
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOrganic semiconductor nanoparticles for visible-light-driven CO2 conversion†
Mariia
Ferree
 a,
Jan
Kosco
a,
Nisreen
Alshehri
ad,
Lingyun
Zhao
b,
Catherine S. P.
De Castro
a,
Christopher E.
Petoukhoff
a,
Iain
McCulloch
c,
Martin
Heeney
a and
Frédéric
Laquai
*a
a,
Jan
Kosco
a,
Nisreen
Alshehri
ad,
Lingyun
Zhao
b,
Catherine S. P.
De Castro
a,
Christopher E.
Petoukhoff
a,
Iain
McCulloch
c,
Martin
Heeney
a and
Frédéric
Laquai
*a
aPhysical Science and Engineering Division, KAUST Solar Centre (KSC), King Abdullah University of Science and Technology (KAUST), Thuwal 23955-6900, Kingdom of Saudi Arabia. E-mail: frederic.laquai@kaust.edu.sa
bImaging and Characterisation Core Lab, King Abdullah University of Science and Technology (KAUST), Thuwal 23955-6900, Kingdom of Saudi Arabia
cDepartment of Chemistry, Chemistry Research Laboratory, University of Oxford, Oxford, OX1 3TA, UK
dPhysics and Astronomy Department, College of Sciences, King Saud University, Riyadh 12372, Kingdom of Saudi Arabia
First published on 15th April 2024
Abstract
We present an experimental proof-of-concept study of organic semiconductor nanoparticles (NPs) for visible-light-driven carbon dioxide (CO2) conversion. Donor:acceptor NPs, consisting of the conjugated polymer donor PM6 and small molecule electron acceptors, namely, PC71BM or Y6, decorated with silver as cocatalyst, produce CH4 with reaction rates of (3.6 ± 0.8) and (4.2 ± 1.4) μmol g−1 h−1, respectively. NPs consisting of PCE10 as donor polymer and ITIC as small molecule acceptor and decorated with gold as cocatalyst, exhibit CO production rates of (4.7 ± 1.2) μmol g−1 h−1. Importantly, the synergetic effect of efficient cocatalyst deposition and charge carrier generation within the NPs determines their photocatalytic activity.
Photocatalytic water splitting and CO2 reduction1,2 driven by abundant solar energy3 can support the urgently needed transition to a carbon-neutral society, while simultaneously addressing the challenge of intermittency of solar energy by producing energy carriers, namely, solar fuels, compatible with existing energy conversion technologies.4,5
Recently, organic semiconductors such as π-conjugated polymers and small molecules have been designed specifically for photocatalytic CO2 conversion into value-added products, most commonly CO and CH4.6–10 The ability to tune their energy levels to the targeted redox half-reactions, their strong absorption in the visible to near-infrared (NIR) spectral region, and the solution processibility of these materials make them promising candidates for the task.11,12 Yet, short electron–hole pair (exciton) diffusion lengths in organic semiconductors and their intrinsically high exciton binding energies due to the low dielectric constant of organic materials often limit charge separation and therefore, the photocatalytic activity.13,14
The general approach to overcome the exciton binding energy (about 0.3–0.5 eV) in organic semiconductors is to fabricate so-called type-II heterojunctions from blends of electron donor and electron acceptor semiconductors with offset ionization energies and electron affinities.15 In fact, the energy offsets provide the required driving force at the donor:acceptor (D:A) heterojunction to separate bound excitons into spatially separated electron and hole polarons.16 Water-processable organic “bulk heterojunction” nanoparticles (NPs) have already been demonstrated as state-of-the-art particulate visible-light-driven photocatalysts for H2 evolution from water.17–19 However, their potential for photocatalytic CO2 conversion has not yet been explored.
The work presented here highlights an experimental proof-of-concept study using recently reported19 and newly designed water-processable organic semiconductor NPs as visible-light active heterogeneous photocatalysts in visible-light-driven CO2 conversion. We demonstrate that PM6:PC71BM and PM6:Y6 NPs functionalised with Ag as cocatalyst facilitate CO2 conversion to CH4 with generation rates of (3.6 ± 0.8) and (4.2 ± 1.4) μmol g−1 h−1, respectively. Additionally, PCE10:ITIC NPs functionalised with Au as cocatalyst generate CO with a reaction rate of (4.7 ± 1.2) μmol g−1 h−1. Ag and Au are used as cocatalysts since they are known to poorly stabilise H* reaction intermediates and therefore, limit the competing H2 evolution process, while facilitating the CO2 reduction reaction.20,21 Furthermore, we demonstrate that the photocatalytic activity of the NPs and the final reaction products strongly depend on the chosen metal cocatalyst and its particle size and distribution, as well as on the efficiency of free charge carrier generation within the light-harvesting NP.
The NPs were fabricated by a previously reported mini-emulsion process.17,22,23 The donor polymer PBDB-T-2F (PM6) was blended with the fullerene acceptor [6,6]-phenyl C71 butyric acid methyl ester (PC71BM) or with the small molecule non-fullerene acceptor BTP-4F (Y6), while the donor polymer PTB7-Th (PCE10) was combined with the non-fullerene acceptor, ITIC. The D![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :A weight ratio was 1:1 in all NP preparation protocols. Sodium 2-(3-thienyl)ethyloxybutyl-sulfonate (TEBS) was used as a stabilising surfactant.24,25 The average hydrodynamic diameter (Zavg) of all NP batches was 80–100 nm as determined by dynamic light scattering (DLS) experiments (Fig. S1 and Table S1, ESI†).
:A weight ratio was 1:1 in all NP preparation protocols. Sodium 2-(3-thienyl)ethyloxybutyl-sulfonate (TEBS) was used as a stabilising surfactant.24,25 The average hydrodynamic diameter (Zavg) of all NP batches was 80–100 nm as determined by dynamic light scattering (DLS) experiments (Fig. S1 and Table S1, ESI†).
Visible light makes up more than 40% of solar energy,26 but often remains largely unused in photocatalytic CO2 conversion due to the use of wide-bandgap (typically metal oxides or nitrides) absorbers. The key advantage of the chosen organic semiconductors is their strong visible light absorption (Fig. S2, ESI†). Additionally, the D:A pairs were chosen to ensure a sufficiently large energetic offset in the blends (Fig. 1), which drives exciton dissociation at the heterojunction and consequently, facilitates efficient generation of free charge carriers.16 At the same time, the electron affinity of the acceptors compared to the required half-cell electrochemical potentials of CO2 reduction half-reactions is predicted to provide sufficient driving force for the CO2 conversion to CO and CH4.27 Importantly, energy levels of organic semiconductors remain virtually constant regardless of the solution's pH.28,29 However, the redox half-reaction potentials typically exhibit the Nernstian shift. Therefore, the driving force for CO2 reduction increases with the solution's acidity (lower pH); pH = 3.6 ± 0.1 in the suspensions used here.
