
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Enhanced deep potential model for fast and accurate molecular dynamics: application to the hydrated electron
Ruiqi
Gao
 a,
Yifan
Li
b and
Roberto
Car
*b
a,
Yifan
Li
b and
Roberto
Car
*b
aDepartment of Electrical and Computer Engineering, Princeton University, Princeton, USA
bDepartment of Chemistry, Princeton University, Princeton, USA. E-mail: rcar@princeton.edu
First published on 23rd August 2024
Abstract
In molecular simulations, neural network force fields aim at achieving ab initio accuracy with reduced computational cost. This work introduces enhancements to the Deep Potential network architecture, integrating a message-passing framework and a new lightweight implementation with various improvements. Our model achieves accuracy on par with leading machine learning force fields and offers significant speed advantages, making it well-suited for large-scale, accuracy-sensitive systems. We also introduce a new iterative model for Wannier center prediction, allowing us to keep track of electron positions in simulations of general insulating systems. We apply our model to study the solvated electron in bulk water, an ostensibly simple system that is actually quite challenging to represent with neural networks. Our trained model is not only accurate, but can also transfer to larger systems. Our simulation confirms the cavity model, where the electron's localized state is observed to be stable. Through an extensive run, we accurately determine various structural and dynamical properties of the solvated electron.
1 Introduction
Molecular dynamics (MD) simulations provide insights for physical and chemical processes at the atomic level and have wide applications. To perform a simulation under Newtonian motion for the atoms, one needs to calculate the forces, which, under the Born–Oppenheimer approximation, are many-body functions of the atomic coordinates. Non-empirical quantum mechanical methods such as density functional theory (DFT) can in principle obtain these forces with good accuracy in many situations, but the high computational cost limits such methods to small systems and short time scales. There also exist classical force fields that are empirical, simple approximations to the many-body force function, which are many orders of magnitude faster to compute and scale linearly with the system size, but often fall short on the accuracy side. In recent years, machine learning (ML) force fields have become a promising direction to combine the advantages of both sides. That is, they are trained for ab initio level of accuracy, while achieving a linear scaling speed. They can still be somewhat slower than classical force fields, but they are much more scalable and faster than DFT and have been widely used in large-scale simulations.There has been a lot of development of ML force fields over the years.1–11 With the popularity of accuracy benchmarking, more recent models7,10,11 generally follow a trend of increasing accuracy at the cost of increasing model and computational complexity. However, in MD simulations, equilibrium and dynamical properties may require timescales of nanoseconds or even microseconds, corresponding to millions to billions of steps for sufficiently large system sizes. To this end, more lightweight and faster models are required. Earlier models like the Behler–Parrinello neural network (BPNN)1 and Deep Potential (DP)4,12–15 model are relatively small and fast, but may be inadequate for accuracy-sensitive systems.
This work is focused on developing a model that runs fast while being accurate enough for MD simulations. It is based on the DP model and we have made various enhancements to it. The most important is the incorporation of a message passing (MP) mechanism, so we call it DP-MP. This enables a richer representation that learns features on top of features, and also effectively increases DP's cutoff radius of the local receptive field. Using most of the building blocks of the existing DP model, we propagate both scalar and vector features for each atom and retain the model's invariance to translation, rotation, and permutation. We also incorporate second-order tensor information in the final features. These enhancements are designed to significantly boost the accuracy of DP without incurring much computational cost.
To make it faster and more flexible, we implement the new scheme with JAX,16 a Python-based autograd and machine learning framework that is optimized on GPUs.† The MD part can be seamlessly connected with frameworks like JAX-MD,17 enabling an end-to-end GPU workflow in Python. We perform a simple benchmark on a water system. Combined with the new implementation gains, the new model is around two orders of magnitude faster than other models achieving similar accuracy.
Additionally, in this work, we also present a new method for the prediction of the Wannier centers, i.e., the centers of maximally localized Wannier distributions.18 Wannier centers can be seen as representing the centers of the charge associated to individual electrons. So far, the Wannier centers can be predicted by a similar neural network like the DP model, which is called the Deep Wannier (DW) model.19 But this scheme is limited to systems where the Wannier centers can be uniquely associated with individual atoms, which precludes modeling electron transfer processes. In the present approach, we encapsulate a prediction model in an iterative refinement process, and it is called DWIR (Deep Wannier Iterative Refinement). With DWIR, one can keep track of the electrons in an atomic simulation, even when they are not uniquely associated to individual atoms.
To illustrate the capabilities of our enhanced models, we apply them to the study of e−(aq), the solvated electron in bulk water. e−(aq) plays an important role in radiation chemistry and biology,20 and despite its apparent simplicity, it has undergone much research effort before the cavity model became well-established: The electron creates a localized quasi-spherical cavity with a shell of surrounding water molecules.20,21 This system poses considerable challenges for ML models since they only see the atoms and not the excess electron, and the structure is quite complex and sensitive compared to bulk water. There have been efforts to learn an ML model of e−(aq),22 but it remains difficult to obtain a sufficiently accurate and robust model.22,23 Also, there has not been a model that can be transferred to larger systems, which is actually a requirement for many applications and technically possible given the localized nature of the electron.
In this work, we perform a DFT simulation of a periodic box of 128 H2O molecules plus one e−, and use the DP-MP scheme to successfully learn a model of e−(aq). We demonstrate its transferability to a larger system of 256 H2O molecules and one e−. In the DFT calculations, we adopt a hybrid functional PBEh(α) with 40% exact exchange and rVV10 van der Waals correction, which has been suggested to be able to reproduce well several experimental properties for water and e−(aq).21,24 We perform a nanosecond-long DP-MP run to collect sufficiently converged statistics, and learn an additional DWIR model to track the position of e−(aq). We calculate various structural and dynamical properties including the size, radial distribution functions, and diffusion mechanism. Our calculation confirms the cavity model and the stability of the localized state. We also identify a form of H–e− bond around the electron that is similar to the H-bond in water, and whose forming and breaking gives rise to the rapid diffusion of e−(aq).
2 Methodology
In this section we describe our new models. In Section 2.1 we give a recap on the DP model. We introduce the DP-MP model in Section 2.2, and the Wannier-center model DWIR in Section 2.3.2.1 Structure of the DP model
Given a system of N atoms with coordinates {ri}Ni = 1, the DP model represents the potential energy surface (PES) as a sum of atomic contributions, each term depending only on the atom's neighboring environment within a cutoff radius rc:where ω represents all the learnable parameters of the model, rij is the relative displacement between atoms i and j, and
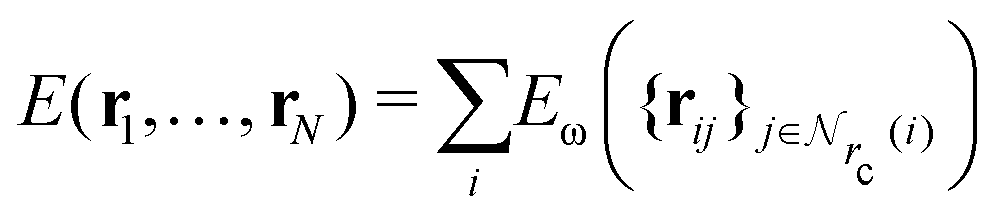
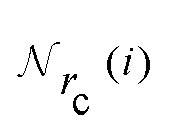 is the set of neighboring atoms j for which rij < rc. The forces are subsequently derived as the gradient of the energy. Each term Eω is computed as follows:
is the set of neighboring atoms j for which rij < rc. The forces are subsequently derived as the gradient of the energy. Each term Eω is computed as follows:
1. Compute a smooth function s(rij) that approximates 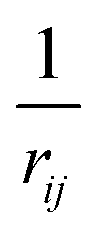 , except that it is modified to become zero when rij ≥ rc.
