 83 Applications of the NTPA-catalysed Mukaiyama aldol in the synthesis of natural products. (a) Towards a total synthesis of metasequirin B. Enantioselective synthesis of a suitable precursor. (b) Enantioselective synthesis of (−)-5-epi-eupomatilone-6, reported by the group of Wu. | ||
Reaction of 7.6 and fluorenyl glyoxylate with the optimised reaction conditions afforded product 7.7 in 87% yield, with 10:1 dr and 93% ee. Then, Baeyer–Villiger oxidation furnishes 7.8, which after removal of the benzyl group and reduction of the ester delivers product 7.9. After a Hiyama coupling reaction, triol 7.10 is obtained. Such triol can serve as a suitable precursor en route to metasequirin B. With two chiral centres induced enantioselectively, the remaining ones can be installed diastereoselectively through substrate control.
In addition, the group of Wu reported an enantioselective synthesis of (−)-5-epi-eupomatilone-6, 7.15, using the Au(I)/NTPA-catalysed Mukaiyama aldol cascade as a key step (Scheme 83b).153 Starting from aryl iodide 7.12, silanol 7.13 was obtained in four steps in 82% yield in a 5-gram scale. Then, reaction with florenyl glyoxylate 2.25 using the Au(I)-mukaiyama aldol conditions, key intermediate 7.14 was obtained in 74% yield, with 10:1 dr and 89% ee. This transformation was achieved in a 6.6-gram scale, using only 1 mol% catalyst loading of the gold catalyst and 5 mol% of (S)-NTPA-26. Such a key step is a good example in which NTPA-catalysed reactions can be scaled-up, making NTPA suitable catalysts for large-scale operations. Finally, after several steps, (−)-5-epi-eupomatilone-6, 7.15, was obtained in 9% overall yield.
Later, in 2015, the groups of Sai and Yamamoto reported an enantioselective Mukaiyama aldol and an enantioselective Hosomi–Sakurai allylation (Scheme 84).154 Their report features an impressive catalyst engineering approach. Commencing with the Mukaiyama aldol reaction (Scheme 84a), the optimisation started with 4-chlorobenzaldehyde 7.16 and silyl enol ether 7.17 in order to afford aldol product 7.18. An initial study revealed that PA, NTPA and the corresponding selenophosphoramides were silylated by 7.17. Thus, shifting the reaction mechanism from Brønsted acid to Lewis acid catalysis. In sharp contrast, thiophosphoramides (TPA) kept themselves as Brønsted acids. The catalyst design and screening is summarised in Scheme 84b. TPA-[A] promoted the reaction effectively, in good yield and with modest diastereocontrol, albeit with low enantioselectivity. Stereocontrol was further increased when the heavily congested TBDPS group replaced the para-iso-propyl substituent in the aryl scaffolds. The most dramatic enhancement in stereoselectivity was achieved when the N-triflyl moiety was substituted by a longer, perfluoro butyl, chain. Extending the perfluorinated chain to perfluoro octyl in the active site led to better enantiocontrol.
 | ||
| Scheme 84 (a) Enantioselective Mukaiyama aldol reaction developed by the groups of Sai and Yamamoto. (b) Catalyst design and screening. (c) Extract from the substrate scope for the Mukaiyama aldol reaction. (d) Enantioselective Hosomi–Sakurai reaction. (e) Extract from the substrate scope. | ||
This asymmetric Mukaiyama aldol reaction features a wide substrate scope (Scheme 84c). Electron-rich aryl products 7.19a and 7.17b were obtained in 98% ee and 95% ee, respectively. The reaction worked equally well for electron-poor aryls (7.19c and 7.19d). Product 7.19e, bearing a cyclobutanone motif was obtained in 91% ee and excellent syn-selectivity. Consistently, the reaction with cyclic silyl enol ethers afforded 1,3-syn adducts with impressive diastereo- and enantiocontrol. Contrariwise, branched silyl enol ethers got 1,3-anti adducts regardless the cis/trans geometry of the enol ether, still with excellent enantiocontrol (7.19f, anti:syn 81:19, 91% ee). Several silyl enol ether were found to be suitable substrates. Unbranched product 7.19g was obtained with excellent enantiocontrol (91% ee). The methodology also works to afford carbocyclic and heterocyclic products 7.19h, 7.19i and 7.19j, albeit with diminished enantioselectivities.
Within the same publication, the newly designed thiophosphoramide was also able to catalyse an asymmetric Hosomi–Sakurai reaction (Scheme 84d). Aldehydes 7.20 are activated by the same catalyst and allow a 1,2-addition of the reactive allylsilane 7.21 in an enantioselective fashion. Homoallylic alcohols 7.22 are obtained in good yields, although the enantioselectivities were not as high as for the Mukaiyama aldol. The methodology presents a good substrate scope, albeit limited to aryl aldehydes (Scheme 84e). Product 7.22a was obtained in good yield and acceptable enantioselectivity (83% ee). Electron-donating groups, as in product 7.22b, are also tolerated even though enantioinduction was diminished (70% ee). Homoalllylic alcohol 7.22c bearing a strong electron-withdrawing motif was afforded in low yield but with good enantiocontrol (38%, 81% ee). On the other hand, product 7.22d having a trifluoromethyl group was obtained with improved yield and excellent enantioselectivity (50%, 96% ee).
The report by the groups of Sai and Yamamoto also points out that every part of the catalyst plays an important role in stereoinduction. Generally, the 3,3′ substituents are regarded as the key feature to be modified in order to increase enantiocontrol. However, not only these scaffolds, but also the active site of the catalyst, both the phosphoryl moiety and the N-electron-withdrawing group, play a crucial role.
Although aldol reactions are mainly in the domain of Lewis acid catalysis, there is plenty of opportunity for Brønsted acid catalysis to be exploited in the aldol and related reactions. Nevertheless, as we have been pointing out throughout the review, catalyst designs and optimisation will still be the most time-consuming part in the reaction discovery process.
The last reaction in this section is not strictly an aldol reaction. However, it headlines an asymmetric addition to an activated carbonyl. The group of Terada developed an enantioselective addition of olefins 7.23 to ethyl glyoxylate (Scheme 85).77 This transformation features perfluorinated BINOL-backbone NTPA, which we discussed earlier (cf.Scheme 69). The reaction affords products 7.24 with good diastereocontrol, albeit with modest yields and enantioselectivities. Nevertheless, the catalyst design and screening offer an alternative viewpoint to the catalyst's backbone and active site. Herein, the focus of catalyst design is the perfluorinated sulfonamide motif, while keeping the perfluorinated BINOL-backbone. Catalyst [A] provided a modest yield with good diastereocontrol (90:10 dr) and with moderate enantiocontrol (69% ee). A pentafluorophenyl group, [B], resulted in a dramatic decrease in yield and diminished enantioselectivity. On the other hand, a longer, perfluorobutyl chain, [C], afforded the best enantioinduction (81% ee), at the cost of yield. Diastereoselectivity, though, seemed to be independent from the fluorinated motif in the active site.
 | ||
| Scheme 85 (a) Enantioselective addition of olefins to ethyl glyoxylate, developed by the group of Terada. (b) Catalyst screening and design with focus on the active site. | ||
The authors did not further elaborate in explaining the differences of reactivity and enantioselectivities with the different active sites. Thus, these reactions serve as a suitable set to develop computational models that can explain the dependence of enantioselectivity on the sulfonamide motif, and not only the role of the trifluoromethyl group in standard NTPA.155
8. Addition reactions to double bonds
Another relatively less exploited area in enantioselective NTPA-catalysed reactions is additions to double bonds. This might be due to the mild but fair nucleophilic character of double bonds to be protonated by a highly acidic catalyst (cf.Scheme 68a). Furthermore, most of the activation of π systems rely on an adjacent electron-withdrawing motif (e.g. a carbonyl group), which makes the electrophile an efficient Michael acceptor. Reactions in which the double bond participates without the need of an adjacent activating moiety are scarce.In the last section, we examined the importance of the active site in enantiocontrol. The group of Terada reported another example in which the active site is thoroughly engineered.75 They developed an enantioselective intramolecular SN2′ reaction of bis-trichloromethylimidates 8.1 to yield heterocycles 8.2 (Scheme 86a). The transformation features the induction of a quaternary stereocentre. The catalyst design is summarised in Scheme 86b. Using a standard NTPA, product 8.2a was obtained in good yield and enantioselectivity (94%, 80% ee). Extending the perfluoroalkyl chain in the active site to perfluorobutyl lead to decreased yield and enantioinduction (79%, 65% ee). Using a perfluorophenyl motif in the sulfonamide component increased enantioselectivity slightly (83% ee). The best result was obtained with a larger perfluoro-2-naphthyl substituent (96%, 89% ee). PA afforded only trace amounts of product.
 | ||
| Scheme 86 (a) Enantioselective intramolecular SN2′ reaction. (b) Catalyst design. | ||
Through experimental and computational studies, the authors concluded that the reaction goes through an anti-SN2′ pathway (Scheme 87). Alternative mechanisms were also evaluated computationally and were found to be energetically disfavoured. Experimental studies suggested that an allyl cation is not involved. One of the trichloroacetimidate motifs acts as the incoming nucleophile while the other is protonated by the catalyst and serves as a leaving group. Initially, the catalyst protonates one of the nitrogen atoms. Then, as the authors propose, a three-point H-bonded transition state is established. The protonated nitrogen atom engages in H-bonding with the oxygen atom in the phosphoryl moiety, ion pairing might also be plausible as an additional stabilising feature. The phosphoryl oxygen also interacts through a non-classical H-bond with the C–H moiety in the π bond. The third contact point is established with one of the oxygen atoms in the sulfonamide motif and the N–H group from the incoming nucleophile.
 | ||
| Scheme 87 Proposed activation mode for the enantioselective intramolecular SN2′ reaction. Several experiments and DFT calculations support this model. All these interactions can effectively lock the substrate inside the catalyst chiral pocket in order to achieve a rigid transition state that accounts for the high levels of enantiocontrol. | ||
The groups of Tan and Liu developed an enantioselective hydroamination reaction.156 For this reaction, alkenes 8.3 with a thiourea handle were found to be suitable substrates (Scheme 88a). Treatment with SPIRO-backbone catalyst (R)-S-NTPA-5 leads to intramolecular cyclisation to afford pyrrolidines 8.4 in high yields and excellent enantioselectivities. The corresponding PA gave lower yield and enantioselectivity. The reaction works well with various substrates having different electronic and steric properties (Scheme 88b). Product 8.4a was obtained in 95% yield and 94% ee. The reaction also works with electron-rich (8.4b, 91% ee) and electron-poor (8.4c, 85% ee) substituents in the thiourea motif. Spiro product 8.4d was also obtained with excellent enantiocontrol (94% ee). In addition, acid-sensible groups could be included, as in the case of acetal 8.4e or alkyne 8.4f, without decrease in enantioselectivity (93% ee and 94% ee, respectively).
 | ||
| Scheme 88 (a) Enantioselective hydroamination reaction of alkenes, developed by the groups of Tan and Liu. (b) Extract from the substrate scope. | ||
The authors propose an activation mode in which the thiourea handle engages in multiple H-bonding with the catalyst (Scheme 89). This model could provide a suitable rigid chiral environment to deliver effective enantiocontrol. It is proposed that the hydroamination goes through a six-membered chair transition state. The authors also suggest two possible ways in which the thiourea interacts with the NTPA active site. Either the thiourea itself (Scheme 89a) or its tautomeric form (Scheme 89b) are suitable substrates for the reaction.
 | ||
| Scheme 89 Suitable transition states for the hydroamination reaction. The substrate can be activated whether through the thiourea (a) or its tautomeric form (b). | ||
The next example has two enantioselective addition reactions, developed by the group of Shi: an asymmetric oxy-sulfenylation and an oxy-selenylation (Scheme 90).157 The authors regard these reactions as desymmetrisations of cyclic olefins. However, we decided to include them within this section, as the reactions are additions to a double bond.
 | ||
| Scheme 90 (a) Asymmetric oxy-sulfenylation and (b) asymmetric oxy-selenylation reactions developed by the group of Shi. | ||
For the oxy-sulfenylation reaction (Scheme 90a), olefins 8.5 react with both 8.6 and benzoic acid 8.7 in the presence of catalyst (S)-NTPA-26. Products 8.8 are obtained in good yields but with low to modest enantiocontrol (25–50% ee). The substrate scope is mainly oriented to cyclic alkenes, with just a couple of examples regarding acyclic olefins. On the other hand, the oxy-selenylation reaction proved to be more efficient (Scheme 90b). In that case, higher enantiocontrol was achieved. Selenylating reagent 8.9 was found to be best for this reaction. Products 8.10 are obtained in 67–84% ee. The better enantiocontrol in the oxy-selenylation reaction, over the corresponding oxy-sulfenylation, might be attributed to the larger size of the selenonium ion involved in the enantiodetermining step.