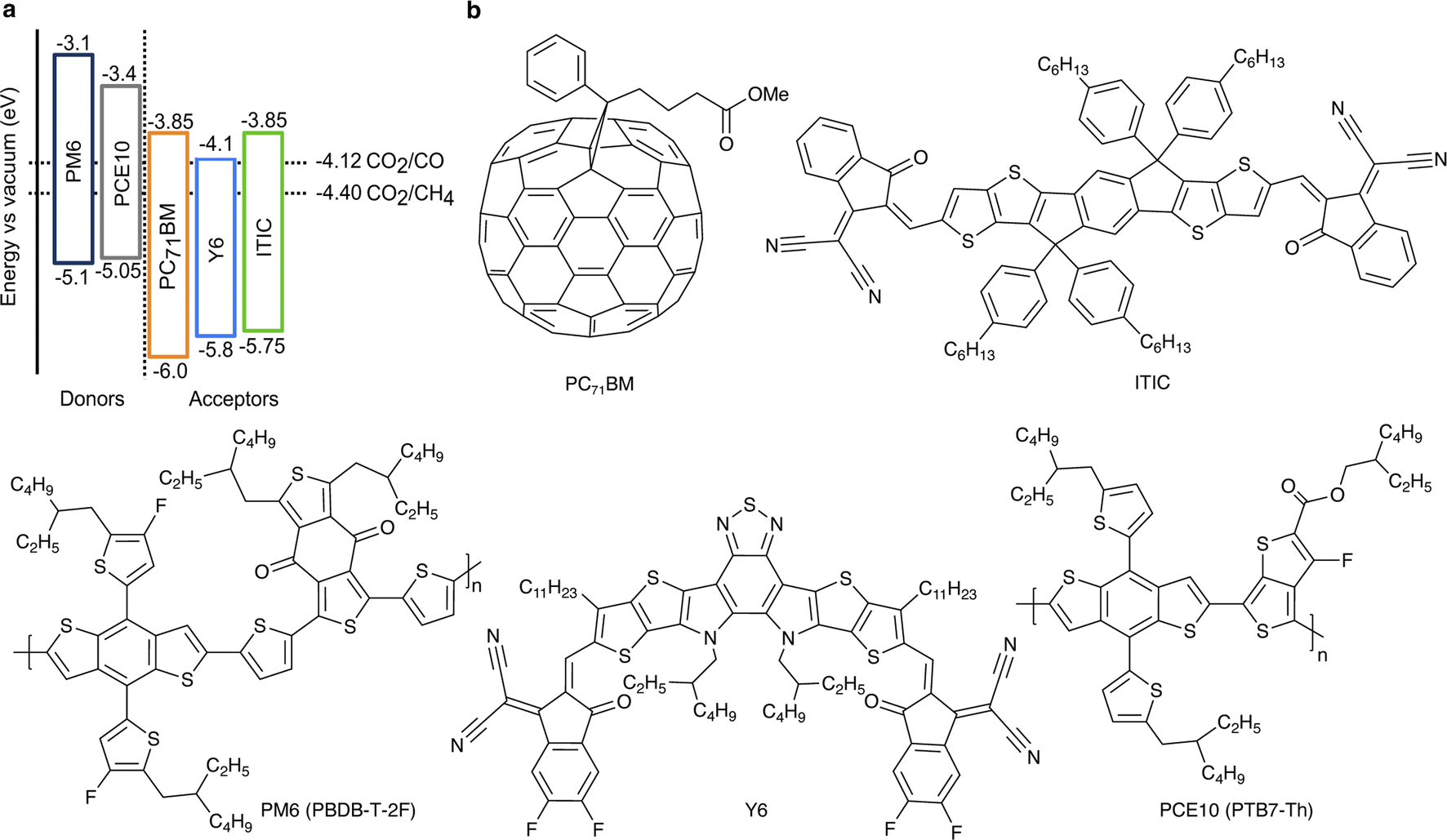 | ||
| Fig. 1 (a) The organic semiconductors' energy levels compared to the reduction half-reaction potentials of CO2 conversion into CO and CH4 at pH = 3.6.27 Ionization energies were determined by ultraviolet photoelectron spectroscopy (UPS) and electron affinities by inverse photoemission spectroscopy (IPES).16 All energy levels and electrochemical potentials are expressed on the absolute electrochemical scale (0 V vs. SHE = −4.44 V vs. vacuum). (b) Chemical structures of the selected organic semiconductors. | ||
Ag or Au was photodeposited (targeted fraction: 10 wt%) as a cocatalyst onto the surface of the organic NPs. Ascorbic acid (AA, 0.1 M aqueous solution) was used as a sacrificial electron donor to facilitate regeneration of the oxidized donor and to inhibit self-oxidation.30
PM6:PC71BM/Ag and PM6:Y6/Ag NPs demonstrated CH4 production rates of (3.6 ± 0.8) and (4.2 ± 1.4) μmol g−1 h−1, respectively (Fig. 2a and Table S2, ESI†). Both photocatalysts also generated traces of carbon monoxide (CO). PCE10:ITIC/Au produced primarily CO with a rate of (4.7 ± 1.2) μmol g−1 h−1 and in addition traces of CH4 (Fig. 2b and Table S2, ESI†). Importantly, the reaction rates are comparable and even exceed the rates of prototypical photocatalysts such as titanium dioxide (TiO2 (P25)) and graphitic carbon nitride (g-C3N4) functionalised with Ag and Au cocatalysts, which were measured under identical conditions in our home-built CO2 reduction setup (Fig. 2 and S3, ESI†).