, except that it is modified to become zero when rij ≥ rc.
2. An embedding neural network G takes each s(rij) as input and outputs the feature G(s(rij)), a vector of length M1.
3. Average over neighboring atoms to obtain a scalar (T(1)) and vector (T(3)) feature of length M1 for each atom i:

where Nnbr is a precomputed constant that stands for the average number of neighbors, and
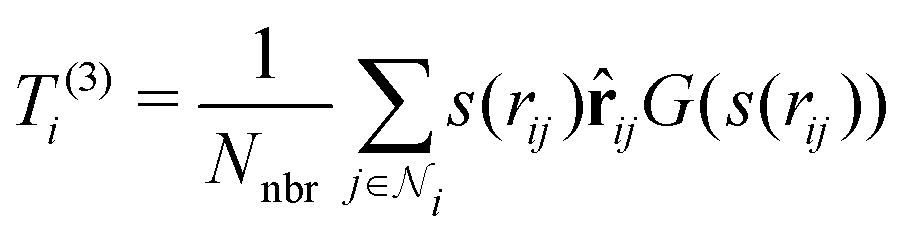
![[r with combining circumflex]](https://www.rsc.org/images/entities/b_char_0072_0302.gif) ij is the normalized rij.
ij is the normalized rij.
4. Obtain an invariant feature Di of size M1M2: this is done by taking a subset of M2 (<M1) “axis” features from T, and for each m1 in {1,…,M1} and m2 in the subset we compute

5. Apply a fitting neural network F that yields the atomic energy
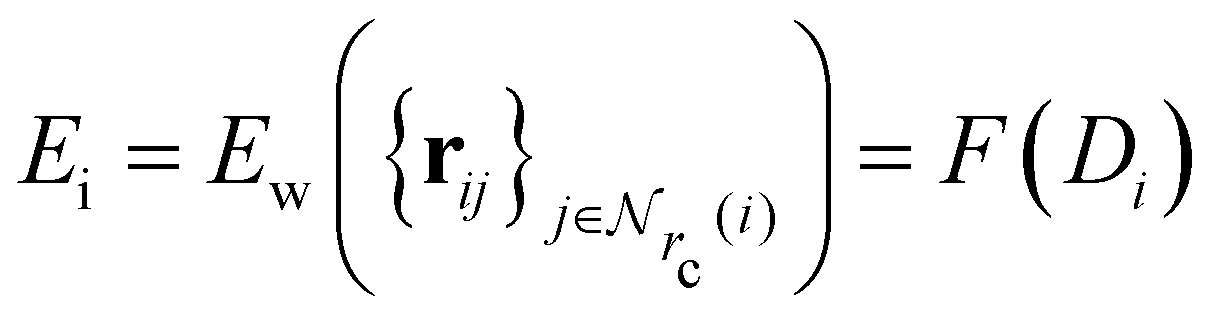
The model designed this way preserves the translational, rotational, and permutational symmetry of the energy function. All trainable parameters lie in the embedding network G and fitting network F, which are both multi-layer fully-connected residual networks (ResNets).25 In practice, depending on the chemical species, an embedding network is trained for each type-pair of (i,j) and a fitting network is trained for each type of i. But for simplicity we omit the atomic type in the formulas.
The calculation of the embedding network in step 2 is performed for pairs of atoms, making it the most time-consuming step. Thankfully, the input s(rij) is one dimensional, so it can be approximated by a piecewise polynomial at inference time, or referred to as compressed.26 With DP's simple design as well as compression, it is very fast compared to recent ML force fields.
DP has found many successful applications in systems like water, silicon, metals, metal oxides and so on, and has been applied to many studies including the phase diagram, and processes involved in crystal nucleation, combustion, interfacial systems etc.27–32 However, being a simple model, the expressive power of DP is somewhat limited. In addition, in more complex systems such as e−(aq), the radius of its influence most probably extends beyond the usual cutoff where DP is seen to perform well (like 6 Å in water). This brings us to the enhanced design, described in the next subsection.
2.2 Enhanced DP model with message passing
Now we describe our enhanced DP model with message passing, or DP-MP. The architecture is illustrated in Fig. 1 and 2. Message passing is a common design in Graph Neural Networks, which allows one to learn features on top of features iteratively. When applied to the DP model, the idea is simple: After computing the embedding network and summing over the neighbors, we obtain per-atom features T(1)i,T(3)i and Di from step 3 and 4. This can be used as a starting point for a new round of embedding calculations for each neighbor pair (i,j). The only difference is that, for the first round of embedding network in step 2, the input is only a scalar s(rij). But now we have much more information related to a pair (i,j) like rij,G(s(rij)),Ti,Di,Tj,Dj and so on. Among them, we make use of the invariant features G(s(rij)),Di,Dj, as well as create a new set of invariant features: 〈T(3)i,rij〉3 and 〈Tj(3),rij〉3. These invariant features are concatenated.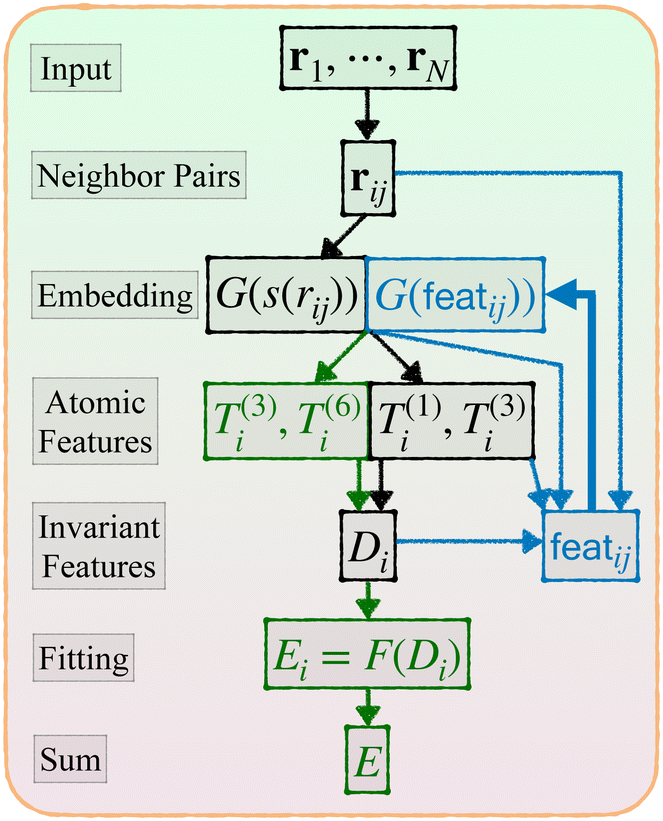 | ||
| Fig. 1 Architecture of DP-MP. The blue parts indicate the message passing steps, and the green parts indicate the final loop. | ||
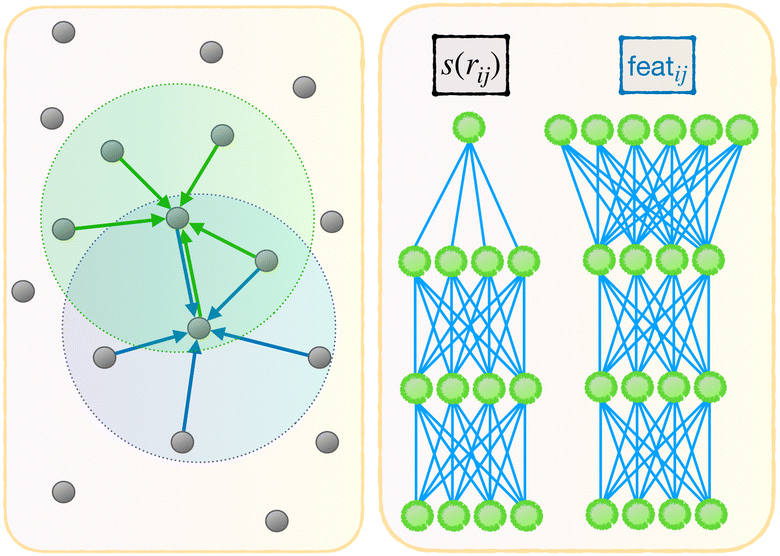 | ||
| Fig. 2 Illustration of the message-passing mechanism. Left: The features are aggregated from neighboring features repeatedly, effectively increasing the cutoff radius of the model. Right: Comparing the embedding network of the first pass and the MP pass, with the input of the latter being a multidimensional feature associated to an atom pair i,j. | ||
featij = concatenate (G(s(rij)),Di,Dj, 〈T(3)i,rij〉3, 〈T(3)j,rij〉3)
| as the new input to the embedding network. |
This process can be iterated: after each embedding network pass, we aggregate features from neighbors by step 3. We obtain new T and D atomic features from step 3 and 4, which are used in the input to the new embedding pass starting from step 2. After a few loops we can terminate and enter the previous fitting process described in step 5.