Regarding the substrate scope for these two reactions, some interesting trends are noticeable (Scheme 91). In terms of ring size from the starting olefin, low enantioselectivities are obtained with five-membered rings. Presumably, the molecules might be small enough to fit loosely inside the catalyst's cavity, thus, yielding diminished enantio-differentiation. For six-membered rings, the enantioselectivites were the highest, suggesting a more ordered transition state. The lowest enantiocontrol is obtained for seven-membered products. These rings may deliver relatively more flexible transition states. For the larger eight-membered rings, enantioinduction increases again.
 | ||
| Scheme 91 Enantioselectivity trends with varying ring sizes in the oxy-sulfenylation and oxy-selenylation reactions. Yields are grayed-out for clarity. These series of examples provide a platform of reactions in order to systematically study these trends through computational chemistry. | ||
Reactivity of NTPA can not only be enhanced by modifying the active site: combination with Lewis acids can lead to acidity enhancement as well. In this context, the group of Zhao reported an enantioselective intermolecular hydroalkoxylation reaction of unactivated alkenes 8.11 to afford chiral tetrahydrofurans 8.12 (Scheme 92a).158 Although good yields and modest enantiocontrol were achieved, the reaction scope is somehow limited. Gem-diaryl substituents in the starting material were found to be crucial for reactivity, indicating a strong Thorpe–Ingold effect on the cyclisation. Mechanistically, the reaction is promoted by the combination of (R)-NTPA-6 and TiCl4, each in 10 mol%. As shown in Scheme 92b, the authors suggest that the combination of these acids might afford a Ti(IV)–NTPA complex 8.13. This complex catalyst, benefits from enhanced Brønsted acidity in the NTPA part. On the other hand, the authors argue that a Lewis acid-catalysed mechanism, in which the NTPA acts as a chiral ligand, cannot be ruled out.
 | ||
| Scheme 92 (a) Enantioselective hydroalkoxylation reaction reported by the group of Zhao. (b) Proposed Ti(IV)–NTPA complex as a stronger Brønsted acid. | ||
9. Rearrangements, desymmetrisations and enantioselective protonations
In this section, we account for those reactions, which we believe best fit into a separate category regarding rearrangements and desymmetrisations. Nevertheless, it may not always be clear whether a reaction ought to be classified as a rearrangement or a desymmetrisation.The reactions covered herein are closely related, and in some cases, the enantiodetermining step is a rearrangement involving the desymmetrisation of the starting material. So far, we have attempted to classify NTPA-catalysed transformations according to a specific reaction intermediate or to the activated electrophile, or to reaction types. The classification we have made is not absolute and some of the readers may think that a certain reaction would belong to a different group instead. As we mentioned before, our main criterion of classification is the enantiodetermining step or the species involved directly on it. We summarise each reaction and highlight what we think are key features or important details in the discovery and development of NTPA-catalysed reactions.
We commence this section with an enantioselective desymmetrisation of azetidines, reported by the groups of Lin and Sun.159 Azetidines 9.1 react with 2-mercaptobenzothiazole 9.2 to yield the desymmetrised product 9.3 (Scheme 93a). The reaction screening started with the search of a suitable catalyst as well as a good N-protecting group. The latter needed to both activate the substrate and increase the leaving group character of the nitrogen atom. During initial studies, at room temperature, PA were tried, but unsuccessfully. The reaction improved when (R)-NTPA-26 was used as Brønsted acid; in addition, the reaction was performed at a higher temperature. The N-protecting group was found to have a profound impact in enantiocontrol (Scheme 93b). Tosyl and acetyl groups showed good reactivity but low enantiocontrol was observed (27% ee and 43% ee, respectively). Benzoyl protecting groups, either electron-poor, electron-rich or neutral, increased the enantioselectivities up to 81% ee. The best results were obtained with the electron-rich 3,4,5-trimethoxy benzoyl protecting group (88% ee).
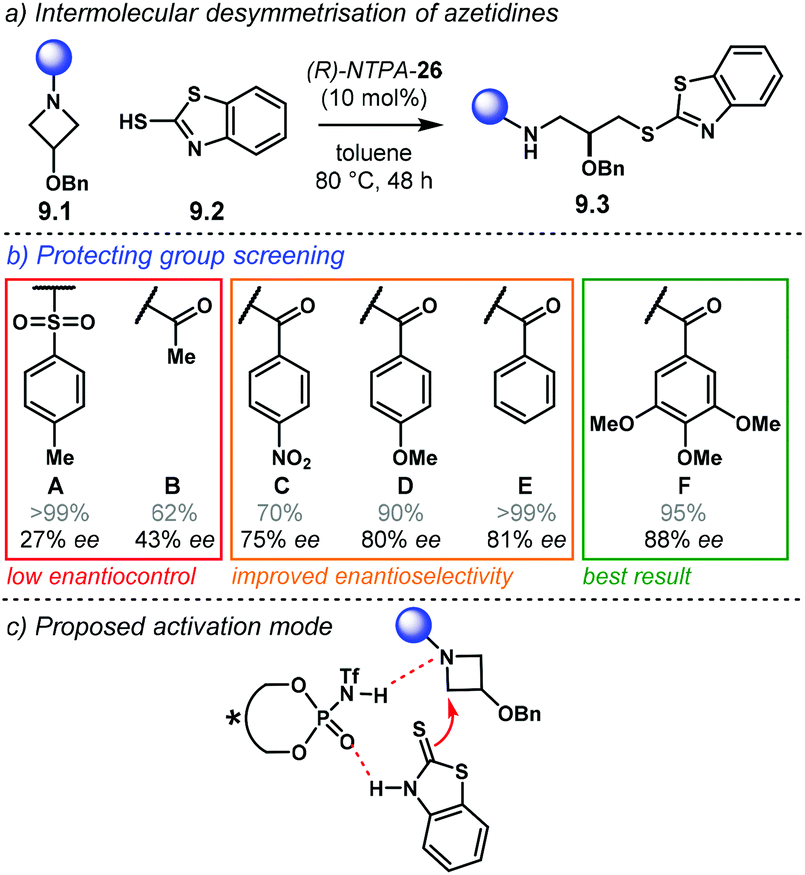 | ||
| Scheme 93 (a) Enantioselective desymmetrisation of azetidines, reported by the groups of Lin and Sun. (b) Substrate screening. (c) Proposed bifunctional activation mode. | ||
The initial screening proved that NTPA were suitable catalysts for this desymmetrisation reaction. Surprisingly, after further studies, it was found that the corresponding PA was indeed a better catalyst in terms of enantioselectivity. Thus, the substrate scope and the rest of their report concerns with the PA-catalysed reaction. The mechanism and activation mode for the NTPA-catalysed reaction is very likely to resemble the one the authors propose for the PA-catalysed reaction. The azetidine is activated through a strong H-bond between the N atom and the acidic H atom in the catalyst. In addition, the nucleophile is H-bonded and directed to the reaction site thus establishing a bifunctional activation mode (Scheme 93c).
The group of Njardarson reported a copper-catalysed ring opening reaction of oxetanes.160 Their study mostly focuses on the copper-catalysed process to yield racemic products. However, they did report one of such reactions using chiral Brønsted acids (Scheme 94). Oxetane 9.4 reacts with chiral Brønsted acids to yield enantioenriched product 9.5. This transformation represents an example in which Brønsted acids with different acidities can perform similarly. (S)-PA-7 afforded the heterocyclic product in modest yield and enantioselectivity (51%, 71% ee). Using the corresponding NTPA, enantiocontrol was improved slightly, at the expense of yield (38%, 73% ee). It might have been possible that the higher acidity of the NTPA lead to product or starting material degradation. The slightly less acidic N-tosylphosphoramide (S)-NTsPA-7 significantly enhanced enantioinduction, albeit with a modest yield (41%, 90% ee).
 | ||
| Scheme 94 Chiral Brønsted acid catalysed ring opening reaction of oxetanes, reported by the group Njardarson. | ||
In another desymmetrisation-rearrangement sequence, the groups of Tu and Zhang reported an intramolecular Schmidt reaction (Scheme 95).161 In this NTPA-promoted reaction, meso-diketones 9.6 afforded enantioenriched bicyclic amides 9.7. However, the reaction is modest-yielding and utilises a stoichiometric amount of (R)-NTPA-22 (1.5 equivalents). In addition, enantiocontrol was moderate (41 to 59% ee).
 | ||
| Scheme 95 Desymmetrisative Schmidt rearrangement. | ||
Regarding the reaction mechanism, diketone 9.6 is activated through a strong H-bond to the triflamide motif as shown in 9.8. Then, the azide moiety attacks in an enantioselective fashion. The authors propose that a stabilising n → π* interaction between the phosphoryl oxygen and the diazo motif might be one factor controlling the desymmetrisation process. Intermediate 9.9 undergoes a ring expansion/contraction process along with nitrogen extrusion to afford product 9.7.
The use of a stoichiometric amount of the Brønsted acid might be due to protonation of the final product. The amide motif might be basic enough to inactivate the catalyst and prevent it from turn over. However, this catalyst inactivation drawback was latter resolved.
One of the first reactions using an NTPA with a SPINOL-backbone was reported in 2013 by the group of Du.162 Their report describes an enantioselective 1,2-rearrangement of racemic epoxides (Scheme 96). Staring materials 9.10 were converted to aldehydes 9.11. Via an NTPA-catalysed protonation-ring opening sequence, intermediate 9.13 is formed. Subsequent 1,2-rearrangement delivers aldehyde 9.11. During the reaction optimisation, the authors found that the aldehyde was unstable. Therefore, one-pot subsequent reduction with NaBH4 was needed. Overall, this sequence affords alcohols 9.12 in moderate yields and modest enantiocontrol (up to 50% ee). PA showed low reactivity. In addition, the reaction was unsuitable for the rearrangement of trisubstituted epoxides.
 | ||
| Scheme 96 Asymmetric rearrangement of racemic epoxides. | ||
Another enantioselective 1,2-shift was creatively exploited in an acyloin rearrangement, reported by the group of Zhu (Scheme 97).163 Acetals 9.14 react with (S)-NTPA-26 to afford α-alkoxy ketones 9.15 in excellent yields and remarkable enantiocontrol for such a difficult transformation (up to 92% ee). PA turned out to be suitable catalysts to induce chirality, but were not reactive enough for good yields (8%, 60% ee). The reaction offers a wide substrate scope, mainly substituted aryl rings. In addition, the alkoxy motif can be varied without loss of enantioselectivity; ethyl, iso-propyl, butyl, cyclopentyl or cycloheptyl acetals performed equally well.
 | ||
| Scheme 97 Enantioselective acyloin rearrangement, reported by the group of Zhu. | ||
The reaction starts with protonation of acetal 9.14. An equilibrium is established. Elimination of cyclohexanol from 9.16 delivers oxy-carbenium ion 9.17. The authors propose that this species is highly stabilised by tight ion pairing. In addition, the phosphoryl moiety in the catalyst engages in H-bond with the –OH motif in the intermediate. This establishes an H-bond/ion-pairing bifunctional activation mode. Then, enantioselective 1,2-migration of one aryl motif takes place. Simultaneous regeneration of the catalyst delivers enantioenriched product 9.15.