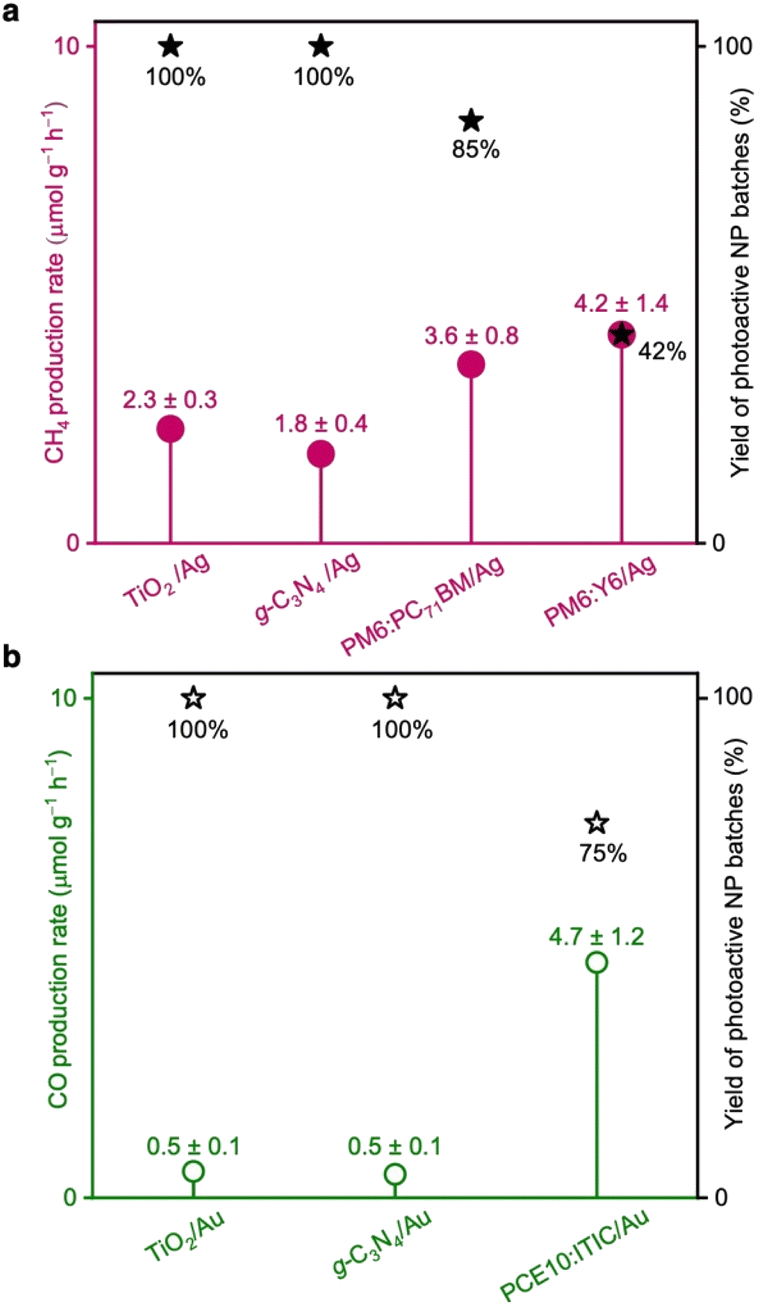 | ||
| Fig. 2 (a) CH4 production rates in CO2 reduction reactions with PM6:PC71BM/Ag and PM6:Y6/Ag NPs. (b) CO production rate in CO2 reduction reactions with PCE10:ITIC/Au NPs. The results are compared to the rates obtained under identical conditions with the benchmark photocatalysts TiO2 (P25) and g-C3N4, functionalised with Ag or Au cocatalysts. The yield of photoactive NP batches indicates the reproducibility of the production rates across 8 or 10 reactions. | ||
No reproducible photocatalytic activity was observed for PCE10:ITIC/Ag, PM6:PC71BM/Au, and PM6:Y6/Au photocatalysts. We attribute this to inefficient cocatalyst photodeposition, as discussed further below. Furthermore, no CO2 reduction products were detected in the reactions where NPs without cocatalysts were used (Fig. S4, ESI†). We attribute this to inefficient charge extraction from organic NPs and consequently, insufficient charge accumulation for these multi-electron reactions, as well as to a weak adsorption of CO2 molecules at the organic NP's surface, and the absence of suitable reaction centres that could facilitate the multi-electron CO2 reduction reaction.
Occasionally, we observed that functional NP batches exhibited poor photocatalytic activity towards CO2 conversion. Therefore, besides the reaction rates, we also determined the yield of fully-functional NP batches. In other words, the yield of photoactive NPs corresponds to the fraction of separately prepared NP batches that showed photocatalytic activity above a certain threshold. All NP batches that exhibited CO2 conversion with generation rates less than half of the maximum rates determined for each photocatalyst were considered non-functional. The NP batches showing reactions with rates above the threshold were all considered photoactive and functional. The yield of active PM6:PC71BM/Ag and PM6:Y6/Ag NP batches that generated CH4 was determined to 85% and 42%, respectively, while the yield of active PCE10:ITIC/Au NP batches that generated CO was 75% (Fig. 2).
Additionally, the NPs' re-usability was tested in separate 12 hour-long reaction cycles. PM6:Y6/Ag could be reused two times before the material aggregated, yet a decrease in the CH4 production rate was observed between cycles (Fig. S5a, ESI†). PM6:PC71BM/Ag could only be reused once, while PCE10:ITIC/Au aggregated during washing already after the first batch reaction (Fig. S5b, ESI†).
There is a need to improve the yield (reproducibility) of photoactive NP batches and their re-usability, possibly by further optimization of the NP preparation conditions and of the cocatalyst deposition, both part of ongoing works.
Importantly, a series of control experiments were performed to verify the photocatalytic activity of the organic NPs towards CO2 reduction and to rule out that the obtained products originated from NP degradation (Fig. S4, ESI†). Firstly, no carbon-based products were observed when the reactions were carried out in the dark. Secondly, no carbon-based products were observed in reactions with NP/cocatalyst in an inert (N2) atmosphere, indicating that no degradation of organic semiconductors due to self-oxidation/reduction under illumination occurred. Thirdly, the competing process of H2 evolution was strongly suppressed (Fig. S6, ESI†), demonstrating cocatalyst selectivity towards carbon-based products.
Lastly, 13C-isotope labelling was employed to determine the origin of the observed CO and CH4 products. The products of 13CO2 conversion were analysed by gas chromatography coupled with mass spectrometry (GC/MS). In the photoreactions with PM6:PC71BM/Ag and PM6:Y6/Ag as photocatalysts GC/MS spectra revealed the presence of ions with m/z = 17 at the retention time (RT = 1.8 ± 0.3 min) corresponding to 13CH4 (Fig. S7a and b, ESI†). In the case of 13CO2 conversion with PCE10:ITIC/Au as photocatalyst, ions with m/z = 29 (RT = 7.5 ± 0.3 min) were detected, corresponding to 13CO (Fig. S7c, ESI†). Thus, the 13C-isotope labelling experiments together with the control reactions confirmed that the products, here, CO and CH4, were generated by photocatalytic conversion of CO2 with the aid of organic semiconductor NPs functionalised with either Au or Ag cocatalysts and did not originate from degradation of the organic materials themselves.
To further investigate the observed photocatalysts' activity in relation to their morphology and particle dimensions, the NPs were imaged by cryogenic transmission electron microscopy (cryo-TEM). As reported earlier, PM6:PC71BM NPs with a D:A ratio of 1:1 exhibited a core/shell-type structure (Fig. 3a).19,31 Here, PC71BM formed the amorphous core and PM6 the semi-uniform shell, as determined from the 2.2 nm lattice spacing observed in the shell, which corresponds to the typical PM6 lamellar stacking distance (Fig. S8, ESI†).32 In accordance with inductively coupled plasma optical emission spectroscopy (ICP-OES) results (Table S3, ESI†), a higher ratio of Ag particles per organic NP (9.3 ± 0.3 wt%) was observed in cryo-TEM images when compared to Au particles (8.5 ± 0.3 wt%) (Fig. 3b and c). Moreover, Au particles were mostly deposited as large clusters, 15–20 nm in size, whereas Ag particle sizes were below 10 nm and the particles were more evenly distributed across the NP surface (Fig. S9, ESI†).
