
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Structural and biophysical insights into the mode of covalent binding of rationally designed potent BMX inhibitors†
João D.
Seixas‡
*a,
Bárbara B.
Sousa‡
ab,
Marta C.
Marques
a,
Ana
Guerreiro
a,
Rui
Traquete
a,
Tiago
Rodrigues
 a,
Inês S.
Albuquerque
a,
Marcos F. Q.
Sousa
bc,
Ana R.
Lemos
bc,
Pedro M. F.
Sousa
bc,
Tiago M.
Bandeiras
bc,
Di
Wu
d,
Shelby K.
Doyle
e,
Carol V.
Robinson
d,
Angela N.
Koehler
e,
Francisco
Corzana
f,
Pedro M.
Matias
bc and
Gonçalo J. L.
Bernardes
*ag
a,
Inês S.
Albuquerque
a,
Marcos F. Q.
Sousa
bc,
Ana R.
Lemos
bc,
Pedro M. F.
Sousa
bc,
Tiago M.
Bandeiras
bc,
Di
Wu
d,
Shelby K.
Doyle
e,
Carol V.
Robinson
d,
Angela N.
Koehler
e,
Francisco
Corzana
f,
Pedro M.
Matias
bc and
Gonçalo J. L.
Bernardes
*ag
aInstituto de Medicina Molecular João Lobo Antunes, Faculdade de Medicina, Universidade de Lisboa, Avenida Professor Egas Moniz, 1649-028 Lisboa, Portugal. E-mail: gbernardes@medicina.ulisboa.pt; joaoseixas@medicina.ulisboa.pt
bInstituto de Tecnologia Química e Biológica António Xavier, Universidade Nova de Lisboa, Av. da República, EAN, 2780-157 Oeiras, Portugal
cIBET - Instituto de Biologia Experimental e Tecnológica, Av. da República, EAN, 2780-157 Oeiras, Portugal
dDepartment of Chemistry, University of Oxford, South Parks Road, Oxford, OX1 3QZ, UK
eDavid H. Koch Institute for Integrative Cancer Research, Massachusetts Institute of Technology, Cambridge, MA 02142, USA
fDepartamento de Química, Universidad de La Rioja, Centro de Investigación en Síntesis Química, 26006 Logroño, Spain
gDepartment of Chemistry, University of Cambridge, Lensfield Road, Cambridge, CB2 1EW, UK. E-mail: gb453@cam.ac.uk
First published on 28th August 2020
Abstract
The bone marrow tyrosine kinase in chromosome X (BMX) is pursued as a drug target because of its role in various pathophysiological processes. We designed BMX covalent inhibitors with single-digit nanomolar potency with unexploited topological pharmacophore patterns. Importantly, we reveal the first X-ray crystal structure of covalently inhibited BMX at Cys496, which displays key interactions with Lys445, responsible for hampering ATP catalysis and the DFG-out-like motif, typical of an inactive conformation. Molecular dynamic simulations also showed this interaction for two ligand/BMX complexes. Kinome selectivity profiling showed that the most potent compound is the strongest binder, displays intracellular target engagement in BMX-transfected cells with two-digit nanomolar inhibitory potency, and leads to BMX degradation PC3 in cells. The new inhibitors displayed anti-proliferative effects in androgen-receptor positive prostate cancer cells that where further increased when combined with known inhibitors of related signaling pathways, such as PI3K, AKT and Androgen Receptor. We expect these findings to guide development of new selective BMX therapeutic approaches.
Introduction
Over recent years, the development of covalent kinase inhibitors has gained more traction both in academia and pharmaceutical industry.1–3 Historically, irreversible covalent inhibitors were considered unsafe because of their lack of selectivity and concomitant undesired engagement of off-targets. However, these potential liabilities can be overcome and the development of covalent small molecule kinase inhibitors has recently seen renewed interest. Irreversible covalent inhibitors can display higher efficacy, since they achieve high target occupancy and a prolonged pharmacodynamic effect, depending on the de novo re-synthesis rate of the target protein.4,5 Supporting the value and “renaissance” of covalent inhibitors, since October 2018 six kinase-targeting small molecule covalent inhibitors6 were approved by the FDA for clinical use: the EGFR inhibitors Afatinib®, Neratinib®, Osimertinib® and Dacomitinib® and the BTK inhibitors Ibrutinib® and Acalabrutinib®.7–11 However, not all kinases are accessible for covalent binding since the covalent bond formation depends on the nature and positioning of the target amino acid.12–15 One such kinase of interest is the epithelial and endothelial tyrosine kinase, commonly known as bone marrow tyrosine kinase in chromosome X (BMX). BMX is a major member of the TEC family of non-receptor tyrosine kinases, together with ITK, TEC, BTK and TXK [reviewed in ref. 16 and 17]. TEC kinases are activated by many cell-surface receptor-associated signaling complexes and are recruited to the plasma membrane or specific micro-environments by a variety of lipids and proteins. Through this mechanism, they are involved in signal transduction in response to a myriad of extracellular stimuli, including those mediated by growth-factor receptors, cytokine receptors, G-protein coupled receptors, antigen-receptors, integrins and death receptors. Moreover, TEC kinases regulate many of the major signaling pathways, such as those of PI3K, PKC, PLCγ, AKT, STAT3 and p21-activated kinase 1 (PAK1)18,19 and are responsible for a variety of cell processes, including regulation of gene expression, calcium mobilization, actin reorganization/motility and survival/apoptosis.16,17BMX is widely expressed in granulocytes, monocytes, cells of epithelial and endothelial lineages, as well as brain, prostate, lung and heart.19–22 It is specifically involved in tumorigenicity, adhesion, motility, angiogenesis, proliferation and differentiation. Moreover, it has been found to be overexpressed in numerous cancer types, such as breast,23–25 prostate,26,27 colon28 and cervical carcinoma,29 which suggests that elevated levels of BMX increase cancer-cell survival. BMX is also required for stem-cell maintenance and survival22 and its up-regulation provides a survival benefit to both primary tumors and cancer stem cells that are highly resistant to apoptosis and many chemotherapeutic agents.
Homozygous BMX knockout mice have a normal life span without any obvious altered phenotype, which suggests that therapies based on BMX inhibition might have few side effects30 and although BMX is a key regulator it might not represent a fundamental effector. Therefore, by considering the existence of multiple downstream target proteins, the integration in multiple and diverse signaling pathways, and the fact that it regulates proliferation, migration and has an anti-apoptotic effect, BMX emerges as a potential target for multiple aspects of cancer therapy. Recent studies also highlighted that modulation of BMX activity sensitizes cells to therapeutic agents to improve response to chemotherapy DNA damaging agents or radiation. These studies show strong evidence that both direct inhibition of BMX and modulation of related pathways result in increased therapeutic efficacy.28,31,32
BMX-IN-1 is one of the most potent BMX inhibitors (IC50: 8.0 nM) reported in the literature, which also binds to BTK with very high affinity (IC50: 10.4 nM).33 Like other BMX covalent inhibitors, it reacts with a cysteine residue (Cys496) in the ATP binding site. This residue is a unique occurrence found in the ATP binding pocket and is present in all five members of the TEC family kinase members. Therefore, by virtue of structural homology these compounds could also be covalent inhibitors of the other kinases in the TEC family.
In this study, we describe the discovery of JS24–JS27, which are among the most potent covalent inhibitors of BMX reported to date and possess topological pharmacophoric features not exploited in the BMX inhibitors’ chemical space. We asserted the selectivity against a panel of 36 kinases that possess an equally placed cysteine or up- and downstream regulators of the BMX signaling pathway. We further demonstrated that the lead compounds have the potential to inhibit proliferation of androgen-receptor-positive prostate-cancer cells (LNCaP) and their inhibitory potential is enhanced in a co-treatment regimen with known PI3K, AKT and androgen receptor inhibitors (LY294002, AKT1/2 and Flutamide, respectively). As part of our efforts to explore this scaffold to identify regions of the molecule amenable to conjugation we also report the first X-ray structure of BMX with a covalent inhibitor as well as MD simulations on two complexes with this receptor, which provide insight into the mode of binding and will contribute towards the future development of inhibitors with improved efficacy and selectivity.