At the final loop at step 3, a slightly different feature set is employed: instead of T(1)i and T(3)i, we use T(3)i and T(6)i, where T(6)i is a set of 6-vectors defined by
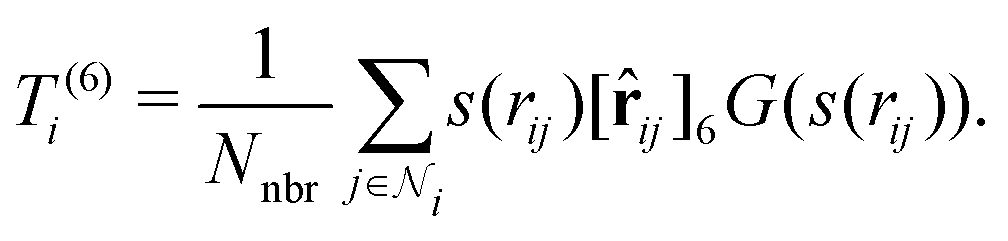
 , a 6-vector that incorporates 0th and 2nd order tensorial information. The subsequent step 4 is computed by
, a 6-vector that incorporates 0th and 2nd order tensorial information. The subsequent step 4 is computed bystill invariant under rotation. We find this to be a good balance between improving the expressive power of the model and not incurring much computational cost. In fact, these T features can be mathematically interpreted as a subset of the complete equivariant representation.10
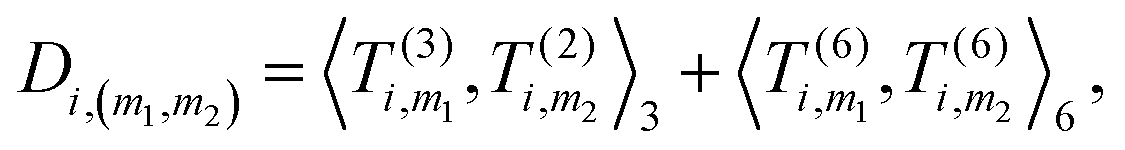
There are certain implementation details that we did not dive into for the sake of clarity. Firstly, in calculating s(rij), we use a slightly simpler function than the original DP
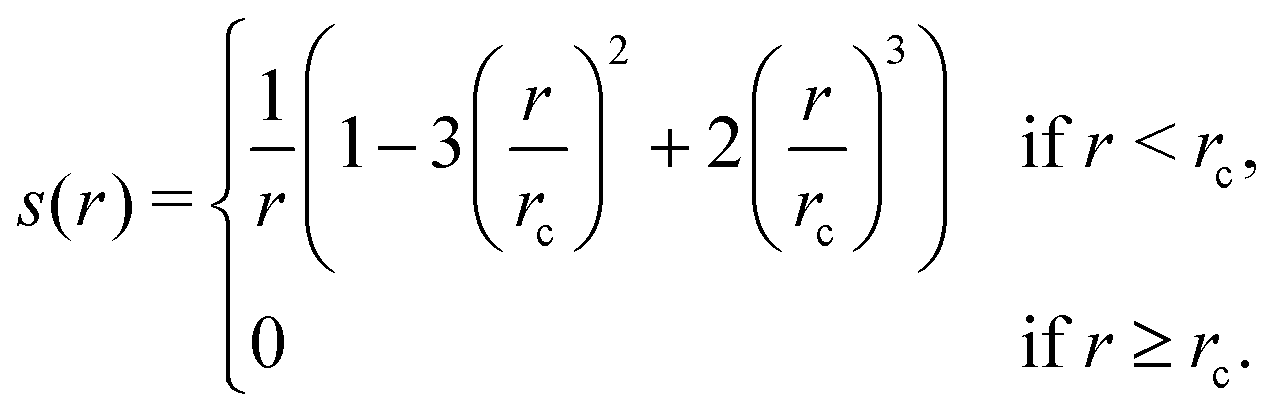 ij and concatenation in featij, which gives the equivalent math with reduced computational cost. For more details, we refer to the published code.
ij and concatenation in featij, which gives the equivalent math with reduced computational cost. For more details, we refer to the published code.
2.3 Iterative Wannier center prediction
Maximally localized Wannier functions give a well-defined alternative representation of the Bloch wave functions for the valence electrons in insulators. They are localized in space, and their distributional centers, short as Wannier centers (WCs), can be seen as representing the centers of charge of individual electrons. WCs are connected to the local and global polarization of the system.18,19 They are also used to explicitly calculate the long-range dipole–dipole Coulomb interactions,33 which is important in the study of charged systems.The previous DP model has been used to predict the Wannier centroid, defined as the average position of WCs associated with a certain atom. For example, in an H2O molecule, there are 4 WCs associated with it. Each one of them represents a pair of electrons with opposite spin, with two of them for the bonding pairs and two for the lone pairs. The dipole moment is determined by the average of the 4 WCs or the Wannier centroid. The Wannier centroid obtained from DFT calculations can be learned by a separate neural network in DP, sometimes called the Deep Wannier (DW) model.19 Compared to the standard DP model which predicts a scalar energy for each atom and then sums them up, DW predicts a vector for each Oxygen atom, representing its relative position to the Wannier centroid. This is achieved by modifying the final step 5 where one changes the fitting network's output F(Di) to be a feature of length M1, and the relative displacement from the i-th oxygen atom is expressed by 〈F(Di),T(3)i〉M1.
DW works well on predicting Wannier centroids in water, or more generally, insulating systems where you can assign the WCs to individual atoms. However, in general, WCs are not unambiguously associated with certain atoms. Examples include e−(aq), as well as more complex reactions involving electron transfer. Still, WCs are functions of the atomic coordinates under the Born–Oppenheimer approximation. This calls for a new scheme to predict the WCs without anchoring them to given atoms.