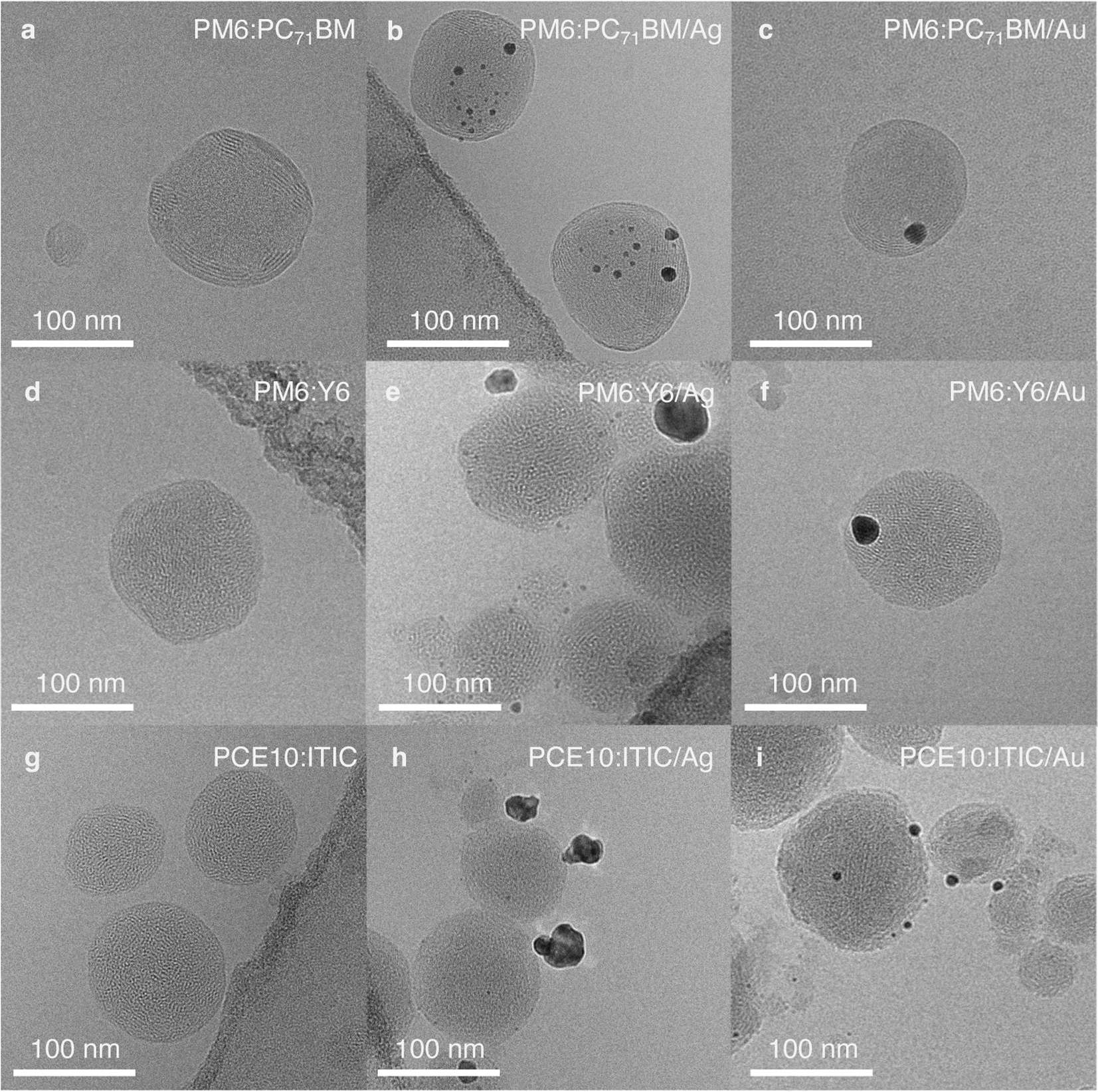 | ||
| Fig. 3 Bright-field cryo-TEM images of NPs showing their structures and the differences in cocatalyst deposition: (a), (d) and (g), pristine organic NPs of different composition with D:A weight ratio 1:1; (b), (e) and (h), corresponding NPs with Ag cocatalyst; (c), (f) and (i), corresponding NPs with Au cocatalyst. Note: the dark contrast in the corners of the images (b), (d), (e), and (g) arises from the carbon TEM grid. Additional images and their analysis are shown in Fig. S8–S16.† | ||
PM6:Y6 NPs exhibit an intermixed structure with randomly distributed semi-crystalline domains (Fig. 3d, S10a and b, ESI†).19 The areas of enhanced crystallinity with the lattice spacing of ∼2 nm correspond to Y6 diffraction peaks observed in thin films (Fig. S11, ESI†).32 Ag cocatalyst photodeposition on the surface of PM6:Y6 NPs resulted in many very small, 1–3 nm, particles (9.9 ± 0.4 wt%). However, large clusters, up to 30 nm, were also observed occasionally (Fig. 3e, S10c and d, ESI†). In the case of Au deposition, predominantly single large particles, 15–20 nm in size, were obtained on the organic NPs surface (Fig. 3f, S10e and f, ESI†). The total amount of the deposited Au cocatalyst measured by ICP-OES was 8.5 ± 0.3 wt%, which is slightly less than the intended 10 wt%.
PCE10 and ITIC are highly miscible and do not show pronounced phase segregation.33 Hence, PCE10:ITIC NPs exhibited an intimately mixed structure (Fig. 3g). In some NPs, areas of enhanced crystallinity with 1.9 nm lattice spacing could be observed, which corresponds to the ITIC lamellar stacking distance (Fig. S12, ESI†),34,35 while in other NPs, PCE10 lamellar stacking with a lattice spacing of 2.2 nm was found (Fig. S13–S15, ESI†).34,36 During photodeposition, Ag formed mostly large clusters (up to 25 nm, 10.0 ± 3.3 wt%) on the PCE10:ITIC surface (Fig. 3h, S14, S16a and b, ESI†). On the other hand, deposition of Au resulted in small particles, typically below 10 nm in size, distributed across the surface of the organic NPs, resulting in a larger cocatalyst surface area available to facilitate CO2 reduction than in the case of Ag cocatalyst (Fig. 3i, S15, S16c and d, ESI†). Here, the actual Au cocatalyst concentration as determined by ICP-OES was 8.1 ± 0.2 wt%.
Importantly, the concentration, size, and shape of the cocatalyst affect the reactivity of its active sites and can even alter selectivity towards particular reaction products.37,38 The surface energy of Au is larger than that of Ag, which makes Au nanoparticles more prone to sintering induced by irradiation with light.39,40 Moreover, it has been demonstrated for electrocatalysts that optimal binding and stabilisation of reaction intermediates is possible on the surface of metal nanoparticles less than 10 nm in size.41,42 These findings should also apply to cocatalysts in photoreactions. Thus, the negligible photocatalytic activity of PM6:PC71BM/Au, PM6:Y6/Au and PCE10:ITIC/Ag could be attributed to unfavourable cocatalyst deposition, such as single large metal clusters (above 10 nm in size) on the organic NPs' surface. In fact, PM6:PC71BM/Ag, PM6:Y6/Ag and PCE10:ITIC/Au, which exhibit smaller and more evenly distributed cocatalyst particles across the surface of the organic NPs, successfully perform CO2 conversion to CH4 and CO, respectively. Nevertheless, the relatively large variance in the generation rates (as indicated by the errors of the rates shown in Fig. 2) appears to originate from the differences in cocatalyst loading, distribution, and particle size.