Results and discussion
Discovery of a single-digit nanomolar BMX inhibitor
To evaluate substituent tolerability at each position and to establish an optimal vector through positioning of different functionalities, a structure–activity relationship (SAR) study was used to establish the limitations of the tool chemotype BMX-IN-1. A total of 24 analogues were synthesized in an attempt to both enhance potency and optimize physicochemical properties within the allowable SAR study (Fig. S1, ESI†). Upon systematic evaluation, we found that analogues with substituents in position R3 (Fig. 1a) of the quinoline moiety had only marginal effects on BMX inhibition Furthermore, any change of the electrophilic warhead (position R1, Fig. 1a) for cysteine covalent ligation resulted in loss of potency as illustrated for instance by the introduction of the enamide substituents in JS10 and JS11 (Fig. S1, ESI†). The substituents in the aromatic ring bearing the amide functionality play an unexpected relevant role for the activity, affording different reactivity patterns arising from non-covalent interactions. Introducing a strong electron-donating group such as a methoxy group (OMe) (JS9E) in R2 decreases potency by 4-fold. In contrast, the use of a weak electron-donating group, such as a methyl substituent, has different effects depending on the positioning. Thus, moving the methyl group to the ortho position abolishes target inhibition (JS9C), while a methyl positioning in the meta position slightly improved inhibition by 2-fold. Even more striking is the effect of no substituent in the ring (JS9D), which increased inhibition by 6-fold. Since the electronic influence of the methyl substituents in the different positions is not expected to account for these differences, we consider that a conformational effect may play an important role. The ortho substitution may increase the constraints for fitting into the pocket, while the removal of the methyl groups affords less spatial restriction.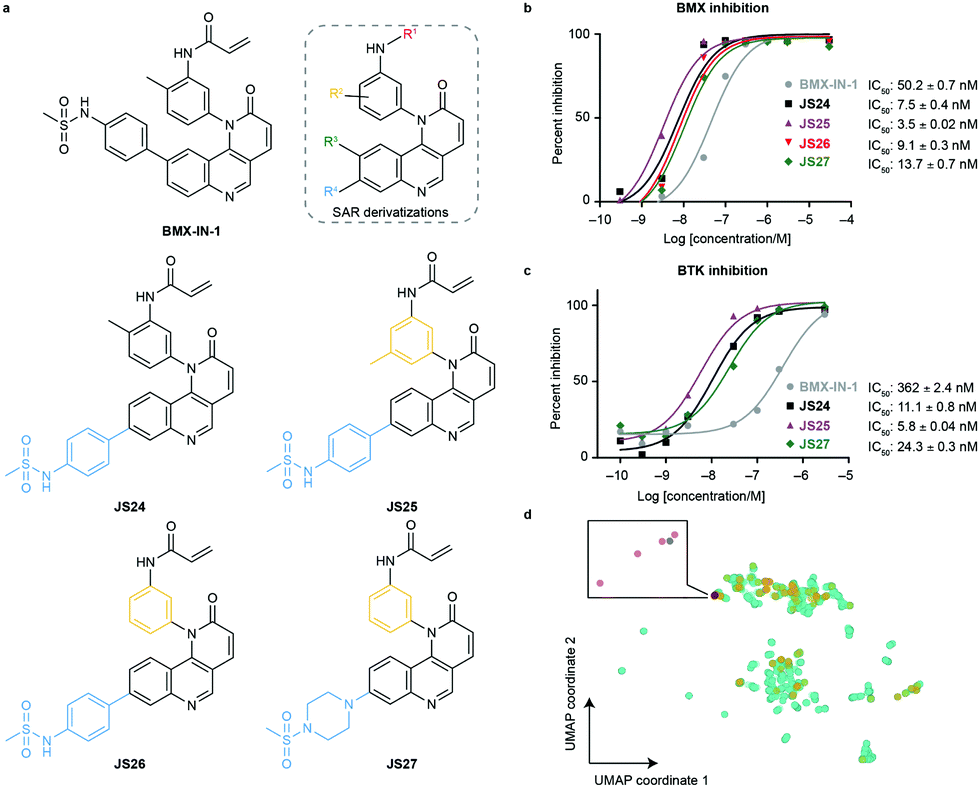 | ||
| Fig. 1 Compound structures and biochemical characterization of their inhibitory effects on BMX and BTK. (a) The structures of BMX-IN-1, the SAR explored (shown in Fig. S1, ESI†) and JS24–JS27 leads generated in this study. (b) Eurofins DiscoverX in vitro BMX activity evaluation by measuring the phosphorylation of a biotinylated peptide with human recombinant enzyme expressed in insect cells and HTRF detection method, tested in duplicate, showing mean ± S.D. Cells were treated for 1 h and IC50 values were calculated and plotted by using GraphPad Prism 8 based on a sigmoidal dose response curve. (c) Eurofins DiscoverX in vitro BTK activity evaluation by measuring scintillation count with a radiometric assay tested in duplicate, showing mean ± S.D. Cells were treated for 1 h and IC50 values were calculated and plotted by using GraphPad Prism 8 based on a sigmoidal dose response curve. (d) Projection of BMX and BTK inhibitor space with a multidimensional scaling algorithm. Green: BTK inhibitors; orange: BMX inhibitors; grey: BMX-IN-1; red: JS24–J27. | ||
Interestingly, substituents in position R4 (Fig. 1a and Fig. S2, ESI†) were found to significantly enhance BMX inhibition relative to BMX-IN-1 (Fig. 1a and Fig. S2, ESI†). With this SAR information in hand, we decided to prepare JS24–JS27 that were designed to include substituents at position R4 of the quinoline and incorporated features from previous analogues that afforded a preferred overall profile, varying methyl positioning in R2 and retaining the acrylamide electrophilic warhead for cysteine covalent binding. Details for the synthesis of JS24–JS27 can be found in the ESI† (Schemes S1–S4). Compound JS24, which features a methyl sulfonamide at position 7, showed considerable gain of inhibition potency from IC50 50 to 7.5 nM relative to the parent molecule BMX-IN-1 (Fig. 1a and b). Further derivatives that presented changes in the aniline core (R2) with a methyl in the meta position (JS25) and without any substituent in this position (JS26), but that features the same methyl sulfonamide at R4, were designed and prepared. Both compounds showed significantly improved activity (IC50 of 3.5 and 9.1 nM, respectively; Fig. 1a and 1b).
Similarly, in JS27 we installed a substituted piperazine in the R4 position that affords less restraint relative to the aromatic phenyl-sulfonamide ring, but which renders it the least active analogue of the series (13.7 nM) albeit considerably more potent (≈4-fold) relative to BMX-IN-1 (Fig. 1a and b).
Modulation of physicochemical profile, BTK inhibition and pharmacophore diversity
Taking into account that BMX-IN-1 does not exhibit an optimal physicochemical profile, we aimed at lowering the lipophilicity and increasing water solubility within the established SAR. Initial investigations showed an improved profile when the sulfonamide aromatic ring was replaced by cyclic aliphatic amines (Table S1, ESI†). These observations prompted us to use 1-(methylsulfonyl)piperazine in compound JS27. Consequently, we were able to obtain the analogue with the best in silico lipophilicity and water solubility profile (c![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) logP = 2.32 and logS = −4.36; Table S1, ESI†). Interestingly, compound JS26 shows a slightly improved reduction of clogP. The presence of a methyl group (compounds JS24 and JS25) increases hydrophobicity and, consequently, removal of this group (JS26–JS27) decreases the hydrophobicity and the partition coefficient.
logP = 2.32 and logS = −4.36; Table S1, ESI†). Interestingly, compound JS26 shows a slightly improved reduction of clogP. The presence of a methyl group (compounds JS24 and JS25) increases hydrophobicity and, consequently, removal of this group (JS26–JS27) decreases the hydrophobicity and the partition coefficient.
We anticipated that some analogues could have limited membrane permeability, which is of utmost importance for any drug, in particular if a molecule is targeting cytoplasmic proteins. For assessment of drug permeability, we relied on parallel artificial membrane permeability assay (PAMPA) performed at Pion Inc. with the PAMPA Evolution™ instrument (Table S1, ESI†). We observed that compounds JS24 (6.8 × 10−6 cm s−1) and JS25 (3.8 × 10−6 cm s−1) show a lower PAMPA permeability relative to BMX-IN-1 (8.9 × 10−6 cm s−1), whereas compounds with the unsubstituted backbone aniline (JS26 and JS27) show increased permeability (19 × 10−6 cm s−1 and 12 × 10−6 cm s−1, respectively). As shown in Fig. 1a, the four leads share a similar scaffold and only compound JS27 displays a distinct structural feature. Because other analogues display similar clogP values with improved PAMPA permeability (up to 45 × 10−6 cm s−1; Table S1, ESI†), the observed increased permeability may be mostly a result of intramolecular interactions, such as hydrogen bonding, more than the lipophilic contribution, because the algorithm for clogP calculation does not consider 3D conformations. In addition, we measured particle sizes by using dynamic light scattering (DLS). Up to 95% of false positive readouts in high-throughput screens originate from colloidal aggregation.34 This phenomenon is driven by the physicochemical properties of the small molecule and buffer conditions. Generally, aggregates bind non-specifically to proteins, sequestering and denaturating them. Our data shows that, regardless of their limited solubility, compounds JS24–JS27 do not form aggregates at the relevant inhibitory concentrations, which rules out unspecific binding to BMX (Table S1, ESI†).
To date, all the reported BMX inhibitors also display the ability to inhibit Bruton's tyrosine kinase (BTK). To determine if our leads were selective binders of BMX, we evaluated inhibitory capacity of our compounds against BTK. For the BTK IC50 assay (KinaseProfiler by Eurofins DiscoverX), we selected BMX-IN-1, the two analogues with higher BMX inhibitory capacity (JS24 and JS25) and JS27, which presents the best in silico physicochemical profile, and also offers the possibility of derivatization. The results showed that all the leads are also potent BTK inhibitors, in the low nanomolar range (Fig. 1c). The same inhibitory trend is observed with an increase of 62-, 33- and 15-fold potency gain with JS25, JS24 and JS27, respectively, relative to BMX-IN-1. Interestingly, in this assay BMX-IN-1 displays 7-fold higher IC50 against BTK than BMX.
We aimed to modulate the physicochemical properties of the molecules to enhance the overall “drug-likeness” profile of the ligands. Ligand efficiency (LE) and lipophilic efficiency (LipE) are two important metrics that are associated with improved prospects for good drug properties (e.g. bioavailability) and are used as criteria for progression of the most promising candidates across drug discovery pipelines.35 LE is used to compare binding efficacy of inhibitors/ligands relative to their size, and LipE is used to compare binding efficacy by taking into consideration the lipophilicity of the molecules. With regards to BMX inhibition, compound JS27 displayed a major LipE improvement relative to BMX-IN-1 (5.54 versus 3.36) empowered by the drastic reduction in clogP as a result of the introduction of the aliphatic sulfonamide. Analogues JS24–JS26 displayed mild LipE improvement (4.17, 4.43 and 4.45), which is mainly due to their structural similarity with BMX-IN-1. However, the LE improvement is mostly driven by the increased potency of all analogues rather than a decrease in the molecules’ size (Table 1). Similarly, the designed ligands offer a greater improvement of LE and LipE metrics with respect to BTK binding, relative to BMX-IN-1, driven also by a drastic potency gain.
| Compound | LE (BMX) | LipE (BMX) | LE (BTK) | LipE (BTK) |
|---|---|---|---|---|
| LE – ligand efficiency; LipE – lipophilic efficiency; Nd – not defined. | ||||
| BMX-IN-1 | 0.26 | 3.36 | 0.23 | 2.50 |
| JS24 | 0.29 | 4.17 | 0.29 | 4.00 |
| JS25 | 0.30 | 4.43 | 0.30 | 4.22 |
| JS26 | 0.30 | 4.45 | Nd | Nd |
| JS27 | 0.30 | 5.54 | 0.29 | 5.29 |
Finally, we analyzed the pharmacophore diversity of the designed ligands against that of known BMX and BTK inhibitors. Ligand data was collected from ChEMBL v24, pre-processed as previously described36 and projected to the plane by means of a learning algorithm (Fig. 1d). It is apparent that BTK has been more often interrogated with small molecules (green) and that the studied chemotypes are more diverse in regard to topological pharmacophore arrangements relative to previously described BMX modulators (blue). Compounds JS24–JS27 (yellow) focus on unexplored regions in BMX inhibitor space but overlaps with previously studied BTK chemotypes. Indeed, our compounds have shown potent activities against BTK, which is fully in line with the output of the learning algorithm. Altogether, our data shows that compounds JS24–JS27 explore a new chemical space and provides a rationale to re-investigate and potentially repurpose BTK inhibitors as leads for future development of BMX ligands and vice versa.