Here we introduce the new Deep Wannier iterative refinement (DWIR) model (Fig. 3). The idea is simple: now the WCs are anchored to themselves, and we predict only a correction displacement on top of a given prediction. Suppose the atomic coordinates are {ri}Ni=1, and we have some initial guess of the WCs 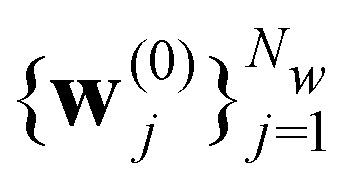 , where Nw is the total number of WCs. The initial guess is subject to errors, but we use a DW-like model to correct it iteratively:
, where Nw is the total number of WCs. The initial guess is subject to errors, but we use a DW-like model to correct it iteratively:
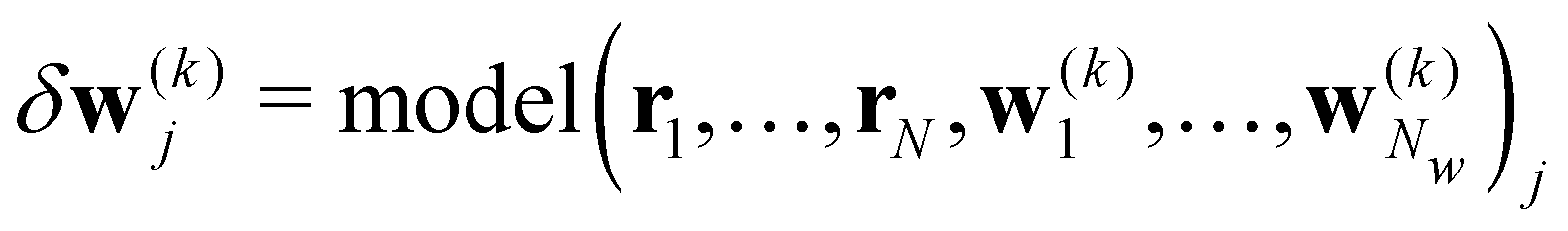
| w(k+1)j = w(k)j + δw(k)j. |
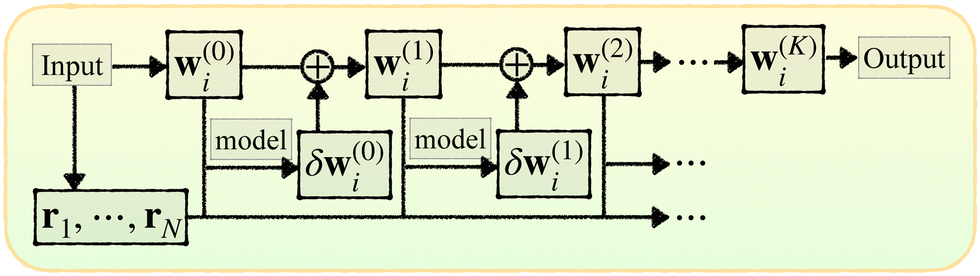 | ||
| Fig. 3 Illustration of DWIR, where the model takes the current WC prediction as part of the input and predicts an update. | ||
Starting from k = 0, the model is reused to iterate K times, and we obtain the final prediction  . We train the model with a loss function
. We train the model with a loss function

![[small script l]](https://www.rsc.org/images/entities/i_char_e146.gif) is a loss function for an individual prediction, γ > 1 is some fixed constant, and w* stands for the true WCs, whose permutation is determined by a greedy pairing with the predicted WCs based on a closest-distance principle. The scheme penalizes errors at later iterations, encouraging the process to converge to a fixed point that equals the true WCs in just a few iterations.
is a loss function for an individual prediction, γ > 1 is some fixed constant, and w* stands for the true WCs, whose permutation is determined by a greedy pairing with the predicted WCs based on a closest-distance principle. The scheme penalizes errors at later iterations, encouraging the process to converge to a fixed point that equals the true WCs in just a few iterations.
In a model's architecture, the WCs are treated just as a different kind of point particle, so any existing model can be used here, such as using DP-MP for improved accuracy. Also, DWIR can work with either spin-saturated or spin-polarized calculations. The latter is used in the e−(aq) system where one WC represents one electron instead of a pair.
The initial guess, while not important for the final result, should not deviate too much from the true WCs, otherwise the model will have a hard time converging. For example, in water, one can initialize 4/8 (spin-saturated/spin-polarized) random WCs around each Oxygen atom during training. In e−(aq), one can initialize the excess electron's WC to be within 1 Å of the true WC during training. In a simulation, one simply uses the previous step's prediction as the initial guess for the next step.
Again, there are certain implementation details. For example, s(r) is modified to be finite at r = 0 to handle potentially overlapping particle positions. For more details, we refer to the published code.
3 Benchmark results on water
In this section, we present a simple benchmark result of our enhanced models on a water system. Our dataset consists of some short DFT simulation trajectories of a periodic box of 128 H2O molecules totaling a few picoseconds, which are split into a training set of 7797 configurations and a validation set of 1501 configurations. We use the same DFT functional as in the e−(aq) simulation (described in Section 4) apart from doing a spin-saturated calculation without the excess electron.We use a cutoff radius of 6 Å and find that one MP pass in the DP-MP model is best in achieving a good accuracy while offering a significant speed advantage over other models. We use the default network width (number of neurons in each layer) of (32,32) for the initial embedding network, (64,32,64) for the MP embedding network, and (64,64,64,1) for the fitting network. We also benchmark the newly implemented DP model (referred to as DP(JAX)) with the default embedding network width (32,32,64).‡ Apart from the implementation, it differs from the original DP34(implemented in TensorFlow, referred to as DP(TF)) in that the per-atom features in step 3 are (T(3)i, T(6)i) as in the final loop of in DP-MP.
We measure the root mean square error (RMSE) or mean absolute error (MAE) of force predictions on the validation set. We also measure the speed of the models in simulations of a system of 128 H2O molecules on a single NVIDIA A100 GPU. The results are summarized in Table 1. We also compare with other neural network force fields including SchNet,35 NequIP,11 and MACE.36§¶ We plot the accuracy-speed trade-off in Fig. 4. It can be seen that DP-MP is almost two orders of magnitude faster compared to other models with similar accuracy. This is largely due to the design of the model itself, but various implementation gains|| play an important role as well. Together, the new enhanced models (DP-MP and DP(JAX)) achieve a great balance between accuracy and speed and offer a good choice for large-scale simulations.
| Model | Force RMSE | Force MAE | Cost |
|---|---|---|---|
| DP(TF) | 35 | 27 | 3.25 |
| DP(JAX) | 23 | 18 | 1.99 |
| DP-MP | 9.3 | 6.5 | 6.77 |
 | ||
| Fig. 4 Accuracy-speed trade-off on the PBEh(0.4) water system. | ||
4 Simulating the solvated electron in water
The solvated electron in water, also called the hydrated electron, is a byproduct of water radiolysis, a simple and potent reducing agent, and the culprit for DNA damage in biological systems. It has been attracting interest for decades of studies.20–22,38–49 Upon being created by ionizing radiation, it occupies a delocalized state as a quasi-free electron. Then, on a picosecond timescale, it thermalizes by creating a cavity in the surrounding water molecules, and localizes into a stable state. It is now generally agreed that the localized state is a quasi-spherical cavity model with a shell of surrounding water molecules.20,21We will only focus on studying the localized state. One reason is that non-adiabatic effects can be present in the delocalized state, which are not captured by electronic ground state simulations. Another reason would be that ML models are agnostic to the total number of electrons. If a model were to be transferable to larger systems, it should work for both e−(aq) and normal bulk water. Upon creating a delocalized electron in a finite box, the atoms are still at a bulk water configuration, indistinguishable to the ML model, but the atomic forces become different, making the forces ill-defined if these states are to be included.