To confirm the type-II heterojunction formation in the D:A NPs and to monitor the exciton quenching following photoexcitation, time-resolved photoluminescence (TRPL) spectroscopy was used. Upon photoexcitation of PM6:PC71BM NPs at 450 nm, both the donor and the acceptor, are simultaneously excited (Fig. S17, ESI†). The observed PL quenching efficiency (PLQE) was modest for the donor (44.7 ± 1.8)% when measured relative to neat PM6 NPs, and larger for the acceptor (75.3 ± 7.9)% when measured relative to neat PC71BM NPs (Fig. S18 and Table S4, ESI†). The PM6:PC71BM NPs functionalisation with Ag as cocatalyst along with the addition of the sacrificial electron donor AA enhanced the PM6 PLQE to (72.4 ± 2.6)%, implying a more efficient PM6 regeneration or reductive quenching of the exciton via electron transfer from the AA. However, it did not significantly change the observed acceptor's PL lifetime (PLQEPCBM = 86.2 ± 9.0%), possibly because exciton quenching in D:A blends occurs faster (ps timescale) and more efficiently than electron transfer to the metal and subsequently to the reactants. Moreover, the absence of electron-accepting reactants in the dispersion (in N2 atm) and the generally low efficiency of photoreduction half-reactions did not have a noticeable impact on the acceptor's exciton lifetimes determined by TRPL measurements.
Since charge carrier dynamics in organic semiconductors are typically elusive in TRPL experiments and are observed on longer timescales, transient absorption spectroscopy (TAS) on the nanosecond timescale was employed to investigate the charge carrier dynamics in the photocatalytic NPs. Fig. 4a shows the TAS spectra of PM6:PC71BM/Ag after pulsed (sub-ns) laser excitation at 532 nm. The TAS spectra exhibit PM6 ground-state bleaching (GSB) between 550–650 nm and a broad PM6 hole polaron-induced (PM6+) absorption at 650–1000 nm. The PC71BM spectral signatures could not be seen in the TAS measurements as they are typically present at higher probe photon energies and generally have significantly lower excited state absorption cross-sections. The corresponding PM6+ (cation) kinetics revealed the presence of long-lived free charge carriers with an average lifetime of τavg = (54.9 ± 5.3) ns. The presence of a metal cocatalyst did not alter the observed charge carrier dynamics on the nanosecond timescale (Fig. S19, ESI†).
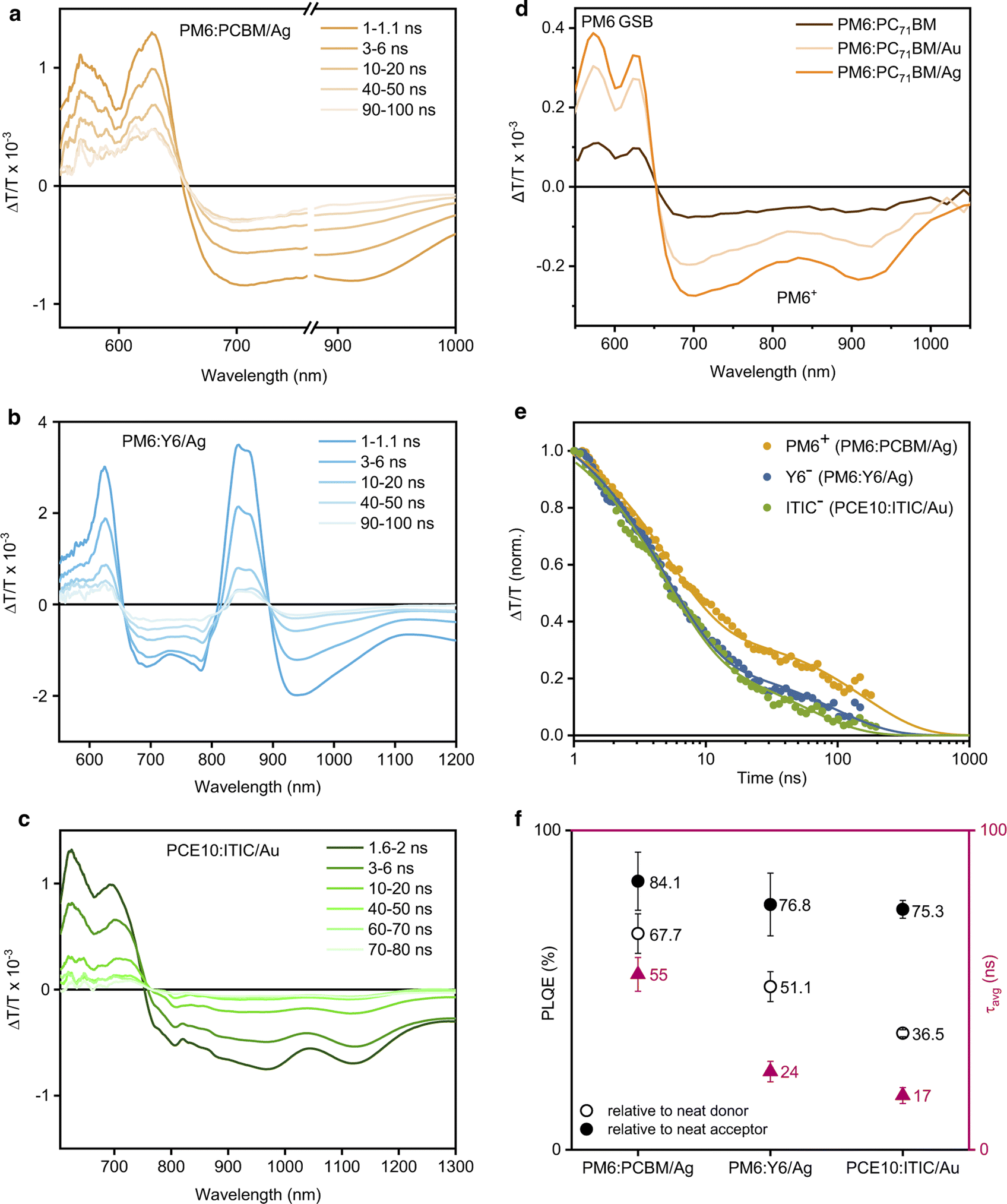 | ||
| Fig. 4 Photophysical characterisation of D:A/cocatalyst NP dispersions. (a)–(c) Transient absorption spectra at different time delays following 532 nm photoexcitation of PM6:PC71BM/Ag (fluence 11.7 μJ cm−2), PM6:Y6/Ag (fluence 31 μJ cm−2), and PCE10:ITIC/Au (fluence 11.7 μJ cm−2) respectively. (d) Comparison of the PIA spectra of PM6:PC71BM NPs suspensions with and without metal cocatalysts (absorbance 0.28, 1 mm pathlength) following 515 nm photoexcitation (1.93 W cm−2). (e) Comparison of the transient absorption decay dynamics of the corresponding photocatalysts, probed at 757–758 nm for PM6+ in PM6:PC71BM/Ag, at 920–950 nm for Y6− in PM6:Y6/Ag, and at 770–890 nm for ITIC− in PCE10:ITIC/Au.46–48 (f) Comparison of the charge carrier lifetimes (τavg) and PL quenching efficiencies (PLQE) of the corresponding photocatalysts. | ||
Subsequently, the presence of longer-lived excited states with lifetimes of tens to hundreds of microseconds was investigated by quasi steady-state photoinduced absorption (PIA) spectroscopy. NP dispersions were photoexcited with an electronically modulated laser beam and a white light probe source (100 W) was used to measure the changes in transmission induced by the presence of photoexcited states.43–45 Following 515 nm photoexcitation, an increased PIA signal amplitude was observed for PM6+ in PM6:PC71BM/Ag when compared to the PM6+ signal in PM6:PC71BM NPs and in PM6:PC71BM/Au (Fig. 4d). We note that the PIA signal amplitude is determined by the concentration of free charge carriers and their respective lifetimes. Hence, the increased signal amplitude indicates that in the case of a uniformly deposited Ag cocatalyst, charge generation in PM6:PC71BM NPs is facilitated and/or charge carrier lifetime is prolonged compared to the bulky non-uniformly deposited Au cocatalyst. That, along with a larger cocatalyst surface area available for CO2 adsorption and reduction appears to increase the photocatalytic activity of PM6:PC71BM/Ag when compared to PM6:PC71BM/Au.