Another closely related rearrangement, involving a 1,2-migration is the pinacol, and semi-pinacol, rearrangement. These reactions have been widely studied in the field of organocatalysis. Several of these transformations have been reported using PA. In addition, NTPA are also suitable catalysts for these rearrangement reactions.
The group of Zhu, developed an enantioselective 1,4-migration reaction; a vinylogous pinacol rearrangement (Scheme 98a).164 (E)-1,4-Diols 9.18 undergo such rearrangement to afford ketones 9.19 in good yields and with excellent enantiocontrol (up to 96% ee). BINOL-backbone catalyst (S)-NTPA-26 was the best one for getting high yields and enantioselectivities. PA provided modest reactivity but low enantioinduction. The reaction afforded the best enantiocontrol in MTBE as solvent. The 1,4-rearrangement did not work with the corresponding (Z)-1,4-diols.
 | ||
| Scheme 98 (a) Enantioselective vinylogous pinacol rearrangement, reported by the group of Zhu. (b) Proposed bifunctional activation mode: H-bonding/ion-pairing. | ||
The reaction proceeds via a protonation–dehydration mechanism. This delivers carbocation intermediate 9.20 (Scheme 98b). The electron-donating group in the aryl motifs controls regioselectivity in the dehydration step. The carbocation is formed adjacent to the most electron rich groups. In addition, electron-rich heterocycles, like thiophene, were suitable to control regiochemistry. Allylic carbocation 9.20 is proposed to be stabilised through H-bonding and tight ion pairing. These interactions would provide a rigid bifunctional activation mode in order to account for the high levels of enantiocontrol.
Using the protonation–dehydration strategy, the group of Zhu developed a set of two complementary methodologies involving an enantioselective 1,2-migration rearrangement.165 These reactions grant access to an all-carbon quaternary stereocentre.
The first of these reactions comprises an enantioselective pinacol rearrangement (Scheme 99a). Diols 9.21 undergo a protonation–dehydration sequence to afford carbocation 9.22. The alkyne and the electron-rich groups favour site-selective dehydration. Both H-bonding and tight ion pairing activate this intermediate. Such a bifunctional mode can provide a rigid transition state inside the catalyst chiral pocket. Then, 1,2-migration expands the five-membered ring to deliver cyclohexanone 9.23 with an all-carbon quaternary stereocentre. (S)-NTPA-26 was the best catalyst for this transformation. PA were also tried but yielded low enantioselectivities. For this pinacol rearrangement, the authors propose that an intermediate pQM 9.24 might be plausible (Scheme 99b). In that case, a bifunctional activation is also possible, presumably via a double H-bond; however, it was found that a free –OH moiety in the substrate is not necessary to achieve high enantioinduction. Therefore, an H-bonding/ion-pairing bifunctional mode might be operational.
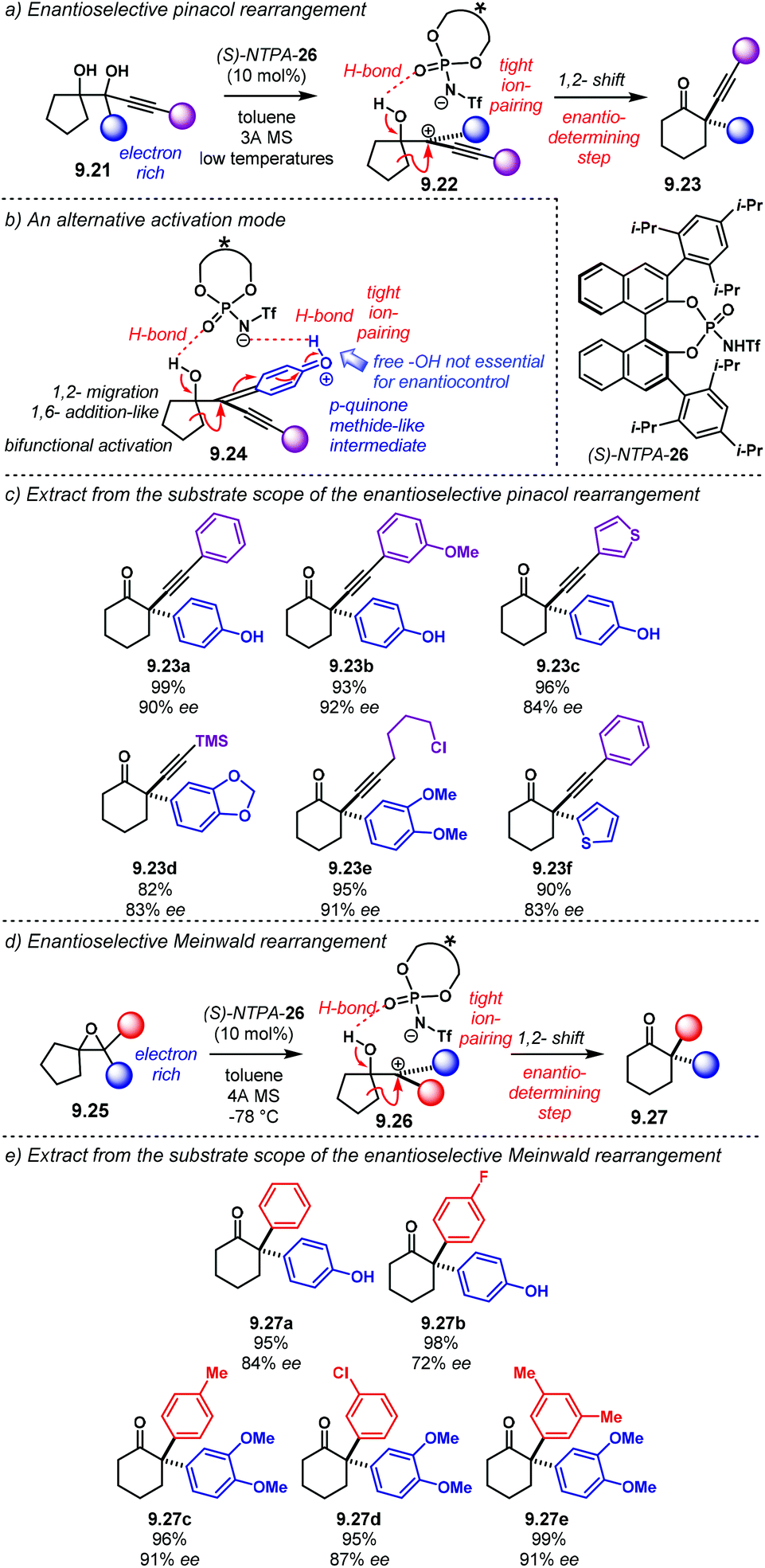 | ||
| Scheme 99 1,2-Rearrangement reactions reported by the group of Zhu. (a) Enantioselective pinacol rearrangement. (b) Alternative activation mode proposed by the authors. A pQM intermediate might be a likely intermediate. (c) Extract from the substrate scope for the pinacol reaction. (d) Enantioselective Meinwald rearrangement. (e) Extract from the substrate scope for the Meinwald reaction. | ||
The reaction shows a broad substrate scope, delivering enantioenriched cyclohexanones in high yields and excellent enantiocontrol (Scheme 99c). Substitution in the alkyne is tolerated as well. Products 9.23a–c are obtained with good enantiocontrol (90% ee, 92% ee and 84% ee, respectively). Relatively acid-sensitive groups like TMS in 9.23d were tolerated, yielding good enantioselectivity (83% ee). Functionalised alkyl chains, like in 9.23e were also suitable (91% ee). In addition, heterocyclic product 9.23f was obtained with good enantiocontrol (83% ee).
The second reaction comprises an enantioselective Meinwald rearrangement of spiro epoxides 9.25 (Scheme 99d). Protonation and subsequent ring opening furnishes intermediate 9.26, similar to 9.22. The electron-rich groups in the substrate drive ring-opening selectivity. A bifunctional H-bonding/ion-pairing activation mode is likely to be operating. Enantioselective 1,2-migration delivers cyclohexanones 9.27. Because the size of the aryl substituents is similar, enantioselectivity seems to be dependent on electronics. Enantiomeric excesses for this methodology are also remarkable, albeit the substrate is not as broad as for the pinacol reaction (Scheme 99e). Products 9.27a–e are obtained in good yields and enantiocontrol (up to 91% ee).
This pinacol rearrangement was remarkably useful for a short total synthesis of (+)-mesembrane 9.32 (Scheme 100). Starting from acyl chloride 9.28 and 1,2-dimethoxy benzene 9.29, key 1,2-diol precursor 9.30 was synthesized. Then, treatment with (S)-NTPA-26 followed by removal of the TMS group afforded key intermediate 9.31 in 92% yield and 90% ee. After further steps, including hydroformylation and an intramolecular reductive amination, (+)-mesembrane 9.32 was obtained.
 | ||
| Scheme 100 A short asymmetric total synthesis of (+)-mesembrane. | ||
The group of Sun developed a similar enantioselective House–Meinwald rearrangement using PA.166 Products similar to those presented in Scheme 99e were obtained as single regioisomers, in good yields and excellent enantioselectivities. However, the substrate needs a free –OH moiety in order to work with PA. The development of the methodology reported by the group of Sun mainly uses PA. Nevertheless, two examples stand out regarding NTPA and are presented in Scheme 101. In those reactions, the free –OH motif is methylated. In the first example, (Scheme 101a) epoxide 9.33 undergoes a House–Meinwald rearrangement to furnish product 9.34, enantioselectively. (R)-S-PA-12 afforded the product with full conversion, albeit with 39% ee. On the other hand, (R)-[H8]-NTPA-14 gave a 90% yield with 78% ee and opposite sense of enantioinduction. Secondly, for the acyclic substrate 9.35 (Scheme 101b), (S)-PA-4 delivered product 9.36 with 76% conversion and a low 28% ee. Using the more acidic (S)-S-NTPA-11, 82% yield and 72% ee were achieved.
 | ||
| Scheme 101 House–Meinwald rearrangements reported by the group of Sun, using (a) cyclic substrates and (b) acyclic substrates. | ||
In these reactions, enantioselectivity seems to be more dependent on the Brønsted acid counter anion (Scheme 102). This suggests that two strong H-bonds are participating in a bifunctional activation mode, 9.37, when a PA is used as catalyst. In contrast, if a second H-bond is not possible to be formed, the second interaction is more likely to be ion pairing, as in 9.38. A similar bifunctional H-bond/ion-pairing activation mode is expected for NTPA, 9.39.
 | ||
| Scheme 102 Bifunctional activation modes proposed for the House–Meinwald rearrangement reported by the group of Sun. | ||
These two examples provide another set of reactions to develop computational models to explain enantioselectivity for an H-bonding/ion-pairing activation mode. In addition, the authors do not discuss the differences in the two catalysts: the chiral backbones and the substituents in the 3,3′ positions. Therefore, a computational model can be extremely helpful to understand the ways in which different chiral frameworks and scaffolds transfer the chiral information to the products.