4.1 Setup for DFT simulation
We first perform a DFT simulation in a periodic box of 128 H2O molecules plus one e−, with the NVT ensemble at experimental density. We use the CP2K software50 and adopt a setting described as follows, which has been used in previous works and well-tested for the description of e−(aq).21,24,51 We use the hybrid functional PBEh(α), with the fraction of Fock exchange α set to 0.4. The van der Waals correction is included by the rVV10 functional where the parameter b is set to 5.3. We use the triple-ζ polarized (TZP) basis and Goedecker–Teter–Hutter pseudopotentials. The charge density, expanded in a plane-wave basis, has an 800 Ry cutoff. We use a spin-polarized calculation with an added uniform background charge to neutralize the system. The temperature is maintained at 350 K via the use of a Nosé–Hoover thermostat in order to ensure a frank diffusive motion.21 Starting from the initial equilibrated bulk water configuration, it takes around 0.2–0.4 ps for the excess electron to localize, and the initial configurations that are not fully localized are not used in model training.4.2 Setup for model training and simulation
We train a DP-MP model to simulate the solvated electron. We use a cutoff radius of 6 Å, same as previous DP models for bulk water, and employ a single MP pass. The loss function is a sum of energy and force terms similar to that used in the training of DP. The model is trained with a batch size of 1 for 500![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 batches using the Adam optimizer with an exponentially decaying learning rate from 2 × 10−3 to 10−6. The training takes around 1.5 hours on an NVIDIA A100 GPU. An active learning procedure, DP-GEN,52 is followed to improve the model's robustness. This involves training several initial models with different random seeds, performing a simulation with one of the trained models, and sampling a small set of extra configurations from the trajectory based on a model deviation metric. These extra configurations are then labeled by DFT calculations and added to the training set. The DFT ab initio molecular dynamics (AIMD) trajectories have a total length of around 15 ps. With some extra configurations from DP-GEN, the final training set has around 30000 configurations.
000 batches using the Adam optimizer with an exponentially decaying learning rate from 2 × 10−3 to 10−6. The training takes around 1.5 hours on an NVIDIA A100 GPU. An active learning procedure, DP-GEN,52 is followed to improve the model's robustness. This involves training several initial models with different random seeds, performing a simulation with one of the trained models, and sampling a small set of extra configurations from the trajectory based on a model deviation metric. These extra configurations are then labeled by DFT calculations and added to the training set. The DFT ab initio molecular dynamics (AIMD) trajectories have a total length of around 15 ps. With some extra configurations from DP-GEN, the final training set has around 30000 configurations.
To keep track of the electron's position, we also train a DWIR model. We use the same DP-MP base architecture. The configurations used for training are also the same, with the WCs calculated from the Kohn–Sham orbitals of the DFT calculations. Since we're only interested in the excess electron here, only one WC per configuration needs to be predicted by the model, though the DWIR model can equally well keep track of all the electrons. We perform K = 4 iterations of refinement. We use a batch size of 64 and trained for 50000 batches using Adam with an exponentially decaying learning rate from 10−2 to 10−4.
The DP-MP model achieves a root mean square validation error of 12 meV Å−1 for the forces. The DWIR model achieves a root mean square validation error of 0.025 Å for the WC. The DP-MP model is then used to perform a 1 ns-long simulation** in the same NVT ensemble as the DFT simulation. To show the transferability of the model, we also perform a simulation of a larger system of 256 H2O molecules plus one e−, with the same DP-MP model. The DWIR model is used to predict the WCs after the simulation, and the WCs are then used to calculate various properties of the solvated electron.
4.3 Results on the solvated electron
Our simulation confirms the cavity model, where the electron has been observed to remain stably localized as depicted in Fig. 5, both in DFT and DP-MP simulations. The structural results are shown in Fig. 6. Our results are in general consistent with the DFT results from the previous literature,21 but certain sensitive numbers vary because DP-MP based long trajectories give more converged statistics than previous AIMD trajectories.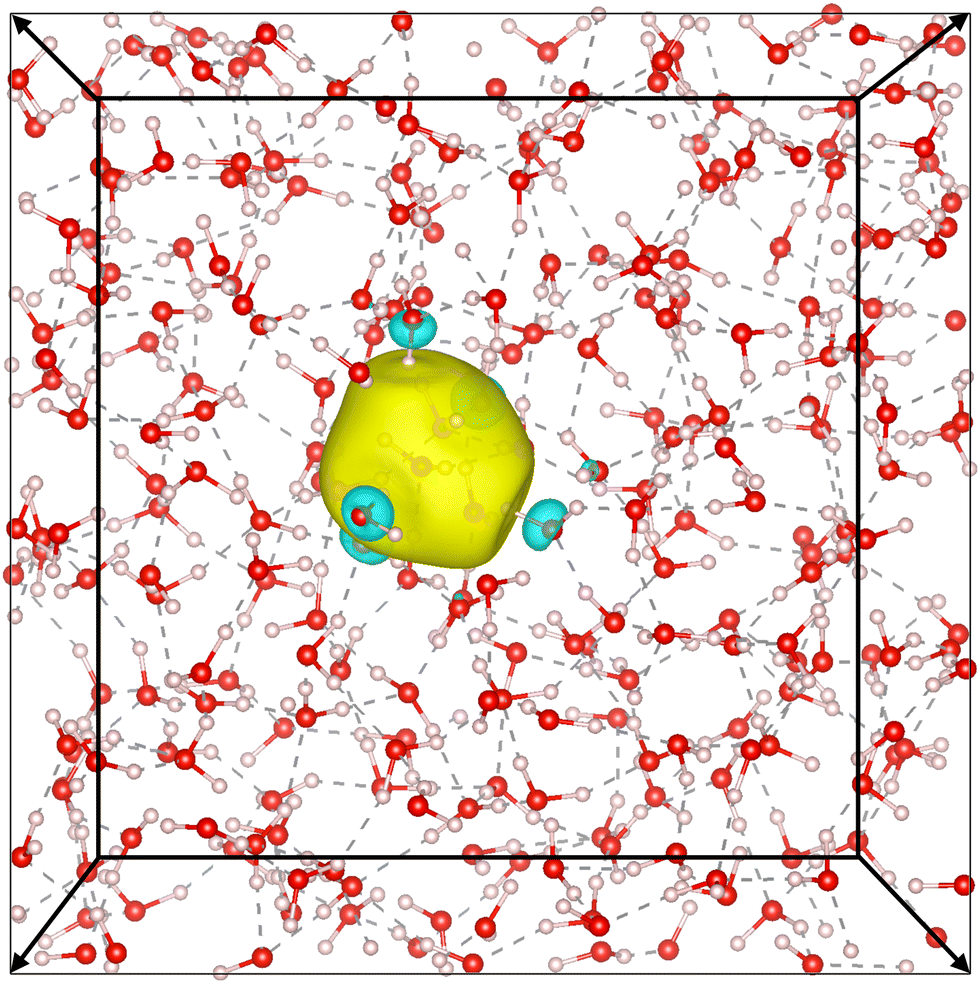 | ||