Next, we turn to the photophysical properties of the PM6:Y6 NP photocatalyst. Upon selective photoexcitation of Y6 at 780 nm (Fig. S17, ESI†), the Y6 emission was observed to be quenched significantly (77.6 ± 10.1)% in PM6:Y6 NPs, indicating efficient hole transfer from Y6 to the PM6 donor polymer (Fig. S20 and Table S5, ESI†). Photoexcitation at 532 nm predominantly excited PM6 rather than the acceptor Y6 (Fig. S17, ESI†). For PM6:Y6 NPs, excitation at 532 nm yielded a PLQE of (44.9 ± 4.2)% relative to the neat PM6, indicating moderate exciton quenching efficiency by energy/electron transfer from PM6 to the Y6 acceptor. The addition of Ag cocatalyst and AA did not alter the observed exciton dynamics significantly. This is likely a consequence of the rapid charge/energy transfer between the donor and the acceptor, and hence fast exciton quenching, whereas electron transfer to Ag and regeneration of the ground state by AA occur on a much longer timescale and possibly with lower reaction yield.
Thus, we used ns-TAS to investigate the charge carrier dynamics in PM6:Y6 NPs. The TAS spectra of PM6:Y6/Ag exhibit the GSB of the donor polymer PM6 at 550–650 nm and the acceptor Y6 at 850 nm (Fig. 4b). The broad signal at 670–790 nm was assigned to photoinduced absorption from PM6+ hole and Y6− electron polarons, along with the overlapping photoinduced absorption of PM6+ and Y6− polarons at 920 nm.46 The corresponding Y6− dynamics in PM6:Y6/Ag were shorter-lived (τavg = 24.5 ± 3.2 ns) than the dynamics of Y6− in PM6:Y6 and PM6:Y6/Au (Fig. S21, ESI†). We assign this to the additional electron transfer from the organic NPs to the Ag cocatalyst, which competes with carrier recombination in PM6:Y6.
Additionally, PIA spectra indicate that in the presence of a metal cocatalyst, photogenerated electrons are transferred from the acceptor to the metal cocatalyst, since a decrease in the GSB signal amplitude of the Y6 acceptor is observed (Fig. S22, ESI†).47 It appears that the generation of long-lived free charge carriers and their accumulation in the Ag cocatalyst, along with the efficient Ag photodeposition onto the PM6:Y6 NP surface determines the photocatalytic activity of the PM6:Y6/Ag system.
Finally, we performed a spectroscopic characterisation of the PCE10:ITIC photocatalyst. Since both, PCE10 and ITIC, strongly absorb in the 500–800 nm region, selective photoexcitation of either the donor or the acceptor is impossible (Fig. S2, ESI†). Following PCE10:ITIC NP photoexcitation at 650 nm, a low PLQE of (12.3 ± 0.3)% and a moderate PLQE of (65.9 ± 2.3)% were observed relative to the neat PCE10 and neat ITIC NPs, respectively (Fig. S23 and Table S6, ESI†). The addition of the Au cocatalyst and AA facilitated exciton dissociation possibly due to electron transfer to Au and hole extraction by AA: consequently, the PLQE reached (49.7 ± 1.5)% and (80.4 ± 3.0)% relative to the neat PCE10 and ITIC, respectively.
The TAS spectra of PCE10:ITIC/Au NPs exhibit a broad PCE10 and ITIC GSB at 600–750 nm, together with photoinduced absorption of ITIC anions (ITIC−) at 810 nm and PCE10 cations (PCE10+) at 1120 nm, as well as the overlapping photoinduced absorption from ITIC− electron and PCE10+ hole polarons at 870–1030 nm (Fig. 4c).48 The corresponding ITIC− (anion) kinetics revealed the presence of free charge carriers with an average lifetime of (17.0 ± 2.5) ns (Fig. S24, ESI†). In this case, the presence of a metal cocatalyst did not have any effect on the charge carrier kinetics observed in TAS.
However, the PCE10+ (cation) PIA signal amplitude of PCE10:ITIC/Au was larger than that of PCE10:ITIC and PCE10:ITIC/Ag, indicative of enhanced hole transfer from the ITIC acceptor to the PCE10 donor in the presence of Au (Fig. S25, ESI†). Thus, it appears that the favourable Au deposition onto the PCE10:ITIC NPs' surface enables efficient CO2 adsorption, facilitates charge transfer, and hence, determines the photocatalytic activity of PCE10:ITIC/Au.
It has previously been shown that the photocatalytic activity of the NPs is determined by the presence of free charge carriers in the NP bulk rather than by the density of catalytically active sites.49 Importantly, Ag and Au cocatalysts have demonstrated the ability to capture and store electrons, which causes the Fermi level to shift.50 The smaller the metal particle size, the more the Fermi level shifts toward more negative potentials (vs. NHE) and the higher the semiconductor's photocatalytic activity becomes.51 CO2 reduction to CH4 requires 8 electrons and 8 protons, while CO2 reduction to CO is a less demanding process with only 2 electrons and 2 protons.27 Therefore, in the photocatalytic reactions with PM6:PC71BM/Ag and PM6:Y6/Ag NPs, we attribute the CH4 production to the more efficient generation of long-lived free charge carriers along with efficient electron transfer and charge accumulation at the surface reaction sites by the Ag cocatalyst. However, even though Au has been shown to release accumulated electrons faster than Ag,52 the modest exciton quenching efficiency in PCE10:ITIC NPs leads to moderate charge carrier generation and thus, limits the photocatalytic activity of this system (Fig. 4d and e). However, it appears that the amount of electrons accumulated in the cocatalyst of PCE10:ITIC/Au NPs is sufficient to drive the 2-electron CO2 reduction reaction to CO, consequently, resulting in CO as a major product.