JS24–JS27 show strong binding interactions with BMX
We further characterized the binding interaction between JS24–JS27 and BMX by using differential scanning fluorimetry (DSF) and surface plasmon resonance (SPR). Purified recombinant human BMX protein (hBMX) was first subjected to thermal scanning in the absence and presence of JS24–JS27, and the respective protein melting temperature (Tm) calculated. As shown in Table 2, BMX-IN-1 increases the Tm value by 8.04 °C. Among the lead inhibitors, JS24 displayed the highest stabilization, with an increase in Tm of 11.34 °C. Compounds JS27 (10.81 °C), JS26 (9.34 °C) and JS25 (9.30 °C) showed high stabilization of the protein, which also suggests direct binding to BMX with a higher affinity relative to parent scaffold BMX-IN-1 (Table 2 and Table S2, ESI†). The binding of JS24–JS27 to BMX immobilized surfaces was then monitored in real-time by SPR assays (Table 2 and Fig. S3, Table S3, ESI†). BMX-IN-1 was shown to bind to BMX with an affinity of KD = 69 nM. It is important to note that it was not possible to accurately fit the dissociation rate constant of BMX-IN-1 interaction with BMX because of the initial non-covalent binding event. In contrast, JS24–JS27KD values could not be determined due to the even higher prolonged off-rates, which were outside the range of the instrument specifications. The results suggest that compounds JS24–JS27 have higher affinity interactions with BMX, showing comparable association rates (kon from 5.4 × 104–1.4 × 105 M−1s−1) but, most importantly, very slow dissociation rates (koff < 1 × 10−4 s−1), which is in agreement with the covalent nature of the interaction (Table 2).| Compound | Aveg. Tm (°C) | apo-BMX Tm (°C) | ΔTm (°C) | k on/M−1 s−1 | k off/s−1 | K D/M |
|---|---|---|---|---|---|---|
| Nd – not determined.a KD not measured due to very prolonged off-rates (outside instrument specifications). | ||||||
| BMX-IN-1 | 60.17 ± 0.32 | 52.13 ± 0.11 | 8.04 ± 0.32 | 7.4 × 103 | 5.10 × 10−4 | 6.9 × 10−8 |
| JS24 | 63.57 ± 0.01 | 52.23 ± 0.06 | 11.34 ± 0.01 | 1.4 × 105 | <1 × 10−4 | Nda |
| JS25 | 61.43 ± 0.48 | 52.13 ± 0.11 | 9.30 ± 0.48 | 5.4 × 104 | <1 × 10−4 | Nda |
| JS26 | 61.47 ± 0.21 | 52.13 ± 0.11 | 9.34 ± 0.21 | 7.2 × 104 | <1 × 10−4 | Nda |
| JS27 | 62.94 ± 0.06 | 52.13 ± 0.11 | 10.81 ± 0.06 | 9.9 × 104 | <1 × 10−4 | Nda |
JS24–JS27 displays greater irreversible binding efficacy relative to BMX-IN-1
The inactivation of BMX occurs in a two-step process that is governed by two parameters: the affinity of the initial non-covalent binding, KI, and the rate of the subsequent covalent bond-forming reaction with the thiol of the cysteine residue, kinact. The rate of inactivation (kinact/KI) is a second-order event, which describes the efficiency of covalent bond formation,.37 Therefore, we evaluated the irreversible binding efficiency of our rationally designed compounds, as previously described.38 The kinetic analysis presented in Table 3, reveals that compound JS25 exhibits the best binding fit with the target, with an inhibition rate constant of 323 pM. This represents an increase in excess of 10-fold relative to BMX-IN-1 (KI: 4.07 nM). The other leads display a similar binding affinity among themselves (1.93–2.52 nM), lower than JS25 and approximately 2-fold higher than BMX-IN-1. However, the rate of covalent bond formation of the bound inhibitor (determined by kinact) shows that compounds JS24, JS25, and JS26 showed slightly improved efficiency (0.335, 0.378 and 0.443 min−1, respectively) in comparison to BMX-IN-1 (0.217 min−1) and JS27 (0.166 min−1). Consequently, the irreversible binding efficiency of JS25 (19.4 μM−1 s−1) is the highest of the series, whereas BMX-IN-1 shows the lowest result (0.89 μM−1 s−1) relative to the remaining inhibitors. Overall, these results provide quantitative evidence that the improved activity is mostly driven by changes in the binding complementarity between the compound and target rather than faster rate of covalent binding. Thus, taking into account that all the analogues have the same Michael acceptor moiety, the enhanced activity must be a result of the structural modifications introduced in the scaffold.| Compound | K I [nM] | k inact [min−1] | k inact/KI [μM−1 s−1] |
|---|---|---|---|
| a Results tested in duplicate, showing mean ± S.D. b Value with a 0.06 μM−1 s−1 deviation from published results.38 | |||
| JS25 | 0.32 ± 0.05 | 0.378 ± 0.034 | 19.4 ± 1.55 |
| JS26 | 1.93 ± 0.18 | 0.443 ± 0.003 | 3.86 ± 0.34 |
| JS24 | 2.52 ± 0.01 | 0.335 ± 0.001 | 2.22 ± 0.01 |
| JS27 | 2.15 ± 0.13 | 0.166 ± 0.003 | 1.29 ± 0.10 |
| BMX-IN-1 | 4.07 ± 0.06 | 0.217 ± 0.005 | 0.89 ± 0.20b |
JS24 covalently modifies cysteine 496 in BMX
Mass spectrometry (MS) studies confirmed the covalent binding of JS24 at cysteine 496 of BMX. The truncated hBMX was analyzed by native MS and the protein mass found was 30899 Da (Fig. 2a). The protein was then treated with 2.5 mM of JS24 at room temperature for 30 min in PBS pH 7.4 and directly analyzed by denaturing MS. The protein was fully denatured and cleaned on a reverse phase column, discarding any non-specific binding, to retain only any covalently linked compound. The mass found upon incubation with JS24 is 31424 Da, which is 525 Da larger than the apo-form of hBMX (Fig. 2b). This result suggests covalent conjugation of a single molecule of JS24 to hBMX. Furthermore, MS/MS analysis after digestion of the drug conjugated hBMX indicates the covalent modification at cysteine residue 496 (Fig. 2c).
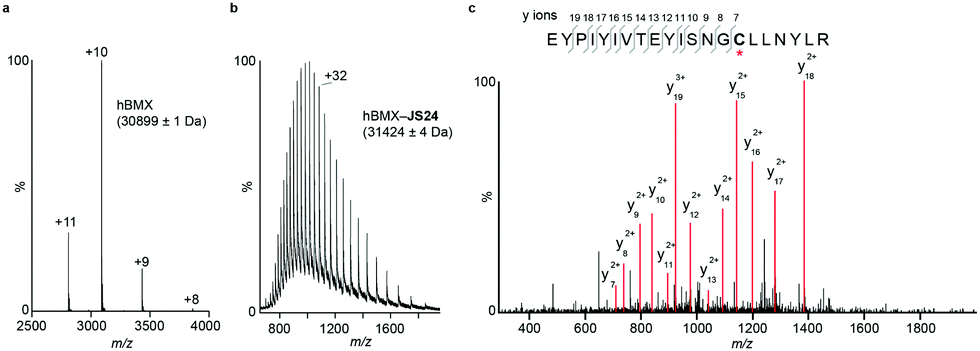 | ||
| Fig. 2 Mass spectrometric analysis of BMX and JS24 conjugated to BMX. (a) Native MS analysis of hBMX. The measured molecular weight is indicated. (b) Denaturing MS analysis of drug-conjugated hBMX. The measured molecular weight is indicated. (c) Tandem MS analysis of the drug conjugated tryptic peptide of hBMX labelled on the sequence (top) and MS/MS mass spectrum (bottom). The red asterisk indicates the Cys site to which JS24 is covalently linked. | ||
The first X-ray structure of BMX with covalent inhibitor
To characterize the inhibition mechanism and binding mode of JS24 to BMX at the atomic level, we tested a variety of commercial crystallization screens to obtain a protein crystal suitable for X-ray diffraction. Crystals were grown through co-crystallization of BMX protein at a final concentration of 10 mg mL−1, with a 2-fold molar excess concentration of inhibitor JS24, in a lead condition that consists of 0.2 M imidazole-malate buffer, pH 5.5, and 42% v/v PEG 600. The X-ray crystal structure of BMX in complex with inhibitor JS24 was determined at 2.0 Å resolution (PDB ID: 6I99) with a well-defined electron density map around the BMX ATP binding pocket in which the inhibitor is bound (Fig. 3a). The values of the equivalent isotropic atomic displacement parameters for the ligand atoms within the pocket are comparable to those of the protein atoms they are interacting with, an indication of full ligand occupancy of the binding site. Not surprisingly, an increase is observed in the sulfonamide aromatic ring because this group is more exposed to the solvent and hence more mobile. | ||
| Fig. 3 Crystal structure of the BMX-JS24 complex (a). Representation of the co-crystal structure of BMX catalytic domain in complex with JS24 (PDB code 6I99). The panel shows the well-defined electron density around the inhibitor, which is bound to BMX ATP binding pocket through a covalent bond with a cysteine residue (Cys496). (b) Non-bonding interactions of JS24 in the ATP binding pocket. (c) BMX DFG-motif adopting the out-like conformation. (d) Analysis of the BMX-DFGout-like motif conformation and BTK DFGin. (e) Analysis of the BMX DFGout-like conformation with BTK DFGout. | ||
The crystal structure shows the expected covalent binding between the acrylamide warhead and Cys496. Other major interactions of the inhibitor with the enzyme active site are mediated through hydrogen bonds between the nitrogen in the quinoline ring and Ile492, and between the Lys445 and the oxygen located in the fused pyridinone ring (Fig. 3b). The hydrogen bond between JS24 and Lys445 is actually one of the key points to regulate BMX activity. The conserved β3 Lys interacts with αC-helix Glu residue to form a salt bridge required for ATP catalysis. The binding of JS24 to Lys445 alters this interaction between the β3 Lys and the αC-helix Glu and consequently inactivates BMX. The aromatic rings of JS24 are engaged in CH/π interactions with the side chains of Tyr491, Ala443, Val431, and Leu543 (Fig. S4, ESI†). Compound JS24 is further stabilized by a hydrogen bond between a water molecule (W1) and the carbonyl oxygen of the acrylamide group. A second water molecule (W2) stabilizes the first (W1) through a hydrogen bond, and forms hydrogen bonds with the peptide nitrogen of Cys496 and the terminal amide group of Asn499 (Fig. 3b).