To finalise this section, we include an example of an enantioselective protonation reaction, developed by the groups of Zhu and Zhou.167 The reaction features a sulfa-Michael addition reaction to exo-cyclic ene-ones 9.40 (Scheme 103). Thiols 9.41, with a wide variety of substitution, including aryl and alkyl groups, were suitable nucleophiles. α-chiral ketones 9.42 were obtained in excellent yields and with remarkable enantiocontrol (up to 97% ee). (R)-S-NTPA-8, having a highly congested active site, was found to be the best catalyst for this transformation.
 | ||
| Scheme 103 Sulfa-Michael addition/enantioselective protonation reaction, developed by the groups of Zhu and Zhou. | ||
Mechanistically, the NTPA locks both substrates through H-bonding, as shown in 9.43. This provides a bifunctional activation mode. However, during the 1,4-addition step, no chiral centres are induced. Then, enol intermediate 9.44 is protonated enantioselectively to deliver the final product. This enantioselective protonation step is likely to happen through a mono-activation mode with the electrophile as the acidic proton of the catalyst itself. The authors propose an eight-membered-ring transition state. This process regenerates the catalyst at the same time the enol is protonated.
In addition, this reaction can also be suitable for DFT studies principally to understand how the catalyst transfers the chiral information to the product. Since a proton is the electrophile, both the SPINOL-backbone and the bulky scaffolds are playing important roles for enantio-differentiation.
10. Mnemonics and overview of mechanistic analyses
The detailed mechanisms of the reactions discussed throughout the review and the factors which determine their regio- and stereoselectivity are complex and diverse. In this section, we outline some mnemonics which summarise the results of a large number of these results. These mnemonics are not the firmly-established conclusions of exhaustive computational work. They simplify a complex landscape of reactivity and selectivity. However, we present them as useful, qualitative, guides which can aid in understanding, which can provide a fast, initial analysis of a reaction, and can make it possible to distinguish results which fit well into established reactivity patterns and ones which fall outside these frameworks.We represent the chiral NTPA catalysts as illustrated in Fig. 3. This simplified form illustrates how the flat naphthyl groups create a chiral cavity around the central H–N–PO atoms which form interactions with the reaction substrates. PA behave in a similar way to NTPA, except that they are slightly weaker acids and slightly less effective hydrogen-bond acceptors. The triflyl group may accept hydrogen bonds through the sulfone oxygens, and has a small steric presence in the chiral cavity.
 | ||
| Fig. 3 Three representations of NTPA. The left-hand form is commonly used to record the structure unambiguously. The centre representation illustrates the molecule with the central N–PO unit in the plane of the paper. The right-hand form, which simplifies the naphthyl groups and omits the 3,3′ substituents, looks along the carbon–carbon bond linking the two naphthyl groups and is a useful basis for mnemonics. SPINOL-derived catalysts, such as S-NTPA-11 (cf.Scheme 13), may also be represented in the right-hand simplified form. | ||
We have used this representation to illustrate the selectivity of a number of reactions with chiral PA, including asymmetric allylboration of aldehydes and ketones,168,169 addition reactions of imines,114,116,170–172 Friedel–Crafts reactions of indole with imines,115 propargylation of aldehydes and ketones,173,174 boronate additions,175 and Pictet–Spengler reactions.176
The same mnemonics can be used to outline the diverse reactions we have recounted in sections 2–9 which all have NTPA catalysts and can be divided into groups which can mainly be described using appropriate diagrams of this type. These diagrams can be sketched by hand without a computer or the need of sophisticated calculations. Many of the reactions in this review can be explained by assigning them to one of two classes:
(A) The catalyst forms two hydrogen bonds with one of the substrates.
(B) The catalyst interacts with two different substrates and helps them interact in a specific way (bifunctional activation).
These classes of mechanisms, or activation modes, are directly involved in the enantiodetermining step and, therefore, control the stereochemical output of a reaction. These mnemonics are illustrated in Fig. 4. Still, there are reactions which do not seem to fit neatly into these classes, and there may well be other origins of the selectivity in such cases. For example, it may be that the NTPA catalyst coordinates to one part of the molecule and then acts as an internal base to remove a specific proton from the complex and trigger an enantioselective rearrangement.177 Nonetheless, classes A and B have substantial explanatory power.
 | ||
| Fig. 4 Mnemonics for two ways of controlling stereochemistry. (a) Class A. Two point binding/activation of an electrophile. An aldehyde (left) and a Z-imine (right) coordinating in the catalyst active site. The imine's thermodynamic configuration may be E, but the Z geometry coordinates better to the catalyst. The sp2 carbon which is attacked by the nucleophile has its left-hand face (as drawn) shielded, and its right-hand face exposed. The preferred approach of a nucleophile will be from the right-hand side, at the front of the structure. (b) Class B. Bifunctional activation. Herein, the catalyst locks both substrates inside the chiral cavity and provides a rigid, asymmetric environment to achieve enantioinduction; not only imines, but also other electrophiles prone to form hydrogen bonds fit in this model as well. | ||
Our mnemonics can be used to help remember the outcomes of many reactions, and they may also give some insights into the reasons for the stereoselectivity. However, rigorous computational analysis, which would be expensive and time-consuming, is needed to provide quantitative analyses. The rest of this section shows how classes (A) and (B) from Fig. 4 can be used to account for many of the reactions in this review.
10.1. Type A: the catalyst forms two hydrogen bonds with one of the substrates
The first group is reactions for which one of the substrates forms two hydrogen bonds with the catalyst, and the other component of the reaction then attacks this complex. These reactions often involve an aldehyde, imine or iminium unit, and the other components of the reaction are unlikely to form hydrogen-bonding interactions with the catalyst. In addition, these reactions can be inter- or intramolecular. The catalyst–substrate complex is illustrated in Fig. 4a.Fig. 5 applies this idea to summarise the enantioselectivity observed in reactions from Schemes 8, 11, 81, 82 and 85, for aldehydes. In these cases, two hydrogen-bonding interactions are formed with the aldehyde. The first of which, between the acidic proton on the catalyst and the oxygen atom in the aldehyde. The second, between a Brønsted basic site in the catalyst and the formyl C–H moiety.
 | ||
| Fig. 5 Applications of the mnemonic to aldehyde-based reactions, with the conformation constrained by a formyl hydrogen-bond. In these examples, the nucleophile approaches the unhindered face of the substrate-catalyst complex. | ||
Fig. 6 applies our mnemonics for the imine-based reactions presented in Schemes 9, 10, 38, 41, 44, 48, 49, 51 and 53. Similar to the activation of aldehydes, iminium ions are locked into the catalyst's active site through two hydrogen bond interactions. One between the N–H motif and the second one with the formyl-like C–H moiety. However, for that case, the imine must have a (Z) configuration. Although the thermodynamic configuration may be (E), (E)/(Z) isomerisation is possible in acidic media.178,179 In addition, the polarity of the reaction media can influence the (E)/(Z) ratio in aldimines.180
 | ||
| Fig. 6 Applications of the mnemonic to imine-based reactions. | ||
Additions to aldehydes and to imines are the most common reactions in our review. However, there are also molecular fragments which may have similar arrangements. The carbonyl oxygen or imine nitrogen could form a hydrogen bond with the N–H moiety of the triflimide and an adjacent hydrogen from the substrate forms an interaction with the PO motif. This means that enones, quinones or hydrazones might be able to coordinate with a defined geometry that will influence the selectivity of nucleophilic attack, just as it can with aldehydes and imines. This idea, using our mnemonics is illustrated in Fig. 7.
 | ||
| Fig. 7 Applications of the mnemonic to reactions involving (a) enones, α-hydrogen coordination; (b) quinones, β-hydrogen coordination; (c) hydrazones. | ||
Some molecules will be able to coordinate in different ways. Acrolein (CH2CHCHO), for example, could form a formyl hydrogen-bond or have α-hydrogen coordination, and these two modes would lead to opposite selectivity. In general, we might expect that the most energetically-favourable mode would be selected, and so acrolein would form a formyl hydrogen bond rather than showing α-hydrogen coordination. However, α-hydrogen coordination would be favoured over β-hydrogen coordination, for molecules which can show both.
Fig. 8a applies this construction to summarise the enantioselectivity observed in Schemes 7, 19 and 80 for coordination to an α-hydrogen. Fig. 8b does the same thing for the β-hydrogen-coordinated processes for reactions in Schemes 19, 21, 22, 57, 73 and 103. In addition, as shown in the example from Scheme 5, more distant coordination may also be possible.
 | ||
| Fig. 8 Applications of the mnemonic to reactions which can be explained by (a) α-hydrogen coordination, (b) β-hydrogen coordination, and (c) Mnemonic suggests an explanation for a reverse in selectivity: a switch from α-hydrogen coordination to β-hydrogen coordination, driven by the need to accommodate the methyl group on the imine nitrogen. | ||
Comparison of the reactions in Schemes 19 and 21 gives a surprising reverse in selectivity for two closely related processes. The reactions are compared in Fig. 8c. If these two processes had similar transition states, the reverse in enantioselectivity would be hard to explain. The mnemonic suggests an explanation, however. Coordination between the chiral catalyst and the α-hydrogen is consistent with the observed selectivity for the reaction from Scheme 19. Such coordination is hard for an imine (reaction from Scheme 21), because the alkyl group on the imine nitrogen would be forced to be close to the adjacent double bond. To avoid this, β-hydrogen coordination may be preferred. This inverts the orientation of the molecule in the catalyst chiral pocket and naturally leads to the inverse enantioselectivity. The reverse in selectivity changing from a carbonyl to an imine is switched back if the imine reacts with ortho-hydroxy styrene (cf.Scheme 20). This is explained in the next section.
Finally, Fig. 9 utilises our mnemonics for the two hydrazone processes that were presented in Scheme 12, in which the substrate forms two hydrogen bonds with the catalyst.
 | ||
| Fig. 9 Hydrazones can also form two hydrogen-bonding interactions with the catalyst. | ||
10.2. Type B: catalyst interacts with different substrates and helps them interact in a specific way (bifunctional activation)
Chiral BINOL-derived PA control many reactions by coordinating to one reactant with its P–O–H hydrogen-bond donor and the other with its PO hydrogen-bond acceptor. This insight enabled us to account for the selectivity of the Hantzsch-ester hydrogenation of imines.117 In addition, we have built on this to construct general models of imine reductions by this class of asymmetric catalysts.59,114,171 BINOL-derived NTPA fit into these models, with the P–O–H hydrogen bond donor replaced by P–N–H moiety. These models can readily explain the outcomes of many reactions. Fig. 10 shows mnemonics for other reactions from this review, which fit into a similar scheme.
 | ||
| Fig. 10 Reactions for which the catalyst brings together two components in a defined orientation to react. Bifunctional activation. | ||
In these examples, the largest species occupies the empty space of the chiral cavity, either at the front or at the back, in order to avoid steric clash with the catalyst's substituents. Therefore, it is sensible that the way in which the bulkiest component is arranged inside the chiral pocket dictates the sense of stereoinduction. We have shown that this is true for PA-catalysed reactions of imines and it is likely to occur for NTPA as well.181,182
This bifunctional model can be used to account for several experimental observations. For example, the reverse in selectivity changing from a carbonyl to an imine (cf.Fig. 8c) is switched back if the imine reacts with ortho-hydroxy styrene (cf.Scheme 20). The β-hydrogen coordination suggested for the imine reactions in Fig. 8c is not very strong. The presence of a hydroxy group in the other reactive species makes it possible to form a stronger hydrogen-bonding interaction so that the catalyst coordinates to both components of the reaction (Fig. 11). This brings them together in a rigid conformation, which accounts for the experimentally observed selectivity. In these mnemonics, the bulkiest part of the imine component, the PMP group, allocates in the empty space of the chiral cavity.
 | ||
| Fig. 11 If the second component of the reaction contains a good hydrogen good donor, then it may displace coordination to α-hydrogen or β-hydrogen coordination. | ||
In a slightly different type of activation, the mnemonics are also helpful to explain stereoselectivity for reactions which do not strictly form hydrogen bonds to the reacting species. One example is the reaction that was presented on Scheme 47 (Fig. 12). In this reaction, it is proposed that both catechol borane components are locked by the catalyst's active site. In the mnemonics, these motifs occupy the empty space of the chiral cavity, accounting overall for the observed enantioselectivity.