Conclusion
We have successfully demonstrated that organic semiconductor heterojunction NPs photocatalytically reduce CO2 into CO and CH4 in suspension under visible light irradiation. PM6:PC71BM/Ag and PM6:Y6/Ag NPs achieved CH4 generation rates of (3.6 ± 0.8) and (4.2 ± 1.4) μmol g−1 h−1, respectively. The PCE10:ITIC/Au photocatalyst exhibited a CO generation rate of (4.7 ± 1.2) μmol g−1 h−1. Efficient cocatalyst deposition is critical to obtain high photocatalytic activity. Smaller, uniformly distributed cocatalyst nanoparticles provided a larger surface area for CO2 adsorption and possibly stabilised the reaction intermediates. Photophysical characterisation using TRPL, TA, and PIA spectroscopies revealed that the photogenerated excitons are quenched efficiently due to the D:A heterojunction in these NPs, generating charge carriers with lifetimes on the microsecond timescale, which is sufficiently long to drive photoredox reactions. The charges, in turn, are transferred to the metal cocatalysts and to the surface reaction sites where they drive CO2 reduction reactions. The yield of products obtained in CO2 conversion reactions seems to depend on the number of free charge carriers photogenerated in the organic NPs and accumulated at the surface reaction sites of the metal cocatalysts. Thus, further enhancing the efficiency with which charges are transferred to the metal cocatalysts seems crucial for the advancement of photocatalytic organic D:A NPs for CO2 conversion.Author contributions
M. F., J. K. conceived the idea. M. F., N. A., L. Z., C. S. P. C. performed the experiments. C. E. P. contributed to data analysis. I. M., M. H., F. L. supervised the project. M. F. wrote the first draft of the paper and all authors contributed to the final version.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This publication is based upon work supported by King Abdullah University of Science and Technology (KAUST) under Award No. ORA-CRG9-2020-4384. C. E. P. acknowledges support from King Abdullah University of Science and Technology (KAUST) Global Fellowship Program under Award No. ORA-2022-5002. C. S. P. C. acknowledges support from King Abdullah University of Science and Technology (KAUST) under Award No. OSR-CRG8-2019-4025. The authors are sincerely grateful to Dr C. Combe for his assistance in the initial development of the experimental setup. The authors also thank Dr C. Rossi De Oliveira for providing the necessary equipment for GC/MS analysis of 13C-isotope labelled products.References
- K. Stolarczyk, S. Bhattacharyya, L. Polavarapu and J. Feldmann, ACS Catal., 2018, 8, 3602–3635 CrossRef.
- X. G. Yang and D. W. Wang, ACS Appl. Energy Mater., 2018, 1, 6657–6693 CrossRef CAS.
- G. W. Crabtree and N. S. Lewis, Phys. Today, 2007, 60, 37–42 CrossRef CAS.
- O. S. Bushuyev, P. De Luna, C. T. Dinh, L. Tao, G. Saur, J. van de Lagemaat, S. O. Kelley and E. H. Sargent, Joule, 2018, 2, 825–832 CrossRef CAS.
- J. Mertens, C. Breyer, K. Arning, A. Bardow, R. Belmans, A. Dibenedetto, S. Erkman, J. Gripekoven, G. Léonard, S. Nizou, D. Pant, A. S. Reis-Machado, P. Styring, J. Vente, M. Webber and C. J. Sapart, Joule, 2023, 7, 442–449 CrossRef.
- X. X. Yu, S. Y. Tian, F. T. Zhang, G. P. Gao, C. Zhang, Y. Han, S. Y. Ji, H. Guo and X. H. Jin, ACS Sustainable Chem. Eng., 2022, 10, 16182–16188 CrossRef.
- Z. W. Fu, A. Vogel, M. A. Zwijnenburg, A. I. Cooper and R. S. Sprick, J. Mater. Chem. A, 2021, 9, 4291–4296 RSC.
- S. H. Wang, P. Z. Chen, Y. Y. Chen, F. Khurshid, C. W. Cai, Y. Y. Lai, P. W. Chung, R. J. Jeng, S. P. Rwei and L. Y. Wang, ACS Appl. Mater. Interfaces, 2022, 14(38), 43109–43115 CrossRef CAS PubMed.
- Y. F. Diao, S. Jung, M. Kouhnavard, R. Woon, H. R. Yang, P. Biswas and J. M. D'arcy, ACS Cent. Sci., 2021, 7, 1668–1675 CrossRef CAS PubMed.
- F. Moruzzi, W. M. Zhang, B. Purushothaman, S. Gonzalez-Carrero, C. M. Aitchison, B. Willner, F. Ceugniet, Y. B. Lin, J. Kosco, H. Chen, J. F. Tian, M. Alsufyani, J. S. Gibson, E. Rattner, Y. Baghdadi, S. Eslava, M. Neophytou, J. R. Durrant, L. Steier and I. McCulloch, Nat. Commun., 2023, 14, 3443 CrossRef CAS PubMed.
- X. Chi, Z. A. Lan, Q. Chen, X. R. Zhang, X. Chen, G. G. Zhang and X. C. Wang, Angew. Chem., Int. Ed., 2023, 62, e202303785 CrossRef CAS PubMed.
- Y. P. Wang, G. P. Ji, W. Q. Ye, F. T. Zhang, Y. D. Wang, Y. F. Zhao and Z. M. Liu, ACS Sustainable Chem. Eng., 2022, 10, 9460–9468 CrossRef CAS.
- Y. Firdaus, V. M. Le Corre, S. Karuthedath, W. L. Liu, A. Markina, W. T. Huang, S. Chattopadhyay, M. M. Nahid, M. I. Nugraha, Y. B. Lin, A. Seitkhan, A. Basu, W. M. Zhang, I. McCulloch, H. Ade, J. Labram, F. Laquai, D. Andrienko, L. J. A. Koster and T. D. Anthopoulos, Nat. Commun., 2020, 11, 5220 CrossRef CAS PubMed.
- Y. Tamai, H. Ohkita, H. Benten and S. Ito, J. Phys. Chem. Lett., 2015, 6, 3417–3428 CrossRef CAS PubMed.
- A. Wadsworth, Z. Hamid, J. Kosco, N. Gasparini and I. McCulloch, Adv. Mater., 2020, 32, 2001763 CrossRef CAS PubMed.
- S. Karuthedath, J. Gorenflot, Y. Firdaus, N. Chaturvedi, C. S. P. De Castro, G. T. Harrison, J. I. Khan, A. Markina, A. H. Balawi, T. A. D. Pena, W. N. Liu, R. Z. Liang, A. Sharma, S. H. K. Paleti, W. M. Zhang, Y. B. Lin, E. Alarousu, D. H. Anjum, P. M. Beaujuge, S. De Wolf, I. McCulloch, T. D. Anthopoulos, D. Baran, D. Andrienko and F. Laquai, Nat. Mater., 2021, 20, 378–384 CrossRef CAS PubMed.
- J. Kosco, M. Bidwell, H. Cha, T. Martin, C. T. Howells, M. Sachs, D. H. Anjum, S. Gonzalez Lopez, L. Y. Zou, A. Wadsworth, W. M. Zhang, L. S. Zhang, J. Tellam, R. Sougrat, F. Laquai, D. M. DeLongchamp, J. R. Durrant and I. McCulloch, Nat. Mater., 2020, 19, 559–565 CrossRef CAS PubMed.
- Y. J. Yang, D. H. Li, J. L. Cai, H. Wang, C. H. Guo, S. Wen, W. Li, T. Wang and D. Liu, Adv. Funct. Mater., 2023, 33, 2209643 CrossRef CAS.
- J. Kosco, S. Gonzalez-Carrero, C. T. Howells, T. Fei, Y. F. Dong, R. Sougrat, G. T. Harrison, Y. Firdaus, R. Sheelamanthula, B. Purushothaman, F. Moruzzi, W. D. Xu, L. Y. Zhao, A. Basu, S. De Wolf, T. D. Anthopoulos, J. R. Durrant and I. McCulloch, Nat. Energy, 2022, 7, 340–351 CrossRef CAS.
- D. W. S. Kref, H. Junge and M. Beller, EnergyChem, 2020, 2, 100044 CrossRef.
- D. Voiry, H. S. Shin, K. P. Loh and M. Chhowalla, Nat. Rev. Chem, 2018, 2, 0105 CrossRef CAS.