The crystal structure also shows that the DFG-motif adopts an out-like conformation (Fig. 3c) in which the Asp554 side chain is positioned in the back cleft, away from the ATP binding pocket, and the Phe555 aromatic ring points up into the gatekeeper region blocking the β3 Lys445-αC Glu460 ion pair formation. Both the activation loop and the DFG-out-like conformation are similar to what is observed in the only reported BMX crystal structure with non-covalent inhibitors Dasatinib and PP2.39 The positioning of the BMX DFG-motif is reminiscent of an inactive conformation or DFG-out, typically found in BTK and other kinases inactive structures,40 and it is also commonly observed in type II inhibitor complexes.41 In the apo-BTK (PDB: 3P08), the DFG-in Asp539 rotates towards the ATP binding pocket to chelate magnesium and the DFG-in Phe540 is positioned in the back cleft to allow the formation of the β Lys430-αC Glu445 ion pair, which is important for catalysis. In BMX/JS24 complex the DFG-out-like Asp554 points down and away from the ATP binding pocket and the Phe555 swings up to block the ion pair formation (Fig. 3d). However, relative to the DFG-out-like structure in BMX/JS24 complex with a BTK DFG-out structure (PDB: 3PIY), both structures display complete rotation of the DFG-aspartate residue away from the ATP binding pocket. Only the BTK DFG-out Phe540 residue rotates away from the core of the protein and towards the ATP binding pocket to create a back pocket capable of accepting an aromatic moiety (Fig. 3e).
Finally, the positioning of the sulfonamide aromatic ring is also of utmost importance. This group does not interact with any important residue and it is in fact pointing out of the ATP pocket (Fig. S4, ESI†). Interestingly, this feature would allow for the installation of a linker or chemical handle in this region of the molecule without altering significantly the inhibitor binding capacity of the lead compound.
Molecular dynamics (MD) simulations on BMX covalently linked to JS24 and JS27
We performed then 0.5 μs MD simulations on BMX covalently linked to JS24 (Fig. 4a and b). Computational details can be found in the ESI.† According to these calculations, the binding mode found in the X-ray structure is retained in solution. Both hydrogen bonds releveled by the crystallographic studies, one between the backbone of Ile492 and the nitrogen atom of the quinoline and the other one between the side chain of Lys445 and the carbonyl oxygen of the quinoline ring, are populated in the complex (Fig. 4c). Also, as it occurs in the X-ray structure, JS24 is engaged in CH/π interactions with the side chains of Val431, Ala443, Tyr491 and Leu543 (Fig. S5, ESI†). The MD calculations show that the DFG-motif also adopts an out-like conformation in solution (Fig. 4a). Thus, Phe555 precludes the ion pair formation between Lys445 and Glu460, which is supported by the distance between the side chains of Lys455 and Glu460 (Fig. 4d). Asp554 side chain is located away from the ATP binding pocket (Fig. 4a). The good agreement between the X-ray and the structure proposed by the simulations prompted us to propose a 3D-model for BMX linked to ligand JS27. To this purpose, JS27 covalently linked to Cys496 was superimpose on the X-ray structure of the BMX/JS24 complex. and used as starting structure in the simulations. The simulations show that JS27 adopts a similar pose in the binding site than JS24 (Fig. 4b). Thus, JS27 is engaged in the same hydrogen bonds with the receptor as in BMX/JS24 complex, with populations close to 61.0% and 34.6% for the interactions involving Lys445 and Ile492, respectively. The aromatic system of the ligand interacts with the hydrophobic residues Val431, Ala443, Tyr491 and Leu543 through hydrophobic interactions (Fig. S5, ESI†). As in BMX/JS24 complex, the binding of JS27 impedes the ion pair Lys445-Gl460 formation (Fig. 4d). Significantly, in both complexes, the sulfonamide group of the ligands does not stablish any contact with the receptor and is exposed to the solvent.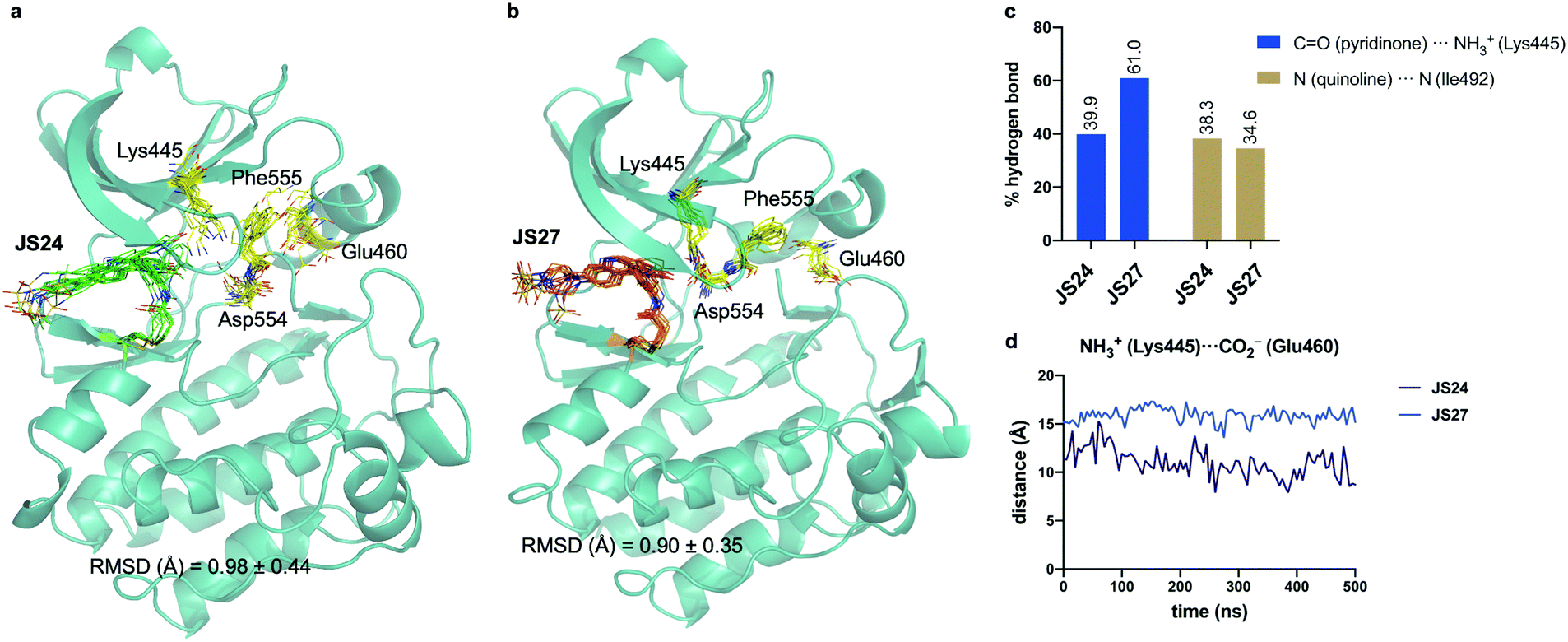 | ||
| Fig. 4 (a and b) Structural ensembles derived from 0.5 μs MD simulations on BMX covalently linked to JS24 and JS27. (c) Population of the most relevant hydrogen bonds between the ligands and BMX derived from the MD simulations. (d) Evolution of the Lys445–Glu460 distance along the MD simulations of both complexes. See also Videos 1 and 2 (ESI†). JS24, JS27, Lys445, Glu460, Asp554 and Phe555 are shown as sticks. The protein is shown as ribbons and only the first conformer (time = 0 ns) is shown for clarity. | ||
JS25 is a selective TEC family inhibitor
Most of BMX inhibitors reported to date offer poor selectivity because they are both BMX and BTK inhibitors. Their cellular effect is often attributed to off-target activity either upstream or downstream of BMX signaling pathways. To investigate in which targets the new inhibitors could have an effect we tested potent inhibitor JS25 against a panel of 36 BMX-related kinases in the Eurofins DiscoverX's KINOMEscan™ platform at a concentration of 1 μM.It is important to note that from the extensive number of accessible cysteine residues distributed across the whole kinome not all are available for covalent modification.12–14 BMX belongs to a restricted group that includes 10 other kinases that share an equivalently placed cysteine in the ATP binding pocket. This group comprises members from the TEC family (BTK, ITK, TXK and TEC), the EGFR family (EGFR, Her2, Her4), JAK3, BLK and dual specificity mitogen-activated protein kinase 7 (MAP2K7). Therefore, we included the whole TEC, EGFR and JAK family in our screening, and the Src family and Lkb1, which also have a cysteine within the same sequence alignment. We also included kinases involved in upstream (Src, FAK, PI3K, mTOR, PDK1) and downstream (Akt, PAK1, TAM) regulation of BMX signaling pathway and non-receptor tyrosine protein kinase Abl. The KinomeScan platform is a binding assay and the screening showed that JS25 displays a strong binding affinity against all the members of TEC family that share an equivalently placed cysteine and within these, higher affinity is observed towards BMX, BTK and TEC (Table 4).