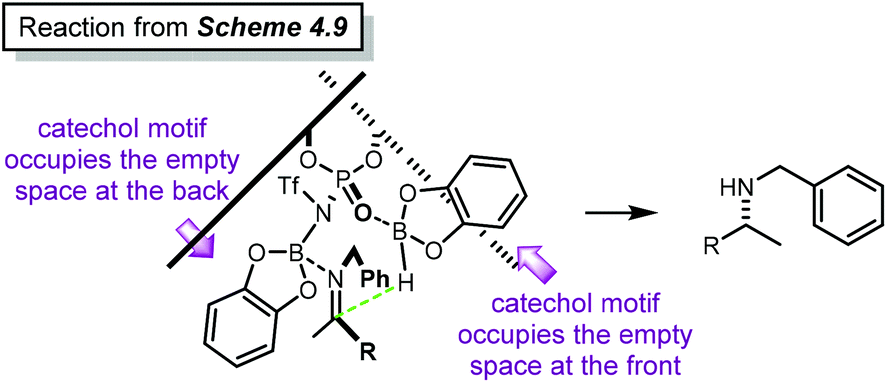 | ||
| Fig. 12 In this example, the hydrogen of the hydrogen bond donor is replaced by boron. | ||
One of the main advantages of our mnemonics is that they can be used to predict the sense of enantioinduction in reactions where this has not been experimentally determined. However, this does not provide a substitute for rigorous experimental determination of the absolute stereochemistry of a product, but it provides a useful and qualitative answer. One example is the reaction presented on Scheme 15, for which the authors did not report what enantiomer was obtained.
Fig. 13 illustrates how our mnemonics can be used to predict stereochemistry. In this case, the electrophilic substrate allocates the bulkiest substituents on the empty space at the back of the chiral cavity. Herein, the N–H moiety from the indole forms a hydrogen bond with the triflyl motif, as it was shown by the authors that such interaction was essential for reactivity. Therefore, the alkene counterpart has to be locked in the front of the catalyst's active site. This model predicts the major enantiomer to have an (R) stereochemistry. If the (S) enantiomer was to be obtained, the electrophile would have to allocate the phenyl groups next to the catalyst's scaffold. In this case, steric clash would disfavour this approach.
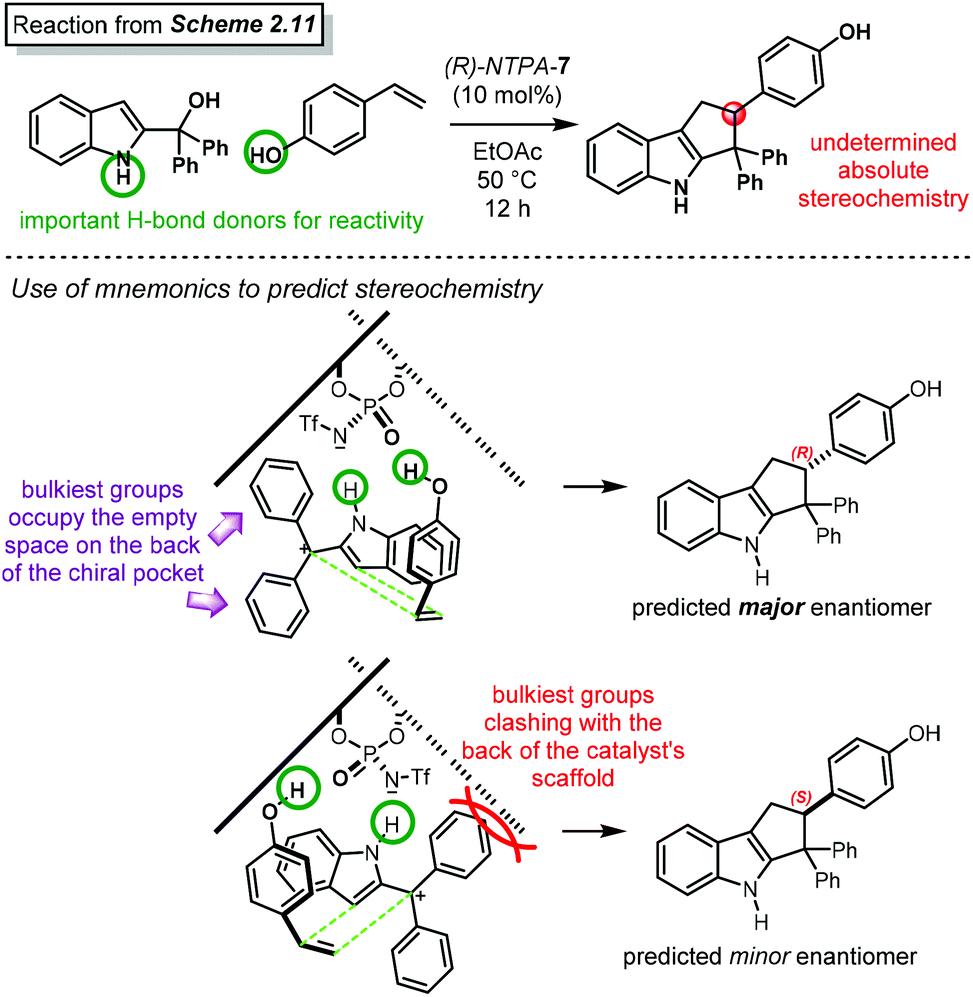 | ||
| Fig. 13 Prediction of stereochemistry using the mnemonic system. In order to account for enantiocontrol, the bulkiest group in the cationic intermediate is placed at the back of the chiral cavity to avoid steric clash with the 3,3′ substituents. | ||
10.3. Bifunctional activation through ion-pairing/hydrogen bonding
The substrates from the reactions discussed in the past section are activated through hydrogen bonding. Therein, these interactions bring together two reactive species in an enantioselective fashion. However, it is also possible that one of the reagents is locked to the active site of the catalyst through ion pairing.183–186 In order to build up the mnemonics, we propose a bifunctional activation mode in which one of the substrates is anchored through ion-pairing and the other via hydrogen bonding. In this case, a cationic intermediate interacts with the anion of the NTPA catalyst. This is illustrated in Fig. 14 for the case of a benzylic carbocation. | ||
| Fig. 14 Bifunctional activation through tight ion-pairing/H-bonding. The left picture shows a proposed interaction between the π density of a phenyl ring with the triflyl motif of the NTPA. The representation in the centre simplifies the aforementioned ion-pair, as shown in the aromatic group highlighted in blue. The picture on the right shows the bifunctional ion-pairing/hydrogen bonding activation mode in the mnemonics. | ||
Most of cationic intermediates bear an aromatic ring, in order to stabilise such an intermediate. Therefore, we propose that a non-covalent interaction is established between the π density of the aromatic group and the triflyl anion in the catalyst's active site. In this case, the bulkiest substituent of the carbocation is sit on the empty space in the back of the chiral cavity (Fig. 14, right picture). Once this ion-pair is established, the nucleophile forms a hydrogen bond with the phosphoryl moiety in the catalyst. These two interactions, then, brings together both reactive species, again, in an enantioselective fashion.
Several of the reactions discussed in section 6 involve a carbocation stabilised by an adjacent aryl group. Using our mnemonics, we can provide a qualitative model to account for stereocontrol. Fig. 15 shows the mnemonics to explain enantioselectivity for reactions from Schemes 66, 68, 70, 74 and 75. In these transformations, the bulkiest groups occupy the empty space in the chiral cavity, whether at the front or at the back of the catalyst's framework.
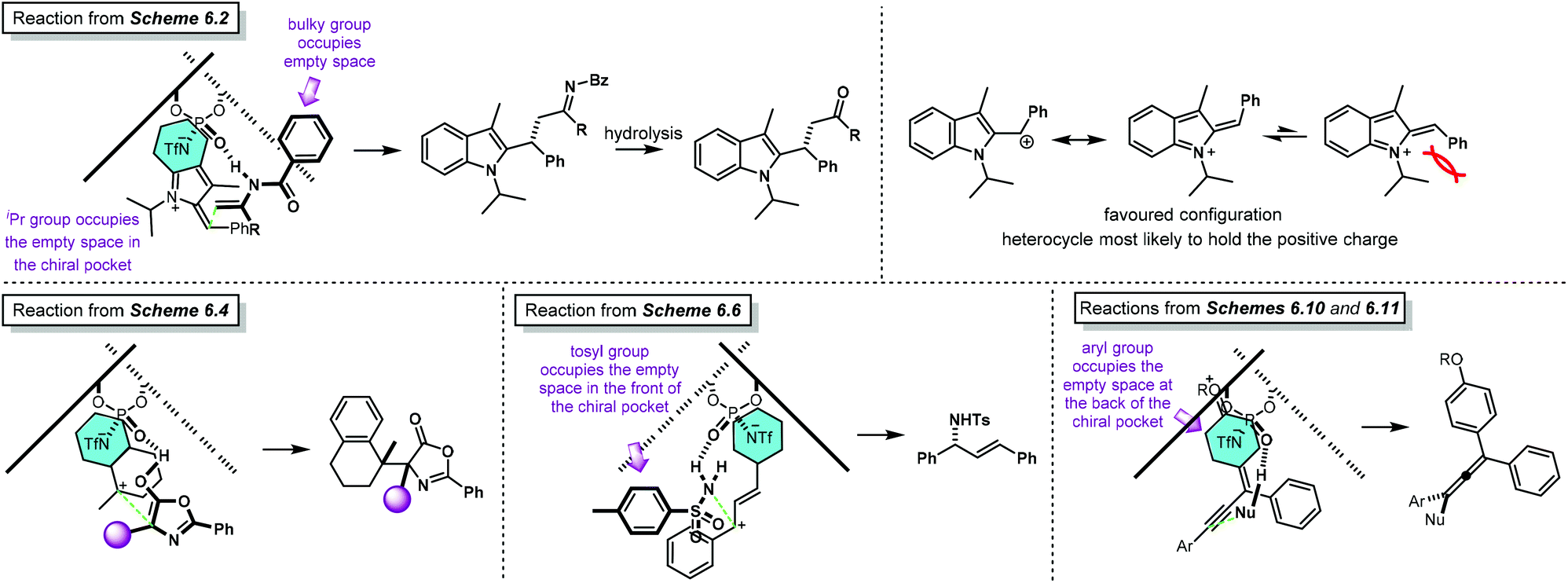 | ||
| Fig. 15 Mnemonics for a bifunctional ion-pairing/hydrogen bonding activation mode. In all of these reactions, a positive charge delocalised over an aromatic ring (coloured blue) provides a favourable interaction with the anionic triflyl group. In the reaction from Scheme 66, the reaction is reported with an iso-propyl group on the indolyl nitrogen. The bulk of this group is sufficient to make the intermediate cation prefer the E configuration. | ||
10.4. Alternative activation modes
In the vast majority of the reactions summarised in this review, the NTPA catalyst activates the substrates through non-covalent interactions, hydrogen bonding or ion-pairing. However, in a couple of cases, the catalyst forms a covalent bond with one of the reagents (cf.Schemes 38 and 47). It is also possible to account for stereocontrol using our mnemonics system. This idea was originally illustrated in Fig. 12 and the mnemonics can be adapted for the electrocyclic reaction described in Scheme 38, in which the active catalyst is the brominated thio-NTPA.Fig. 16 shows the transition states for the enantioselective bromination of the double bond with the active catalyst—the brominated thio-NTPA. Therein, in the favoured TSs, leading to the major enantiomer, the R group occupies the empty space of the chiral cavity in order to avoid steric clash with the catalyst's scaffolds. On the other hand, the disfavoured TSs sit this group in such a way that clashes with the 3,3′ substituents. Once the bromonium ion is formed in an enantioselective fashion, the electrocyclisation is triggered to yield the final product.