| Fig. 5 Picture of the positive and negative isosurfaces of the Wannierized wave function of e−(aq). Surrounding water molecules point toward the electron through one hydrogen atom, resembling an H-bond. The solvated electron causes disturbances to the H-bond network in water. The two boxes represent training on a smaller system and transferring to a larger system. | ||
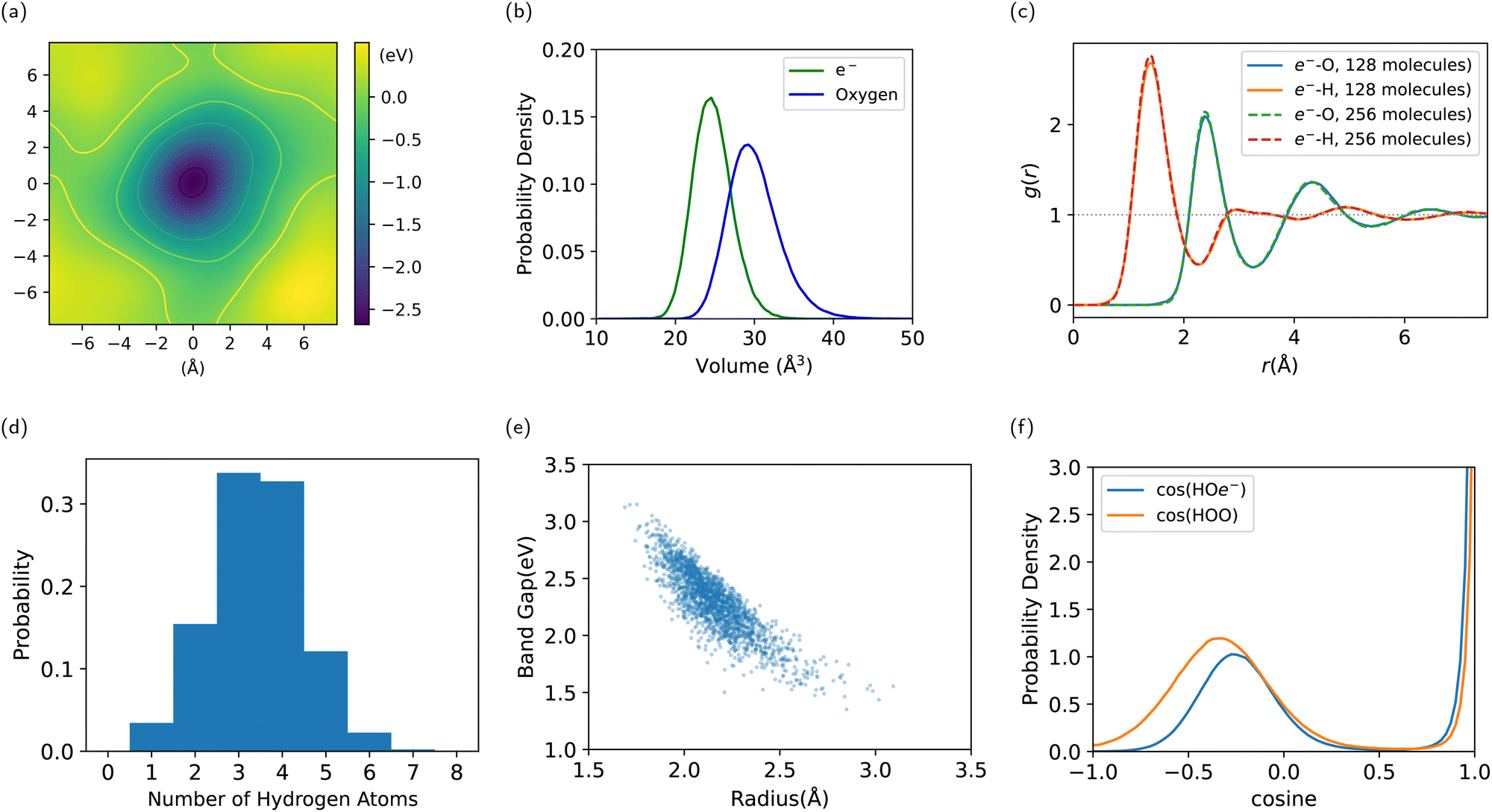 | ||
| Fig. 6 Results on the structure of e−(aq). (a) Qualitative electrostatic potential of the system minus the excess electron. (b) Distribution of the Voronoi volume of e− and water molecules. (c) Radial distribution function between e− and O/H atoms. (d) Distribution of the number of hydrogen atoms in the first shell of e−. (e) Radius of gyration of the Wannierized state of e−, plotted against the Kohn–Sham band gap. (f) Distribution of the cosine angle of H–O–e− and H–O–O, showing the resemblance of the H–e− bond to the H-bond. | ||
First off, to qualitatively see that WCs are intuitively representative of the charge distribution, we take a random snapshot of the system where we calculated the (negative) electrostatic potential of the system minus the excess e−, shown in Fig. 6a. This calculation is based on an approximation of spherical Gaussian distribution of positive charge at atomic cores and negative charge at WCs of the system except that of e−, with the same inverse spread β = 0.4 Å−1 as suggested by Zhang et al.33, followed by a particle–particle particle-mesh (PPPM) calculation. This is sufficient to get the long range electrostatic potential up to dipole contributions. We plot the horizontal slice of the periodic box with the WC of e− centered at the origin. It can be seen that the WC agrees with the minimum of the quasi-spherical potential well created by the surrounding molecules.
We conduct a Voronoi analysis on the WC of e− and all oxygen atoms. The respective volume distributions are shown in Fig. 6b. The volume of e− is smaller than that occupied by a water molecule. From the average volume we deduce a radius of 1.80 Å compared to 1.92 Å for a water molecule.
In Fig. 6c, we show the radial distribution function between the WC of e− and O/H atoms. The first peak is at 1.4 Å for e−–H, and 2.4 Å for e−–O. The first minimum is at 2.3 Å for e−–H, and 3.3 Å for e−–O. We obtain the result for the 256-molecule system as well. It is slightly more structured and localized, which alludes to the importance of using a large enough box to simulate e−(aq), where a smaller box lowers the energy barrier for delocalization. But the difference between system size 128 and 256 is already tiny, indicating valid results with 128 molecules.
We compute the coordination number of hydrogen atoms within the first minimum 2.3 Å, resulting in a mean of 3.4†† and a high standard deviation of 1.1, where the distribution spans from 1 to 7 as shown in Fig. 6d. This is due to a highly volatile H-bond network in the vicinity of the electron.
By sampling 2000 configurations from the trajectory and conducting DFT calculations again, we compute the radius of gyration from the spread of the Wannierized wave function of e−. The average number is 2.16 ҇ with a standard deviation of 0.18 Å, which, compared to the radius inferred from the Voronoi volume, indicates that the electronic density extends into the first shell. It is still localized but much less localized than the Wannierized valence-band electrons in water. The radius of gyration is plotted against the Kohn–Sham band gap in Fig. 6e, showing a clear negative correlation, where a larger radius corresponds to a smaller band gap and a state of higher energy. A lack of an extended tail at the bottom-right is an indication of stable localization.
To examine the nature of the interaction between e− and surrounding water molecules, we compute the distribution of the cosine angle of H–O–e−, conditioning on H–O being covalently bonded, as well as the distance between O and e− being within 3.0 Å. This is compared with the angle of H–O–O, or H–O1–O2, where H–O1 is covalently bonded and O1 –O2 is within 3.0 Å. In Fig. 6f, the latter shows a strong peak at 0°, which corresponds to the H-bond angle, with another soft peak that stands for the other H atom covalently bonded to O1. The cosine of H–O–e− also shows the similar two peaks, which indicates that H–e− bonds are similar to H-bonds, with one hydroxyl group pointing to the electron as indicated in Fig. 4. The soft peak for water is at around −0.33, corresponding to a tetrahedral H-bond network, while the soft peak for e− is at around −0.27, much closer to the cosine angle of the water molecule itself. This indicates that the H-bond network is disturbed by the excess electron.