| Family | Target | % Ctrl | Family | Target | % Ctrl |
|---|---|---|---|---|---|
| TEC | BMX | 1.3 | Src | FYN | 99 |
| BTK | 0 | SRC | 92 | ||
| ITK | 4.7 | YES1 | 85 | ||
| TEC | 0.4 | BLK | 16 | ||
| TXK | 3.4 | FGR | 93 | ||
| EGFR | EGFR | 87 | LCK | 80 | |
| ERBB2 | 89 | HCK | 95 | ||
| ERBB3 | 91 | LYN | 100 | ||
| ERBB4 | 66 | mTOR | MTOR | 100 | |
| JAK | JAK1(JH1domain-catalytic) | 93 | Liver Kinase B1 | STK11 | 52 |
| JAK2(JH1domain-catalytic) | 81 | Pkb | AKT1 | 100 | |
| JAK3(JH1domain-catalytic) | 21 | AKT2 | 94 | ||
| TYK2(JH1domain-catalytic) | 100 | AKT3 | 99 | ||
| FAK | PTK2 | 93 | PAK1 | PAK1 | 100 |
| PI3K | PIK3CA | 79 | TAM | AXL | 93 |
| PIK3CB | 89 | MERTK | 89 | ||
| PIK3CG | 64 | Abl | ABL1-phosphorylated | 100 | |
| PIK3CD | 100 | PDPK1 | PDPK1 | 92 |
As stated above, the TEC family has high sequence similarity and in particular residues in the ATP binding kinase domain share 40–65% identity and 60–80% similarity. The ATP binding sites are also highly conserved between the TEC and Src families with 14 identical residues out of 18 that comprise the ATP binding pocket. More specifically, BMX shares a 57% similarity to Src and most importantly, one of the key determinants of kinase selectivity – the gatekeeper residue – is a Thr in both the Src family and the TEC family members except ITK.39 It is therefore not surprising that JS25 also binds Blk (and JAK3) whereas no affinity was observed with other potential targets. These results reveal JS25 as a selective probe for TEC kinases and suggest that any cellular activity mediated by JS25 is probably a result of inhibition of any of the TEC kinases rather than any off-target inhibition of upstream and downstream BMX regulators.
Intracellular BMX inhibition and degradation by JS25
To validate target affinity and identification for JS25, we performed an intracellular target engagement kinase assay with HEK293 cells expressing NanoLuc®-BMX fusion vector with Promega's NanoBRET™ TE Intracellular Kinase Assay. The cell proliferation depends on BMX kinase activity that was used to monitor the cellular activity of the compounds (IC50). As shown in Fig. 5a, the IC50 determination showed the inhibitory capacity of JS25 (IC50: 44.8 nM) is 10 times greater than BMX-IN-1 (IC50: 495 nM), which aligns with the previous observations of an increased biochemical potency with similar activity difference. We next investigated whether the treatment of wild-type PC3 cells with JS25, as well as BMX-IN-1 (as a control), would induce degradation of BMX. After 24 and 72 h, it was possible to verify that the level of expressed BMX protein in PC3 cells decreased upon treatment with JS25 as well as with the control molecule (Fig. 5b and Fig. S11, ESI†). This data indicates that JS25 is able both inhibit catalytic activity and degrade BMX it in cells.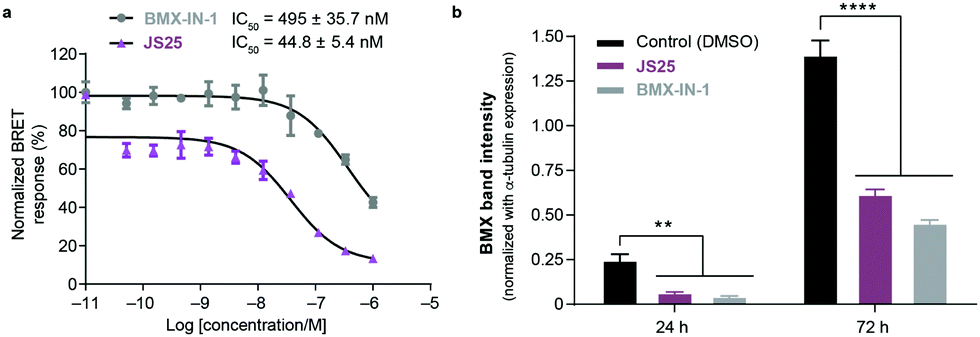 | ||
| Fig. 5 (a) Intracellular target engagement in HEK293 cells transiently transfected with BMX expressing NanoLuc®-BMX fusion vector with Promega's NanoBRET™ TE Intracellular Kinase Assay. Assay performed at Reaction Biology Corporation (USA), with concentrations tested in duplicate, showing mean ± S.D. Cells were treated for 1 h and IC50 values were calculated and plotted by using GraphPad Prism 8 based on a sigmoidal dose response curve. (b) JS25 (10 μM) and BMX-IN-1 (10 μM) induce degradation of wild-type BMX in PC3 cells. Sampling was taken after 24 h and 72 h of incubation with JS25 and BMX-IN-1. Band intensity was measured using ImageJ and normalized with α-Tubulin band intensity. Differences between groups were revealed through 2way-ANOVA. Data are mean ± standard deviation obtained from at least three independent measurements (n = 3). See ESI† for additional data and analysis. | ||
Cancer cell growth inhibition by BMX inhibitors
The role of BMX in different pathologies is not yet fully validated. Nevertheless, it has been implicated in many regulatory mechanisms and despite the absence of a BMX dependent disease model, prostate cancer cell lines have been used to evaluate anti-proliferative effects of the inhibitors in a cellular context. In a previous experiment (unpublished results) we screened several inhibitors from Table S1 (ESI†) in a collection of cell lines representing prostate, brain, blood, breast, ovary, lung, bone marrow and lymphoid tumour tissues. Compounds were incubated with cells for 72 h in a 386 well-plate format to monitor dose-dependent impact on viable cell growth by using the CellTiter-Glo® luminescent assay, which quantifies ATP and the presence of metabolically active cells. The study included JS24, BMX-IN-1 and the structurally similar compounds JS10 and JS11 which do not bind to BMX (Fig. S1 and Table S1, ESI†).The results presented in Table 5 show that JS10 and JS11 (non-binders) have little or no effect on viable cell growth of the majority of the tested cell lines. BMX-IN-1 demonstrated more potent inhibitory effects relative to JS24 in the four prostate cancer cell lines that were included in the panel, 22RV1, PC3, LNCaP and DU145, particularly in those dependent on androgen receptor signaling (LNCaP and 22RV1). In contrast, androgen receptor negative cells (DU145 and PC3) were overall more resistant to treatment. In addition, JS24 showed potent inhibitory effects against LNCaP and 22RV1 but also against PC3, which are androgen receptor negative cells. Furthermore, both compounds were also potent inhibitors of viable cell growth for RS4 (11) (lymphoblast) and DAUDI (T-lymphoblast) cells, in which BTK is highly overexpressed. Altogether, these results demonstrate BMX inhibition impacts viable cell growth of prostate cancer cells and prompted us to investigate further the importance of the androgen receptor and related BMX pathways in these cell lines.