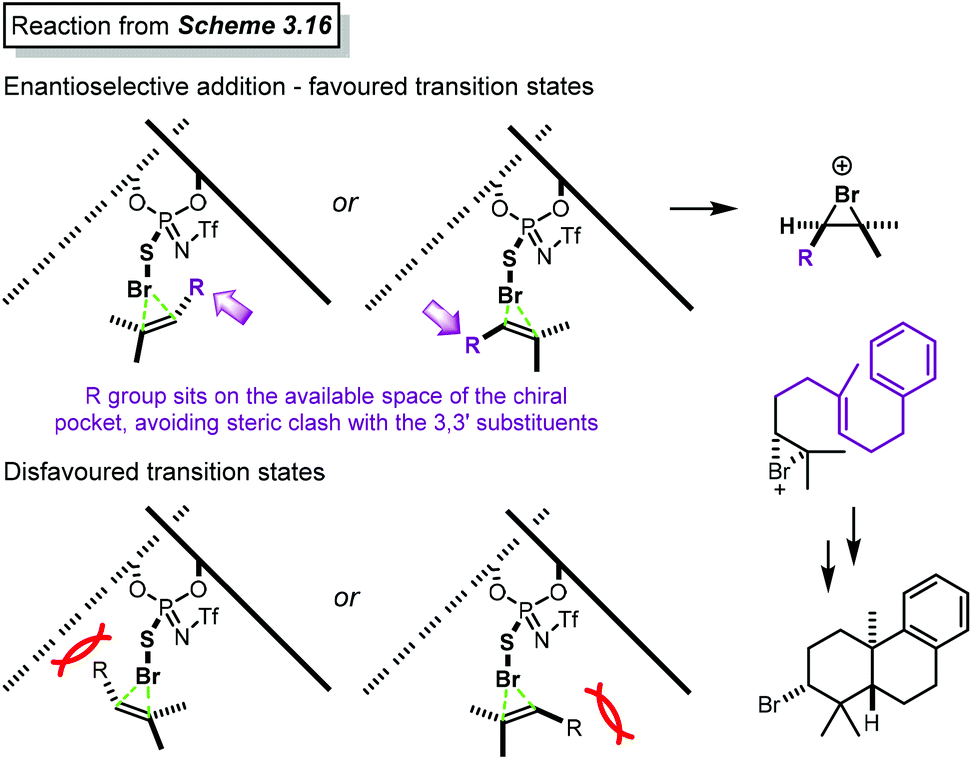 | ||
| Fig. 16 Activation through a covalently bound reagent. In the favoured transition state, the R group sits in the empty space of the chiral pocket to avoid steric clash with the 3,3′ substituents. | ||
Despite the fact that the previously discussed reaction is not strictly catalysed by the Brønsted acid per se, it provides an example in which our mnemonics fit to explain enantiocontrol, therefore, disclosing the predictive and explanatory power of our qualitative diagrams.
11. Conclusions and outlook
Enantioselective organocatalysis is a challenging and constantly growing field. NTPA are shown to be efficient and versatile catalysts for asymmetric transformations. This is shown by the large number of reactions reported in the past decade using this type of chiral Brønsted acids.In this review, we presented those reactions developed from 2011 to early 2020. These transformations were discussed in detail and allocated into one of the following categories. The enantiodetermining step was considered as key criterion. This classification is not absolute and some reactions can fit into another or in several groups.
• Cycloadditions
• Electrocyclisations
• Addition reactions to imines
• Electrophilic aromatic substitution reactions
• Addition reactions to carbocations
• Aldol and related reactions
• Addition reactions to double bonds
• Rearrangements, desymmetrisations and enantio-selective protonations
In addition, the available experimental methodologies to install the active site of NTPA were discussed, highlighting potential advantages and drawbacks from each method. The construction of the catalyst's chiral scaffold was also briefly covered. The successful application of NTPA as catalysts in key steps in total syntheses was highlighted as well.
NTPA outperform the corresponding PA in the majority of the reactions we presented, mainly due to their higher acidity. However, in limited cases better reactivity and/or stereocontrol was achieved with PA. Furthermore, in some reactions both PA and NTPA perform equally well. Even though catalyst screening is a trial and error process, we suggest the following cases in which NTPA ought to be tried straightway as catalysts in the development of new asymmetric methodologies. This, mostly regarding their high acidity.
• Reactions that involve a protonation–dehydration sequence to produce a reactive intermediate (carbocations or quinone methides)
• Reactions involving a direct protonation of a double bond
• Reactions using carbonyl compounds as starting materials (aldehydes and ketones) as well as Michael acceptors
• Reactions in which the nucleophile is a rather unreactive (hetero)aryl group
Finally, throughout the review, the modes in which NTPA activate the reacting species were discussed in detail. In the last section, we presented a mnemonic system to explain and account for the stereochemical output in NTPA-catalysed transformations which fits most of the reactions we covered.
Choosing the right catalyst for a particular transformation is not always an easy task and we hope that the analysis and mnemonics in this review will be helpful for researchers in this area, both to develop new enantioselective methodologies and to design more efficient and sustainable catalysts.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
GC thanks CONACYT and the Cambridge Trust for providing the funding with a CONACYT Cambridge scholarship (reference 600435/438243).Notes and references
- D. Stevenson and G. A. Williams, in Chiral Separations, Springer, Boston, MA, 1988, pp. 1–9 Search PubMed.
- N. A. McGrath, M. Brichacek and J. T. Njardarson, J. Chem. Educ., 2010, 87(12), 1348–1349 CrossRef CAS.
- L. Bernardi, M. Fochi, M. C. Franchini and A. Ricci, Org. Biomol. Chem., 2012, 10, 2911–2922 RSC.
- J. Seayad and B. List, Org. Biomol. Chem., 2005, 3, 719–724 RSC.
- T. Akiyama, Chem. Rev., 2007, 107, 5744–5758 CrossRef CAS PubMed.
- M. Mahlau and B. List, Angew. Chem., Int. Ed., 2013, 52, 518–533 CrossRef CAS PubMed.
- G. Zhan, W. Du and Y. C. Chen, Chem. Soc. Rev., 2017, 46, 1675–1692 RSC.
- I. Agranat, H. Caner and J. Caldwell, Nat. Rev. Drug Discovery, 2002, 1, 753–768 CrossRef CAS PubMed.
- H. Caner, E. Groner, L. Levy and I. Agranat, Drug Discovery Today, 2004, 9, 105–110 CrossRef CAS PubMed.
- D. M. Walden, O. M. Ogba, R. C. Johnston and P. H. Y. Cheong, Acc. Chem. Res., 2016, 49, 1279–1291 CrossRef CAS PubMed.
- T. Akiyama, J. Itoh, K. Yokota and K. Fuchibe, Angew. Chem., Int. Ed., 2004, 43, 1566–1568 CrossRef CAS PubMed.
- D. Uraguchi and M. Terada, J. Am. Chem. Soc., 2004, 126, 5356–5357 CrossRef CAS PubMed.
- M. Yamanaka, J. Itoh, K. Fuchibe and T. Akiyama, J. Am. Chem. Soc., 2007, 129, 6756–6764 CrossRef CAS PubMed.
- I. D. Gridnev, M. Kouchi, K. Sorimachi and M. Terada, Tetrahedron Lett., 2007, 48, 497–500 CrossRef CAS.
- D. Parmar, E. Sugiono, S. Raja and M. Rueping, Chem. Rev., 2014, 114, 9047–9153 CrossRef CAS PubMed.
- J. M. M. Verkade, L. J. C. Van Hemert, P. J. L. M. Quaedflieg and F. P. J. T. Rutjes, Chem. Soc. Rev., 2008, 37, 29–41 RSC.
- J. Shen and C. H. Tan, Org. Biomol. Chem., 2008, 6, 3229–3236 RSC.
- F. E. Held, D. Grau and S. B. Tsogoeva, Molecules, 2015, 20, 16103–16126 CrossRef CAS PubMed.
- J. Yu, F. Shi and L.-Z. Gong, Acc. Chem. Res., 2011, 44, 1156–1171 CrossRef CAS PubMed.
- M. Rueping, A. Kuenkel and I. Atodiresei, Chem. Soc. Rev., 2011, 40, 4539–4549 RSC.
- S. J. Connon, Angew. Chem., Int. Ed., 2006, 45, 3909–3912 CrossRef CAS PubMed.
- P. Renzi, J. Hioe and R. M. Gschwind, Acc. Chem. Res., 2017, 50, 2936–2948 CrossRef CAS PubMed.
- M. Terada, Chem. Commun., 2008, 4097 RSC.
- A. Zamfir, S. Schenker, M. Freund and S. B. Tsogoeva, Org. Biomol. Chem., 2010, 8, 5262–5276 CAS.
- A. K. Mutyala and N. T. Patil, Org. Chem. Front., 2014, 1, 582–586 RSC.
- D. Nakashima and H. Yamamoto, J. Am. Chem. Soc., 2006, 128, 9626–9627 CrossRef CAS PubMed.
- M. Rueping, B. J. Nachtsheim, W. Ieawsuwan and I. Atodiresei, Angew. Chem., Int. Ed., 2011, 50, 6706–6720 CrossRef CAS PubMed.
- M. Rueping, W. Ieawsuwan, A. P. Antonchick and B. J. Nachtsheim, Angew. Chem., Int. Ed., 2007, 46, 2097–2100 CrossRef CAS PubMed.
- D. Enders, A. A. Narine, F. Toulgoat and T. Bisschops, Angew. Chem., Int. Ed., 2008, 47, 5661–5665 CrossRef CAS PubMed.
- M. Rueping, B. J. Nachtsheim, S. A. Moreth and M. Bolte, Angew. Chem., Int. Ed., 2008, 47, 593–596 CrossRef CAS PubMed.
- M. Zeng, Q. Kang, Q. L. He and S. L. You, Adv. Synth. Catal., 2008, 350, 2169–2173 CrossRef CAS.
- P. Jiao, D. Nakashima and H. Yamamoto, Angew. Chem., Int. Ed., 2008, 47, 2411–2413 CrossRef CAS PubMed.
- H. C. Cheol and H. Yamamoto, J. Am. Chem. Soc., 2008, 130, 9246–9247 CrossRef PubMed.
- M. Rueping, T. Theissmann, A. Kuenkel and R. M. Koenigs, Angew. Chem., Int. Ed., 2008, 47, 6798–6801 CrossRef CAS PubMed.
- M. Rueping and W. Ieawsuwan, Adv. Synth. Catal., 2009, 351, 78–84 CrossRef CAS.
- M. Rueping and B. J. Nachtsheim, Synlett, 2010, 119–122 CrossRef CAS.
- D. Enders, M. Seppelt and T. Beck, Adv. Synth. Catal., 2010, 352, 1413–1418 CrossRef CAS.
- M. Rueping and M.-Y. Lin, Chem. – Eur. J., 2010, 16, 4169–4172 CrossRef CAS PubMed.
- C. H. Cheon and H. Yamamoto, Org. Lett., 2010, 12, 2476–2479 CrossRef CAS PubMed.
- M. Rueping, E. Merino and R. M. Koenigs, Adv. Synth. Catal., 2010, 352, 2629–2634 CrossRef CAS.
- K. Kaupmees, N. Tolstoluzhsky, S. Raja, M. Rueping and I. Leito, Angew. Chem., Int. Ed., 2013, 52, 11569–11572 CrossRef CAS PubMed.
- R. Maji, S. C. Mallojjala and S. E. Wheeler, Chem. Soc. Rev., 2018, 47, 1142–1158 RSC.