To understand the diffusion properties of e−(aq), additional NVE simulations are performed at the same temperature. By fitting the mean square displacement to the Einstein relation, the diffusion coefficient of e−(aq) is calculated as 0.33 ± 0.01 Å2 ps−1. As a comparison, the diffusion coefficient for bulk water molecules under the same setting is 0.24 ± 0.01 Å2 ps−1. The absolute value of the diffusion coefficient depends on various factors like the DFT functional and the system size, but the relative value indicates that the solvated electron is more mobile than water molecules. The fast diffusion is the result of the frequent entry and exit of water molecules into the shell surrounding the electron. The solvated electron acts as a H-bond acceptor but not a donor, disturbing the H-bond network in water. This is already implied by the high variance of the e−–H coordination number. To see it more, we calculate the survival time of the hydogen bonds as well as the H–e− bond, defined by a unified geometric criterion due to their resemblance: The distance between the donor(O) and acceptor(O or e−) is less than 3.3 Å, and the H–O–O/H–O–e− angle is less than 30°. The bond is deemed broken if the H atom forms a different bond according to this criterion, or a weaker criterion of 3.6 Å/60° is violated. The average survival time is calculated to be 0.81 ps for H-bonds, and 0.58 ps for H–e− bonds. The relative value directly indicates that the H–e− bond is less stable than H-bonds, consistent with the increased diffusion.
4.4 Remarks
Some additional remarks are in order. The localization of the solvated electron in water depends crucially on the adopted density functional approximation. In fact, standard semi-local functionals in water typically do not support the presented localized electron configurations, in contrast to experiment. Hybrid functionals that include a substantial fraction of exact exchange are necessary to reconcile theory and experiment. Here we adopted the functional suggested by Ambrosio et al.21, which is found to agree with experiment on several important properties of the hydrated electron. With this functional the electron is always well localized on the time scale of our simulations, and the cavity is very stable and never deforms to such an extent as to split in two cavities between which the electron can tunnel. Such large fluctuations of the charge distribution of e−(aq) may occur with different models, with insufficient system size,53 or with nuclear quantum effects.22 Additionally, works on anions showed that these species are quite sensitive to the density functional used,54 where tuning the fraction of exchange for different anionic species may lead to more accurate results. However, a study of the dependence of the behavior of e−(aq) on the adopted functional approximation is not within the scope of the present work, which is, more modestly, just to assess the fact that the adopted ML model can describe the e−(aq) when this is described as in the work of Ambrosio et al.215 Conclusion
In this work, we have enhanced the design of Deep Potential models, providing an excellent balance between accuracy and speed. We have also introduced a new DWIR model to predict Wannier centers without relying on atom anchoring. These models have been applied to simulate the solvated electron in water, providing new insights into the structure and dynamics of the system. We expect the new DP and DP-MP models to set a new standard for the simulation of complex systems. We also expect our models to be a useful tool to simulate more complex electron transfer reactions in future work.Data availability
Code for the enhanced DP models is available at https://github.com/SparkyTruck/deepmd-jax. The DFT dataset for the solvated electron configurations is available at https://doi.org/10.5281/zenodo.12614014.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge Yixiao Chen, Linfeng Zhang, Han Wang, and Duo Zhang for very useful discussions during the development of the model. This work is supported by the Computational Chemical Sciences Center “Chemistry in Solution and at Interfaces” funded by the U.S. Department of Energy under Award No. DE-SC0019394. This research mainly uses resources of the National Energy Research Scientific Computing Center (NERSC), which is supported by the U.S. Department of Energy (DOE), Office of Science under Contract No. DE-AC02-05CH11231. We also acknowledge Princeton Research Computing at Princeton University for providing resources.References
- J. Behler and M. Parrinello, Phys. Rev. Lett., 2007, 98, 146401 Search PubMed.
- K. T. Schütt, F. Arbabzadah, S. Chmiela, K. R. Müller and A. Tkatchenko, Nat. Commun., 2017, 8, 13890 Search PubMed.
- J. Gilmer, S. S. Schoenholz, P. F. Riley, O. Vinyals and G. E. Dahl, Int. Conf. Mach. Learn., 2017, 1263–1272 Search PubMed.
- J. Han, L. Zhang and R. Car, et al., arXiv, 2017, preprint, arXiv:1707.01478 DOI:10.48550/arXiv.1707.01478.
- J. Gasteiger, J. Groß and S. Günnemann, arXiv, 2020, preprint, arXiv:2003.03123 DOI:10.48550/arXiv.2003.03123.
- W. Hu, M. Shuaibi, A. Das, S. Goyal, A. Sriram, J. Leskovec, D. Parikh and C. L. Zitnick, arXiv, 2021, preprint, arXiv:2103.01436 DOI:10.48550/arXiv.2103.01436.
- J. Gasteiger, F. Becker and S. Günnemann, Adv. Neural Inf. Proc. Syst., 2021, 34, 6790–6802 Search PubMed.
- C. Ying, T. Cai, S. Luo, S. Zheng, G. Ke, D. He, Y. Shen and T.-Y. Liu, Adv. Neural Inf. Proc. Syst., 2021, 34, 28877–28888 Search PubMed.
- Y. Liu, L. Wang, M. Liu, Y. Lin, X. Zhang, B. Oztekin and S. Ji, International Conference on Learning Representations, 2022.
- S. Batzner, A. Musaelian, L. Sun, M. Geiger, J. P. Mailoa, M. Kornbluth, N. Molinari, T. E. Smidt and B. Kozinsky, Nat. Commun., 2022, 13, 2453 CrossRef CAS PubMed.
- Y.-L. Liao and T. Smidt, The Eleventh International Conference on Learning Representations, 2022.
- L. Zhang, J. Han, H. Wang, R. Car and E. Weinan, Phys. Rev. Lett., 2018, 120, 143001 CrossRef CAS PubMed.
- L. Zhang, J. Han, H. Wang, W. Saidi and R. Car, et al., Advances in neural information processing systems, 2018, vol. 31 Search PubMed.
- W. Jia, H. Wang, M. Chen, D. Lu, L. Lin, R. Car, E. Weinan and L. Zhang, SC20: International conference for high performance computing, networking, storage and analysis, 2020, pp. 1–14.
- D. Lu, H. Wang, M. Chen, L. Lin, R. Car, E. W. einan, W. Jia and L. Zhang, Comput. Phys. Commun., 2021, 259, 107624 CrossRef CAS.
- J. Bradbury, R. Frostig, P. Hawkins, M. J. Johnson, C. Leary, D. Maclaurin, G. Necula, A. Paszke, J. VanderPlas, S. Wanderman-Milne and Q. Zhang, JAX: composable transformations of Python + NumPy programs, 2018, https://github.com/google/jax.
- S. Schoenholz and E. D. Cubuk, Adv. Neural Inf. Process Syst., 2020, 33, 11428–11441 Search PubMed.
- N. Marzari, A. A. Mostofi, J. R. Yates, I. Souza and D. Vanderbilt, Rev. Mod. Phys., 2012, 84, 1419 CrossRef CAS.
- L. Zhang, M. Chen, X. Wu, H. Wang, E. Weinan and R. Car, Phys. Rev. B, 2020, 102, 041121 CrossRef CAS.
- J. M. Herbert and M. P. Coons, Annu. Rev. Phys. Chem., 2017, 68, 447–472 CrossRef CAS PubMed.
- F. Ambrosio, G. Miceli and A. Pasquarello, J. Phys. Chem. Lett., 2017, 8, 2055–2059 CrossRef CAS PubMed.