| Tissue | Cell line | BMX-IN-1 | JS24 | JS10 | JS11 |
|---|---|---|---|---|---|
| a Viable cell growth was measured after 72 h incubation in 386 well-plate format. GI50 values were tested in triplicate and are reported as the mean ± SD in μM. ND, non-determined, no growth inhibition observed within the concentrations tested. NC, non-calculated. When ambiguous fit was observed curves were top (100%) and bottom (0%) constrained and GI50 was determined with 4-P least squares fit. In these cases, SD is not calculated by GraphPad Prism 8. | |||||
| Prostate | LNCaP | 1.81 ± 0.05 | 4.4 ± NC | 9.7 ± NC | 10.41 ± NC |
| 22 RV1 | 2.07 ± 0.06 | 6.66 ± 0.09 | 4.86 ± 0.11 | 7.3 ± NC | |
| PC3 | 10.98 ± 1.13 | 4.8 ± NC | ND | 20.12 ± NC | |
| DU145 | 17.7 ± NC | ND | ND | ND | |
| Brain | U-87MG | 5.33 ± 0.19 | 5.04 ± 0.01 | ND | ND |
| SK-N-MC | 2.36 ± NC | 8.53 ± 0.44 | 11.19 ± NC | 8.24 ± NC | |
| Blood | Jurkat | 5.99 ± NC | 5.48 ± ND | 9.71 ± 1.48 | 6.36 ± 0.17 |
| Kasumi | 3.13 ± 0.06 | 5.12 ± 0.12 | 4.37 ± 0.04 | 10.14 ± 0.07 | |
| Breast | MDA-MB-231 | 23.61 ± 0.48 | ND | ND | ND |
| Ovary | CAOV3 | 7.68 ± 0.13 | 8.56 ± NC | 17.30 ± NC | 19.31 ± NC |
| OVCAR3 | ND | ND | ND | ND | |
| Bone marrow | H1299 | ND | 7.28 ± 0.37 | 19.42 ± NC | ND |
| Lung | RS4(11) | 1.176 ± 0.06 | 2.09 ± NC | 5.06 ± NC | 6.66 ± NC |
| Lymphoid | DAUDI | 1.68 ± 0.07 | 1.27 ± 0.05 | 2.57 ± 0.09 | 4.57 ± 0.12 |
Androgen-receptor positive prostate-cancer cells are sensitive to JS24–JS27
Based on the results showed in the previous section, we tested the ability of compounds JS24–JS27 to inhibit the proliferation of LNCaP and PC3 prostate cancer cell lines by using CellTiter-Glow®. The androgen-receptor negative PC3 cells are resistant to the treatment, with no significant anti-proliferative effect at the maximum concentration tested (10 μM). With androgen-receptor positive cells LNCaP a different profile was observed. BMX-IN-1 and JS24 showed a GI50 of 1.7 and 1.5 μM, respectively. Compound JS27 was the least active (GI50 9.3 μM), and JS25 and JS26 inhibited proliferation with a GI50 of 6.6 and 7.7 μM, respectively, as shown in Fig. 6.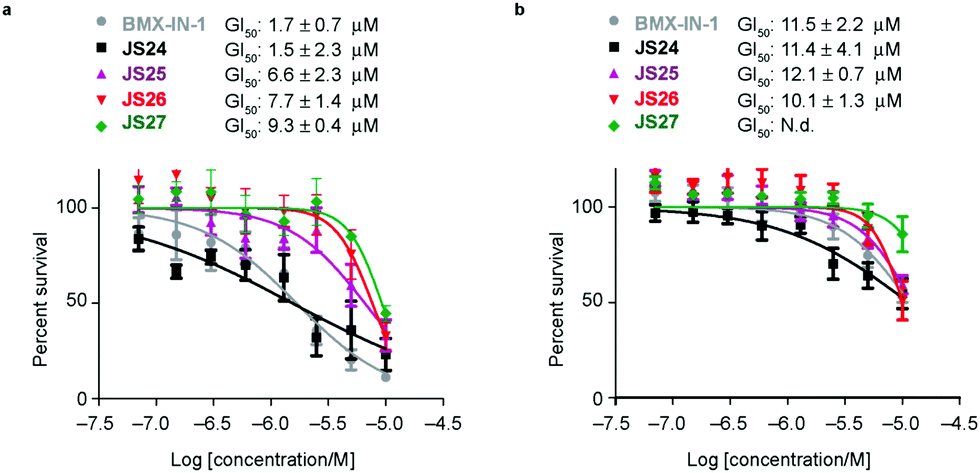 | ||
| Fig. 6 Anti-proliferative activity of compounds BMX-IN-1 and JS24–JS27 against LNCaP (a) and PC3 (b) prostate-cancer cell lines. Briefly, proliferation in LNCaP and PC3 cells was measured after 96 h incubation, in 96-well plates with BMX-IN-1 and JS24–JS27. GI50 values are reported in μM and are the mean of three individual experiments performed in triplicate. N.d., non-determined, no growth inhibition observed within the concentrations tested. | ||
To determine whether the growth inhibition was due to apoptosis, we carried out fluorescence-assisted cell sorting analysis with propidium iodide staining. LNCaP cells were incubated with BMX-IN-1 and JS24–JS27 for 64h, at 10 μM and results showed that no marked differences in the percentage of necrotic events relative to the vehicle control, which suggests that in these conditions these molecules do not enhance cell death (Fig. S6, ESI†). It is not surprising that all these compounds show only moderate proliferation inhibitory potential in prostate-cancer cell lines and it remains questionable whether modulation of BMX per se is relevant or not, towards anti-proliferative effects.33 In fact, a large body of evidence in the literature shows that selective or dual BMX/BTK inhibitors have poor anti-proliferative effects in BMX-dependent models, most probably from dynamic compensation of signaling mechanisms. Focus has been placed on the modulation of BMX activity to sensitize prostate-cancer cells to other therapeutic agents because anti-proliferative effects are only observed in combination with inhibitors of related pathways.42BMX-IN-1 growth inhibition of RV-1 cells could only be potentiated with the Akt inhibitor MK2206;33 ABT-737, a non-covalent inhibitor only induces apoptosis upon co-treatment with PI3K inhibitors;28 the dual BMX/BTK inhibitor CTN06 requires co-treatment with autophagy inhibitor chloroquine (CQ) or docetaxel to inhibit PC3 cells growth43 and a similar profile is observed with the dual BMX/Src inhibitor CTA095 to synergize with CQ and paclitaxel.44
The activation of BMX in response to PI3K signaling is just one of the mechanisms through which the levels of BMX become increased in prostate cancer.43,45 A very recent study46 showed that BMX expression in prostate cancer is suppressed directly through androgen receptor as a result of binding to BMX. Consequently, BMX expression rapidly increases in response to androgen deprivation therapy which enhances tyrosine kinase signaling and the subsequent emergence of castration-resistant prostate cancer. This study further highlights the potential use of BMX inhibitors in combination therapy, in this case in combination with AR targeting. To further validate this hypothesis and assess the effect of our inhibitors with other drugs, we performed a co-treatment regimen with known inhibitors of related upstream and downstream pathways.
Co-treatment of LNCaP cells with JS24–JS26 and androgen receptor antagonist, PI3K and AKT inhibitors
As shown above, BMX inhibition alone induces limited cell death in BMX-expressed cell lines owing to the existence of compensatory mechanisms in signaling pathways. To evaluate whether our BMX inhibitors could potentially be used in combination treatment regimens, we sought to look at the synergistic anti-proliferative effects of BMX inhibitors when combined with other therapeutic agents, which pre-sensitize prostate cancer cells. For this purpose, LNCaP cells were co-treated in a combinatorial fashion with compounds JS24–26, AKT1/2 (AKT inhibitor), Flutamide (androgen receptor antagonist) and LY294002 (PI3K inhibitor). Cell viability was evaluated after 5 days with CellTiter-Glo® and compared with the overall anti-proliferative effects of the compounds alone. An optimization study was performed by screening several concentrations (Fig. S7, ESI†) to determine the ideal conditions to obtain initial viability above 80% with the individual inhibitors alone (Fig. S8, ESI†). Based on these results, we tested JS24 (at 3 μM), JS25 (5 μM) and JS26 (6 μM) with AKT1/2 (1 μM), Flutamide (50 μM) and LY294002 (3 μM). Results are shown in Fig. 7.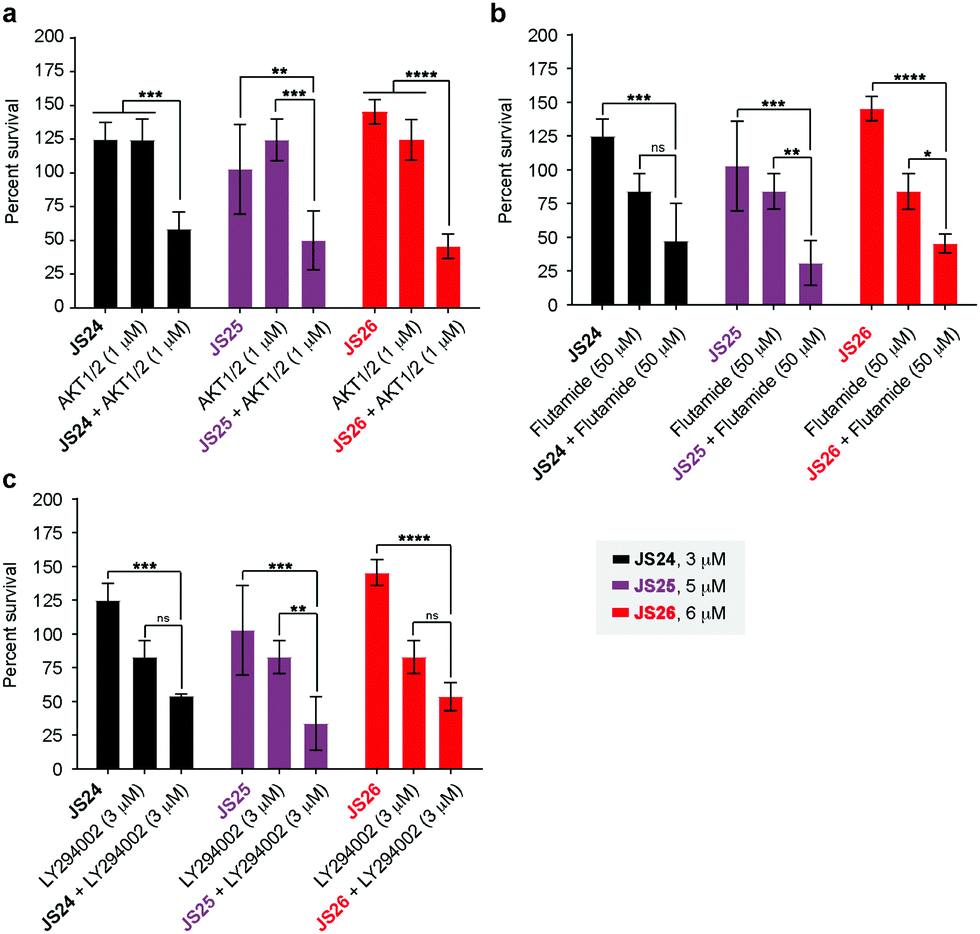 | ||
| Fig. 7 Anti-proliferative activity in LNCaP cells of compounds JS24–JS26 in combination with AKT1/2 (AKT inhibitor), Flutamide (androgen receptor antagonist) and LY293002 (PI3K inhibitor). (a) Cells co-treated with JS24 (3 μM), JS25 (5 μM) and JS26 (6 μM) with AKT1/2 (1 μM); (b) JS24 (3 μM), JS25 (5 μM) and JS26 (6 μM) with Flutamide (50 μM); (c) JS24 (3 μM), JS25 (5 μM) and JS26 (6 μM) with LY294002 (3 μM). Values are reported in % cell viability normalized to DMSO controls and are the mean of three individual experiments performed in triplicate. Determined P-values are illustrated as ns (P > 0.05), * (P ≤ 0.05), ** (P ≤ 0.01), *** (P ≤ 0.001) and **** (P ≤ 0.0001). | ||
Although the control concentrations of JS24–26 and the inhibitors did not have an effect on reducing cell viability upon co-treatment, a marked viability decrease was observed in all tested conditions. With AKT1/2 a decrease in cell viability ranging from 48% (with JS24) to 63% (with JS26) was observed, relative to control AKT1/2. With Flutamide, the most effective combination was with compound JS25 (63% cell viability reduction) and the least effective with JS24 (44% reduction). Finally, co-treatment with LY294002 decreased cell viability by 35% (with JS24 and JS26) and 59% (with JS25). Overall, these results demonstrate a synergistic effect between JS24-26 and AKT1/2, Flutamide and LY294002 in cancer cell proliferation capable of overcoming the compensatory mechanisms of BMX inhibition, and open the possibility of becoming useful molecules for drug combination approaches.