- A. Stapleton, B. Vaughan, B. F. Xue, E. Sesa, K. Burke, X. J. Zhou, G. Bryant, O. Werzer, A. Nelson, A. L. D. Kilcoyne, L. Thomsen, E. Wanless, W. Belcher and P. Dastoor, Sol. Energy Mater. Sol. Cells, 2012, 102, 114–124 CrossRef CAS.
- N. P. Holmes, S. Chambon, A. Holmes, X. Xu, K. Hirakawa, E. Deniau, C. Lartigau-Dagron and A. Bousquet, Curr. Opin. Colloid Interface Sci., 2021, 56, 101511 CrossRef CAS.
- S. Subianto, R. Balu, L. de Campo, A. Sokolova, N. K. Dutta and N. R. Choudhury, ACS Appl. Mater. Interfaces, 2018, 10, 44116–44125 CrossRef CAS PubMed.
- A. Dolan, J. M. de la Perrelle, T. D. Small, E. R. Milsom, G. F. Metha, X. Pan, M. R. Andersson, D. M. Huang and T. W. Kee, ACS Appl. Nano Mater., 2022, 5, 12154–12164 CrossRef CAS.
- Q. Fu, Encyclopedia of Atmospheric Sciences, 2003, pp. 1859–1863 Search PubMed.
- A. J. Bard, R. Parsons and J. Jordan, Standard Potentials in Aqueous Solutions, CRC Press, New York, 1985 Search PubMed.
- P. Bornoz, M. S. Prévot, X. Y. Yu, N. Guijarro and K. Sivula, J. Am. Chem. Soc., 2015, 137, 15338–15341 CrossRef CAS PubMed.
- L. Yao, Y. P. Liu, H. H. Cho, M. Xia, A. Sekar, B. P. Darwich, R. A. Wells, J. H. Yum, D. Ren, M. Grätzel, N. Guijarro and K. Sivula, Energy Environ. Sci., 2021, 14, 3141–3151 RSC.
- X. Y. Wang, L. J. Chen, S. Y. Chong, M. A. Little, Y. Z. Wu, W. H. Zhu, R. Clowes, Y. Yan, M. A. Zwijnenburg, R. S. Sprick and A. I. Cooper, Nat. Chem., 2018, 10, 1180–1189 CrossRef CAS PubMed.
- I. Persson, H. Laval, S. Chambon, G. Bonfante, K. Hirakawa, G. Wantz, B. Watts, M. A. Marcus, X. Xu, L. Ying, G. Lakhwani, M. R. Andersson, J. M. Cairney and N. P. Holmes, Nanoscale, 2023, 15, 6126–6142 RSC.
- J. Yuan, Y. Zhang, L. Zhou, G. Zhang, H.-L. Yip, T.-K. Lau, X. Lu, C. Zhu, H. Peng, P. A. Johnson, M. Leclerc, Y. Cao, J. Ulanski, Y. Li and Y. Zou, Joule, 2019, 3, 1140–1151 CrossRef CAS.
- L. Zhong, L. Gao, H. Bin, Q. Hu, Z.-G. Zhang, F. Liu, T. P. Russell, Z. Zhang and Y. Li, Adv. Energy Mater., 2017, 7, 1602215 CrossRef.
- Y. Wu, Q. Hu, P. Fan, Y. F. Jiang, W. K. Zhong, W. G. Hu and T. P. Russell, J. Phys. Chem. C, 2021, 125, 15863–15871 CrossRef CAS.
- J. L. Wang, J. J. Peng, X. Y. Liu and Z. Q. Liang, ACS Appl. Mater. Interfaces, 2017, 9, 20704–20710 CrossRef CAS PubMed.
- L. Q. Huang, G. Wang, W. H. Zhou, B. Y. Fu, X. F. Cheng, L. F. Zhang, Z. B. Yuan, S. X. Xiong, L. Zhang, Y. P. Xie, A. D. Zhang, Y. D. Zhang, W. Ma, W. W. Li, Y. H. Zhou, E. Reichmanis and Y. W. Chen, ACS Nano, 2018, 12, 4440–4452 CrossRef CAS PubMed.
- H. Mistry, R. Reske, Z. H. Zeng, Z. J. Zhao, J. Greeley, P. Strasser and B. Roldan Cuenya, J. Am. Chem. Soc., 2014, 136, 16473–16476 CrossRef CAS PubMed.
- K. Li, T. Y. Peng, Z. H. Ying, S. S. Song and J. Zhang, Appl. Catal., B, 2016, 180, 130–138 CrossRef CAS.
- Q. Fu and T. Wagner, Surf. Sci. Rep., 2007, 62, 431–498 CrossRef CAS.
- M. Dilla, A. Pougin and J. Strunk, J. Energy Chem., 2017, 26, 277–283 CrossRef.
- W. L. Zhu, R. Michalsky, Ö. Metin, H. F. Lv, S. J. Guo, C. J. Wright, X. L. Sun, A. A. Peterson and S. H. Sun, J. Am. Chem. Soc., 2013, 135, 16833–16836 CrossRef CAS PubMed.
- A. Salehi-Khojin, H. R. M. Jhong, B. A. Rosen, W. Zhu, S. C. Ma, P. J. A. Kenis and R. I. Masel, J. Phys. Chem. C, 2013, 117, 1627–1632 CrossRef CAS.
- F. Le Formal, E. Pastor, S. D. Tilley, C. A. Mesa, S. R. Pendlebury, M. Grätzel and J. R. Durrant, J. Am. Chem. Soc., 2015, 137(20), 6629–6637 CrossRef CAS PubMed.
- G. Boschloo and A. Hagfeldt, Inorg. Chim. Acta, 2008, 361, 729–734 CrossRef CAS.
- H. Kallel, G. Latini, F. Paquin, R. Rinfret, N. Stingelin and C. Silva, arXiv, 2010, preprint, arXiv:1007.3035, DOI:10.48550/arXiv.1007.3035.
- J. Grüne, G. Londi, A. J. Gillett, B. Stähly, S. Lulei, M. Kotova, Y. Olivier, V. Dyakonov and A. Sperlich, Adv. Funct. Mater., 2023, 33, 2212640 CrossRef.
- S. Shoaee, H. M. Luong, J. Song, Y. Zou, T.-Q. Nguyen and D. Neher, Adv. Mater., 2023, 2302005 CrossRef PubMed.
- M. A. Alamoudi, J. I. Khan, Y. Firdaus, K. Wang, D. Andrienko, P. M. Beaujuge and F. Laquai, ACS Energy Lett., 2018, 3, 802–811 CrossRef CAS.
- M. O'Connor, T. J. Aubry, O. G. Reid and G. Rumbles, Adv. Mater., 2023, 2210481 CrossRef PubMed.
- P. V. Kamat, J. Phys. Chem. Lett., 2012, 3, 663–672 CrossRef CAS PubMed.
- V. Subramanian, E. E. Wolf and P. V. Kamat, J. Am. Chem. Soc., 2004, 126, 4943–4950 CrossRef CAS PubMed.
- A. Wood, M. Giersig and P. Mulvaney, J. Phys. Chem. B, 2001, 105, 8810–8815 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4se00196f |
| This journal is © The Royal Society of Chemistry 2024 |