- J. Lan, V. Kapil, P. Gasparotto, M. Ceriotti, M. Iannuzzi and V. V. Rybkin, Nat. Commun., 2021, 12, 766 CrossRef CAS PubMed.
- J. Lan, V. V. Rybkin and A. Pasquarello, Angew. Chem., Int. Ed., 2022, 61, e202209398 CrossRef CAS PubMed.
- F. Ambrosio, G. Miceli and A. Pasquarello, J. Phys. Chem. B, 2016, 120, 7456–7470 CrossRef CAS PubMed.
- K. He, X. Zhang, S. Ren and J. Sun, Proceedings of the IEEE conference on computer vision and pattern recognition, 2016, pp. 770–778.
- D. Lu, W. Jiang, Y. Chen, L. Zhang, W. Jia, H. Wang and M. Chen, J. Chem. Theory Comput., 2022, 18, 5559–5567 CrossRef CAS PubMed.
- L. Bonati and M. Parrinello, Phys. Rev. Lett., 2018, 121, 265701 CrossRef CAS PubMed.
- T. E. Gartner III, L. Zhang, P. M. Piaggi, R. Car, A. Z. Panagiotopoulos and P. G. Debenedetti, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 26040–26046 CrossRef PubMed.
- H. Niu, L. Bonati, P. M. Piaggi and M. Parrinello, Nat. Commun., 2020, 11, 2654 CrossRef CAS PubMed.
- M. F. C. Andrade, H.-Y. Ko, L. Zhang, R. Car and A. Selloni, Chem. Sci., 2020, 11, 2335–2341 RSC.
- J. Zeng, L. Cao, M. Xu, T. Zhu and J. Z. Zhang, Nat. Commun., 2020, 11, 5713 CrossRef CAS PubMed.
- L. Zhang, H. Wang, R. Car and E. Weinan, Phys. Rev. Lett., 2021, 126, 236001 CrossRef CAS PubMed.
- L. Zhang, H. Wang, M. C. Muniz, A. Z. Panagiotopoulos and R. Car, et al., The, J. Chem. Phys., 2022, 156, 124107 CrossRef CAS PubMed.
- J. Zeng, D. Zhang, D. Lu, P. Mo, Z. Li, Y. Chen, M. Rynik, L. Huang, Z. Li and S. Shi, et al., The, J. Chem. Phys., 2023, 159, 054801 CrossRef CAS PubMed.
- K. Schütt, P.-J. Kindermans, H. E. Sauceda Felix, S. Chmiela, A. Tkatchenko and K.-R. Müller, Advances in neural information processing systems, 2017 Search PubMed.
- I. Batatia, D. P. Kovacs, G. N. C. Simm, C. Ortner and G. Csanyi, Advances in neural information processing systems, 2022 Search PubMed.
- X. Fu, Z. Wu, W. Wang, T. Xie, S. Keten, R. Gomez-Bombarelli and T. Jaakkola, arXiv, 2022, preprint, arXiv:2210.07237 DOI:10.48550/arXiv.2210.07237.
- E. J. Hart and J. W. Boag, J. Am. Chem. Soc., 1962, 84, 4090–4095 CrossRef CAS.
- J. Schnitker and P. J. Rossky, J. Chem. Phys., 1987, 86, 3471–3485 CrossRef CAS.
- L. Turi, W.-S. Sheu and P. J. Rossky, Science, 2005, 309, 914–917 CrossRef CAS PubMed.
- L. Turi and P. J. Rossky, Chem. Rev., 2012, 112, 5641–5674 CrossRef CAS PubMed.
- M. Boero, M. Parrinello, K. Terakura, T. Ikeshoji and C. C. Liew, Phys. Rev. Lett., 2003, 90, 226403 CrossRef PubMed.
- R. E. Larsen, W. J. Glover and B. J. Schwartz, Science, 2010, 329, 65–69 CrossRef CAS PubMed.
- F. Uhlig, O. Marsalek and P. Jungwirth, J. Phys. Chem. Lett., 2012, 3, 3071–3075 CrossRef CAS PubMed.
- J. Savolainen, F. Uhlig, S. Ahmed, P. Hamm and P. Jungwirth, Nat. Chem., 2014, 6, 697–701 CrossRef CAS PubMed.
- F. Uhlig, J. M. Herbert, M. P. Coons and P. Jungwirth, J. Phys. Chem. A, 2014, 118, 7507–7515 CrossRef CAS PubMed.
- E. Alizadeh, T. M. Orlando and L. Sanche, Annu. Rev. Phys. Chem., 2015, 66, 379–398 CrossRef CAS PubMed.
- J. M. Herbert, Phys. Chem. Chem. Phys., 2019, 21, 20538–20565 RSC.
- V. Svoboda, R. Michiels, A. C. LaForge, F. Stienkemeier, P. Slavíček and H. J. Wörner, Sci. Adv., 2020, 6, eaaz0385 CrossRef CAS PubMed.
- T. D. Kühne, M. Iannuzzi, M. Del Ben, V. V. Rybkin, P. Seewald, F. Stein, T. Laino, R. Z. Khaliullin, O. Schütt and F. Schiffmann, et al. , J. Chem. Phys., 2020, 152, 194103 CrossRef PubMed.
- M. Pizzochero, F. Ambrosio and A. Pasquarello, Chem. Sci., 2019, 10, 7442–7448 RSC.
- Y. Zhang, H. Wang, W. Chen, J. Zeng, L. Zhang, H. Wang and E. Weinan, Comput. Phys. Commun., 2020, 253, 107206 CrossRef CAS.
- S. J. Park and B. J. Schwartz, J. Chem. Theory Comput., 2022, 18, 4973–4982 CrossRef CAS PubMed.
- L. N. Anderson, M. B. Oviedo and B. M. Wong, J. Chem. Theory Comput., 2017, 13, 1656–1666 CrossRef CAS PubMed.
Footnotes |
| † Code available at https://github.com/SparkyTruck/deepmd-jax. |
| ‡ The embedding network of the DP model, as well as the initial embedding network of the DP-MP model, is compressed by default. |
| § We employ the training parameters of SchNet and NequIP from Fu et al.37 and MACE from the official documentation with a small(64-0) and large(128-2) model. Since our dataset is larger than the examples in these references we reduce the number of training epochs accordingly but ensure that further training does not improve the validation error. |
| ¶ While there are presumably more accurate models, they tend to be even slower and are not included in the comparison. |
| || Firstly, JAX tends to be somewhat faster than other packages like PyTorch or TensorFlow with which the other models are implemented. Also, we connect the model with JAX-MD, enabling an end-to-end GPU workflow. The simulation of other models either uses the LAMMPS(DP(TF)) or ASE(SchNet, NequIP, MACE) interface. While the simulation part is not the computational bottleneck compared to the evaluation of the neural network, such an interface can still cause some overhead. In addition, we use 32-bit floating point accuracy in DP-MP and DP(JAX) by default, which we find to have no impact on the prediction accuracy compared to 64-bit. |
| ** In practice, the lifetime of e−(aq) may not be this long due to the reaction with other species like the hydronium ion, but our simulation aims at collecting the statistics of e−(aq) itself. |
| †† This result is smaller than previous results21 because the coordination number is quite sensitive to the measurement of the minimum of the radial distribution function, where we give a slightly smaller 2.3 Å. |
| ‡‡ This radius is slightly smaller than previous estimates21 based on the Bloch state, but gives the same qualitative picture since the state of e− does not mix much with the valence band during Wannierization. |
| This journal is © the Owner Societies 2024 |