Conclusions
We explored a chemical scaffold that contains an archetypal tricyclic core of a quinoline with a fused pyridinone, which is present in BMX covalent inhibitor BMX-IN-1. We sought to introduce a chemical handle that may be used for further derivatization whilst simultaneously tuning the physicochemical properties. We found that rational modification introduced at the position 7 of the quinoline moiety leads to potent, single-digit nanomolar inhibition of BMX and BTK. Topological pharmacophoric features are outside the chemical space previously explored in BMX inhibition, and afford molecules with more favorable physicochemical profiles with a reduced clogP and increased permeability (JS26 and JS27). We also unveiled the X-ray crystal structure of BMX with a covalent inhibitor (JS24), which shows the protein with the “DFG-out like” motif typical of an inactive conformation. The crystal structure also shows that the “tail substituent”, opposite to the acrylamide warhead points outside the ATP pocket, which suggests an exit vector for further derivatizations. A comparable pose for ligands JS24 and JS27 were proposed by extensive MD simulations performed on the complexes with BMX.
We determined the rate of covalent modification and, to our knowledge, this is the highest value reported in the literature. The kinetic analysis showed that this is mostly driven from the potency of the first reversible binding event (KI = 323 pM) and shows that our rational design afforded a preferred fit for the BMX binding pocket. In a cellular context, most potent compound JS25 also showed low nanomolar potency in a target engagement assay (45 nM) in BMX-dependent cells (transfected HEK293) which is 10-fold superior to the reference ligand. Treatment of PC3 cells with JS25 also led to degradation BMX. Furthermore, all lead compounds displayed anti-proliferative effects in androgen-receptor positive prostate cancer cells that where further increased when combined with known inhibitors of related signaling pathways, further highlighting the potential of combinatorial effects with BMX-related pathways.
As stated above, selectivity among members of the TEC family is hard to achieve. Interestingly, available data shows that therapeutically active drugs are not selective molecules. Ibrutinib, developed as a covalent BTK inhibitor, has been approved by the FDA for the treatment of chronic lymphocytic leukemia, mantle cell leukemia (MCL) and Waldenström macroglobulinemia and is currently in multiple clinical trials because it has proved efficacy in different indications, such as non-small cell lung cancer and autoimmune diseases.7,47,48 With a broad selectivity profile, Ibrutinib inhibits the whole TEC family, EGFR, JAK3, Her2, Blk and Itk kinases. Acalabrutinib, a second generation BTK inhibitor that was also granted FDA approval for MCL is more selective, and only inhibits BTK, TEC, BMX and TXK.49 Other BTK inhibitors in clinical development (Spebrutinib, Zanubrutinib and Tirabrutinib) also display a broad selectivity for kinases with a cysteine as the Cys496 residue in BMX,50 consequently, it is reasonable to extrapolate that the compounds developed here can become therapeutically useful as BMX inhibitors and find application in other TEC-related B-cell malignancies. The most potent compound JS25 also has a multi-target profile and is active against all five TEC kinases, JAK3 and BLK. As such, we are currently evaluating the utility of these new molecules in B-cell related lymphocytic diseases where TEC-kinases play a prominent role.17,51
Conflicts of interest
J. D. S. and G. J. L. B. are inventors in a patent application related to the findings reported in this manuscript. Other authors declare no competing interests.Acknowledgements
Funded under the Royal Society (URF to G. J. L. B., URF\R\180019), FCT Portugal (iFCT to G. J. L. B., IF/00624/2015, Postdoctoral Fellowship SFRH/BPD/95253/2013 to J. D. S., project PTDC/MED-QUI/28764/2017 to J. D. S., 02/SAICT/2017 grant 28333 to T. R., doctoral studentship SFRH/BD/143583/2019 to B. B. S., and DL 57/2016/CP1451/CT0025 to J. D. S.). This project has received funding from the European Union's Horizon 2020 research and innovation programme under grant agreements No. 807281 and 702428. The authors acknowledge the Wellcome Trust (104633/Z/14/Z, to D. W. and C. V. R.), Ministerio de Economía y Competitividad (project RTI2018-099592-B-C21 to F. C.), the Royal G. and Mae H. Westaway Family Memorial Fund (A. N. K.), National Science Foundation Graduate Research Fellowship (Grant No 1122374 to S. K. D.) and Ludwig Center at MIT's Koch Institute (S. K. D.). We also thank the High-Throughput Science Core at MIT supported by the Koch Institute Cancer Center Support Core Grant (P30-CA14051 to A. N. K.). The NMR spectrometers are part of the National NMR Facility, supported by Fundação para a Ciência e Technologic (RECI/BBB-BQB/0230/2012). The authors also thank ESRF and the ID30A-3 beamline staff for support with the X-ray data collection. iNOVA4Health – UID/Multi/04462/2013, a program financially supported by Fundação para a Ciência e Technologic/Ministério da Educação e Ciência, through national funds and co-funded by FEDER under the PT2020 Partnership Agreement, and MOSTMICRO (project LISBOA-01-0145-FEDER-007660) co-funded by FCT/MCTES and FEDER funds through COMPETE2020—Programa Operacional Competitividade e Internacionalização (POCI) are also acknowledged. The authors thank Dr Vikki Cantrill for her help with the preparation and editing of this manuscript.Notes and references
- J. Singh, R. C. Petter, T. A. Baillie and A. Whitty, Nat. Rev. Drug Discovery, 2011, 10, 307–317 CrossRef CAS PubMed.
- Z. Zhao and P. E. Bourne, Drug Discovery Today, 2018, 23, 727–735 CrossRef CAS PubMed.
- A. Chaikuad, P. Koch, S. A. Laufer and S. Knapp, Angew. Chem., Int. Ed., 2018, 57, 4372–4385 CrossRef CAS PubMed.
- T. Barf and A. Kaptein, J. Med. Chem., 2012, 55, 6243–6262 CrossRef CAS PubMed.
- R. Lagoutte, R. Patouret and N. Winssinger, Curr. Opin. Chem. Biol., 2017, 39, 54–63 CrossRef CAS PubMed.
- A. Abdeldayem, Y. S. Raouf, S. N. Constantinescu, R. Moriggl and P. T. Gunning, Chem. Soc. Rev., 2020, 49, 2617–2687 RSC.
- L. A. Honigberg, A. M. Smith, M. Sirisawad, E. Verner, D. Loury, B. Chang, S. Li, Z. Pan, D. H. Thamm, R. A. Miller and J. J. Buggy, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 13075–13080 CrossRef CAS PubMed.
- J. C. Byrd, B. Harrington, S. O’Brien, J. A. Jones, A. Schuh, S. Devereux, J. Chaves, W. G. Wierda, F. T. Awan, J. R. Brown, P. Hillmen, D. M. Stephens, P. Ghia, J. C. Barrientos, J. M. Pagel, J. Woyach, D. Johnson, J. Huang, X. Wang, A. Kaptein, B. J. Lannutti, T. Covey, M. Fardis, J. McGreivy, A. Hamdy, W. Rothbaum, R. Izumi, T. G. Diacovo, A. J. Johnson and R. R. Furman, N. Engl. J. Med., 2015, 374, 323–332 CrossRef PubMed.
- V. A. Miller, V. Hirsh, J. Cadranel, Y. M. Chen, K. Park, S. W. Kim, C. Zhou, W. C. Su, M. Wang, Y. Sun, D. S. Heo, L. Crino, E. H. Tan, T. Y. Chao, M. Shahidi, X. J. Cong, R. M. Lorence and J. C. Yang, Lancet Oncol., 2012, 13, 528–538 CrossRef CAS PubMed.
- J.-C. Soria, Y. Ohe, J. Vansteenkiste, T. Reungwetwattana, B. Chewaskulyong, K. H. Lee, A. Dechaphunkul, F. Imamura, N. Nogami, T. Kurata, I. Okamoto, C. Zhou, B. C. Cho, Y. Cheng, E. K. Cho, P. J. Voon, D. Planchard, W.-C. Su, J. E. Gray, S.-M. Lee, R. Hodge, M. Marotti, Y. Rukazenkov and S. S. Ramalingam, N. Engl. J. Med., 2017, 378, 113–125 CrossRef PubMed.
- S. K. Rabindran, C. M. Discafani, E. C. Rosfjord, M. Baxter, M. B. Floyd, J. Golas, W. A. Hallett, B. D. Johnson, R. Nilakantan, E. Overbeek, M. F. Reich, R. Shen, X. Shi, H.-R. Tsou, Y.-F. Wang and A. Wissner, Cancer Res., 2004, 64, 3958–3965 CrossRef CAS PubMed.
- J. Zhang, P. L. Yang and N. S. Gray, Nat. Rev. Cancer, 2009, 9, 28–39 CrossRef CAS PubMed.
- Q. Liu, Y. Sabnis, Z. Zhao, T. Zhang, S. J. Buhrlage, L. H. Jones and N. S. Gray, Chem. Biol., 2013, 20, 146–159 CrossRef CAS PubMed.