- R. B. Sunoj, Acc. Chem. Res., 2016, 49, 1019–1028 CrossRef CAS PubMed.
- J. M. Brunel, Chem. Rev., 2005, 105, 857–897 CrossRef CAS PubMed.
- F. Xu, D. Huang, C. Han, W. Shen, X. Lin and Y. Wang, J. Org. Chem., 2010, 75, 8677–8680 CrossRef CAS PubMed.
- T. Akiyama and K. Mori, Chem. Rev., 2015, 115, 9277–9306 CrossRef CAS PubMed.
- C. H. Cheon and H. Yamamoto, Chem. Commun., 2011, 47, 3043–3056 RSC.
- T. James, M. Van Gemmeren and B. List, Chem. Rev., 2015, 115, 9388–9409 CrossRef CAS PubMed.
- L. Ratjen, M. Van Gemmeren, F. Pesciaioli and B. List, Angew. Chem., Int. Ed., 2014, 53, 8765–8769 CrossRef CAS PubMed.
- M. C. Benda and S. France, Org. Biomol. Chem., 2020, 18, 7485–7513 RSC.
- A. Berkessel, P. Christ, N. Leconte, J.-M. Neudörfl and M. Schäfer, Eur. J. Org. Chem., 2010, 2010, 5165–5170 CrossRef.
- L. Schreyer, R. Properzi and B. List, Angew. Chem., Int. Ed., 2019, 58, 12761–12777 CrossRef CAS PubMed.
- K. M. Diemoz and A. K. Franz, J. Org. Chem., 2019, 84, 1126–1138 CrossRef CAS PubMed.
- K. Rothermel, M. Melikian, J. Hioe, J. Greindl, J. Gramüller, M. Žabka, N. Sorgenfrei, T. Hausler, F. Morana and R. M. Gschwind, Chem. Sci., 2019, 10, 10025–10034 RSC.
- P. Christ, A. G. Lindsay, S. S. Vormittag, J. M. Neudörfl, A. Berkessel and A. C. O'Donoghue, Chem. – Eur. J., 2011, 17, 8524–8528 CrossRef CAS PubMed.
- C. Yang, X. S. Xue, J. L. Jin, X. Li and J. P. Cheng, J. Org. Chem., 2013, 78, 7076–7085 CrossRef CAS PubMed.
- C. Yang, X. S. Xue, X. Li and J. P. Cheng, J. Org. Chem., 2014, 79, 4340–4351 CrossRef CAS PubMed.
- J. P. Reid and J. M. Goodman, J. Am. Chem. Soc., 2016, 138, 7910–7917 CrossRef CAS PubMed.
- J. P. Reid, L. Simón and J. M. Goodman, Acc. Chem. Res., 2016, 49, 1029–1041 CrossRef CAS PubMed.
- A. Rahman and X. Lin, Org. Biomol. Chem., 2018, 16, 4753–4777 RSC.
- M. Rueping, B. J. Nachtsheim, R. M. Koenigs and W. Ieawsuwan, Chem. – Eur. J., 2010, 16, 13116–13126 CrossRef CAS PubMed.
- J. H. Tay, A. J. Arguelles and P. Nagorny, Org. Lett., 2015, 17, 3774–3777 CrossRef CAS PubMed.
- K. Maruoka, T. Itoh, Y. Araki, T. Shirasaka and H. Yamamoto, Bull. Chem. Soc. Jpn., 1988, 61, 2975–2976 CrossRef CAS.
- V. B. Birman, A. L. Rheingold and K. C. Lam, Tetrahedron: Asymmetry, 1999, 10, 125–131 CrossRef CAS.
- P. C. Knipe and M. D. Smith, Org. Biomol. Chem., 2014, 12, 5094–5097 RSC.
- Y. Hatanaka, S. Nantaku, Y. Nishimura, T. Otsuka and T. Sekikaw, Chem. Commun., 2017, 53, 8996–8999 RSC.
- L. Villar, U. Uria, J. I. Martínez, L. Prieto, E. Reyes, L. Carrillo and J. L. Vicario, Angew. Chem., Int. Ed., 2017, 56, 10535–10538 CrossRef CAS PubMed.
- A. Jolit, C. F. Dickinson, K. Kitamura, P. M. Walleser, G. P. A. Yap and M. A. Tius, Eur. J. Org. Chem., 2017, 2017, 6067–6076 CrossRef CAS.
- S. Lee, P. S. J. Kaib and B. List, Synlett, 2017, 1478–1480 CAS.
- P. S. J. Kaib, L. Schreyer, S. Lee, R. Properzi and B. List, Angew. Chem., Int. Ed., 2016, 55, 13200–13203 CrossRef CAS PubMed.
- L. Liu, H. Kim, Y. Xie, C. Fares, P. S. J. Kaib, R. Goddard and B. List, J. Am. Chem. Soc., 2017, 139, 13656–13659 CrossRef CAS PubMed.
- L. L. Tolstikova, A. V. Bel'skikh and B. A. Shainyan, Russ. J. Gen. Chem., 2009, 79, 1752–1754 CrossRef CAS.
- L. L. Tolstikova, A. V. Bel'skikh and B. A. Shainyan, Russ. J. Gen. Chem., 2010, 80, 1258–1262 CrossRef CAS.
- E. Kasprzycka, V. A. Trush, V. M. Amirkhanov, L. Jerzykiewicz, O. L. Malta, J. Legendziewicz and P. Gawryszewska, Chem. – Eur. J., 2017, 23, 1318–1330 CrossRef CAS PubMed.
- M. Shimizu, J. Kikuchi, A. Kondoh and M. Terada, Chem. Sci., 2018, 9, 5747–5757 RSC.
- J. Kikuchi and M. Terada, Angew. Chem., Int. Ed., 2019, 58, 8458–8462 CrossRef CAS PubMed.
- J. Kikuchi, Y. Aizawa and M. Terada, Org. Chem. Front., 2020, 1383–1387 RSC.
- A. Gade and N. Patil, Synlett, 2017, 1096–1100 CAS.
- A. Borovika and P. Nagorny, Tetrahedron, 2013, 69, 5719–5725 CrossRef CAS.
- M. Klussmann, L. Ratjen, S. Hoffmann, V. Wakchaure, R. Goddard and B. List, Synlett, 2010, 2189–2192 CrossRef CAS.
- M. Hatano, K. Moriyama, T. Maki and K. Ishihara, Angew. Chem., Int. Ed., 2010, 49, 3823–3826 CrossRef CAS PubMed.
- M. Terada and K. Kanomata, Synlett, 2011, 1255–1258 CrossRef CAS.
- K. Mori, A. Miyake and T. Akiyama, Chem. Lett., 2014, 43, 137–139 CrossRef CAS.
- L. Kong, X. Han and P. Jiao, Chem. Commun., 2014, 50, 14113–14116 RSC.
- Z. Y. Han, D. F. Chen, Y. Y. Wang, R. Guo, P. S. Wang, C. Wang and L. Z. Gong, J. Am. Chem. Soc., 2012, 134, 6532–6535 CrossRef CAS PubMed.
- C. C. J. Loh and D. Enders, Chem. – Eur. J., 2012, 18, 10212–10225 CrossRef CAS PubMed.
- S. M. Inamdar, A. Konala and N. T. Patil, Chem. Commun., 2014, 50, 15124–15135 RSC.
- W. Xiang, L. Mingli and G. Liuzhu, Acta Chim. Sin., 2013, 71, 1091 CrossRef.
- D.-F. Chen, Z.-Y. Han, X.-L. Zhou and L.-Z. Gong, Acc. Chem. Res., 2014, 47, 2365–2377 CrossRef CAS PubMed.
- S. M. Inamdar, R. G. Gonnade and N. T. Patil, Org. Biomol. Chem., 2017, 15, 863–869 RSC.
- M. Hatano, Y. Goto, A. Izumiseki, M. Akakura and K. Ishihara, J. Am. Chem. Soc., 2015, 137, 13472–13475 CrossRef CAS PubMed.
- M.-H. Cao, N. J. Green and S.-Z. Xu, Org. Biomol. Chem., 2017, 15, 3105–3129 RSC.
- M. G. Memeo and P. Quadrelli, Chem. – Eur. J., 2012, 18, 12554–12582 CrossRef CAS PubMed.
- K. A. Jørgensen, Angew. Chem., Int. Ed., 2000, 39, 3558–3588 CrossRef.
- N. Li, D. F. Chen, P. S. Wang, Z. Y. Han and L. Z. Gong, Synthesis, 2014, 46, 1335–1361 Search PubMed.
- M. Rueping, M. S. Maji, H. B. Küçük and I. Atodiresei, Angew. Chem., Int. Ed., 2012, 51, 12864–12868 CrossRef CAS PubMed.
- X. Hong, H. B. Küçük, M. S. Maji, Y. F. Yang, M. Rueping and K. N. Houk, J. Am. Chem. Soc., 2014, 136, 13769–13780 CrossRef CAS PubMed.
- M. M. Xu, H. Q. Wang, Y. Wan, S. L. Wang and F. Shi, J. Org. Chem., 2017, 82, 10226–10233 CrossRef CAS PubMed.
- C. C. Hsiao, S. Raja, H. H. Liao, I. Atodiresei and M. Rueping, Angew. Chem., Int. Ed., 2015, 54, 5762–5765 CrossRef CAS PubMed.
- L. Z. Li, C. S. Wang, W. F. Guo, G. J. Mei and F. Shi, J. Org. Chem., 2018, 83, 614–623 CrossRef CAS PubMed.
- H. H. Liao, C. C. Hsiao, I. Atodiresei and M. Rueping, Chem. – Eur. J., 2018, 24, 7718–7723 CrossRef CAS PubMed.
- M. Kretzschmar, F. Hofmann, D. Moock and C. Schneider, Angew. Chem., Int. Ed., 2018, 57, 4774–4778 CrossRef CAS PubMed.
- J. P. Lovie-Toon, C. M. Tram, B. L. Flynn and E. H. Krenske, ACS Catal., 2017, 7, 3466–3476 CrossRef CAS.
- J. Jin, Y. Zhao, A. Gouranourimi, A. Ariafard and P. W. Hong Chan, J. Am. Chem. Soc., 2018, 140, 5834–5841 CrossRef CAS PubMed.
- M. Rueping and W. Ieawsuwan, Chem. Commun., 2011, 47, 11450–11452 RSC.
- S. Raja, W. Ieawsuwan, V. Korotkov and M. Rueping, Chem. - Asian J., 2012, 7, 2361–2366 CrossRef CAS PubMed.
- D. J. Kerr and B. L. Flynn, Org. Lett., 2012, 14, 1740–1743 CrossRef CAS PubMed.
- A. Jolit, P. M. Walleser, G. P. A. Yap and M. A. Tius, Angew. Chem., Int. Ed., 2014, 53, 6180–6183 CrossRef CAS PubMed.
- B. M. Yang, P. J. Cai, Y. Q. Tu, Z. X. Yu, Z. M. Chen, S. H. Wang, S. H. Wang and F. M. Zhang, J. Am. Chem. Soc., 2015, 137, 8344–8347 CrossRef CAS PubMed.
- Y. Cai, Y. Tang, I. Atodiresei and M. Rueping, Angew. Chem., Int. Ed., 2016, 55, 14126–14130 CrossRef CAS PubMed.
- J. Jin, Y. Zhao, E. M. L. Sze, P. Kothandaraman and P. W. H. Chan, Adv. Synth. Catal., 2018, 360, 4744–4753 CrossRef CAS.
- L. Fan, C. Han, X. Li, J. Yao, Z. Wang, C. Yao, W. Chen, T. Wang and J. Zhao, Angew. Chem., Int. Ed., 2018, 57, 2115–2119 CrossRef CAS PubMed.