- Z. Zhao, Q. Liu, S. Bliven, L. Xie and P. E. Bourne, J. Med. Chem., 2017, 60, 2879–2889 CrossRef CAS PubMed.
- B. R. Lanning, L. R. Whitby, M. M. Dix, J. Douhan, A. M. Gilbert, E. C. Hett, T. O. Johnson, C. Joslyn, J. C. Kath, S. Niessen, L. R. Roberts, M. E. Schnute, C. Wang, J. J. Hulce, B. Wei, L. O. Whiteley, M. M. Hayward and B. F. Cravatt, Nat. Chem. Biol., 2014, 10, 760–767 CrossRef CAS PubMed.
- C. I. Smith, T. C. Islam, P. T. Mattsson, A. J. Mohamed, B. F. Nore and M. Vihinen, BioEssays, 2001, 23, 436–446 CrossRef CAS PubMed.
- N. J. Horwood, A. M. Urbaniak and L. Danks, Int. Rev. Immunol., 2012, 31, 87–103 CrossRef CAS PubMed.
- J. S. Jarboe, S. Dutta, S. E. Velu and C. D. Willey, Recent Pat. Anticancer Drug Discovery, 2013, 8, 228–238 CrossRef CAS PubMed.
- Y. Qiu and H. J. Kung, Oncogene, 2000, 19, 5651–5661 CrossRef CAS PubMed.
- X. Wen, H. H. Lin, H. M. Shih, H. J. Kung and D. K. Ann, J. Biol. Chem., 1999, 274, 38204–38210 CrossRef CAS PubMed.
- O. A. Guryanova, Q. Wu, L. Cheng, J. D. Lathia, Z. Huang, J. Yang, J. MacSwords, C. E. Eyler, R. E. McLendon, J. M. Heddleston, W. Shou, D. Hambardzumyan, J. Lee, A. B. Hjelmeland, A. E. Sloan, M. Bredel, G. R. Stark, J. N. Rich and S. Bao, Cancer Cell, 2011, 19, 498–511 CrossRef CAS PubMed.
- J. Kaukonen, I. Lahtinen, S. Laine, K. Alitalo and A. Palotie, Br. J. Haematol., 1996, 94, 455–460 CrossRef CAS PubMed.
- R. Bagheri-Yarmand, M. Mandal, A. H. Taludker, R. A. Wang, R. K. Vadlamudi, H. J. Kung and R. Kumar, J. Biol. Chem., 2001, 276, 29403–29409 CrossRef CAS PubMed.
- K. Y. Chen, L. M. Huang, H. J. Kung, D. K. Ann and H. M. Shih, Oncogene, 2004, 23, 1854–1862 CrossRef CAS PubMed.
- I. Cohen, M. Maoz, H. Turm, S. Grisaru-Granovsky, B. Maly, B. Uziely, E. Weiss, R. Abramovitch, E. Gross, O. Barzilay, Y. Qiu and R. Bar-Shavit, PLoS One, 2010, 5, e11135 CrossRef PubMed.
- B. Dai, H. Chen, S. Guo, X. Yang, D. E. Linn, F. Sun, W. Li, Z. Guo, K. Xu, O. Kim, X. Kong, J. Melamed, S. Qiu and Y. Qiu, Cancer Res., 2010, 70, 5587–5596 CrossRef CAS PubMed.
- B. Dai, O. Kim, Y. Xie, Z. Guo, K. Xu, B. Wang, X. Kong, J. Melamed, H. Chen, C. J. Bieberich, A. D. Borowsky, H. J. Kung, G. Wei, M. C. Ostrowski, A. Brodie and Y. Qiu, Cancer Res., 2006, 66, 8058–8064 CrossRef CAS PubMed.
- D. S. Potter, P. Kelly, O. Denneny, V. Juvin, L. R. Stephens, C. Dive and C. J. Morrow, Neoplasia, 2014, 16, 147–157 CrossRef CAS PubMed.
- Y. Li, N. Cui, P. S. Zheng and W. T. Yang, Oncotarget, 2017, 8, 49238–49252 CrossRef PubMed.
- I. Rajantie, N. Ekman, K. Iljin, E. Arighi, Y. Gunji, J. Kaukonen, A. Palotie, M. Dewerchin, P. Carmeliet and K. Alitalo, Mol. Cell. Biol., 2001, 21, 4647–4655 CrossRef CAS PubMed.
- D. S. Potter, M. Galvin, S. Brown, A. Lallo, C. L. Hodgkinson, F. Blackhall, C. J. Morrow and C. Dive, Mol. Cancer Ther., 2016, 15, 1248–1260 CrossRef CAS PubMed.
- J. L. Fox and A. Storey, Cancer Res., 2015, 75, 1345–1355 CrossRef CAS PubMed.
- F. Liu, X. Zhang, E. Weisberg, S. Chen, W. Hur, H. Wu, Z. Zhao, W. Wang, M. Mao, C. Cai, N. I. Simon, T. Sanda, J. Wang, A. T. Look, J. D. Griffin, S. P. Balk, Q. Liu and N. S. Gray, ACS Chem. Biol., 2013, 8, 1423–1428 CrossRef CAS PubMed.
- D. Reker, G. J. L. Bernardes and T. Rodrigues, Nat. Chem., 2019, 11, 402–418 CrossRef CAS PubMed.
- M. M. Hann and G. M. Keserü, Nat. Rev. Drug Discovery, 2012, 11, 355–365 CrossRef CAS PubMed.
- T. Rodrigues, M. Werner, J. Roth, E. H. G. da Cruz, M. C. Marques, P. Akkapeddi, S. A. Lobo, A. Koeberle, F. Corzana, E. N. da Silva Júnior, O. Werz and G. J. L. Bernardes, Chem. Sci., 2018, 9, 6899–6903 RSC.
- J. M. Strelow, SLAS Discovery, 2016, 22, 3–20 Search PubMed.
- B. Wang, Y. Deng, Y. Chen, K. Yu, A. Wang, Q. Liang, W. Wang, C. Chen, H. Wu, C. Hu, W. Miao, W. Hur, W. Wang, Z. Hu, E. L. Weisberg, J. Wang, T. Ren, Y. Wang, N. S. Gray, Q. Liu and J. Liu, Eur. J. Med. Chem., 2017, 137, 545–557 CrossRef CAS PubMed.
- J. Muckelbauer, J. S. Sack, N. Ahmed, J. Burke, C. Y. Chang, M. Gao, J. Tino, D. Xie and A. J. Tebben, Chem. Biol. Drug Des., 2011, 78, 739–748 CrossRef CAS PubMed.
- M. M. Sultan, R. A. Denny, R. Unwalla, F. Lovering and V. S. Pande, Sci. Rep., 2017, 7, 15604 CrossRef PubMed.
- Z. Zhao, H. Wu, L. Wang, Y. Liu, S. Knapp, Q. Liu and N. S. Gray, ACS Chem. Biol., 2014, 9, 1230–1241 CrossRef CAS PubMed.
- J. L. Fox, F. Ismail, A. Azad, N. Ternette, S. Leverrier, M. J. Edelmann, B. M. Kessler, I. M. Leigh, S. Jackson and A. Storey, EMBO J., 2010, 29, 3853–3868 CrossRef CAS PubMed.
- W. Guo, R. Liu, G. Bhardwaj, J. C. Yang, C. Changou, A. H. Ma, A. Mazloom, S. Chintapalli, K. Xiao, W. Xiao, P. Kumaresan, E. Sanchez, C. T. Yeh, C. P. Evans, R. Patterson, K. S. Lam and H. J. Kung, Cell Death Dis., 2014, 5, e1409 CrossRef CAS PubMed.
- W. Guo, R. Liu, G. Bhardwaj, A. H. Ma, C. Changou, J. C. Yang, Y. Li, C. Feng, Y. Luo, A. Mazloom, E. Sanchez, Y. Wang, W. Huang, R. Patterson, C. P. Evans, K. S. Lam and H. J. Kung, PLoS One, 2013, 8, e70910 CrossRef CAS PubMed.
- C. H. Chau, K. Y. Chen, H. T. Deng, K. J. Kim, K. Hosoya, T. Terasaki, H. M. Shih and D. K. Ann, Oncogene, 2002, 21, 8817–8829 CrossRef CAS PubMed.
- S. Chen, C. Cai, A. G. Sowalsky, H. Ye, F. Ma, X. Yuan, N. I. Simon, N. S. Gray and S. P. Balk, Cancer Res., 2018, 78, 5203–5215 CrossRef CAS PubMed.
- C. S. Lee, M. A. Rattu and S. S. Kim, J. Oncol. Pharm. Pract., 2016, 22, 92–104 CrossRef PubMed.
- B. Xia, F. Qu, T. Yuan and Y. Zhang, Oncol. Lett., 2015, 10, 3339–3344 CrossRef CAS PubMed.
- T. Barf, T. Covey, R. Izumi, B. van de Kar, M. Gulrajani, B. van Lith, M. van Hoek, E. de Zwart, D. Mittag, D. Demont, S. Verkaik, F. Krantz, P. G. Pearson, R. Ulrich and A. Kaptein, J. Pharmacol. Exp. Ther., 2017, 363, 240–252 CrossRef CAS PubMed.
- A. Kaptein, G. de Bruin, M. Emmelot-van Hoek, B. van de Kar, A. de Jong, M. Gulrajani, D. Demont, T. Covey, D. Mittag and T. Barf, Blood, 2018, 132, 1871 CrossRef.
- M. J. de Bruijn, J. Rip, E. K. van der Ploeg, L. W. van Greuningen, V. T. Ta, L. P. Kil, A. W. Langerak, G. F. Rimmelzwaan, W. Ellmeier, R. W. Hendriks and O. B. Corneth, J. Immunol., 2017, 198, 3058–3068 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Methods and materials, supporting figures and tables, synthesis and characterisation including NMR spectra, and references. Two supporting videos are also available. See DOI: 10.1039/d0cb00033g |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2020 |