- R. C. Samanta and H. Yamamoto, J. Am. Chem. Soc., 2017, 139, 1460–1463 CrossRef CAS PubMed.
- L. Simón and J. M. Goodman, J. Org. Chem., 2011, 76, 1775–1788 CrossRef PubMed.
- L. Simón and J. M. Goodman, J. Org. Chem., 2010, 75, 589–597 CrossRef PubMed.
- L. Simón and J. M. Goodman, J. Am. Chem. Soc., 2009, 131, 4070–4077 CrossRef PubMed.
- L. Simón and J. M. Goodman, J. Am. Chem. Soc., 2008, 130, 8741–8747 CrossRef PubMed.
- J. B. Sweeney, in Aziridines and Epoxides in Organic Synthesis, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, FRG, 2006, pp. 117–144 Search PubMed.
- L. Degennaro, P. Trinchera and R. Luisi, Chem. Rev., 2014, 114, 7881–7929 CrossRef CAS PubMed.
- J. M. de los Santos, A. M. Ochoa de Retana, E. Martínez de Marigorta, J. Vicario and F. Palacios, ChemCatChem, 2018, 10, 5092–5114 CrossRef CAS.
- H. Pellissier, Adv. Synth. Catal., 2014, 356, 1899–1935 CrossRef CAS.
- T. Hashimoto, H. Nakatsu, K. Yamamoto and K. Maruoka, J. Am. Chem. Soc., 2011, 133, 9730–9733 CrossRef CAS PubMed.
- S. P. Bew, J. Liddle, D. L. Hughes, P. Pesce and S. M. Thurston, Angew. Chem., Int. Ed., 2017, 56, 5322–5326 CrossRef CAS PubMed.
- D. Enders, A. Rembiak and M. Seppelt, Tetrahedron Lett., 2013, 54, 470–473 CrossRef CAS.
- J. F. Bai, H. Sasagawa, T. Yurino, T. Kano and K. Maruoka, Chem. Commun., 2017, 53, 8203–8206 RSC.
- F. Zhou and H. Yamamoto, Angew. Chem., Int. Ed., 2016, 55, 8970–8974 CrossRef CAS PubMed.
- Z. Wei, J. Zhang, H. Yang and G. Jiang, Org. Lett., 2019, 21, 2790–2794 CrossRef CAS PubMed.
- Y. Wang, L. Cui, Y. Wang and Z. Zhou, Tetrahedron: Asymmetry, 2016, 27, 85–90 CrossRef CAS.
- S. G. Wang, L. Han, M. Zeng, F. L. Sun, W. Zhang and S. L. You, Org. Biomol. Chem., 2012, 10, 3202–3209 RSC.
- E. Aranzamendi, N. Sotomayor and E. Lete, ACS Omega, 2017, 2, 2706–2718 CrossRef CAS PubMed.
- F. Bartoccini, M. Mari, M. Retini, S. Bartolucci and G. Piersanti, J. Org. Chem., 2018, 83, 12275–12283 CrossRef CAS PubMed.
- H. H. Liao, A. Chatupheeraphat, C. C. Hsiao, I. Atodiresei and M. Rueping, Angew. Chem., Int. Ed., 2015, 54, 15540–15544 CrossRef CAS PubMed.
- M. Zeng, W. Zhang and S. You, Chin. J. Chem., 2012, 30, 2615–2623 CAS.
- J. W. Zhang, Q. Cai, X. X. Shi, W. Zhang and S. L. You, Synlett, 2011, 1239–1242 Search PubMed.
- Q. Cai, Z.-A. Zhao and S.-L. You, Angew. Chem., Int. Ed., 2009, 48, 7428–7431 CrossRef CAS PubMed.
- Z. Wei, J. Zhang, H. Yang and G. Jiang, Adv. Synth. Catal., 2019, 361, 3694–3697 CrossRef CAS.
- F. Rabasa-Alcañiz, M. Sánchez-Roselló, S. Fustero and C. Del Pozo, J. Org. Chem., 2019, 84, 10785–10795 CrossRef PubMed.
- S. Qi, C. Y. Liu, J. Y. Ding and F. S. Han, Chem. Commun., 2014, 50, 8605–8608 RSC.
- D. Blanco-Ania and F. P. J. T. Rutjes, Beilstein J. Org. Chem., 2018, 14, 2568–2571 CrossRef CAS PubMed.
- C. Y. Liu and F. S. Han, Chem. Commun., 2015, 51, 11844–11847 RSC.
- G. Caballero-García, G. Mondragón-Solórzano, R. Torres-Cadena, M. Díaz-García, J. Sandoval-Lira and J. Barroso-Flores, Molecules, 2018, 24, 79 CrossRef PubMed.
- J. Sandoval-Lira, G. Mondragón-Solórzano, L. I. Lugo-Fuentes and J. Barroso-Flores, J. Chem. Inf. Model., 2020, 60, 1445–1452 CrossRef CAS PubMed.
- M. Zhuang and H. Du, Org. Biomol. Chem., 2014, 12, 4590–4593 RSC.
- J. Zhou and H. Xie, Org. Biomol. Chem., 2018, 16, 380–383 RSC.
- M. Rueping, U. Uria, M.-Y. Lin and I. Atodiresei, J. Am. Chem. Soc., 2011, 133, 3732–3735 CrossRef CAS PubMed.
- C. C. Hsiao, H. H. Liao and M. Rueping, Angew. Chem., Int. Ed., 2014, 53, 13258–13263 CrossRef CAS PubMed.
- D. Qian, L. Wu, Z. Lin and J. Sun, Nat. Commun., 2017, 8, 1–9 CrossRef CAS PubMed.
- L. Zhang, Y. Han, A. Huang, P. Zhang, P. Li and W. Li, Org. Lett., 2019, 21, 7415–7419 CrossRef CAS PubMed.
- J. F. Bai, K. Yasumoto, T. Kano and K. Maruoka, Angew. Chem., Int. Ed., 2019, 58, 8898–8901 CrossRef CAS PubMed.
- Y. Cui, L. A. Villafane, D. J. Clausen and P. E. Floreancig, Tetrahedron, 2013, 69, 7618–7626 CrossRef CAS PubMed.
- J. Liu, L. Zhou, C. Wang, D. Liang, Z. Li, Y. Zou, Q. Wang and A. Goeke, Chem. – Eur. J., 2016, 22, 6258–6261 CrossRef CAS PubMed.
- P. S. Wang, K. N. Li, X. Le Zhou, X. Wu, Z. Y. Han, R. Guo and L. Z. Gong, Chem. – Eur. J., 2013, 19, 6234–6238 CrossRef CAS PubMed.
- X. Wu, M. L. Li and P. S. Wang, J. Org. Chem., 2014, 79, 419–425 CrossRef CAS PubMed.
- M. Sai and H. Yamamoto, J. Am. Chem. Soc., 2015, 137, 7091–7094 CrossRef CAS PubMed.
- L. Zhu, H. Yuan and J. Zhang, J. Catal., 2020, 383, 230–238 CrossRef CAS.
- J. S. Lin, P. Yu, L. Huang, P. Zhang, B. Tan and X. Y. Liu, Angew. Chem., Int. Ed., 2015, 54, 7847–7851 CrossRef CAS PubMed.
- H. Guan, H. Wang, D. Huang and Y. Shi, Tetrahedron, 2012, 68, 2728–2735 CrossRef CAS.
- P. Zhao, A. Cheng, X. Wang, J. Ma, G. Zhao, Y. Li, Y. Zhang and B. Zhao, Chin. J. Chem., 2020, 38, 565–569 CrossRef CAS.
- Z. Wang, F. K. Sheong, H. H. Y. Sung, I. D. Williams, Z. Lin and J. Sun, J. Am. Chem. Soc., 2015, 137, 5895–5898 CrossRef CAS PubMed.
- B. Guo, G. Schwarzwalder and J. T. Njardarson, Angew. Chem., Int. Ed., 2012, 51, 5675–5678 CrossRef CAS PubMed.
- M. Yang, Y. M. Zhao, S. Y. Zhang, Y. Q. Tu and F. M. Zhang, Chem. - Asian J., 2011, 6, 1344–1347 CrossRef CAS PubMed.
- M. Zhuang and H. Du, Org. Biomol. Chem., 2013, 11, 1460–1462 RSC.
- H. Wu, Q. Wang and J. Zhu, Angew. Chem., Int. Ed., 2017, 56, 5858–5861 CrossRef CAS PubMed.
- H. Wu, Q. Wang and J. Zhu, Angew. Chem., Int. Ed., 2016, 55, 15411–15414 CrossRef CAS PubMed.
- H. Wu, Q. Wang and J. Zhu, J. Am. Chem. Soc., 2019, 141, 11372–11377 CrossRef CAS PubMed.
- D. Ma, C. B. Miao and J. Sun, J. Am. Chem. Soc., 2019, 141, 13783–13787 CrossRef CAS PubMed.
- Y. P. Li, S. F. Zhu and Q. L. Zhou, Org. Lett., 2019, 21, 9391–9395 CrossRef CAS PubMed.
- M. N. Grayson, S. C. Pellegrinet and J. M. Goodman, J. Am. Chem. Soc., 2012, 134, 2716–2722 CrossRef CAS PubMed.
- R. S. Paton, J. M. Goodman and S. C. Pellegrinet, Org. Lett., 2009, 11, 37–40 CrossRef CAS PubMed.
- J. P. Reid and J. M. Goodman, Org. Biomol. Chem., 2017, 15, 6943–6947 RSC.
- J. P. Reid and J. M. Goodman, Chem. – Eur. J., 2017, 23, 14248–14260 CrossRef CAS PubMed.
- J. P. Reid, K. Ermanis and J. M. Goodman, Chem. Commun., 2019, 55, 1778–1781 RSC.
- M. N. Grayson and J. M. Goodman, J. Am. Chem. Soc., 2013, 135, 6142–6148 CrossRef CAS PubMed.
- M. N. Grayson and J. M. Goodman, J. Org. Chem., 2013, 78, 8796–8801 CrossRef CAS PubMed.
- M. N. Grayson and J. M. Goodman, J. Org. Chem., 2015, 80, 2056–2061 CrossRef CAS PubMed.
- L. M. Overvoorde, M. N. Grayson, Y. Luo and J. M. Goodman, J. Org. Chem., 2015, 80, 2634–2640 CrossRef CAS PubMed.
- B. N. Falcone, M. N. Grayson and J. B. Rodriguez, J. Org. Chem., 2018, 83, 14683–14687 CrossRef CAS PubMed.
- J. E. Johnson, N. M. Morales, A. M. Gorczyca, D. D. Dolliver and M. A. McAllister, J. Org. Chem., 2001, 66, 7979–7985 CrossRef CAS PubMed.
- M. E. Belowich and J. F. Stoddart, Chem. Soc. Rev., 2012, 41, 2003–2024 RSC.
- J. Bjørgo, D. R. Boyd, C. G. Watson, W. B. Jennings and D. M. Jerina, J. Chem. Soc., Perkin Trans. 2, 1974, 1081–1084 RSC.
- J. P. Reid and J. M. Goodman, Chem. – Eur. J., 2017, 23, 14248–14260 CrossRef CAS PubMed.
- J. P. Reid, K. Ermanis and J. M. Goodman, Chem. Commun., 2019, 55, 1778–1781 RSC.
- M. Mahlau and B. List, Angew. Chem., Int. Ed., 2013, 52, 518–533 CrossRef CAS PubMed.
- S. Mayer and B. List, Angew. Chem., Int. Ed., 2006, 45, 4193–4195 CrossRef CAS PubMed.
- S. Liao and B. List, Adv. Synth. Catal., 2012, 354, 2363–2367 CrossRef CAS.
- K. Brak and E. N. Jacobsen, Angew. Chem., Int. Ed., 2013, 52, 534–561 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2021 |